[level-membership-for-pathology-category]
Orbit
Normal Anatomy
I. The orbital volume is approximately 30 ml, and the orbital depth (anterior to posterior) is approximately 4.5 cm (Figs. 14.1–14.5).
A. The medial orbital wall is quite thin (<0.5 mm) and transparent.
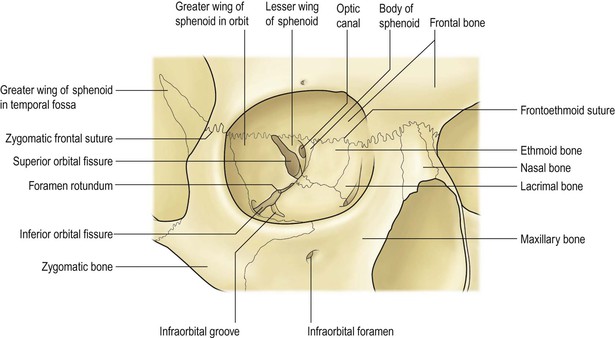
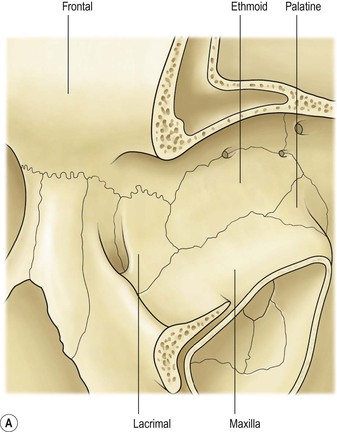
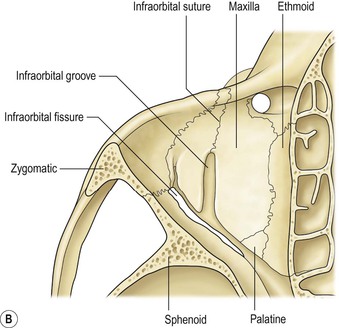
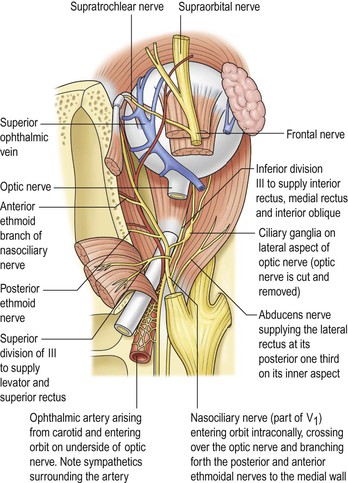
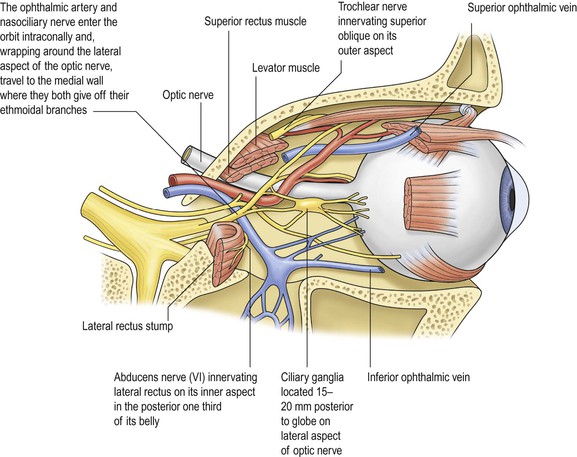
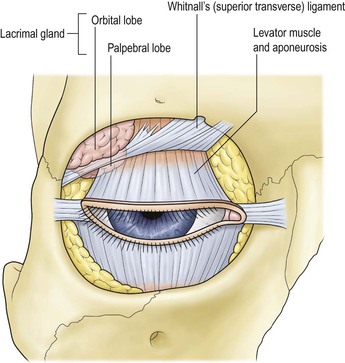
II. In addition to the bony walls, the eye, and the optic nerve, the orbit contains many soft-tissue structures such as fat, muscle (striated and nonstriated), cartilage, bone, fibrous tissue, nerves, and blood vessels.
B. All orbital structures may be involved in disease processes.
Exophthalmos
I. The main clinical manifestation of orbital disease is exophthalmos (ocular proptosis), the extent and direction of which depend on a number of factors1: (1) size of lesion, (2) character of lesion (expansile vs. infiltrative growth, rapid vs. slow growth), (3) location of the lesion in the orbit (small lesion in muscle cone causes more exophthalmos than lesion of same size outside the muscle cone; lesions anterior to septum orbitale do not produce exophthalmos unless they also grow posteriorly), and (4) lesion’s effect on the extraocular muscles (complete paralysis of all muscles by itself can cause 2 mm of exophthalmos).
Developmental Abnormalities
Developmental Abnormalities of Bony Orbit
Developmental abnormalities are usually associated with abnormalities of the cranial and facial bones such as tower skull or hypertelorism.
