[level-membership-for-pathology-category]1
Pathologic reactions in the CNS
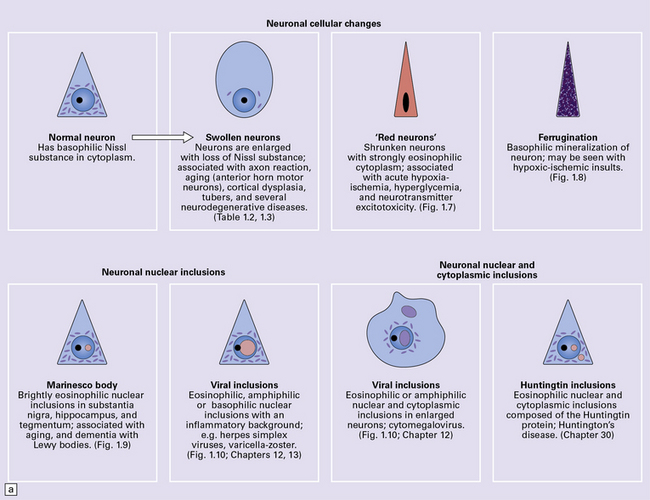
PATHOLOGIC RESPONSES IN NEURONS
NORMAL NEURONAL CYTOLOGY AND STAINING
The cytologic appearance of neurons varies depending upon their location. In general, neurons have moderate to abundant cytoplasm and a relatively large round nucleus with a prominent nucleolus. Projecting from the neuronal cell body are branching dendrites and a single axon. A range of tinctorial and immunohistochemical staining techniques are used for demonstrating neurons and their processes (Table 1.1).
Table 1.1
Histologic demonstration of neurons: techniques and comments concerning their applications
Conventional staining
Hematoxylin and eosin
Good for assessing general cytoarchitecture
Nissl stain (e.g. cresyl fast violet)
Good for assessing general cytoarchitecture. Allows estimation of cell density in thick sections; may be combined with stains for myelin (e.g. Luxol fast blue).
Axon silver impregnation techniques
Good for demonstration of axons and some neuronal inclusions. Used in conjunction with myelin stains to distinguish between demyelination and fiber degeneration
Golgi stain
Allows visualization of fine detail of neuronal cell processes. A technically difficult stain to perform which depends on block impregnation
Immunohistochemistry
Neurofilament proteins
Strongly expressed in perikaryal and axonal cytoplasm (in normal neurons, perikaryal neurofilament proteins are non-phosphorylated and axonal neurofilament proteins phosphorylated)
NeuN
Neuron-specific nuclear protein, strongly expressed in neuronal nuclei. NeuN antibodies also show weak labeling of the neuronal cytoplasm
Neuron specific enolase
Strongly expressed in neuronal and axonal cytoplasm
Synaptophysin
Antibodies to synaptophysin detect neurosecretory vesicles, which are mainly located at synapses, with little staining of neuronal cell bodies
PGP9.5
An antibody to neuron-specific ubiquitin C-terminal hydrolase, which is abundant in neurons and neuronal cell processes
Chromogranin A
Antibodies detect dense-core neurosecretory vesicles, which are sparsely distributed within the perikaryon of some neurons and concentrated at the synapses. Moderate staining of neuronal cell bodies
Neurotransmitter-related
Antibodies allow detection of neurotransmitter substance or enzyme involved in its biosynthesis
ABNORMALITIES OF NEURONAL MORPHOLOGY
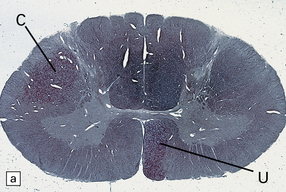
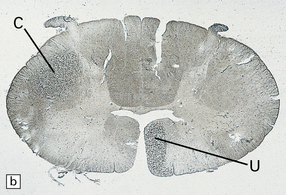
Axonal degeneration inevitably follows the death of the neuron of which it is a part. A severed or severely damaged axon undergoes distal degeneration without usually provoking death of the proximal part of the neuron, although this does undergo a series of structural and metabolic changes (i.e. axon reaction and chromatolysis, see below). Within days, the distal part of the axon fragments and the surrounding myelin sheath breaks up into ovoids. Over the next 3 weeks or so, the axon and myelin debris are taken up by macrophages, which infiltrate the degenerating fiber tracts. Several methods can be used to demonstrate degenerating nerve fibers in the CNS. These include specialized silver impregnation techniques and stains for degenerating myelin and for lipid (Fig. 1.1).
AXON REACTION AND CHROMATOLYSIS
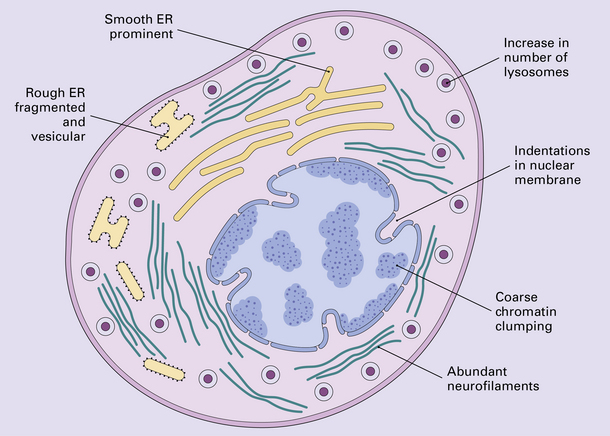
Damage to the axon provokes a series of morphologic and biochemical changes in the neuronal cell body. These are collectively referred to as the axon reaction (Figs 1.2, 1.3). The changes include disruption and dispersion of Nissl bodies (chromatolysis) associated with rearrangement of the cytoskeleton and marked accumulation of intermediate filaments. The axon reaction is conspicuous in large neurons with axons that project into the peripheral nervous system and in some of the larger neurons with central projections. Chromatolysis is not visible on conventional light microscopy of small neurons or certain large neurons such as the cerebellar Purkinje cells, but changes can be demonstrated in these cells by electron microscopy and immunohistochemistry.
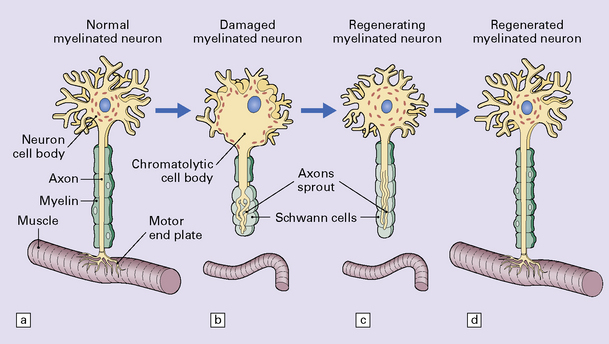
1.2 Axon reaction.
The changes that constitute the axon reaction have been most extensively investigated in anterior horn cells after peripheral nerve injury, as depicted in this figure, but neurons undergo similar alterations after injury to axons in the CNS. (a) A normal neuronal cell body is characterized by well-organized dendritic branches making synaptic contact with other neurons. Nissl bodies are prominent. (b) Following axonal damage there is degeneration of the peripheral axon. Dendrites that normally form synapses retract and synaptic contacts are lost. The rough endoplasmic reticulum becomes reorganized into small vesicular elements. The neuron inactivates genes coding for high molecular weight neurofilament protein (NFH) and switches on genes for peripherin. Neurofilaments and peripherin accumulate in the neuronal cell body. At this stage the neuronal cell body is swollen, shows only weak acidophilia, and lacks Nissl bodies. Denervated muscle fibers atrophy. (c) The axon regenerates in the peripheral nervous system (but not within the CNS). NFH synthesis is re-established with axonal elongation. (d) If the target tissue is reinnervated, the neuronal dendrites re-establish synaptic contact with other neurons and the normal cytologic appearance of the cell body is regained.
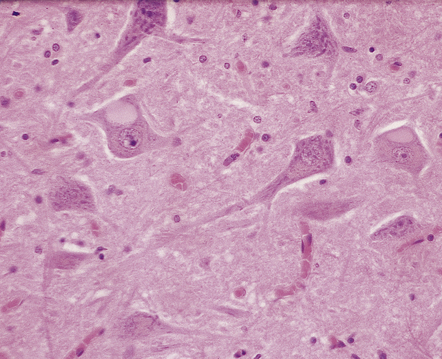
SWOLLEN NEURONS
Swollen or ballooned neurons are a feature both of the axon reaction and of a variety of diseases in which perikaryal changes occur independently of axonal damage (Table 1.2). Histologically, they appear as distended, weakly-staining cells with large, relatively clear nuclei (Fig. 1.4). Occasionally the cells contain small vacuoles. In conventionally processed tissues an artefactual lacuna is often present around the abnormal cell. Swollen or ballooned neurons can be demonstrated with several immunohistochemical markers (Fig. 1.5, Table 1.3).
Table 1.2
Causes of ballooning of neurons
Physiologic
Anterior horn motor neurons, with age
Nutritional
Pellagra (niacin deficiency): spinal, brain stem, and cortical neurons
Developmental
Localized cortical dysplasia with cytomegaly
Cortical dysplasia with hemimegalencephaly
Tuberous sclerosis
Neurodegenerative
Several types of frontotemporal lobar degeneration, including Pick’s disease and FTDP-17: neocortical and some basal neurons
Corticobasal degeneration: neocortical and some basal neurons
Swollen neurons can also occur in Alzheimer disease, argyrophilic grain disease, progressive supranuclear palsy and several other neurodegenerative diseases
Prion disease
Cortical and some basal neurons
Table 1.3
Immunohistochemical profile of swollen neurons
Neurofilament protein
Accumulation of highly phosphorylated high-molecular-weight neurofilament protein, which is normally restricted to the axon
αB-crystallin
Accumulation of αB crystallin, which is not normally expressed by neurons
Tau protein
Accumulation of abnormally phosphorylated tau protein by a proportion of swollen neurons in corticobasal degeneration and Pick’s disease
Ubiquitin
Variably increased immunoreactivity for ubiquitin-protein conjugates
PGP9.5
Variably increased immunoreactivity for this ubiquitin C-terminal hydrolase
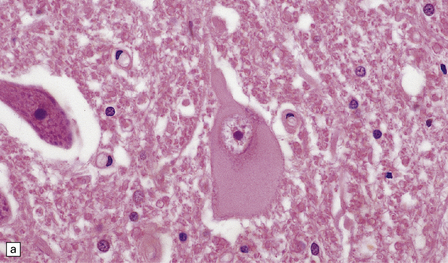
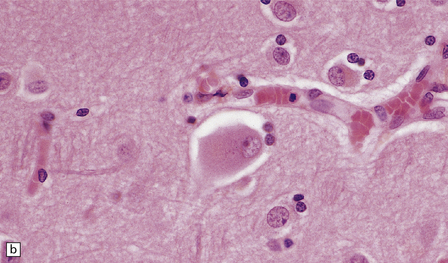
1.4 Swollen neurons.
The cytoplasm lacks Nissl bodies and is weakly eosinophilic. The nucleus is large and vesicular. (a) Example from the anterior horn of the spinal cord. (b) Example from the cerebral cortex in corticobasal degeneration (see Chapter 28).
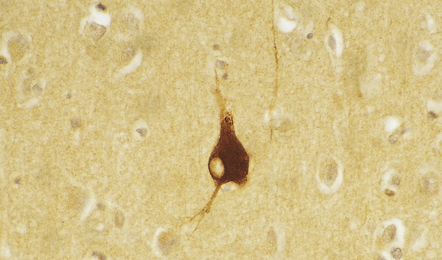
1.5 αB-crystallin immunoreactivity.
This is a characteristic feature of swollen neurons in most conditions. The protein is not, however, expressed by swollen neurons in pellagra (see Chapter 21), suggesting that neuronal swelling in this disorder has a different mechanism.
TRANS-SYNAPTIC NEURONAL DEGENERATION AND OLIVARY HYPERTROPHY
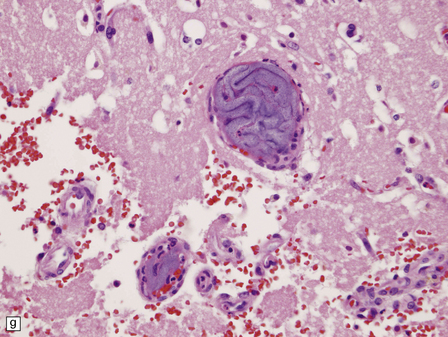
Neurons within some nuclei in the CNS atrophy and degenerate in response to deafferentation. Examples are neurons in the lateral geniculate nucleus, which degenerate after optic nerve or tract lesions, and neurons in the pontine nuclei, which degenerate after interruption of descending frontopontine afferent fibers. Neurons in the inferior and accessory olivary nuclei undergo an unusual form of trans-synaptic degeneration after a destructive lesion (such as an infarct) of the ipsilateral central tegmental tract (Fig. 1.6). The olivary ribbon as a whole becomes thickened and neurons show marked enlargement, cytoplasmic vacuolation, and some dispersion of Nissl bodies. Olivary hypertrophy is associated with the development of palatal myoclonus in some patients.
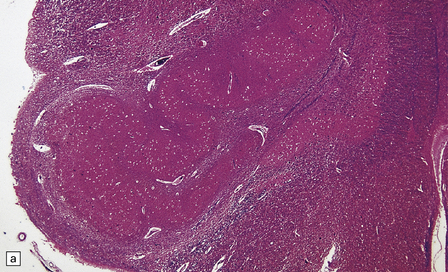
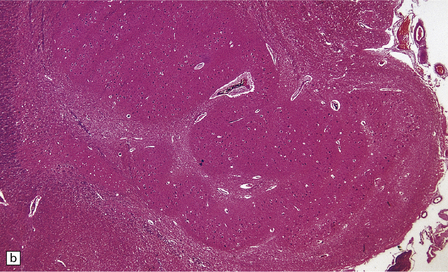
1.6 Hypertrophy of the left olive due to a lesion involving the left central tegmental tract.
(a) Normal right olive. (b) Hypertrophy of the left olive. The lesion involving the left central tegmental tract is not shown. Compare the normal-sized neurons in the right olive with the enlarged vacuolated neurons in the left olive. (c) Normal neurons of the right inferior olivary nucleus stained with cresyl violet. (d) Neurons of the hypertrophic left inferior olive are enlarged and vacuolated.
