CHAPTER 5 Pathology of the marrow
General considerations and infections/reactive conditions
Principles of bone marrow examination
The bone marrow may be examined after aspiration through a special wide-bore needle. The aspirate and trephine biopsy specimens are complementary and when both are obtained, they permit a comprehensive evaluation of the bone marrow. Guidelines based on preferred best practices have been published by the International Council for Standardization in Hematology.1 The usual sites of aspiration are the posterior or anterior iliac crest or manubrium sterni in adults, the posterior superior iliac spine in infants and children and the medial aspect of the upper end of the tibia in neonates. The aspirate, which consists of fragments of marrow tissue, individual nucleated marrow cells and a variable quantity of blood, is spread on glass slides and examined after fixation and staining. Examination of marrow smears allows a detailed analysis of the morphology and cytochemistry of cells and the percentages of various cell types (see Chapter 2 for details). However, differential counts performed on marrow smears do not give accurate data on the prevalence of some cell types, such as megakaryocytes and macrophages, which are relatively resistant to aspiration or have a tendency to remain attached to the aspirated marrow fragments during the preparation of the smears. This drawback may be overcome, to a large extent, by examining preparations obtained by crushing marrow fragments between two glass slides and pulling the slides apart. Apart from its use for cytologic studies with the light microscope, aspirated BM can be used for immunophenotypic, cytogenetic, molecular genetic, biochemical and electron microscope studies.
The histology of the BM is investigated by examining sections of aspirated marrow particles (either in clotted aspirates or after concentration of the particles in various ways) or by examining sections of a core of bone obtained using a specially-constructed trephine needle. The usual site of trephine biopsy is the posterior superior iliac spine. Unlike marrow smears, histologic sections of trephine biopsies permit an appreciation of intercellular relationships and particularly of the relationship between hemopoietic cells, non-hemopoietic cells, the blood vessels and bone (see Chapter 3). Such sections are therefore useful for the detection of granulomas and focal accumulations of malignant cells. They also allow a study of the quantity and distribution of reticulin and collagen in marrow and make possible the diagnosis of myelofibrosis. Histologic sections are usefully stained with hematoxylin and eosin (H&E), Giemsa stain and a reticulin stain. Some laboratories also stain for hemosiderin routinely but an alternative is to apply this stain selectively, only when an aspirate for a Perls stain with an adequate number of fragments is not available.
Alterations of the marrow in disease
Alterations in cellularity
The method of assessing the proportion of the marrow tissue that is composed of hemopoietic cells as opposed to fat cells (percentage cellularity) and the normal values for this parameter are discussed in Chapters 2 and 3. Hypocellularity (i.e. cellularity <25%) is seen in the acquired aplastic or hypoplastic anemias, Fanconi anemia, paroxysmal nocturnal hemoglobinuria, rare cases of acute leukemia, and in normal adults over the age of 70 years. Hypercellularity (cellularity >75%) may be seen in a variety of conditions, including hemolytic anemia, hemorrhage, megaloblastic and sideroblastic anemias, the congenital dyserythropoietic anemias, polycythemia vera and other chronic myeloproliferative neoplasms (MPN), infections, malignant disease, myelodysplastic syndromes (MDS), the leukemias and in normal infants and young children.
Alterations in the frequency and morphology of various types of bone marrow cells
Erythroblasts and neutrophil precursors
Myeloid : erythroid ratio
The myeloid : erythroid ratio (M : E ratio) is often defined as the ratio between the number of cells of the neutrophil granulocyte series (including mature granulocytes) and the number of erythroblasts. The normal range for this ratio in adults has been reported as 2.0–8.3 and 1.1–5.2 in BM smears and 1.5–3.0 in histologic sections.1,2 Some hematologists include eosinophils, basophils and monocytes, and their precursors in the ‘myeloid’ figure but this has only a slight effect on the values for the M : E ratio in normal subjects. The M : E ratio can be used as an index of total erythropoietic activity in patients in whom there is reason to assume that the total number of marrow granulocytes and their precursors is normal (e.g. in patients with normal counts of circulating granulocytes). Conversely, the M : E ratio may be used as an index of total granulocytopoietic activity, provided that the total number of erythroblasts in the body can be assumed to be normal. Some causes of a reduced or increased M : E ratio are listed in Box 5.1.
Morphologic changes in erythroblasts
Most of the erythroblasts in normal BM are uninucleate, show synchrony between nuclear and cytoplasmic maturity and do not display any peculiar cytologic features. In a study of normal BM, only 0.31% of erythroblasts were binucleate (range 0–0.57%), 0.24% (range 0–0.91%) of normal erythroblasts showed basophilic stippling of the cytoplasm, up to 0.7% had vacuolated cytoplasm, 2.38% (range 0.72–4.77%) had intererythroblastic cytoplasmic bridges and 0.22% (range 0–0.55%) possessed markedly irregular or karyorrhectic nuclei.3 Howell–Jolly bodies were found in 0.18% (range 0–0.39%) and asynchrony between nuclear and cytoplasmic maturation was found very infrequently. In various diseases accompanied by a disturbance of erythropoiesis, the frequency of such cytologic features (’dysplastic’ changes) is increased; hence these cytologic ‘aberrations’ are described as dyserythropoietic changes. Morphologic abnormalities that are not encountered in normal BM, such as internuclear chromatin bridges and giant erythroblasts, may also be found in a few diseases (Table 5.1). Dyserythropoiesis occurs in a number of congenital and acquired disorders. The congenital disorders include some thalassemia syndromes, homozygosity for hemoglobin C or E, some unstable hemoglobins, hereditary sideroblastic anemias, thiamine-responsive anemia, homozygosity for pyruvate kinase deficiency, and the congenital dyserythropoietic anemias. The acquired disorders include vitamin B12 and folate deficiency, iron deficiency anemia, alcohol abuse, MDS, acute myeloid leukemia (AML), aplastic anemia, paroxysmal nocturnal hemoglobinuria, acquired immunodeficiency syndrome (AIDS), Plasmodium falciparum and P. vivax malaria, kala azar and liver disease. Dyserythropoietic changes have also been reported after excess ingestion of kelp tablets (containing arsenic) and after BM transplantation.
Table 5.1 Morphologic abnormalities in erythroblasts that may be detected in pathologic states using the light microscope
| Feature | Abnormality | Examples of causative disorder |
|---|---|---|
| Cell size | Large | Megaloblastic anemia Parvovirus B19 infection Congenital dyserythropoietic anemia |
| Small (micronormoblast) |
Iron deficiency anemia Thalassaemia Anemia of chronic disease (when severe) Congenital sideroblastic anemia |
|
| Nuclei | N : C asynchrony | Megaloblastic anemia Myelodysplastic syndrome and erythroleukemia |
| Irregular shape | Myelodysplastic syndrome Non-neoplastic dysplasia Congenital dyserythropoietic anemia |
|
| Nuclear bridges | Myelodysplastic syndrome Congenital dyserythropoietic anemia |
|
| Multinuclearity | Myelodysplastic syndrome and erythroleukaemia Congenital dyserythropoietic anemia |
|
| Karyorrhexis | Myelodysplastic syndrome Non-neoplastic dysplasia |
|
| Cytoplasm | Vacuolation | Alcohol Sideroblastic anemia Chloramphenicol Copper deficiency |
| Cytoplasmic bridging | Myelodysplastic syndrome Congenital dyserythropoietic anemia |
|
| Basophilic stippling | Myelodysplastic syndrome Thalassaemia |
|
| Sideroblasts | Reduced | Iron deficiency Anemia of chronic disease |
| Increased | Iron overload Megaloblastic anemia Myelodysplastic syndrome |
|
| Ring sideroblasts | Congenital sideroblastic anemia Mitochondrial cytopathy (Pearson syndrome) Myelodysplastic syndrome Lead poisoning Drug-induced sideroblastic anaemia |
Increased vacuolation of the cytoplasm of erythropoietic cells has been observed during treatment with chloramphenicol and as an effect of taking excess ethanol. It has also been reported in aplastic anemia associated with glue sniffing, in protein-energy malnutrition, riboflavin and phenylalanine deficiency, AML and hyperosmolar diabetic coma.4 Increased vacuolation of both erythroid cells and granulocyte precursors is observed in Pearson syndrome (a mitochondrial cytopathy) and in copper deficiency.
When there is asynchrony between nuclear and cytoplasmic maturation in a substantial proportion of erythroblasts, erythropoiesis is described as being megaloblastic. A detailed description and the causes of megaloblastic erythropoiesis are given in Chapter 12.
Morphologic changes in the neutrophil series
Morphological abnormalities in the neutrophil series may be of cell size, or shape of nucleus or cytoplasm. There may be abnormalities in the differentiation pathway with left shift or maturation arrest. These changes may be inherited or acquired; the latter may be the result of hematinic deficiencies, infections, drugs, cytokine administration or malignancy. Cytoplasmic abnormalities include the absence of specific granules in the myelocytes and metamyelocytes in acute leukemia and MDS, the reduction of nuclear segmentation in the marrow granulocytes in cases of the inherited and acquired Pelger–Huët anomaly and the formation of giant metamyelocytes and macropolycytes in vitamin B12 or folate deficiency. Giant metamyelocytes may also be found, usually in small numbers, in the absence of evidence of vitamin B12 or folate deficiency, in iron deficiency, infections, malignant disease, falciparum malaria (especially chronic falciparum malaria) and protein-energy malnutrition. They are seen quite often in the BM of patients with AIDS.5 Macropolycytes are also not specific for vitamin B12 or folate deficiency, being found in infections, myeloproliferative neoplasms, drug-induced marrow damage and protein-energy malnutrition. Increased numbers of binucleate cells of the neutrophil series occur in protein-energy malnutrition and to a lesser extent in vitamin B12 or folate deficiency. Macropolycytes and binucleate cells are sometimes seen in MDS. An increased proportion of neutrophils with ring- or doughnut-shaped nuclei may be seen in chronic myelogenous leukemia (CML), AML, MDS, AIDS and falciparum malaria.