Microphthalmos with Cyst
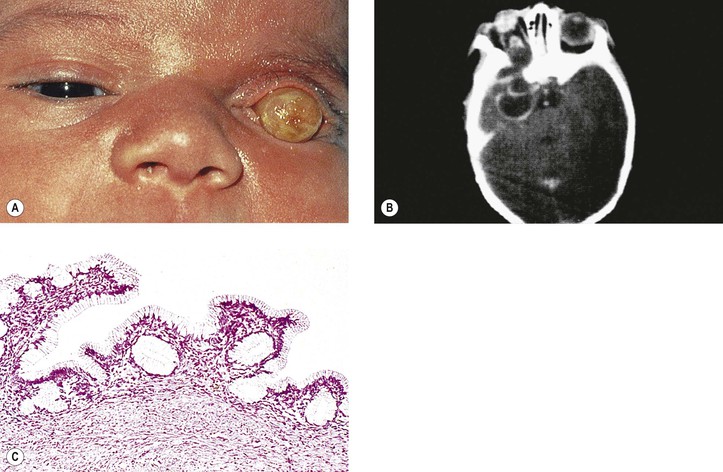
I. Microphthalmos with cyst (see Fig. 2.10) is usually a unilateral condition, but it may be bilateral.
II. The cyst may be so large as to obscure the microphthalmic eye.
IV. The condition is caused by incomplete closure of the fetal cleft.
Orbital Inflammation
Acute
I. Nonsuppurative (see section Nonsuppurative, Chronic Nongranulomatous Uveitis and Endophthalmitis in Chapter 3)—orbital cellulitis is most commonly caused by extension of an inflammation from the paranasal sinuses (Fig. 14.6).