HYPOXIC CELL CHANGE
Neurons are especially vulnerable to damage from hypoxia, which causes the following distinctive histologic changes (Fig. 1.7):
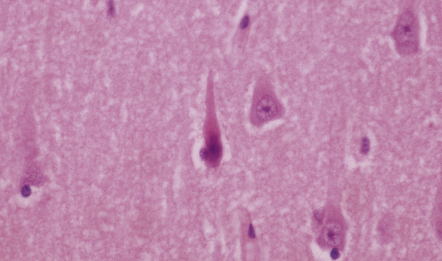
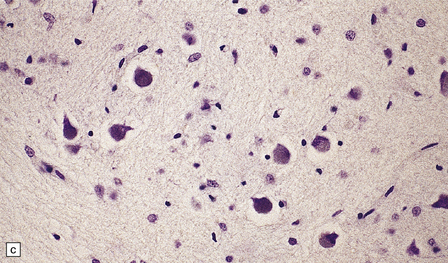
1.7 Hypoxic cell change in neurons.
Affected neurons become shrunken and eosinophilic. The nuclei become condensed and lose their crisp contours. In this illustration the neuron in the center is affected while surrounding neurons appear normal.
 Microvacuolation (visible only at an ultrastructural level), due to swelling of endoplasmic reticulum and mitochondria.
Microvacuolation (visible only at an ultrastructural level), due to swelling of endoplasmic reticulum and mitochondria.
 Shrinkage of the cell body and increasing cytoplasmic acidophilia.
Shrinkage of the cell body and increasing cytoplasmic acidophilia.


Neurons in certain parts of the brain that are especially vulnerable to hypoxic damage are:
 pyramidal neurons in the CA1 field of the hippocampus
pyramidal neurons in the CA1 field of the hippocampus


The pattern of regional susceptibility to hypoxia differs between infants and adults (see Chapters 2 and 8).
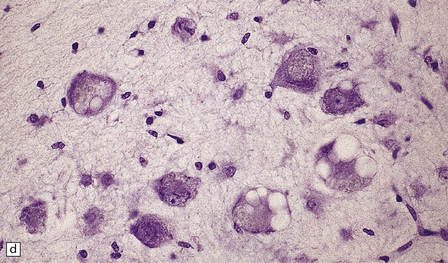
FERRUGINATION
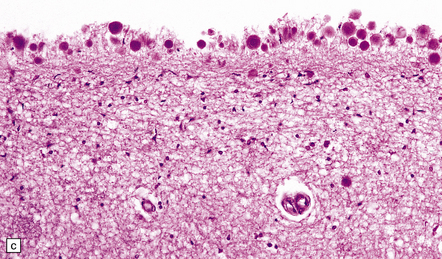
Although dead neurons usually undergo liquefaction or are removed by phagocytosis, they may instead become encrusted and replaced by mineral salts, a process termed ferrugination. This is particularly prominent in the infant brain in response to hypoxic–ischemic damage (Fig. 1.8).
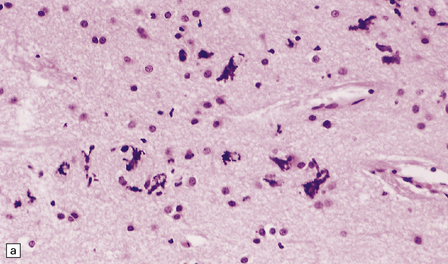
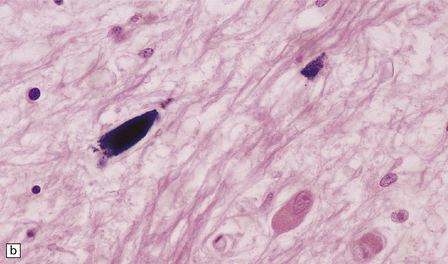
1.8 Ferruginated neurons.
Ferruginated neurons become encrusted in mineral deposits and appear dark purple in sections stained with hematoxylin and eosin. The mineral deposits contain both calcium and iron and can be stained by Perls’ method and von Kossa’s technique. (a) Many ferruginated neurons in the cortex of a neonate who had hypoxic brain damage. Astroglial cells are not affected. (b) Higher power illustration of neurons replaced by mineral deposits.
NUCLEAR INCLUSIONS
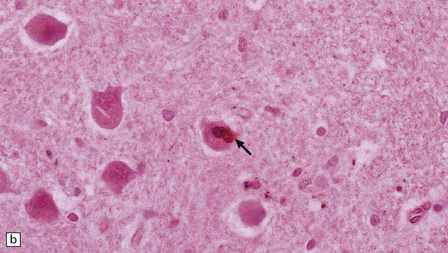
MARINESCO BODIES
These are small spherical nuclear inclusions that are brightly eosinophilic and are often seen in neurons of the adult substantia nigra (Fig. 1.9a). They may also occur in other neurons, such as the pyramidal cells of the hippocampus and neurons in the tegmentum of the brain stem. Marinesco bodies are most common in the elderly and in dementia with Lewy bodies. They have an increased prevalence in neurons containing Lewy bodies.
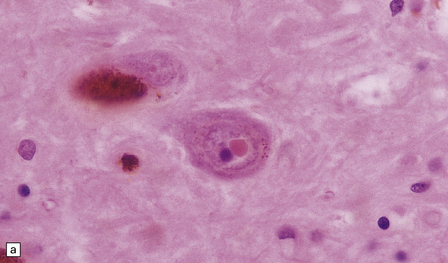
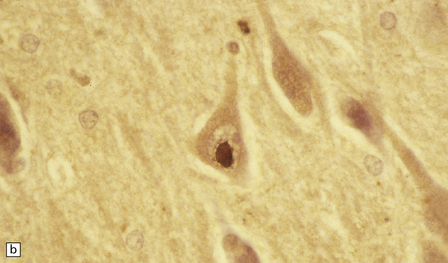
1.9 Marinesco bodies.
These intranuclear inclusions are visible in several neuronal groups. (a) Marinesco bodies are brightly eosinophilic intranuclear inclusions about the same size as a large nucleolus. (b) As shown here, they are strongly immunoreactive for ubiquitin.
Ultrastructurally, Marinesco bodies are composed of filaments with the same diameter as intermediate filaments, and may be derived from the nuclear lamins. They are immunoreactive for ubiquitin, an 8 kD polypeptide involved in the degradation of many abnormal or short-lived proteins (Fig. 1.9b).
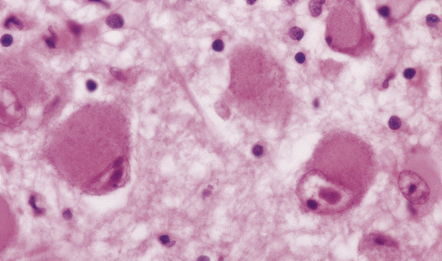
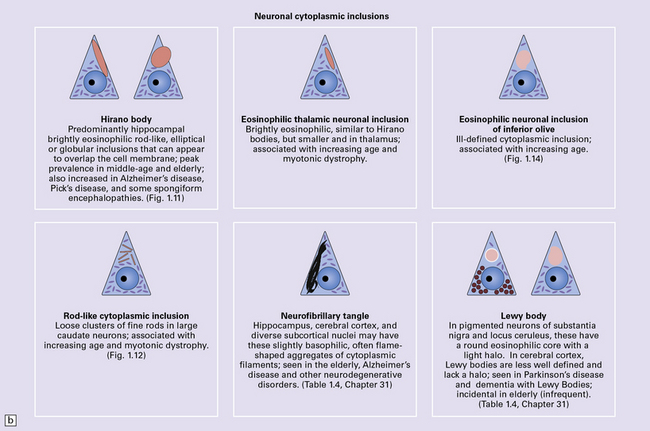
NEURONAL CYTOPLASMIC INCLUSIONS
CYTOSKELETAL AND FILAMENTOUS INCLUSIONS
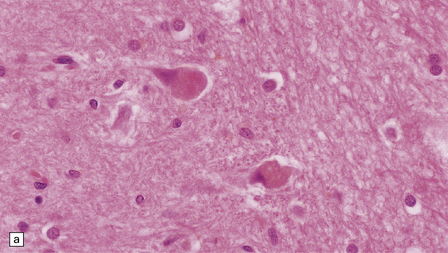
Hirano bodies
These are brightly eosinophilic rod-shaped or elliptical cytoplasmic inclusions that may appear to overlap the edge of a neuron (Fig. 1.11). They are immunoreactive for actin, actin-associated proteins, and caspase-cleaved TDP43.
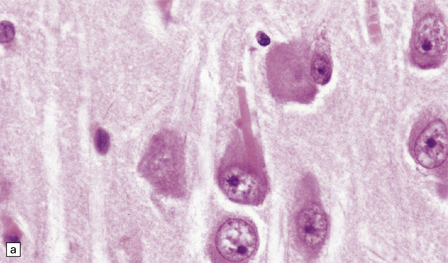
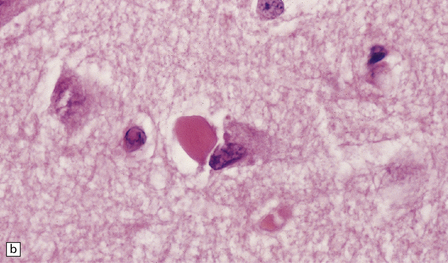
ROD-LIKE CYTOPLASMIC INCLUSIONS
These eosinophilic cytoplasmic inclusions occur in large neurons of the caudate nucleus. The inclusions resemble loose bundles of twigs and, like the thalamic neuronal inclusions described above, are common in patients with myotonic dystrophy. However, they are sometimes an incidental finding in elderly patients who do not have neurologic or muscular disease. The rod-like inclusions stain strongly with phosphotungstic acid/hematoxylin (Fig. 1.12).
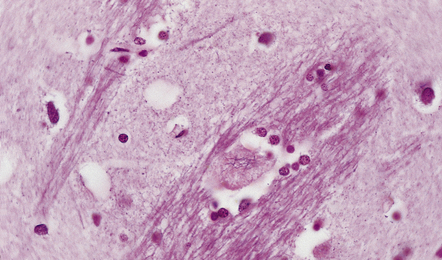
1.12 Rod-like cytoplasmic inclusions in the caudate nucleus.
These occur exclusively in large neurons in the caudate nucleus. The inclusions may be an incidental finding, particularly in the elderly, but are most common in myotonic dystrophy. (Courtesy of Dr D A Hilton, Derriford Hospital, Plymouth.)
OTHER FILAMENTOUS NEURONAL INCLUSIONS
There are several other types of neuronal inclusion that comprise or include elements of the cytoskeleton and usually occur in the context of specific neurodegenerative diseases (Table 1.4). These inclusions are described in more detail and illustrated in the sections concerned with the relevant diseases.
Table 1.4
Examples of inclusion bodies in specific conditions and diseases
| Inclusion | Association | Main constituents |
| Lewy bodies | Aging, Parkinson’s disease, dementia with Lewy bodies | α-synuclein, neurofilament protein, and ubiquitin |
| Neurofibrillary tangles | Aging, Alzheimer disease, progressive supranuclear palsy, post-encephalitic parkinsonism, Guam parkinsonism–dementia, myotonic dystrophy, subacute sclerosing panencephalitis, Niemann–Pick disease type C, other rare disorders | Phosphorylated tau protein (3R, 4R), ubiquitin |
| Pick bodies | Pick’s disease | Neurofilament protein, phosphorylated tau protein (3R), ubiquitin |
| MND inclusions | Motor neuron disease/amyotrophic lateral sclerosis | TAR DNA-binding protein 43 (sporadic cases) or superoxide dismutase 1 (some familial cases) or fused-in-sarcoma protein (other familial cases) ubiquitin, p62 |
CYTOSOLIC INCLUSIONS
These are composed of polyglucosans (polymers of sulfated polysaccharides) and are similar to corpora amylacea in composition and staining characteristics (see below). They are present in large numbers in Lafora’s disease (see Chapter 7), both in the CNS and in certain peripheral tissues such as sweat glands, liver, and skeletal muscle. Lafora body formation has been linked to aberrant glycogen hyperphosphorylation. The inclusions usually have a round core that is intensely periodic acid–Schiff (PAS)-positive (Fig. 1.13). Spicules of the core may radiate outwards, into a surrounding zone of less intensely PAS-positive material.
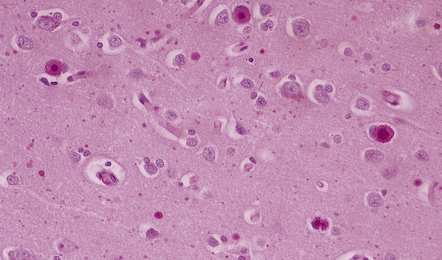
EOSINOPHILIC INCLUSIONS IN THE INFERIOR OLIVES
A characteristic feature of aging is the development of ill-defined eosinophilic inclusions in the neurons of the inferior olivary nuclei. These inclusions are intensely immunoreactive with antibodies to ubiquitin (Fig. 1.14) and should not be confused with lipofuscin, which also accumulates with age (see below) and is particularly abundant in neurons of the inferior olivary nuclei.
MEMBRANE-BOUND CYTOPLASMIC INCLUSIONS
Colloid inclusions are round eosinophilic inclusions that usually occur in neurons in the hypoglossal nuclei (Fig. 1.15), but may be seen in other large neurons, particularly in the elderly. Electron microscopy shows that these inclusions result from dilatation of the endoplasmic reticulum by amorphous material. The importance of recognizing colloid inclusions, which do not have any clinical significance, is that they are occasionally confused with inclusions that are clinically significant such as Lewy bodies, pale bodies, and hyaline inclusions of motor neuron disease (see Chapter 27).
