Chapter 4 Parkinsonism
Clinical features and differential diagnosis
Introduction
Parkinsonism is a syndrome manifested by a combination of the following six cardinal features: tremor-at-rest, rigidity, bradykinesia, loss of postural reflexes, flexed posture, and freezing (motor blocks). A combination of these signs is used to clinically define definite, probable, and possible parkinsonism (Table 4.1). The most common form of parkinsonism is the idiopathic variety known as Parkinson disease (PD), first recognized as a unique clinical entity by James Parkinson in 1817, who in his An Essay on the Shaking Palsy identified six cases, three of whom he personally examined and the others he observed on the streets of London (Parkinson, 1817). Previously referred to as “paralysis agitans,” Charcot later in the nineteenth century gave credit to Parkinson by referring to the disease as “maladie de Parkinson” and pointed out that slowness of movement should be distinguished from weakness; he also recognized non-tremulous forms of PD (Kempster et al., 2007). With the recognition of marked clinical-pathologic heterogeneity of parkinsonism due to a single mutation and some uncertainty whether PD should be defined clinically, pathologically, or genetically, a variety of other names have been proposed for this neurodegenerative disorder, including “Parkinson complex” and “Parkinson Lewy disease” (Langston, 2006), but it is unlikely that these names will replace the traditional name “Parkinson disease.” Some have argued that PD is not a single entity, a notion supported by genetic forms of parkinsonism with variable clinical and pathologic features (Weiner, 2008).
It was not until 100 years after Parkinson’s landmark paper that the loss of dopamine-containing cells in the substantia nigra (SN) was recognized (Tretiakoff, 1919). In 1960, Ehringer and Hornykiewicz (1960) first noted that the striatum of patients with PD was deficient in dopamine, and the following year, Birkmayer and Hornykiewicz (1961) injected levodopa in 20 patients with PD and postencephalitic parkinsonism and noted marked improvement in akinesia but not in rigidity. Later in the same decade, Cotzias and colleagues (1967, 1969) are credited with making levodopa clinically useful in patients with PD. The recent disclosure of the diagnosis of PD in several public figures has contributed to increased awareness about the disease, which should translate into greater research funding.
Clinical features
There are dozens of symptoms and signs associated with PD, and the clinician must become skilled in eliciting the appropriate history and targeting the neurologic examination in a way that will bring out and document the various neurologic signs (Jankovic and Lang, 2008; Tolosa et al., 2006; Jankovic, 2007, 2008). The manifestation of PD may vary from a barely perceptible tremor to a severe disability during the end-stage of the disease. Rest tremor in the hands or in the lips might be not just socially embarrassing but may cause a severe handicap in people whose occupation depends on a normal appearance. Therefore, it is important that the severity of the disease be objectively assessed in the context of the individual’s goals and needs. In some cases, unintended movements accompanying voluntary activity in homologous muscles on the opposite side of the body, the so-called mirror movements, may occur even in early, asymmetric PD (Espay et al., 2005; Li et al., 2007), although one study showed that mirror movements actually occur less frequently in PD patients than in healthy controls (29% vs. 71%, P < 0.0001) (Ottaviani et al., 2008).
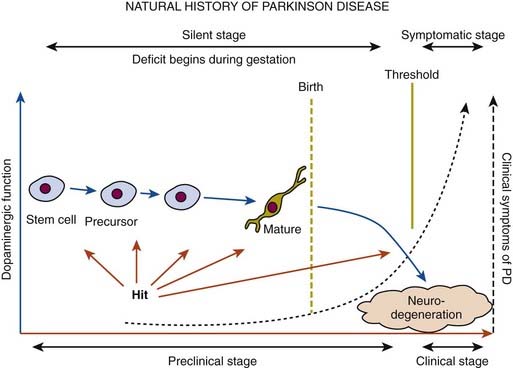
In a retrospective study of patients with PD, early nonspecific symptoms that were reported included generalized stiffness, pain or paresthesias of the limbs, constipation, sleeplessness, and reduction in volume of the voice (Przuntek, 1992). More specific complaints that were elicited on a detailed history as the disease progressed included problems with fine motor skills, decreased sense of smell, loss of appetite, and a tremor occurring with anxiety. Family members retrospectively reported decreased arm swing on the affected side, decreased emotional expression, and personality changes, including more introversion and inflexibility. Using strict criteria for asymmetry, 46% of patients with PD had characteristic asymmetric presentation that correlated with handedness (Uitti et al., 2005). Handedness, however, did not predict the onset of PD motor symptoms in another study (Stochl et al., 2009). The mechanisms of the observed asymmetry of PD symptoms and signs are not well understood, but the side of predominant involvement may be stochastically determined, similar to other complex diseases such as cancer (Djaldetti et al., 2006). The notion that the side of predominant involvement is merely coincidental and determined by chance alone is supported by seemingly random right and left distribution without correlation to hand dominance, lack of concordance for the affected side within family members of genetically determined PD, and the frequent presence of asymmetric involvement in drug-induced parkinsonism. In some cases, parkinsonism may remain confined to one side and may be associated with hemiatrophy. In one study, the mean age at onset of the 30 patients who satisfied the criteria for hemiparkinsonism–hemiatrophy was 44.2 (15–63) years with a mean duration of symptoms of 9.7 (2–20) years (Wijemanne and Jankovic, 2007) (Table 4.2). Half of all patients had dystonia at onset and dystonia was present in 21 (70%) of all patients during the course of the syndrome. The majority of patients were responsive to levodopa, and perinatal and early childhood cerebral injury appeared to play an important role in about half of the cases. This syndrome of hemiparkinsonism–hemiatrophy also suggests that some cases of PD may start prenatally, and as a result of the low number of dopaminergic neurons from birth and subsequent age-related attrition, develop PD symptoms in middle age (Le et al., 2009) (Fig. 4.1). The asymmetrical lateral ventricular enlargement that is associated with PD motor asymmetry (Lewis et al., 2009) may represent a nonspecific marker of underlying neurodegeneration or may suggest an early insult, similar to what has been postulated in hemiparkinsonism–hemiatrophy (Wijemanne and Jankovic, 2007).
Clinical heterogeneity of PD and the rich phenomenology associated with the disease are well recognized. In a survey of 181 treated PD patients, Bulpitt and colleagues (1985) found at least 45 different symptoms that were attributable to the disease, nine of which were reported by the patients with more than fivefold excess compared with a control population of patients randomly selected from a general practice. These common symptoms included being frozen or rooted to a spot, grimacing, jerking of the arms and legs, shaking hands, clumsy hands, salivation, poor concentration, severe apprehension, and hallucinations. Hallucinations, although usually attributed to dopaminergic therapy, may be part of PD, particularly when there is a coexistent dementia and depression (Fenelon et al., 2006; Marsh et al., 2006). However, even these frequent symptoms are relatively nonspecific and do not clearly differentiate PD patients from diseased controls. Gonera and colleagues (1997) found that 4–6 years prior to the onset of classic PD symptoms, patients experience a prodromal phase characterized by more frequent visits to general practitioners and specialists in comparison to normal controls. During this period, PD patients, compared to normal controls, had a higher frequency of mood disorder, fibromyalgia, and various pains (Defazio et al., 2008), particularly shoulder pain (Stamey et al., 2008; Madden and Hall, 2010). In one study of 25 PD patients and 25 controls, PD patients had 21 times the odds of having shoulder pain compared with those without PD (Madden and Hall, 2010).
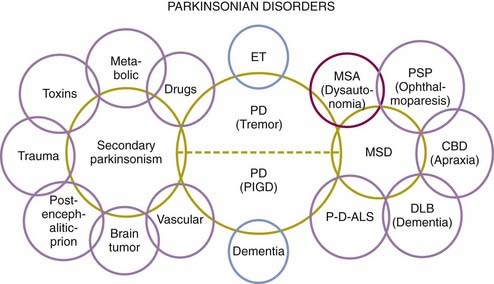
Because of the marked heterogeneity of clinical symptoms and natural progression, several studies have attempted to identify clinical subtypes of PD. Using cluster analysis, a systematic review of the literature confirmed the existence of the following disease subtypes: (1) young age at onset and slow disease progression, (2) old age at onset and rapid disease progression, (3) tremor-dominant, and (4) postural instability and gait difficulty (PIGD) (Jankovic et al., 1990) dominated by bradykinesia and rigidity (van Rooden et al., 2009). Patients who manifest predominantly axial symptoms, such as dysarthria (Ho et al., 1999), dysphagia, loss of equilibrium, and freezing of gait, are particularly disabled by their disease in comparison to those who have predominantly limb manifestations (Jankovic et al., 1990; Muslimovic et al., 2008). The poor prognosis of patients in whom axial symptoms predominate, many of whom have either the PIGD form of PD or some atypical parkinsonism (Fig. 4.2), is partly due to a lack of response of these symptoms to dopaminergic drugs (Jankovic et al., 1990; Kompoliti et al., 2000). In this regard, one study suggested that an abnormal tandem gait (the “ten steps” test) is much more common in patients with atypical parkinsonism than in those with typical PD and this test seems to differentiate the two groups with 82% sensitivity and 92% specificity (Abdo et al., 2006). In another classification of PD subtypes, the differences are largely driven by the severity of “nondopaminergic” features and levodopa-related motor complications (van Rooden et al., 2011).
Bradykinesia
Bradykinesia, the most characteristic clinical hallmark of PD, may be initially manifested by slowness in activities of daily living and slow movement and reaction times (Cooper et al., 1994; Touge et al., 1995; Giovannoni et al., 1999; Jankovic et al., 1999a; Rodriguez-Oroz et al., 2009). In addition to whole-body slowness, bradykinesia is often manifested by impairment of fine motor movement, demonstrated on examination by slowness in rapid alternating movements. Although speed and amplitude are usually assessed together on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III, there is some evidence that amplitude is disproportionately more affected than speed in patients with PD and may be due to different motor mechanisms and should probably be assessed separately (Espay et al., 2009). Other manifestations of bradykinesia include drooling due to failure to swallow saliva (Bagheri et al., 1999; Lal and Hotaling, 2006), monotonic and hypophonic dysarthria, loss of facial expression (hypomimia), and reduced arm swing when walking (loss of automatic movement). Micrographia has been postulated to result from an abnormal response due to reduced motor output or weakness of agonist force coupled with distortions in visual feedback (Teulings et al., 2002). Bradyphrenia refers to slowness of thought. Bradykinesia, like other parkinsonian symptoms, is dependent on the emotional state of the patient. With a sudden surge of emotional energy, the immobile patient may catch a ball or make other fast movements. This curious phenomenon, called kinesia paradoxica, demonstrates that the motor programs are intact in PD but that patients have difficulty utilizing or accessing the programs without the help of an external trigger. Therefore, parkinsonian patients are able to make use of prior information to perform an automatic or preprogrammed movement, but they cannot use this information to initiate or select a movement. Although PD represents the most common form of parkinsonism, there are many other causes of bradykinesia, the parkinsonian clinical hallmark (Table 4.2).
The pathophysiology of bradykinesia is not well understood, but it is thought to result from failure of basal ganglia output to reinforce the cortical mechanisms that prepare and execute the commands to move (Jankovic, 2007). This is manifested by slowness of self-paced movements and prolongation of reaction and movement time. Evarts and colleagues (1981) first showed that both reaction (RT) and movement (MT) times are independently impaired in PD. The RT is influenced not only by the degree of motor impairment but also by the interaction between the cognitive processing and the motor response. This is particularly evident when choice RT is used and compared to simple RT. Bradykinetic patients with PD have more specific impairment in choice RT, which involves a stimulus categorization and a response selection and reflects disturbance at more complex levels of cognitive processing. Ward and colleagues (1983b) found that of the various objective assessments of bradykinesia, the MT correlates best with the total clinical score, but it is not as sensitive an indicator of the overall motor deficit as the clinical rating.
Reduced dopaminergic function has been hypothesized to disrupt normal motor cortex activity, leading to bradykinesia. In recordings from single cortical neurons in free-moving rats, a decrease in firing rate correlated with haloperidol-induced bradykinesia, demonstrating that reduced dopamine action impairs the ability to generate movement and causes bradykinesia (Parr-Brownlie and Hyland, 2005). The premovement EEG potential (Bereitschaftspotential) is reduced in PD, probably reflecting inadequate basal ganglia activation of the supplementary motor area (Dick et al., 1989). On the basis of electromyographic (EMG) recordings in the antagonistic muscles of parkinsonian patients during a brief ballistic elbow flexion, Hallett and Khoshbin (1980) concluded that the most characteristic feature of bradykinesia was the inability to energize the appropriate muscles to provide a sufficient rate of force required for the initiation and maintenance of a large, fast (ballistic) movement. Therefore, PD patients need a series of multiple agonist bursts to accomplish a larger movement. Thus, the amount of EMG activity in PD is underscaled (Berardelli et al., 2001). Although many patients with PD complain of “weakness,” this subjective symptom is probably due to a large number of factors including bradykinesia, rigidity, fatigue, and also reduced power due to muscle weakness, particularly when lifting heavy objects (Allen et al., 2009).
Of the various parkinsonian signs, bradykinesia correlates best with a reduction in the striatal fluorodopa uptake measured by positron emission tomography (PET) scans and in turn with nigral damage (Vingerhoets et al., 1997). This is consistent with the finding that decreased density of SN neurons correlates with parkinsonism in the elderly, even without PD (Ross et al., 2004). PET scans in PD patients have demonstrated decreased 18F-fluorodeoxyglucose uptake in the striatum and accumbens-caudate complex roughly proportional to the degree of bradykinesia (Playford and Brooks, 1992). Studies performed initially in monkeys made parkinsonian with the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Bergman et al., 1990) and later in patients with PD provide evidence that bradykinesia results from excessive activity in the subthalamic nucleus (STN) and the internal segment of globus pallidus (GPi) (Dostrovsky et al., 2002). Thus, there is both functional and biochemical evidence of increased activity in the outflow nuclei, particularly subthalamic nucleus and GPi, in patients with PD.
Tremor
By using the term shaking palsy, James Parkinson in his An Essay on the Shaking Palsy (1817) drew attention to tremor as a characteristic feature of PD. Indeed, some parkinsonologists regard rest tremor as the most typical sign of PD, and its absence should raise the possibility that the patient’s parkinsonism is caused by a disorder other than PD. The typical rest tremor has a frequency between 4 and 6 Hz, and the tremor is almost always most prominent in the distal part of an extremity. In the hand, the tremor has been called a “pill-rolling tremor.” In the head region, tremor occurs most commonly in the lips, chin and jaw, but while a common manifestation of essential tremor, head tremor is rare in PD (Roze et al., 2006; Gan et al., 2009). Some patients with PD complain of an internal, not visible, tremor, called “inner tremor.” Rest tremor of PD is often exacerbated during potential provocations, such as walking and counting backwards (Raethjen et al., 2008).
As pointed out below, presentation with tremor as the initial symptom often confers a favorable prognosis with slower progression of the disease and some have suggested the term “benign tremulous parkinsonism” for a subset of patients with minimal progression, frequent family history of tremor, and poor response to levodopa (Josephs et al., 2006; O’Suilleabhain et al., 2006). Rajput and colleagues (1991) noted that 100% of 30 patients with pathologically proven PD experienced some degree of rest tremor at some time during the course of their disease. However, in another clinical-pathologic study, only 76% of pathologically proven cases of PD had tremor (Hughes, et al., 1992b). In an expanded series of 100 pathologically proven cases of PD, tremor was present at onset in 69%; 75% had tremor during the course of the illness, and 9% lost their tremor late in the disease (Hughes et al., 1993).
Although rest tremor is a well-recognized cardinal feature of PD, many PD patients have a postural tremor that is more prominent and disabling than the classic rest tremor. Postural tremor without parkinsonian features and without any other known etiology is often diagnosed as essential tremor (ET), but isolated postural tremor may be the initial presentation of PD, and it may be found with higher-than-expected frequency in relatives of patients with PD (Brooks et al., 1992b; Jankovic et al., 1995; Jankovic, 2002; Louis et al., 2003). Jankovic and colleagues (1995) and others (Louis et al., 2003) have shown that relatives of patients with tremor-dominant PD have a significantly higher risk of having action tremor than relatives of patients with the PIGD form of PD, but it is not yet clear whether the isolated tremor in the relatives is ET or whether it represents an isolated manifestation of PD. The two forms of postural tremor, ET and PD, can be differentiated by a delay in the onset of tremor when arms assume an outstretched position. Most patients with PD tremor have a latency of a few seconds (up to a minute) before the tremor reemerges during postural holding, hence the term reemergent tremor (Jankovic et al., 1999b) (Video 4.1). In contrast, postural tremor of ET usually appears immediately after arms assume a horizontal posture. Since the reemergent tremor has a frequency similar to that of rest tremor and both tremors generally respond to dopaminergic drugs, reemergent tremor most likely represents a variant of the more typical rest tremor. In addition to the rest and postural tremors, a kinetic tremor, possibly related to enhanced physiologic tremor, may also impair normal reach-to-grasp movement (Wenzelburger et al., 2000).
While bradykinesia and rigidity are most likely associated with nigrostriatal dopaminergic deficit, the pathophysiology of PD rest tremor is probably more complicated and most likely results from dysfunction of both the striato-pallidal-thalamocortical and the cerebello-dentato-thalamocortical circuits (Boecker and Brooks, 2011). The pallidum, in particular, appears to play a fundamental role in generation of tremor as suggested by a 4-8 Hz GPi neuronal firing in primate models of parkinsonism, correlation of tremor severity with pallidal (but not striatal) dopamine depletion, and complete abolition or a marked improvement of tremor with GPi ablation or DBS (Helmich et al., 2011).
As a result of the abnormal neuronal activity at the level of the GPi, the muscle discharge in patients with PD changes from the normal high (40 Hz) to pulsatile (10 Hz) contractions. These muscle discharges, which may be viewed as another form of PD-associated tremor, can be auscultated with a stethoscope (Brown, 1997). 
