Chapter 21 Overview of Disorders of Brain Development
Introduction
 largely (but not exclusively) represent defects during the earliest stages of brain development, and thus reflect the underlying embryology and developmental genetics of the nervous system
largely (but not exclusively) represent defects during the earliest stages of brain development, and thus reflect the underlying embryology and developmental genetics of the nervous system


Thus, an understanding of brain malformations is important in assessing almost all types of neurological disorders in children. On a practical basis, we have separated brain malformations into disorders involving development of the neural tube (Chapter 22), forebrain (Chapter 23), and mid-hindbrain (Chapter 24); disorders of cortical development, separated into those of brain size (Chapter 25) and those of migration and later cortical development (Chapter 26); disorders predisposing to hydrocephalus (Chapter 27); and disorders of skull development (Chapter 28). We end with an overview of prenatal diagnosis for all brain malformations (Chapter 29). Here, we will address a few topics relevant to brain malformations generally, including epidemiology, classification, clinical recognition, relationship to other neurological disorders and selected environmental factors, and genetic counseling.
Epidemiology
The incidence of brain malformations has been estimated to be approximately 3.32 per 1000 and the prevalence approximately 2.21 per 1000 at age 14 years from studies of a 1-year birth cohort from northern Finland [Von Wendt and Rantakallio, 1986]. These are much higher rates than were recognized in the era before magnetic resonance imaging (MRI) and recent increases in surgical treatment of hydrocephalus and epilepsy [Kuzniecky et al., 1993; Massimi et al., 2009; Warkany et al., 1981]. Not surprisingly, the incidence is much higher (i.e., approximately 88 per 1000) in studies of children with cerebral palsy [Rankin et al., 2010]. This is an important point. To emphasize this, a boy was recently evaluated with apparent cerebral palsy attributed to prematurity at approximately 29 weeks’ gestation. But his brain MRI revealed mild callosal and cerebellar vermis hypoplasia, and chromosome microarray revealed a small deletion in 22q11.2, which implies either a genetic or a mixed pathogenesis.
Classification
While the chapters that follow review many different malformations, they are not complete, as the number of recognized malformations continues to grow steadily. Presenting these data is also complicated by the tendency for malformations to co-occur in some patients. For example, Figure 21-1 shows a striking example of a boy with malformations of the forebrain (agenesis of the corpus callosum), mid-hindbrain (severe cerebellar hypoplasia and mega-cisterna magna), brain size (megalencephaly), neuronal migration (periventricular nodular heterotopia), and cortical organization (polymicrogyria overlying the heterotopia).
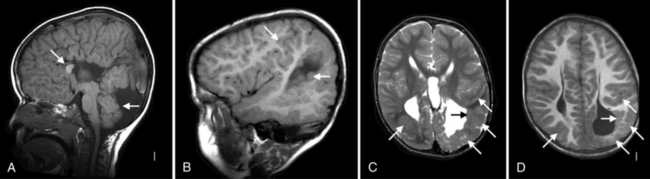
Fig. 21-1 Brain images from a single patient showing multiple malformations.
(Courtesy of WB Dobyns, research subject LR00-086.)
From time to time, flexible classification schemes have been constructed for many of these malformations that primarily rely on traditional concepts such as embryology and anatomy, with a contribution from genetic discoveries. While recent discoveries are leading us to think more of genes and gene pathways than of embryology and anatomy, a more traditional classification scheme is presented in this and the following chapters. The most current outline for brainstem and cerebellar (mid-hindbrain) malformations is shown in Box 21-1, and an outline for cortical malformations in Box 21-2. Further details regarding most subgroups of malformations and the basis for the classification are given in the primary references [Barkovich et al., 2005, 2009]. These schemes were constructed relying on – in decreasing order of priority – the underlying genetic basis when known, the relevant embryology, brain imaging features, and miscellaneous other clinical features.