BUNINA BODIES
These are small beaded eosinophilic inclusions seen in motor neurons in motor neuron disease. Cystatin C, transferrin, and peripherin have been detected in Bunina bodies. Ultrastructurally, they appear as electron-dense membrane-bound bodies (see Chapter 27).
INCLUSIONS DERIVED FROM THE ACID VESICLE SYSTEM
The acid vesicle system consists of endosomes, lysosomes, and lysosome-derived dense bodies.
Lipofuscin (Fig. 1.16) is produced by oxidation of lipids and lipoproteins within the lysosomal system. It appears as orange-brown granular material in sections stained with hematoxylin and eosin, and is acid-fast (as demonstrated by the long Ziehl–Neelsen method) and autofluorescent under ultraviolet light. The granules are also sudanophilic and stain with PAS and Schmorl’s stain. Lipofuscin accumulates with aging in neurons and glia, particularly in:
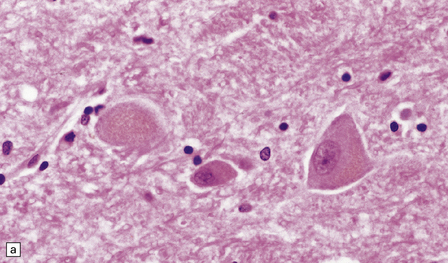
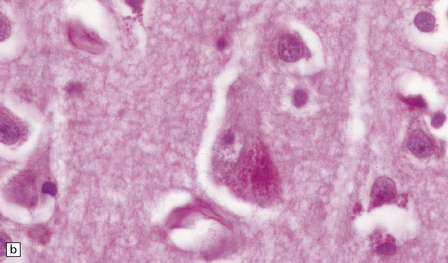
1.16 Lipofuscin.
This accumulates with age in neurons and glia. (a) It has a golden brown color in sections stained with hematoxylin and eosin. (b) Lipofuscin stains strongly with PAS.


 Pyramidal neurons in the cerebral cortex and hippocampus.
Pyramidal neurons in the cerebral cortex and hippocampus.


Granulovacuolar degeneration. This term describes the accumulation of vacuoles containing small round dense bodies (granulovacuoles) (Fig. 1.17). The dense bodies are ubiquitinated and react with antibodies to some epitopes of the microtubule-associated tau protein. The immunohistochemical data have been interpreted as suggesting that dense bodies are derived from partial degradation of tau protein within lysosomes. Although anti-phosphoTDP43 antibody is sensitive for detecting granulovacuolar degeneration, TDP43 proteinopathies are not associated with an increase in granulovacuoles. Granulovacuolar degeneration is seen in normal aging after the sixth decade, predominantly in the hippocampal formation. Neurons in the CA1 field are most severely affected and, in descending order of severity, those in the prosubiculum, CA2, CA3, and CA4 fields. The density of hippocampal neurons showing granulovacuolar degeneration is increased in patients with Alzheimer disease and Pick’s disease, in whom granulovacuolar degeneration may also occur in neurons in the subcortical nuclei.
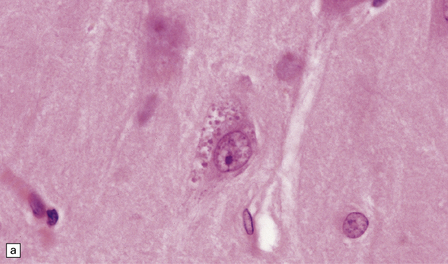
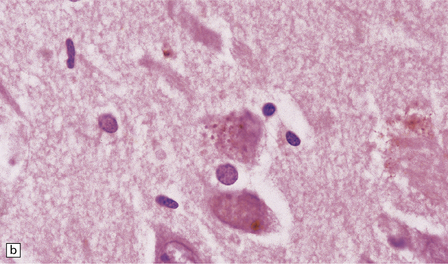
1.17 Granulovacuolar degeneration.
(a) These are small round basophilic bodies within otherwise clear vacuoles. They are mainly found in pyramidal neurons of the hippocampus. (b) The granules are variably immunoreactive for ubiquitin.
Storage products in neurometabolic diseases. The accumulation of material within the acid vesicle system is a feature of many neurometabolic diseases including the lysosomal storage disorders, as well as certain other metabolic disorders caused by non-lysosomal enzyme defects. These disorders are considered in Chapter 23.
STRUCTURAL ABNORMALITIES OF AXONS
AXONAL SPHEROIDS AND DYSTROPHIC
AXONAL SWELLINGS
Spheroids are axonal swellings. They are composed of neurofilaments, organelles, and other material that is normally conveyed along the axon by anterograde transport systems, but accumulates focally when these are impaired (Fig. 1.18a,b). Spheroids are a feature of axonal damage by diverse extrinsic insults, especially trauma (see Chapter 11) and infarcts. There may be numerous axonal spheroids around the edge of an infarct.
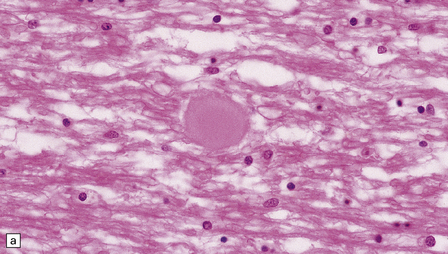
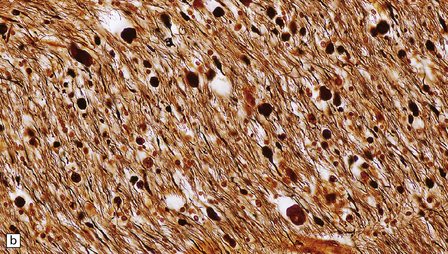
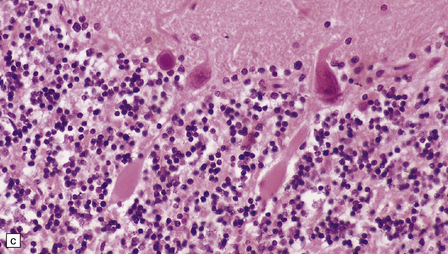
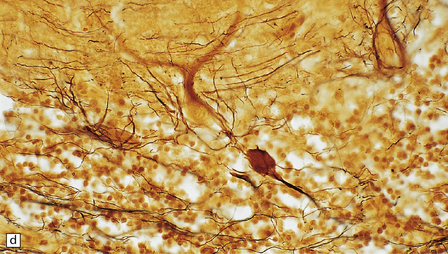
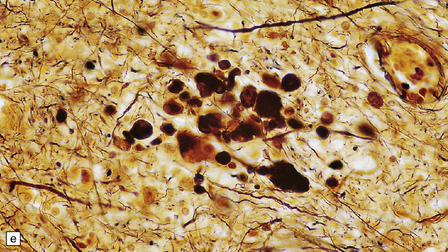
1.18 Axonal spheroids.
(a) These are homogeneous or finely granular, rounded, eosinophilic bodies. (b) They can be demonstrated with antibodies to neurofilament proteins or by axonal silver impregnation, as in this section through the centrum semiovale after head trauma that resulted in diffuse axonal injury with numerous spheroids. (c) Torpedoes are Purkinje cell axonal swellings seen in the granule cell layer of the cerebellum. They have a characteristic fusiform appearance, as shown here, and occur in conditions that cause cerebellar cortical atrophy with degeneration of Purkinje cells. (d) Torpedoes with a characteristic fusiform appearance. (e) Dystrophic axonal swellings are a feature of some metabolic diseases, as illustrated in this section of putamen from a patient with Niemann–Pick disease type C.
The term ‘torpedo’ is applied to Purkinje cell axonal swellings, which are a feature of a wide range of metabolic and degenerative cerebellar diseases. These swellings appear as fusiform eosinophilic structures in the cerebellar granule cell layer (Fig. 1.18c,d). They are well demonstrated by silver impregnation (see chapter 29).
The axonal swellings that develop when axonal transport is disrupted by neuronal metabolic dysfunction are usually termed dystrophic (Fig. 1.18e). These occur in certain nutritional deficiencies (particularly vitamin E deficiency, see Chapter 21) and inherited metabolic diseases (e.g. Niemann–Pick disease type C). Dystrophic axonal swellings are the principal pathologic abnormality seen in a group of conditions termed the neuroaxonal dystrophies (see Chapter 33).
The term ‘dystrophic neurite’ is also used to describe neuronal processes within the gray matter that are distended by tau protein or other abnormal ubiquitinated material. These occur in several neurodegenerative diseases (Table 1.5).
Table 1.5
Dystrophic axons in aging and neurodegenerative diseases
| Condition | Dystrophic processes |
| Normal aging | Neuronal processes accumulate dot-like ubiquitin- immunoreactive material (see Fig. 1.24) |
| Alzheimer disease | The swollen neuronal processes around amyloid plaques are termed dystrophic neurites. Many contain ubiquitinated proteins, with or without accumulation of tau protein (Chapter 31) |
| Lewy body disease | Abnormal neurites in affected brain regions can be detected by α-synuclein, ubiquitin or p62 immunohistochemistry (Chapter 28) |
| Huntington disease | Abnormal neurites in the cerebral cortex can be detected by ubiquitin or p62 immunohistochemistry (Chapter 30) |
| Frontal lobe dementia | Abnormal neurites in the cerebral cortex can be detected by TDP-43, ubiquitin or p62 immunohistochemistry (Chapter 31) |
In brains from people over 60 years of age, antibodies to ubiquitin label smaller dot-like structures in the neuropil of the cerebral cortex and in the white matter. These correspond to membrane-bound dense bodies in dystrophic neurites and foci of granular degeneration in myelin sheaths (Fig. 1.19).
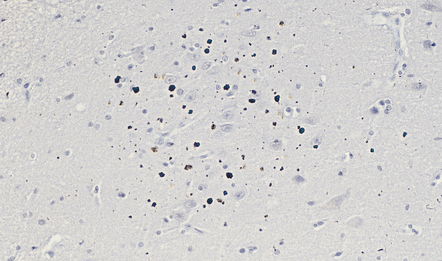
1.19 Dot-like ubiquitin immunoreactivity in the temporal cortex in one of the pre-α cell clusters.
The frequency and density of ubiquitin-immunoreactive dot-like bodies increases with age in both gray and white matter. This example shows dot-like bodies in the temporal cortex in one of the pre-α cell clusters.
PATHOLOGIC REACTIONS OF ASTROCYTES
Astrocytes are vital support cells of the nervous system. They can be demonstrated by a range of tinctorial stains and immunohistochemical techniques (Fig. 1.20) and undergo a range of structural changes in reaction to CNS disease.
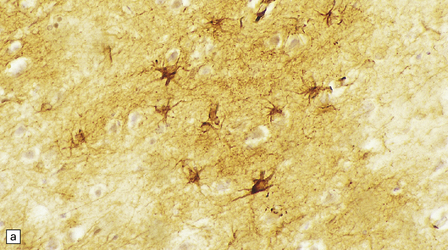
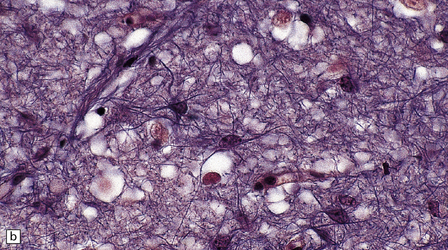
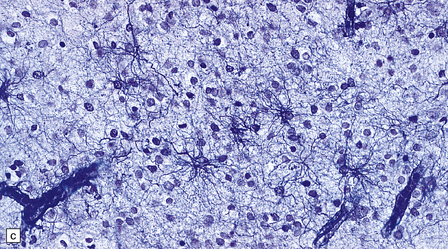
1.20 Stains demonstrating astrocytes.
(a) Astrocytes are often demonstrated by immunohistochemical staining for glial fibrillary acidic protein (GFAP). This reveals their stellate morphology as well as the blush-like staining of very fine processes in the neuropil. (b) Phosphotungstic acid–hematoxylin stain demonstrating astrocytes. (c) Holzer stain. This also demonstrates astrocytes, but is seldom used now because of the potential carcinogenicity of some of the reagents used in its preparation.
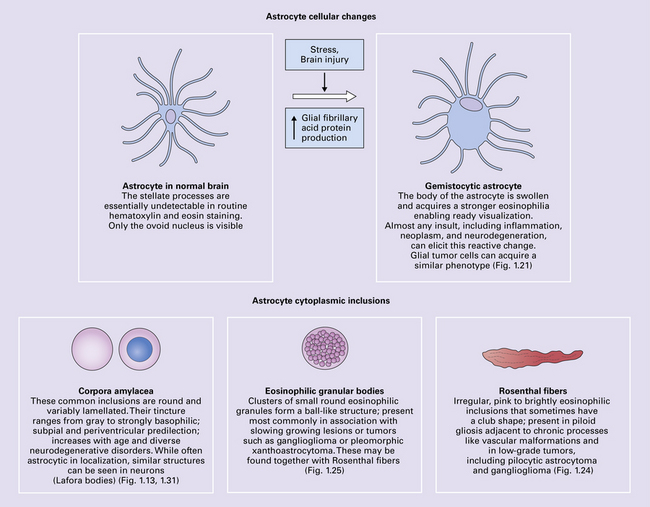
SWELLING OF ASTROCYTES
This is a relatively rapid response to a wide range of stimuli. Acutely reactive astrocytes are swollen by hyaline eosinophilic cytoplasm and have enlarged vesicular nuclei (Fig. 1.21).
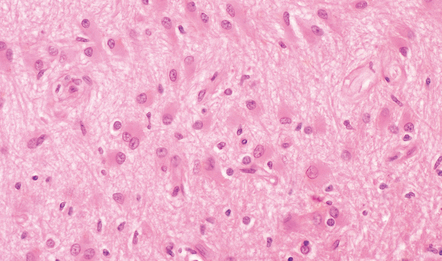
FIBRILLARY GLIOSIS
 Isomorphic fibrillary gliosis in which the alignment of astrocyte processes conforms to the architecture of previously normal local tissues (Fig. 1.22). This occurs mainly in chronic degenerative conditions.
