[level-membership-for-neurology-category]
Chapter 50 Pediatric Epilepsy
An Overview
After a first unprovoked seizure in childhood, the risk of a recurrence is about 50 percent, and after the second the risk is about 80 percent. By convention, epilepsy may be diagnosed only after a child has had two or more unprovoked seizures [Camfield et al., 1985a; Shinnar et al., 2000]. The term unprovoked implies that there has been no closely associated concurrent illness, fever, or acute brain injury. Recurrent seizures immediately after a head injury or associated with drug intoxication do not qualify for the diagnosis of epilepsy. Some specific provoking factors leading to reflex seizures are permitted, such as seizures provoked by patterns and flashes from video terminals for children with photosensitive epilepsy. Seizures are still viewed as unprovoked if they occur with stresses related to personal activity, such as sleep deprivation or severe emotional distress, unless these stresses are extreme.
Seizure Type and Epilepsy Syndrome
The definition of epilepsy allows for a tremendous variety of disorders. The seizure types, age of onset, cause, severity, comorbid conditions, response to medication, and clinical course vary widely. There are two main ways of grouping patients to bring an ordered approach to classification, treatment, and prognosis: seizure type and epilepsy syndrome. Both concepts continue to evolve. The International League against Epilepsy defined the seizure types listed in Box 50-1 [Commission on Classification and Terminology of the International League against Epilepsy, 1981]. There is a basic distinction between seizures with a generalized onset that seem to arise from everywhere in the cortex at once, and seizures with a partial or focal onset that begin in a defined area of cortex. A proposed update in the classification of epileptic seizures suggested that the distinction between simple partial and complex partial seizures is often difficult to assess and not needed [Engel, 2001].
Box 50-1 International Classification of Seizure Type



















Unclassifiable Epileptic Seizures
(Adapted from the Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22:489.)
A given seizure type may occur with many different associations. For example, a 2-year-old child with severe mental handicap may have generalized tonic-clonic seizures that are completely resistant to medication, or a normal teenager may have the same seizure type that is completely suppressed by medication. Factors beyond seizure type allow a more comprehensive diagnosis – an epilepsy syndrome diagnosis. Epileptic syndromes may have one or more seizure types, often with a characteristic interictal electroencephalogram (EEG). Each syndrome has a typical age of onset, a defined group of causes, sometimes a clear response to specific treatments, and sometimes a defined clinical course and prognosis. As with seizure types, syndromes have been classified as localization-related (focal) or generalized, and then subdivided into idiopathic (when microdysgenesis or a biochemical etiology is likely), symptomatic (when a diffuse or localized brain abnormality is known), and cryptogenic (when a diffuse or localized brain abnormality is suspected but unproven; Table 50-1). The term cryptogenic has been discouraged, as the detection of a structural lesion is dependent on the evolving quality of brain imaging and detection of a biochemical abnormality is dependent on the evolving understanding of genomic and proteomic abnormalities. In adults, localization-related epilepsy syndromes are most common, usually symptomatic, and caused by a localized, structural abnormality. Children have similar disorders, but there are many other important syndromes. The idiopathic partial and generalized syndromes all begin in childhood, and genetic influences are significant. The generalized symptomatic syndromes of childhood account for most intractable epilepsy in children [Arzimanogolou et al., 2004; Camfield and Camfield, 2003].
Table 50-1 Proposal from the International Classification of Epilepsies and Epileptic Syndromes and Related Seizure Disorders of 1989 and the Listing of Epilepsy Syndromes Recognized in the Proposed 2001 ILAE Classification Scheme
| 1989 ILAE Classification* | 2001 ILAE Proposal† |
|---|---|
| Localization-Related (Local, Focal, Partial Epilepsies and Syndromes | |
| Idiopathic (with Age-Related Onset) | |
| Benign childhood epilepsy with centrotemporal spikes | Benign childhood epilepsy with centrotemporal spikes |
| Childhood epilepsy with occipital paroxysms | Early-onset benign childhood occipital epilepsy (Panayiotopoulos type) (new) Late-onset childhood occipital epilepsy (Gastaut type) (new) Idiopathic photosensitive occipital lobe epilepsy (new) |
| Primary reading epilepsy | Primary reading epilepsy Now called Reflex epilepsies, which also includes: |
(Diagnosis of epilepsy not now required)
* Modified from the 1989 International League Against Epilepsy (ILAE) Proposal.
† Modified from the 2001 ILAE Proposal. In this proposal there are no headings or subheadings, only a listing of epilepsy syndromes by specific syndrome name. Our format of presentation has indicated where syndromes are removed or renamed from the 1989 ILAE proposal or newly recognized in the 2001 proposal. Each syndrome was “forced” into the 1989 proposed schema.
The International League Against Epilepsy (ILAE) classified febrile seizures as a special syndrome. Febrile seizures are the most common convulsive event in the human species. Because these seizures are provoked by fever, febrile seizures do not fit well with the usual definition of epilepsy, and few children with febrile seizures later develop unprovoked seizures. The disorder merits special consideration, as reviewed in Chapter 57.
The 1989 ILAE syndrome classification scheme is still in wide use, and most patients can be “forced” into one of the broad categories, but many children, especially those with symptomatic generalized epilepsy, do not meet the criteria for a more specific syndrome [Commission on Classification and Terminology of the International League Against Epilepsy, 1989]. There are many recently described epilepsy syndromes, but a newer, comprehensive syndrome classification system has been elusive. Advances in genetics have revealed that the same mutation may cause a variety of epilepsy syndromes, including partial and generalized epilepsies, even within the same family (see Chapter 52). This finding has been particularly well illustrated in large families with mutations in the gene coding for neuronal sodium channel – SCN1A.
Another evolving classification is “epilepic encephalopathy,” defined as an epilepsy syndrome where the seizures and/or interictal EEG discharge have permanent deleterious effects on brain development. Examples would be West’s syndrome with infantile spasms and hypsarrhythmia on EEG, Lennox–Gastaut syndrome with mixed generalized seizures and nearly continuous slow spike wave EEG discharge, or Landau–Kleffner syndrome with relatively mild clinical seizures but very active bitemporal epileptic EEG discharge (see Chapter 56).
