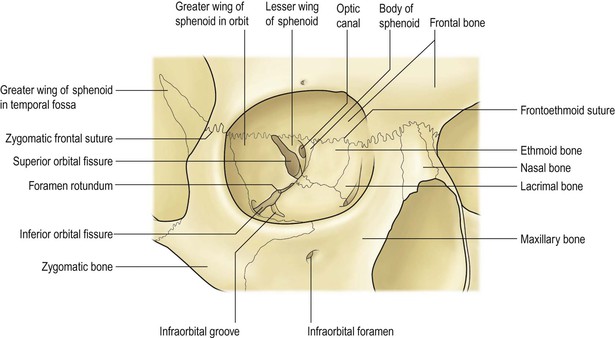
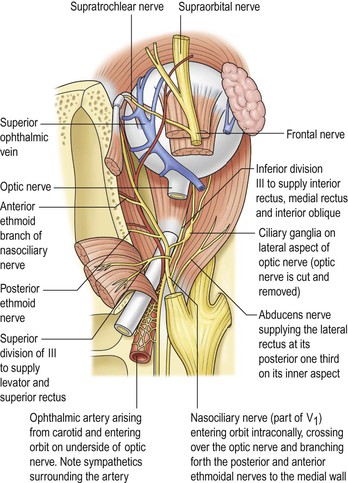
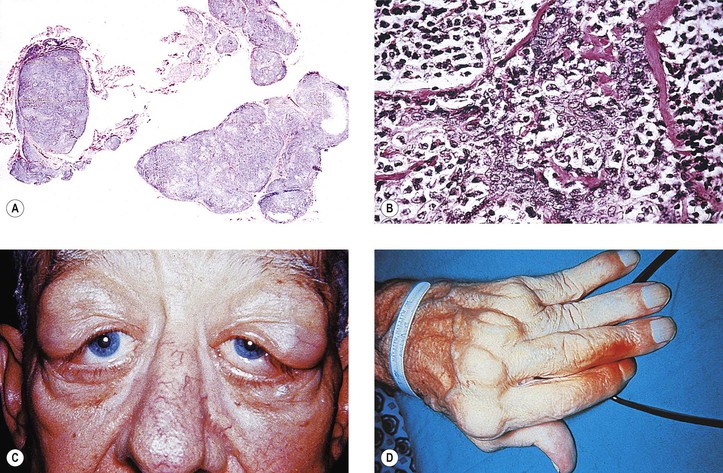
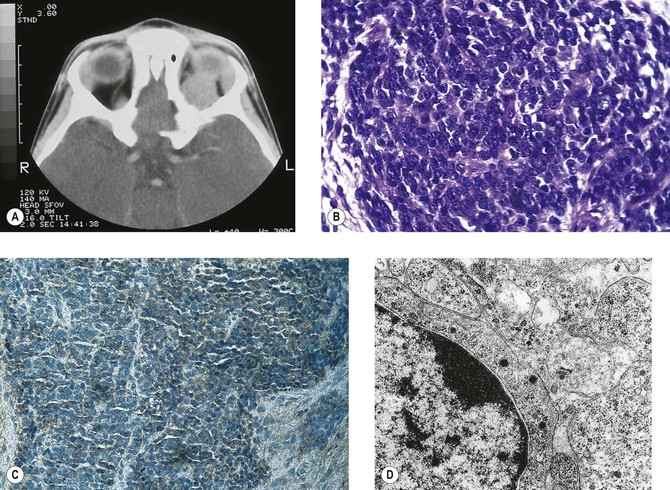
I. The main clinical manifestation of orbital disease is exophthalmos (ocular proptosis), the extent and direction of which depend on a number of factors1: (1) size of lesion, (2) character of lesion (expansile vs. infiltrative growth, rapid vs. slow growth), (3) location of the lesion in the orbit (small lesion in muscle cone causes more exophthalmos than lesion of same size outside the muscle cone; lesions anterior to septum orbitale do not produce exophthalmos unless they also grow posteriorly), and (4) lesion’s effect on the extraocular muscles (complete paralysis of all muscles by itself can cause 2 mm of exophthalmos).
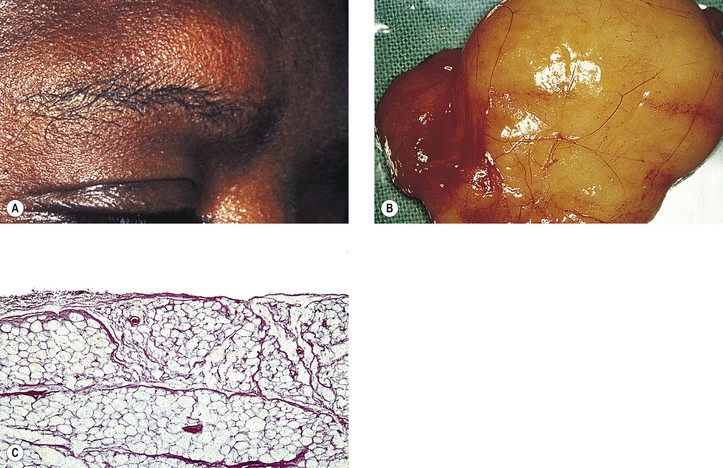
Although dermoids are one of the most common orbital tumors, if not the most common, they rarely cause exophthalmos because of their position, which is usually anterior to the septum orbitale.
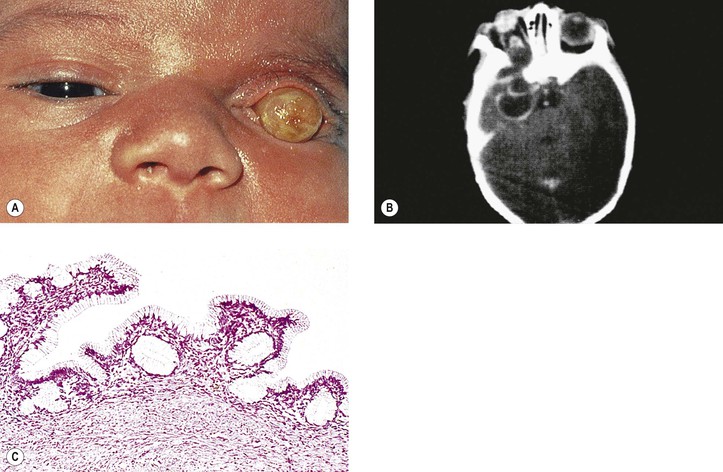
Although microphthalmos with cyst usually has no known cause, it may be associated with the 13q deletion or chromosome 18 deletion defect (partial 18 monosomy). A congenital cystic eye may also be associated with contralateral persistent hyperplastic vitreous and cerebrocutaneous abnormalities, called cranial ectodermopathy.