Box 21-1 Classification Scheme for Mid-Hindbrain Malformations
I Malformations Secondary to Early Anteroposterior and Dorsoventral Patterning Defects, or to Mis-Specification of Mid-Hindbrain Germinal Zones
II Malformations Associated with Later Generalized Developmental Disorders that Significantly Affect the Brainstem and Cerebellum (and have Pathogenesis at Least Partly Understood)
III Localized Brain Malformations that Significantly Affect the Brainstem and Cerebellum (Pathogenesis Partly or Largely Understood; Includes Local Proliferation, Cell Specification, Migration, and Axonal Guidance)
Box 21-2 Classification Scheme for Malformations of Cortical Development
I Malformations Due to Abnormal Neuronal and Glial Proliferation or Apoptosis
Brain Imaging Recognition
The improved quality of brain imaging studies, especially advances in MRI technology, has led directly to increased recognition and more accurate classification of brain malformations. Still, several recurrent types of classification errors continue to occur, based on studies sent to the authors for review. First, pachygyria appears to be the best known of the severe cortical malformations, and accordingly all types of severe cortical malformations are often interpreted as “pachygyria.” The prime examples of malformations mistaken for pachygyria include severe congenital microcephaly (but here the cortex is thin rather than thick), polymicrogyria, and cobblestone malformations (for these, the cortex is moderately thick but the surface and cortex–white matter interface are irregular rather than smooth). This unfortunately often leads to testing of the “lissencephaly” genes in patients with these other cortical malformations, with uniformly negative results. Second, a thin corpus callosum may result from reduced volume of white matter due to abnormal development of white matter, progressive white-matter dysgenesis, or white-matter injury. However, this appearance is sometimes interpreted as agenesis of the corpus callosum. Next, diverse causes of cerebellar hypoplasia are often interpreted as Dandy–Walker malformation (when associated with a large posterior fossa and an enlarged fourth ventricle) or the so-called “Dandy–Walker variant” (equated with isolated cerebellar vermis hypoplasia). The latter is so overused and misapplied that the authors have abandoned the term. Finally, enlarged fluid collections below and especially behind the cerebellum are interpreted as arachnoid cysts or as “mega-cisterna magna,” considering the latter a nonpathogenic variant. In the authors’ experience, mega-cisterna magna with fluid both below and behind the cerebellum sometimes represents a developmental disorder that belongs in the Dandy–Walker spectrum [Aldinger et al., 2009], and may be incorrectly interpreted as an arachnoid cyst.
Relationships to Other Neurologic Disorders
The close connection between brain malformations and other classes of developmental disorders is conceptually important. A few examples include agenesis of the corpus callosum associated with nonketotic hyperglycinemia [Dobyns, 1989], cerebellar vermis hypoplasia or heterotopia with multiple acyl-CoA dehydrogenase deficiency, also known as glutaric aciduria type 2 [Bohm et al., 1982; Lehnert et al., 1982], cobblestone malformations and cerebellar hypoplasia with congenital disorders of glycosylation [Aronica et al., 2005; Morava et al., 2009; Van Maldergem et al., 2008], pachygyria variants with severe peroxisomal disorders such as Zellweger’s syndrome [Barkovich and Peck, 1997; Van Der Knaap and Valk, 1991], and cerebellar hypoplasia or agenesis of the corpus callosum with either autism or infantile spasms [Chugani and Conti, 1996; Courchesne et al., 1988; Kato et al., 2004; Manes et al., 1999; Schiffmann et al., 1993]. Further, most malformations of cortical development are associated with epilepsy, which may be severe [Guerrini, 2005; Leventer et al., 2008]. Observations in these and many other disorders collectively lead us to hypothesize that brain malformations represent the most severe expression or “tip of the iceberg” of a host of developmental brain disorders. Of course, this is much more than a hypothesis, as it has already been proven for disorders reviewed in chapters throughout this text.
Relationship to Environmental Factors
The genetic basis for many brain malformations has been known for years, and new genes are constantly being discovered. The question arises: are all brain malformations genetic? While easy to overlook, substantial data exist to support environmental (extrinsic) causes for several brain malformations. Both microcephaly and hydrocephalus can result from numerous prenatal and early-life diseases, such as intraventricular hemorrhage in premature infants, other causes of intracranial bleeding, hypoxic-ischemic injury, central nervous system infections, and a host of other disorders reviewed throughout this text. Holoprosencephaly has been associated with pregestational diabetes and with structural analogs of cholesterol that interfere with cholesterol metabolism or uptake in humans and animals [Edison and Muenke, 2004; Haas and Muenke, 2010; Johnson and Rasmussen, 2010; Lipinski et al., 2010]. Periventricular nodular heterotopia has been seen in mice and rats following prenatal exposure to high-dose ionizing radiation, and possibly in humans as well [Barth, 1987; Dekaban, 1968; Ferrer et al., 1993]. Schizencephaly and polymicrogyria have been associated with second trimester (13–21 weeks’ gestation) prenatal vascular disruption and with intrauterine cytomegalovirus infections [Barth and Van Der Harten, 1985; Curry et al., 2005; Marques Dias et al., 1984; Mcbride and Kemper, 1982; Pati and Helmbrecht, 1994; Sherer and Salafia, 1996; Suchet, 1994].