Rigidity and flexed posture
Rigidity, tested by passively flexing, extending, and rotating the body part, is manifested by increased resistance throughout the range of movement. Cogwheeling is often encountered, particularly if there is associated tremor or an underlying, not yet visible, tremor. In 1926 Froment and Gardere published a series of papers based on their studies of parkinsonian rigidity, including the observation of enhanced resistance to passive movement of a limb about a joint detected during voluntary movement of a contralateral limb (“Froment’s maneuver”) (Broussolle et al., 2007). Rigidity may occur proximally (e.g., neck, shoulders, and hips) and distally (e.g., wrists and ankles). At times, it can cause discomfort and actual pain. Painful shoulder, possibly due to rigidity but frequently misdiagnosed as arthritis, bursitis, or rotator cuff, is one of the most frequent initial manifestations of PD (Riley et al., 1989; Stamey et al., 2008). In a prospective, longitudinal study of 6038 individuals, mean age 68.5 years, who participated in the Rotterdam study and had no dementia or parkinsonian signs at baseline, subjective complaints of stiffness, tremor, and imbalance were associated with increased risk of PD with hazard ratios of 2.11, 2.09, and 3.47, respectively (de Lau et al., 2006). During the mean 5.8 years of follow-up, 56 new cases of PD were identified.
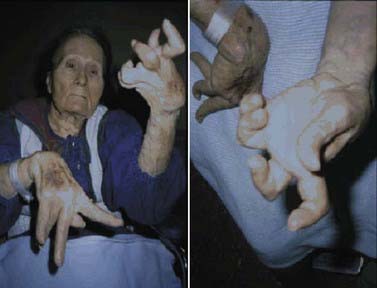
Rigidity is often associated with postural deformity resulting in flexed neck and trunk posture and flexed elbows and knees. But rigidity is a common sign in early PD, whereas flexed posture occurs later in the disease. Some patients develop “striatal hand” deformity, characterized by ulnar deviation of hands, flexion of the metacarpophalangeal joints, and extension of the interphalangeal joints (Fig. 4.3), and there may be extension of the big toe (“striatal toe”) or flexion of the other toes, which can be confused with arthritis (Jankovic and Tintner, 2001; Ashour et al., 2005; Ashour and Jankovic, 2006; Jankovic, 2007). Striatal toe was found to be present in 13 of 62 (21%) of patients with clinically diagnosed PD (Winkler et al., 2002).
Other skeletal abnormalities include extreme neck flexion (“dropped head” or “bent spine”) (Oerlemans and de Visser, 1998; Askmark et al., 2001; Ashour and Jankovic, 2006; Kashihara et al., 2006; Gdynia et al., 2009; Oyama et al., 2009) and truncal flexion (camptocormia) (Djaldetti et al., 1999; Umapathi et al., 2002; Azher and Jankovic, 2005; Tiple et al., 2009; Sako et al., 2009). Askmark and colleagues (2001) found 7 patients out of 459 with parkinsonism who had a head drop attributed to neck extensor weakness. Myopathic changes on EMG were noted in all seven, and five patients who consented had abnormal muscle biopsy, with mitochondrial abnormalities in two. Isolated neck extensor myopathy was reported in other patients with anterocollis associated with parkinsonism (Lava and Factor, 2001; van de Warrenburg et al., 2007; Gdynia et al., 2009), although its true frequency in patients with PD, multiple system atrophy (MSA), and other parkinsonian disorders is unknown. The following etiologies have also been identified in various series of patients with head drop (head ptosis or anterocollis), bent spine, or camptocormia: dystonia, disproportionately increased tone in the anterior neck muscles resulting in fibrotic and myopathic changes, amyotrophic lateral sclerosis, focal myopathy, inclusion body myositis, polymyositis, nemaline myopathy, facioscapulohumeral dystrophy, myasthenia gravis, encephalitis, dopamine agonists (Uzawa et al., 2009), and valproate toxicity (Umapathi et al., 2002; Gourie-Devi et al., 2003; Schabitz et al., 2003; Azher and Jankovic, 2005; van de Warrenburg et al., 2007).
Camptocormia is characterized by extreme flexion of the thoracolumbar spine that increases during walking and resolves in supine position (Videos 4.2, 4.3, 4.4). It appears to be more common in patients with more severe PD and if they had prior vertebral surgery (Tiple et al., 2009). The term was coined during World War I when young soldiers who were apparently attempting to escape the stress of battle developed this peculiar posture, perhaps promoted by a stooped posture when walking in the trenches. There appear to be two possible mechanisms of camptocormia: (1) dystonia due to a central disorder and (2) extensor trunkal muscle myopathy (Bloch et al., 2006; Lepoutre et al., 2006; Melamed and Djaldetti, 2006; Gdynia et al., 2009). 
Other truncal deformities include scoliosis and tilting of the trunk, referred to as the Pisa syndrome (Villarejo et al., 2003) (Fig. 4.4). The axial dystonias resulting in scoliosis and camptocormia may improve with botulinum toxin injections into the paraspinal or rectus abdominis muscles (Azher and Jankovic, 2005; Bonanni et al., 2007). In some cases, dystonia may be the presenting symptom of PD, particularly the early-onset Parkinson disease (EOPD) variety such as is seen in patients with the parkin mutation (Lücking et al., 2000; Jankovic and Tintner, 2001; Hedrich et al., 2002). In addition, several autosomal recessive disorders, including PANK2, PLA2G6, ATP13A2, FBX07, TAF1, and PRKRA-associated neurodegeneration, are manifested by the dystonia–parkinsonism combination (Schneider et al., 2009). Another form of dystonia associated with PD is paroxysmal exercise-induced foot dystonia, which may be the presenting feature of young-onset PD (YOPD) (Bozi and Bhatia, 2003).
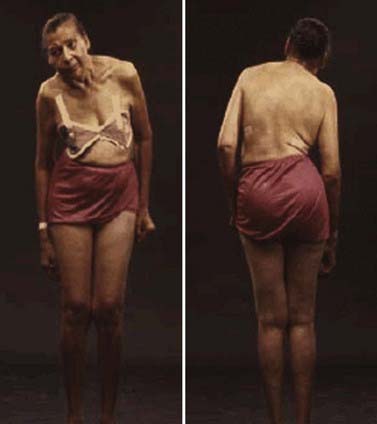
Loss of postural reflexes
Loss of postural reflexes is a characteristic feature in PD patients who exhibit the PIGD phenotype and usually occurs in more advanced stages of the disease along with freezing of gait and other symptoms that often lead to falling. One of the distinguishing features of PD fallers is their tendency to overestimate balance performance on functional reach testing compared to controls, and this overestimation worsens with worsening disease severity and when concurrently performing complex motor (e.g., carrying a tray) and cognitive tasks (e.g., performing mental arithmetic). In contrast to controls, PD patients are willing to sacrifice motor performance to complete competing tasks and make significantly more motor errors when performing a complex motor-cognitive task, whereas controls were more likely to preserve motor performance while sacrificing cognitive accuracy (Bloem et al., 2006). The loss of protective reactions further contributes to fall-related injuries. In one study, a fall in the past year, abnormal axial posture, cognitive impairment, and freezing of gait were independent risk factors for falls and predicted 38/51 fallers (75%) and 45/62 non-fallers (73%) (Latt et al., 2009). Additional measures contributing to falls include frontal impairment, poor leaning balance, and leg weakness. In another study, female gender, symmetrical onset, postural and autonomic instability appear to be the most reliable predictors of falls in PD (Williams et al., 2006). Using a battery of neurologic and functional tests in 101 patients with early PD and in an optimally medicated state, 48% reported a fall and 24% more than one fall in a prospective follow-up over 6 months (Kerr et al., 2010). The following measures provided the best sensitivity (78%) and specificity (84%) for predicting falls: UPDRS total score, total freezing of gait score, occurrence of symptomatic postural orthostasis, Tinetti total score, and extent of postural sway in the anterior-posterior direction.
The average period from onset of symptoms to the first fall in progressive supranuclear palsy (PSP) is 16.8 months, as compared to 108 months in PD, 42 months in MSA, 54 months in dementia with Lewy bodies, and 40.8 months in vascular parkinsonism. Many patients with postural instability, particularly when associated with flexed truncal posture (camptocormia), have festination, manifested by faster and faster walking as if chasing their center of gravity to prevent falling. When combined with axial rigidity and bradykinesia, loss of postural reflexes causes the patient to collapse into the chair when attempting to sit down. The pull test (pulling the patient by the shoulders) is commonly used to determine the patient’s degree of retropulsion or propulsion (see above) (Visser et al., 2003; Hunt and Sethi, 2006; Valkovic et al., 2008). Alterations in cholinergic rather than dopaminergic neurotransmission have been implicated in disturbed balance and falls associated with PD, partly because of evidence that gait control depends on cholinergic system-mediated higher-level cortical and subcortical processing, including pedunculopontine nucleus (PPN) function. This is supported by a cross-sectional study of 44 patients with PD without dementia and 15 control subjects who underwent a clinical assessment and [11C]methyl-4-piperidinyl propionate (PMP) acetylcholinesterase (AChE) and [11C]dihydrotetrabenazine (DTBZ) vesicular monoamine transporter type 2 (VMAT2) brain PET imaging (Bohnen et al., 2009). The study found reduced cortical AChE hydrolysis rates demonstrated in the PD fallers (−12.3%) compared to PD nonfallers (−6.6%) and control subjects (P = 0.0004). In another study, involving 22 normal controls, 12 patients with PD, 13 with MSA-P, and 4 with PSP, PET with [11C]PMP showed a significant decrease in AChE activity in most cerebral cortical regions in PD and MSA-P, and a nonsignificant decrease in PSP. On the other hand, subcortical cholinergic activity was significantly more decreased in MSA-P and PSP than in PD. The authors suggested that the more substantial decrease in subcortical AChE in MSA-P and PSP reflects greater impairment in the pontine cholinergic group (PPN) and may account for the greater gait disturbances in the early stages of these two disorders compared to PD (Gilman et al., 2010). Thus cholinergic hypofunction, possibly related to PPN degeneration, may be contributing to falls in patients with PD (Thevathasan and Aziz, 2010).
Freezing
One of the most disabling symptoms of PD is freezing, also referred as motor blocks, considered by some as a form of akinesia (loss of movement) (Giladi et al., 1997, 2001; Giladi and Nieuwboer, 2008; Morris et al., 2008) (Videos 4.5, 4.6). Although it most often affects the legs when walking, it can also involve upper limbs and the eyelids (apraxia of eyelid opening or eyelid closure) (Boghen, 1997). Freezing consists of sudden, transient (a few seconds) inability to move. It typically causes start hesitation when initiating walking and the sudden inability to move feet (as if glued to the ground) when turning or walking through narrow passages (such as the door or the elevator) (Almeida and Lebold, 2010), when crossing streets with heavy traffic, or when approaching a destination (target hesitation). Freezing is the most common cause of falls in patients with PD that can result in injuries, including hip fractures. Patients often adopt a variety of cues or tricks to overcome the freezing attacks: marching to command (“left, right, left, right”), stepping over objects (the end of a walking stick, a pavement stone, cracks in the floor, etc.), walking to music or a metronome, shifting body weight, rocking movements, and others (Dietz et al., 1990; Fahn, 1995; Marchese et al., 2001; Rubinstein et al., 2002; Suteerawattananon et al., 2004; Nieuwboer, 2008). “Off” gait freezing was found to correlate with dopa-responsive abnormal discriminatory processing as determined by abnormally increased temporal discrimination threshold (Lee et al., 2005). Freezing may be a manifestation of the “off” phenomenon in PD patients who fluctuate but may also occur during “on” time (“on freezing”), independent of bradykinesia and tremor (Bartels et al., 2003). Based on responses by 6620 patients to a questionnaire sent to 12 000 members of the German Parkinson Association, 47% of patients reported freezing, and it was present more frequently in men than women and less frequently in patients who considered tremor as their main symptom (Macht et al., 2007). When freezing occurs early in the course of the disease or is the predominant symptom, a diagnosis other than PD should be considered. Disorders associated with prominent freezing include progressive supranuclear palsy (PSP), MSA, and vascular (lower body) parkinsonism (FitzGerald and Jankovic, 1989; Elble et al., 1996; Winikates and Jankovic, 1999; Jankovic et al., 2001). Freezing has been thought to be related to noradrenergic deficiency as a result of degeneration of the locus coeruleus (Zarow et al., 2003), as suggested by possible response to noradrenergic agents such as L-threo-dihydroxy-phenylserine, or DOPS (Narabayashi, 1999). Neurophysiologic studies in monkeys treated with MPTP found that dopamine depletion is associated with impaired selection of proprioceptive inputs in the supplementary motor area, which could interfere with motor planning and may be related to motor freezing (Escola et al., 2002). Integrating EMG signals over real time while recording EMG activity from lower extremities before and during freezing, Nieuwboer and colleagues (2004) showed significantly abnormal timing in the tibialis anterior and gastrocnemius muscles, although reciprocity is preserved. Thus, before freezing, the tibialis anterior and gastrocnemius contract prematurely, and the duration of contraction is shortened in the tibialis anterior, but the amplitude of the EMG burst is increased (probably a compensatory strategy pulling the leg into swing), whereas the contraction is prolonged in the gastrocnemius during the actual swing phase. Isolated freezing usually suggests a diagnosis other than PD and may be present in atypical forms of parkinsonism or brainstem strokes (Kuo et al., 2008). The pathologic involvement of brainstem in patients with pure akinesia and gait freezing is suggested by decreased glucose metabolism on PET scans in the midbrain of such patients, similar to the findings in patients with PSP (Park et al., 2009). Neither medical (Giladi, 2008) nor surgical (Ferraye et al., 2008; Nashatizadeh and Jankovic, 2008) treatments generally provide satisfactory control of freezing. 
Other motor abnormalities
Some patients exhibit the reemergence of primitive reflexes attributed to a breakdown of the frontal lobe inhibitory mechanisms that are normally present in infancy and early childhood, hence the term release signs (Vreeling et al., 1993; Thomas, 1994; Rao et al., 2003). The glabellar tap reflex, also known as Meyerson sign, has often been associated with PD. However, its diagnostic accuracy has not been subjected to rigorous studies. The glabellar tap reflex is elicited through repeated stimuli to the glabellar region of the forehead, inducing concomitant blinking with each tap. In the normal subject, the reflex blinking habituates or the subject stops blinking with each stimulus tap after the second to fifth tap. Brodsky and collegues (2004) examined the glabellar reflex and the palmomental reflex in 100 subjects, which included patients with PD (n = 41), patients with PSP (n = 12), patients with MSA (n = 7), and healthy, age-matched controls (n = 40). Using a standardized protocol and a “blinded” review of videotapes, we found that (1) both reflexes were present significantly more frequently in patients with PD as compared to normal controls; (2) glabellar, but not palmomental, reflex was more frequently present in patients with PSP than in controls; (3) there was no difference in the frequency of these reflexes between normal controls and patients with MSA; (4) the two reflexes occurred with similar frequency among the three parkinsonian disorders; (5) glabellar, but not palmomental, reflex correlated with parkinsonian motor deficit; and (6) the primitive reflexes correlated with mental deficit. While relatively sensitive signs of parkinsonian disorders, particularly PD, these primitive reflexes lack specificity, as they do not differentiate among the three most common parkinsonian disorders (Brodsky et al., 2004). Abnormal spontaneous blinking, particularly the longer pauses between closing and opening phase, compared to normal controls suggests that the decreased blinking in PD reflects underlying bradykinesia (Agostino et al., 2008). In addition to these primitive reflexes, there are other “frontal” and “cortical disinhibition” signs, such as the applause sign (Wu et al., 2008), but none of them are specific for PD.
Besides the classic cardinal signs, there are many other motor abnormalities that may be equally or even more disabling. One of the most prominent features of motor impairment in PD is the inability to perform multiple tasks simultaneously. Using functional magnetic resonance imaging (fMRI), Wu and Hallett (2008) found that during dual task execution, greater activity was recorded in the precuneus region, cerebellum, premotor area, and parietal and prefrontal cortex. They concluded that difficulties in dual task performance in PD were associated with limited attentiveness and defective central executive function, and that training may improve the performance. The bulbar symptoms (dysarthria, hypophonia, dysphagia, and sialorrhea) are thought to result from orofacial-laryngeal bradykinesia and rigidity (Hunker et al., 1982). PD-associated speech and voice impairment, often referred to as hypokinetic dysarthria, is characterized by low volume (hypophonia), uniform (monotonous) loudness and pitch (aprosody), imprecise consonants, hesitation, and short rushes of speech (tachyphemia). Other speech characteristics include a variable (abnormally slow or increased) speech rate, palilalia, and stuttering. PD patients have been found to have higher speech acceleration than controls and a significant reduction in the number of pauses, indicating abnormal speech rate and rhythm (Skodda and Schlegel, 2008). A history of childhood stuttering that had remitted can subsequently recur with onset of PD, suggesting an involvement of the dopaminergic system in this speech disorder (Shahed and Jankovic, 2001). When speech therapy designed to stimulate increased vocal fold adduction with instructions to “think loud, think shout,” the Lee Silverman Voice Treatment (Ramig et al., 2001), was compared with “speak loud and low,” the Pitch Limiting Voice Treatment (de Swart et al., 2003), the two methods produced the same increase in loudness, but the latter method was found to prevent strained voicing. Other treatment strategies for PD-related dysarthria include the use of various verbal cues to regulate speech volume (Ho et al., 1999), but deep brain stimulation has a variable effect (Pinto et al., 2004). The low-volume voice in PD has been attributed in part to vocal fold bowing due to loss of muscle mass and control (Schulz et al., 1999), and augmentation of vocal folds with collagen injections provides improvement in voice quality and has a significantly beneficial impact on quality of life (Hill et al., 2003). Respiratory difficulties result from a variety of mechanisms, including a restrictive component due to rigid respiratory muscles and levodopa-induced respiratory dyskinesias (Rice et al., 2002).
In addition to categorization of patients into clinical subtypes, there is a growing appreciation for differences in clinical presentation depending on genetic background. Thus patients with parkin mutations (PARK2), who account for nearly a third of patients with early-onset PD, tend to develop levodopa-induced dyskinesias and hallucinations relatively early in the course of the disease. They also may present with dystonic gait, cervical dystonia, dopa-responsive dystonia, hemiparkinsonism–hemiatrophy, freezing, festination, retropulsion, leg tremor at rest and on standing, marked sleep benefit, hyperreflexia, ataxia, peripheral neuropathy, and dysautonomia (Klein and Lohmann, 2009). Carriers of LRRK2 G2019S mutation are more likely to manifest the PIGD subtype of PD rather than the tremor-dominant phenotype, although in contrast to the PIGD in patients with sporadic PD, LRRK2 patients tend to have a much slower, less aggressive course (Alcalay et al., 2009; Dächsel and Farrer, 2010). Other studies have confirmed that in comparison with genetically undefined patients, LRRK2 mutation carriers had more severe motor symptoms, a higher rate of dyskinesia, and less postural tremor, whereas PINK1 mutation carriers have younger age at onset and slower progression, but similar to LRRK2 PD patients have an increased rate of drug-induced dyskinesia and a lower rate of postural tremor (Nishioka et al., 2010).