A new proposal for syndrome classification in 2001 is based on a five-axis system [Engel, 2001]. Axis 1 concerns ictal phenomenology (i.e., what the seizure looks like). Axis 2 is the specific seizure type. Axis 3 is the specific epilepsy syndrome, when it can be defined; axis 4 is the cause; and axis 5 describes the degree of impairment associated with epilepsy. This proposal comes with a more comprehensive list of epilepsy syndromes, although the system is less categorical than the 1989 classification [Commission on Classification and Terminology of the International League against Epilepsy, 1989]. The 2001 proposal has not been widely accepted, although it does serve well to alert the clinician to the many aspects of the epileptic diathesis in an individual patient. Table 50-1 highlights the differences between the two classification schemes by using each to characterize some accepted epileptic syndromes. A clinician interested in epilepsy should be conversant with the 1989 and 2001 classification schemes and must anticipate additional revisions.
Incidence
In developed countries, the overall incidence of childhood epilepsy from birth to 16 years is approximately 40 cases in 100,000 children per year [Camfield et al., 1996b; Hauser et al., 1993]. The incidence in the first year of life is about 120 in 100,000. Between 1 and 10 years of age, the incidence plateaus at 40–50 cases in 100,000 children, and then drops further in the teenage years to about 20 in 100,000. The details of incidence by year of life from the Nova Scotia childhood epilepsy study are illustrated in Figure 50-1. Hauser estimates that about 1 percent of all children will have at least one afebrile seizure by age 14 years, and that 0.4–0.8 percent will have epilepsy by age 11 years [Hauser and Hesdorffer, 1990]. In less developed countries, there is suggestive evidence for a higher incidence of childhood epilepsy, possibly related to higher incidence of trauma and central nervous system infections.
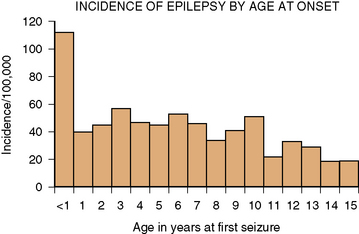
Fig. 50-1 Incidence of epilepsy by age of onset.
(From Camfield CS et al. Incidence of epilepsy in childhood and adolescents: A population based study in Nova Scotia from 1977–1985. Epilepsia 1996b;37:19.)
Epilepsy types vary in incidence. Syndromes dominated by generalized tonic-clonic or partial seizures account for 75 percent of childhood epilepsy. Syndromes dominated by absence seizures account for approximately 15 percent, and the secondary generalized epilepsies account for 10 percent. The latter group includes most of the “catastrophic” epilepsy syndromes, such as West and Lennox–Gastaut syndromes [Camfield et al., 1996b]. Prevalence data for seizure type or epilepsy syndrome are not easily collated because of various definitions of active epilepsy. When active epilepsy was defined as receiving daily antiepileptic drugs or a seizure within the past 5 years, the prevalence of epilepsy in children was 4.3–9.3 in 1000 [Hauser and Hesdorffer, 1990]. Because some epileptic syndromes rarely remit, they contribute more to prevalence of epilepsy than its incidence. The relative prevalence of symptomatic generalized epilepsies is higher than their relative incidence. This finding means that physicians who focus on newly diagnosed children will encounter a predominance of benign epilepsy syndromes. Those primarily treating chronic cases will observe a higher proportion of more malignant seizure disorders. A population-based prevalence study from Finland found that the main seizure types for each patient were focal in 43 percent, with complex partial and partial with secondary generalization most common. For 44 percent, generalized seizures were dominant, with generalized tonic-clonic seizures most common. Overall, 45 percent had localization-related epilepsy syndromes, and 48 percent had generalized syndromes [Eriksson and Koivikko, 1997].
Differential Diagnosis
The diagnosis of epilepsy is based almost exclusively on a clinical history of two or more unprovoked seizures. Parents vary in their capacity to describe these frightening events, and physicians vary in their ability to ask good questions. Some children are misdiagnosed. A Dutch group of neurologists shared case descriptions of childhood seizure disorders and were able to agree about most diagnoses of epilepsy; however, in 207 cases of possible first seizures, there was disagreement in 35 percent [Van Donselaar et al., 1989] – even experts sometimes disagree. Studies of interobserver reliability have demonstrated important differences in observations when families and neurologists have reviewed the same video seizures, and neurologists often disagree with each other. Neurology residents and faculty often classify seizures differently based on written histories [Camfield and Camfield, 2003]. When the history is not clear-cut, it is better to wait for additional attacks before diagnosing epilepsy. A list of disorders in children that are frequently confused with epilepsy is found in Box 50-2. Reflex anoxic seizures associated with pallid or vasodepressor syncope or with cyanotic breath-holding are particularly likely to be misinterpreted [Stephenson, 1991] (see Chapter 64).

















The EEG can be used to make the diagnosis of epilepsy only if a seizure is recorded – a rare event on routine EEG acquisition because most children with epilepsy have infrequent seizures. A small percentage of normal children have epileptiform activity on EEG but never have a seizure [Petersen and Eeg-Olofsson, 1968], and up to 40 percent of children with chronic epilepsy never demonstrate epileptiform discharges on interictal EEG [Camfield et al., 1995]. EEG findings may change significantly over time and reveal conflicting findings [Camfield et al., 1995; Trojaborg, 1968]. An interictal EEG is often useful for syndrome clarification but must be interpreted in the clinical context.
Natural History of Childhood Epilepsy
Recurrence after a First Seizure
Many children are seen by a physician after a first unprovoked generalized tonic-clonic seizure, a few after a first complex partial seizure, but almost none after a single absence or myoclonic seizure. For children presenting after a single unprovoked seizure, there is an immediate question: Will it happen again? Berg and Shinnar’s meta-analysis of the recurrence risk after a first seizure concluded that, overall, about 40 percent will have a second seizure [Berg and Shinnar, 1991; Shinnar et al., 1996]. There is much less information about the further recurrence risk after a second seizure, especially if medication is withheld. Current data suggest that the risk of a third seizure after a second is at least 80 percent [Camfield et al., 1985b; Shinnar et al., 2000; Hauser et al., 1998]. These observations validate the restriction of the diagnosis of epilepsy to those with two or more unprovoked seizures, because the concept of epilepsy implies a chronic, recurring disorder.