Genetic Counseling
While the genetic basis for more and more brain malformations and malformation syndromes is being uncovered, few studies examining the overall contribution of genetic disorders to brain malformations have been reported. Accordingly, only partial and selective information about the genetic recurrence risk for different brain malformations is available. While the authors have conducted only a few formal studies of recurrence risk, we collectively have several decades of experience with these disorders and can provide some general guidelines, which are listed in Table 21-1.
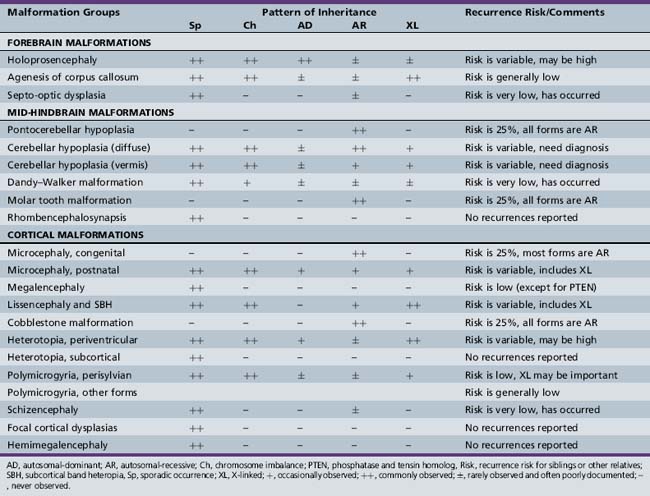
For example, the recurrence risk for holoprosencephaly reported in the literature is approximately 6 percent [Roach et al., 1975]. This report dates from more than 35 years ago, and we are now aware of frequent mild expression or “formes frustes” of this malformation, and accordingly we suggest using a higher recurrence risk of 13 percent for isolated holoprosencephaly without known chromosome imbalances [Mercier et al., 2010]. For several malformations such as agenesis of the corpus callosum and polymicrogyria, single-gene inheritance has been reported, but only rarely, and no formal studies are available. We therefore suggest a “generally low” risk, and counseling for some uncertainty, given that the experience is limited and exceptions occur. For some other malformations, such as rhombencephalosynapsis, hemimegalencephaly, and focal cortical dysplasia, no examples of familial recurrence have ever been reported, despite clinical recognition for decades. We therefore suggest that the recurrence risk is very low. The most difficult malformations are those with significantly different recurrence risks for different subtypes and syndromes, such as diffuse and vermis-predominant cerebellar hypoplasia. These estimates are largely anecdotal in origin, so treating physicians and genetic counselors are encouraged to review information on the specific disorder at hand when counseling families.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aldinger K.A., Lehmann O.J., Hudgins L., et al. Foxc1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 dandy-walker malformation. Nat Genet. 2009;41(9):1037-1042.
Aronica E., van Kempen A.A., van der Heide M., et al. Congenital disorder of glycosylation type Ia: A clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol. 2005;109(4):433-442.
Barkovich A.J., Kuzniecky R.I., Jackson G.D., et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65(12):1873-1887.
Barkovich A.J., Millen K.J., Dobyns W.B. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132(Pt 12):3199-3230.
Barkovich A.J., Peck W.W. MR of Zellweger syndrome. Am J Neuroradiol. 1997;18(6):1163-1170.
Barth P.G. Disorders of neuronal migration. Can J Neurol Sci. 1987;14(1):1-16.
Barth P.G., van der Harten J.J. Parabiotic twin syndrome with topical isocortical disruption and gastroschisis. Acta Neuropathol. 1985;67(3–4):345-349.
Bohm N., Uy J., Kiessling M., et al. Multiple acyl-CoA dehydrogenation deficiency (glutaric aciduria type II), congenital polycystic kidneys, and symmetric warty dysplasia of the cerebral cortex in two newborn brothers. II. Morphology and pathogenesis. Eur J Pediatr. 1982;139(1):60-65.
Chugani H.T., Conti J.R. Etiologic classification of infantile spasms in 140 cases: Role of positron emission tomography. J Child Neurol. 1996;11(1):44-48.
Courchesne E., Yeung-Courchesne R., Press G.A., et al. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N Engl J Med. 1988;318(21):1349-1354.
Curry C.J., Lammer E.J., Nelson V., et al. Schizencephaly: Heterogeneous etiologies in a population of 4 million California births. Am J Med Genet A. 2005;137(2):181-189.
Dekaban A.S. Abnormalities in children exposed to x-radiation during various stages of gestation: Tentative timetable of radiation injury to the human fetus. I. J Nucl Med. 1968;9(9):471-477.