Nonmotor manifestations
Although James Parkinson in his original description focused on the motor symptoms, he also drew attention to several nonmotor features, including problems associated with sleep and gastrointestinal function (Parkinson, 1817). Traditionally viewed as primarily a motor disorder, there is growing recognition that nonmotor symptoms of PD, which occur in 88% of all patients, are as troublesome if not more so than the classic motor features (Simuni and Sethi, 2008). The nonmotor manifestations and fluctuations in nonmotor symptoms have been found to be more disabling than the motor symptoms in 28% of PD patients (Witjas et al., 2002). These nonmotor, nondopaminergic symptoms have been largely ignored, but several recent studies have highlighted their frequency and their serious impact on quality of life, particularly in more advanced stages of the disease (Lang and Obeso, 2004; Chaudhuri et al., 2006a, 2006b, 2007; Ahlskog, 2007; Martinez-Martin et al., 2007; Pfeiffer, 2007; Lim et al., 2009). The Sydney Multicenter Study showed that PD patients treated with “modern” initial therapy continue to die at a rate in excess of their peers, with only one-third of original study subjects remaining alive at 15 years after diagnosis, and most were disabled more by their nonmotor than motor symptoms: 84% experienced cognitive decline with 48% meeting diagnostic criteria for dementia, 58% were unable to live alone, and 40% were in long-term care facilities (Hely et al., 2005). After 20 years’ follow-up, only 36 (26%) survived and the standardized mortality ratio reached 3.1 (Hely et al., 2008). Of the 30 included in this longitudinal study, 100% had levodopa-induced dyskinesia and end of dose failure, dementia was present in 83%, and 48% were in nursing homes. Other problems included excessive daytime sleepiness in 70%, falls in 87%, freezing in 81%, fractures in 35%, symptomatic postural hypotension in 48%, urinary incontinence in 71%, moderate dysarthria in 81%, choking in 48%, and hallucinations in 74%. Of the 87 patients followed prospectively in the Sydney study whose brain was examined at autopsy, the final diagnosis was PD in 29, PD with dementia in 52, and dementia with Lewy bodies (DLB) in 6 (Halliday et al., 2008). The clinical-pathologic correlations suggested that there were three groups of patients: (1) younger-onset patients with a typical PD clinical course; brainstem Lewy bodies predominate in those surviving to 5 years, and by 13 years, 50% of cases have a limbic distribution of Lewy bodies; (2) older-onset PD cases with shorter survival and with higher Lewy body loads and additional plaque pathology; and (3) early malignant, dementia-dominant syndrome and severe neocortical disease, consistent with DLB. In a multicenter study of 1072 consecutive patients with PD in 55 Italian centers, the so-called Priamo study, 98.6% of patients reported a mean of 7.8 (range 0–32) nonmotor symptoms, such as fatigue (58%), anxiety (56%), leg pain (38%), insomnia (37%), urinary urgency and nocturia (35%), drooling of saliva (31%) and difficulties in maintaining concentration (31%) (Barone et al., 2009). Apathy was the symptom associated with worse PDQ-39 score but presence of fatigue, attention/memory, and psychiatric symptoms also had a negative impact on quality of life. The nonmotor features associated with PD are presumably related to involvement of the nondopaminergic systems and even pathology outside the central nervous system (Djaldetti et al., 2009).
Autonomic dysfunction
Autonomic failure is typically associated with MSA and may be the presenting feature of that disease, but it may also herald the onset of PD (Kaufmann et al., 2004; Mostile and Jankovic, 2009). In contrast to PD associated with dysautonomia, due to predominantly peripheral (ganglionic and postganglionic) involvement, in MSA the primary lesion is preganglionic; also dysautonomic symptoms are more severe at baseline and become more global in MSA as compared to PD (Lipp et al., 2009). Rating scales for dysautonomia associated with PD have been developed (Evatt et al., 2009). Dysautonomia, such as orthostatic hypotension, sweating dysfunction, sphincter dysfunction, and sexual impotence occur frequently in patients with PD (Senard et al., 1997; Swinn et al., 2003). In one study, 7 of 51 (14%) patients with early, untreated PD had a decrease of more than 20 mmHg in systolic blood pressure (Bonuccelli et al., 2003). Another community-based study of a cohort of PD patients showed that 42 of 89 (47%) met the diagnostic criteria for orthostatic hypotension (Allcock et al., 2004). Orthostatic hypotension, however, is not often detected in the clinic. Although the symptom of orthostatic lightheadedness has a relatively high specificity, it seems to have low sensitivity in predicting orthostatic hypotension, partly because it is more likely to occur after tilting than on standing, and is often delayed by longer than the recommended 3 minutes (Jamnadas-Khoda et al., 2009). Autonomic symptoms, particularly orthostatic hypotension, seem to be more common in the PIGD form of PD (Allcock and et al., 2006). Autonomic symptom severity was associated with more motor dysfunction, depressive symptoms, cognitive dysfunction, psychiatric complications, nighttime sleep disturbances, and excessive daytime sleepiness (P < 0.01) (Verbaan et al., 2007a). While dysautonomia is typically associated with MSA, it may also be prominent in PD, although autonomic testing might not always differentiate between PD and MSA (Riley and Chelimsky, 2003).
Orthostatic hypotension in patients with PD has been traditionally attributed to dopaminergic therapy, but recent studies have provided evidence that orthostatic hypotension in PD is due to failure of reflexive sympathetically mediated cardiovascular stimulation from sympathetic denervation, as demonstrated by markedly decreased 6-[18F]-fluorodopamine-derived radioactivity in septal and ventricular myocardium (Goldstein et al., 2002). This sympathetic nervous system deficit involved postganglionic catecholaminergic, not cholinergic, nerves (Sharabi et al., 2003).
Sweating dysfunction, hyperhidrosis, and to a lesser extent hypohidrosis, were reported by 64% of patients with PD as compared to 12.5% of controls (P < 0.005) (Swinn et al., 2003). These symptoms did not correlate with the severity of the disease but occurred most frequently during the “off” periods and during “on with dyskinesia” periods. Because sudomotor skin response was reduced in the palms, the axial hyperhidrosis has been suggested to be a compensatory phenomenon for reduced sympathetic function in the extremities (Schestatsky et al., 2006). Sweating may be a particularly troublesome symptom during wearing off (Pursiainen et al., 2007). The presence of α-synuclein deposits in the dermis of a patient with pure autonomic failure provides evidence that this disorder as well as other disorders associated with autonomic failure (e.g., PD, DLB, and MSA) should be viewed as variant synucleinopathies (Kaufmann and Goldstein, 2010; Shishido et al., 2010).
Bladder and other urologic symptoms are frequent in PD and are among the most common complaints requiring medical attention (Blackett et al., 2009; Sakakibara et al., 2010). One survey found that over one-fourth of men with PD had urinary difficulty, most often causing urinary urgency (Araki and Kuno, 2000). In one study, urge episodes and urge incontinence were observed in 53% and 27% of the patients with PD, respectively, and detrusor overactivity in 46% of the patients with PD, which was less prevalent than in patients with dementia with Lewy bodies and Alzheimer disease, while mean voided volume, free flow, cystometric bladder capacity, and detrusor pressor were similar in the groups (Ransmayr et al., 2008).
Despite the possibility of hypersexuality, usually related to dopaminergic drugs, many patients with PD have sexual dysfunction (Celikel et al., 2008; Meco et al., 2008; Hand et al., 2010). In a review of sexual functioning of 32 women and 43 men with PD, women reported difficulties with arousal (87.5%), reaching orgasm (75.0%), and sexual dissatisfaction (37.5%) (Bronner et al., 2004). Men reported erectile dysfunction (68.4%), sexual dissatisfaction (65.1%), premature ejaculation (40.6%), and difficulties reaching orgasm (39.5%). Reduced sexual drive and dissatisfaction with orgasm was particularly common in female PD patients (Celikel et al., 2008). Among 90 patients with PD, loss of libido was reported by 65.6%, and 42.6% of men also complained of erectile dysfunction (Kummer et al., 2009). Aging, female gender, lower education, and depression were significantly associated with decreased sexual desire. Premorbid sexual dysfunction may contribute to cessation of sexual activity during the course of the disease (among 23.3% of men and 21.9% of women). Associated illnesses, use of medications, motor difficulties, depression, anxiety, and advanced stage of PD contributed to sexual dysfunction.
Drooling (sialorrhea) is one of the most embarrassing symptoms of PD (Chou et al., 2007). While some studies have shown that PD patients actually have less saliva production (Proulx et al., 2005) than normal controls, others have suggested that the excessive drooling is due to a difficulty with swallowing (Bagheri et al., 1999). Salivary sympathetic denervation, however, could not be demonstrated by 6-[18F]-fluorodopamine scanning (Goldstein et al., 2002). Dysphagia (Hunter et al., 1997) along with delayed gastric emptying (Hardoff et al., 2001) and constipation (Ashraf et al., 1997; Bassotti et al., 2000; Winge et al., 2003; Cersosimo and Benarroch, 2008) represent the most frequent gastrointestinal manifestations of PD. In addition to constipation, PD patients often experience pharyngeal and esophageal dysphagia and 60% have evidence of delayed gastric emptying (Krygowska-Wajs et al., 2009).
When PD patients were compared to healthy controls, those with PD were found to swallow significantly more often during inhalation, at low tidal volumes, and exhibited significantly more post-swallow inhalation (Gross et al., 2008). Impaired coordination of breathing and swallowing may contribute to the high frequency of aspiration pneumonia in PD.
Gastrointestinal dysfunction in PD has been attributed to many mechanisms such as involvement, including neurodegeneration and the presence of Lewy bodies, of the dorsal motor nucleus of the vagus, paravertebral sympathetic ganglia, and intrinsic neurons of the enteric nervous system (Cersosimo and Benarroch, 2008). On the basis of information on the frequency of bowel movements in 6790 men in the Honolulu Heart Program, Abbott and colleagues (2001) concluded that infrequent bowel movements are associated with increased risk for future PD. Based on the observation from the Honolulu-Asia Aging Study that bowel frequency was lower in subjects who were found to have incidental Lewy bodies in their brains at postmortem examination than in controls, the investigators suggested that constipation was one of the earliest symptoms of PD (Abbott et al., 2007). Also, constipation was associated with low SN neuron density (Petrovitch et al., 2009). Based on a review of Mayo Clinic medical records of 196 case-control pairs (N = 392), constipation preceding PD was more common in cases than in controls (odds ratio 2.48; P = 0.0005) (Savica et al., 2009). Constipation in patients with PD is associated with slow colonic transit, weak abdominal strain, decreased phasic rectal contraction, and paradoxical sphincter contraction on defecation (Sakakibara et al., 2003). Dermatologic changes such as seborrhea, hair loss, and leg edema may represent evidence of peripheral involvement in PD, although some of these changes may be exacerbated by anti-PD drugs (Tabamo and Di Rocco, 2002; Tan and Ondo, 2000).
Autonomic complications, coupled with motor and mental decline, contribute to a higher risk of hospitalization and nursing home placement. Examination of hospital records of 15 304 cases of parkinsonism and 30 608 age- and sex-matched controls showed that PD patients are six times more likely to be admitted to hospital with aspiration pneumonia than are nonparkinsonian controls (Guttman et al., 2004). Other comorbid medical conditions significantly more common in patients with PD include fractures of the femur, urinary tract disorders, septicemia, and fluid/electrolyte disorders. But similarly to other reports (Jansson and Jankovic, 1985; Gorell et al., 1994; Vanacore et al., 1999; Inzelberg and Jankovic, 2007), this study showed that cancer might be less common in patients with PD, with the major exception being malignant melanoma with an almost twofold increased risk (Olsen et al., 2005) and a higher risk of family history of melanoma (Gao et al., 2009).
Cognitive and neurobehavioral abnormalities
Cognitive and neuropsychiatric disturbances have as much impact on the quality of life of a patient with PD as the motor symptoms (Aarsland et al., 2009). Cognitive deficits have been found in 30% of patients with early PD (Elgh et al., 2009). The Sydney Multicenter Study showed that after 15 years of follow-up, 84% have cognitive decline and 48% meet diagnostic criteria for dementia, 58% were unable to live alone, and 40% were in long-term care facilities (Hely et al., 2005). A long-term follow-up study of 233 subjects in Norway found that 60.1% of subjects by 12 years into the course of the disease had evidence of dementia (Buter et al., 2008). Based on this study, a 70-year-old man with PD but without dementia has a life expectancy of 8 years, 3 of which will be marked by coexistent dementia. In 537 patients with dementia associated with PD (PDD), 58% had associated depression, 54% apathy, 49% anxiety, and 44% hallucinations (Aarsland et al., 2007). A structured interview of 50 patients with PD found that anxiety (66%), drenching sweats (64%), slowness of thinking (58%), fatigue (56%), and akathisia (54%) were the most frequent nonmotor fluctuations. Many patients, for example, exhibit neurobehavioral disturbances, such as depression, dementia, tip-of-the-tongue phenomenon and other word-finding difficulties (Matison et al., 1982), various psychiatric symptoms, and sleep disorders (van Hilten et al., 1994; Aarsland et al., 1999; Pal et al., 1999; Tandberg et al., 1999; Olanow et al., 2000; Wetter et al., 2000; Ondo et al., 2001; Emre, 2003; Grandas and Iranzo, 2004; Adler and Thorpy, 2005; Goetz et al., 2008a). Although neurobehavioral abnormalities are often considered late features of PD, cognitive impairment affecting attention, psychomotor function, episodic memory, executive function, and category fluency (Elgh et al., 2009) may be detected even in early stages of the disease, and depression (Alonso et al., 2009) may be one of the earliest symptoms of PD.
A variety of instruments have been developed, designed to assess behavioral and cognitive impairments associated with PD (Goetz et al., 2008a). On the basis of the most frequently affected cognitive domains in PD, Marinus and colleagues (2003) proposed the SCOPA-COG (Scales for Outcomes of Parkinson’s disease – cognition). Using this scale and the search of the literature, they concluded that the cognitive functions that are most frequently affected in PD include attention, active memory, executive, and visuospatial functions, whereas verbal functions, thinking, and reasoning are relatively spared. PD patients may have a limited perception of large spatial configurations (seeing trees but not the forest) (Barrett et al., 2001). Aarsland and colleagues (2001) found in a community-based, prospective study that patients with PD have an almost six-fold increased risk of dementia. In an 8-year prospective study of 224 patients with PD, they found that 78.2% fulfilled the DSM-III criteria for dementia (Aarsland et al., 2003). The mean annual decline on Mini-Mental State Examination (MMSE) in patients with PD is 1 point, in patients with PD and dementia, it is 2.3 points (Aarsland et al., 2004). While the MMSE has been used traditionally to screen for cognitive deficits, it often fails to detect early cognitive decline because of its ceiling effect, and, therefore, the Montreal Cognitive Assessment (MoCA) has been developed to detect mild cognitive impairment in PD (Gill et al., 2008). In a study designed to compare the two scales in 88 patients with PD, the percentage of subjects scoring below a cutoff of 26/30 (used by others to detect mild cognitive impairment) was higher on the MoCA (32%) than on the MMSE (11%) (P < 0.000002), suggesting that the MoCA is a more sensitive tool to identify early cognitive impairment in PD (Zadikoff et al., 2007). Of the various scales specifically designed to assess cognitive impairment in PD the SCOPA-COG, which mainly assesses “frontal-subcortical” cognitive defects, and the PD-CRS (Parkinson’s Disease – Cognitive Rating Scale), which assesses “instrumental-cortical” functions, have been most rigorously validated (Kulisevksy and Pagonabarraga, 2009). The MMP (Mini-Mental Parkinson) and PANDA (Parkinson Neuropsychiatric Dementia Assessment) are brief screening tests that still require more extensive clinimetric evaluations.
A variety of measures have been investigated for their predictability of cognitive impairment. In the DATATOP study of patients with early PD, cumulative incidence of cognitive impairment, defined as scoring 2 standard deviations below age- and education-adjusted MMSE norms, was 2.4% (95% confidence interval 1.2–3.5%) at 2 years and 5.8% (3.7–7.7%) at 5 years (Uc et al., 2009a). Risk factors for cognitive impairment in this group of 740 patients was older age, hallucinations, male gender, increased symmetry of parkinsonism, increased severity of motor impairment (except for tremor), speech and swallowing impairments, dexterity loss, and presence of gastroenterologic/urologic disorders at baseline.
Functional imaging has been also used to study risk factors for cognitive decline in PD. Temporoparietal cortical hypometabolism is present in patients with PD and may be a useful predictor of future cognitive impairment (Hu et al., 2000). Another predictor of cognitive dysfunction appears to be reduced 18F-fluorodopa uptake in the caudate nucleus and frontal cortex (Rinne et al., 2000) as well as in the mesolimbic pathways (Ito et al., 2002). Using event-related fMRI to compare groups of cognitively impaired and unimpaired patients, Lewis and colleagues (2003) showed a significant signal intensity reduction during a working-memory paradigm in specific striatal and frontal lobe sites in PD patients with cognitive impairment. These studies indicate that cognitive impairments in early PD are related to reductions in activity of frontostriatal neural circuitry. In a PET study of brain activation during frontal tasks, such as trial-and-error learning, Mentis and colleagues (2003) found that even in early PD when learning is still relatively preserved, PD patients had to activate four times as much neural tissue as the controls in order to achieve learning performance equal to controls. Although the sequence learning is impaired even in early PD, this learning deficit does not appear to reflect impairments in motor execution or bradykinesia and may be related to reduced attention (Ghilardi et al., 2003).
Patients with PD have nearly twice the risk for developing dementia as controls, and siblings of demented PD patients have an increased risk for Alzheimer disease (Marder et al., 1999). In addition to the MMSE, other tests (e.g., the Frontal Assessment Battery) have been developed and validated to assess the cognitive and frontal lobe function (Dubois et al., 2000) in patients with dementia with or without parkinsonism. In agreement with basal forebrain cholinergic denervation even in early PD, prominent and widespread reduction in cortical, particularly the medial occipital secondary visual cortex (Brodmann area 18), acetylcholinesterase can be demonstrated using N-[11C]methyl-4-piperidyl acetate PET (Shimada et al., 2009). These changes were more pronounced but similar in patients with PDD and DLB.