Vacuolation of the neutrophil precursors, usually from the promyelocyte/myelocyte stage onwards, may be seen in patients with acute alcoholic intoxication, severe infections, drug-induced marrow damage (e.g. chloramphenicol toxicity), protein-energy malnutrition and certain rare conditions such as the Chédiak–Higashi syndrome, severe congenital neutropenia, hereditary transcobalamin II deficiency,6 neutral lipid storage disease with and without ichthyosis and carnitine deficiency. The term Jordan’s anomaly is sometimes used to designate familial vacuolation of leukocytes; Jordan’s original patient may have had carnitine deficiency.7 Giant metamyelocytes, whatever the condition with which they are associated, may also be vacuolated.
Eosinophil series
An increase of eosinophils and their precursors, sometimes without an associated eosinophilia in the peripheral blood (PB), may be seen in parasitic infections, allergic disorders, certain skin diseases, Hodgkin lymphoma, non-Hodgkin lymphoma, acute lymphoblastic leukemia (ALL), carcinoma, sarcoma and collagen vascular diseases. A BM and PB eosinophilia is also seen in CML, eosinophilic leukemias including those associated with rearrangement of PDGFRA, PDGFRB and FGFR1 (see Chapter 25), other chronic myeloproliferative neoplasms, occasional cases of AML and the idiopathic hypereosinophilic syndrome.
Mast cells
Mast cells may be increased in the marrow in infection and inflammation, renal failure, aplastic anemia, paroxysmal nocturnal hemoglobinuria, lymphoplasmacytic lymphoma including Waldenström macroglobulinemia, chronic lymphocytic leukemia, non-Hodgkin lymphoma, MDS, scleroderma, systemic mastocytosis (in which the mast cells are neoplastic; see Chapter 26), some chronic eosinophilic leukemias (see Chapter 25) and less often in a variety of other conditions. Mast cells are difficult to recognize in sections stained with H&E but are detectable when sections are stained with a Giemsa stain, which stains the granules purple. The granules also stain by the periodic acid-Schiff (PAS) reaction, are α-naphthyl AS-D chloroacetate esterase-positive (positive Leder stain) and stain metachromatically with toluidine blue. They are readily identified with an immunohistochemical stain for mast cell tryptase and may also express mast cell chymotryptase, CD68 and CD117.
Megakaryocytes
Conditions associated with an increased number of megakaryocytes include CML, polycythemia vera, essential thrombocythemia, primary myelofibrosis, infections, chronic alcoholism, reactive changes due to generalized malignant disease, Hodgkin lymphoma, non-Hodgkin lymphoma and hemorrhage. Megakaryocytes are also increased in diseases such as ‘idiopathic’ (autoimmune) thrombocytopenic purpura and thrombotic thrombocytopenic purpura in which thrombocytopenia is primarily caused by a reduced platelet life span (see Chapter 33).
A decreased number of megakaryocytes is seen in acute leukemia, Fanconi anemia and other constitutional aplastic anemias, the syndrome of thrombocytopenia with absent radii, and acquired aplastic anemia. The morphological abnormalities that may affect megakaryocytes in HIV infection are described later in this chapter. Those found in inherited disorders are reviewed in Chapter 32 and those in the myeloproliferative neoplasms in Chapters 22–27.
Lymphocytes
Lymphocytes are more numerous in the BM in children than in adults (Chapters 2 and 3). An artifactual increase in the percentage of lymphocytes occurs if a BM aspirate is much diluted with PB. BM lymphocytes are increased in many reactive and malignant conditions in which there is a PB lymphocytosis (e.g. infectious mononucleosis, chronic lymphocytic leukemia). In addition, BM lymphocytes may be increased in the absence of a PB lymphocytosis in non-Hodgkin lymphoma. The majority of lymphocytes in normal BM are T cells, but BM infiltration is more likely to be seen in B-lymphoproliferative disorders.
A trephine biopsy will show whether a lymphocytic infiltrate is diffuse or focal and whether any focal infiltration is paratrabecular or nodular (with or without follicle formation) (see Chapter 3). In normal BM, lymphocytes are spread diffusely through the marrow but small aggregates or nodules also develop, and rarely they may have germinal centers.8 The incidence of such lymphoid aggregates rises with age, and an increased incidence is seen in pernicious anemia, chronic myeloproliferative neoplasms, hemolytic states, inflammatory reactions and autoimmune conditions such as rheumatoid arthritis and secondary to immunotherapy (e.g. rituximab treatment of lymphoma patients). BM biopsy specimens showing lymphoid aggregates are more likely than other BM specimens to show lipid granulomas and plasmacytosis. A malignant lymphocytic infiltrate may be diffuse or focal and occasionally a nodular pattern is seen (detailed description in Chapters 28 and 29).
Plasma cells
There are less than 1–2% of plasma cells in normal BM. An increased percentage of BM plasma cells may be found in a wide range of pathologic conditions (Box 5.2). In various non-neoplastic conditions, up to 50% of nucleated BM cells may be plasma cells (reactive plasmacytosis). It is sometimes difficult to distinguish between reactive plasmacytosis and multiple myeloma on the basis of the morphologic characteristics of the plasma cells and definitive diagnosis requires immunohistochemistry.9 Some features useful in differential diagnosis are listed in Table 5.2. Russell bodies, Mott cells (plasma cells containing multiple Russell bodies) and apparently intranuclear inclusions resembling Russell bodies (Dutcher–Fahey bodies) may be seen in both reactive conditions and multiple myeloma but apparently intranuclear inclusions (which represent invagination of a cytoplasmic inclusion into the nucleus) are more often seen in myeloma (see Chapter 30). Cells with flaming cytoplasm may be found in both reactive plasmacytosis and myeloma but a substantial proportion of such cells is more likely in myeloma. Examination of the distribution of plasma cells in histologic sections of BM is of considerable value in elucidating the cause of plasmacytosis since homogeneous nodules of these cells, with little supporting stroma, are found in myeloma but not in reactive plasmacytosis (Table 5.2). Myeloma cells may show hemophagocytosis. Hemosiderin-containing granules are sometimes found in the cytoplasm of plasma cells in alcoholics and in copper deficiency, porphyria cutanea tarda, megaloblastic anemia, refractory normoblastic anemia and iron overload.
Box 5.2 Causes of plasmacytosis in the bone marrow
Table 5.2 Characteristics of bone marrow plasma cells in reactive plasmacytosis and multiple myeloma
| Reactive plasmacytosis | Multiple myeloma | |
|---|---|---|
| Number | Up to 10–20%, rarely up to 50% | Usually 30–90% |
| Cytology | Most cells are mature and look like normal plasma cells Majority of cells are mononucleate; four nuclei per cell rare Nucleoli only in occasional cells Nucleocytoplasmic asynchrony usually not a prominent feature |
More cells show features of immaturity; cells are either pleomorphic or monomorphic; occasionally lymphoid Multinuclearity common Nucleoli common Nucleocytoplasmic asynchrony common |
| Distribution | Interstitial infiltrate, especially perivascular; some cells may be aggregated around macrophages Small clusters of plasma cells may be present but large homogeneous nodules are absent Broad band-like infiltrates very rare |
Commonly near endosteal surface as well as perivascular Large homogeneous nodules of plasma cells with little intervening hemopoietic tissue common Broad band-like infiltrates common |
| Immunocytochemistry | κ : λ ratio about 2 : 1 CD19 positive CD56 negative |
Monotypic κ- or λ-chains CD19 negative CD56 positive (90% cases) |
Macrophages (histiocytes)
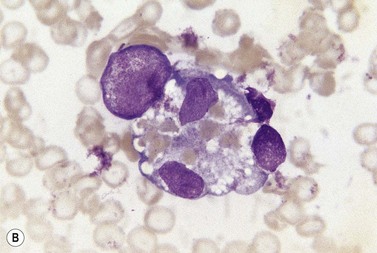
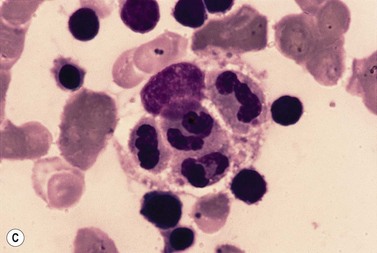
An increase of BM macrophages is common in a wide variety of hematologic and non-hematologic conditions. These include various infective and inflammatory disorders, conditions associated with increased blood cell destruction or increased ineffective hemopoiesis and post-granulocyte-macrophage colony-stimulating factor (GM-CSF) therapy. The macrophages range from immature cells showing little phagocytic activity to mature cells containing phagocytosed material or having foamy cytoplasm. In most instances the increase in macrophages is reactive. However, in the rare neoplastic condition designated malignant histiocytosis the increase results from the proliferation of cells of a neoplastic clone.10 The histiocytic disorders are defined by their constitutive cell type11 (Box 5.3). The two morphological features that distinguish malignant histiocytosis from reactive macrophage hyperplasia are: 1) pleomorphism of macrophages, with immature and atypical features such as a prominent nucleolus, distinct and thick nuclear membrane, irregular nuclear chromatin and multinuclearity; and 2) the presence among the macrophages and large multinucleate cells of many monoblasts and promonocytes12 (Fig. 5.1). The number of malignant cells in the BM varies from 5 to 90%12 and the infiltration of the marrow may be focal or diffuse (Fig. 5.2). The demonstration of positivity for CD163 (to differentiate from other mesenchymal tumors that may express CD68 shared by monocytes/macrophages) and a clonal cytogenetic abnormality in BM cells provides strong supporting evidence. Cytochemical reactions of the malignant cells are similar to those of monocytes and macrophages; they are positive for nonspecific esterase, acid phosphatase and lysozyme. Immunohistochemical analysis distinguishes tumors derived from Langerhans cells (Langerhans cell histicytosis and sarcoma), follicular dendritic cell sarcoma and interdigitating cell sarcoma from true histocytic sarcoma and malignant histiocytosis.13 Primary bone marrow involvement in these tumors is relatively rare. The blood count may show anemia, leukopenia, thrombocytopenia and eosinophilia and the PB film may contain macrophages and small numbers of monoblasts. A minor degree of hemophagocytosis is common. It is now apparent that the majority of patients initially described as having malignant histiocytosis actually had a reactive hemophagocytic syndrome, which could have been associated with malignant lymphoma and/or Epstein–Barr virus (EBV) infection.14–16
Box 5.3 Classification of benign histiocytic disorders
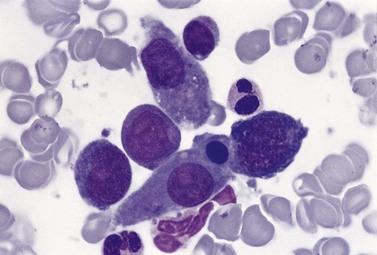
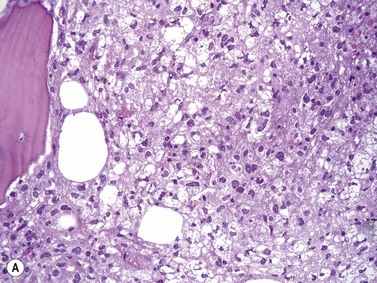
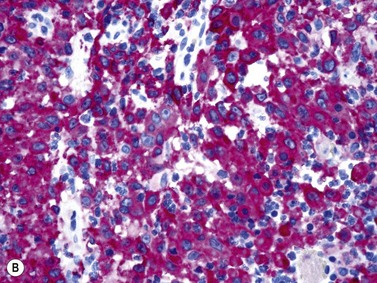
Hemophagocytic syndromes
The hemophagocytic syndrome, also called hemophagocytic lymphohistiocytosis (HLH), is a collection of non-malignant but frequently life-threatening disorders, associated with an ever growing list of genetic and acquired causes17,18 (Box 5.4). In HLH, there is a deregulation of T-lymphocytes and excessive production of cytokines leading to macrophage hyperplasia, enhanced macrophage activity and increased phagocytosis by macrophages of red cells, granulocytes, platelets and hemopoietic cells. The clinico-pathologic features of the syndrome include fever, hepatosplenomegaly, lymphadenopathy, skin rash, neurologic abnormalities, cytopenias, hypertriglyceridemia, high serum ferritin level and coagulopathy. In the ‘primary’ hemophagocytic syndrome (familial hemophagocytic lymphohistiocytosis), there is an inherited immune defect. Many cases of acquired hemophagocytic syndrome have an underlying predisposing condition leading to immunosuppression such as HIV infection, renal transplantation, malignant disease and autoimmune disease. The most frequent cause of a virus-associated hemophagocytic syndrome is EBV. The many other causes of infection-associated hemophagocytic syndrome include tuberculosis and P. falciparum malaria (Fig. 5.3). Reactive macrophages containing phagocytosed blood or BM cells can be distinguished on the basis of cytologic and cytochemical characteristics from other malignant cells showing hemophagocytic activity (Box 5.4).