Dobyns W.B. Agenesis of the corpus callosum and gyral malformations are frequent manifestations of nonketotic hyperglycinemia. Neurology. 1989;39(6):817-820.
Edison R.J., Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med. 2004;350(15):1579-1582.
Ferrer I., Santamaria J., Alcantara S., et al. Neuronal ectopic masses induced by prenatal irradiation in the rat. Virchows Arch A Pathol Anat Histopathol. 1993;422(1):1-6.
Guerrini R. Genetic malformations of the cerebral cortex and epilepsy. Epilepsia. 2005;46(Suppl 1):32-37.
Haas D., Muenke M. Abnormal sterol metabolism in holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):102-108.
Johnson C.Y., Rasmussen S.A. Non-genetic risk factors for holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):73-85.
Kato M., Das S., Petras K., et al. Mutations of arx are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23(2):147-159.
Kuzniecky R., Murro A., King D., et al. Magnetic resonance imaging in childhood intractable partial epilepsy: Pathologic correlations. Neurology. 1993;43:681-687.
Lehnert W., Wendel U., Lindenmaier S., et al. Multiple acyl-CoA dehydrogenation deficiency (glutaric aciduria type II), congenital polycystic kidneys, and symmetric warty dysplasia of the cerebral cortex in two brothers. I. Clinical, metabolical, and biochemical findings. Eur J Pediatr. 1982;139(1):56-59.
Leventer R.J., Guerrini R., Dobyns W.B. Malformations of cortical development and epilepsy. Dialogues Clin Neurosci. 2008;10(1):47-62.
Lipinski R.J., Godin E.A., O’Leary-Moore S.K., et al. Genesis of teratogen-induced holoprosencephaly in mice. Am J Med Genet C Semin Med Genet. 2010;154C(1):29-42.
Manes F., Piven J., Vrancic D., et al. An MRI study of the corpus callosum and cerebellum in mentally retarded autistic individuals. J Neuropsychiatry Clin Neurosci. 1999;11(4):470-474.
Marques Dias M.J., Harmant-van Rijckevorsel G., Landrieu P., et al. Prenatal cytomegalovirus disease and cerebral microgyria: Evidence for perfusion failure, not disturbance of histogenesis, as the major cause of fetal cytomegalovirus encephalopathy. Neuropediatrics. 1984;15(1):18-24.
Massimi L., Paternoster G., Fasano T., et al. On the changing epidemiology of hydrocephalus. Childs Nerv Syst. 2009;25(7):795-800.
McBride M.C., Kemper T.L. Pathogenesis of four-layered microgyric cortex in man. Acta Neuropathol. 1982;57(2–3):93-98.
Mercier S., Dubourg C., Belleguic M., et al. Genetic counseling and “Molecular” Prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med Genet. 2010;154C(1):191-196.
Morava E., Wevers R.A., Willemsen M.A., et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, debre type. Neurology. 2009;73(14):1164-1165.
Pati S., Helmbrecht G.D. Congenital schizencephaly associated with in utero warfarin exposure. Reprod Toxicol. 1994;8(2):115-120.
Rankin J., Cans C., Garne E., et al. Congenital anomalies in children with cerebral palsy: A population-based record linkage study. Dev Med Child Neurol. 2010;52(4):345-351.
Roach E., Demyer W., Conneally P.M., et al. Holoprosencephaly: Birth data, benetic and demographic analyses of 30 families. Birth Defects Orig Artic Ser. 1975;11(2):294-313.
Schiffmann R., Mannheim G.B., Stafstrom C.E., et al. Posterior fossa abnormalities in children with infantile spasms. J Child Neurol. 1993;8(4):360-365.
Sherer D.M., Salafia C.M. Midtrimester genetic amniocentesis of a twin gestation complicated by immediate severe fetal bradycardia with subsequent associated fetal anomalies. Am J Perinatol. 1996;13(6):347-350.
Suchet I.B. Schizencephaly: Antenatal and postnatal assessment with colour-flow doppler imaging. Can Assoc Radiol J. 1994;45(3):193-200.
van der Knaap M.S., Valk J. The MR spectrum of peroxisomal disorders. Neuroradiology. 1991;33(1):30-37.
Van Maldergem L., Yuksel-Apak M., Kayserili H., et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, debre type. Neurology. 2008;71(20):1602-1608.
von Wendt L., Rantakallio P. Congenital malformations of the central nervous system in a 1-year birth cohort followed to the age of 14 years. Childs Nerv Syst. 1986;2(2):80-82.
Warkany J., Lemire R.J., Cohen M.M.Jr. Mental retardation and congenital malformations of the central nervous system. Chicago: Year Book Medical Publishers; 1981.