There are several reasons why patients with PD have an associated dementia. Pathologically, dementia correlates with cortical pathology, including Lewy bodies (Hughes et al., 1993; Hurtig et al., 2000), especially in the cingulate and entorhinal cortex (Kovari et al., 2003). But the significance of cortical Lewy bodies is not clear, since most patients with PD have some detectable Lewy bodies in the cerebral cortex, and patients with PD with no dementia during life have been found to have neuropathologic findings diagnostic of Lewy body dementia (Colosimo et al., 2003). In contrast to earlier studies showing relatively low frequency of dementia in PD, more recent studies suggest that the cumulative prevalence may be as high as 78%, correlating best with cortical and limbic Lewy bodies (Emre, 2004).
Depression is a common comorbid condition in patients with PD, with clinically significant depression present in about a third of all PD patients (Reijnders et al., 2008; Stella et al., 2008; Pankratz et al., 2008) and it may precede other symptoms of signs of PD (Alonso et al., 2009). Death or suicide ideation has been reported in 28% and 11% respectively, and 4% of PD patients have a lifetime suicide attempt, correlated with severity of depression, impulse control disorder, and psychosis (Nazem et al., 2008; Weintraub, 2008). The severity and impact of depression in PD may be assessed by several instruments, but the Hamilton Depression Scale (HAM-D), Beck Depression Inventory (BDI), Hospital Anxiety and Depression Scale (HADS), Montgomery-Asperg Depression Rating Scale (MADRS), and Geriatric Depression Scale (GDS) have been found particularly useful for screening purposes and HAM-D, MADRS, BDI, and the Zung Self-Rating Depression Scale (SDS) have been recommended for assessment of severity (Schrag et al., 2007a). While the HADS and the GDS may be particularly useful in measuring severity of depression, these scales are not apparently sensitive enough to detect a change in patients with severe depression. In addition to instruments used to assess depression, scales for apathy and anhedonia (Leentjens et al., 2008a) and for anxiety (Leentjens et al., 2008b) associated with PD have been developed and validated. Even without these tools, using DSM-IV-TR and a diagnostic examination by psychiatrists, a 49% lifetime prevalence of anxiety was found in a physician-based sample of 127 patients with PD (Pontone et al., 2009).
A community-based study showed that 7.7% of PD patients met the criteria for major depression, 5.1% met those for moderate to severe depression, and another 45.5% had mild depressive symptoms (Tandberg et al., 1996). Depression in PD, clearly a multifactorial disorder, has a major impact on the quality of life (Schrag, 2006). In 139 patients with PD, Aarsland and colleagues (1999) found at least one psychiatric symptom in 61% of the patients. These included depression (38%), hallucinations (27%), and a variety of other behavioral and cognitive changes. In a study of 114 PD patients, 27.6% screened positive for depression during the average 14.6 months of follow-up; 40% were neither treated with antidepressants nor referred for further psychiatric evaluation (Ravina et al., 2007a). Furthermore, depression, as assessed by the GDS-15, correlated with impairment in activities of daily living (ADLs) (P < 0.0001). Subsequent analysis showed that increasing severity of depressive symptoms, older age, and longer PD duration predicted a lower likelihood of symptom resolution (Ravina et al., 2009). Patients with depression may be three times more likely to later develop PD (Schuurman et al., 2002). In one study, depression was found in 15% of patients with PD, and it had more impact on the ADLs than on the motor subscale of UPDRS (Holroyd et al., 2005). Anhedonia is another frequent symptom of PD, which is independent from depression or motor deficits (Isella et al., 2003). Despite the high frequency of depression, patients with PD appear to have higher levels of anger control, consistent with the recognized stoic personality trait (Macías et al., 2008). Stage of illness, motor impairment, and functional disability clearly correlate with depressive symptoms (Pankratz et al., 2008).
Using [11C]RTI-32 PET as a marker of both dopamine and norepinephrine transporter binding in 8 PD patients with and 12 without depression, Remy and colleagues (2005) showed significantly lower binding of this ligand in the locus coeruleus and various limbic regions in depressed and anxious patients compared to those without these psychiatric symptoms. In a group of 94 patients with primary depression, Starkstein and colleagues (2001) found that 20% of patients had parkinsonism that was reversible on treatment of the depression. Blunted reactivity to aversive (pleasant and unpleasant) stimuli has been found in a group of nondemented PD patients (Bowers et al., 2006). Some investigators have attributed the various nonmotor symptoms associated with PD, such as depression, anxiety, lack of energy, and sexual dysfunction, to comorbid testosterone deficiency (found in 35% of PD patients) and suggested that testosterone treatment may be the appropriate therapy for these patients (Okun et al., 2002) and may also improve apathy associated with PD (Ready et al., 2004; Kirsch-Darrow et al., 2006). However, in a subsequent control clinical trial, testosterone was not found to be beneficial in men with PD (Okun et al., 2006).
Psychosis has been long recognized to complicate the course of PD and several scales have been developed to assess this symptom (Fernandez et al., 2008). Diagnostic criteria for psychosis in PD emphasize primarily the presence of paranoid delusions, visual hallucination, illusions, and false sense of presence in contrast to auditory hallucinations and thought disorder typically seen in patients with schizophrenia (Ravina et al., 2007b). Several studies have shown that the occurrence of psychosis is frequently associated with other psychiatric comorbidities, especially depression, anxiety, and apathy (Marsh et al., 2004), and with dementia (Factor et al., 2003). One study concluded that the presence of hallucinations is the strongest predictor of nursing home placement and death (Aarsland et al., 2000). The prognosis of PD-associated psychosis, however, has improved with the advent of atypical neuroleptics in that the incidence of death within 2 years of nursing home placement decreased from 100% to 28%. Minor hallucinations may occur in as many as 40% of patients with PD, illusions in 25%, formed visual hallucinations in 22%, and auditory hallucinations in 10% (Fénelon et al., 2000). Risk factors for hallucinations include older age, duration of illness, depression, cognitive disorder, daytime somnolence, poor visual acuity, family history of dementia (Paleacu et al., 2005), and dopaminergic drugs (Barnes and David, 2001; Goetz et al., 2001; Holroyd et al., 2001). Hallucinations seem to correlate with daytime episodes of rapid eye movement (REM) sleep as well as daytime non-REM and nocturnal REM sleep, suggesting that hallucinations and psychosis may represent a variant of narcolepsy-like REM sleep disorder (Arnulf et al., 2000) and that dream imagery plays an important role in visual hallucinations (Manni et al., 2002). Other studies, however, have found no correlation between hallucinations and abnormal sleep patterns (Goetz et al., 2005). The sleep abnormalities observed in patients with PD may possibly be related to a 50% loss of hypocretin (orexin) neurons (Fronczek et al., 2007; Thannickal et al., 2007).
Besides the cardinal motor signs, there are many behavioral and cognitive symptoms associated with PD, such as depression, sleep disorders, and fatigability, that can adversely influence the overall quality of life in patients with PD (Karlsen et al., 1999). In one study, 50% of patients with PD had significant fatigue that had a major impact on health-related quality of life (Herlofson and Larsen, 2003). PD-related fatigue contributes to poor functional capacity and physical function (Garber and Friedman, 2003; Chaudhuri and Behan, 2004; Friedman et al., 2007). A 16-item self-report instrument designed to measure fatigue associated with PD has been developed (Brown et al., 2005). One study showed that depression, postural instability, and cognitive impairment have the greatest influence on quality of life (Schrag et al., 2000). In a prospective longitudinal study of 111 patients followed for 4 years, Karlsen and colleagues (2000) showed significantly increased distress, based on health-related quality of life, not only due to motor symptoms but also because of pain, social isolation, and emotional reactions.
Variants of bradyphrenia (slowness of thought), such as abulia (severe apathy and lack of initiative and spontaneity) as well as akinetic mutism and catatonia (immobility, mutism, refusal to eat or drink, staring, rigidity, posturing, grimacing, negativism, waxy flexibility, echophenomenon, and stereotypy), have been recognized in patients with parkinsonism. Apathy in PD appears to be related to the underlying disease process rather than being a psychologic reaction to disability or to depression (Kirsch-Darrow et al., 2006) and is closely associated with cognitive impairment (Pluck and Brown, 2002). Various studies have reported that 32–54% exhibit apathy (Aarsland et al., 2007; Dujardin et al., 2007; Aarsland et al., 2009). Although depression and dementia are the most frequent comorbidities associated with apathy, about 13% of patients with PD exhibit apathy alone (Starkstein et al., 2009). Whether these symptoms represent a continuum of bradykinesia–bradyphrenia or different disorders is not easy to answer with the current rudimentary knowledge of these disorders (Muqit et al., 2001).
There have been several studies attempting to address the question of “premorbid parkinsonian personality.” Twin and other studies have suggested that since childhood, PD patients tend to be more introverted, cautious, socially alert, tense, nervous, and rigid compared to controls (Ishihara and Brayne, 2006). Some have found PD patients, even before onset of motor symptoms, to often avoid risk-seeking behavior, such as smoking (Ward et al., 1983a), and to exhibit lower impulsive and less novelty-seeking behavior (Menza et al., 1993; Fujii et al., 2000; Evans et al., 2004). Since dopamine is involved in the reward system, presymptomatic dopamine deficiency may predispose some individuals to exhibit a “non-smoking personality,” thus accounting for the lower frequency of smokers among PD patients (Wirdefeldt et al., 2005). Many studies have demonstrated that even before they first develop any motor symptoms, PD patients tend to have relatively characteristic personality traits, such as industriousness, seriousness, inflexibility, and a tendency to be “honest” (Przuntek, 1992; Macías et al., 2008; Abe et al., 2009). One study of 32 patients, using F-fluorodeoxyglucose PET scans, showed that PD patients are indeed “honest” and have difficulties making deceptive responses, and that this personality trait might be derived from dysfunction of the prefrontal cortex (Abe et al., 2009). Many patients with PD also develop obsessive-compulsive behavior, addictive personality, and impulse control disorder, particularly exemplified by compulsive gambling and shopping, hypersexuality, hoarding and other compulsive behaviors (Molina et al., 2000; Alegret et al., 2001; Geschwandtner et al., 2001; Driver-Dunckley et al., 2003; Ondo and Lai, 2008; Stamey and Jankovic, 2008; Mamikonyan et al., 2008; Ferrara and Stacy, 2008; Robert et al., 2009; O’Sullivan et al., 2010). In one study at Baylor College of Medicine, 300 consecutive patients taking dopamine agonists for PD (n = 207), restless legs syndrome (n = 89), or both (n = 4), 19.7% reported increased impulsivity: 30 gambling, 26 spending, 11 sexual activity, and 1 wanton traveling, but only 11/59 (18.6%) felt the change was deleterious (Ondo and Lai, 2008). Increased impulsivity correlated with a younger age (P = 0.01) and larger doses of dopamine agonist. Using perfusion TC99m single-photon emission computed tomography (SPECT) to study brain activity, PD patients with pathologic gambling have been found to have resting state dysfunction of the mesocorticolimbic network involved in addictive behavior (Cilia et al., 2008). Thus it is postulated that in such patients there is lack of the usual reduction in cortical perfusion typically associated with the neurodegenerative process and that pathologic gambling results from abnormal drug-induced overstimulation of the relatively spared mesocorticolimbic dopamine system. Increased striatal dopamine release has been postulated in PD patients with pathologic gambling, based on findings from raclopride PET scans (Steeves et al., 2009).
Various intrusive cognitive events with associated repetitive behaviors, representing the spectrum of obsessive-compulsive disorder in PD, include the following domains: (1) checking, religious, and sexual obsessions; (2) symmetry and ordering; (3) washing and cleaning; and (4) punding. Punding is characterized by intense fascination with repetitive handling, examining, sorting, and arranging of objects, inordinate writing, doodling, painting, collecting things, shuffling through personal papers, journaling/blogging, internet play, excessive cleaning or gardening or sorting household objects, humming or singing, and reciting long, meaningless soliloquies without an audience (Evans et al., 2004; Voon, 2004; Silveira-Moriyama et al., 2006). The behavior may be based on one’s past experiences and hobbies or may be more related to obsessive-compulsive disorder features such as gambling, which in turn may be exacerbated by dopaminergic drugs (Kurlan, 2004). In a survey of 373 consecutive patients with PD, only 1.4% exhibited punding behavior (Miyasaki et al., 2007). Compulsive singing may be another variant of punding (Bonvin et al., 2007). Pathologic gambling has been attributed most frequently to the use of dopamine agonists (Kurlan, 2004; Dodd et al., 2005; Stamey and Jankovic, 2008), but levodopa and even subthalamic nucleus deep brain stimulation have been also reported to cause pathologic gambling. The obsessive-compulsive disorder that is associated with PD has been reported to improve with high-frequency stimulation of the subthalamic nucleus (Mallet et al., 2002).
Another behavioral abnormality, possibly related to underlying obsessive-compulsive disorder, is “hedonistic homeostatic dysregulation.” This behavior is seen particularly in males with young-onset or early-onset PD (YOPD) who misuse and abuse dopaminergic drugs and develop cyclic mood disorder with hypomania or manic psychosis (Giovannoni et al., 2000; Pezzella et al., 2005). Other behavioral symptoms associated with dopamine dysregulation syndrome include compulsive dopaminergic replacement (Lawrence et al., 2003), craving, binge eating, compulsive foraging, euphoria, dysphoria, hypersexuality, pathologic gambling, compulsive shopping, aggression, insulting gestures, paranoia, jealousy, phobias, impulsivity, and other behaviors (Evans and Lees, 2004; Isaias et al., 2008; Mamikonyan et al., 2008; Stamey and Jankovic, 2008; Weintraub, 2008). Dopaminergic drugs, particularly dopamine agonists, have been demonstrated to precipitate or exacerbate behavioral symptoms of impulse control disorder and the symptoms usually improve with reduction of dosage or cessation of the offending drug (Mamikonyan et al., 2008).
Sleep disorders
Sleep disorders are being increasingly recognized as a feature of PD. An instrument consisting of 15 questions for assessing sleep and nocturnal disability has been described (Chaudhuri et al., 2002). While most studies have attributed the excessive daytime drowsiness and irresistible sleep episodes (sleep attacks) to anti-PD medications (Ondo et al., 2001), some authors believe that these sleep disturbances are an integral part of PD and are age-related (Gjerstad et al., 2006). Increasing the nighttime sleep with antidepressants or benzodiazepines may not necessarily alleviate daytime drowsiness (Arnulf et al., 2002). In a study of 303 PD patients, 63 (21%) had symptoms of restless legs syndrome, possibly associated with low ferritin levels, but there was no evidence that restless legs syndrome leads to PD (Ondo et al., 2002). These results are nearly identical to another study that found restless legs syndrome in 22% of 114 patients with PD (Gomez-Esteban et al., 2007). In another study, 10 of 126 (7.9%) patients with PD and 1 of 129 (0.8%) controls had symptoms of restless legs syndrome (Krishnan et al., 2003). Tan and colleagues (2002) found motor restlessness in 15.2% of their patients with PD, but the prevalence of restless legs syndrome, based on diagnostic criteria proposed by the International Restless Legs Syndrome Study Group, in the PD population was the same as that in the general or clinic population. Degeneration of the diencephalospinal dopaminergic pathway has been postulated to be the mechanism of restless legs syndrome in patients with PD (Nomura et al., 2006).
Several studies have provided evidence that up to 50% of patients presenting with idiopathic rapid eye movement (REM) sleep behavior disorder (RBD) eventually developed parkinsonism and idiopathic RBD is now considered to represent a pre-parkinsonian state (Schenk et al., 1996; Plazzi et al., 1997; Comella et al., 1998; Wetter et al., 2000; Ferini-Strambi and Zucconi, 2000; Matheson and Saper, 2003; Gagnon et al., 2006; Iranzo et al., 2006; Postuma et al., 2006; Boeve et al., 2007; Britton and Chaudhuri, 2009; Postuma et al., 2009a; Postuma et al., 2010). RBD seems to be predictive of PD-associated dysautonomia, particularly orthostatic hypotension (Postuma et al., 2009b). The RBD Screening Questionnaire (RBDSQ) seems to be a sensitive instrument that captures most of the characteristics of RBD and may be useful as a screening tool (Stiasny-Kolster et al., 2007). In one study, 86% of patients with RBD had associated parkinsonism (PD: 47%; MSA: 26%; PSP: 2%) (Olson et al., 2000). RBD was found in 11 of 33 (33%) patients with PD, and 19 of 33 (58%) had REM sleep without atonia (Gagnon et al., 2002). In another study, RBD preceded the onset of parkinsonism in 52% of patients with PD (Olson et al., 2000). The strong male preponderance that is seen in patients with RBD is much less evident in patients who eventually develop MSA. Although considered a parkinsonian symptom, RBD is often exacerbated by dopaminergic therapy (Gjerstad et al., 2008). [11C]-dihydrotetrabenazine PET (Albin et al., 2000) and [123I]-iodobenzamide SPECT (Eisensehr et al., 2000) found evidence of significantly reduced dopaminergic terminals and striatal D2 receptor density, respectively, in patients with RBD, though not to the same degree as in patients with PD. Patients with RBD are significantly less likely to have the tremor-dominant form of PD, have higher frequency of falls, and are less responsive to medications than PD patients without RBD (Postuma et al., 2008). Furthermore, patients with RBD have been found to have a high incidence of mild cognitive impairment (Gagnon et al., 2009), impaired color discrimination, olfactory dysfunction, and dysautonomia, in addition to motor impairment (Postuma et al., 2006). RBD has been also found to predict cognitive impairment in PD patients without dementia (Vendette et al., 2007). Sleep walking has been also demonstrated in PD patients with RBD (Poryazova et al., 2007). Only two cases with RBD have been examined at autopsy, showing evidence of Lewy body disease associated with degenerative changes in the SN and locus coeruleus in one case, but not in the other case (Boeve et al., 2007). Thus individuals with symptoms of RBD have a significantly increased risk of developing parkinsonism, particularly if functional imaging shows decreased nigrostriatal dopaminergic activity, over the next decade, but progression to neurodegenerative disease is not inevitable (Britton and Chaudhuri, 2009; Postuma et al., 2009a).