II. Suppurative (see section Suppurative Endophthalmitis and Panophthalmitis in Chapter 3)
A. Purulent infection (e.g., with Staphylococcus) occurs commonly after trauma.
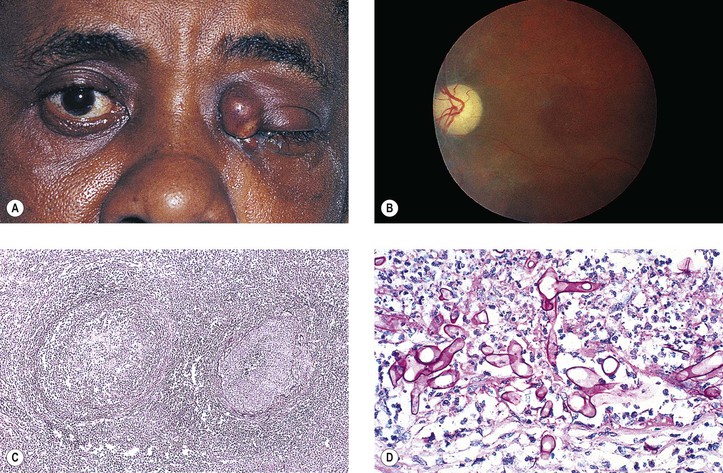
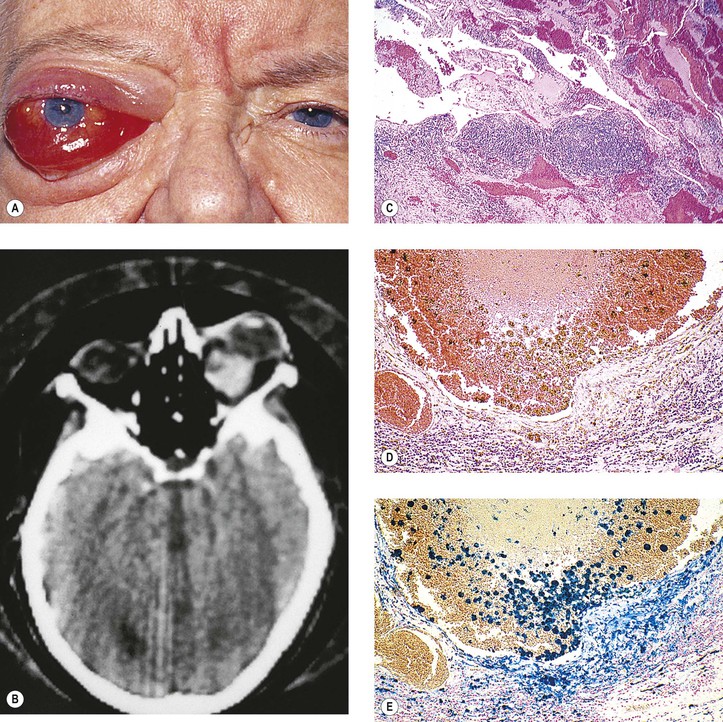
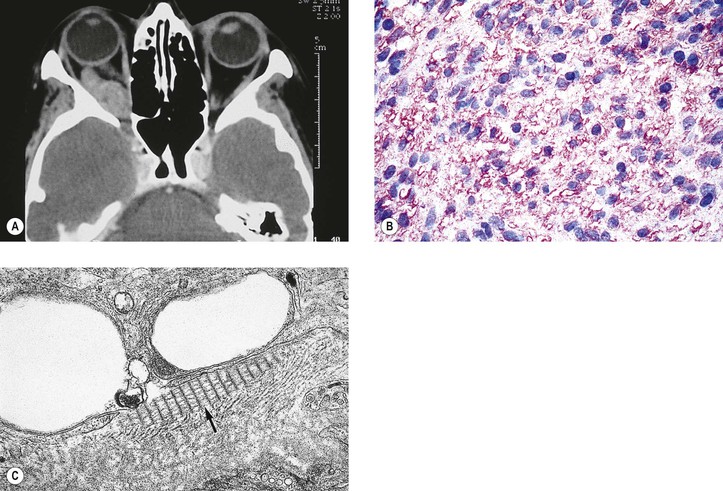
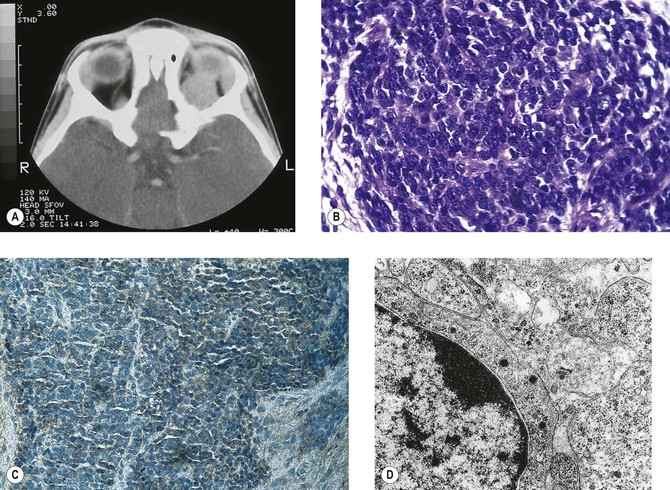
B. Phycomycosis (mucormycosis) is a devastating cause of suppurative orbital inflammation (Fig. 14.7; see Chapter 4).
Chronic
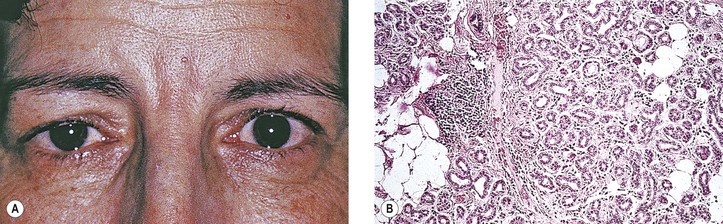
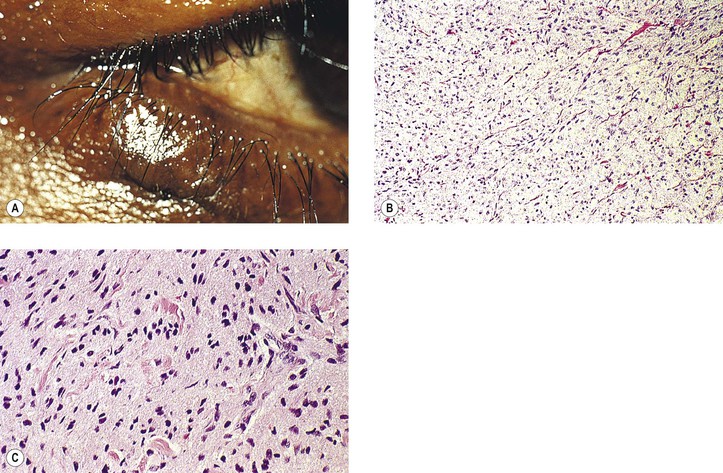
B. A rare cause is the benign lymphoepithelial lesion of Godwin2 (Fig. 14.8).
2. It may be part of Sjögren’s syndrome (see later).
3. Rarely, it may become malignant.
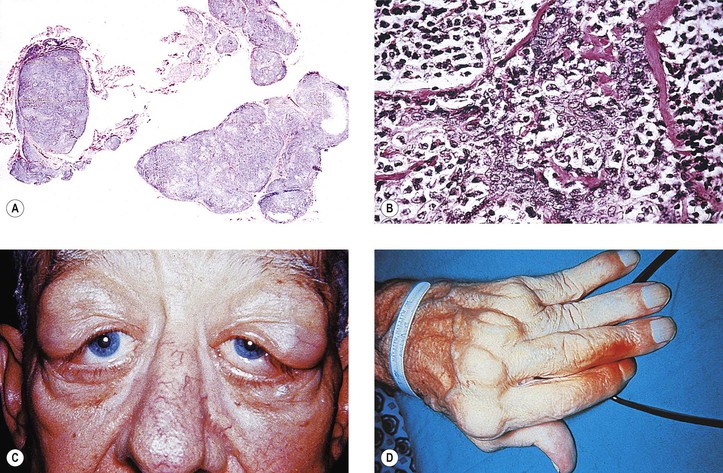
C. Sjögren’s syndrome (see Fig. 14.8)
2. The tissue destruction results in the symptoms of keratoconjunctivitis sicca and xerostomia.
3. Indirect support exists for a putative role of the Epstein–Barr virus (see Chapter 3) in the pathogenesis of the disease.
D. Inflammatory pseudotumor (see later in this chapter)
II. Granulomatous
A. Granulomatous inflammations rarely involve the orbit.
4. Histologically, a granulomatous reaction surrounds cholesterol crystals and altered blood.
Injuries
Penetrating Wounds
I. Direct effect on whatever tissue may be wounded by the injury
II. Complications (indirect effect)
A. Infection
1. Organisms may be introduced at the time of injury.
a. Bacteria cause an acute purulent inflammation.
b. Fungi cause a delayed, chronic, granulomatous inflammation.