Box 5.4 Classification of hemophagocytic syndromes
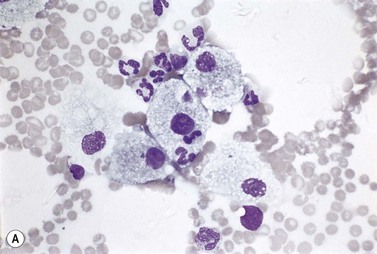
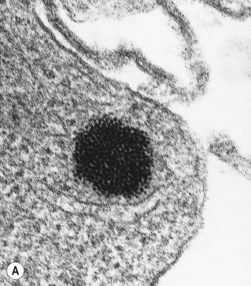
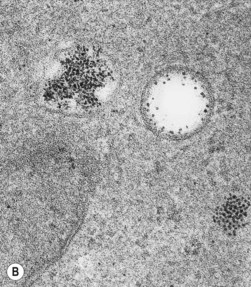
In lysosomal storage diseases, BM macrophages are laden with lipid or mucopolysaccharide.
Changes in iron stores and intraerythroblastic iron
Iron stores
In the BM, storage iron is normally present in the form of ferritin and hemosiderin and is mainly within the macrophages but also in endothelial cells. The stores of hemosiderin can be assessed by examining BM smears or histologic sections of either trephine biopsy sections or aspirated marrow fragments. In unstained smears and sections and in H&E-stained sections, hemosiderin granules appear as golden-yellow or brown refractile particles. In preparations stained by Perls acid ferrocyanide method (Prussian blue method), the hemosiderin appears as blue or bluish-black granules that may vary considerably in size. Various methods of grading hemosiderin stores semiquantitatively have been employed by different authors but in practice, it is adequate to grade hemosiderin iron as absent (or greatly reduced), present or increased.1 In examining BM smears it is necessary to examine a minimum of seven particles before concluding that hemosiderin is absent.19 BM hemosiderin stores are either absent or virtually absent in iron deficiency anemia from any cause. Rarely, patients recently treated with large doses of iron dextran develop iron deficiency anemia in the presence of stainable BM iron; in this situation the stainable iron is in a form that is unavailable for rapid mobilization.
There is a reasonable correlation between the cytochemical assessment of iron stores in stained preparations of marrow and biochemical determinations such as the iron content of the marrow or liver or, with certain exceptions, the serum ferritin level. The causes of alterations in the serum ferritin level are given elsewhere (Chapter 11).
Alterations in stainable non-hemoglobin iron within erythroblasts
When normal BM smears are stained by Perls acid ferrocyanide method and examined at high magnification (e.g. as high as × 950) using an oil immersion lens, 20–90% of the polychromatic erythroblasts are sideroblasts containing one or a few (up to five) very small (usually barely visible) blue-staining siderotic granules randomly distributed in the cytoplasm.20 Ultrastructural studies indicate that the siderotic granules present in normal erythroblasts correspond to intracytoplasmic aggregates of altered ferritin molecules (siderosomes or ferritin bodies) that may be membrane-bound (Fig. 5.4). In iron deficiency anemia and the anemia of chronic diseases, the percentage of sideroblasts is decreased. By contrast, in a wide variety of diseases associated with an increase in the percentage saturation of transferrin (e.g. hemolytic anemia, megaloblastic anemia, thalassemia, hereditary and secondary hemochromatosis), the number (per erythroblast) and size of siderotic granules are increased, but the granules remain randomly distributed. Erythroblasts showing this phenomenon are described as abnormal sideroblasts. Most of the siderotic granules of such erythroblasts also consist of siderosomes (albeit abnormally large ones) but a few consist of iron-laden mitochondria. In the sideroblastic anemias there is an increase in both the coarseness and number (per erythroblast) of siderotic granules but additionally the majority of the granules tend to be distributed in either a partial or complete perinuclear ring; cells showing such perinuclear rings are termed ring sideroblasts; ring sideroblasts have been defined as erythroblasts containing at least five perinuclear granules.21 Ultrastructural studies indicate that most of the siderotic granules within a ring sideroblast consist of iron-laden mitochondria. The sideroblastic anemias are discussed in Chapter 14.
Infections and the bone marrow
In microbial infections associated with monocytosis, the marrow may show an increased proportion of cells of the mononuclear phagocyte system. In occasional patients with some types of infection the macrophages show prominent hemophagocytosis (see Box 5.4 and Fig. 5.3). Certain infections are characterized by granulomatous lesions in the BM (see below).
Microscopic examination of BM smears and sections after staining with specific stains may be useful in diagnosing certain mycobacterial and fungal infections, especially but not exclusively in patients with granulomas. Fungi are usually seen in the marrow in immunocompromised patients and may be found extracellularly and within macrophages. In Whipple’s disease, Tropheryma whipplei (previously Tropheryma whippelii), which stain violet with Romanowsky stains and black with methenamine silver, may be seen within BM macrophages.22
In certain bacterial and fungal infections, the organisms can be cultured from marrow. In histoplasmosis, cryptococcosis, candidiasis, blastomycosis and coccidioidomycosis organisms may be demonstrated both by microscopy of smears or histologic sections and by culture.23
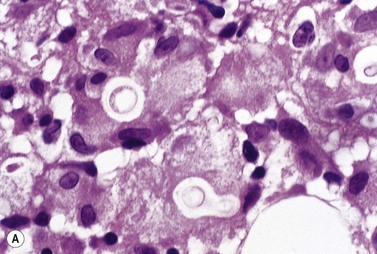
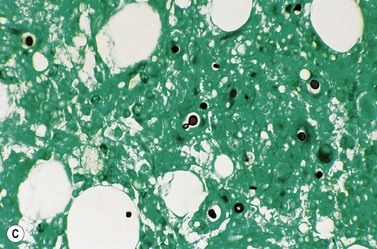
Chronic Plasmodium falciparum malaria in young children is associated with a marked increase of dyserythropoiesis and ineffective erythropoiesis.24 Dyserythropoiesis is also seen in severe acute falciparum malaria, especially cerebral malaria. BM aspiration is not usually performed for the diagnosis of malaria and there is limited information on its value. A BM aspirate is helpful in making a retrospective diagnosis of Plasmodium falciparum malaria in an undiagnosed but treated patient; in this situation, malarial pigment will be present in BM macrophages (Fig. 5.5). In post-mortem examinations of patients who have had recurrent attacks of falciparum malaria, malarial pigment is found in the marrow, spleen and liver.
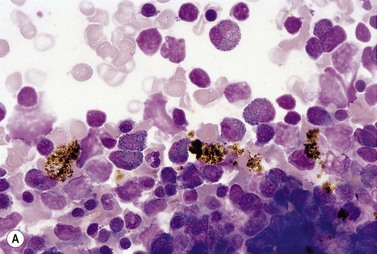
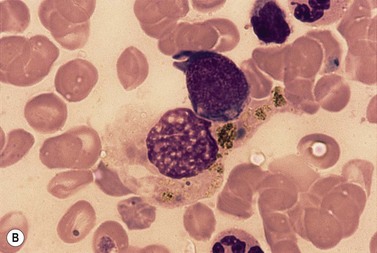
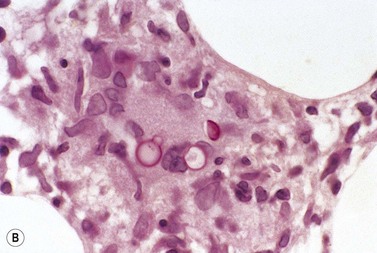
Leishmaniasis may be diagnosed by studies of BM smears or sections although culture of the BM aspirate is more sensitive (Fig. 5.6); occasionally, the organisms may also be seen in PB monocytes. As in the case of chronic falciparum malaria, the erythroblasts of patients with leishmaniasis show nonspecific morphologic abnormalities indicative of dyserythropoiesis25 (Fig. 5.7).