There is a growing body of evidence supporting the notion that dopamine activity is normally influenced by circadian factors (Rye and Jankovic, 2002). For example, tyrosine hydroxylase falls several hours before the person wakes, and its increase correlates with motor activity. It has been postulated that low doses of dopaminomimetic drugs stimulate D2 inhibitory autoreceptors located on cell bodies of neurons in the ventral tegmental area resulting in sedation. This is consistent with the findings that local (ventral tegmental area) application of D2 antagonists causes sedation while administration of amphetamines initiates and maintains wakefulness. Although the cerebrospinal fluid levels of hypocretin (Sutcliffe and de Lecea, 2002) have been reported to be normal in three PD patients with excessive daytime drowsiness, further studies are needed to explore the relationship between hypocretin and sleep disorders associated with PD (Overeem et al., 2002). Since the loss of dopamine in PD generally progresses from putamen to the caudate and eventually to the limbic areas, it has been postulated that it is loss of dopamine in these latter circuits, most characteristic of advanced disease, that is a potential factor in the expression of excessive daytime drowsiness and sleep-onset REM in PD (Rye and Jankovic, 2002).
Sensory abnormalities
In addition to motor and behavioral symptoms, PD patients often exhibit a variety of sensory deficits. Sensory complaints, such as paresthesias, akathisia, and oral and genital pain (Comella and Goetz, 1994; Ford et al., 1996; Djaldetti et al., 2004; Tinazzi et al., 2006; Defazio et al., 2008) are frequently not recognized as parkinsonian symptoms and result in an inappropriate and exhaustive diagnostic evaluation.
Olfactory function is typically impaired in 90% of PD cases, but hyposmia may be present in 25% of controls due to head trauma, rhinitis, and other causes. Several studies have demonstrated that hyposmia may be present even in very early stages of PD (Stern et al., 1994; Katzenschlager and Lees, 2004; Lee et al., 2006; Silveira-Moriyama et al., 2009b), and may predate other clinical symptoms of PD by at least 4 years (Ross et al., 2008). One study showed that idiopathic olfactory dysfunction (hyposmia) may be associated with a 10% increased risk of developing PD (Ponsen et al., 2004). Camicioli and colleagues (2001) found that a combination of finger tapping, olfaction ability (assessed by the University of Pennsylvania Smell Identification Test, or UPSIT), and visual contrast sensitivity, or Paired Associates Learning, discriminated between PD patients and controls with 90% accuracy. A color-coded probability scale has been developed to interpret UPSIT in patients with suspected parkinsonism (Silveira-Moriyama et al., 2009a). In one study, after 5 years of prospective follow-up 5/40 (12.5%) hyposmic first-degree relatives of patients with PD fulfilled clinical diagnostic criteria for PD; none of the other 349 relatives available for follow-up developed PD (Ponsen et al., 2010). All hyposmic individuals developing PD had an abnormal baseline 2beta-carboxymethoxy-3-beta-(4[123I]iodophenyl)tropane (β-CIT) SPECT scan. Thus a two-step approach using olfactory testing followed by dopamine transporter (DAT) SPECT scanning in hyposmic individuals appears to have a high sensitivity and specificity in detecting PD. The mechanism of olfactory loss in PD is not well understood, but it does not appear to be due to damage to the olfactory epithelium, but rather results from abnormalities in central regions involved in odor perception (Witt et al., 2009). In fact, recent studies indicate that cholinergic denervation of the limbic archicortex is a more important determinant of hyposmia than nigrostriatal dopaminergic denervation in patients with PD and that deficits in odor identification are associated with greater cognitive impairment (Bohnen et al., 2010). These findings are based on a study of 58 patients with PD who underwent PET scans using [11C]PMP acetylcholinesterase as a cholinergic ligand. The investigators found that odor identification test scores correlated positively with acetylcholinesterase activity in the hippocampal formation (r = 0.56, P < 0.0001), amygdala (r = 0.50, P < 0.0001) and neocortex (r = 0.46, P = 0.0003) and with cognitive measures such as episodic verbal learning (r = 0.30, P < 0.05).
To determine which signs in very early, presymptomatic or prodromal phase of the disease predict the subsequent development of PD, Montgomery and colleagues (1999) studied 80 first-degree relatives of patients with PD and 100 normal controls using a battery of tests of motor function, mood, and olfaction (Tissingh et al., 2001; Double et al., 2003). They found that 22.5% of the relatives and only 9% of the normal controls had abnormal scores. It is, of course, not known how many of the relatives with abnormal scores will develop PD; therefore, the specificity and sensitivity of the test battery in predicting PD cannot be determined. UPSIT administered to 62 twin pairs who were discordant for PD showed that smell identification was reduced in the twins affected with PD in comparison to those without symptoms (Marras et al., 2005a). After a mean interval of 7.3 years, 19 of the twins were retested. Neither of two twins who developed new PD had had impaired smell identification at baseline, although their UPSIT scores declined more than those of the other 17 twins. The authors concluded that “smell identification ability may not be a sensitive indicator of future PD 7 or more years before the development of motor signs.” In another study of 295 PD patients and 150 controls, olfactory impairment was found to be an independent feature of PD, unrelated to other PD symptoms (Verbaan et al., 2008). Furthermore, parkin and DJ-1 mutation carriers had normal olfaction. Impaired olfaction correlates well with decreased β-CIT uptake and may precede the onset of motor symptoms of PD (Berendse et al., 2002; Siderowf et al., 2005) and with decreased 123I-metaiodobenzylguanidine (MIBG) cardiac uptake (Lee et al., 2006). Reduced olfaction in PD may be related to a neuronal loss in the corticomedial amygdala (Harding et al., 2002) or to an increase of dopaminergic neurons in the olfactory bulb (dopamine inhibits olfactory transmission), as determined by increased tyrosine hydroxylase-reactive neurons (Huisman et al., 2004). It is important to point out, however, that olfaction is impaired in the elderly population. Using the San Diego Odor Identification Test and self-report, Murphy and colleagues (2002) found that 24.5% of people between the ages of 53 and 97 have impaired olfaction, and the incidence is 62.5% in people over age 80. Loss of smell is characteristic of PD and Alzheimer disease but is usually not present in ET, PSP, corticobasal degeneration, vascular parkinsonism (Katzenschlager et al., 2004), or parkinsonism due to parkin mutation (Khan et al., 2004). Within PD, patients with the tremor-dominant form with family history were found to have less olfaction loss than those without family history, suggesting that the familial tremor-dominant form of PD might be a different disease from sporadic PD (Ondo and Lai, 2005). In 19 patients with PARK8 (LRRK2 G2019S mutations), the mean UPSIT scores were significantly lower than in healthy controls (P < 0.001) and similar to that of patients with PD, but the score was normal in two asymptomatic carriers (Silveira-Moriyama et al., 2008). α-Synuclein pathology, including Lewy bodies, was found in the rhinencephalon of four brains of the LRRK2 patients who had hyposmia. Biopsy of olfactory nasal neurons does not aid in differentiating PD and Alzheimer disease from the other neurodegenerative disorders (Hawkes, 2003). Some investigators think that olfactory testing is comparable to other diagnostic tests, such as MRI, SPECT, and neuropsychologic testing, in differentiating PD from other parkinsonian disorders and in early detection of PD (Katzenschlager and Lees, 2004). Olfactory impairment early in the course of PD has led to the hypothesis for the pathogenesis of PD suggesting that some infectious, prion-like process, or environmental agent enters the brain via the olfactory route (Doty, 2008; Lerner and Bagic, 2008). The prodromal phase, often associated with olfactory deficit, dysautonomia, and sleep disorder, lasts months or years before the onset of typical motor features of PD (Hawkes, 2008).
There are other sensory abnormalities in patients with PD. One study showed that patients with PD who experience pain have increased sensitivity to painful stimuli (Djaldetti et al., 2004). Using quantitative sensory testing with thermal probes, laser-evoked potentials (LEPs), and laser-induced sudomotor skin responses (1-SSRs) in “off” and “on” conditions, Schestatsky et al. (2007) found lower heat pain and laser pinprick thresholds, higher LEP amplitudes, and less habituation of sympathetic sudomotor responses to repetitive pain stimuli in patients with PD who complained of primary central pain as compared with PD patients without pain and control subjects, suggesting an abnormal control of the effects of pain inputs on autonomic centers. Joint position has been found to be impaired in some patients with PD (Zia et al., 2000).
There is some evidence that while the visual acuity in PD is usually spared, some patients experience progressive impairment of color discrimination, contrast sensitivity (especially in the blue-green axis), visual speed, visual construction, and visual memory (Bodis-Wollner, 2002; Diederich et al., 2002; Uc et al., 2005). A review of 81 PD patients found nonmotor tasks were affected by visual or visuospatial impairment (Davidsdottir et al., 2005). Motor disturbances were directly attributed to visual hallucinations, double vision, and estimating spatial relations, and most often produced freezing of gait. Although color visual discrimination, as measured by the Farnsworth Munsell 100 Hue Test, has been found to be abnormal in some patients with PD, this does not appear to be an early marker for PD (Veselá et al., 2001). It is not known whether this visual dysfunction is due to retinal or postretinal abnormality. In their review of ophthalmologic features of PD, Biousse and colleagues (2004) noted that any of the following may contribute to the ocular and visual complaints in patients with PD: decreased blink rate, ocular surface irritation, altered tear film, visual hallucinations, blepharospasm, decreased blink rate, and decreased convergence. Electrophysiologic testing has revealed prolonged visual evoked potential latencies and abnormal electroretinographic patterns, suggesting retinal ganglion cell impairment playing a role in the loss of acuity in PD subjects (Sartucci et al., 2003). Disturbances in visual pathway from the retina to the occipital cortex have been demonstrated and may account for a large variety of visual disturbances experienced by patients with PD (Archibald et al., 2009). In one study, levodopa was found to significantly increase response time for reflexive (stimulus driven) prosaccades and reduced error rate for voluntary (internally guided) antisaccades, suggesting that medicated patients are better able to plan and execute voluntary eye movements, mediated by the frontostriatal system (Hood et al., 2007).
Motor fluctuations related to levodopa therapy are well recognized, but what is not readily appreciated is that many patients also experience nonmotor fluctuations, such as sensory symptoms, dyspnea, facial flushing, hunger (and sweet cravings), and other symptoms (Hillen and Sage, 1996). Weight loss is another, though poorly understood, typical manifestation of PD (Jankovic et al., 1992; Ondo et al., 2000; Chen et al., 2003; Lorefalt et al., 2004; Bachmann and Trenkwalder, 2006; Uc et al., 2006b; Barichella et al., 2009). In one study, PD patients lost 7.7% ± 1.5% of body weight over a mean of 7.2 years of follow-up, as compared to only 0.2% ± 0.7% over a mean of 10 years (55.6% of PD patients vs. 20.5% of controls lost >5% of weight, P < 0.001) (Uc et al., 2006b). The weight loss correlated with worsening of parkinsonism, age at diagnosis, visual hallucinations, and possibly dementia. In Huntington disease, weight loss has been attributed to higher sedentary energy expenditure (Pratley et al., 2000), but the mechanism of weight loss in PD is not well understood, though it is not thought to be due to reduced energy intake (Chen et al., 2003). It is important to note that these nonmotor symptoms may be as disabling as the classic motor symptoms or even more so (Lang and Obeso, 2004).
While most animal models of PD have focused on the motor symptoms, there is growing interest in developing models with both motor and nonmotor features to better simulate the human condition. Mice genetically engineered to have vesicular monoamine 2 (VMAT2) deficiency, in addition to progressive loss of striatal dopamine, levodopa-responsive motor deficits, α-synuclein accumulation, and nigral dopaminergic cell loss, also display progressive deficits in olfactory discrimination, delayed gastric emptying, altered sleep latency, anxiety-like behavior, and age-dependent depressive behavior (Taylor et al., 2009). Restoring monoamine function in these animals (and patients with PD) may be beneficial in treating the disease.
Clinical-pathologic correlations
The clinical heterogeneity in parkinsonian patients suggests that there is variable involvement of the dopaminergic and other neurotransmitter systems. Alternatively, the subgroups might represent different clinical-pathologic entities, thus indicating that PD is not a uniform disease but a syndrome. By using statistical cluster analysis of 120 patients with early PD, four main subgroups were identified: (1) young-onset, (2) tremor-dominant, (3) non-tremor-dominant with cognitive impairment and mild depression, and (4) rapid disease progression but no cognitive impairment (Lewis et al., 2005). A systematic review of 242 pathologically verified cases of PD showed that the cases were segregated into earlier disease onset (25%), tremor-dominant (31%), non-tremor-dominant (36%), and rapid disease progression without dementia (8%) subgroups (Selikhova et al., 2009). As noted before, the non-tremor cases were more likely to have cognitive disability, the earlier disease onset group had the longest duration to death and greatest delay to the onset of falls and cognitive decline, and rapid disease progression was associated with older age, early depression, and early midline motor symptoms. Furthermore, the non-tremor dominant subgroup had significantly more cortical Lewy bodies, amyloid-beta plaque load, and cerebral amyloid angiopathy than early disease onset and tremor-dominant groups.
Highly predictive diagnostic criteria are essential to select an appropriate patient population for genetic studies and clinical trials (Gelb et al., 1999; Dickson et al., 2009). In support of the notion that the PIGD subgroup represents a distinct disorder, separate from PD, is the finding that only 27% of patients with the PIGD form of idiopathic parkinsonism have Lewy bodies at autopsy (Rajput et al., 1993). In the London brain bank series, only 11% of the 100 pathologically proven cases of PD had tremor-dominant disease, and 23% had “akinetic/rigid” disease; the rest (64%) were diagnosed as having a “mixed pattern” (Hughes et al., 1993). In contrast to the 76–100% occurrence of tremor in PD, only 31% of those with atypical parkinsonism (progressive supranuclear palsy, or PSP; striatonigral degeneration, or SND; Shy–Drager syndrome, or SDS; and the combination of SND and olivopontocerebellar atrophy, or OPCA) had rest tremor (Rajput et al., 1991), and 50% of the 24 cases with non-PD parkinsonism in the London series had tremor, type not specified (Hughes et al., 1992b). Women tend to have the tremor-dominant form of PD, which has a slower progression than the non-tremor forms.
In a clinical-pathologic study, Hirsch and colleagues (1992) have demonstrated that patients with PD and prominent tremor have degeneration of a subgroup of midbrain (A8) neurons, whereas this area is spared in PD patients without tremor. Other clinical-pathologic studies have confirmed that the tremor-dominant type of PD shows more damage to the retrorubral field A8, containing mainly calretinin-staining cells but only a few tyrosine hydroxylase and dopamine transporter immunoreactive neurons (Jellinger, 1999). Also the tremor-dominant PD seems to be associated with more severe neuron loss in medial than in lateral zona compacta of SN. The ventral (rostral and caudal) GPi seems to be relatively spared in tremor-dominant PD as the dopamine levels in this area are essentially normal, but are markedly decreased in the other pallidal regions (Rajput et al., 2008). In contrast, A8 is rather preserved in the PIGD, rigid-akinetic PD, possibly owing to the protective role of calcium-binding protein. Using voxel-based morphometry of 3 teslas, T1-weighted MR images in 14 patients with tremor-dominant PD and 10 PD patients without rest tremor, decreased gray matter volume in the cerebellum was associated with parkinsonian rest tremor (Benninger et al., 2009). These findings support the hypothesis that differential damage of subpopulations of neuronal systems is responsible for the diversity of phenotypes seen in PD and other parkinsonian disorders. Detailed clinical-pathologic-biochemical studies will be required to prove or disprove this hypothesis.
Using 18F-6-fluorodopa, Vingerhoets and colleagues (1997) demonstrated that bradykinesia is the parkinsonian sign that correlates best with nigrostriatal deficiency. In contrast, patients with the tremor-dominant PD have increased metabolic activity in the pons, thalamus, and motor association cortices (Antonini et al., 1998). The presence of tremor in PD also seems to correlate with serotonergic dysfunction as suggested by a 27% reduction in the midbrain raphe 5-HT1A binding demonstrated by 11C-WAY 100635 PET scans (Doder et al., 2003). In contrast to dopamine, which is reduced by >80% in the caudate and >98% in the putamen of brains of patients with PD, serotonin markers are reduced by 30–66% (Kish et al., 2008). While the reduction of serotonin in the caudate nucleus has been suggested to be associated with “associative-cognitive problems,” the clinical significance of relative serotonin preservation in the putamen is not known. The role of serotonin in motor dysfunction, levodopa-induced dyskinesias, mood, and psychosis associated with PD has been recently reviewed, but because of lack of data no definite conclusions can be made (Fox et al., 2009).
There is a growing appreciation not only for the clinical heterogeneity of PD, but also for genetic heterogeneity (Tan and Jankovic, 2006). As a result, the notion of PD is evolving from the traditional view of a single clinical-pathologic entity to “Parkinson diseases” with different etiologies and clinical presentations. For example, the autosomal recessive juvenile parkinsonism (AR-JP) due to mutation in the parkin gene on chromosome 6q25.2–27 (PARK2) may present with a dystonic gait or camptocormia during adolescence or early adulthood (or even in the sixth or seventh decade) and with levodopa-responsive dystonia and may be characterized by symmetric onset, marked sleep benefit, early levodopa-induced dyskinesias, hemiparkinsonism–hemiatrophy, hyperreflexia, and “slow” orthostatic tremor. Patients with parkin mutation seem to have a slower disease course (Lücking et al., 2000; Rawal et al., 2003; Schrag and Schott, 2006). Furthermore, similar to EOPD, patients with parkin mutations show marked decrease in striatal 18F-FDOPA PET, but in contrast to PD, parkin patients show additional reductions in caudate and midbrain as well as significantly decreased raclopride binding in striatal, thalamic, and cortical areas (Scherfler et al., 2004). PARK2 (due to parkin mutations) is the most common cause of parkinsonism (EOPD), followed by DJ-1, and PINK1 mutations, although these genetic causes accounted for only 9% of all cases of EOPD (Macedo et al., 2009). Other causes of levodopa-responsive juvenile parkinsonism or EOPD include dopa-responsive dystonia, spinocerebellar atrophy type 2 (SCA2), SCA3, and other causes (Paviour et al., 2004).