Autoantibodies to the ribonucleoprotein (RNP) particles SS-A (also called Ro RNA particle) and SS-B (also called La snRNA) are produced systemically. The immune response to 120-kDa α-fodrin may be important in the initial development of Sjögren’s syndrome.
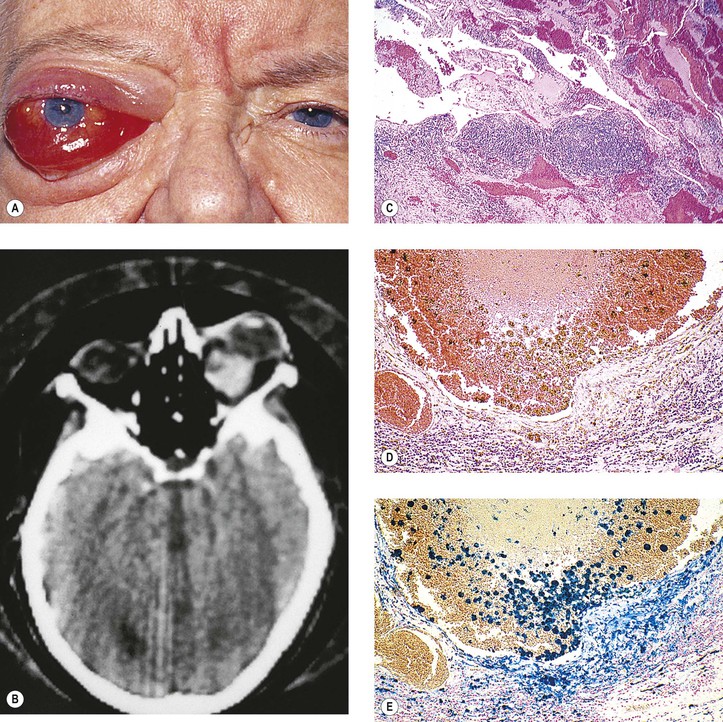
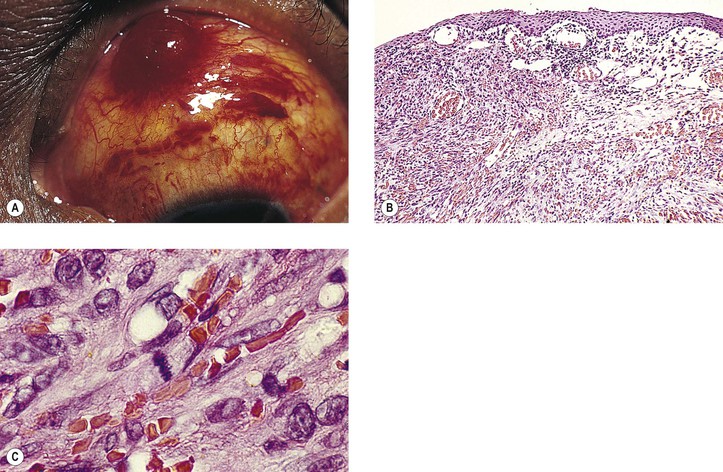
Some authors incorrectly use the term cholesteatoma interchangeably with epidermoid cyst. The term epidermoid cyst should not be used, or should it be restricted to postinflammatory tumors that contain squamous epithelium and keratin debris; cholesterol granuloma is never associated with any epithelial elements.
The effects of nonpenetrating wounds are those secondary to contusion and concussion, mainly hemorrhage, secondary muscle palsies, and infraorbital nerve involvement.
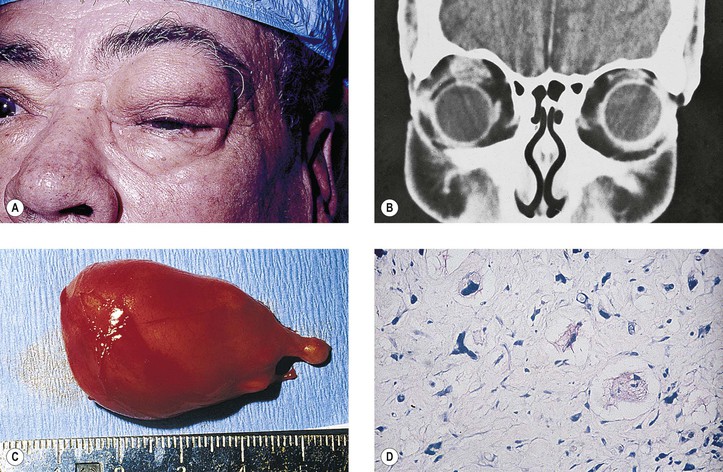
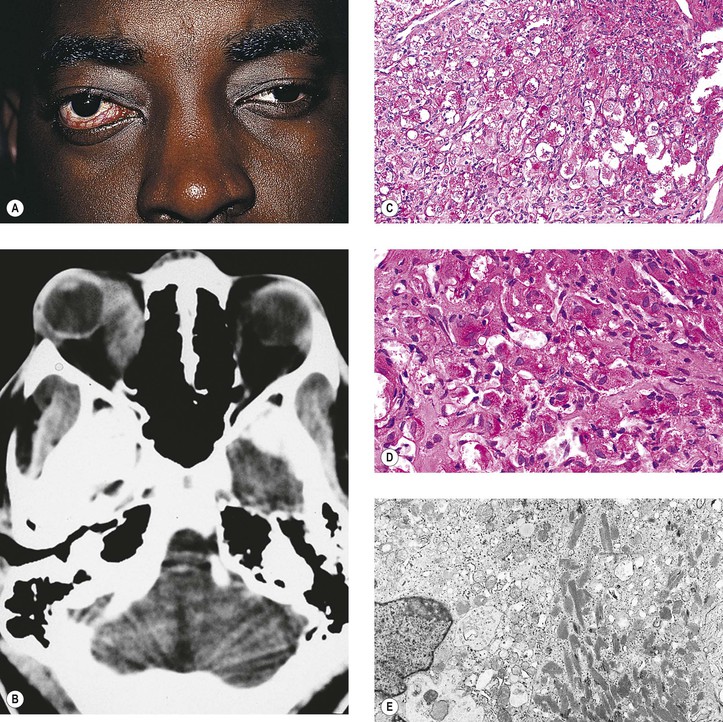
Orbital varix, also called distensible venous malformation (Fig. 14.9), may occur anterior to the septum orbitale and not cause exophthalmos, or it may occur posterior to the septum orbitale, causing exophthalmos. The exophthalmos may be acute if the varix undergoes thrombosis.