B. Inflammation
1. The inflammation may be secondary to toxic products of tissue destruction.
2. Orbital thrombophlebitis may result.
3. A retained intraorbital foreign body may induce inflammation.
Nonpenetrating Wounds
The effects of nonpenetrating wounds are those secondary to contusion and concussion, mainly hemorrhage, secondary muscle palsies, and infraorbital nerve involvement.
Vascular Disease
Primary
I. Primary orbital vascular disease is rare.
II. Causes include varices, arteriovenular aneurysm, thrombophlebitis, and cavernous sinus thrombosis.
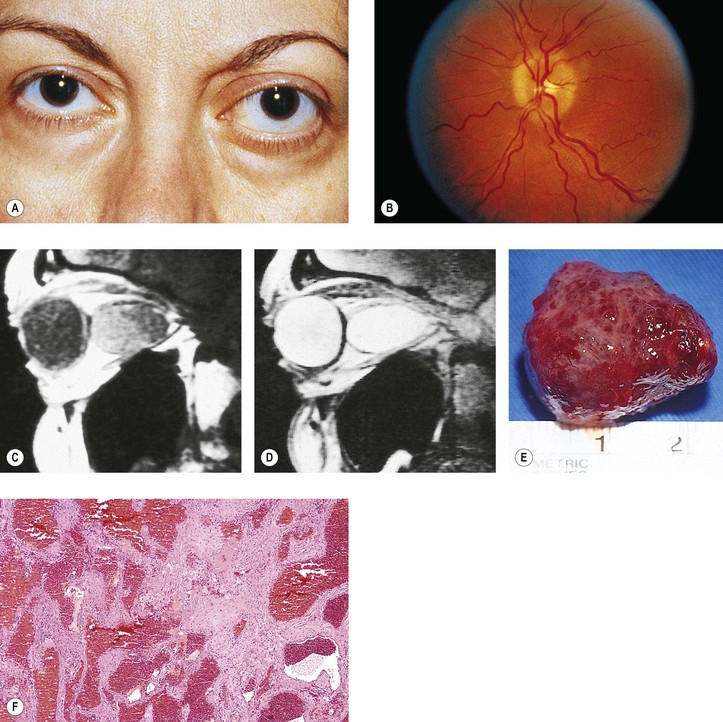
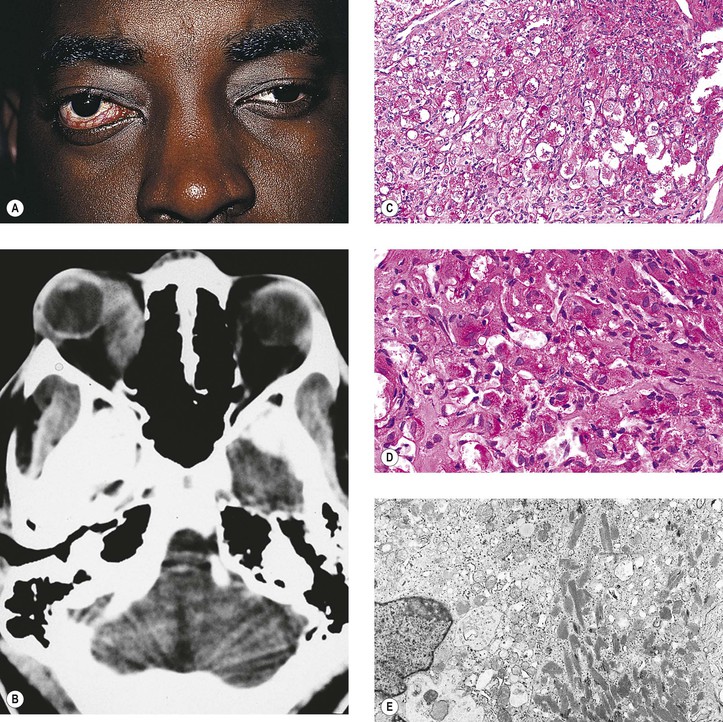
Orbital varix, also called distensible venous malformation (Fig. 14.9), may occur anterior to the septum orbitale and not cause exophthalmos, or it may occur posterior to the septum orbitale, causing exophthalmos. The exophthalmos may be acute if the varix undergoes thrombosis.