If a child has a single seizure, a small number of clinical features are helpful in predicting the risk of a second seizure. These include remote symptomatic cause, partial seizures, presence of intellectual or mental handicap, and possibly EEG spike discharge [Berg and Shinnar, 1991]. A normal child with a first generalized tonic-clonic seizure and a normal EEG has a recurrence risk of 20–30 percent, whereas a child with mental handicap, a partial seizure, and spike discharge on the EEG has a recurrence risk of 80–90 percent [Berg and Shinnar, 1991; Camfield et al., 1985b]. The value of MRI lesions in predicting recurrence after a first seizure has not been extensively evaluated [Arthur et al., 2008]. If a second seizure is to occur, it usually happens shortly after the first. At least 75 percent of recurrences happen within 6 months of the first, and most occur within a few weeks. Few recurrences are observed after 2 years.
Starting Medication Treatment
There is little justification for beginning daily medication after a child’s first unprovoked seizure because 60 percent of these children will never have another attack [Van Donselaar et al., 1989; Hirtz et al., 2003]. Population-based studies have found that medication prescription after the first seizure does not alter the recurrence rate, probably because of poor compliance in taking the medication [Camfield et al., 1985b; Hauser et al., 1982; Hirtz et al., 1984]. An open-label, randomized trial of medication versus no medication after a first seizure in 397 children and adults demonstrated a significant reduction in recurrences for those on medication over a 2-year treatment period [First Seizure Trial Group, 1993]. The risk of recurrence without treatment was 51 percent by 24 months, but there were still relapses in 25 percent of those randomized to medication (i.e., 2.8 times higher; 95 percent confidence interval [CI] = 1.9–4.2). When this cohort was followed for several more years, the eventual remission rate was the same for those treated or untreated after the first seizure [Musicco et al., 1997].
It has become common practice to prescribe medication after a second seizure. There is no evidence that the prescription of medication alters the long-term outlook of childhood epilepsy. There is convincing information that delaying antiepileptic drug treatment until the child has had up to 10 seizures does not alter the ease of seizure control or the long-term remission rate [Camfield et al., 1996a; Arts and Geerts, 2009]. In other words, if a child has few seizures, there is no evidence that each seizure facilitates the next. The main reasons to treat children with antiepileptic drugs are the avoidance of bodily injury from seizures and improvement in psychosocial function. Results from several studies indicate that injury is uncommon with most seizure types in children. Peters et al. [2001] described 79 children with benign rolandic epilepsy who together had more than 900 seizures over an 8.5-year period. There were no significant injuries. In a group of 59 children with generalized absence seizures followed over 15 years, Wirrell et al. [1997] observed that 16 had a serious physical injury as the result of a seizure. Kirsch et al. [2001] compared 31 cognitively normal children with epilepsy with best-friend controls and found no significant differences in injury number or severity. Hodgman et al. [1979] described 25 adolescents with generalized tonic-clonic epilepsy and observed that those with poorer seizure control were better able to communicate about seizures and had a better self-image. The “hidden handicap” for children with controlled seizures may have important effects on social adjustment. Epilepsy in children and adults has attracted social stigma in most cultures. Helping children deal with perceived and actual stigma may be an important part of treatment [Bandstra et al., 2008]. Once medication is started, most children still have more seizures. Only about 20 percent have “smooth-sailing epilepsy,” meaning that they start medication, become immediately seizure-free, and later are able to discontinue medication successfully without ever having another seizure [Camfield et al., 1993]. Only 50 percent of children continue to receive the same medication 1 year after initiating treatment [Camfield et al., 1985b; Canadian Childhood Epilepsy Study Group, 1996; Verity et al., 1995]. The decision to start medication is often not the end of the seizure problem.
Long-Term Remission
For many children, epilepsy is transient, and with maturation, the problem seems to vanish. At the time of diagnosis, it is possible to predict that at least 50 percent of children will outgrow their disorder and be able to discontinue medication [Camfield et al., 1993]. The Dutch Study of Epilepsy in Childhood followed 453 newly diagnosed children and found that 64 percent were no longer receiving medication 5 years later [Arts et al., 2004; Arts and Geerts, 2009]. The longer the follow-up, the higher the proportion with remission of symptoms. In Rochester, Minnesota, 115 children with epilepsy beginning before age 10 years were followed through the Mayo Clinic record linkage system. Ten years later, 75 percent had been seizure-free for at least 5 years, and 51 percent no longer received medication [Hauser et al., 1993]. At the end of a remarkable 30-year follow-up study of children with epilepsy from a population-based sample in Turku, Finland, Sillanpaa [1993] found that 76 percent of survivors had been seizure-free for at least 3 years.
Across most studies, factors that predict which children will outgrow their epilepsy have included normal intelligence, normal neurologic examination findings, relatively small numbers of seizures at diagnosis (which makes complex partial seizures unlikely), age of the first seizure below about 12 years, and absence of a remote symptomatic cause (an identified brain problem that preceded the onset of epilepsy). In an 8-year follow-up study of a population-based study of 504 children with epilepsy from Nova Scotia, a predictive scoring system was developed; it is outlined in Table 50-2 [Sillanpaa et al., 1995]. Those with a good prognosis had an 80 percent chance of remission (i.e., seizure-free and no longer receiving medication). For those with one or more of these adverse factors, the chance of remission was less but still about 40 percent. The Dutch Study of Epilepsy in Childhood found comparable predictive variables and supplemented these with the clinical course in the first 6 months of treatment. Rates of correct prediction were similar. Based on clinical features present at the time of diagnosis, it is possible to predict the long-term outcome with moderate accuracy. Surprisingly, most epilepsy syndromes do not have a definite outcome; however, a few specific syndromes allow an accurate prognosis [Camfield et al., 2003]. Epilepsy is always outgrown in cases of benign rolandic epilepsy, benign myoclonic epilepsy of infancy, and benign occipital epilepsy, early-onset type. Epilepsy is never outgrown by children with Rasmussen’s syndrome, Lennox–Gastaut syndrome, or Dravet’s syndrome. The Dutch and Nova Scotia studies were combined to allow study of 1055 newly diagnosed patients with at least 5 years of follow-up [Geelhoed et al., 2005]. Multivariate analysis that took into account clinical factors, syndrome diagnosis, and response to treatment in the first 6 months revealed that, at the time of diagnosis, a prognostic scoring scheme could correctly predict remission or no remission in about 70 percent. This means that, at the time of diagnosis, if parents ask “will the seizures be outgrown?”, the physician will give the wrong answer in about one-third of cases.