Isomorphic fibrillary gliosis in which the alignment of astrocyte processes conforms to the architecture of previously normal local tissues (Fig. 1.22). This occurs mainly in chronic degenerative conditions.
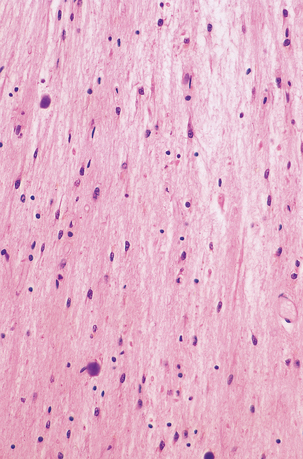
1.22 Isomorphic astrocytic gliosis.
The orientation of astrocyte processes conforms to that of cell processes in the underlying tissue. This pattern is characteristic of chronic degenerative conditions.
 Anisomorphic fibrillary gliosis in which there is a haphazard deposition of astrocyte processes (Fig. 1.23). This is typical of the gliosis that occurs in relation to destructive lesions.
Anisomorphic fibrillary gliosis in which there is a haphazard deposition of astrocyte processes (Fig. 1.23). This is typical of the gliosis that occurs in relation to destructive lesions.
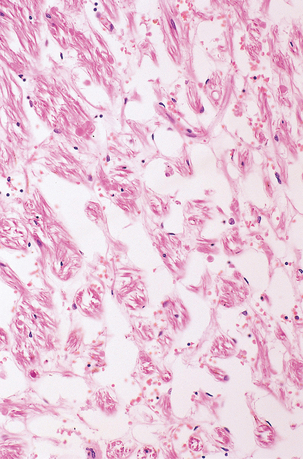
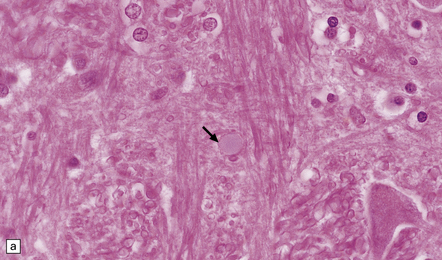
ROSENTHAL FIBERS
Rosenthal fibers are inclusions that develop in astrocytes in chronic reactive and neoplastic proliferations. They are brightly eosinophilic structures of variable size and shape (Fig. 1.24a,b). On electron microscopy they contain admixed amorphous granular material and 10 nm diameter filaments (Fig. 1.24c). Immunohistochemistry reveals GFAP, αB-crystallin, 27 kD heat-shock protein, and ubiquitin at the periphery of the Rosenthal fibers (Fig. 1.24d–f), while the central hyaline region is generally unlabeled.
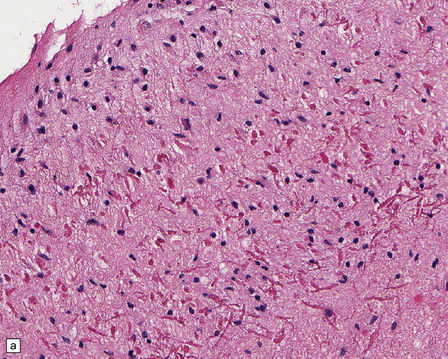
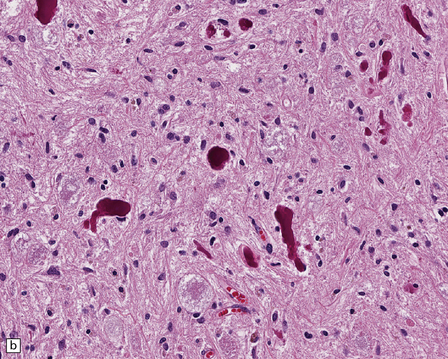
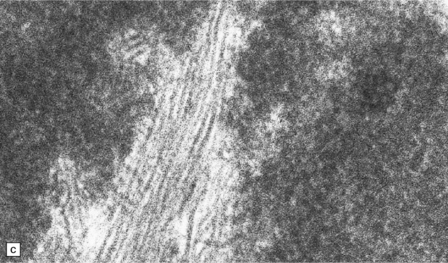
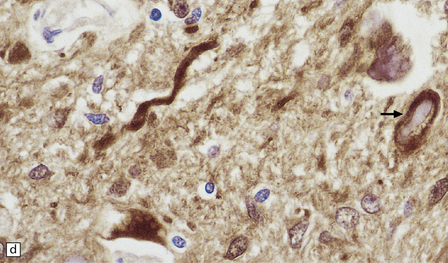
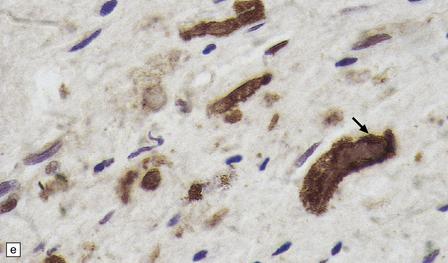
1.24 Rosenthal fibers.
(a,b) These fibers appear as brightly eosinophilic, rounded, elongated or carrot-shaped structures in hematoxylin- and eosin-stained sections. Their shape ranges from slender and small (a) to thick and large (b). (c) Electron microscopy revealing amorphous granular material and 10 nm diameter filaments within a Rosenthal fiber. (d) Immunostain showing peripheral labeling (arrow) for GFAP. (e) Immunostain showing peripheral labeling (arrow) for ubiquitin. (f) Immunostain showing labeling for αB-crystallin.
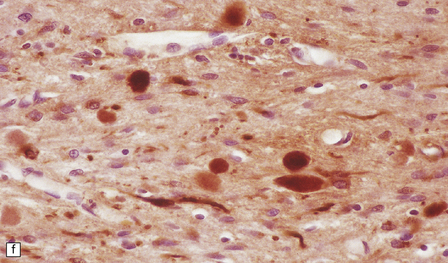
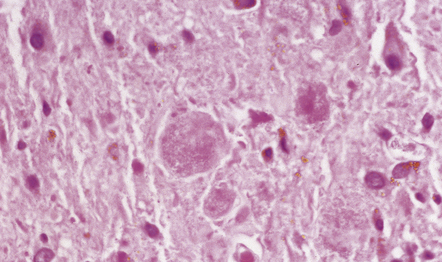
GRANULAR BODIES
Granular bodies are found in regions of chronic astrocytic gliosis, usually in conjunction with Rosenthal fibers. They are clusters of round eosinophilic granules within the cytoplasm of astrocytes (Fig. 1.25) and express GFAP as well as αB-crystallin. Ultrastructural examination reveals membrane-bound dense bodies.
TAU-IMMUNOREACTIVE GLIA
Tau protein has long been known to be the main constituent of neurofibrillary tangles, which are neuronal inclusions in Alzheimer disease and several other disorders (see Chapters 28, 29, and 31), but the recognition that glial cells can accumulate tau protein is a relatively recent finding. Several different types of tau-immunoreactive inclusions have been described (Fig. 1.26, Table 1.6). See also Pathologic reactions of oligodendroglia, p.16.
Table 1.6
Tau-reactive glial abnormalities
| Type | Association(s) | Immunoreactivity |
| Glial fibrillary tangles | PSP, CBD, Pick’s disease | Tau |
| Tufted astrocytes | PSP, Pick’s disease | Tau |
| Thorn-shaped astrocytes | Several degenerative diseases | Tau |
| Interfascicular threads | PSP and CBD white matter (oligodendroglial inclusions) | Tau |
| Coils and threads | PSP, CBD, FTDP-17 | Tau |
| Astrocytic plaques | CBD | Tau |
| Glial cytoplasmic inclusions | Multiple system atrophy, CBD | α-synuclein, tau, ubiquitin |
PSP, progressive supranuclear palsy; CBD, corticobasal degeneration.
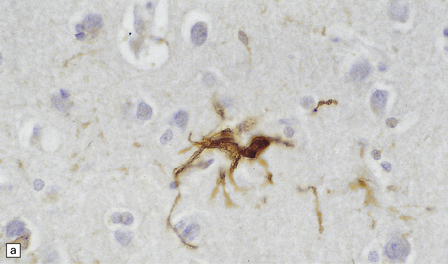
1.26 Tau immunoreactivity in astrocytes.
This is usually seen in neurodegenerative diseases in which there is also a prominent accumulation of tau protein in neurons as inclusions. (a) Glial fibrillary tangles are stellate cells with an astroglial morphology. (b) Tufted astrocytes are a particular feature of progressive supranuclear palsy and corticobasal degeneration and are mainly seen in the cerebral cortex. (c) Thorn-shaped astrocytes have more abundant immunostained cytoplasm and blunt thorn-shaped processes and are found in many tau-related neurodegenerative diseases.
CORPORA AMYLACEA
Corpora amylacea are spherical inclusions, predominantly in astrocyte processes (Fig. 1.27a,b), although they occasionally occur within axons. They range from 10 μm to 50 μm in diameter, consist largely of polyglucosans, and stain with hematoxylin, PAS, and methyl violet. Minor constituents include ubiquitin, heat-shock proteins, tau protein, complement proteins, and some oligodendrocyte proteins (i.e. myelin basic protein, proteolipid protein, galactocerebroside, and myelin oligodendrocyte glycoprotein). Ultrastructurally, corpora amylacea consist of densely packed 6–7 nm filaments that may be admixed with amorphous granular material and are not membrane bound.
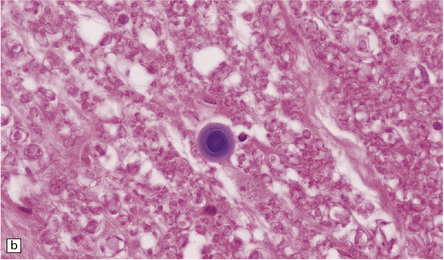
1.27 Corpora amylacea.
These are basophilic round bodies. (a) Some are only weakly basophilic (arrow). (b) Some, possibly more mature corpora amylacea, are more strongly basophilic and may consist of concentric lamellae of varying staining intensity. Some corpora amylacea contain tiny eosinophilic pockets of incorporated glial cytoplasm. (c) Corpora amylacea are relatively numerous in the subpial region.
Corpora amylacea increase in number with normal aging, particularly in subpial (Fig. 1.27c) and subependymal regions, around subcortical blood vessels, and in the spinal white matter. Their increase in number is greater in conditions where there has been atrophy and gliosis, including Alzheimer disease and other neurodegenerative disorders. Transglutaminase 1-mediated cross-linking and polymerization of cytoskeletal or cytoskeletal-associated proteins has been postulated as a mechanism of corpora amylacea formation.
NUCLEAR CHANGES IN ASTROCYTES
Alzheimer type II astrocytes are seen in cortical and subcortical gray matter regions in liver failure. They have an enlarged vesicular nucleus with marginated chromatin, scanty cytoplasm, and little or no demonstrable GFAP (Figs 1.28–1.32) (see also Chapter 22).
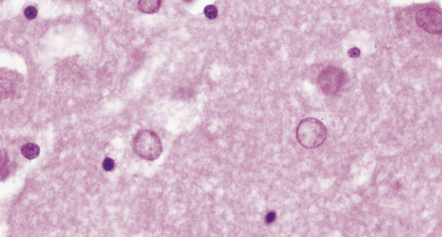
1.28 Alzheimer type II astrocytes.
These have enlarged empty-looking nuclei and virtually no discernible cytoplasm. The nuclei of Alzheimer type II astrocytes in the globus pallidus, dentate nucleus, and brain stem are often irregularly lobulated.
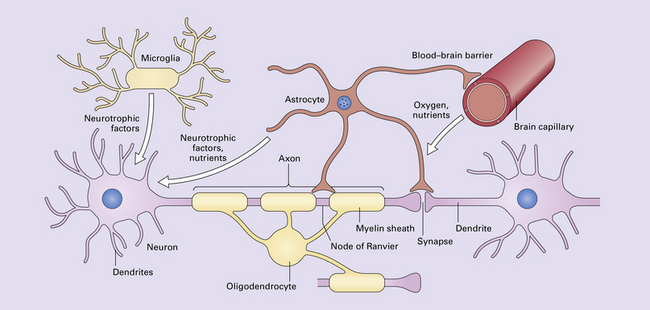
1.29 Normal neural function requires neurons, glia, and vasculature.
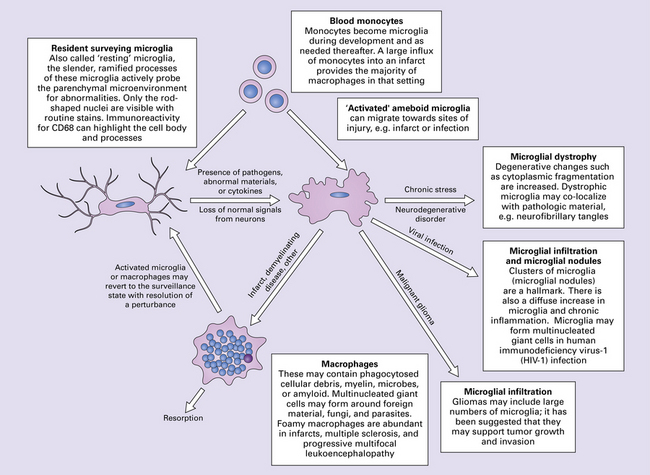
Networks and circuits of neurons, through the electrochemical signaling of axonal action potentials and synaptic neurotransmitters, are the central components of neural communication. Less appreciated is the importance of the other 90% of brain cells which are glia, mostly astrocytes and oligodendrocytes. Each oligodendrocyte maintains the myelin sheaths that surround several adjacent axons, enabling rapid saltatory conduction and supporting the integrity of the axons. Astrocytes modulate synaptic transmission and vasodilation, facilitate synaptogenesis and synaptic plasticity, maintain the blood–brain barrier, regulate extracellular ion concentrations, support neuronal intermediary metabolism, provide neuroprotective factors, support oligodendrocyte myelination, and are part of glial-neuronal signaling networks. They also provide structure and are responsible for the CNS equivalent of scarring: gliosis. Microglia provide immunosurveillance, antigen-presentation, phagocytic capabilities, other pro-inflammatory functions, and neurotrophic support for neurons. Glia are also critical for brain development. In this tightly intertwined neuroglial-vascular network that sustains, protects and optimizes neural processing, derangement of any one component can adversely affect brain function, resulting in neurologic disease.