Part of Systemic Disease
Ocular Muscle Involvement in Systemic Disease
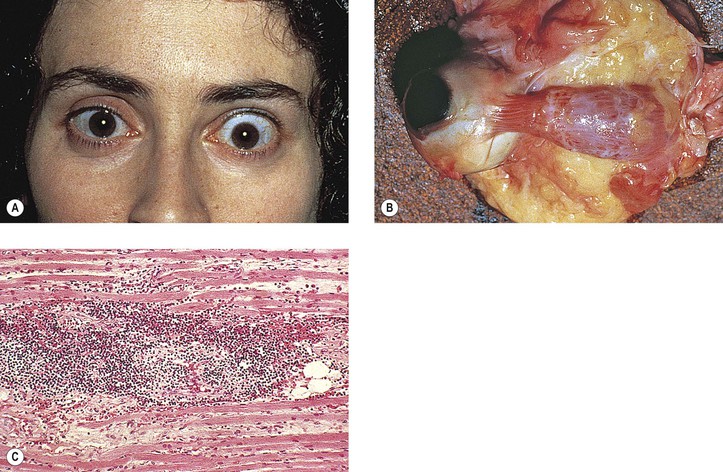
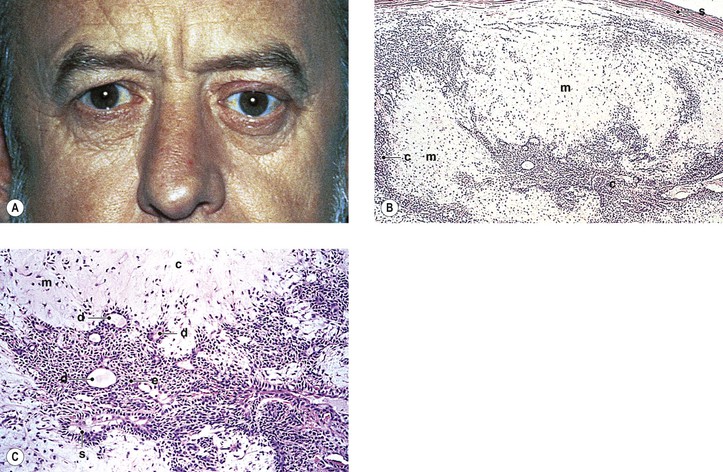
Graves’ Disease (Fig. 14.10)
I. Classification of eye changes of Graves’ disease (Box 14.1)
II. Mild form (“thyrotoxic” exophthalmos)
B. It may present initially with unilateral involvement but usually becomes bilateral.
C. Clinically and chemically, the patient is hyperthyroid.
D. Lid retraction, the most common clinical sign, may simulate exophthalmos.
E. Occasionally exophthalmos is present.
F. Prognosis for vision is good.
III. Severe form (“thyrotropic” or “malignant” exophthalmos; thyroid ophthalmopathy; thyroid orbitopathy)
A. The severe form is an autoimmune disease that affects people in middle age (average age, 50 years).
1. The disease is characterized by an increased percentage of suppressor/cytotoxic T lymphocytes.
2. Circulating T cells are directed against thyroid follicular cell antigens.
C. Clinically and chemically, the patient may be hyperthyroid, hypothyroid, or euthyroid.
E. Prognosis for vision is poor.
F. Histologically, the orbital tissue is characterized predominantly by extraocular and periorbital muscle involvement by edema, lymphocytic infiltration (mainly CD4+ and CD8+ T cells along with some focal aggregates of B cells, plasma cells, and mast cells), endomysial fibrosis, and mucopolysaccharide deposition.
Myotonic Dystrophy (Myotonia Dystrophica; Steinert’s Disease)
A. Its onset is between 20 and 30 years of age, with patients rarely living beyond 40 or 50 years.
B. Frontal baldness and endocrinopathy, especially testicular atrophy, are common.
II. Ocular findings
B. Foveal dystrophy that hardly affects vision and pigmentary retinopathy
III. Histologically, selective atrophy of muscle fibers is seen.
Mitochondrial Myopathies
A. Leber’s hereditary optic atrophy (see Chapter 13)
B. CPEO
2. Ptosis, external ophthalmoplegia, and often a pigmentary retinopathy can be seen.
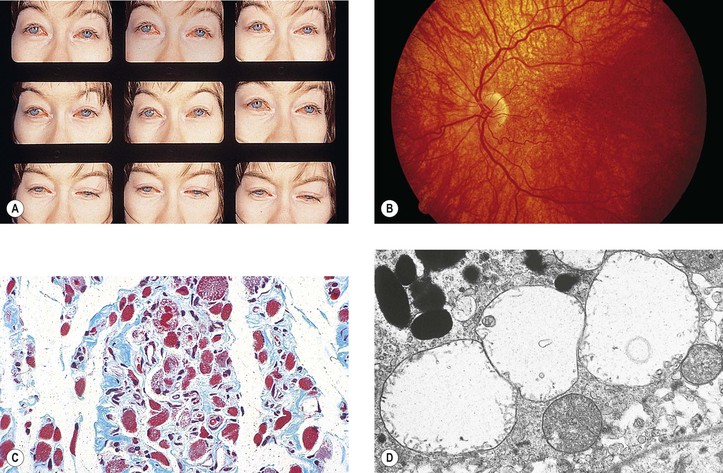
C. KSS
2. Patients have short stature and increased serum and cerebrospinal fluid lactate levels.
5. Histologically, along with the characteristic ragged-red appearance seen under light microscopy with the modified trichrome stain, electron microscopy shows an increased number of mitochondria containing abnormal cristae.
a. The eyes show abnormalities of the macular photoreceptor–retinal pigment epithelium (RPE)–choriocapillaris complex, namely absent or degenerated outer segments and hyperpigmented and hypopigmented RPE.
1) The cytoplasm of RPE cells show ballooned, structurally abnormal (“giant”) mitochondria.
Dermatomyositis
Neoplasms and Other Tumors3
See Box 14.2 for classification of neoplasms and other tumors.
Box 14.2
Classification of Neoplasms and Other Tumors

† The tumor is common, important, or both.
‡ The tumor is uncommon, unimportant, or both.