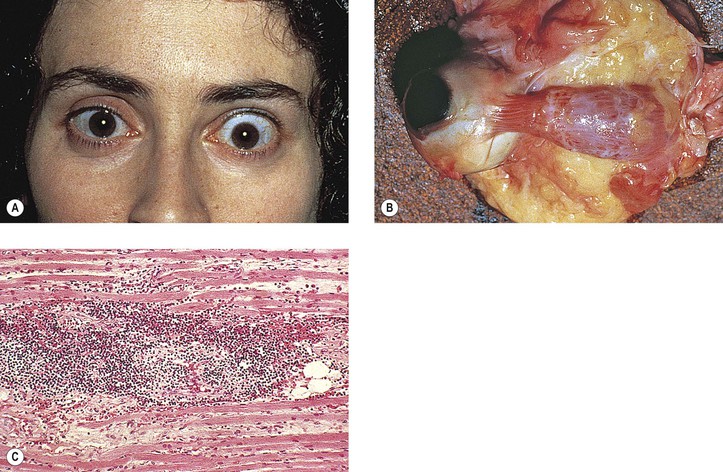
Smokers have an increased risk for development of both Graves’ disease and thyroid ophthalmopathy. Temporally, the diagnosis of Graves’ ophthalmopathy tends to follow the diagnosis of hyperthyroidism. Treatment of hyperthyroidism with iodine-131 does not seem to alter the course of Graves’ ophthalmopathy.
The term euthyroid Graves’ disease describes ocular manifestations of Graves’ disease in patients who are “euthyroid” and have no past history suggesting hyperthyroidism. The eye signs are frequently asymmetric. The patients may have a family history of thyroid disease or pernicious anemia. All of the euthyroid patients, however, do show some mild thyroid abnormality (e.g., thyroid autoantibodies, negative thyrotropin-releasing hormone test, negative triiodothyronine suppression test, and goiter).
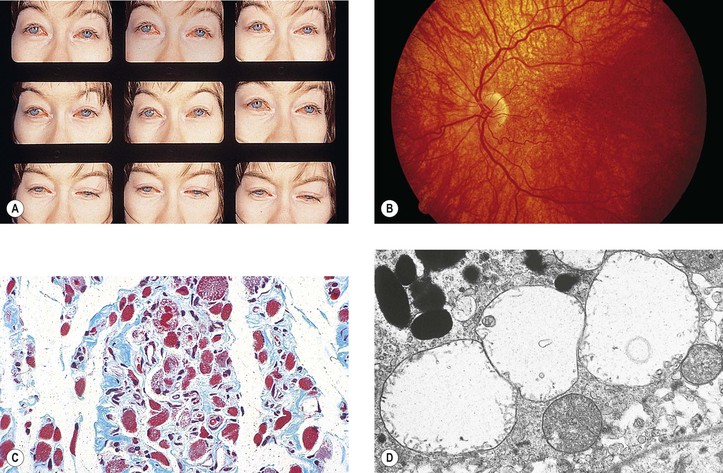
As a rough test, the ptosis can often be aggravated by having the patient raise and lower the eyes 10–30 times in rapid succession. The normal patient can do this easily with no ptosis afterward.
Myotonic dystrophy, like Huntington’s disease, is caused by an expansion of a repeated sequence of three nucleotides. In myotonic dystrophy, the expansion occurs in the 3′ untranslated region of the DM gene. Perhaps disrupted activity of a CUG-binding protein induced by repeats in RNA prevents the protein from doing its normal job of splicing a certain family of genes.
The inheritance of these point mutations of mitochondrial DNA is from mothers alone because the mitochondrial contribution to the embryo comes only from the maternal ovum.
Mitochondria of selective muscle fibers appear abnormal in size, shape, number, and internal structure, whereas others seem normal, resembling the changes found in Leber’s hereditary optic atrophy. Because the modified trichrome stain colors the abnormal muscle fibers red, the fibers have been called ragged-red fibers.
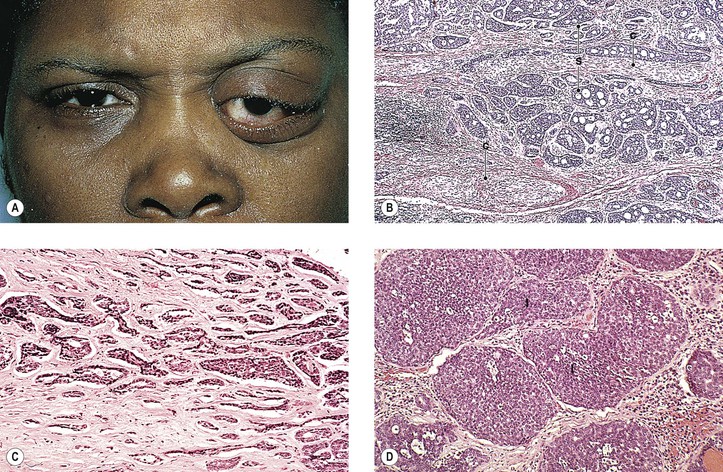
Erythematous, edematous patches of skin are frequently seen. A predilection for the face and periorbital region exists, where a violaceous heliotrope may occur around the eyes, on the lids, and on the cheeks.