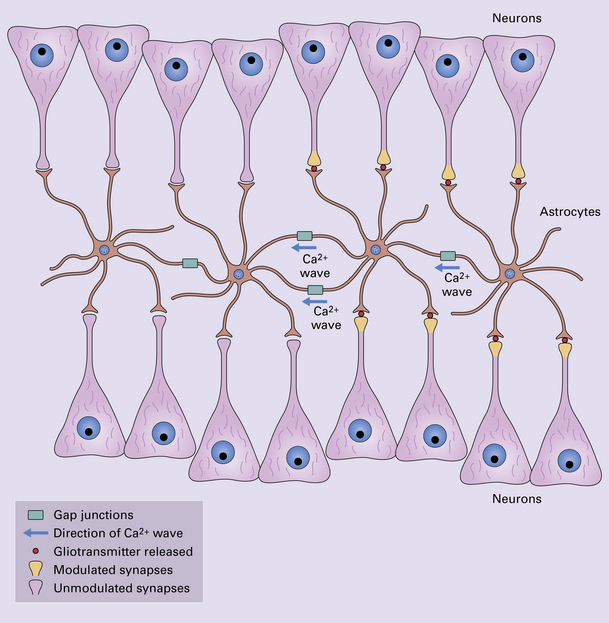
1.32 Glial-neuronal networks.
Neural processing may be better understood in the context of glial-neuronal networks rather than neuronal networks alone. Populations of astrocytes, linked by gap junctions, can form a glial syncytium. When a Ca2 + wave is propagated through the astrocytic population via these junctions, the astrocytes release gliotransmitters such as glutamate or ATP. As each astrocyte contacts multiple neurons and many thousands of synapses, the astrocytes can modulate synaptic transmission in large groups of neurons. Through their metabolic, vasomodulative, trophic, and structural support for neurons, astrocytes contribute to a higher order organization of neural communications.
PATHOLOGIC REACTIONS OF OLIGODENDROGLIA
IMMUNOHISTOCHEMICAL FEATURES
DEMYELINATION AND REMYELINATION
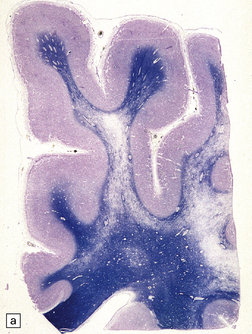
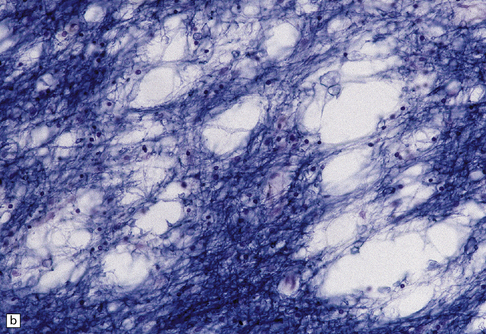
Demyelination, or loss of normal myelin with relative preservation of axons, may result from selective destruction of the myelin sheath, or may be secondary to damage to the oligodendrocyte cell body. Demyelination is often immunologically mediated, as in multiple sclerosis (see Chapter 19) and acute disseminated encephalomyelitis (see Chapter 20), but can also be caused by toxic and metabolic insults (see Chapters 22 and 25), viral infection (see Chapter 13), compression (see Chapter 20), and ischemia (see Chapter 9). Areas of demyelination show loss of staining with Luxol fast blue, or solochrome cyanin (Fig. 1.33). Electron microscopy demonstrates the absence of myelin sheaths. Remyelinated nerve fibers have myelin sheaths that are abnormally thin in relation to the caliber of the axon (Fig. 1.33). Groups of remyelinated fibers are visible histologically as discrete areas of white matter in which staining of myelin is present but reduced (Fig. 1.33).
1.33 Demyelination and remyelination.
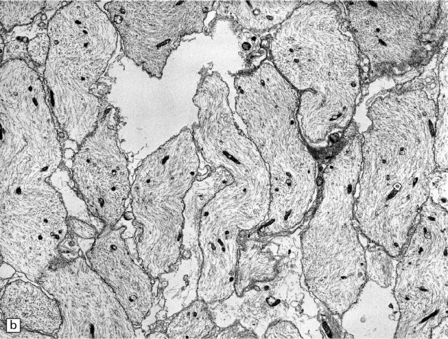
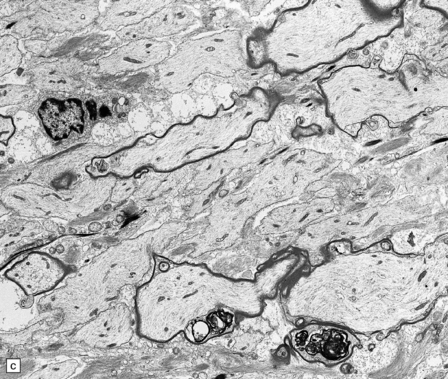
(a) Section of brain in multiple sclerosis. Normal myelin has been stained deep blue, with Luxol fast blue/cresyl violet. The absence of Luxophilic staining from the periventricular region (arrowhead) corresponds to a chronic plaque of demyelination. The white matter also includes areas of remyelination, known as shadow plaques (arrow), which have stained only weakly with the Luxol fast blue. (b) Electron microscopy of demyelinated white matter (in this case due to vascular compression of the trigeminal nerve root; see Chapter 20) shows axons lacking myelin sheaths. (c) Electron microscopy shows this plaque, from a patient with multiple sclerosis, to comprise a mixture of demyelinated and thinly-remyelinated nerve fibers. Some of the oligodendrocytes contain small amounts of myelin debris.
DYSMYELINATION
This term is sometimes used to describe the failure to form and/or maintain myelin sheaths that occurs in the leukodystrophies, a heterogeneous collection of genetically-determined disorders of white matter. Most of these dysmyelinating disorders are due to defects of peroxisomal or lysosomal metabolism. The leukodystrophies are considered in Chapters 5 and 23.
INTRAMYELINIC VACUOLATION
Several disorders of the CNS cause a distinctive pattern of edema in which the initial accumulation of fluid is in ‘vacuoles’ formed by separation of the layers of the myelin sheath along intraperiod lines. On light microscopy, this intramyelinic vacuolation gives the white matter a spongy appearance (Fig. 1.34). The causes include some mitochondrial encephalopathies (particularly Kearns–Sayre syndrome; see Chapter 24), Canavan’s disease and several disorders of amino acid metabolism (see Chapter 5), vitamin B12 deficiency (see Chapter 21), various toxins (see Chapter 25), and retroviral infections – vacuolar myelopathy due to HIV, and myelopathy due to HTLV-1 (see Chapter 13).
AXONAL DEGENERATION
As noted above, axonal degeneration, of whatever etiology, leads to fragmentation and degeneration of myelin, and its subsequent phagocytosis and degradation by macrophages. The degradation products can be detected by Marchi’s method, and oil red O or other stains for neutral lipid (Fig. 1.1). The infiltrating macrophages are well demonstrated with specific immunohistochemical markers (e.g. antibody to CD68).
INCLUSIONS
Oligodendrocytes accumulate cytoplasmic aggregates of filamentous or microtubular material in several neurodegenerative diseases. Inclusions that consist largely of tau protein are a feature of progressive supranuclear palsy, corticobasal degeneration, and frontotemporal lobar degeneration linked to chromosome 17 (FTDP-17). In these diseases (discussed in detail in Chapters 28 and 31), tau inclusions occur in the perinuclear oligodendrocyte cytoplasm where they often partially encircle the nucleus and are called ‘coiled bodies’ (Fig. 1.35). In progressive supranuclear palsy, delicate strands of aggregated tau also occur in the in the inner and outer loops of the myelin sheath, forming inclusions know as ‘glial threads’ or ‘interfascicular threads’. In multiple system atrophy (see Chapter 28), oligodendrocytes form ‘glial cytoplasmic inclusions’ (GCIs) – curved, filamentous, flame- or sickle-shaped inclusions that often partly encircle the nucleus (Fig. 1.35). They are readily demonstrable by Gallyas silver impregnation, and immunohistochemically with antibodies to α-synuclein (Fig. 1.35). They can also be labeled with antibodies to ubiquitin, and some antibodies to tau. Electron microscopy shows GCIs to consist of microtubular structures with a coating of granular material.
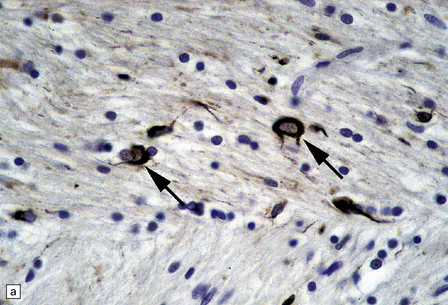
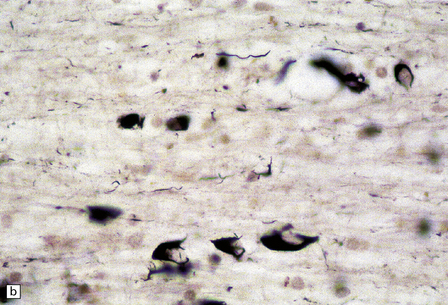
PATHOLOGIC REACTIONS OF EPENDYMA
RESPONSES TO INJURY
Ependymal cells are usually columnar with basal nuclei and prominent cilia (Fig. 1.36). A distinct ependymal lining is present around the central canal of the spinal cord in utero and during infancy, but with age this cavity is largely obliterated, leaving discontinuous stretches of canal and disorganized clusters of ependymal cells in later life (Fig. 1.37). Enlargement of an intact central canal (hydromyelia) is usually an incidental finding at necropsy (Fig. 1.37). It has been speculated that in some patients with hydromyelia, progressive enlargement of the central canal may lead to injury and sloughing of ependyma, with progressive enlargement, and transformation into syringomyelia (see Chapter 3).
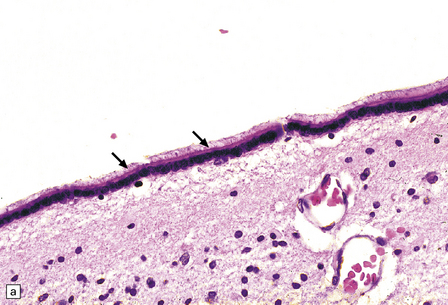
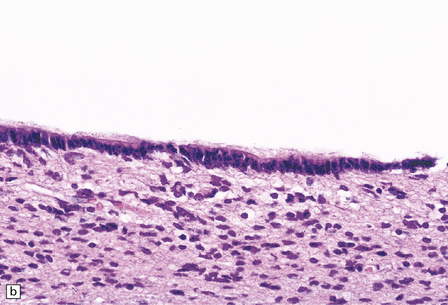
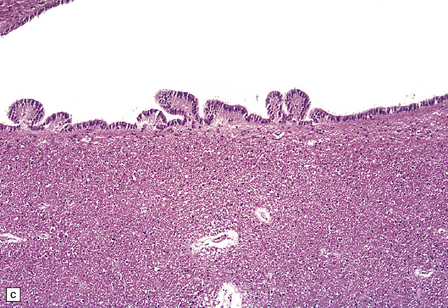
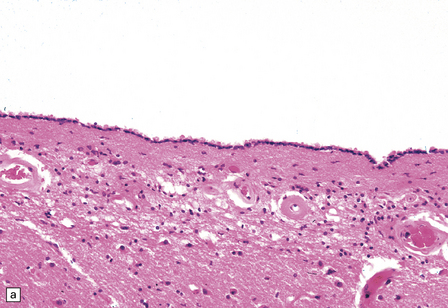
1.36 Normal ependyma.
(a,b) Note variable density of subependymal astrocytes. (a) ependyma adjacent to hippocampus; (b) ependyma adjacent to basal ganglia. The ependymal cells are columnar or cuboidal, with basal nuclei. The ventricular surface of ependyma has prominent cilia (arrows in a). (c) Ependyma can sometimes appear slightly folded, with protrusions into the ventricular cavity; contrast this appearance with that of subventricular glial nodules in Fig. 1.39.
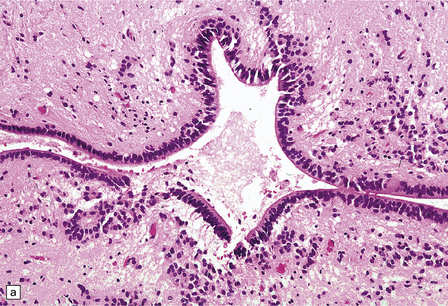
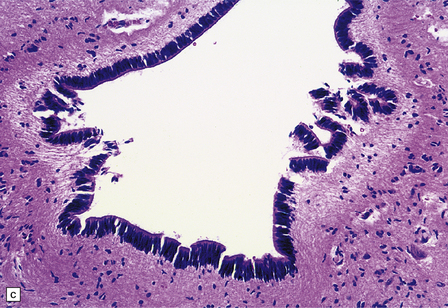
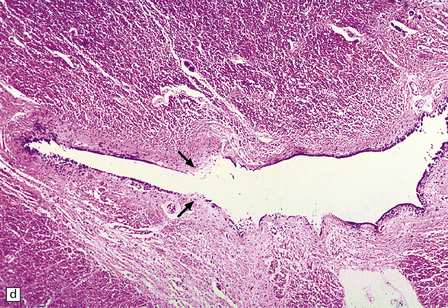
1.37 Central canal of the spinal cord.
(a) In transverse sections through the cord in infants and children this appears as a circular, oval or ‘forked’ cavity surrounded by ciliated columnar ependymal cells similar to those illustrated in Fig. 1.32. (b) With age, the central lumen of the canal tends to disappear, leaving a variably disorganized meshwork of ependymal cells. (c) and (d) enlargement of central canal. In (d) there is focal loss of ependyma (arrows), possibly representing an early stage of syringomyelia. Note the prominent astrocytosis underlying the regions of ependymal loss.