Primary Orbital Tumors
I. Choristomas—these are congenital tumors not normally present at the involved site.
1. An epidermoid cyst is composed of epidermis (i.e., stratified squamous epithelium with no epidermal appendages in the wall of the cyst) and contains desquamated keratin in the cyst, which appears as a cheesy material.
b. A much rarer type of congenital epithelial cyst is called primary nonkeratinized epithelial cyst (“conjunctival cyst”; see Figs. 14.12D and 14.12E). It is usually found in the superonasal aspect of the orbit, is lined by nonkeratinizing epithelium that resembles conjunctival epithelium, contains no adnexal structures in its wall, and is filled with clear fluid.
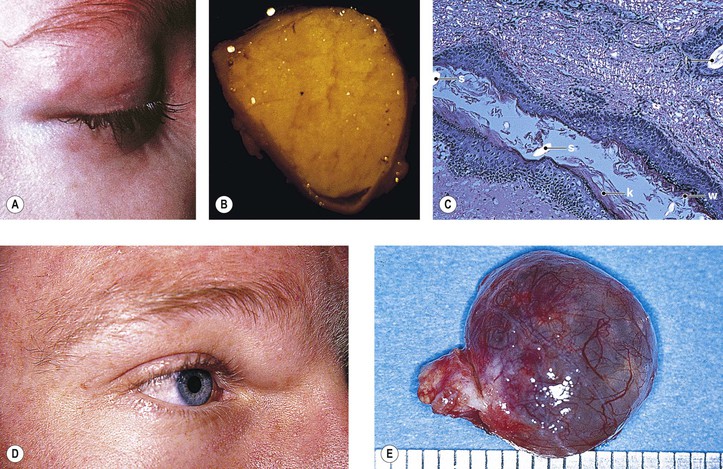
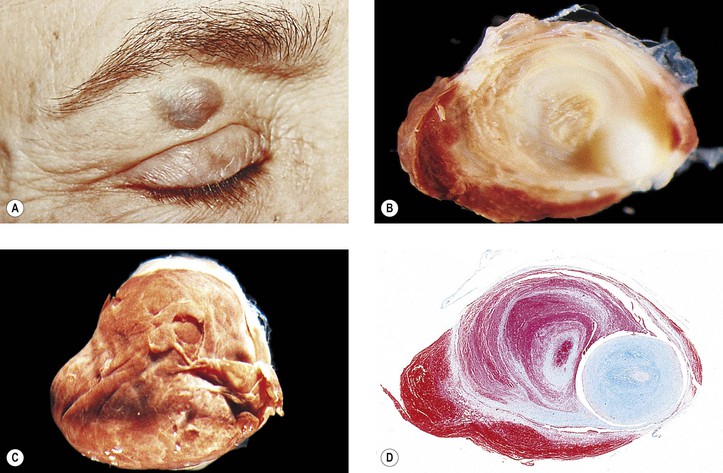
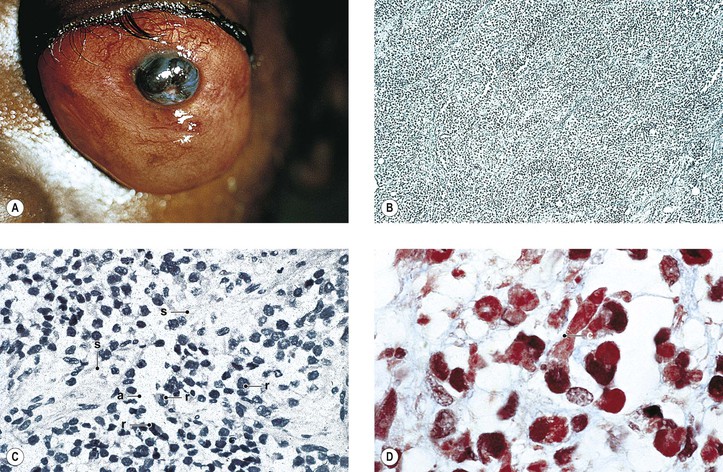
B. Dermoid cyst† (Fig. 14.12)
1. A dermoid cyst is probably a result of the sequestration of surface ectoderm pinched off at bony suture lines or along lines of embryonic closure.
2. Histologically, a dermoid cyst, derived from ectoderm, is composed of a wall surrounding a cavity.
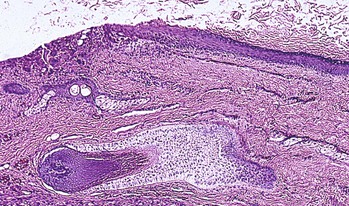
Rupture of a dermoid cyst can cause a chronic granulomatous inflammatory reaction (Fig. 14.13). Rarely, squamous cell carcinoma may develop in an epidermoid or dermoid cyst.
b. The cavity contains desquamated keratin, hair shafts, and debris.
C. Teratoma‡ (Fig. 14.14)
3. It has malignant potential and may also involve the eye.
D. Cholesterol granuloma and cholesteatoma (see previously in this chapter)
E. Ectopic lacrimal gland‡ (Fig. 14.15)
2. It usually causes symptoms only when inflamed; the origin of the inflammation is unknown.
II. Hamartomas—these are congenital tumors normally found at the involved site.
A. Phakomatoses (see Chapter 2)
B. Hemangioma
1. Capillary hemangioma (“cherry” hemangioma)† (Fig. 14.16)
c. A form of capillary hemangioma is found in angiomatosis retinae (see Chapter 2).
d. A variant of capillary hemangioma, often called hemangioendothelioma (Fig. 14.17), is observed at birth (20%), infancy, or early childhood. Typically, it appears suddenly, grows rapidly, and is characterized by relatively solid cords of round, multilayered endothelial cells with little or no evidence of lumen.