Rarely, an epidermoid cyst may originate in the diploic space of the orbital bone, called an intradiploicepidermoid cyst. Also rarely, squamous cell carcinoma may develop in an epidermoid or dermoid cyst.
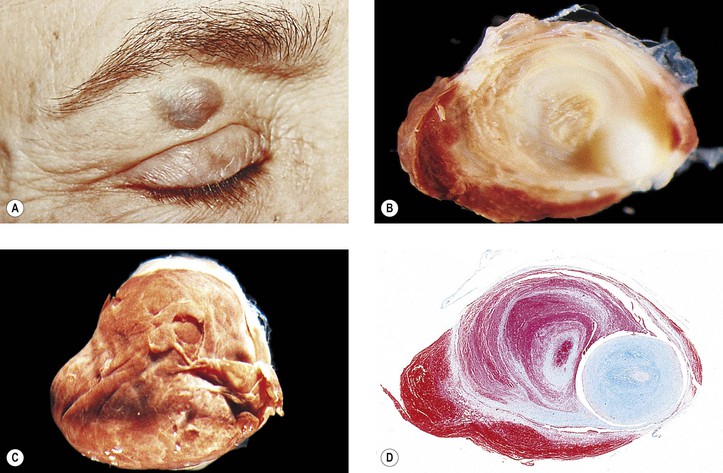
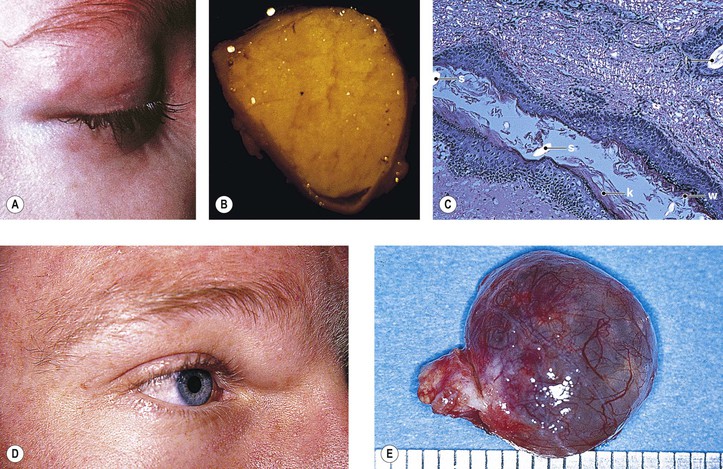
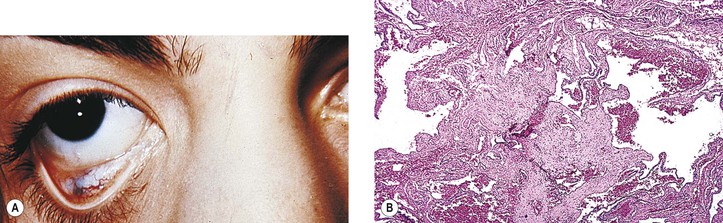
Primary nonkeratinized epidermoid cysts probably represent developmental sequestrations of forniceal or caruncular conjunctival epithelium. They are found in the superonasal quadrant (see Figs. 14.12D and 14.12E) and are not associated with an osseous defect. They constitute approximately 75% of the superonasal dermoids, the other 25% being the typical dermoids lined by keratinizing squamous epithelium.
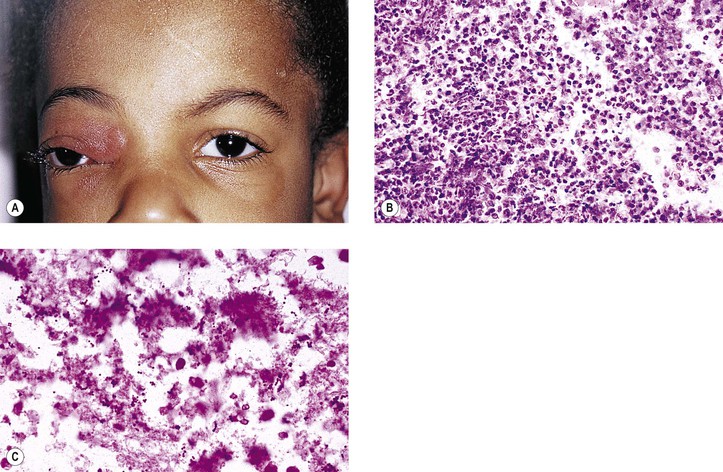
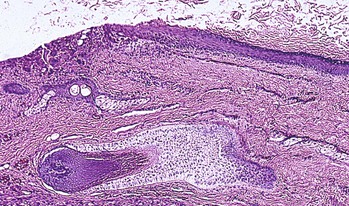
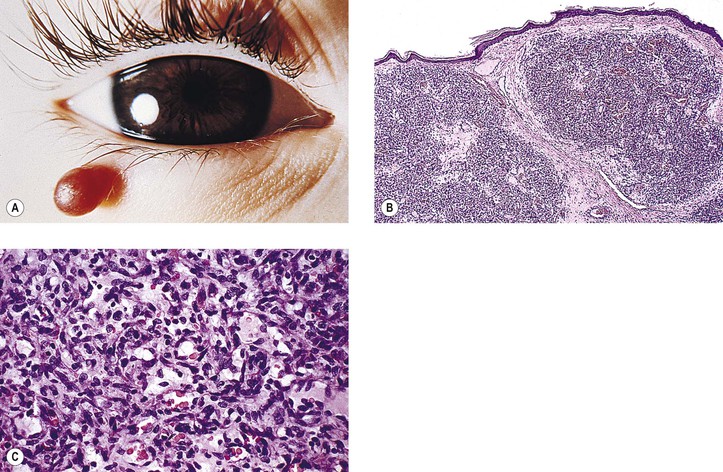
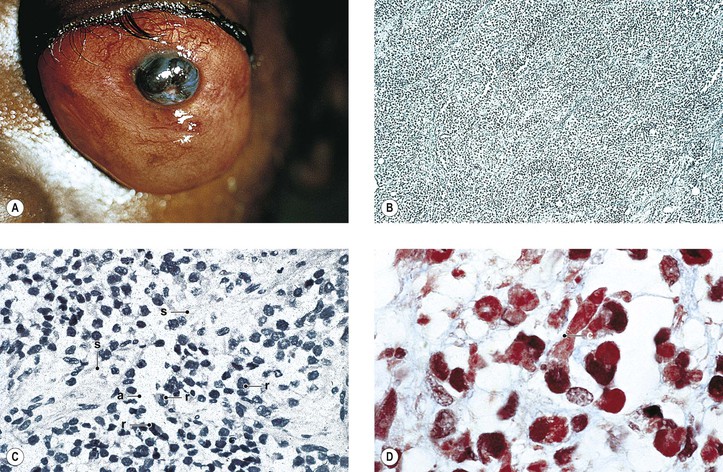
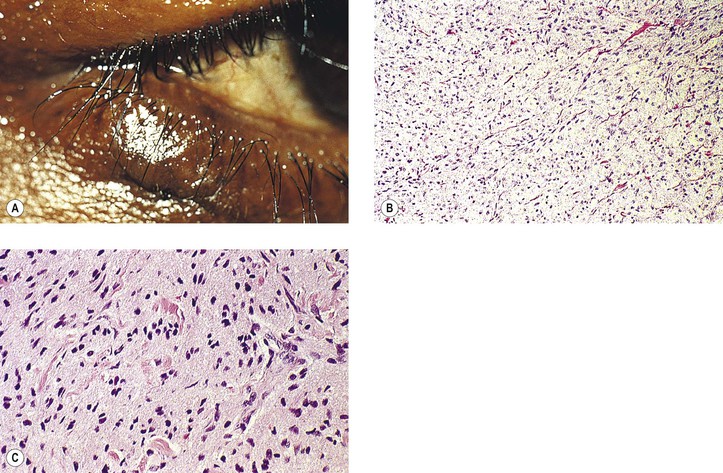
Rupture of a dermoid cyst can cause a chronic granulomatous inflammatory reaction (Fig. 14.13). Rarely, squamous cell carcinoma may develop in an epidermoid or dermoid cyst.
A rare teratoma has been reported in an intraocular location. A teratoma can also have both an orbital and an extraorbital (limited intracranial extension) or periorbital (beneath the skin and scalp) location.