Subtypes and natural history of Parkinson disease
The rich and variable clinical expression of PD has encouraged a search for distinct patterns of neurologic deficits that may define parkinsonian subtypes and predict the future course (Jankovic, 2005; Le et al., 2009). On the basis of an analysis of a cohort of 800 PD patients, two major subtypes were identified: one characterized by tremor as the dominant parkinsonian feature and the other dominated by PIGD (Zetusky et al., 1985; Jankovic et al., 1990; McDermott et al., 1995). The mean tremor score was defined as the mean of the sum of the baseline tremor (UPDRS Part II) and tremor scores (UPDRS Part III) for face, right and left hand, right and left foot, and right and left hand action tremor. The mean PIGD score was defined as the sum of an individual’s baseline falling, freezing, walking, gait, and postural stability UPDRS scores divided by five. Patients were categorized as having tremor-dominant PD if the ratio of the mean tremor score to the mean PIGD score was ≥1.50 and as PIGD dominant if the ratio was ≤1.00 (Jankovic et al., 1990). The tremor-dominant form of PD seems to be associated with a relatively preserved mental status, earlier age at onset, and a slower progression of the disease than the PIGD subtype, which is characterized by more severe bradykinesia and a more rapidly progressive course (Post et al., 2007). Furthermore, several studies have demonstrated that patients with the PIGD form of PD have more cognitive impairment than those with the tremor-dominant form of PD (Verbaan et al., 2007b). A presentation with bradykinesia and the PIGD type of PD seems to be associated with a relatively malignant course, whereas PD patients who are young and have tremor at the onset of their disease seem to have a slower progression and a more favorable prognosis. In a meta-analysis of 1535 titles and abstracts, of which 27 fulfilled a set of predetermined criteria, higher age at onset and higher PIGD score provided the strongest evidence of poor prognosis (Post et al., 2007). In one study, the relative risk of death in patients with the tremor-dominant form of PD was significantly lower than in PD patients without rest tremor (1.52 vs. 2.04, P < 0.01) (Elbaz et al., 2003). Using the SPES/SCOPA rating scale (Marinus et al., 2004) in 399 PD patients, four distinct motor patterns were identified: tremor-dominant, bradykinetic-rigid, and two types of axial patterns: (a) rise, gait, postural instability (similar to PIGD) and (b) freezing, speech, and swallowing, the latter related to complications of dopaminergic therapy (van Rooden et al., 2009). In a random sample of 173 patients, four different subtypes were identified: rapid disease progression subtype, young-onset subtype, non-tremor-dominant subtype (associated with hypokinesia, rigidity, postural instability and gait disorder, cognitive deterioration, depressive and apathetic symptoms, and hallucinations), and a tremor-dominant subtype (Reijnders et al., 2009). The AAN Practice Parameter on diagnosis and prognosis of new-onset PD made the following conclusions: (1) Early falls, poor response to levodopa, symmetry of motor manifestations, lack of tremor, and early autonomic dysfunction are probably useful in distinguishing other parkinsonian syndromes from PD. (2) Levodopa or apomorphine challenge and olfactory testing are probably useful in distinguishing PD from other parkinsonian syndromes. (3) Predictive factors for more rapid motor progression, nursing home placement, and shorter survival time include older age at onset of PD, associated comorbidities, presentation with rigidity and bradykinesia, and decreased dopamine responsiveness (Suchowersky et al., 2006).
The more favorable course of the tremor-dominant form of PD is also supported by the finding that the reduction in FDOPA uptake, as measured by PET and expressed as Ki, was 12.8% over a 2-year period in PD patients with severe tremor compared with a 19.4% reduction in the mild or no tremor group (P = 0.04) (Whone et al., 2002). Furthermore, Hilker and colleagues (2005) provided evidence for a variable progression of the disease based on the clinical phenotype. Similar to other studies (Jankovic and Kapadia, 2001), they showed that patients with the tremor-dominant form of PD progressed at a slower rate than patients with the other PD subtypes (Figs 4.5 and 4.6). A clinicopathologic study of 166 patients with PD followed for over 39 years (1968–2006) showed that the age at onset was significantly younger, progression to Hoehn and Yahr stage 4 was slower, and dementia was least common in the tremor-dominant cases (Rajput et al., 2009). In addition, the tremor-dominant form of PD is associated with a more frequent family history of tremor, and it is more likely to have coexistent ET (Jankovic et al., 1995; Shahed and Jankovic, 2007). In 22 patients with PD with family history of ET, 90% (20 of 22) had the tremor-predominant subtype of PD (Hedera et al., 2009). Axial impairment, probably mediated predominantly by nondopaminergic systems, is associated with incident dementia (Levy et al., 2000). While executive function was found to be impaired in both familial and sporadic PD, explicit memory recall is more impaired in the sporadic form of PD (Dujardin et al., 2001).
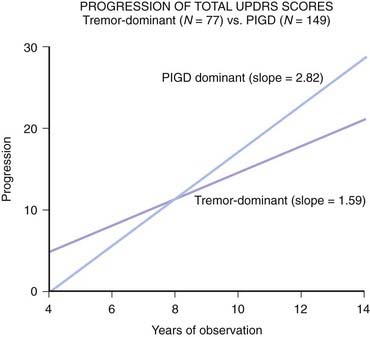
Figure 4.5 More rapid progression of PIGD versus tremor-dominant form of PD.
Data from Jankovic J, Kapadia AS. Functional decline in Parkinson’s disease. Arch Neurol 2001;58:1611–1615.
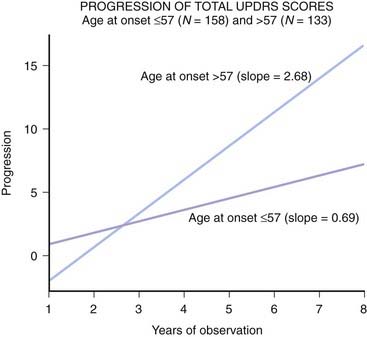
Figure 4.6 Young-onset PD progresses at a slower rate than late-onset PD.
Data from Jankovic J, Kapadia AS. Functional decline in Parkinson’s disease. Arch Neurol 2001;58:1611–1615.
To determine whether age at onset is a predictor of the future course and response to levodopa, 48 patients with YOPD (onset between 20 and 40 years of age) were compared to 123 late-onset PD (LOPD) patients (onset at 60 years of age or older) (Jankovic et al., 1997). YOPD patients presented more frequently with rigidity, while LOPD presented more frequently with PIGD; there was no difference in the occurrence of tremor at onset. YOPD patients generally respond to levodopa better but are more likely to develop dyskinesias and “wearing-off” (Quinn et al., 1987; Jankovic et al., 1997; Schrag et al., 1998; Kumar et al., 2005; Schrag and Schott, 2006; Wickremaratchi et al., 2009). Furthermore, the YOPD subtype is characterized by slower progression of disease, increased rate of dystonia and levodopa-induced dyskinesia, and less motor and cognitive disability (Graham and Sagar, 1999; Diederich et al., 2003; Wickremaratchi et al., 2009). Many, if not most, YOPD patients have been found to have parkin mutations (PARK2) or other mutations in other gene loci (PARK6 and PARK7), but their clinical characteristics and 18F-FDOPA uptake are similar to those in other YOPD patients without mutations (Thobois et al., 2003). PD seems to have a much greater psychosocial impact on YOPD patients in terms of loss of employment, disruption of family life, perceived stigmatization, and depression than on LOPD patients (Schrag et al., 2003).
There is growing evidence that the progression of PD is not linear and that the rate of deterioration is much more rapid in the early phase of the disease (Jankovic, 2005; Schapira and Obeso, 2006; Lang, 2007; Maetzler et al., 2009). This is also supported by functional imaging (FDOPA PET) (Brück et al., 2009) and postmortem pathologic studies (Fearnley and Lees, 1991) (see below). To study the overall rate of functional decline and to assess the progression of different signs of PD, 297 patients (181 males) with clinically diagnosed PD for at least 3 years were prospectively followed (Jankovic and Kapadia, 2001) (Figs 4.5 and 4.6). Data from 1731 visits over a period of an average of 6.36 years (range: 3–17) were analyzed. The annual rate of decline in the total UPDRS scores was 1.34 units when assessed during “on” and 1.58 when assessed during “off.” Patients with older age at onset had a more rapid progression of disease than those with younger age at onset. Furthermore, the older-onset group had significantly more progression in mentation, freezing, and Parts I and II UPDRS subscores. Handwriting was the only component of UPDRS that did not significantly deteriorate during the observation period. Regression analysis of 108 patients, whose symptoms were rated during their “off” state, showed a faster rate of cognitive decline as the age at onset increased. The slopes of progression in UPDRS scores, when adjusted for age at initial visit, were steeper for the PIGD group of patients than in the tremor-dominant group. In a study of 573 patients with newly diagnosed PD, PIGD, cognitive impairment, and hallucinations were among the most reliable predictors of high mortality (Lo et al., 2009).
These results are similar to the 1.5-point annual decline, based on longitudinal assessments using the motor function (Part III) portion of the UPDRS, reported by Louis and colleagues (1999) in a community-based study of 237 patients with PD who were followed up prospectively for a mean of 3.30 years. Another prospective study, involving 232 patients with PD, showed annual decline in UPDRS motor score of 3.3 points (range 0–108; 3.1%) and 0.16 points in Hoehn–Yahr stage (range 0–5; 3.2%), with slower and more restricted decline in YOPD cases (Alves et al., 2005). In a prospective study of 145 clinic-based patients followed for 1 year and 124 community-based patients followed for 4 years, the annual mean rate of deterioration in motor and disability scores ranged between 2.4% and 7.4% (Schrag et al., 2007b). These findings, based on longitudinal follow-up data, provide evidence for a variable course of progression of the different PD symptoms, thus implying different biochemical or degenerative mechanisms for the various clinical features associated with PD. Although the deterioration in motor scores seems to flatten in more advanced stages of the disease, disability scores continued to progress, probably because of emergence of nonmotor symptoms. The study by Greffard and colleagues (2006) has also suggested that the rate of progression might not be linear and that the disease might progress more rapidly initially (about 8–10 UPDRS points in the first year) and the rate of deterioration slows in more advanced stages of the disease. This is supported by the findings in moderately advanced cases of PD requiring levodopa treatment compared with patients in early stages of the disease such as those enrolled in the DATATOP study (Parkinson Study Group, 1998). In that study of early, previously untreated patients, the rate of annual decline in the total UPDRS score was 14.02 ± 12.32 (mean ± SD) in the placebo-treated group. This is nearly identical to the 1 UPDRS unit of decline per month in the ELLDOPA study (Fahn et al., 2004). In contrast, in a group of 238 patients treated with levodopa, bromocriptine, or both in whom progression was estimated on the basis of a retrospectively determined duration of the symptoms, the annual rate of decline in bradykinesia score was 3.5% during the first year but was estimated to be only 1.5% in the tenth year (Lee et al., 1994). More rapid progression in the early stages than in more advanced stages of the disease is also suggested by the finding of mean of 0.5 annual decline in FDOPA influx constant in the contralateral putamen in the first 2 years and only 0.2 during the subsequent 3 years (Brück et al., 2009).
Interestingly, in a study of 787 older (mean age at baseline: 75.4 years) Catholic clergy without clinically diagnosed PD who were prospectively followed for up to 7 years, the average decline in UPDRS units was 0.69 per year (Wilson et al., 2002). In those subjects who had some worsening of their global UPDRS score (79% of all subjects), the risk of death was 2.93 times the rate in those without progression (21%). The risk of death was associated with worsening of gait and posture but not with rigidity or postural reflex impairment, even though the latter two signs (but not bradykinesia or tremor) also worsened. The average reported rate of decline in total UPDRS is about 8 units per year. A systematic review of 13 studies investigating predictors of prognosis concluded that greater baseline impairment, early cognitive disturbance, older age, and lack of tremor at onset are relatively predictive of a poor prognosis (Marras et al., 2002). The aging process has been found to contribute particularly to the axial (gait and postural) impairment in PD (Levy et al., 2005) and advancing age, rather than duration of the disease, seems to be the most important determinant of clinical progression (Levy, 2007).
The natural history of PD appears to be influenced not only by the age at onset and the clinical presentation, but also by a number of other factors, such as stress (Tanner and Goldman, 1996), pregnancy (Shulman et al., 2000), intercurrent illness (Onofrj and Thomas, 2005), and therapy. Infection, gastrointestinal disorder, and surgery are among the most common causes of the syndrome of acute akinesia, a sudden deterioration in motor performance that usually last 4–26 days and represents a life-threatening complication of PD, usually requiring hospitalization (Onofrj and Thomas, 2005). Although therapeutic advances have had a positive impact on the quality of life, epidemiologic studies have not been able to demonstrate that levodopa significantly prolongs life (Clarke, 1995). Several studies, however, have concluded that PD patients have a nearly normal life expectancy (Lilienfeld et al., 1990; Clarke, 1995; Parkinson Study Group, 1998). In a prospective study of 800 patients who were followed longitudinally from the early stages of their disease for an average of 8.2 years, the overall death rate was 2.1% per year, which was similar to that of an age- and gender-matched US population without PD (Parkinson Study Group, 1998). In a 10-year Sydney multicenter follow-up of 149 patients with PD initially enrolled in a double-blind study of levodopa-carbidopa versus bromocriptine, the standardized mortality ratio (SMR) was 1.58, which was significantly higher than that of the Australian population (P < 0.001) (Hely et al., 1999). In a subsequent report, based on a population-based study, Morgante and colleagues (2000) showed a relative risk of death in patients with PD of 2.3 (95% confidence interval 1.60–3.39). In another study of 170 elderly patients with PD, with a mean age at death of 82 years, who were followed for a median of 9.4 years, the relative risk of death compared to referent subjects was 1.60 (95% confidence interval 1.30–1.8), and the mean duration of illness was 12.8 years (Fall et al., 2003). Pneumonia was the most frequent cause of death in both studies. The SMR was reported to be 1.52 in a community-based study of a Norwegian population (Herlofson et al., 2004). The hazard ratio for mortality was 1.64 for patients with PD compared to controls, but the mortality increased if there was associated dementia, depression, or both (Hughes et al., 2004). In a 15-year follow-up of patients who were originally enrolled in the Sydney Multicenter Study of PD comparing levodopa with low-dose bromocriptine, the SMR was 1.86 (Hely et al., 2005). In a comprehensive review of the literature, SMR has been reported to range between 1 and 3.4 (Ishihara et al., 2007). On the basis of 296 deaths in a cohort of patients who were originally enrolled in the DATATOP study and followed for 13 years, survival was found to be strongly related to response to levodopa (Marras et al., 2005b). SMR based on a cohort analysis of 22 071 participants in the Physicians’ Health Study, with 560 incident cases of PD, was found to be 2.32 (95% CI 1.85–2.92) (Driver et al., 2008). Age-specific life expectancy was found to be reduced in patients with PD, particularly those with young-onset PD (Ishihara et al., 2007). In a 20-year follow-up study of 238 consecutive patients with PD, the SMR was 0.9 by 10 years and 1.3 by 20–30 years (Diem-Zangerl et al., 2009). The authors concluded that when PD patients are “Under regular specialist care using all currently available therapies, life expectancy in PD does not appear seriously compromised, but male gender, gait disorder, and absent rest tremor at presentation are associated with poorer long-term survival.”
Differential diagnosis
Causes of parkinsonism other than PD can be classified as secondary, multiple system degeneration, or the parkinsonism-plus syndromes and heredodegenerative disorders (Stacy and Jankovic, 1992) (Table 4.3). Features that are found to be particularly useful in differentiating PD from other parkinsonian disorders include absence or paucity of tremor, early gait abnormality (such as freezing), postural instability, pyramidal tract findings, and poor response to levodopa (Tables 4.1 and 4.4). Although good response to levodopa is often used as an index of well-preserved postsynaptic receptors, supporting the diagnosis of PD, only 77% of pathologically proven cases had “good” or “excellent” initial levodopa response in the London series (Hughes et al., 1993). Furthermore, two patients with pathologically proven Lewy body parkinsonism but without response to levodopa have been reported (Mark et al., 1992). Therefore, while improvement with levodopa supports the diagnosis of PD, response to levodopa cannot be used to reliably differentiate PD from other parkinsonian disorders (Parati et al., 1993). Subcutaneous injection of apomorphine, a rapidly active dopamine agonist, has been used to predict response to levodopa and thus to differentiate between PD and other parkinsonian disorders (Hughes et al., 1990; D’Costa et al., 1991; Bonuccelli et al., 1993). Although PD patients are much more likely to improve with apomorphine, this test is cumbersome, and it does not reliably differentiate PD from the atypical parkinsonian disorders. Furthermore, response to apomorphine test is not superior to chronic levodopa therapy in diagnosis of PD; therefore, this test adds little or nothing to the diagnostic evaluation (Clarke and Davies, 2000). The differences in response to dopaminergic drugs may be partly explained by differences in the density of postsynaptic dopamine receptors. These receptors are preserved in PD, in which the brunt of the pathology is in the SN, whereas they are usually decreased in other parkinsonian disorders in which the striatum is additionally affected.
Table 4.3 Classification of parkinsonism (see also Table 9.1 for a more complete list)
| I. Primary (idiopathic) parkinsonism |
Reprinted with permission from Jankovic J, Lang AE: Classification of movement disorders. In Germano IM (ed): Surgical Treatment of Movement Disorders. Lebanon, NH, American Association of Neurological Surgeons, 1998, pp. 3–18.
Table 4.4 Motor and nonmotor symptoms associated with PD
| Motor | Nonmotor |
|---|---|
| Tremor, bradykinesia, rigidity, postural instability | Behavioral: depression, apathy, anhedonia, pseudobulbar effect, cautious personality, fatigue |
| Hypomimia, dysarthria, dysphagia, sialorrhea | Cognitive: bradyphrenia, tip-of-the-tongue, dementia |
| Microphagia, difficulties cutting food, feeding, dressing, hygiene; slow ADL | Sensory: anosmia, ageusia, impaired visual acuity, contrast, and color sensitivity, paresthesias, pain (shoulder) |
| Decreased arm swing, shuffling gait, freezing, festination, difficulty rising from chair, turning in bed | Dysautonomia: orthostatic hypotension, constipation, urinary and sexual dysfunction, abnormal swelling, seborrhea, rhinorrhea, weight loss |
| Glabellar reflex, blepharospasm, dystonia, skeletal deformities, striatal hand/foot | Sleep disorders: RBD, vivid dreams, daytime drowsiness, sleep fragmentation, restless legs syndrome? |
Perhaps expression analysis of genes in brains of patients with various neurodegenerative disorders, and recognizing disease-specific patterns will in the future assist in differentiating PD from other parkinsonian disorders. For example, using microarray technology in SN samples from six patients with PD, two with PSP, one with frontotemporal dementia–parkinsonism (FTDP), and five controls, Hauser and colleagues (2005) found 142 genes that were differentially expressed in PD cases and controls, 96 in the combination of PSP-FTDP, and 12 that were common to all three disorders. Further studies are needed to confirm this intriguing finding.