The following histopathologic responses may occur:
ATROPHY
There is loss of cytoplasm in ependymal cells, which become flattened. Nuclei lose their basal position and encroach upon the rest of the cytoplasm in affected ependymal cells (Fig. 1.38). Atrophy usually occurs in association with hydrocephalus or cerebral atrophy (e.g. in encephalopathies of childhood, age-related dementias).
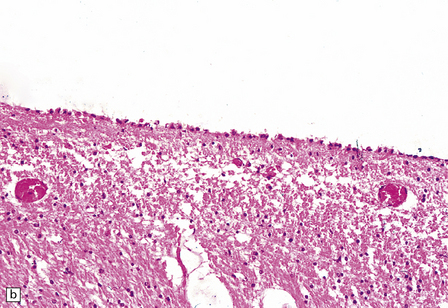
1.38 Atrophy of ependyma.
(a) Contrast appearance with that of normal ependyma, shown in Fig. 1.36. (b) Focal discontinuities of ependyma, possibly a sequel of severe atrophy.
SUBVENTRICULAR GLIOSIS (GRANULAR EPENDYMITIS, EPENDYMAL GRANULATIONS)
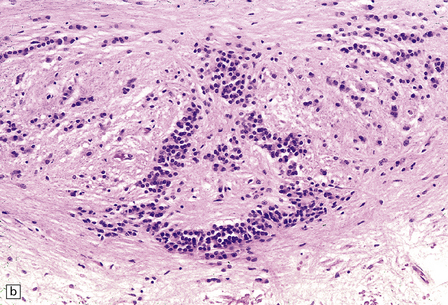
This probably occurs within 1–2 weeks of ependymal tearing or other injury. Proliferation of astrocytes occurs in the subventricular region, and astrocytic processes produce a dense meshwork that extends along the denuded ventricular lining and may form nodular protrusions into the ventricular cavity (Fig. 1.39). The formation of these subventricular glial nodules is sometimes termed ‘granular ependymitis’, although it is not often part of an inflammatory process. When subventricular gliosis is associated with ventricular hemorrhage, hemosiderin-laden macrophages are present.
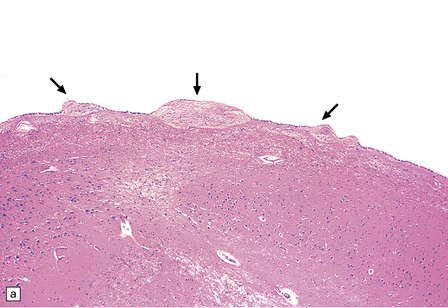
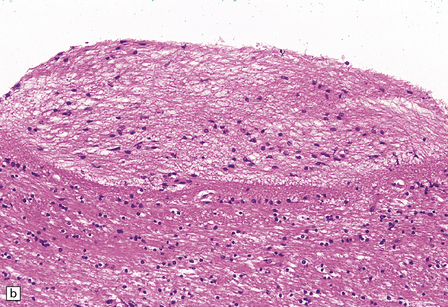
1.39 Subventricular glial nodules.
(a,b) These result from focal loss of ependyma, and subsequent proliferation of subventricular astrocytes. Multiple foci can often be seen along a length of ependyma (arrows in a). (c) Sometimes sheaf-like bundles of glial fibers are prominent within these nodules (arrow). Glial nodules are also referred to as ‘granular ependymitis’.
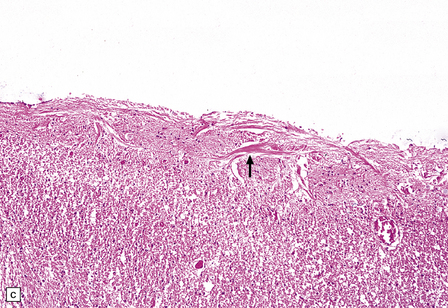
INCLUSIONS IN EPENDYMAL CELLS AND CHOROID PLEXUS EPITHELIUM
These are intracellular accumulations of amyloid fibrils in ependymal cells and choroid plexus epithelium. The number of Biondi bodies increases with age, and they are consistently present in large numbers in the elderly. In choroid plexus epithelium the fibrils may be arranged in wispy strands or large cytoplasmic rings, which are also known as Biondi rings (Fig. 1.40). In ependymal cells the fibrils appear as irregular strands rather than rings. The fibrils consist of tightly packed bundles of straight 10 nm filaments, which stain with thioflavin S and less intensely with Sirius red or Congo red. They have been reported to react with antibody to Aβ-amyloid.
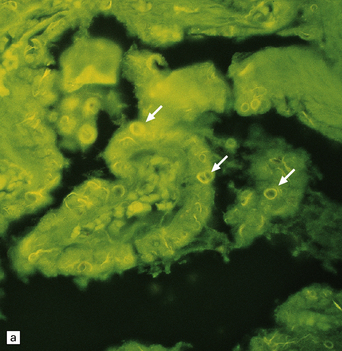
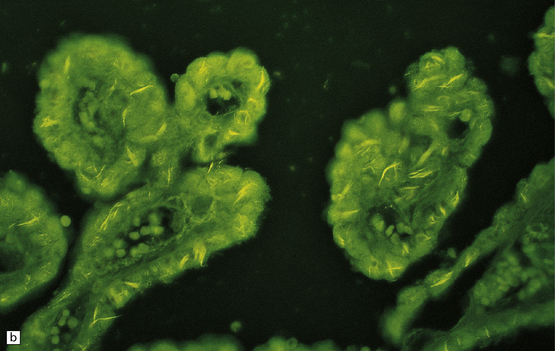
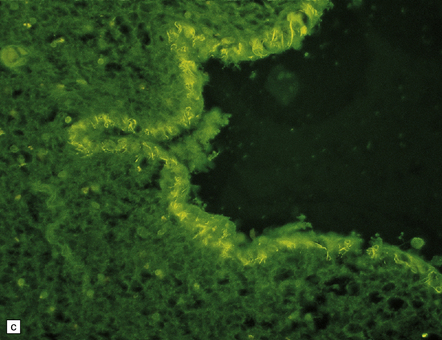
PATHOLOGIC REACTIONS OF MICROGLIA
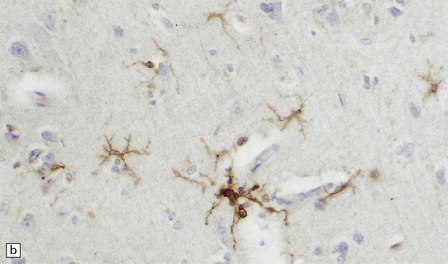
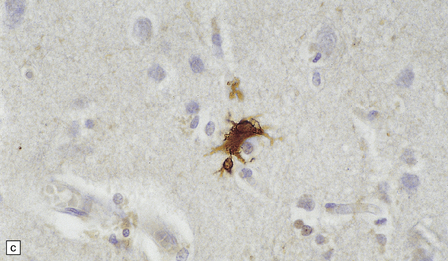
MICROGLIAL ACTIVATION AND ROD CELL FORMATION
Microglia are cells of monocyte lineage and are inconspicuous in the normal brain. They can be demonstrated by silver impregnation (Fig. 1.41a), but this has been largely superseded by immunohistochemical and lectin-binding techniques. The antibodies and lectins that label microglia are mostly those that react with monocyte–macrophage markers (Fig. 1.41b). Microglia in normal brain have been subdivided into:
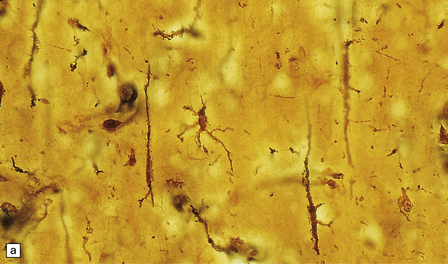
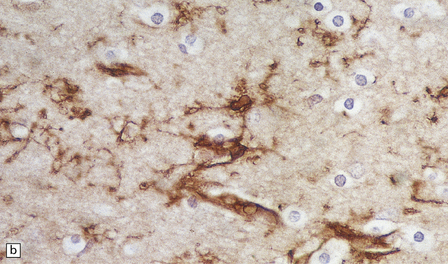
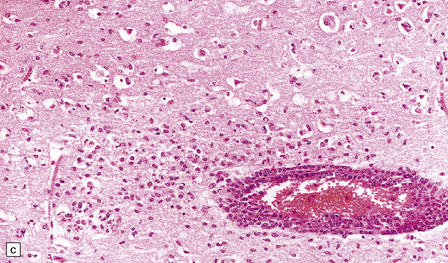
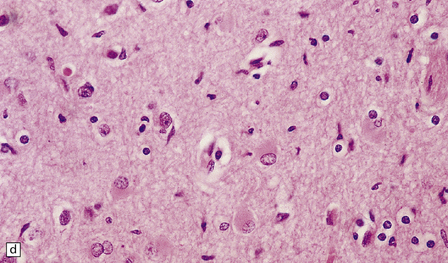
1.41 Microglia.
(a) Microglia can be detected by silver impregnation, as shown in this silver carbonate preparation from a patient with syphilitic general paresis. (b) The silver impregnation technique has been largely superseded by immunostaining for monocyte–macrophage markers such as CD68 or the lectin Ricinus communis agglutinin, as shown here. (c) There is perivascular cuffing and infiltration of the brain by hematogenous monocytes. (d) At higher magnification the infiltrating monocytes or microglia can be recognized by their rod-shaped nuclei.
 Resident microglia, which undergo little turnover with hematogenous monocytes and occur within the CNS parenchyma, though they may abut blood vessels.
Resident microglia, which undergo little turnover with hematogenous monocytes and occur within the CNS parenchyma, though they may abut blood vessels.

 Increased entry of hematogenous monocytes into the CNS (Fig. 1.41c).
Increased entry of hematogenous monocytes into the CNS (Fig. 1.41c).


In sections stained with hematoxylin and eosin, infiltrating monocytes or microglia can be recognized by their rod-shaped nucleus (Fig. 1.41d). These ‘rod cells’ are a prominent finding in chronic infections such as general paresis (see Chapter 16) and chronic viral encephalitides (see Chapter 13).
In regions of tissue damage hematogenous monocytes infiltrate the CNS and phagocytose dead cells and necrotic debris. The accumulation of lipid material by phagocytic cells gives their cytoplasm a foamy appearance in paraffin-embedded material and they are therefore often described as ‘foam cells’ or ‘foamy macrophages’ (Fig. 1.42).
1.42 Microglia and macrophages.
Microglia can release pro-inflammatory factors, reactive oxygen species and nitric oxide, which are injurious to pathogens and potentially to neurons. The secretion of neurotrophic factors and phagocytosis of harmful material can be beneficial. Depending on the stimulus and the balance of these potential responses, activated microglia may have neuroprotective or neurotoxic effects. In some animal models of demyelinating disease, depletion of microglia can reduce disease severity, and in models of infarction, microglia have been reported to minimize infarct size and neuronal apoptosis.
MINERALIZATION IN THE BRAIN
Small mineral deposits can be found in most parts of the brain. They are deeply basophilic, and stain for calcium and, variably, for iron. Early mineralization takes the form of rows of small calcospherites lying along capillaries (Fig. 1.43a). These small deposits may enlarge to form large perivascular concretions (Fig. 1.43b). Larger deposits also occur within the media of small and medium-sized arteries and veins (Fig. 1.43c). Large foci of mineralization are presumed to result from coalescence of smaller deposits (Fig. 1.43d).