Table 50-2 Scoring System for Remission of Childhood Epilepsy at the Time of Diagnosis
| Variable | Score* |
|---|---|
| Age of first seizure (months) | |
| <12 | 99 |
| 12–144 | 142 |
| >144 | 0 |
| Intelligence | |
| Normal | 111 |
| Retardation | 0 |
| Previous neonatal seizures | |
| No | 218 |
| Yes | 0 |
| Number of seizures before starting medication | |
| 1 or 2 | 72 |
| 3–20 | 123 |
| >20 | 0 |
* Add the scores from this column. If the total score is greater than 495, the child is predicted to have remission of epilepsy.
(Adapted from Sillanpaa M et al. Predicting long term outcome of childhood epilepsy in Nova Scotia, Canada, and Turku: Validation of a simple scoring system. Arch Neurol 1995;52:589.)
Stopping Medication
About 60–70 percent of children with epilepsy who have become seizure-free for 1–2 years can successfully stop medication treatment [Berg and Shinnar, 1991; Dooley et al., 1996]. The rate of success is no greater if medication is continued for up to 5 years seizure-free [Hollowach-Thurston et al., 1982]. Factors that predict successful discontinuation of medication include generalized seizures, age of onset before 10–12 years, normal neurologic examination, and in some studies, resolution of interictal EEG spike discharges. Children with no adverse factors may have an 80–90 percent success rate. Each factor has an additive effect and those with all of the adverse factors may have only a 10–20 percent success rate.
If an initial discontinuation trial is unsuccessful, medication is usually restarted. About 50 percent of children again become seizure-free for sufficient time to try to discontinue medication a second time, with a 70 percent success rate [Camfield et al., 1993]. The remission rate for juvenile myoclonic epilepsy (JME) is low, although a recent population-based study suggested that eventually 40 percent will no longer require daily medication [Camfield et al., 2009]. Still, most experts suggest that medication for JME should be continued lifelong and that further attempts to discontinue medication after an initial failure are probably not warranted [Delgado-Escueta and Enrile-Bacsal, 1984].
A Scandinavian study randomized 207 children at the time of diagnosis of epilepsy to receive 1 or 3 years of treatment [Braathen and Melander, 1997]. If the child was seizure-free for the last 6 months of study, medication was discontinued. This practice meant that some of the children in the 1-year treatment group had been seizure-free for only 6 months before medication was stopped. The success rate for those in the short-treatment group was significantly less than for the 3-year group (53 percent vs. 71 percent). A Dutch study randomized 161 children with epilepsy that was controlled within 2 months of starting treatment to 6 months or 1 year of treatment. The 6-month group had a higher relapse rate when medication was stopped; however, by 2 years later, the rates of remission were identical [Peters et al., 1998]. None the less, for a substantial number of children, epilepsy was a short-lived disorder requiring only short-term medication use. Medication treatment for benign rolandic epilepsy often is not needed [Peters et al., 2001], and further studies will likely identify other children who do not require drug treatment.
Intractability
Defining intractable epilepsy is difficult, and many definitions have been suggested [Berg et al., 2006a]. For a study of 613 children with epilepsy from Connecticut, Berg and Shinnar [2001] defined intractability as a “failure, for lack of seizure control, of more than 2 first-line antiepileptic drugs with an average of more than one seizure per month for 18 months and no more than 3 consecutive months seizure free during that interval.” There were 60 intractable cases in the first 24 months after diagnosis. The proportions of intractable patients in each major syndrome grouping were as follows: cryptogenic/symptomatic generalized, 34.6 percent; idiopathic generalized, 2.7 percent; other localization-related, 10.7 percent; and unclassified, 8.2 percent. Sillanpaa’s [1993] follow-up study from Turku, Finland, defined intractability as one or more seizures per year in the past 10 years of follow-up. After 20 years of follow-up, 22 percent of the subjects met these criteria. Predictors of intractability included poor initial response to medication, remote symptomatic cause, and status epilepticus. The Dutch Study of Childhood Epilepsy (using Berg and Shinnar’s definition of intractability), with 453 children followed for 5 years, found that 6 percent were intractable [Arts et al., 2004]. Camfield et al. [1993] defined intractability as “at least one seizure each three months for the last year of follow-up, with failure of at least 3 antiepileptic drugs.” For those with partial and generalized tonic-clonic epilepsies, 8 percent of 511 children became intractable during an average of 8 years of follow-up. After 20 years of follow-up, 51 percent of 75 patients with childhood-onset symptomatic generalized epilepsy had intractable seizures. In this study, the major predictor was severe neurologic deficit at the time of diagnosis.
An important issue in the definition of intractability is the length of follow-up. In the clinical setting, many children with intractable epilepsy eventually become well controlled. In a series of 145 children with intractible seizures (i.e., less than one per month for at least 2 years) followed over 18 years, Huttenlocker and Hapke observed that 75 percent of normally intelligent children and 30 percent with mental handicap had complete or nearly complete seizure remission (i.e., less than one seizure per year) [Huttenlocker and Hapke, 1990]. In the Nova Scotia cohort, 39 patients with partial and generalized tonic-clonic epilepsies had intractable epilepsy after 7 years’ follow-up. After 12.5 years of follow-up, 7 (18 percent) of 39 became seizure-free [Camfield et al., 1996a].
There is controversy about how the number of drug failures required before intractability can be declared. In a Scottish study of 470 newly treated adults, 47 percent became seizure-free with their first antiepileptic drug [Kwan and Brodie, 2000]. Only 11 percent gained control with subsequent medications. In children, the failure of a first drug is important but far less ominous [Camfield et al., 1997]. Of 417 eligible patients with partial and generalized tonic-clonic epilepsies with at least 4 years of follow-up, 83 percent were successfully treated with a single antiepileptic drug during the first year of treatment. Of these, 61 percent eventually had remission of epilepsy, and 4 percent had intractable seizures. Among the 17 percent who failed treatment with their first antiepileptic drug, 42 percent eventually had complete remission of epilepsy, although 29 percent developed intractable seizures. Children with absence epilepsies had nearly identical findings [Wirrell et al., 2001].
Psychosocial Outcome for Children with Epilepsy
Children with epilepsy have high rates of behavior and cognitive problems that contribute to social dysfunction in childhood and in later adulthood, even if the epilepsy resolves. These issues are reviewed in more detail in Chapter 51. Austin et al. [2002] reported that, at the time of diagnosis, 40 percent of children were at significant risk of behavior problems, as judged by scores on the Child Behavior Checklist. Those with multiple recurrent seizures were at even higher risk. The Isle of Wight study found that children with epilepsy had high rates of behavioral disorders [Rutter et al., 1970], which might have had their origins in social stigmatization, comorbid cognitive disorders, or medication effects. Oostrom et al. [2003] demonstrated greater cognitive and behavioral problems and the need for special educational assistance among 51 outpatient children with idiopathic or cryptogenic epilepsy compared with their classmate controls. Trostle [1988] reported that parents indicate a reluctance to have their normal children play with a child with epilepsy. Less than one-third of 20,000 U.S. high-school students indicated that they would date a person with epilepsy [Austin et al., 2002]. These behavioral, cognitive, and social stigma problems in childhood all point to grave concerns for the social adjustment and success of children with epilepsy after they reach adulthood.