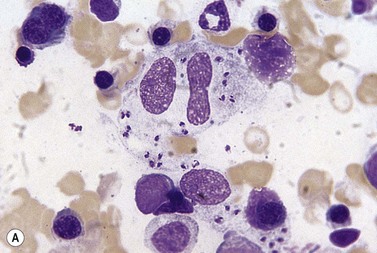
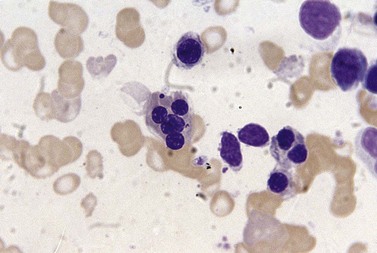
In viral infections, the BM contains an increased number of normal or atypical lymphocytes. Especially in herpesvirus infections, macrophages may show hemophagocytosis. In cytomegalovirus (CMV) infection the marrow may contain the typical giant cells with eosinophilic intranuclear inclusions.26 Infection by parvovirus B19 causes transient red cell aplasia and, consequently, severe anemia occurs in patients with an underlying hemolytic state (e.g. sickle cell anemia, thalassemia intermedia, hereditary spherocytosis and pyruvate kinase deficiency).27 Chronic pure red cell aplasia with resultant anemia can occur in parvovirus B19-infected patients with congenital or acquired immune deficiency, who are unable to mount an immune response that is adequate to clear the virus. In some cases of the congenital rubella syndrome, thrombocytopenia is at least partly due to reduced numbers of megakaryocytes in the marrow. In other cases the thrombocytopenia is mainly caused by a decreased platelet life span and is associated with normal or increased numbers of BM megakaryocytes.
HIV infection
A variety of hematologic abnormalities may be found in HIV infection,28 especially at the later stages of infection. These include cytopenias, dysplastic changes affecting all hemopoietic cell lineages and changes in the BM resulting from opportunistic infections.
Changes in the peripheral blood
During the phase of clinically latent infection that follows the primary infection there is a slowly progressive CD4+ lymphopenia. The total lymphocyte count may be initially normal because of a CD8+ lymphocytosis. The prevalence of various cytopenias increases with progression of infection (i.e. increasing viral load). In a study of over 32 000 HIV-infected patients in the USA, an Hb <10 g/dl was found in 37% of patients with AIDS and 12% of patients without AIDS but with a CD4+ lymphocyte count <0.2 × 109/l.29 When isolated thrombocytopenia occurs during the clinically latent phase, platelet-associated immune-complexes are frequently present and the antibody in some such complexes may have specificity against HIV antigens. Rarely, an immune thrombocytopenia and autoimmune hemolysis may develop as may a microangiopathic hemolytic anemia and thrombocytopenia resembling that seen in the hemolytic uremic syndrome or thrombotic thrombocytopenic purpura.30
The blood film in AIDS may show various changes. The red cells are normocytic and normochromic or macrocytic; macrocytosis may occur even in the absence of zidovudine therapy. There may be reticulocytopenia, monocytopenia and atypical lymphocytes with lobulated nuclei. Some neutrophils may show various dysplastic changes including Howell–Jolly-body-like nuclear fragments, hypogranularity, the acquired Pelger–Huët anomaly, a high nucleocytoplasmic ratio, bizarre nuclear shapes and binuclearity.5,31 There may also occasionally be circulating giant metamyelocytes and giant neutrophils. Neutrophils may also show changes related to infection, such as toxic granulation, vacuolation, Döhle bodies and a ‘left-shift’.
Changes in the bone marrow
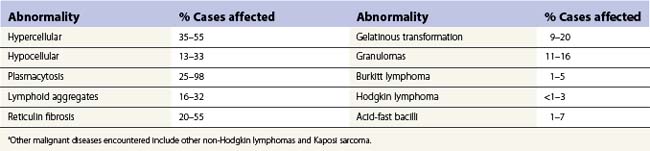
The various abnormalities found in trephine biopsy sections from patients with AIDS and their prevalence are shown in Table 5.3. The BM is hypercellular at the early stages and hypocellular in advanced AIDS. Polymorphous lymphoid aggregates are seen in the absence of lymphoma or opportunistic infections and appear to be at least partly a manifestation of the HIV infection itself; they are aggravated by opportunistic infections. The reticulin fibrosis is usually mild or moderate and leads to the marrow sinusoids being held open in paraffin-embedded trephine biopsy sections.
Opportunistic infections in AIDS32–35 may be due to:
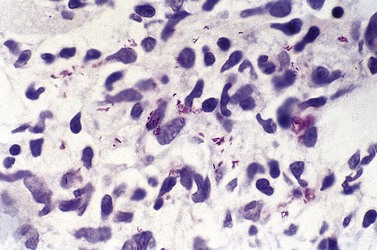
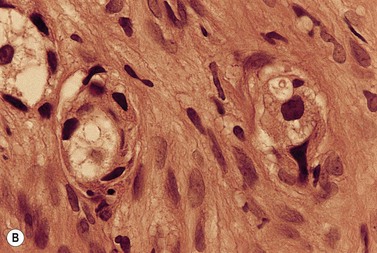
In keeping with the virulence of Mycobacterium tuberculosis, this organism becomes disseminated in patients whose CD4+ lymphocyte count is not severely reduced and, consequently, well-formed granulomas may be found in the marrow. However, less-virulent opportunistic organisms (e.g. atypical mycobacteria and fungi) generally provoke poorly formed granulomas (Fig. 5.8). BM trephine biopsies are more useful than aspirates in detecting such infections. Sections should be stained by the Ziehl–Neelsen stain to look for mycobacteria (Fig. 5.9), by the PAS stain or Grocott’s methenamine silver stain to look for fungi such as Cryptococcus neoformans (Fig. 5.10), Histoplasma capsulatum and Candida albicans and the Giemsa stain, to look for protozoa such as Toxoplasma gondii and Leishmania donovani. These organisms may be found within poorly formed granulomas or diffusely in the marrow, within macrophages.
A number of different mechanisms interact to cause the hematologic abnormalities in HIV infection5,36–38 and the relative importance of the different mechanisms may vary from patient to patient. The possible mechanisms are:
Bone marrow granulomas
A granuloma is a compact collection of mature cells of the mononuclear phagocyte system. The types of monocyte-derived cells that may be found in granulomas include epithelioid cells, macrophages, Langhans-type giant cells (containing numerous small nuclei situated around the periphery of the cell) and foreign body-type giant cells (containing a smaller number of nuclei scattered throughout the cell). Granulomas may also contain lymphocytes, plasma cells, neutrophils, eosinophils, fibroblasts and necrotic or caseating areas. BM granulomas are seen in many conditions characterized by the formation of granulomas in other tissues39 (Box 5.5). Immunodeficient patients may fail to generate granulomas in response to organisms that evoke granuloma formation in immunocompetent subjects. This is because the development of granulomas requires normal lymphocyte functions; in experimental animals granuloma formation is suppressed by neonatal thymectomy and antilymphocyte serum.
Box 5.5 Causes of bone marrow granulomas
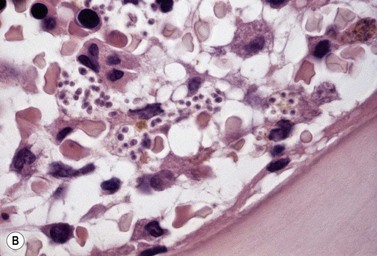
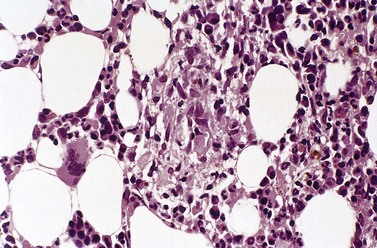
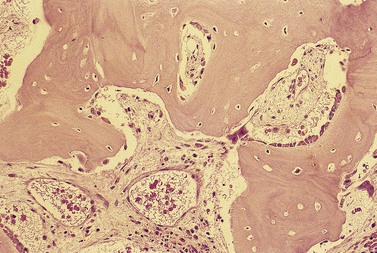
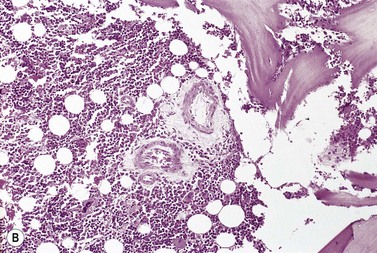
Epithelioid cells may rarely be seen in Romanowsky-stained BM smears suggesting the possibility of granuloma formation. In BM smears, epithelioid cells tend to occur in groups and have abundant blue-gray to dark blue cytoplasm and round, oval or reniform nuclei. However, BM granulomas are best detected in histologic sections of trephine biopsy specimens or clot sections of aspirated marrow (Fig. 5.11).
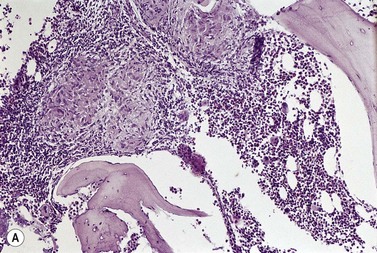
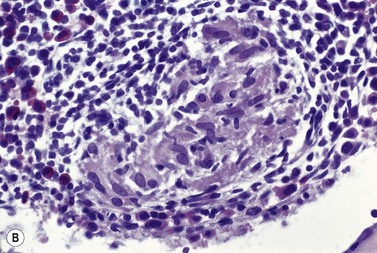
Fig. 5.11 (A, B) Trephine biopsy sections of bone marrow showing sarcoid granulomas. H&E. (A) × 94; (B) × 375.