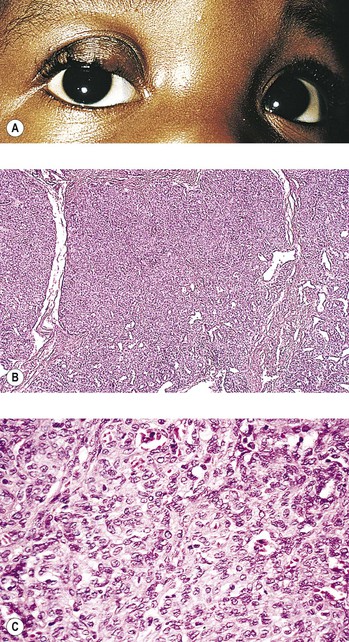
The tumor has also been called benign hemangioendothelioma and strawberry, infantile, and juvenile hemangioma. The lesion, although benign, may arise in a number of areas simultaneously and thereby simulate invasion and malignancy. The tumor almost always regresses spontaneously. Histologically, it consists mainly of plump endothelial cells, some of which form a capillary lumen.
Rarely, the tumor may arise in the orbital bones. Even more rarely, the tumor may be associated with the blue rubber bleb nevus syndrome (multiple cutaneous and visceral bluish-red, rubbery hemangiomas; may be autosomal dominant but most cases are sporadic). If bilateral orbital hemangiomas are present, they may be part of the blue rubber bleb nevus syndrome or Maffuci’s syndrome (nonhereditary disease characterized by hemangiomas and enchondromas).
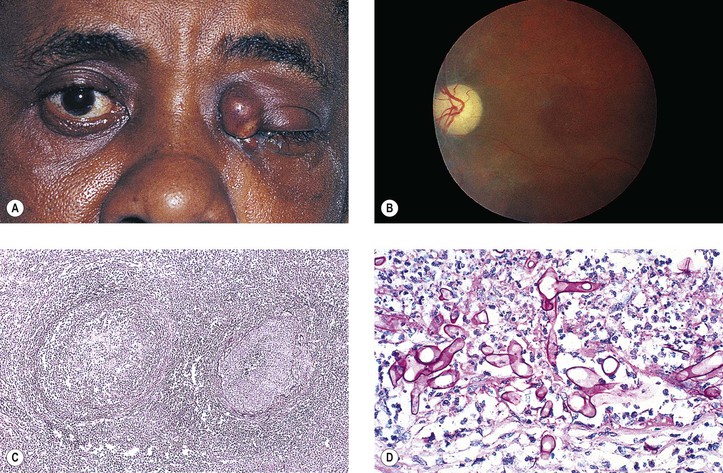
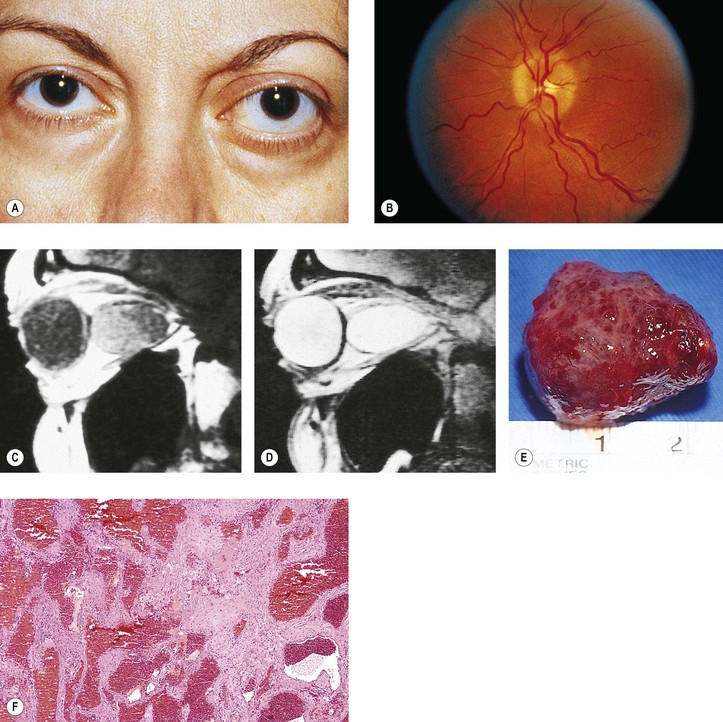
Rarely, a cavernous hemangioma may bleed and give rise to a hematic cyst. The cyst contains birefringent crystals and altered blood that seem to initiate a granulomatous inflammation (similar to a cholesterol granuloma; see previously in this chapter). Other causes of hematic cyst include blunt trauma, spontaneous orbital hemorrhage, blood dyscrasias, vascular disease, and lymphangioma.