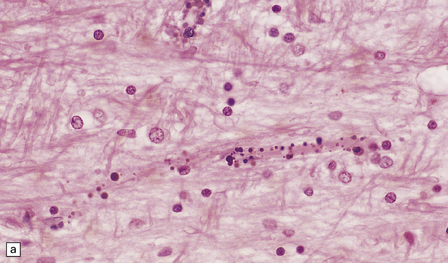
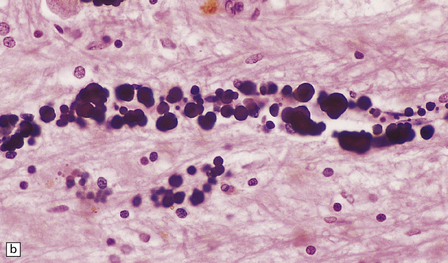
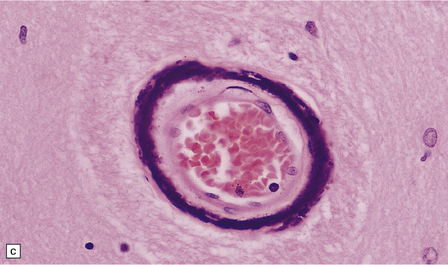
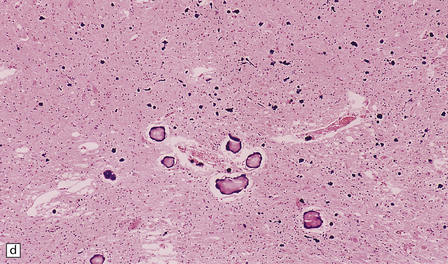
PATHOLOGIC REACTIONS OF BLOOD VESSELS
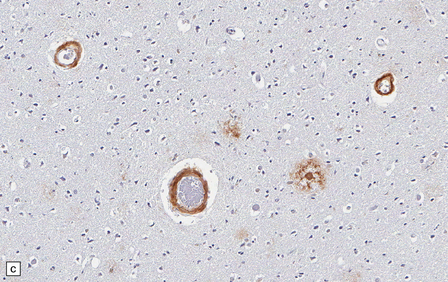
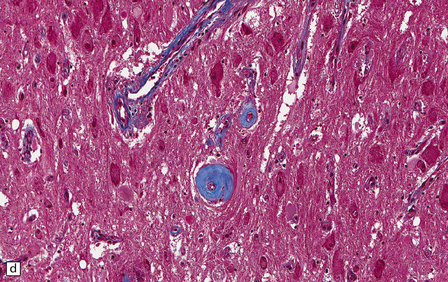
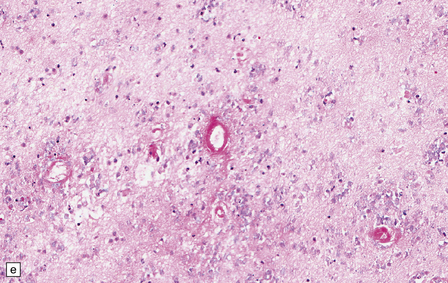
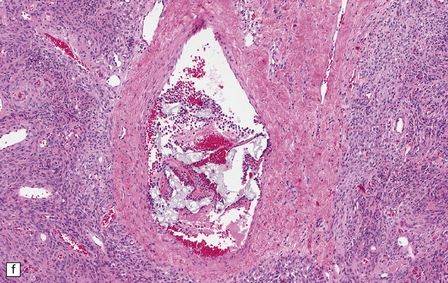
The central nervous system vasculature may demonstrate a broad range of pathologic changes. These are discussed in greater detail in Chapters 7–10. Atherosclerosis, thromboembolic disease, vascular malformations, aneurysms, vasculitides, infections, and other vasculopathies may involve the blood vessels of the brain. Highlighted here are some entities that are distinctive or that can cause diagnostic confusion. In cerebral amyloid angiopathy (Chapter 31), the amyloid is rarely that seen in systemic amyloidoses (Fig. 1.44a–c). Amyloid-β (Aβ), the same protein as that in the plaques of Alzheimer disease, is the most common vascular deposit in elderly patients. The involved blood vessels are typically leptomeningeal or cortical; not surprisingly therefore, rupture of Aβ-laden blood vessels usually results in superficial lobar hemorrhage. Hypertension can result in arteriosclerosis, including arteriolosclerosis (Chapter 31). Hypertensive hemorrhages often involve deep gray matter and may extend into the ventricles. With routine stains, the collagen of arteriolosclerosis can be difficult to distinguish from amyloid angiopathy but a combination of amyloid and collagen stains can be helpful (Fig. 1.44a–d). Amyloid deposition and atherosclerosis may co-exist. Fibrinoid necrosis of the blood vessel wall typically occurs in the context of radiation necrosis or vasculitis (Fig. 1.44e) and should not be confused with amyloid angiopathy. The patient’s presentation and other pathologic features usually obviate the necessity for special stains. In addition to thromboemboli, intravascular iatrogenic foreign material can be seen. Meningiomas and vascular malformations are often embolized preoperatively with polyvinyl alcohol or acrylic particles. The embolic material leaves intravascular areas devoid of red blood or white blood cells and fibrin. Intermediate to large-sized blood vessels within the tumor, vascular malformation, or dura are most likely to be affected (Fig. 1.44f). Depending on the type of embolic material, the embolic material may appear amorphous, pigmented material, or as empty spaces if the material is removed during processing. Hydrophilic polymers stripped from the coatings of interventional devices during endovascular procedures can occlude microvessels in the brain parenchyma (Fig. 1.44g) resulting in microinfarcts. The polymers are often gray to basophilic and may be serpentine or needle-shaped. The polymers are not polarizable and may elicit little inflammatory response.
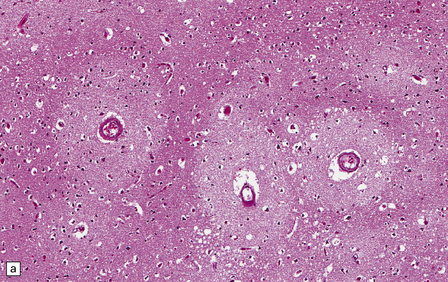
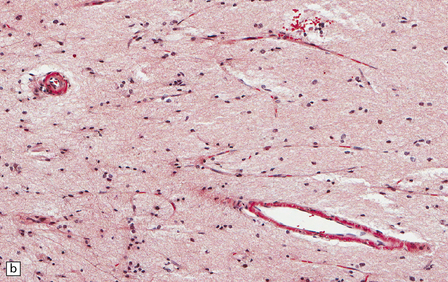
1.44 Vascular pathology.
(a) Slightly smudgy eosinophilic vascular walls suggest amyloid. (b) Congo red stains the amyloid deep pink or red (and shows apple-green birefringence if viewed with polarized light). (c) Antibody to amyloid-β (Aβ) labels the most common form of cerebral amyloid angiopathy. (d) Mural collagen in arteriolosclerosis is well seen with a trichrome stain. (e) Fibrinoid necrosis in radiation necrosis. (f) Amorphous intravascular embolic material in a meningioma forms an irregular meshwork that is devoid of blood. (g) Hydrophilic polymer embolus in a parenchymal microvessel.
REFERENCES
Dickson, D.W. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol.. 2009;3:1–23.
Fuller, G.N., Burger, P.C. Central nervous system. In: Mills S., ed. Histology for pathologists. 3 rd edn. Philadelphia: Lippincott-Williams & Wilkins; 2007:274–320.
Halliday, G.M., Holton, J.L., Revesz, T., et al. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol.. 2011;122:187–204.
Perl, D.P. Neuropathology of Alzheimer’s disease. Mt Sinai J Med.. 2010;77:32–42.
Schott, J.M., Reiniger, L., Thom, M., et al. Brain biopsy in dementia: clinical indications and diagnostic approach. Acta Neuropathol.. 2010;120:327–341.
Pathologic reactions of neurons
Beach, T.G., Walker, D.G., Sue, L.I., et al. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol.. 2004;63:329–337.
Dickson, D.W. α-synuclein and the Lewy body disorders. Curr Opin Neurol.. 2001;14:423–432.
Goedert, M., Spillantini, M.G., Davies, S.W. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol.. 1998;8:619–632.
Hirano, A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol.. 1994;20:3–11.
Mackenzie, I.R. The neuropathology of FTD associated With ALS. Alzheimer Dis Assoc Disord.. 2007;21:S44–S49.
Mackenzie, I.R., Rademakers, R., Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol.. 2010;9:995–1007.
Martinez-Saez, E., Gelpi, E., Rey, M., et al. Hirano body-rich subtypes of Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol.. 2011. [doi:org/10.1111/j.1365–2990.2011.01208.x].
Mizuno, Y., Fujita, Y., Takatama, M., et al. Peripherin partially localizes in Bunina bodies in amyotrophic lateral sclerosis. J Neurol Sci.. 2011;302:14–18.
Okamoto, K., Mizuno, Y., Fujita, Y. Bunina bodies in amyotrophic lateral sclerosis. Neuropathology.. 2008;28:109–115.
Thal, D.R., Del Tredici, K., Ludolph, A.C., et al. Stages of granulovacuolar degeneration: their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol.. 2011;122:577–589.
Turnbull, J., Wang, P., Girard, J.M., et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann Neurol.. 2010;68:925–933.
Pathologic reactions of astrocytes and oligodendrocytes
Ahmed, Z., Doherty, K.M., Silveira-Moriyama, L., et al. Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol.. 2011;122:415–428.
Bradl, M., Lassmann, H. Oligodendrocytes: biology and pathology. Acta Neuropathol.. 2010;119:37–53.
Fellner, L., Jellinger, K.A., Wenning, G.K., et al. Glial dysfunction in the pathogenesis of a-synucleinopathies: emerging concepts. Acta Neuropathol.. 2011;121:675–693.
Iwaki, T., Iwaki, A., Tateishi, J., et al. αB-crystallin and 27-kd heat shock protein are regulated by stress conditions in the central nervous system and accumulate in Rosenthal fibers. Am J Pathol.. 1993;143:487–495.
Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol.. 1999;9:663–679.
Love, S. Demyelinating diseases. J Clin Pathol.. 2006;59:1151–1159.
Matsusaka, H., Ikeda, K., Akiyama, H., et al. Astrocytic pathology in progressive supranuclear palsy: significance for neuropathological diagnosis. Acta Neuropathol.. 1998;96:248–252.
Quinlan, R.A., Brenner, M., Goldman, J.E., et al. GFAP and its role in Alexander disease. Exp Cell Res.. 2007;313:2077–2087.
Sidoryk-Wegrzynowicz, M., Wegrzynowicz, M., Lee, E., et al. Role of astrocytes in brain function and disease. Toxicol Pathol.. 2011;39:115–123.
Takahashi, M., Weidenheim, K.M., Dickson, D.W., et al. Morphological and biochemical correlations of abnormal tau filaments in progressive supranuclear palsy. J Neuropathol Exp Neurol.. 2002;61:33–45.
Takeda, A., Arai, N., Komori, T., et al. Tau immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett.. 1997;234:63–66.
Verkhratsky, A., Parpura, V. Recent advances in (patho)physiology of astroglia. Acta Pharmacol Sin.. 2010;31:1044–1054.
Wakabayashi, K., Takahashi, H. Cellular pathology in multiple system atrophy. Neuropathology.. 2006;26:338–345.
Wilhelmus, M.M., Verhaar, R., Bol, J.G., et al. Novel role of transglutaminase 1 in corpora amylacea formation? Neurobiol Aging.. 2011;32:845–856.
Pathologic reactions of ependyma
Biondi, G. Zur Histopathologie des menschlichen Plexus Chorioideus und des Ependyms. Arch Psychiatr Nervenkr.. 1934;101:666–728.
Bruni, J.E. Ependymal development, proliferation, and functions: a review. Microsc Res Tech.. 1998;41:2–13.
Eriksson, L., Westermark, P. Characterization of intracellular amyloid fibrils in the human choroid plexus epithelial cells. Acta Neuropathol.. 1990;80:597–603.
Sarnat, H.B. Regional differentiation of the human fetal ependyma: immunocytochemical markers. J Neuropathol Exp Neurol.. 1992;51:58–75.
Sarnat, H.B. Ependymal reactions to injury. A review. J Neuropathol Exp Neurol.. 1995;54:1–15.
Vinters, H.V. Neuropathology of syringomyelia. In: Batzdorf U., ed. Syringomyelia: current concepts in diagnosis and treatment. Baltimore: Williams & Wilkins; 1991:35–58.
Wolburg, H., Paulus, W. Choroid plexus: biology and pathology. Acta Neuropathol.. 2010;119:75–88.
Pathologic reactions of microglia
Graeber, M.B., Streit, W.J. Microglia: biology and pathology. Acta Neuropathol.. 2010;119:89–105.
Kato, H., Walz, W. The initiation of the microglial response. Brain Pathol.. 2000;10:137–143.
Streit, W.J., Walter, S.A., Pennell, N.A. Reactive microgliosis. Prog Neurobiol.. 1999;57:563–581.
Wirenfeldt, M., Babcock, A.A., Vinters, H.V. Microglia – insights into immune system structure, function, and reactivity in the central nervous system. Histol Histopathol.. 2011;26:519–530.
Pathologic reactions of vasculature
Fealey, M.E., Edwards, W.D., Giannini, C., et al. Complications of endovascular polymers associated with vascular introducer sheaths and metallic coils in 3 patients, with literature review. Am J Surg Pathol.. 2008;32:1310–1316.
Love, S. Autopsy approach to stroke. Histopathology.. 2011;58:333–351.
Mehta, R.I., Mehta, R.I., Solis, O.E., et al. Hydrophilic polymer emboli: an under-recognized iatrogenic cause of ischemia and infarct. Mod Pathol.. 2010;23:921–930.
Werring D.J., ed. Cerebral microbleeds: Pathophysiology to clinical practice. Cambridge: Cambridge University Press, 2011.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]1
Pathologic reactions in the CNS
PATHOLOGIC RESPONSES IN NEURONS
NORMAL NEURONAL CYTOLOGY AND STAINING
The cytologic appearance of neurons varies depending upon their location. In general, neurons have moderate to abundant cytoplasm and a relatively large round nucleus with a prominent nucleolus. Projecting from the neuronal cell body are branching dendrites and a single axon. A range of tinctorial and immunohistochemical staining techniques are used for demonstrating neurons and their processes (Table 1.1).
Table 1.1
Histologic demonstration of neurons: techniques and comments concerning their applications
Conventional staining
Hematoxylin and eosin
Good for assessing general cytoarchitecture
Nissl stain (e.g. cresyl fast violet)
Good for assessing general cytoarchitecture. Allows estimation of cell density in thick sections; may be combined with stains for myelin (e.g. Luxol fast blue).
Axon silver impregnation techniques
Good for demonstration of axons and some neuronal inclusions. Used in conjunction with myelin stains to distinguish between demyelination and fiber degeneration
Golgi stain
Allows visualization of fine detail of neuronal cell processes. A technically difficult stain to perform which depends on block impregnation
Immunohistochemistry
Neurofilament proteins
Strongly expressed in perikaryal and axonal cytoplasm (in normal neurons, perikaryal neurofilament proteins are non-phosphorylated and axonal neurofilament proteins phosphorylated)
NeuN
Neuron-specific nuclear protein, strongly expressed in neuronal nuclei. NeuN antibodies also show weak labeling of the neuronal cytoplasm
Neuron specific enolase
Strongly expressed in neuronal and axonal cytoplasm
Synaptophysin
Antibodies to synaptophysin detect neurosecretory vesicles, which are mainly located at synapses, with little staining of neuronal cell bodies
PGP9.5
An antibody to neuron-specific ubiquitin C-terminal hydrolase, which is abundant in neurons and neuronal cell processes
Chromogranin A
Antibodies detect dense-core neurosecretory vesicles, which are sparsely distributed within the perikaryon of some neurons and concentrated at the synapses. Moderate staining of neuronal cell bodies
Neurotransmitter-related
Antibodies allow detection of neurotransmitter substance or enzyme involved in its biosynthesis
ABNORMALITIES OF NEURONAL MORPHOLOGY
Axonal degeneration inevitably follows the death of the neuron of which it is a part. A severed or severely damaged axon undergoes distal degeneration without usually provoking death of the proximal part of the neuron, although this does undergo a series of structural and metabolic changes (i.e. axon reaction and chromatolysis, see below). Within days, the distal part of the axon fragments and the surrounding myelin sheath breaks up into ovoids. Over the next 3 weeks or so, the axon and myelin debris are taken up by macrophages, which infiltrate the degenerating fiber tracts. Several methods can be used to demonstrate degenerating nerve fibers in the CNS. These include specialized silver impregnation techniques and stains for degenerating myelin and for lipid (Fig. 1.1).