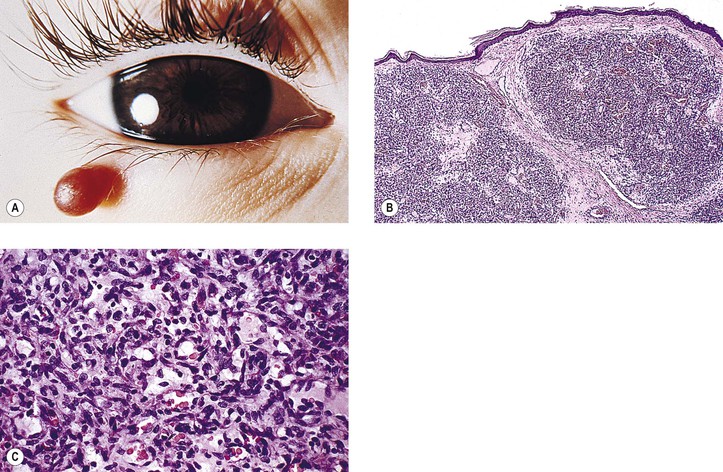
2. Cavernous hemangioma† (orbital cavernoma) (Fig. 14.18)
a. Cavernous hemangioma is the most common primary orbital tumor producing exophthalmos.
b. It may press on the coats of the eye and cause chorioretinal striae.
d. No feeder vessels are demonstrated by dye study techniques.
3. Arteriovenous (AV) communication‡
b. It occurs in AV communication of retina and brain or as an incidental finding.
a. Telangiectasia of the orbit is rare.
b. Telangiectasia is found in meningocutaneous angiomatosis (see Chapter 2), ataxia–telangiectasia (see Chapter 2), and Osler–Rendu–Weber disease (see Chapter 7).
c. Histologically, it is composed of dilated and tortuous capillaries.
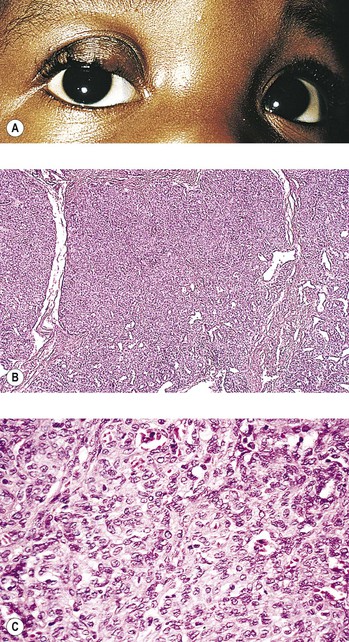
C. Lymphangioma† (Fig. 14.19)
1. Frequently, the clinical onset of lymphangioma is in children younger than 10 years of age.
2. It may diffusely involve the orbit, conjunctiva, and lids, and it tends to be invasive and slow-growing.
b. Although retinal folds may be seen, compressive optic neuropathy is rare.
3. The tumor probably regresses somewhat in time but easily becomes infected.
III. Mesenchymal tumors
A. Vascular
1. Hemangiomas and lymphangioma (see previously under discussion of hamartomas)
a. This rare orbital tumor is most common in the fourth decade.
d. The tumor probably arises from pericytes.
e. Histologically, an increased number of thin-walled vascular channels are separated by tumor cells in a network of extracellular material.
1) Perivascular massing of pericytes is present. The cell morphology is uniform.
2) Silver-stained material reveals reticulin characteristically segregating cells into groups.
3) Focal staining for vimentin, CD34, and factor XIIIa occurs.
3. Glomus tumor (glomangioma‡; Fig. 14.20)
b. Histologically, small vessels lined by a single layer of endothelial cells are surrounded by one or more layers of glomus cells.
1) The cells are round and have small, round nuclei and clear cytoplasm.
4. Hemangiosarcoma‡ (angiosarcoma, malignant hemangioendothelioma)
a. This is a rare orbital tumor.
b. Histologically, it is composed of intercommunicating channels or irregular vascular spaces lined by atypical endothelial cells confined in a thin reticulin network.
2) Marked histologic variation is seen in the tumor and from tumor to tumor.
5. Kaposi’s sarcoma (KS‡; Fig. 14.21)
e. Histologically, it is composed of many foci of capillary clusters in a stroma of malignant spindle cells. KSHV is a reliable marker (by polymerase chain reaction) to distinguish KS, particularly at its early stage, from other vascular lesions.
B. Fatty
1. Lipoma† (Fig. 14.22)
b. Histologically, a lipoma is composed of groups of mature, univacuolar, white, fat cells separated from other groups by delicate fibrovascular septa.
1) Coarser septa divide the tumor into lobules.
2) A true lipoma usually has a thin, fibrous capsule.
2. Liposarcoma‡ (Fig. 14.23)
a. This extremely rare orbital tumor may be primary or secondary to radiation therapy.
c. Histologically, liposarcomas tend to be well differentiated or myxoid.
1) The tumors are composed of univacuolar signet-ring lipoblasts.