Kokkonen et al. [1997] described the social outcome (by interview or questionnaire) of 81 young, “noninstitutionalized” adults from the catchment area of Oulu, Finland, compared with 211 randomly selected controls from the same birth cohort. At least 20 percent of this sample were unable to complete 9 years of education and presumably had severe learning disorders or mental retardation. They accounted for most of the poor social outcome, including a high rate of educational failure, failure to marry, and unemployment.
In Nova Scotia and Finland, children with “epilepsy only” were followed into young adulthood [Camfield et al., 1993; Jalava et al., 1997]. “Epilepsy only” meant having normal intelligence and no other neurologic handicaps. About 30 percent of each cohort had significant social adjustment problems, with decreased rates of stable relationships, marriage, social contacts, job satisfaction, and work achievement. Rates of unemployment or underemployment were high. Social outcome was not clearly related to epilepsy remission, and in the Nova Scotia study, variables related directly to epilepsy, such as age of onset, type of medication treatment, presence or absence of seizure remission, and frequency and severity of seizures, did not appear to predict social outcome in young adulthood [Camfield et al., 1993]. The strongest predictor of poor social outcome was the presence of a learning disorder, although predictive models were inaccurate; the main reasons for the unsatisfactory outcomes were unclear.
Wakamoto et al. [2000] studied 148 normally intelligent young adults (>20 years old) living in a rural district of Japan who had childhood-onset epilepsy. This population-based study found that 72 percent attended regular classes (versus 99 percent of those without epilepsy), 66 percent (versus 97 percent) entered high school, 67 percent (versus 95 percent) had employment, and 23 percent (versus 33 percent) married. There were 49 patients with mental handicap having a less satisfactory outcome: 14 percent attended regular class, 6 percent entered high school, 20 percent were employed, and 2 percent married.
Few studies have addressed the social outcome of children with specific epilepsy syndromes. Several studies have indicated a favorable outcome for adults with previous benign rolandic epilepsy (Loiseau et al., 1983; Peters et al., 2001]. In the Nova Scotia study, 56 children with typical childhood absence epilepsy were followed to young adulthood [Wirrell et al., 1997] and compared with a similar cohort with mild juvenile rheumatoid arthritis. Those with absence had significantly greater problems with impulsive behavior, including a 34 percent risk of an unplanned pregnancy. Educational and work achievement, family and other social relationships, and alcohol abuse were more often unsatisfactory. Those with on-going seizures despite medication had greater problems; however, most of the poor social outcome was unrelated to epilepsy-specific factors. The findings in JME were similar – 24 normally intelligent patients followed for 20–30 years had an unemployment rate of 35 percent with many unplanned pregnancies and social isolation [Camfield and Camfield, 2009]. Therefore, childhood epilepsy may have a lifelong, serious effect on social function, an effect that is greater than some other chronic disorders of childhood. Intervention studies have not been undertaken; however, the role of learning disorders and inadequate education is clear. If the long-term social outcome is to be rectified, physicians must address these areas with an enthusiasm equal to the drug treatment of childhood epilepsy. Early referral to an epilepsy support group may be of benefit.
Mortality in Children with Epilepsy
Children with epilepsy have a 5.3 times higher risk of dying than the general population [Camfield, et al., 2002]. The causes that lead to this excess are direct complications of seizures (e.g., aspiration or cardiac arrhythmia), accidents caused by seizures (e.g., falling into a fire), comorbid conditions (e.g., decompensated hydrocephalus), and suicide or sudden unexpected death in epilepsy (SUDEP). Three large prospective studies have found that children with epilepsy but no neurological deficit have the same survival as the general population. [Callenbach et al., 2001; Berg et al., 2006; Camfield et al., 2002]. SUDEP is very rare (2 of 1777 newly diagnosed cases). The excessive mortality in childhood epilepsy is almost entirely accounted for by comorbid severe neurological problems, especially those that affect bulbar function. The clinician can reassure families of normal children with epilepsy that they do not face an increased risk of untimely death despite the frightening nature of seizures.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Arthur T.M., deGrauw T.J., Johnson C.S., et al. Seizure recurrence following a first seizure in neurologically normal children. Epilepsia. 2008;49:1950-1954.
Arts W.F., Brouwer O.F., Peters A.C., et al. Course and prognosis of childhood epilepsy: 5-year follow-up of the Dutch study of epilepsy in childhood. Brain. 2004;127:1774-1784.
Arts W.F., Geerts A.T. When to start drug treatment for childhood epilepsy: The clinical epidemiological evidence. Eur J Paediatr Neurol. 2009;13:93-101.
Arzimanogolou A., Guerrini R., Aicardi J. Epilepsy in children, ed 3. Philadelphia: Lippincott Williams & Wilkins; 2004.
Austin J.K., Shafer P.O., Deering J.B. Epilepsy familiarity, knowledge, and perceptions of stigma: Report from a survey of adolescents in the general population. Epilepsy Behav. 2002;3:368.
Bandstra N., Camfield C.S., Camfield P.R. Stigma against epilepsy in the community. Can J Neurol Sci. 2008;35:436-440.
Berg A.T., Kelly M.M. Defining intractability: comparisons among published definitions. Epilepsia. 2006;47:431-436.
Berg A.T., Shinnar S. The risk of seizure recurrence following a first unprovoked seizure: A meta-analysis. Neurology. 1991;41:965.
Berg A.T., Shinnar S., Levy S.R., et al. Defining early seizure outcomes in pediatric epilepsy: the good, the bad and the in-between. Epilepsy Res. 2001;43:75-84.
Berg A.T., Vickrey B.G., Testa F.M., et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60:73-79.
Braathen G., Melander H. Early discontinuation of treatment in children with uncomplicated epilepsy: A prospective study with a model for prediction of outcome. Epilepsia. 1997;38:561.