BM granulomas are found in 15–40% of patients with miliary tuberculosis, including some patients with normal chest radiology. In tuberculous granulomas, Langhans-type giant cells are usually found, caseation is present in about 50% of patients and acid-fast bacilli are usually absent or, when present, found in small numbers. In disseminated Mycobacterium avium intracellulare infection, granulomas of variable size and appearance are seen in about half the cases. Giant cells and necrosis are uncommon and macrophages are packed with organisms and may appear foamy. The organisms are best demonstrated using the Ziehl–Neelsen stain; they are acid-fast but longer, more curved and more coarsely beaded than Mycobacterium tuberculosis and unlike the latter are PAS-positive. Mycobacterium tuberculosis and atypical mycobacteria may be cultured from the BM sometimes even in patients in whom the Ziehl–Neelsen stain has not revealed organisms. Patients with hairy cell leukemia and those with AIDS may have absent or impaired granuloma formation with mycobacterial infection of the BM.40
Granulomas with large foamy macrophages may be found in typhoid fever and the bacilli may be seen within macrophages; the organisms can usually be cultured from the BM. In leprosy, the Mycobacterium leprae may appear as bacilliform ‘ghosts’ within macrophages in Romanowsky-stained marrow smears, and the acid-fast organisms can be demonstrated by the Fite stain. The ghosts of mycobacteria have also been observed free and within macrophages in atypical mycobacterial infection in AIDS.41 BM granulomas are frequently found in brucellosis and these are smaller and less distinct than those in tuberculosis and sarcoidosis. Large granulomas, often with Langhans-type giant cells and scanty organisms and sometimes with caseation, may occur in patients with histoplasmosis and reasonably normal immunity. By contrast, patients with immune suppression usually have a marked and diffuse increase in macrophages and BM necrosis. In both types of patient, the yeast form of the organism is found within macrophages. The yeast forms appear blue in Romanowsky-stained films and are 2–5 µm in diameter. In histologic sections, they may be seen after staining with H&E but are best demonstrated when stained by the PAS reaction and by Gömöri’s methenamine silver stain. The fungi can be cultured from BM aspirates in 60–75% of patients with disseminated infection. Granulomas may also be seen in the marrow in disseminated Cryptococcus neoformans infection. The organisms (yeasts) are 5–10 µm in diameter, have a thick capsule that appears as a clear halo in sections stained with H&E and they show unequal budding (see Fig. 5.10). The capsule is PAS-positive and also stains with mucicarmine (red) and Alcian blue.
BM granulomas are found in some cases of sarcoidosis (Fig. 5.11) and in one-third of cases the granulomas contain Langhans-type giant cells.42 It is not always possible to distinguish between granulomas in sarcoidosis and tuberculosis or other microbial infections on histologic features alone. Caseation is characteristic of tuberculosis but is neither invariably present nor restricted to it. Furthermore, non-caseating granulomas with no detectable acid-fast bacilli may sometimes be due to tuberculosis rather than sarcoidosis. Both tuberculous and sarcoid granulomas may have associated eosinophils and lymphocytes and these features are not helpful in making a distinction between them. Although sarcoid granulomas do not caseate they may show eosinophilic coagulative necrosis and, in the healing stage, hyaline fibrosis. They may also show asteroid bodies.43
Lesions seen in the BM in systemic mastocytosis and in angioimmunoblastic T-cell lymphoma need to be distinguished from granulomas. In systemic mastocytosis the lesions are composed of mast cells, eosinophils, lymphocytes and collagen fibers (see Chapter 26) and in angioimmunoblastic T-cell lymphoma of immunoblasts, plasma cells, lymphocytes, histiocytes, eosinophils, arborizing capillaries and reticulin fibers.
Metastatic tumors in bone marrow
Patients with metastatic tumor cells in the BM usually have a normochromic normocytic anemia and, less commonly, a hypochromic microcytic anemia, thrombocytopenia or neutropenia. In less than half the patients with BM metastases, the blood film contains some erythroblasts and neutrophil precursors (leukoerythroblastic anemia)44 and the presence of such cells reflects the extent of myelofibrosis. Circulating malignant cells may occasionally be seen, especially in children with small cell tumors and, more rarely, in adults with carcinoma. The PB may also show abnormalities not directly related to the BM infiltration such as the anemia of chronic disease, iron deficiency anemia, red cell fragmentation, neutrophilia, thrombocytosis, eosinophilia and features of hyposplenism (as a result of splenic infiltration).
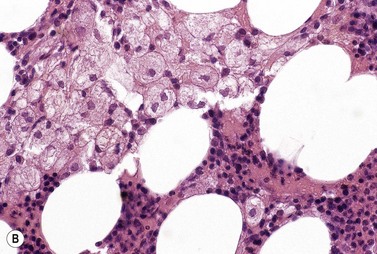
A BM trephine biopsy is generally more useful in detecting metastatic tumor cells in the marrow than a section of aspirated BM and the latter is more useful than a smear.44,45 However, the three procedures should be regarded as complementary. Another advantage of histologic sections (either particle sections or of trephine biopsy specimens) over smears is that they can better reveal intercellular organization such as the formation of rosettes in neuroblastoma or acini in adenocarcinoma (Fig. 5.12A) and can demonstrate fibrosis and osteoblastic reactions that may be associated with tumor metastases (Fig. 5.12B). BM fibrosis in response to tumor metastases is particularly marked in carcinomas of the breast, stomach and prostate and correlates with the occurrence of a leukoerythroblastic anemia. BM fibrosis can make aspiration impossible or lead to only peripheral blood from BM sinusoids being aspirated. In this circumstance, detection of metastases is dependent on the trephine biopsy.
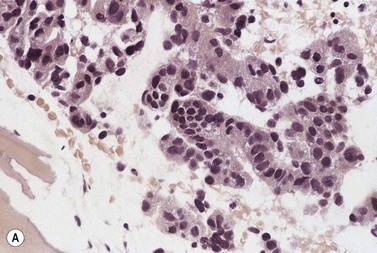
In adults, the tumors that most commonly metastasize to the marrow are carcinomas of the prostate, breast, gastrointestinal tract, lung, thyroid and kidney44 and in children they are neuroblastoma, rhabdomyosarcoma, Ewing tumor and retinoblastoma.46
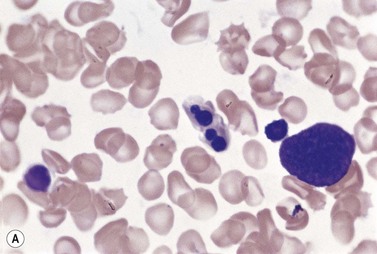
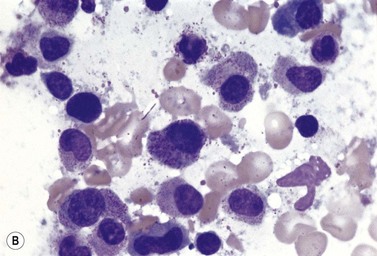
Metastatic carcinoma cells can usually be readily recognized in BM smears because they are larger than all hemopoietic cells other than megakaryocytes and tend to occur in clumps (Fig. 5.13A). Generally, carcinoma cells are markedly pleomorphic and have a moderate quantity of slightly or moderately basophilic cytoplasm, sometimes with vacuoles. Some cells are multinucleate and there may be a high mitotic index. Usually, it is not possible to identify the primary tumor on the basis of the morphologic features of the metastatic cells in BM smears. However, some melanomas can be identified by the presence of intracytoplasmic melanin pigment (which stains positively with the Masson–Fontana or Schmorl stains for melanin) and renal carcinoma may be suspected if the cells have abundant foamy cytoplasm and small nuclei (‘clear cell’ morphology). Mucin-secreting adenocarcinoma cells possess foamy or vacuolated cytoplasm. The mucin may push the nucleus to the periphery, thus giving the cell a ‘signet-ring’ appearance. Stains for mucin (combined diastase-treated PAS/Alcian blue stain) can be used to identify mucin-secreting carcinoma cells. Patients with metastatic carcinoma frequently have increased numbers of macrophages and plasma cells in their BM. When there is associated osteosclerosis, the BM smears may also contain increased numbers of osteoblasts and osteoclasts. Sometimes, there is necrosis of the infiltrated BM and necrotic material may be seen in both smears and histologic sections.
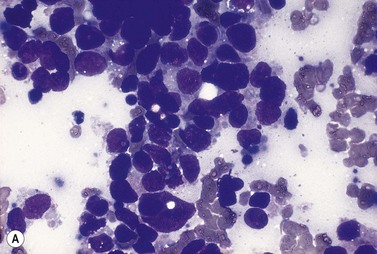
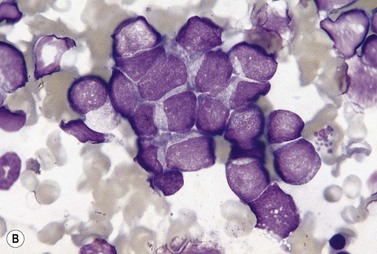
Fig. 5.13 (A, B) Clumps of metastatic tumor cells in bone marrow smears. (A) Carcinoma of the bronchus. (B) Neuroblastoma. May–Grünwald–Giemsa stain.
Information on the nature of malignant cells may be obtained by immunocytochemistry and immunohistochemistry. Monoclonal antibodies against antigens such as human milk fat globulin, epithelial membrane antigen (an antigen found in carcinoma of the breast but also in other adenocarcinomas) or cytokeratin have proved most useful in detecting carcinoma cells either in histologic sections of biopsy specimens or in BM smears.47,48 Antibody against S100 protein reacts with most malignant melanomas, including amelanotic melanomas. The detection of metastatic prostatic carcinoma cells requires the use of antibodies against both prostate-specific antigen and prostatic acid phosphatase.