Often, lymphangiomas contain both venous and lymphatic components; hence, another term for the lesions is combined venous lymphatic malformations. The lesions may be associated with noncontiguous intracranial vascular anomalies.
“Angioblastic type” of meningioma, once thought to be of meningeal origin, is now generally accepted to be a hemangiopericytoma of the central nervous system.
The tumor is more likely to be malignant if the following occur: increased mitotic activity (>4 mitotic figures per 400 field), necrotic foci, pleomorphism, S-phase greater than 9, and a proliferative index greater than 11 (the last two determined by cell cycle analysis). Hemangiopericytoma resembles the vascular form of fibrous histiocytoma (FH; see later in this chapter).
Intravascular papillary endothelial hyperplasia, a benign lesion, has been confused with hemangiosarcoma. Another benign lesion, probably a reactive or immunologic inflammatory process, angiolymphoid hyperplasia with eosinophilia (Kimura’s disease; see later in this chapter), has also been mistaken for hemangiosarcoma.
Approximately 20% of patients who have AIDS also have KS (second only to Pneumocystis carinii infection as a presenting manifestation). In addition to AIDS, an association exists between KS and other cancers such as malignant lymphoma (especially Hodgkin’s disease), leukemia, or a primary carcinoma with a separate histogenesis.
Herpesvirus-like DNA sequences (KSHV) have been found in classic, endemic, and AIDS-associated KS. KSHV is also associated with lesions other than KS in non-AIDS-immunosuppressed patients, and it may be involved in the pathogenesis of the various forms of proliferative skin lesions in organ transplant recipients. The human herpesvirus 8 (HHV-8) is the infectious agent responsible for KS in patients with or without human immunodeficiency virus infection. The latent nuclear antigen-1 (LNA-1) of HHV-8 is a nuclear antigen expressed in all cells latently infected by the virus.
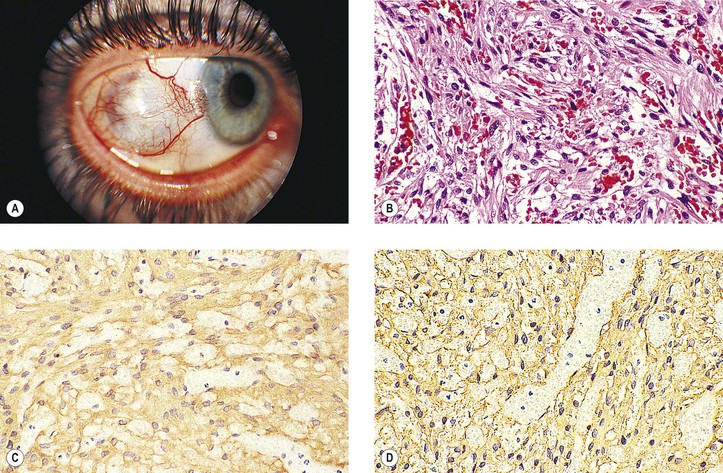
Commonly, the tumor is admixed with lymphocytes, hence the term malignant granulation tissue. The endothelial cells lining well-formed tumor blood vessels give a strong reaction with immunohistochemical staining for FVIII-RAG when the peroxidase–antiperoxidase technique is used. The proliferating spindle cells that form the capillary clusters (“vascular slits”), however, give a negative reaction.
Variants of benign lipoma include angiolipoma, angiomyolipoma, spindle cell lipoma, pleomorphic lipoma, benign lipoblastoma, and hibernoma (multivacuolar brown fat cells).
The group of tumors includes keloids, desmoids, fibromatoses of palmar and plantar fascias and of sternomastoid muscle, radiation fibromatosis, and congenital progressive polyfibromatosis.
FH has been misdiagnosed as hemangiopericytoma, sclerosing hemangioma, dermatofibrosarcoma protuberans (DFSP), and even neurofibroma. When associated with multinucleated giant cells and lipid-filled histiocytes, it has been misdiagnosed as synovial giant cell tumor and villonodular synovitis.