Clinical rating scales and other assessments
Although a variety of neurophysiologic and computer-based methods have been proposed to quantitate the severity of the various parkinsonian symptoms and signs, most studies rely on clinical rating scales, particularly the Unified Parkinson’s Disease Rating Scale (UPDRS), Hoehn–Yahr Staging Scale (Goetz et al., 2004), and Schwab–England Scale of activities of daily living (Fahn et al., 1987; Goetz et al., 1994, 1995; Bennett et al., 1997; Stebbins et al., 1999; Ramaker et al., 2002). The historical section of the UPDRS can be self-administered and reliably completed by nondemented patients (Louis et al., 1996).The Short (0 to 3) Parkinson’s Evaluation Scale (SPES) and the Scale for Outcomes in Parkinson’s Disease (SCOPA) are both short, reliable scales that can be used in both research and practice (Marinus et al., 2004). Although the UPDRS has a number of limitations (Movement Disorder Society Task Force on Rating Scales for Parkinson’s Disease, 2003) such as ambiguities in the written text, inadequate instructions for raters, some metric flaws, and inadequate screening questions for nonmotor symptoms, the scale is the most frequently used instrument in numerous clinical trials. In order to address some of the limitations of the original UPDRS scale, a revised scale, MDS-UPDRS, has been developed (Goetz et al., 2007, 2008b). This new MDS-UPDRS retains the original UPDRS structure of four parts with a total summed score, but the parts have been modified to provide a section that integrates nonmotor elements of PD: I: Nonmotor Experiences of Daily Living; II: Motor Experiences of Daily Living; III: Motor Examination; IV: Motor Complications. All items have five response options with uniform anchors of 0 = normal, 1 = slight, 2 = mild, 3 = moderate, 4 = severe. In some studies, the UPDRS is supplemented by more objective timed tests, such as the Purdue Pegboard test and movement and reaction times (Jankovic and Lang, 2008; Jankovic, 2007). When a particular aspect of parkinsonism requires more detailed study, separate scales should be employed, such as certain tremor scales or the Gait and Balance Scale (GABS) (Thomas et al., 2004). Also, it is important that in performing the UPDRS, the instructions are followed exactly. For example, one study of a pull test, a measure of postural instability (Hunt and Sethi, 2006), in 66 subjects, performed by 25 examiners showed marked variability in the technique among the examiners, and only 9% of the examinations were rated as error-free (Munhoz et al., 2004). Another study showed that the “push and release test” predicts which PD patients will be fallers better than the pull test (Valkovic et al., 2008). The standard pull test consists of a sudden, firm, and quick shoulder pull without prior warning, but with prior explanation, and executed only once (Visser et al., 2003). If the patient takes more than two steps backward, this is considered abnormal. When performing the push and release test, patients are instructed to stand in a comfortable stance with their eyes open while the examiner stands behind them. The patient is then instructed to push backward against the palms of the examiner’s hands placed on the patient’s scapulae while the examiner flexes his elbows to allow slight backward movement of the trunk. The examiner then suddenly removes his hands, requiring the patient to take a backward step to regain balance.
There are also many scales, such as the Parkinson disease questionnaire-39 (PDQ-39) (Hagell and Nygren, 2007) and the Parkinson disease quality-of-life questionnaire (PDQL) (de Boer et al., 1996), that attempt to assess the overall health-related or preference-based quality of life (Marinus et al., 2002; Siderowf et al., 2002; Den Oudsten et al., 2007) and the impact of the disease on the performance of activities of daily living (Lindeboom et al., 2003). The Parkinson’s Disease Quality of Life Scale (PDQUALIF), developed by the Parkinson Study Group, is being used in clinical trials designed to assess the impact of PD on quality of life (Welsh et al., 2003). The briefer version of PDQ-39, the PDQ-8, has been found to be a longitudinally reliable and responsive measure of health-related quality of life (HRQoL) and to estimate the minimally important difference (MID) or minimal clinically important change (MCIC) in response to therapeutic intervention (Schrag et al., 2006; Luo et al., 2009). In addition to these quantitative measures of PD-related disability, screening tools have been developed and validated to enhance early recognition of parkinsonism. One such instrument has used nine questions that were found to reliably differentiate patients with early PD from those without parkinsonism (Hoglinger et al., 2004). The generic 15D instrument has been found to be valid for measuring HRQoL in PD (Haapaniemi et al., 2004). In a study of 227 patients, 82 of whom were followed for up to 8 years, Forsaa et al., (2008) measured changes in HRQoL over time using the Nottingham Health Profile; they found that the steepest progression was in physical mobility, followed by social isolation and emotional reactions. Several instruments have been developed utilizing questionnaires, such as questions about the nonmotor symptoms of PD, including the nonmotor questionnaire or NMS Quest (Barone et al., 2009) and the nonmotor scale or the NMS Scale (Chaudhuri et al., 2006b), or on life satisfaction: “general life satisfaction” (QLSM-A) and “satisfaction with health” (QLSM-G), in which each item is weighted according to its relative importance to the individual. In one study these instruments were validated against the 36-item short form health survey (SF-36) and the EuroQol (EQ-5D) (Kuehler et al., 2003). When the initial questionnaires were reduced to 12 items for a “movement disorder module” (QLSM-MD), and 5 items for a “deep brain stimulation module” (QLSM-DBS), psychometric analysis revealed Cronbach’s α values of 0.87 and 0.73, and satisfactory correlation coefficients for convergent validity with SF-36 and EQ-5D. Other quality-of-life instruments have been used in assessing the response to therapies, particularly surgery as this treatment intervention is especially susceptible to a placebo effect (Martinez-Martin and Deuschl, 2007; Diamond and Jankovic, 2008).
One of the most important factors contributing to quality of life is the ability to drive. Using a standardized open-route method of assigning driving abilities and safety, Wood and colleagues (2005) found that patients with PD are significantly less safe than are controls and, more important, that the driver’s perception of his or her ability to drive correlated poorly with the examiner’s assessment. Distractibility and impaired cognition, visual perception, and motor function, associated with sleepiness, are among the major factors in driving safety errors committed by PD patients (Newman, 2006; Uc et al., 2006a; Singh et al., 2007; Uc et al., 2009c). One study found the following commonest errors committed by PD patients while driving: indecisiveness at T-junctions and reduced usage of rear view and side mirrors (Cordell et al., 2008). Driving simulation under low-contrast visibility conditions, such as fog or twilight, showed that a larger proportion of drivers with PD crashed (76.1% vs. 37.3%, P < 0.0001) and the time to first reaction in response to incursion was longer (median 2.5 vs. 2.0 seconds, P < 0.0001) compared with controls (Uc et al., 2009b). The strongest predictors of poor driving outcomes among the PD cases were worse scores on measures of visual processing speed and attention, motion perception, contrast sensitivity, visuospatial construction, motor speed, and activities of daily living score.
To assess the impact of the various nonmotor symptoms in patients with PD on their quality of life, a 30-item nonmotor symptom screening questionnaire (NMSQuest) was developed, containing nine dimensions: cardiovascular, sleep/fatigue, mood/cognition, perceptual problems, attention/memory, gastrointestinal, urinary, sexual function, and miscellany (Chaudhuri et al., 2007). In 242 patients, mean age 67.2 years and mean duration of symptoms of 6.4 years, the mean score was 56.5 ± 40.7 (range: 0–243); symptoms that were “flagged” by the NMSQuest included: nocturia (61.9%), urinary urgency (55.8%), constipation (52.5%), sad/blues (50.1%), insomnia (45.7%), concentrating (45.7%), anxiety (45.3%), forgetfulness (44.8%), dribbling (41.5%), and restless legs (41.7%) (Martinez-Martin et al., 2007).
Epidemiology
The frequency of PD varies depending on the diagnostic criteria, study population, and epidemiologic methods used, although the prevalence is generally thought to be about 0.3% in the general population and 1% in people over the age of 60 years; the reported incidence figures have ranged from 8 to 18 per 100 000 person-years (de Lau and Breteler, 2006). In a study of 364 incident cases of parkinsonism among residents of Olmsted County, MN, for the period from 1976 through 1990, 154 with PD (42%), 72 with drug-induced parkinsonism (20%), 61 unspecified (17%), 51 with parkinsonism in dementia (14%), and 26 with other causes (7%) were identified (Bower et al., 1999). The average annual incidence rate of parkinsonism (per 100 000 person-years) in the age group 50–99 years was 114.7 and the incidence increased exponentially with age from 0.8 in the age group 0–29 years to 304.8 in the age group 80–99 years. The cumulative incidence of parkinsonism was 7.5% to age 90 years. Men had higher incidence than women at all ages for all types of parkinsonism except drug-induced. In the US studies, African-Americans have been found to be half as likely to be diagnosed with PD as white Americans; these differences could not be explained by differences in age, sex, income, insurance, or access to health care (Dahodwala et al., 2009). Based on a meta-analysis of 29 studies reporting familial aggregation, the relative risk of PD is 2.9 in a first-degree relative, 4.4 in siblings, and 2.7 for a child–parent pair (Thacker and Ascherio, 2008).
Validated screening instruments, designed to detect symptoms of PD with high sensitivity and specificity, are currently lacking. Rest tremor, difficulty walking, difficulty rising from a chair, and walking slowly have been found to be highly specific (93.8–95.9%), but less sensitive (35.9–49.1%) for detecting parkinsonian motor symptoms, while other parkinsonian features such as micrographia and olfactory dysfunction are less specific, but more sensitive (Ishihara et al., 2005). A self-administered, 16-item Baylor Health Screening Questionnaire (BHSQ) is being developed for a web-based use as a potential tool to detect early symptoms of parkinsonism with 91% sensitivity and 92% specificity based on pilot data (Hunter et al., 2008). The instrument used in this study is easy to administer and may be used in mass screenings to identify individuals with undiagnosed PD. If high sensitivity and specificity are confirmed by large prospective studies, this instrument may be used for epidemiologic studies as well as for referrals to appropriate health care or research facilities.
Several diagnostic criteria have been developed for PD, including the UK Parkinson’s Disease Society Brain Bank criteria used in various clinical-pathologic studies (Hughes et al., 1992a, 1992b) (Table 4.5). During a workshop sponsored by the National Institute of Neurological Disorders and Stroke (NINDS), a set of diagnostic criteria for PD was proposed, based on a review of the literature regarding the sensitivity and specificity of the characteristic clinical features (Gelb et al., 1999; Jankovic, 2008). The reliability of the different diagnostic criteria, however, has not been vigorously tested by an autopsy examination, which is commonly considered the gold standard (de Rijk et al., 1997). Early clinical-pathologic series concluded that only 76% of patients with a clinical diagnosis of PD actually met the pathologic criteria; the remaining 24% had evidence of other causes of parkinsonism (Hughes et al., 1992a, 1992b). This study was based on autopsied brains collected from 100 patients who had been clinically diagnosed with PD by the UK Parkinson’s Disease Society Brain Bank (Hughes, et al., 1992a, 1992b). Similar findings were reported in another study, which was based on autopsy examinations of brains from 41 patients who were followed prospectively by the same neurologist over a 22-year period (Rajput et al., 1991). When Hughes et al. (2001) examined the brains of patients diagnosed with PD by neurologists, the diagnostic accuracy increased to 90%; 6% had MSA, 2% had PSP, 1% had neurofibrillary tangles, and 1% had evidence of vascular parkinsonism. In a study of 143 cases of parkinsonism that came to autopsy and had a clinical diagnosis made by neurologists, the positive predictive value of the clinical diagnosis was 98.6% for PD and 71.4% for the other parkinsonian syndromes (Hughes et al., 2002). In the DATATOP study, 800 patients were prospectively followed by trained parkinsonologists from early, untreated stages of clinically diagnosed PD for a mean of 7.6 years (Jankovic et al., 2000). An analysis of autopsy data, imaging studies, response to levodopa, and atypical clinical features indicated an 8.1% inaccuracy of initial diagnosis of PD by Parkinson experts, but the final diagnosis was not based on pathologic confirmation in all cases. In a study of 89 incident patients initially diagnosed with parkinsonism by experienced clinicians, the diagnosis was subsequently changed in 22 (33%) during the median follow-up of 29 months (Caslake et al., 2008). In this cohort, 38% of those initially diagnosed with PD had their diagnosis changed to DLB; other common misdiagnosis was ET in patients initially thought to have PD and vice versa. This and other studies underscore the need for valid diagnostic criteria to be used in assessing patients with initial manifestations of parkinsonism. In a community-based study of 402 patients taking antiparkinsonian medications, parkinsonism was confirmed in 74% and clinically probable PD in 53%. The commonest causes of misdiagnosis were essential tremor (ET), Alzheimer disease, and vascular parkinsonism. Over one-quarter of subjects did not benefit from antiparkinsonian medication (Meara et al., 1999). Parkinsonian signs, including rigidity, gait disturbance, and bradykinesia, may also occur as a consequence of normal aging, although comorbid medical conditions, such as diabetes, may significantly increase the risk of these motor signs (Arvanitakis et al., 2004). There is considerable debate whether levodopa responsiveness should be included among diagnostic criteria for PD. Although nearly all patients with PD do respond, a small minority with “documented” PD have a poor or no response, although levodopa responsiveness has not been well defined in the literature (Constantinescu et al., 2007).
Table 4.5 UK Parkinson’s Disease Society Brain Bank’s clinical criteria for the diagnosis of probable Parkinson disease
Data from Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184; and Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ: What features improve the accuracy of clinical diagnosis in Parkinson’s disease: A clinical pathological study. Neurology 1992;42:1142–1146.
Nearly all epidemiologic studies of PD show that both incidence and prevalence of PD are 1.5–2 times higher in men than in women (Haaxma et al., 2007). While there is no obvious explanation for this observed male preponderance, exposure to toxins, head trauma, neuroprotection by estrogen in women, mitochondrial dysfunction, or X-linked genetic factors have been suggested (Wooten et al., 2004; Haaxma et al., 2007). The most plausible explanation is that symptoms of PD may be delayed in women by higher striatal dopamine levels, possibly due to the effects of estrogen (Haaxma et al., 2007; Taylor et al., 2007), but this would not explain the lack of female preponderance in Asian, particularly Chinese populations (de Lau and Breteler, 2006).
Laboratory tests
Neuroimaging
Although there is no blood or cerebrospinal fluid test that can diagnose PD, certain neuroimaging techniques may be helpful in differentiating PD from other parkinsonian disorders. MRI in patients with typical PD is usually normal; but a high-field-strength (1.5 T) heavily T2-weighted MRI may show a wider area of lucency in the SN that is probably indicative of increased accumulation of iron (Olanow, 1992). Diffusion-weighted imaging (DWI) provides information on neuronal integrity by quantitating motion of water molecules, which is impaired in axonal cell membranes damaged by a neurodegenerative disease, such as PD. Applying this technique to 17 patients with PD, 16 (94%) were correctly discriminated with a sensitivity of 100% and a specificity of 88% (Scherfler et al., 2006). These patients showed significant increases of diffusivity in the region of both olfactory tracts. Using the Spin-Lattice Distribution Index (SI), a measure of MRI signal in the substantia nigra pars compacta (SNc), provides a “highly sensitive” marker for PD (Hutchinson and Raff, 2008). In a study of 14 patients with early, untreated, PD and 14 age- and gender-matched controls using a 3-tesla MRI and high-resolution diffusion tensor imaging (DTI) protocol, fractional anisotropy (FA) was reduced in the SN of subjects with PD compared with controls (P < 0.001), particularly in the caudal SN compared with the rostral region of interest, with 100% sensitivity and specificity for distinguishing patients with PD from healthy subjects (Vaillancourt et al., 2009). The method used apparently corrected for eddy currents-induced distortion but it is not clear that this is the essential element that accounted for the high sensitivity and specificity of this imaging technique or whether the reported decreased FA was a result of impaired water diffusion through iron-induced field gradients in the SN. Although there is a high correlation between DTI findings and number of SNc dopaminergic neurons lost with MPTP intoxication in a murine model of PD, there is no apparent correlation between the FA values and UPDRS motor scores.
By using [18F]-fluorodopa PET scans to assess the integrity of the striatal dopaminergic terminals, characteristic reduction of the [18F]-fluorodopa uptake, particularly in the putamen, can be demonstrated in virtually all patients with PD, even in the early stages (Brooks, 1991). Using [11C]-raclopride to image dopamine D2 receptors, Brooks and colleagues (1992a) showed that in patients with untreated PD, the striatal D2 receptors are well preserved, whereas patients with atypical parkinsonism have a decrease in the density of dopamine receptors. Involvement of the postsynaptic, striatal dopamine receptor-containing neurons in the atypical parkinsonian syndromes is also suggested by decreased binding of iodobenzamide, a dopamine receptor ligand, as demonstrated by SPECT scans (Schwarz et al., 1992). In addition to reduced density of the dopamine receptors, patients with atypical parkinsonism have decreased striatal metabolism as demonstrated by PET scans (Eidelberg et al., 1993). Besides imaging of postsynaptic D2 receptors, SPECT imaging of the striatal dopamine reuptake sites with I-123 labeled β-CIT and of presynaptic vesicles with [11C]-dihydrotetrabenazine may be also helpful in differentiating PD from atypical parkinsonism (Gilman et al., 1996; Marek et al., 1996; Booij et al., 1997). Dopamine transporter (DAT) imaging using DAT SPECT has been found to be a useful tool in reliably differentiating between PD, essential tremor, dystonic tremor, drug-induced, psychogenic, and vascular parkinsonism (Kägi et al., 2010). Although the imaging tests cannot yet be used to reliably differentiate PD from other parkinsonian disorders, future advances in this technology will undoubtedly improve their diagnostic potential.