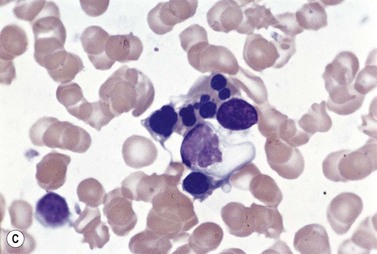
Metastatic tumor cells of neuroblastoma (Fig. 5.13B), medulloblastoma, retinoblastoma, rhabdomyosarcoma and Ewing sarcoma are small and round with relatively little cytoplasm and may be difficult to distinguish from the blast cells of ALL and lymphoblastic lymphoma. In some cases of neuroblastoma, medulloblastoma and rhabdomyosarcoma, the small tumor cells are present in the circulation.49 Metastatic small round cell tumors can sometimes be distinguished from acute leukemia on morphologic criteria alone. However, in other cases this distinction requires cytochemical, ultrastructural and immunochemical studies. In neuroblastoma, both BM smears and sections may show characteristic rosettes of tumor cells near fibrillar extracellular material that stains blue-gray by Romanowsky methods and eosinophilic by H&E.50 However, these features are often absent and the diagnosis may then be made by immunohistochemistry, flow cytometry and/or PCR.51 Rhabdomyosarcoma cells are usually heavily vacuolated, reflecting glycogen in the cytoplasm, and can be identified by electron microscopy on the basis of the presence of cross-striated myofibrils and by the immunohistochemical demonstration of myosin, desmin or myoglobin. In Ewing sarcoma histologic sections may show small numbers of ‘pseudorosettes’ around blood vessels. In medulloblastoma, tumor cells may display hemophagocytosis and autophagocytosis. The malignant cells of ALL give positive reactions with monoclonal antibodies against lymphoid-associated antigens (see Chapter 19) whereas the malignant cells from neuroblastoma, rhabdomyosarcoma and Ewing sarcoma give negative reactions. The blasts of ALL also usually show terminal deoxynucleotidyl transferase (TdT) expression while neuroblastoma and retinoblastoma cells do not.
Bone marrow fibrosis
An increase in the reticulin or reticulin and collagen in the BM is referred to as bone marrow fibrosis.52,53 Reticulin fibers are collagen precursors and are produced by fibroblasts. A scheme for grading bone marrow fibrosis is discussed in Chapter 3. Patients with advanced myelofibrosis may also have osteosclerosis.
Myelofibrosis may be generalized or focal. Generalized myelofibrosis occurs as in so-called ‘primary’ myelofibrosis and also in association with a number of diseases with widely differing etiology (Box 5.6).
Box 5.6 Conditions associated with myelofibrosis
| Generalized: | Focal or localized: |
|---|---|
a There may also be osteosclerosis.
b As this condition is defined in the 2008 WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, reticulin is no more than minimally increased at diagnosis and fibrosis develops rarely.
Storage cells in lysosomal storage diseases
Sphingolipidoses
Gaucher disease
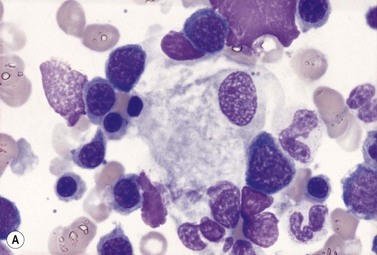
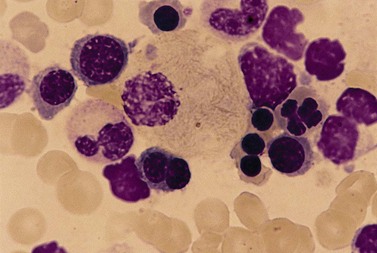
In Gaucher disease, which is usually inherited as an autosomal recessive character, there is defective production of the lysosomal enzyme glucocerebroside β-glucosidase leading to the intracellular accumulation of abnormal quantities of glucocerebrosides.54 Typical storage cells are seen in the BM (Fig. 5.14A) as well as in other tissues; these cells are macrophages distended by glucocerebrosides. Gaucher’s cells are large, round or oval, and have pale blue cytoplasm with a wrinkled appearance due to the presence of many fibrillar structures (Romanowsky stain). The cytoplasm is Sudan Black B- and PAS-positive. Gaucher cells also stain positively for nonspecific esterase and tartrate-resistant acid phosphatase. Stains for iron give weak positive reactions. In histologic sections, Gaucher cells are often found in clumps or sheets and their abundant cytoplasm has a crumpled appearance (Fig. 5.14B). The affected marrow may show an increase in reticulin and collagen. Electron microscopy reveals that the cytoplasm is packed with large elongated sacs containing characteristic tubes, 30–40 nm wide, each of which is made up of spirally arranged fibrils. Occasionally, particularly after splenectomy, Gaucher cells are seen in the PB. Cells resembling Gaucher cells under the light microscope are seen in the marrow in a variety of hematologic disorders including CML, acute leukemia, thalassemia major, the congenital dyserythropoietic anemias (Fig. 5.15), sickle cell anemia, Hodgkin lymphoma, non-Hodgkin lymphoma, multiple myeloma and after many platelet transfusions. However, such cells (pseudo-Gaucher cells) are ultrastructurally and by immunohistochemistry different from Gaucher cells.55 Pseudo-Gaucher cells result from an increased phagocytic load (e.g. of abnormal red cells or erythroblasts or leukemic cells) on the macrophages resulting in the production of lipid in excess of that which can be metabolized, with consequent intracellular accumulation. Morphologically somewhat similar cells can be seen in Mycobacterium avium intracellulare infection but in this instance the abnormal staining characteristics of the macrophages (‘pseudo-pseudo-Gaucher’s cells’) result from the presence of very large numbers of mycobacteria within the macrophages.
Sea-blue histiocyte syndrome
Sea-blue histiocytosis is an inherited group of disorders in which large macrophages containing coarse granules that stain sea-blue or blue-green (Romanowsky stain) are seen in the spleen, liver, BM and other organs.56 The characteristic color of these granules after Romanowsky staining is attributed to the presence of ceroid; when unstained the granules are yellow or brown. The granules stain with oil red O and Sudan Black B; as the pigment ages it develops autofluorescence and, subsequently, PAS-positivity followed by acid-fast positivity. Ceroid is histochemically similar to lipofuscin. Ultrastructural studies have revealed that the granules are pleomorphic and that some of them contain concentric arrangements of membrane (myelin figures). Sea-blue histiocytes have also been observed in the BM in conditions other than the inherited sea-blue histiocyte syndrome (Fig. 5.16). These include acquired disorders such as CML, polycythemia vera, multiple myeloma, lymphoproliferative disorders (including Hodgkin lymphoma), autoimmune thrombocytopenic purpura and rheumatoid arthritis as well as various inherited disorders such as sickle cell anemia, thalassemia, chronic granulomatous disease, Niemann–Pick disease, Tay–Sachs disease, Fabry disease, Hurler disease, Wolman disease, lecithin-cholesterol acyltransferase deficiency, type V hyperlipidemia, and Hermansky–Pudlak syndrome.
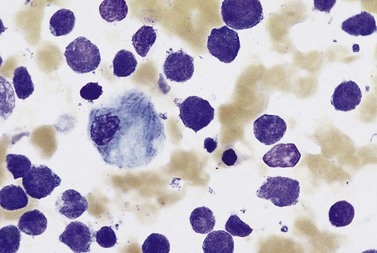
Niemann–Pick disease
Niemann–Pick disease is usually inherited as an autosomal recessive condition and is often due to a deficiency of sphingomyelinase.57 This leads to the accumulation of excess sphingomyelin within the macrophages of the BM and other organs. The cytoplasm of affected macrophages appears foamy, being filled with rounded lipid-containing inclusions (Fig. 5.17). The inclusions stain faint blue with Romanowsky stains and variably with the PAS reaction and lipid stains. Some sea-blue histiocytes are present. The cytoplasm of the foamy cells appears pale-yellow to yellow-brown in sections stained with H&E. The inclusions within these cells vary in their ultrastructure but often show myelin figures towards their periphery. Blood monocytes and lymphocytes in cases of Niemann–Pick disease have lipid-containing inclusions similar to those in macrophages. Foamy macrophages are not specific for Niemann–Pick disease. They are also seen in some other storage diseases and in hyperlipidemias. They may also be found in certain infections, fat necrosis, BM infarction and in Langerhans cell histiocytoses (eosinophilic granuloma, Letterer–Siwe disease and Hand–Schüller–Christian disease).
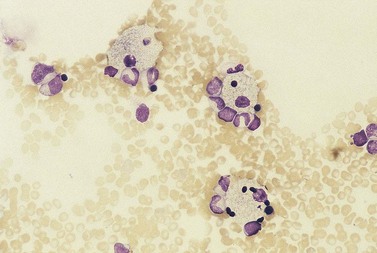
Fabry disease
Fabry disease is an X-linked recessive disorder due to deficiency of α-galactosidase.58 This leads to the accumulation of globotriaosylceramide and other neutral glycolipids in BM macrophages. The cytoplasm of the abnormal macrophages appears foamy, being crowded with small globular structures staining pale blue with Romanowsky stains and strongly with PAS, Sudan Black B, Luxol fast blue, oil red O, and stains for acid phosphatase. In sections stained with H&E, the cytoplasmic globules appear pink.
Mucopolysaccharidoses
In Hurler syndrome and other mucopolysaccharidoses, there is a deficiency of enzymes involved in the metabolism of the carbohydrate component of glycoproteins leading to an accumulation within lysosomes of mucopolysaccharides and glycolipids.59 The abnormal lysosomes inside BM macrophages, plasma cells and lymphocytes may appear as metachromatic granules; the granules stain lilac or purple with Romanowsky stains (Alder–Reilly bodies). The cytoplasm of macrophages may be packed with basophilic inclusions of varying size which are surrounded by a clear halo. In histologic sections, the macrophage cytoplasm appears foamy due to extraction of the mucopolysaccharides.
Bone marrow necrosis
In some pathologic conditions, there is necrosis of both the hemopoietic and the non-hemopoietic cells of the red marrow and there may also be necrosis of adjacent bone.60 BM necrosis is a common finding at autopsy but is less often diagnosed during life. It may be widespread and can recur. The necrosis may be caused by interference with the blood supply or a failure to meet increased metabolic demands from a hypercellular marrow, or both, and may sometimes be related to high concentrations of tumor necrosis factor in the blood.61
The conditions associated with BM necrosis are given in Box 5.7. In sickle cell anemia, Hb SC disease (heterozygous state for both hemoglobin S and hemoglobin C), hemoglobin S/β+-thalassemia and, rarely, heterozygosity for both hemoglobins S and E (plus parvovirus infection), necrosis may occur due to occlusion of the BM microvasculature by sickled cells. In sickle cell disease pregnancy increases the degree of BM hyperplasia and, thereby, further increases the likelihood of BM infarction and also of death from the embolism of necrotic BM to the lungs.