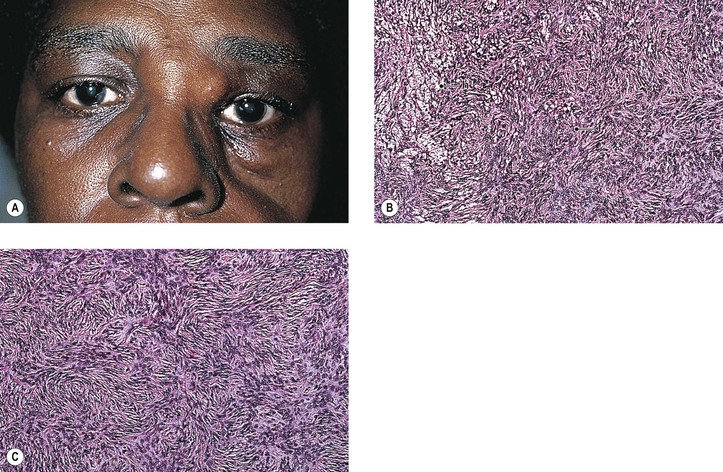
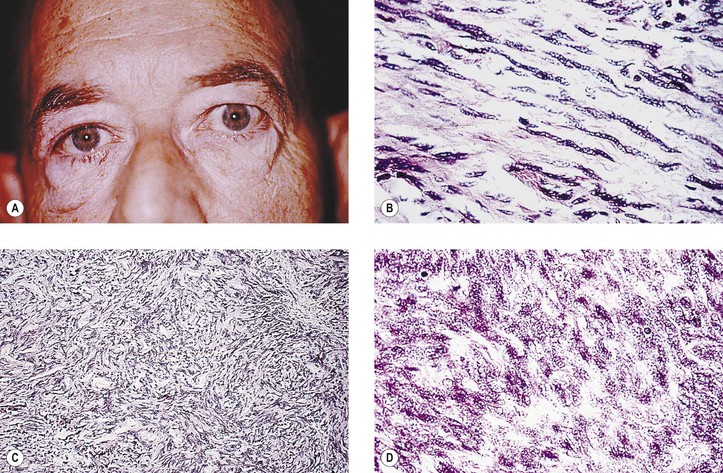
Malignant FH (MFH) is considered to be a sarcoma having an undifferentiated mesenchymal cell origin that differentiates along a broad fibroblastic and histiocytic (fibrohistiocytic) spectrum and usually has a predominant “fibroblastic” component. It may be difficult to differentiate from other pleomorphic soft-tissue tumors, such as pleomorphic liposarcoma, pleomorphic rhabdomyosarcoma, malignant schwannoma, leiomyosarcoma, and epithelioid sarcoma—a very rare orbital malignancy presumably of tendon sheath origin, having both epithelial and mesenchymal features. KP-1 (CD68), a recently described monoclonal antibody to a cytoplasmic epitope present on tissue histiocytes and macrophages, may have specific marker properties to help identify MFH. Vimentin also is usually positive. Finally, molecular assays for specific gene fusion provide a genetic approach to the differential diagnosis of soft-tissue sarcomas.
Although AFX and DFSP can be confused with spindle cell, squamous cell carcinoma and desmoplastic malignant melanoma, the absence of cytokeratin, HMB-45, and HMB-50 staining easily distinguishes AFX and DFSP from the others.
Solitary fibrous tumor, which resembles giant cell angiofibroma, lacks multinucleated giant cells and pseudovascular spaces. Stains for KP1 (CD68) are negative, unlike in FH, in which they are positive.
2. Mesectodermal leiomyoma‡ (see Chapter 9) and leiomyosarcoma‡ are very rare orbital tumors whose origin seems to be from tissue derived from neural crest.
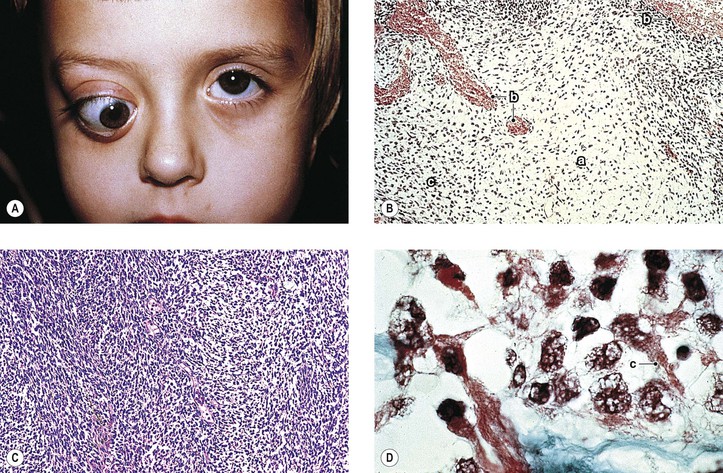
An undifferentiated tumor that resembles rhabdomyosarcoma, but without demonstrable rhabdomyoblasts, should be classified as embryonal sarcoma. Many of the metastases, however, show rhabdomyoblasts with cross-striations; then the classification embryonal rhabdomyosarcoma (or alveolar rhabdomyosarcoma, according to pattern) is appropriate.
Usually, only undifferentiated embryonal cells with large hyperchromic nuclei and a scant amount of cytoplasm are present. In some areas, however, cells with a ribbon of pink cytoplasm can be seen. Cross-striations are most likely to be found in these latter areas.
Insulin-like growth factor-2, which acts as an autocrine growth and motility factor, may be operating in rhabdomyosarcomas. The expression of the myogenic determination gene MyoD, a member of the helix–loop–helix family of transcription factors, is the most sensitive marker for rhabdomyosarcoma. Rhabdomyosarcomas seem to be deficient in a factor required for MyoD activity.
11. Osteoma,‡ osteoblastoma,‡ and osteogenic sarcoma (osteosarcoma)‡
Osteomas may occur in Gardner’s syndrome, an autosomal-dominant disorder characterized by intestinal polyposis, various skin and soft-tissue tumors, retinal pigment epithelial hypertrophy, and osteomas. Osteogenic sarcoma (osteosarcoma)‡ may rarely be primary, may be associated with Paget’s disease of bone, or may follow radiation for retinoblastomas.
Immunohistochemistry may be quite helpful in differentiating the very rare melanotic neurilemmoma from a malignant melanoma, especially if the former arises in the choroid. A rare histologic variant of neurilemoma, called ancient schwannoma, shows distinctive areas of hypercellularity and hyperchromic nuclei suggesting fibrosarcoma, as well as hypocellular areas containing considerable fibrosis; the clinical course, however, tends to be benign.
Mutations in the p53 tumor suppressor gene, located on the short arm of chromosome 17 at position 17p13.1, represent the most frequent genetic alteration detected in human solid malignancies. In approximately half of all cancer cases, p53 is inactivated by mutations and other genomic alterations; in many of the remaining cases, p53 is functionally inactivated by the binding of the cellular MDM2 oncoprotein, a cellular inhibitor of the p53 tumor suppressor. The p53 gene encodes a 53-kDa nucleophosphoprotein that binds DNA and negatively regulates cell division, preventing progression from G1 to S phase. Approximately 25% of adult sarcomas of different types are associated with p53 abnormalities. It also appears to be a marker of tumor progression (i.e., a direct correlation seems to exist between mutations at the p53 locus and increasing histologic grade). This correlation may be especially applicable to malignant peripheral nerve sheath tumors.
Ewing’s sarcoma also has the same genetic abnormality and may represent the opposite end of the same spectrum. However, despite their genetic and antigenic similarity, most authors recognize PNET and extraosseous Ewing’s sarcoma as separate entities, a distinction based primarily on the more neural differentiation of PNET and its graver prognosis.