Besides clinical rating, neuroimaging techniques have been used to assess progression of PD and other neurodegenerative disorders (Antonini and DeNotaris, 2004; Jankovic, 2005; Brooks, 2007; Nandhagopal et al., 2008a; Martin et al., 2008). Abnormal proton transverse relaxation rate (R2*) measured by 3-tesla MRI, consistent with iron deposition in the lateral SNc, seems to correlate with progression of motor symptoms and as such may have potential utility as a biomarker for disease progression (Martin et al., 2008). Neuroimaging techniques can be used not only in diagnosis but also in following the progression of the disease (Wu et al., 2011). Several studies have shown that the annualized rate of reduction in striatal dopaminergic markers, such as uptake of 18F-FDOPA or DAT binding, to range from 4% to 13% for patients with PD and 0% to 2.5% in healthy controls. Jennings and colleagues (2000) found, on the basis of sequential β-CIT and SPECT imaging at intervals ranging from 9 to 24 months, the annual rate of loss of striatal β-CIT uptake to be 7.1% in subjects having a diagnosis of PD for fewer than 2 years compared with a 3.7% rate in those having a diagnosis of PD for longer than 4.5 years. In another study using 18F-FDOPA PET, Nurmi and colleagues (2001) showed a 10.3 ± 4.8% decline in the uptake in the putamen over a 5-year period. Using serial FDOPA PET in a prospective, longitudinal study of 31 patients with PD followed for more than 5 years (mean follow-up: 64.5 ± 22.6 months), Hilker and colleagues (2005) found an annual decline in striatal FDOPA ranging from 4.4% (caudate) to 6.3% (putamen), consistent with most other similar studies (Morrish et al., 1998). They concluded that “the neurodegenerative process in PD follows a negative exponential course,” and in contrast to the long-latency hypothesis, they estimated that the preclinical disease period is relatively short: only about 6 years. Morrish and colleagues (1998), using a similar design, but with an interscan interval of only 18 months, came to the same conclusion. This is similar to the results of other longitudinal studies of PD progression, using imaging ligands either measuring dopamine metabolism (FDOPA PET) or targeting dopamine transporter (β-CIT SPECT), demonstrating an annualized rate of reduction in these striatal markers of about 4–13% in PD patients compared with a 0–2.5% change in healthy controls (Parkinson Study Group, 2002). In a PET follow-up brain graft study of patients with advanced PD, Nakamura and colleagues (2001) found a 4.4% annual decline in the sham operated patients. Thus, longitudinal studies of PD progression, imaging ligands targeting dopamine metabolism ([18F]-dopa) and dopamine transporter density (β-CIT) using PET and SPECT, respectively, have demonstrated an annualized rate of reduction in striatal [18F]-dopa or [123I]β-CIT uptake of about 11.2% (6–13%) in PD patients compared with 0.8% (0–2.5%) change in healthy controls (Marek et al., 2001).
With improved methodology of β-CIT SPECT scans, the annualized rate of decline is now estimated to be 4–8% (Parkinson Study Group, 2002). These imaging studies are consistent with pathologic studies showing that the rate of nigral degeneration in PD patients is eightfold to tenfold higher than that of healthy age-matched controls. The several studies, including the one by Hilker and colleagues (2005), that suggest that the rate of progression of PD is not linear over time, being more rapid initially and slowing in more advanced stages of the disease, argue against the long-latency hypothesis for presymptomatic period in PD (Jankovic, 2005). Finally, on the basis of clinical-pathologic correlation, Fearnley and Lees (1991) suggested that there is a 30% age-related nigral cell loss at disease onset, again indicating rapid decline in nigral dopaminergic cells in the early stages of the disease. Genetic studies have found that the age at onset of PD (and Alzheimer disease) is strongly influenced by a gene on chromosome 10q (Li et al., 2002).
Increased echogenicity on brain parenchyma transcranial sonography (TCS) is an ultrasound sign that has been found to be relatively specific for PD and that has been used to differentiate PD from atypical parkinsonism, mostly MSA (Walter et al., 2003; Berg et al., 2008). The investigators found that 24 of 25 (96%) patients with PD exhibited hyperechogenicity, whereas only 2 of 23 (9%) patients with atypical parkinsonism showed a similar pattern. They concluded that brain parenchyma sonography may be highly specific in differentiating between PD and atypical parkinsonism. In another study the sensitivity of TCS was 90.7% and the specificity was 82.4%; the positive predictive value was 92.9% (Gaenslen et al., 2008). In this study, however, tremor-dominant PD patients were excluded. Furthermore, in about 10% of patients SN cannot be imaged because of inadequate temporal bone window. Since the hyperechogenicity seems to be constant over time, TCS possibly may be used to detect this sign as a marker for PD before the onset of neurologic symptoms.
Presymptomatic diagnosis and biomarkers
One of the most important challenges in PD research is to identify individuals who are at risk for PD and to diagnose the disease even before the initial appearance of symptoms. Searching for sensitive biomarkers, such as clinical, motor, physiologic, and olfactory testing, cerebrospinal fluid proteomics, genetic testing, sleep and autonomic studies, and neuroimaging, that detect evidence of PD even before clinical symptoms first appear, has been the primary focus in many research centers around the world (Michell et al., 2004; Hawkes, 2008; Marek et al., 2008; Halperin et al., 2009; Mollenhauer and Trenkwalder, 2009; Wu et al., 2011) (Fig. 4.7). These biomarkers, if found to be useful, should reliably predict: (1) risk (clinical, genetic, blood/CSF test, imaging), (2) diagnosis, (3) progression (prognosis), and (4) response to treatment. As was noted previously, impaired olfaction is one of the earliest signs of PD, present even before the onset of motor symptoms.
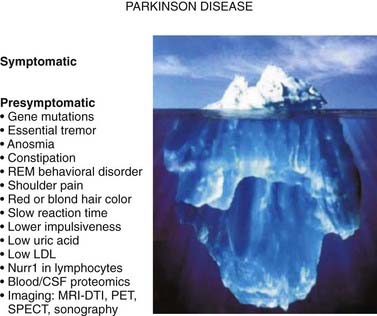
Neuroimaging of the presynaptic nigrostriatal terminals has been suggested as a potential biomarker for diagnosis of early PD and for early differentiation between PD and other parkinsonian disorders. Presymptomatic carriers of the LRRK2 mutation have been shown to have decreased dopaminergic activity and a greater rate of decline in dopaminergic imaging markers, particularly dopamine transporter binding, compared to healthy controls, suggesting that functional neuroimaging may provide a sensitive signal for subclinical dopaminergic deficiency (Nandhagopal et al., 2008b). To evaluate the diagnostic accuracy of dopamine transporter imaging using (123I)β-CIT, Jennings et al. (2004) evaluated 35 patients referred by community neurologists with suspected early PD. The clinical diagnosis was “confirmed” by two movement disorder experts, which represented the diagnostic “gold standard.” A disagreement between the “gold standard” diagnosis and imaging diagnosis occurred in only 8.6% of cases, giving the imaging sensitivity of 0.92 and specificity of 1.00. They concluded that (123I)β-CIT and SPECT imaging is a useful diagnostic tool to differentiate between patients with early PD and other parkinsonian disorders.
Many studies provide evidence suggesting that the latency between the onset of neuronal degeneration (or onset of the disease process) and clinical symptoms might not be as long as was initially postulated (Morrish et al., 1996). On the basis of a study of 36 control and 20 PD brains, Fearnley and Lees (1991) suggested that the presymptomatic phase of PD from the onset of neuronal loss to the onset of symptoms might be only 5 years, thus arguing against aging as an important cause of PD. With advancing age, there is 4.7% per decade rate of loss of pigmented neurons from the SNc, whereas in PD, there is 45% loss in the first decade. Since the rate of progression is so highly variable, it is perhaps not surprising that the estimates of the presymptomatic period vary between 40 years and 3.1 years, depending on the method used (Morrish et al., 1996). The shorter presymptomatic period has been suggested by longitudinal 18F-FDOPA PET studies (Morrish et al., 1996). Although UPDRS has been used in these longitudinal studies as a measure of clinical progression, the instrument is currently being revised to include additional items, including nonmotor experiences of daily living, to capture symptoms that reflect nondopaminergic involvement in PD. Whether the progression as measured with the current or revised UPDRS correlates with nigral and extranigral pathology associated with PD awaits future clinical-pathologic validation.
One of the benefits of longitudinal imaging studies, such as the one by Hilker and colleagues (2005; see also Jankovic, 2005), is that they can be used to estimate duration of the presymptomatic period. Assuming that the threshold at which symptoms are first manifested is at 69% of the normal putaminal FDOPA uptake, Hilker and colleagues (2005) concluded that the preclinical disease period must be relatively short: only about 6 years. This is consistent with other imaging and with autopsy data (Fearnley and Lees, 1991). The 31% loss of striatal dopaminergic terminals needed before onset of symptoms, demonstrated by Hilker and colleagues (2005), is substantially lower than the 60–80% loss of dopaminergic neurons in the SN that is traditionally cited as being required before symptoms of PD first become evident. The difference may be explained by compensatory changes in response to presynaptic dopaminergic loss, such as enhanced synthesis of dopamine in surviving dopaminergic neurons, upregulation of striatal dopa-decarboxylase activity, and increased dopaminergic innervation of the striatum (Jankovic, 2005). Furthermore, there may be functional compensatory changes, as suggested by the finding of increased FDOPA uptake in the globus pallidus interna, in early PD. This enhanced function of the nigropallidal dopaminergic projection maintains a more normal pattern of pallidal output in early stages of the disease, but these compensatory mechanisms eventually fail, and the disease starts to progress. Thus, because of the compensatory changes, FDOPA PET more accurately reflects dopaminergic function at the striatal terminal rather than a cell loss in the SN. These compensatory mechanisms may also explain why despite age-related loss of nigral neurons, there is little or no change in FDOPA uptake with normal aging (Sawle et al., 1990) and why up to 15% of patients with signs of PD, as determined by experienced parkinsonologists, have normal FDOPA or β-CIT scans without evidence of dopaminergic deficit (SWEDD) (Marek et al., 2003; Whone et al., 2003; Clarke, 2004; Fahn et al., 2004; Jankovic, 2005; Scherfler et al., 2007). These SWEDDs might represent patients with PD and compensatory striatal changes or with other disorders. They might also represent false-negative results and therefore highlight the relative lack of sensitivity of these functional neuroimaging studies as potential biomarkers for detection of PD, particularly at early stages of the disease (Michell et al., 2004). Since individuals with SWEDDs fail to develop dopaminergic deficit and fail to show clinical worsening, it is likely that these individuals were incorrectly diagnosed. This is supported by normal olfaction in SWEDD individuals (Silveira-Moriyama et al., 2009b). One possible condition misdiagnosed as PD, but with SWEDD, is adult-onset dystonic tremor, which may present as unilateral or asymmetric rest tremor and decreased arm swing (Schneider et al., 2007).
To the extent that future protective therapies may prevent or even halt the neurodegenerative process, it is essential that they be implemented early in the course of the disease. Therefore, recent clinical and basic studies have focused on a search for presymptomatic biomarkers of PD (Michell et al., 2004). An identification of a disease-specific diagnostic test would be immensely helpful not only in defining the various PD subtypes and in differentiating PD from atypical parkinsonian syndromes, but also, more importantly, in identifying populations that are at increased risk for developing PD. Such potentially vulnerable populations could then be targeted for protective therapy. Novel imaging techniques are being developed not only to monitor the progression of the disease, but also as diagnostic tools in clinically uncertain cases. Using the dopamine transporter ligand [I-123] (N)-(3-iodopropene-2-yl)-2beta-carbomethoxy-3beta-(4-chlorophenyl) tropane (IPT), and SPECT, Schwarz and colleagues (2000) showed a reduction of dopamine transporter binding in patients with early PD, suggesting that this technique has potential in detection of preclinical disease. Comparing inversion recovery MRI and 18F-FDOPA PET in 10 patients with Hoehn and Yahr stage 3 and 4 PD and 8 normal controls, Hu and colleagues (2001) found that discriminant function analysis of the quantified MRI nigral signal correctly classified the combined PD patient/control group, but three patients with PD were incorrectly classified as “normal,” whereas with PET, 100% of PD patients and controls were correctly classified. In a study of 118 patients with clinically uncertain parkinsonian syndromes, all patients with presynaptic parkinsonism had abnormal 123I-ioflupane SPECT (DaTSCAN, Amersham Health), whereas 94% with “nonpresynaptic” parkinsonism had a normal scan (Catafau and Tolosa, 2004). Abnormal echogenicity on transcranial sonography may be detected in early, and possibly even in presymptomatic PD (Weise et al., 2009). Decreased cardiac MIBG uptake was found even in de novo patients with PD, suggesting that this test could be used to detect early or even presymptomatic PD (Oka et al., 2006; Lee et al., 2006). Reduction in myocardial MIBG uptake seems to correlate with presynaptic nigrostriatal dopaminergic deficit as measured by putaminal [123I]FP-CIT SPECT, suggesting that brain and extracranial neurodegeneration in PD are coupled (Spiegel et al., 2007). Cardiac sympathetic degeneration and Lewy body pathology, even in the presymptomatic phase of PD, is likely responsible for these abnormalities, although PD-related clinically evident heart disease has not been demonstrated (Fujishiro et al., 2008).
Besides loss of olfaction, constipation, shoulder pain, RBD, and imaging studies, there are other tests that are being explored as potential biomarkers for early detection of PD. For example, mRNA expression of nuclear receptor related 1 protein (Nurr1) on peripheral lymphocytes has been found to be decreased in patients with PD as compared to other dopaminergic disorders (Pan et al., 2004). Furthermore, mRNA expression of co-chaperone ST13 in peripheral blood, which stabilizes heat-shock protein 70, a modifier of α-synuclein misfolding, has been found to be lower in patients with PD than in controls (P = 0.002) in two independent populations (Scherzer et al., 2007).
Pathologic findings
In the absence of a specific biologic marker or a diagnostic test, the diagnosis of PD can be made with certainty only at autopsy. PD is pathologically defined as a neurodegenerative disorder characterized chiefly by (1) depigmentation of the SN associated with degeneration of melanin- and dopamine-containing neurons, particularly in the SNc and in the norepinephrine-containing neurons in the locus coeruleus, and (2) the presence of Lewy bodies (eosinophilic cytoplasmic inclusions) in the SNc and other brain regions, including the locus coeruleus and some cortical areas. In fact, some studies have found that, despite the universally accepted notion that SN is the site of the brunt of the pathology in PD, neuronal loss in the locus coeruleus is more severe (Zarow et al., 2003). These criteria are open to question, however, since typical cases of levodopa-responsive parkinsonism have been reported without Lewy bodies and with or without neurofibrillary tangles in the SN (Rajput et al., 1991). In contrast, the pathologically typical form of Lewy body parkinsonism has been described with atypical clinical features such as poor response to levodopa (Mark et al., 1992). Both the Canadian (Rajput et al., 1991) and the London Parkinson’s Disease Society Brain Bank study (Hughes et al., 1992b) showed that 24% of patients in each series had a pathologic diagnosis other than PD. Furthermore, in patients with pathologically documented PD, other disorders may be present that can cloud the clinical picture. For example, in 100 cases of pathologically proven PD, Hughes and colleagues (1993) found 34 with coexistent pathology in the striatum and 28 outside the nigrostriatal system; vascular changes involving the striatum were found in 24 patients, Alzheimer changes in 20 (3 had striatal plaques confined to the striatum), and diffuse Lewy body disease or dementia with Lewy bodies in 4. As was noted previously, in a subsequent study, the diagnostic accuracy had improved markedly (Hughes et al., 2002). Until parkinsonian disorders can be differentiated by either disease-specific biologic or etiologic markers, neuroimaging, or other laboratory tests, the separation of the different parkinsonian disorders still depends largely on clinical-pathologic correlations.
While the emphasis in PD research has been on dopaminergic deficiency underlying motor dysfunction, there is a growing body of evidence that the caudal brainstem nuclei (e.g., dorsal motor nucleus of the glossopharyngeal and vagal nerves), anterior olfactory nucleus, and other nondopaminergic neurons might be affected long before the classic loss of dopaminergic neurons in the SN, based on accumulation of Lewy neurites detected by staining for α-synuclein (Braak et al., 2003, 2004; Braak and Del Tredici, 2008). According to the Braak staging, in presymptomatic stage 1, the Lewy neurite pathology remains confined to the medulla oblongata and olfactory bulb. In stage 2, it has spread to involve the pons. In stages 3 and 4, the SN and other nuclear grays of the midbrain and basal forebrain are the focus of initially subtle and then more pronounced changes, at which time the illness reaches its symptomatic phase. In end-stages 5 and 6, the pathologic process encroaches on the telencephalic cortex. A clinical-pathologic study of 129 brains in the UK Brain Bank, focusing on the late phase of PD, indicates that while it takes longer for young-onset patients to reach the end-stage of the disease, marked by more rapid physical and cognitive decline, this terminal stage is rather similar irrespective of age at onset (Kempster et al., 2010). This, according to the authors, supports “a staging system based on the rostral extent and severity of Lewy body pathology, although other pathologies may play a synergistic role in causing cognitive disability”, consistent with the Braak hypothesis.
The Braak staging, however, has been challenged for several reasons, including lack of cell counts to correlate with the described synuclein pathology and no observed asymmetry in the pathologic findings that would correlate with the well-recognized asymmetry of clinical findings. In addition, there is controversy as to the classification of dementia with Lewy bodies; Braak viewed it as part of stage 6, but others suggest that it is a separate entity, since these patients often have behavioral and psychiatric problems before the onset of motor or other signs of PD. This staging proposal, however, has been challenged as there are no cell counts to correlate with the described synuclein pathology, no immunohistochemistry to identify neuronal types, no observed asymmetry in the pathologic findings that would correlate with the well-recognized asymmetry of clinical findings, bulbar symptoms are late not early features of PD despite the suggested early involvement of the dorsal motor nucleus of the vagal nerve, exclusion of cases of dementia with Lewy bodies, the idiopathic Lewy body cases were preselected for the presence of α-synuclein deposition, cases with well-documented α-synuclein inclusions at higher levels in the neuraxis without involvement of caudal brainstem have been reported, the pathologic examination did not include the spinal cord and peripheral autonomic nervous system, and brain synucleinopathy consistent with Braak stages 4 and 6 has been found in individuals without any neurologic signs (Burke et al., 2008).