Box 5.7 Conditions associated with bone marrow necrosis
In mucormycosis, the mucorales invade vessel walls and cause thrombosis and infarction.62 In caisson disease (acute decompression illness) and disseminated intravascular coagulation (DIC) the microvasculature is occluded by bubbles of nitrogen and thrombi, respectively. In acute leukemia, carcinomatous infiltration of the BM and megaloblastic anemia plus infection, the increased metabolic needs of a hyperplastic BM may play a role in the pathogenesis of the BM necrosis. In addition, in leukemia and carcinomatosis, the malignant cells may compress vessels, invade vessel walls and occlude their lumina or cause thrombosis.
BM necrosis is accompanied by bone pain and fever. Extensive necrosis causes a leukoerythroblastic blood picture and pancytopenia. The macroscopic appearance of the aspirated BM fragments may be abnormal with the fragments appearing opaque and white/pale yellow, or plum-colored. In a Romanowsky-stained smear of necrotic BM, little cellular detail is discernible (Fig. 5.18A); the blurred outlines of cells are seen in a background of amorphous pink material. In some patients with BM necrosis secondary to metastatic carcinoma, an aspirate may show a mixture of intact tumor cells and necrotic tumor and hemopoietic cells. If BM aspirated from one site shows only necrosis and leukemia or metastatic carcinoma is suspected, a second aspiration from another site may be useful in demonstrating infiltration by malignant cells. The appearance of the trephine biopsy depends on the time after infarction as well as the underlying disorder. Initially, the cells have indistinct margins, granular cytoplasm and pyknotic nuclei. Later, cell outlines are unrecognizable and there is karyorrhexis. Necrosis of the adjacent bone is common, with loss of osteoclasts, osteoblasts and osteocytes (Fig. 5.18B). Recovery is accompanied by repopulation with hemopoietic tissue but small fibrotic scars or, rarely, large areas of fibrous tissue may develop (Fig. 5.19); new bone is laid down on the spicules of dead bone. Radiologic examination shows no abnormality initially and may show sclerotic changes after some time. However, in the acute phase, bone scanning with 99mTc-sulfur-colloid shows lack of reticuloendothelial function in the infarcted area. The scan gradually returns to normal. Extramedullary hemopoiesis may develop in patients with extensive BM necrosis.
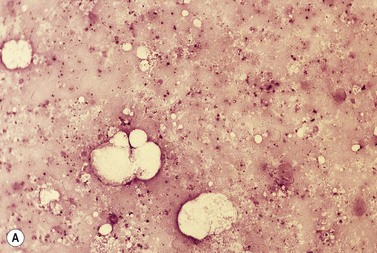
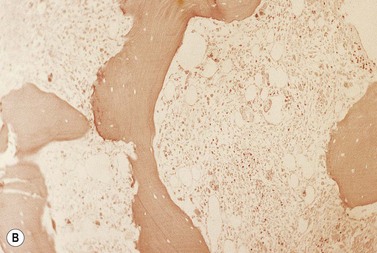
Gelatinous transformation
The BM of most patients with severe malnutrition contains a gelatinous material which consists of acid mucopolysaccharide. This may be seen in severe anorexia nervosa, some patients with cachexia secondary to AIDS and chronic disorders (e.g. tuberculosis, carcinoma) and occasional cases of leukemia post-chemotherapy and of systemic lupus erythematosus (SLE). Gelatinous transformation has also been reported in other conditions including severe hypothyroidism, intestinal lymphangiectasia (Waldman disease) and leishmaniasis.63 The gelatinous material is amorphous, granular or fibrillar, stains pink-purple with Romanowsky stains or H&E (Fig. 5.20A,B) and stains positively with Alcian blue (particularly at a high pH) and the PAS stain. The hemopoietic cells are reduced in number and embedded within the gelatinous material and there is an absence or marked reduction of fat cells. BM fragments showing gelatinous transformation do not smear properly. A deficiency of carbohydrates and calories may underlie the gelatinous transformation and the excessive accumulation of acid mucopolysaccharide may serve to fill the BM space normally occupied by fat cells. Interestingly, young children with protein-energy malnutrition do not show gelatinous transformation.
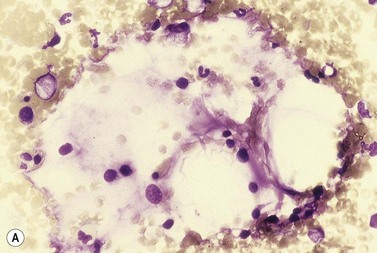
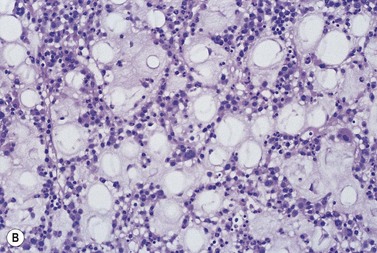
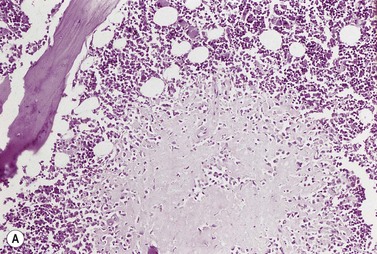
Amyloidosis
Amyloid deposits may be seen in the BM both in smears of aspirated material, infrequently, and in histologic sections. In Romanowsky-stained smears amyloid appears pink to purple, waxy to transparent, and has been described as resembling a cumulus cloud.64 In histologic BM sections, amyloid has the same appearance and staining characteristics as in other tissues (Fig. 5.21). It is seen most often in vessel walls but sometimes in the interstitium. BM amyloid is observed most frequently in light-chain-associated amyloidosis (sometimes referred to as primary amyloidosis) but is also observed in secondary amyloidosis, for example in familial Mediterranean fever and secondary to chronic inflammatory conditions such as rheumatoid arthritis. In light-chain-associated amyloidosis the BM may show, in addition, a slight to moderate increase in monotypic plasma cells or overt multiple myeloma.
Vascular and embolic lesions
The BM may show arteritis and arteriolitis in any form of generalized arteritis, including giant cell arteritis.65 Hypersensitivity reactions to drugs may cause granulomatous vasculitis and in polyarteritis nodosa there may be vasculitic lesions with fibrinoid necrosis.
Trephine biopsy specimens may reveal arteriosclerotic and thromboembolic lesions. Emboli that are acellular or composed of hyaline material or cholesterol crystals may be derived from atheromatous plaques and cause a multisystem disease characterized by anemia, leukocytosis, eosinophilia and elevated erythrocyte sedimentation rate.66 BM emboli are found in 20% of these cases at autopsy and may also be seen in biopsy material. Vessels are partly or completely occluded by acellular material with cholesterol clefts and there may be intimal hyperplasia and infiltration of vessel walls initially with granulocytes and subsequently with mononuclear cells and giant cells.
Aluminum deposition
In patients on hemodialysis and other patients with chronic renal failure aluminum may be deposited both in bone (at the osteoid/mineralized tissue junction) and in BM cells as coarse granules. The aluminum is derived from oral intake and dialysis fluids and may cause dementia.67 Aluminum may be demonstrated by a specific stain (Irwin stain on an undecalcified trephine biopsy). The BM cell that contains the aluminum may be the macrophage.
Post-mortem bone marrow changes
A variety of morphologic changes occur fairly rapidly in marrow cells post mortem68 (Fig. 5.22). This applies particularly to the neutrophil metamyelocytes and granulocytes which are rich in hydrolytic enzymes. Post-mortem BM aspirations and tissue sampling with a trephine needle should therefore be performed as soon after death as possible and preferably not more than 3 h after death. Vacuolation of the cytoplasm of granulocytes may be first seen in BM smears aspirated 1.5–7 h after death and the vacuolation increases progressively thereafter. Swelling of the nuclei of neutrophil metamyelocytes and granulocytes may be detected as early as 2 h after death; the swelling causes the affected metamyelocytes to look like myelocytes. By 8–12 h post mortem, most of the nuclei of the neutrophil metamyelocytes and granulocytes appear rounded, with a loosening of the nuclear chromatin and the appearance of structures resembling small nucleoli. The nuclear membrane is ruptured and many of the cells have indistinct cell membranes. The neutrophil myelocytes begin to lyse after 7–12 h. Karyorrhexis and budding, lobulation or segmentation of erythroblast nuclei may begin as an agonal event or during the first 2 h after death; in occasional cases, over 20% of erythroblasts show nuclear abnormalities 25 min following death, but in others marked changes do not develop in less than 3 h. Macrophages are greatly increased in number in aspirates taken within hours of death, probably mainly because they are released into the aspirate more readily than from living marrow. The post-mortem appearances of marrow aspirated 10–20 h after death differ markedly from those obtained during life and are sufficiently confusing to have led to the incorrect diagnosis of leukemia or malignant infiltration.
1 Lee SH, Erber WN, Porwit A, et al. ICSH guidelines for the standardization of bone marrow specimens and reports. Int J Lab Hematol. 2008;30:349-364.
2 Bain BJ. The bone marrow aspirate of healthy subjects. Br J Haematol. 1996;94:206-209.
3 Wickramasinghe SN, Lee MJ, Furukawa T, et al. Composition of the intra-erythroblastic precipitates in thalassaemia and congenital dyserythropoietic anaemia (CDA): identification of a new type of CDA with intra-erythroblastic precipitates not reacting with monoclonal antibodies to alpha- and beta-globin chains. Br J Haematol. 1996;93:576-585.
4 Lehane DE. Vacuolated erythroblasts in hyperosmolar coma. Arch Intern Med. 1974;134:763-765.
5 Bain BJ. The haematological features of HIV infection. Br J Haematol. 1997;99:1-8.
6 Niebrugge DJ, Benjamin DR, Christie D, Scott CR. Hereditary transcobalamin II deficiency presenting as red cell hypoplasia. J Pediatr. 1982;101:732-735.
7 Oshima Y, Hirota H, Nagai H, et al. Images in cardiovascular medicine. Specific cardiomyopathy caused by multisystemic lipid storage in Jordan’s anomaly. Circulation. 2002;106:280-281.
8 Thiele J, Zirbes TK, Kvasnicka HM, Fischer R. Focal lymphoid aggregates (nodules) in bone marrow biopsies: differentiation between benign hyperplasia and malignant lymphoma – a practical guideline. J Clin Pathol. 1999;52:294-300.