Callenbach P.M., Westendorp R.G., Geerts A.T., et al. Mortality risk in children with epilepsy: the Dutch study of epilepsy in childhood. Pediatrics. 2001;107(6):1259-1263.
Camfield C.S., Camfield P.R. Juvenile myoclonic epilepsy 25 years after seizure onset: a population-based study. Neurology. 2009;73(13):1041-1045. Sep 29
Camfield C.S., Camfield P.R., Gordon K., et al. Outcome of childhood epilepsy: A population-based study with a simple predictive scoring system for those treated with medication. J Pediatr. 1993;122:861.
Camfield C.S., Camfield P.R., Gordon K.G., et al. Does the number of seizures before treatment influence ease of control or remission of childhood epilepsy?. Neurology. 1996;46:41-44.
Camfield C.S., Camfield P.R., Veugelers P.J. Death in children with epilepsy: a population-based study. Lancet. 2002;359(9321):1891-1895. Jun 1
Camfield C.S., Camfield P.R., Wirrell E., et al. Incidence of epilepsy in childhood and adolescents: A population based study in Nova Scotia from 1977–1985. Epilepsia. 1996;37:19.
Camfield P.R., Camfield C.S. Nova Scotia Pediatric Epilepsy Study. In: Jallon P., editor. Prognosis of childhood epilepsy. London: John Libby Publishers, 2003.
Camfield P.R., Camfield C.S., Dooley J.M., et al. Epilepsy after a first unprovoked seizure in childhood. Neurology. 1985;35:1657.
Camfield P.R., Camfield C.S., Smith E., et al. Newly treated childhood epilepsy: A prospective study of recurrences and side effects. Neurology. 1985;35:722.
Camfield P.R., Camfield C.S., Gordon K., et al. If a first antiepileptic drug fails to control a child’s epilepsy, what are the chances of success with the next drug? J Pediatr. 1997;131:821-824.
Camfield P.R., Gordon K., Camfield C.S., et al. EEG results are rarely the same if repeated within six months in childhood epilepsy. Can J Neurol Sci. 1995;22:297.
Canadian Childhood Epilepsy Study Group. Monotherapy clobazam has equivalent efficacy to carbamazepine and phenytoin in childhood epilepsy. Epilepsia. 1996;37:117.
Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489.
Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389.
Delgado-Escueta A.V., Enrile-Bacsal F. Juvenile myoclonic epilepsy of Janz. Neurology. 1984;34:285.
Dooley J.M., Gordon K., Camfield P.R., et al. Discontinuation of anticonvulsant therapy in children free of seizures for 1 year. Neurology. 1996;46:969.
Engel J. International League against Epilepsy (ILAE). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy. Report of the ILAE Taskforce on Classification and Terminology. Epilepsia. 2001;42:796-803.
Eriksson K.J., Koivikko M.J. Prevalence, classification, and severity of epilepsy and epileptic syndromes in children. Epilepsia. 1997;38:1275.
First Seizure Trial Group. Randomized clinical trial on the efficacy of antiepileptic drugs in reducing the risk of relapse after a first unprovoked tonic-clonic seizure. Neurology. 1993;43:478.
Geelhoed M., Boerrigter A., Camfield P.R., et al. The accuracy of outcome prediction models for childhood-onset epilepsy. Epilepsia. 2005;46:526-532.
Hauser W.A., Anderson V.E., Loewneson R.B., et al. Seizure recurrence after a first unprovoked seizure. N Engl J Med. 1982;307:522.
Hauser W.A., Annegers J.F., Kurland L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–84. Epilepsia. 1993;34:453.
Hauser W.A., Hesdorffer D.C. Epilepsy, frequency, causes, and consequences. New York: Demos Publications; 1990.
Hauser W.A., Rich S.S., Lee J.R., et al. Risk of recurrent seizures after two unprovoked seizures. N Engl J Med. 1998;38:429-434.
Hirtz D., Berg A., Bettis D., et al. Practice parameter: Treatment of the child with a first unprovoked seizure: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60:166-175.
Hirtz D.G., Ellenberg J.H., Nelson K.B. The risk of recurrence of non-febrile seizures in children. Neurology. 1984;34:637.
Hodgman C.H., McAnarney E.R., Myers G.J., et al. Emotional complications of adolescent grand mal epilepsy. J Pediatr. 1979;95:309.
Hollowach-Thurston J., Thurston D.L., Hixton B.B., et al. Prognosis of childhood epilepsy: Additional follow-up study of 148 children 15–23 years after withdrawal of anticonvulsant therapy. N Engl J Med. 1982;306:831.
Huttenlocker P.R., Hapke R.J. A follow-up study of intractable seizures in childhood. Ann Neurol. 1990;28:699-705.
Jalava M., Sillanpaa M., Camfield C.S., et al. Social adjustment and competence 35 years after onset of childhood epilepsy: A prospective controlled study. Epilepsia. 1997;38:708.
Kirsch R., Wirrell E. Do cognitively normal children with epilepsy have a higher rate of injury than their nonepileptic peers? Child Neurol. 2001;16:100-104.
Kokkonen J., Kokkonen E.R., Saukkonen A.L., et al. Psychosocial outcome of young adults with epilepsy in childhood. J Neurol Neurosurg Psychiatry. 1997;62:265-268.
Kwan P., Brodie M.J. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314-319.
Loiseau P., Pestre M., Dartigues J.F., et al. Long term prognosis in two forms of childhood epilepsy. Typical absence seizures and epilepsy with rolandic (centrotemporal) EEG foci. Ann Neurol. 1983;13:642-648.
Musicco M., Beghi E., Solari A., et al. Treatment of first tonic-clonic seizure does not improve the prognosis of epilepsy. First Seizure Trial Group (FIRST Group). Neurology. 1997;49:991.
Oostrom K.J., Smeets-Schouten A., Kruitwagen C.L., et al. Not only a matter of epilepsy: early problems of cognition and behavior in children with “epilepsy only” – a prospective, longitudinal, controlled study starting at diagnosis. Pediatrics. 2003;112(Pt 1):1338.
Peters A.C., Brouer O.F., Geerts A.T., et al. Randomized prospective study of early discontinuation of AEDs in children with epilepsy. Neurology. 1998;50:724-730.
Peters J., Camfield C.S., Camfield P.R. Benign rolandic epilepsy can be safely managed without medication: A population-based study. Neurology. 2001;57:537.
Petersen O., Eeg-Olofsson O. Paroxysmal activity in EEG of normal children. In: Kelleway P., Peterson I., editors. Clinical electroencephalography of children. New York: Grune & Stratton, 1968.