AXON REACTION AND CHROMATOLYSIS
Damage to the axon provokes a series of morphologic and biochemical changes in the neuronal cell body. These are collectively referred to as the axon reaction (Figs 1.2, 1.3). The changes include disruption and dispersion of Nissl bodies (chromatolysis) associated with rearrangement of the cytoskeleton and marked accumulation of intermediate filaments. The axon reaction is conspicuous in large neurons with axons that project into the peripheral nervous system and in some of the larger neurons with central projections. Chromatolysis is not visible on conventional light microscopy of small neurons or certain large neurons such as the cerebellar Purkinje cells, but changes can be demonstrated in these cells by electron microscopy and immunohistochemistry.
1.2 Axon reaction.
The changes that constitute the axon reaction have been most extensively investigated in anterior horn cells after peripheral nerve injury, as depicted in this figure, but neurons undergo similar alterations after injury to axons in the CNS. (a) A normal neuronal cell body is characterized by well-organized dendritic branches making synaptic contact with other neurons. Nissl bodies are prominent. (b) Following axonal damage there is degeneration of the peripheral axon. Dendrites that normally form synapses retract and synaptic contacts are lost. The rough endoplasmic reticulum becomes reorganized into small vesicular elements. The neuron inactivates genes coding for high molecular weight neurofilament protein (NFH) and switches on genes for peripherin. Neurofilaments and peripherin accumulate in the neuronal cell body. At this stage the neuronal cell body is swollen, shows only weak acidophilia, and lacks Nissl bodies. Denervated muscle fibers atrophy. (c) The axon regenerates in the peripheral nervous system (but not within the CNS). NFH synthesis is re-established with axonal elongation. (d) If the target tissue is reinnervated, the neuronal dendrites re-establish synaptic contact with other neurons and the normal cytologic appearance of the cell body is regained.
SWOLLEN NEURONS
Swollen or ballooned neurons are a feature both of the axon reaction and of a variety of diseases in which perikaryal changes occur independently of axonal damage (Table 1.2). Histologically, they appear as distended, weakly-staining cells with large, relatively clear nuclei (Fig. 1.4). Occasionally the cells contain small vacuoles. In conventionally processed tissues an artefactual lacuna is often present around the abnormal cell. Swollen or ballooned neurons can be demonstrated with several immunohistochemical markers (Fig. 1.5, Table 1.3).
Table 1.2
Causes of ballooning of neurons
Physiologic
Anterior horn motor neurons, with age
Nutritional
Pellagra (niacin deficiency): spinal, brain stem, and cortical neurons
Developmental
Localized cortical dysplasia with cytomegaly
Cortical dysplasia with hemimegalencephaly
Tuberous sclerosis
Neurodegenerative
Several types of frontotemporal lobar degeneration, including Pick’s disease and FTDP-17: neocortical and some basal neurons
Corticobasal degeneration: neocortical and some basal neurons
Swollen neurons can also occur in Alzheimer disease, argyrophilic grain disease, progressive supranuclear palsy and several other neurodegenerative diseases
Prion disease
Cortical and some basal neurons
Table 1.3
Immunohistochemical profile of swollen neurons
Neurofilament protein
Accumulation of highly phosphorylated high-molecular-weight neurofilament protein, which is normally restricted to the axon
αB-crystallin
Accumulation of αB crystallin, which is not normally expressed by neurons
Tau protein
Accumulation of abnormally phosphorylated tau protein by a proportion of swollen neurons in corticobasal degeneration and Pick’s disease
Ubiquitin
Variably increased immunoreactivity for ubiquitin-protein conjugates
PGP9.5
Variably increased immunoreactivity for this ubiquitin C-terminal hydrolase
1.4 Swollen neurons.
The cytoplasm lacks Nissl bodies and is weakly eosinophilic. The nucleus is large and vesicular. (a) Example from the anterior horn of the spinal cord. (b) Example from the cerebral cortex in corticobasal degeneration (see Chapter 28).
1.5 αB-crystallin immunoreactivity.
This is a characteristic feature of swollen neurons in most conditions. The protein is not, however, expressed by swollen neurons in pellagra (see Chapter 21), suggesting that neuronal swelling in this disorder has a different mechanism.
TRANS-SYNAPTIC NEURONAL DEGENERATION AND OLIVARY HYPERTROPHY
Neurons within some nuclei in the CNS atrophy and degenerate in response to deafferentation. Examples are neurons in the lateral geniculate nucleus, which degenerate after optic nerve or tract lesions, and neurons in the pontine nuclei, which degenerate after interruption of descending frontopontine afferent fibers. Neurons in the inferior and accessory olivary nuclei undergo an unusual form of trans-synaptic degeneration after a destructive lesion (such as an infarct) of the ipsilateral central tegmental tract (Fig. 1.6). The olivary ribbon as a whole becomes thickened and neurons show marked enlargement, cytoplasmic vacuolation, and some dispersion of Nissl bodies. Olivary hypertrophy is associated with the development of palatal myoclonus in some patients.
1.6 Hypertrophy of the left olive due to a lesion involving the left central tegmental tract.
(a) Normal right olive. (b) Hypertrophy of the left olive. The lesion involving the left central tegmental tract is not shown. Compare the normal-sized neurons in the right olive with the enlarged vacuolated neurons in the left olive. (c) Normal neurons of the right inferior olivary nucleus stained with cresyl violet. (d) Neurons of the hypertrophic left inferior olive are enlarged and vacuolated.
HYPOXIC CELL CHANGE
Neurons are especially vulnerable to damage from hypoxia, which causes the following distinctive histologic changes (Fig. 1.7):
1.7 Hypoxic cell change in neurons.
Affected neurons become shrunken and eosinophilic. The nuclei become condensed and lose their crisp contours. In this illustration the neuron in the center is affected while surrounding neurons appear normal.
Microvacuolation (visible only at an ultrastructural level), due to swelling of endoplasmic reticulum and mitochondria.
Shrinkage of the cell body and increasing cytoplasmic acidophilia.
Neurons in certain parts of the brain that are especially vulnerable to hypoxic damage are:
pyramidal neurons in the CA1 field of the hippocampus
The pattern of regional susceptibility to hypoxia differs between infants and adults (see Chapters 2 and 8).
FERRUGINATION
Although dead neurons usually undergo liquefaction or are removed by phagocytosis, they may instead become encrusted and replaced by mineral salts, a process termed ferrugination. This is particularly prominent in the infant brain in response to hypoxic–ischemic damage (Fig. 1.8).
1.8 Ferruginated neurons.
Ferruginated neurons become encrusted in mineral deposits and appear dark purple in sections stained with hematoxylin and eosin. The mineral deposits contain both calcium and iron and can be stained by Perls’ method and von Kossa’s technique. (a) Many ferruginated neurons in the cortex of a neonate who had hypoxic brain damage. Astroglial cells are not affected. (b) Higher power illustration of neurons replaced by mineral deposits.
NUCLEAR INCLUSIONS
MARINESCO BODIES
These are small spherical nuclear inclusions that are brightly eosinophilic and are often seen in neurons of the adult substantia nigra (Fig. 1.9a). They may also occur in other neurons, such as the pyramidal cells of the hippocampus and neurons in the tegmentum of the brain stem. Marinesco bodies are most common in the elderly and in dementia with Lewy bodies. They have an increased prevalence in neurons containing Lewy bodies.
1.9 Marinesco bodies.
These intranuclear inclusions are visible in several neuronal groups. (a) Marinesco bodies are brightly eosinophilic intranuclear inclusions about the same size as a large nucleolus. (b) As shown here, they are strongly immunoreactive for ubiquitin.
Ultrastructurally, Marinesco bodies are composed of filaments with the same diameter as intermediate filaments, and may be derived from the nuclear lamins. They are immunoreactive for ubiquitin, an 8 kD polypeptide involved in the degradation of many abnormal or short-lived proteins (Fig. 1.9b).
NEURONAL CYTOPLASMIC INCLUSIONS
CYTOSKELETAL AND FILAMENTOUS INCLUSIONS
Hirano bodies
These are brightly eosinophilic rod-shaped or elliptical cytoplasmic inclusions that may appear to overlap the edge of a neuron (Fig. 1.11). They are immunoreactive for actin, actin-associated proteins, and caspase-cleaved TDP43.
ROD-LIKE CYTOPLASMIC INCLUSIONS
These eosinophilic cytoplasmic inclusions occur in large neurons of the caudate nucleus. The inclusions resemble loose bundles of twigs and, like the thalamic neuronal inclusions described above, are common in patients with myotonic dystrophy. However, they are sometimes an incidental finding in elderly patients who do not have neurologic or muscular disease. The rod-like inclusions stain strongly with phosphotungstic acid/hematoxylin (Fig. 1.12).
1.12 Rod-like cytoplasmic inclusions in the caudate nucleus.
These occur exclusively in large neurons in the caudate nucleus. The inclusions may be an incidental finding, particularly in the elderly, but are most common in myotonic dystrophy. (Courtesy of Dr D A Hilton, Derriford Hospital, Plymouth.)
OTHER FILAMENTOUS NEURONAL INCLUSIONS
There are several other types of neuronal inclusion that comprise or include elements of the cytoskeleton and usually occur in the context of specific neurodegenerative diseases (Table 1.4). These inclusions are described in more detail and illustrated in the sections concerned with the relevant diseases.
Table 1.4
Examples of inclusion bodies in specific conditions and diseases
| Inclusion | Association | Main constituents |
| Lewy bodies | Aging, Parkinson’s disease, dementia with Lewy bodies | α-synuclein, neurofilament protein, and ubiquitin |
| Neurofibrillary tangles | Aging, Alzheimer disease, progressive supranuclear palsy, post-encephalitic parkinsonism, Guam parkinsonism–dementia, myotonic dystrophy, subacute sclerosing panencephalitis, Niemann–Pick disease type C, other rare disorders | Phosphorylated tau protein (3R, 4R), ubiquitin |
| Pick bodies | Pick’s disease | Neurofilament protein, phosphorylated tau protein (3R), ubiquitin |
| MND inclusions | Motor neuron disease/amyotrophic lateral sclerosis | TAR DNA-binding protein 43 (sporadic cases) or superoxide dismutase 1 (some familial cases) or fused-in-sarcoma protein (other familial cases) ubiquitin, p62 |
CYTOSOLIC INCLUSIONS
These are composed of polyglucosans (polymers of sulfated polysaccharides) and are similar to corpora amylacea in composition and staining characteristics (see below). They are present in large numbers in Lafora’s disease (see Chapter 7), both in the CNS and in certain peripheral tissues such as sweat glands, liver, and skeletal muscle. Lafora body formation has been linked to aberrant glycogen hyperphosphorylation. The inclusions usually have a round core that is intensely periodic acid–Schiff (PAS)-positive (Fig. 1.13). Spicules of the core may radiate outwards, into a surrounding zone of less intensely PAS-positive material.
EOSINOPHILIC INCLUSIONS IN THE INFERIOR OLIVES
A characteristic feature of aging is the development of ill-defined eosinophilic inclusions in the neurons of the inferior olivary nuclei. These inclusions are intensely immunoreactive with antibodies to ubiquitin (Fig. 1.14) and should not be confused with lipofuscin, which also accumulates with age (see below) and is particularly abundant in neurons of the inferior olivary nuclei.
MEMBRANE-BOUND CYTOPLASMIC INCLUSIONS
Colloid inclusions are round eosinophilic inclusions that usually occur in neurons in the hypoglossal nuclei (Fig. 1.15), but may be seen in other large neurons, particularly in the elderly. Electron microscopy shows that these inclusions result from dilatation of the endoplasmic reticulum by amorphous material. The importance of recognizing colloid inclusions, which do not have any clinical significance, is that they are occasionally confused with inclusions that are clinically significant such as Lewy bodies, pale bodies, and hyaline inclusions of motor neuron disease (see Chapter 27).
BUNINA BODIES
These are small beaded eosinophilic inclusions seen in motor neurons in motor neuron disease. Cystatin C, transferrin, and peripherin have been detected in Bunina bodies. Ultrastructurally, they appear as electron-dense membrane-bound bodies (see Chapter 27).
INCLUSIONS DERIVED FROM THE ACID VESICLE SYSTEM
The acid vesicle system consists of endosomes, lysosomes, and lysosome-derived dense bodies.
Lipofuscin (Fig. 1.16) is produced by oxidation of lipids and lipoproteins within the lysosomal system. It appears as orange-brown granular material in sections stained with hematoxylin and eosin, and is acid-fast (as demonstrated by the long Ziehl–Neelsen method) and autofluorescent under ultraviolet light. The granules are also sudanophilic and stain with PAS and Schmorl’s stain. Lipofuscin accumulates with aging in neurons and glia, particularly in:
1.16 Lipofuscin.
This accumulates with age in neurons and glia. (a) It has a golden brown color in sections stained with hematoxylin and eosin. (b) Lipofuscin stains strongly with PAS.
Pyramidal neurons in the cerebral cortex and hippocampus.
[/not-level-membership-for-pathology-category]