9 Ioannou MG, Stathakis E, Lazaris AC, et al. Immunohistochemical evaluation of 95 bone marrow reactive plasmacytoses. Pathol Oncol Res. 2009;15:25-29.
10 Sohn BS, Kim T, Kim JE, et al. A case of histiocytic sarcoma presenting with primary bone marrow involvement. J Korean Med Sci. 2010;25:313-316.
11 Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
12 Lampert IA, Catovsky D, Bergier N. Malignant histiocytosis: a clinico-pathological study of 12 cases. Br J Haematol. 1978;40:65-77.
13 Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002;41:1-29.
14 Shimada A, Kato M, Tamura K, et al. Hemophagocytic lymphohistiocytosis associated with uncontrolled inflammatory cytokinemia and chemokinemia was caused by systemic anaplastic large cell lymphoma: a case report and review of the literature. J Pediatr Hematol Oncol. 2008;30:785-787.
15 Chuang HC, Lay JD, Hsieh WC, Su IJ. Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein–Barr virus-associated T-cell lymphoma: nuclear factor-kappa B pathway as a potential therapeutic target. Cancer Sci. 2007;98:1281-1287.
16 Koh MJ, Sadarangani SP, Chan YC, et al. Aggressive subcutaneous panniculitis-like T-cell lymphoma with hemophagocytosis in two children (subcutaneous panniculitis-like T-cell lymphoma). J Am Acad Dermatol. 2009;61:875-881.
17 Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
18 Tiab M, Mechinaud F, Harousseau JL. Haemophagocytic syndrome associated with infections. Baillière’s Best Pract Res Clin Haematol. 2000;13:163-178.
19 Hughes DA, Stuart-Smith SE, Bain BJ. How should stainable iron in bone marrow films be assessed? J Clin Pathol. 2004;57:1038-1040.
20 Kaplan E, Zuelzer WW, Mouriquand C. Sideroblasts: a study of stainable nonhemoglobin iron in marrow normoblasts. Blood. 1954;9:203-213.
21 Mufti GJ, Bennett JM, Goasguen J, et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93:1712-1717.
22 Krober SM, Kaiserling E, Horny HP, Weber A. Primary diagnosis of Whipple’s disease in bone marrow. Hum Pathol. 2004;35:522-525.
23 Pasternak J, Bolivar R. Histoplasmosis in acquired immunodeficiency syndrome (AIDS): diagnosis by bone marrow examination. Arch Intern Med. 1983;143:2024.
24 Wickramasinghe SN, Abdalla SH. Blood and bone marrow changes in malaria. Baillière’s Best Pract Res Clin Haematol. 2000;13:277-299.
25 Dhingra KK, Gupta P, Saroha V, et al. Morphological findings in bone marrow biopsy and aspirate smears of visceral kala azar: a review. Indian J Pathol Microbiol. 2010;53:96-100.
26 Penchansky L, Krause JR. Identification of cytomegalovirus in bone marrow biopsy. South Med J. 1979;72:500-501.
27 Brown KE. Haematological consequences of parvovirus B19 infection. Baillière’s Best Pract Res Clin Haematol. 2000;13:245-259.
28 Evans RH, Scadden DT. Haematological aspects of HIV infection. Baillière’s Best Pract Res Clin Haematol. 2000;13:215-230.
29 Sullivan PS, Hanson DL, Chu SY, et al. Epidemiology of anemia in human immunodeficiency virus (HIV)-infected persons: results from the multistate adult and adolescent spectrum of HIV disease surveillance project. Blood. 1998;91:301-308.
30 Nair JM, Bellevue R, Bertoni M, Dosik H. Thrombotic thrombocytopenic purpura in patients with the acquired immunodeficiency syndrome (AIDS)-related complex. A report of two cases. Ann Intern Med. 1988;109:209-212.
31 Candido A, Rossi P, Menichella G, et al. Indicative morphological myelodysplastic alterations of bone marrow in overt AIDS. Haematologica. 1990;75:327-333.
32 Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology. 2009;14:474-485.
33 Lawn SD, Churchyard G. Epidemiology of HIV-associated tuberculosis. Curr Opin HIV AIDS. 2009;4:325-333.
34 Cello JP, Day LW. Idiopathic AIDS enteropathy and treatment of gastrointestinal opportunistic pathogens. Gastroenterology. 2009;136:1952-1965.
35 Currier JS, Havlir DV. Complications of HIV disease and antiretroviral therapy. Top HIV Med. 2009;17:57-67.
36 Moses A, Nelson J, Bagby GCJr. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood. 1998;91:1479-1495.
37 Bain BJ. Pathogenesis and pathophysiology of anemia in HIV infection. Curr Opin Hematol. 1999;6:89-93.
38 Bain BJ. Lymphomas and reactive lymphoid lesions in HIV infection. Blood Rev. 1998;12:154-162.
39 Eid A, Carion W, Nystrom JS. Differential diagnoses of bone marrow granuloma. West J Med. 1996;164:510-515.
40 Cohen RJ, Samoszuk MK, Busch D, Lagios M. Occult infections with M. intracellulare in bone-marrow biopsy specimens from patients with AIDS. N Engl J Med. 1983;308:1475-1476.
41 Bain BJ. The importance of a negative image. Am J Hematol. 2008;83:410.
42 Browne PM, Sharma OP, Salkin D. Bone marrow sarcoidosis. JAMA. 1978;240:2654-2655.
43 Bain BJ, Clark DM, Wilkins B. Bone marrow pathology. Oxford: Blackwell Wiley; 2010.
44 Singh G, Krause JR, Breitfeld V. Bone marrow examination: for metastatic tumor: aspirate and biopsy. Cancer. 1977;40:2317-2321.
45 Savage RA, Hoffman GC, Shaker K. Diagnostic problems involved in detection of metastatic neoplasms by bone-marrow aspirate compared with needle biopsy. Am J Clin Pathol. 1978;70:623-627.
46 Anner RM, Drewinko B. Frequency and significance of bone marrow involvement by metastatic solid tumors. Cancer. 1977;39:1337-1344.
47 Bain BJ, Clark DM, Wilkins B. Bone marrow pathology. Oxford: Blackwell Wiley; 2010.
48 Cotta CV, Konoplev S, Medeiros LJ, Bueso-Ramos CE. Metastatic tumors in bone marrow: histopathology and advances in the biology of the tumor cells and bone marrow environment. Ann Diagn Pathol. 2006;10:169-192.
49 Morandi S, Manna A, Sabattini E, Porcellini A. Rhabdomyosarcoma presenting as acute leukemia. J Pediatr Hematol Oncol. 1996;18:305-307.
50 Smith SR, Reid MM. Neuroblastoma rosettes in aspirated bone marrow. Br J Haematol. 1994;88:445-447.
51 Beiske K, Burchill SA, Cheung IY, et al. Consensus criteria for sensitive detection of minimal neuroblastoma cells in bone marrow, blood and stem cell preparations by immunocytology and QRT-PCR: recommendations by the International Neuroblastoma Risk Group Task Force. Br J Cancer. 2009;100:1627-1637.
52 McCarthy DM. Annotation. Fibrosis of the bone marrow: content and causes. Br J Haematol. 1985;59:1-7.
53 Kuter DJ, Bain B, Mufti G, et al. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007;139:351-362.
54 Pastores GM. Gaucher’s Disease. Pathological features. Baillière’s Clin Haematol. 1997;10:739-749.
55 Florena AM, Franco V, Campesi G. Immunophenotypical comparison of Gaucher’s and pseudo-Gaucher cells. Pathol Int. 1996;46:155-160.
56 Varela-Duran J, Roholt PC, Ratliff NBJr. Sea-blue histiocyte syndrome. A secondary degenerative process of macrophages? Arch Pathol Lab Med. 1980;104:30-34.
57 Kolodny EH. Niemann–Pick disease. Curr Opin Hematol. 2000;7:48-52.
58 Mehta AB. Anderson–Fabry disease: developments in diagnosis and treatment. Int J Clin Pharmacol Ther. 2009;47(Suppl 1):S66-S74.
59 Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008;10:e1.
60 Janssens AM, Offner FC, Van Hove WZ. Bone marrow necrosis. Cancer. 2000;88:1769-1780.
61 Knupp C, Pekala PH, Cornelius P. Extensive bone marrow necrosis in patients with cancer and tumor necrosis factor activity in plasma. Am J Hematol. 1988;29:215-221.
62 Caraveo J, Trowbridge AA, Amaral BW, et al. Bone marrow necrosis associated with a Mucor infection. Am J Med. 1977;62:404-408.
63 Bohm J. Gelatinous transformation of the bone marrow: the spectrum of underlying diseases. Am J Surg Pathol. 2000;24:56-65.
64 Stavem P, Larsen IF, Ly B, Rorvik TO. Amyloid deposits in bone marrow aspirates in primary amyloidosis. Acta Med Scand. 1980;208:111-113.
65 Enos WF, Pierre RV, Rosenblatt JE. Giant cell arteritis detected by bone marrow biopsy. Mayo Clin Proc. 1981;56:381-383.
66 Muretto P, Carnevali A, Ansini AL. Cholesterol embolism of bone marrow clinically masquerading as systemic or metastatic tumor. Haematologica. 1991;76:248-250.
67 McClure J, Fazzalari NL, Fassett RG, Pugsley DJ. Bone histoquantitative findings and histochemical staining reactions for aluminium in chronic renal failure patients treated with haemodialysis fluids containing high and low concentrations of aluminium. J Clin Pathol. 1983;36:1281-1287.
68 Findlay AB. Bone marrow changes in the post mortem interval. J Forensic Sci Soc. 1976;16:213-218.
Bain BJ, Clark DM, Wilkins BS. Bone Marrow Pathology, 4th ed. Oxford: Wiley-Blackwell; 2010.
Torlakovic EE, Naresh KN, Brunning RD. Bone Marrow Immunohistochemistry. Chicago: American Society for Clinical Pathology Press; 2009.
Foucar K, Viswanatha DS, Wilson CS. Non-Neoplastic Disorders of Bone Marrow (Atlas of Nontumor Pathology). Washington: AFIP; 2008.