Rutter M., Graham P., Yule W. A neuropsychiatric study in childhood. Clin Dev Med. 1970;35/36:175.
Shinnar S., Berg A.T., Moshe S.L., et al. The risk of seizure recurrence after a first unprovoked afebrile seizure in childhood: An extended follow-up. Pediatrics. 1996;98:216.
Shinnar S., Shinnar S., Berg A.T., et al. Predictors of multiple seizures in a cohort of children prospectively followed from the time of their first unprovoked seizure. Ann Neurol. 2000;48:140-147.
Sillanpaa M. Remission of seizures and predictors of intractability in long-term follow-up. Epilepsia. 1993;34:930.
Sillanpaa M., Camfield P.R., Camfield C.S. Predicting long term outcome of childhood epilepsy in Nova Scotia, Canada, and Turku, Finland: Validation of a simple scoring system. Arch Neurol. 1995;52:589.
Stephenson J.B. Fits and faints. Philadelphia: JB Lippincott; 1991.
Trojaborg W. Changes in spike foci in children. In: Kelleway P., Peterson I., editors. Clinical electroencephalography of children. New York: Grune & Stratton, 1968.
Trostle J.A. Social aspects of epilepsy. In: Hauser W.A., editor. Current trends in epilepsy, Unit 1. Landover, MD: Epilepsy Foundation of America, 1988.
Van Donselaar C.A., Geerts A.T., Meulstee J., et al. Reliability of the diagnosis of a first seizure. Neurology. 1989;39:267-271.
Verity C.M., Hosking G., Easter D.J. A multicentre comparative trial of sodium valproate and carbamazepine in paediatric epilepsy. The Paediatric EPITEG Collaborative Group. Dev Med Child Neurol. 1995;37:97-108.
Wakamoto H., Nagao H., Hayashi M., et al. Long-term medical, educational, and social prognoses of childhood-onset epilepsy: A population-based study in a rural district of Japan. Brain Dev. 2000;22:246.
Wirrell E., Camfield C., Camfield P., et al. Prognostic significance of failure of the initial antiepileptic drug in children with absence epilepsy. Epilepsia. 2001;42:760-763.
Wirrell E., Camfield C.S., Camfield P.R., et al. Long-term psychosocial outcome in typical absence epilepsy: Sometimes a wolf in sheep’s clothing. Arch Pediatr Adolesc Med. 1997;151:152.
Arzimanogolou A., Guerrini R., Aicardi J. Epilepsy in children, ed 3. Philadelphia: Lippincott Williams & Wilkins; 2004.
Freeman J.M., Vining E.P.G., Pillas D.J. Seizures and epilepsy in childhood: A guide, ed 3. Baltimore: Johns Hopkins University Press; 2003.
Devinsky O. Epilepsy patient and family guide, ed 2. Philadelphia: FA Davis; 2002.
Devinsky O., Westbrook L.E. Epilepsy and developmental disabilities. Woburn, MA: Butterworth-Heinemann; 2002.
Jallon P. Prognosis of epilepsies. London: John Libby; 2003.
Roger J., Bureau M., Dravet C., et al. Epileptic syndromes in infancy, childhood and adolescence, ed 3. London: John Libby; 2003.
Wyllie E. The treatment of epilepsy: Principles and practice, ed 3. Philadelphia: Lea & Febiger; 2001.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 50 Pediatric Epilepsy
An Overview
After a first unprovoked seizure in childhood, the risk of a recurrence is about 50 percent, and after the second the risk is about 80 percent. By convention, epilepsy may be diagnosed only after a child has had two or more unprovoked seizures [Camfield et al., 1985a; Shinnar et al., 2000]. The term unprovoked implies that there has been no closely associated concurrent illness, fever, or acute brain injury. Recurrent seizures immediately after a head injury or associated with drug intoxication do not qualify for the diagnosis of epilepsy. Some specific provoking factors leading to reflex seizures are permitted, such as seizures provoked by patterns and flashes from video terminals for children with photosensitive epilepsy. Seizures are still viewed as unprovoked if they occur with stresses related to personal activity, such as sleep deprivation or severe emotional distress, unless these stresses are extreme.
Seizure Type and Epilepsy Syndrome
The definition of epilepsy allows for a tremendous variety of disorders. The seizure types, age of onset, cause, severity, comorbid conditions, response to medication, and clinical course vary widely. There are two main ways of grouping patients to bring an ordered approach to classification, treatment, and prognosis: seizure type and epilepsy syndrome. Both concepts continue to evolve. The International League against Epilepsy defined the seizure types listed in Box 50-1 [Commission on Classification and Terminology of the International League against Epilepsy, 1981]. There is a basic distinction between seizures with a generalized onset that seem to arise from everywhere in the cortex at once, and seizures with a partial or focal onset that begin in a defined area of cortex. A proposed update in the classification of epileptic seizures suggested that the distinction between simple partial and complex partial seizures is often difficult to assess and not needed [Engel, 2001].
Box 50-1 International Classification of Seizure Type
Unclassifiable Epileptic Seizures
(Adapted from the Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22:489.)
A given seizure type may occur with many different associations. For example, a 2-year-old child with severe mental handicap may have generalized tonic-clonic seizures that are completely resistant to medication, or a normal teenager may have the same seizure type that is completely suppressed by medication. Factors beyond seizure type allow a more comprehensive diagnosis – an epilepsy syndrome diagnosis. Epileptic syndromes may have one or more seizure types, often with a characteristic interictal electroencephalogram (EEG). Each syndrome has a typical age of onset, a defined group of causes, sometimes a clear response to specific treatments, and sometimes a defined clinical course and prognosis. As with seizure types, syndromes have been classified as localization-related (focal) or generalized, and then subdivided into idiopathic (when microdysgenesis or a biochemical etiology is likely), symptomatic (when a diffuse or localized brain abnormality is known), and cryptogenic (when a diffuse or localized brain abnormality is suspected but unproven; Table 50-1). The term cryptogenic has been discouraged, as the detection of a structural lesion is dependent on the evolving quality of brain imaging and detection of a biochemical abnormality is dependent on the evolving understanding of genomic and proteomic abnormalities. In adults, localization-related epilepsy syndromes are most common, usually symptomatic, and caused by a localized, structural abnormality. Children have similar disorders, but there are many other important syndromes. The idiopathic partial and generalized syndromes all begin in childhood, and genetic influences are significant. The generalized symptomatic syndromes of childhood account for most intractable epilepsy in children [Arzimanogolou et al., 2004; Camfield and Camfield, 2003].