Ewing’s sarcoma stains positively for periodic acid–Schiff (PAS) stain, vimentin, and especially terminal deoxynucleotidyl transferase and MIC-2 (CD99), a cell surface glycoprotein encoded by genes on chromosomes X and Y. The histologic differentiation includes other small cell tumors such as lymphomas, rhabdomyosarcoma, neuroblastoma, PNET, nephroblastoma, small cell variant of osteosarcoma, and carcinomas with various degrees of neuroendocrine differentiation.
The histogenesis of the tumor is uncertain, and skeletal muscle, fibroblasts, undifferentiated mesenchymal cells, histiocytes, and neural or Schwann cells have all been proposed as cells of origin. Most of the evidence suggests that the Schwann cell is the cell of origin. Rarely, the tumor can occur in the epibulbar region or in the ciliary body.
Another entity, malignant mesenchymoma, a very rare orbital tumor, was thought to be a subtype of alveolar soft-part sarcoma, but it probably represents a separate entity, although this is controversial. The tumor, which usually affects patients older than 60 years, is composed of two or three distinct malignant components (e.g., rhabdomyosarcoma, chondrosarcoma, and osteogenic sarcoma).
The cytoplasmic crystalloids are similar to the rods observed in benign rhabdomyoma cells. The tumor, therefore, has been thought by some to be a unique type of rhabdomyosarcoma instead of being of neural derivation.
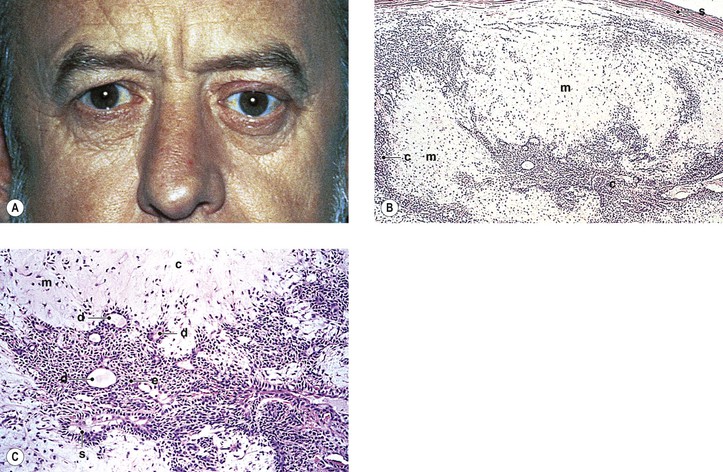
The lymphoid tumors and pseudotumors are identical to those occurring elsewhere in the orbit (see later in this chapter). Küttner tumor consists of a unilateral sclerosing, follicular, lymphoid hyperplasia of the submandibular gland sometimes associated with idiopathic inflammatory masses of the lacrimal gland.
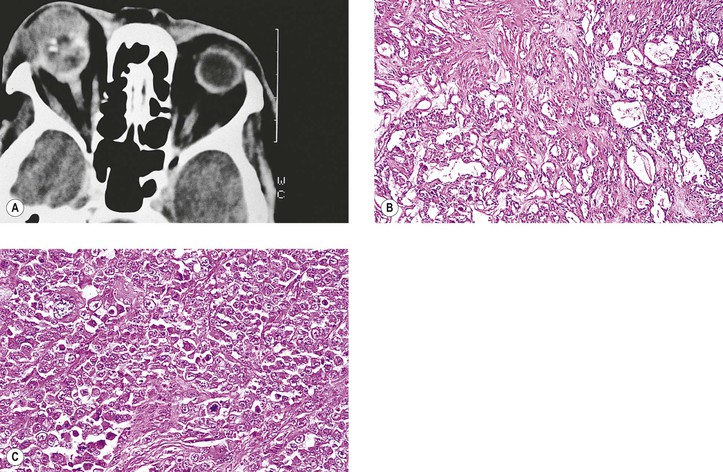
The juxtaposition of highly cellular epithelial areas with the relatively acellular myxomatous areas gives the tumor its characteristic diphasic pattern. The stroma is rich in a hyaluronidase-resistant acid mucopolysaccharide. A pleomorohic adenoma has occurred primarily in the choroid.
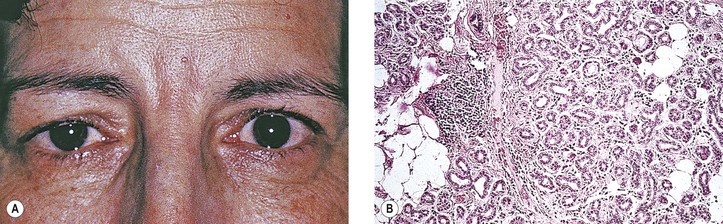
Rarely, the tumor occurs in young people (6.5–18 years of age) and seems to have a more favorable prognosis in this young group. Also, the tumor can occur in the nasal orbit, presumably from ectopic lacrimal gland.
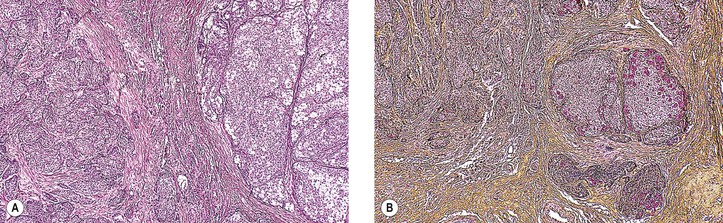
Patients who have a basaloid pattern in their tumor have a five-year survival rate of 21%, compared with a 71% survival rate when no basaloid pattern is present. “Bad” prognostic signs include a basaloid (solid) pattern, the presence of tumor at resection margins, and the presence of abnormal S-phase (proliferative) fraction. A basaloid pattern in an adenoid cystic carcinoma must be differentiated from the entity basal cell adenocarcinoma (see later), which has a lower degree of malignancy and a more favorable prognosis than adenoid cystic carcinoma.
3. Mucoepidermoid carcinoma (Fig. 14.38) is the most common primary carcinoma of the major salivary glands, but it is rare in the lacrimal gland. The tumor contains both epidermoid (squamous) and mucin-producing cells.