Table 50-1 Proposal from the International Classification of Epilepsies and Epileptic Syndromes and Related Seizure Disorders of 1989 and the Listing of Epilepsy Syndromes Recognized in the Proposed 2001 ILAE Classification Scheme
| 1989 ILAE Classification* | 2001 ILAE Proposal† |
|---|---|
| Localization-Related (Local, Focal, Partial Epilepsies and Syndromes | |
| Idiopathic (with Age-Related Onset) | |
| Benign childhood epilepsy with centrotemporal spikes | Benign childhood epilepsy with centrotemporal spikes |
| Childhood epilepsy with occipital paroxysms | Early-onset benign childhood occipital epilepsy (Panayiotopoulos type) (new) Late-onset childhood occipital epilepsy (Gastaut type) (new) Idiopathic photosensitive occipital lobe epilepsy (new) |
| Primary reading epilepsy | Primary reading epilepsy Now called Reflex epilepsies, which also includes: |
(Diagnosis of epilepsy not now required)
* Modified from the 1989 International League Against Epilepsy (ILAE) Proposal.
† Modified from the 2001 ILAE Proposal. In this proposal there are no headings or subheadings, only a listing of epilepsy syndromes by specific syndrome name. Our format of presentation has indicated where syndromes are removed or renamed from the 1989 ILAE proposal or newly recognized in the 2001 proposal. Each syndrome was “forced” into the 1989 proposed schema.
The International League Against Epilepsy (ILAE) classified febrile seizures as a special syndrome. Febrile seizures are the most common convulsive event in the human species. Because these seizures are provoked by fever, febrile seizures do not fit well with the usual definition of epilepsy, and few children with febrile seizures later develop unprovoked seizures. The disorder merits special consideration, as reviewed in Chapter 57.
The 1989 ILAE syndrome classification scheme is still in wide use, and most patients can be “forced” into one of the broad categories, but many children, especially those with symptomatic generalized epilepsy, do not meet the criteria for a more specific syndrome [Commission on Classification and Terminology of the International League Against Epilepsy, 1989]. There are many recently described epilepsy syndromes, but a newer, comprehensive syndrome classification system has been elusive. Advances in genetics have revealed that the same mutation may cause a variety of epilepsy syndromes, including partial and generalized epilepsies, even within the same family (see Chapter 52). This finding has been particularly well illustrated in large families with mutations in the gene coding for neuronal sodium channel – SCN1A.
Another evolving classification is “epilepic encephalopathy,” defined as an epilepsy syndrome where the seizures and/or interictal EEG discharge have permanent deleterious effects on brain development. Examples would be West’s syndrome with infantile spasms and hypsarrhythmia on EEG, Lennox–Gastaut syndrome with mixed generalized seizures and nearly continuous slow spike wave EEG discharge, or Landau–Kleffner syndrome with relatively mild clinical seizures but very active bitemporal epileptic EEG discharge (see Chapter 56).
A new proposal for syndrome classification in 2001 is based on a five-axis system [Engel, 2001]. Axis 1 concerns ictal phenomenology (i.e., what the seizure looks like). Axis 2 is the specific seizure type. Axis 3 is the specific epilepsy syndrome, when it can be defined; axis 4 is the cause; and axis 5 describes the degree of impairment associated with epilepsy. This proposal comes with a more comprehensive list of epilepsy syndromes, although the system is less categorical than the 1989 classification [Commission on Classification and Terminology of the International League against Epilepsy, 1989]. The 2001 proposal has not been widely accepted, although it does serve well to alert the clinician to the many aspects of the epileptic diathesis in an individual patient. Table 50-1 highlights the differences between the two classification schemes by using each to characterize some accepted epileptic syndromes. A clinician interested in epilepsy should be conversant with the 1989 and 2001 classification schemes and must anticipate additional revisions.
Incidence
In developed countries, the overall incidence of childhood epilepsy from birth to 16 years is approximately 40 cases in 100,000 children per year [Camfield et al., 1996b; Hauser et al., 1993]. The incidence in the first year of life is about 120 in 100,000. Between 1 and 10 years of age, the incidence plateaus at 40–50 cases in 100,000 children, and then drops further in the teenage years to about 20 in 100,000. The details of incidence by year of life from the Nova Scotia childhood epilepsy study are illustrated in Figure 50-1. Hauser estimates that about 1 percent of all children will have at least one afebrile seizure by age 14 years, and that 0.4–0.8 percent will have epilepsy by age 11 years [Hauser and Hesdorffer, 1990]. In less developed countries, there is suggestive evidence for a higher incidence of childhood epilepsy, possibly related to higher incidence of trauma and central nervous system infections.
Fig. 50-1 Incidence of epilepsy by age of onset.
(From Camfield CS et al. Incidence of epilepsy in childhood and adolescents: A population based study in Nova Scotia from 1977–1985. Epilepsia 1996b;37:19.)
Epilepsy types vary in incidence. Syndromes dominated by generalized tonic-clonic or partial seizures account for 75 percent of childhood epilepsy. Syndromes dominated by absence seizures account for approximately 15 percent, and the secondary generalized epilepsies account for 10 percent. The latter group includes most of the “catastrophic” epilepsy syndromes, such as West and Lennox–Gastaut syndromes [Camfield et al., 1996b]. Prevalence data for seizure type or epilepsy syndrome are not easily collated because of various definitions of active epilepsy. When active epilepsy was defined as receiving daily antiepileptic drugs or a seizure within the past 5 years, the prevalence of epilepsy in children was 4.3–9.3 in 1000 [Hauser and Hesdorffer, 1990]. Because some epileptic syndromes rarely remit, they contribute more to prevalence of epilepsy than its incidence. The relative prevalence of symptomatic generalized epilepsies is higher than their relative incidence. This finding means that physicians who focus on newly diagnosed children will encounter a predominance of benign epilepsy syndromes. Those primarily treating chronic cases will observe a higher proportion of more malignant seizure disorders. A population-based prevalence study from Finland found that the main seizure types for each patient were focal in 43 percent, with complex partial and partial with secondary generalization most common. For 44 percent, generalized seizures were dominant, with generalized tonic-clonic seizures most common. Overall, 45 percent had localization-related epilepsy syndromes, and 48 percent had generalized syndromes [Eriksson and Koivikko, 1997].
[/not-level-membership-for-neurology-category]