2) Scattered, bizarre, hyperchromatic cells without prominent lipidization may also be present.
C. Fibrous–histiocytic–reactive
1. Nodular fasciitis† (also called subcutaneous pseudosarcomatous fibromatosis, pseudosarcomatous fasciitis, and nodular fibrositis)
b. Histologically, it is composed of nodular proliferations of plump, stellate, or spindle-shaped fibroblasts arranged in parallel bundles or haphazardly (the cells resemble tissue culture fibroblasts).
1) A variable amount of intercellular myxoid ground substance is present.
2) Abundant reticulin fibers and moderate numbers of collagen fibers can be demonstrated.
3) Proliferation of slitlike vascular spaces or well-formed capillaries is characteristic.
2. Juvenile fibromatosis‡ (psammomatoid ossifying fibroma)
a. This is usually seen in children.
b. It may recur after excision and is frequently mistaken for fibrosarcoma.
c. Histologically, it is composed of fibrous tissue interlacing with numerous mature capillaries.
D. Fibrous–histiocytic–neoplastic
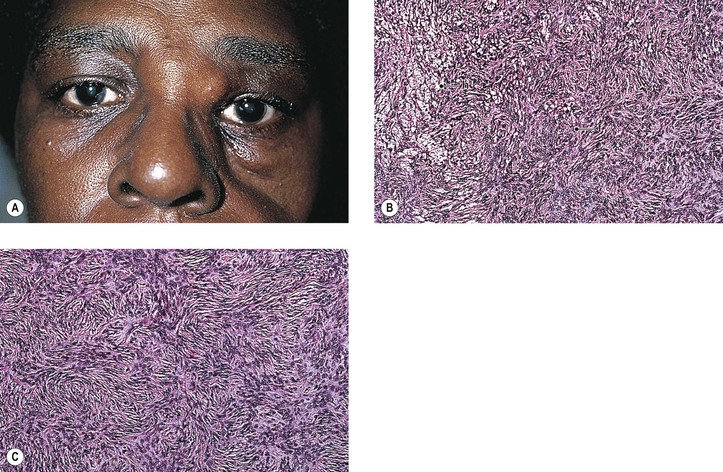
1. Fibrous histiocytoma (FH)† (xanthoma; Fig. 14.24)
a. FH is the most common primary mesenchymal orbital tumor of adults.
e. FH has a malignant potential.
f. Histologically, FH shows a characteristic, diphasic pattern with mainly “storiform” (matted) areas composed of fibrous spindle cells along with scattered areas showing single or grouped foamy histiocytes.
1) FH can be richly vascularized and then easily confused with hemangiopericytoma.
2) MFH shows a mixture of storiform and pleomorphic features.
2. Atypical fibroxanthoma (AFX)‡
a. AFX usually occurs in the sun-damaged skin of the head and neck in the elderly.
b. Histologically, spindle cells and pleomorphic polyhedral cells occur with many giant cells and mitotic figures.
3. Dermatofibrosarcoma (DFSP)‡
a. DFSP usually occurs in young or middle-aged persons as large cutaneous nodules.
b. Histologically, DFSP is composed of relatively uniform spindle cells with a conspicuous storiform pattern (similar to FH except for the lack of a histiocytic component).
2) Immunohistochemically, DFSP and AFX (see previously) stain nearly identically.
a. True orbital fibroma is rare.
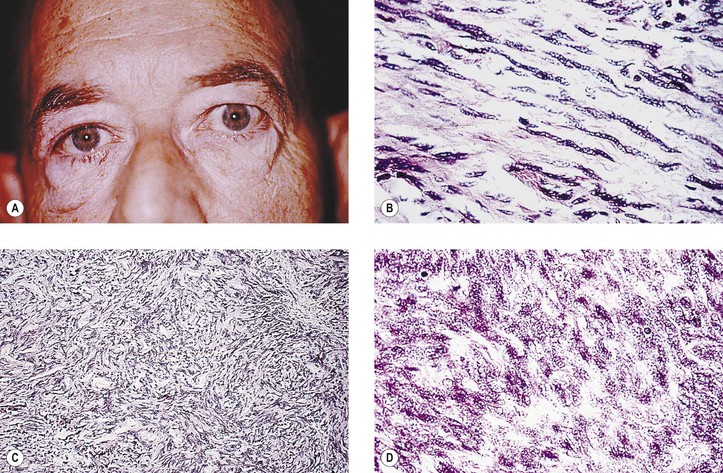
Histologically, orbital fibroma is composed of scattered spindle-shaped cells, sometimes showing a herringbone pattern, but lacking atypia and mitotic figures.
b. Fibrosarcoma (Fig. 14.25) is a very rare, slow-growing, orbital, spindle-cell tumor.
b. Although usually benign, it can recur locally and rarely metastasizes.
6. Fibrous hamartoma of infancy‡
b. Male to female ratio is 2 : 1.
b. Although most occur in the orbit, they can also occur in the lids and, rarely, conjunctiva.
c. Histologically, the tumors are richly vascularized and contain a patternless proliferation of spindle cells, numerous multinucleated giant cells, and some pseudovascular spaces, all embedded in variably collagenized stroma.
1) Myxoid stromal deposition may be present.
2) The spindle cells and multinucleated giant cells stain intensely positive with CD34 and vimentin.
E. Muscle
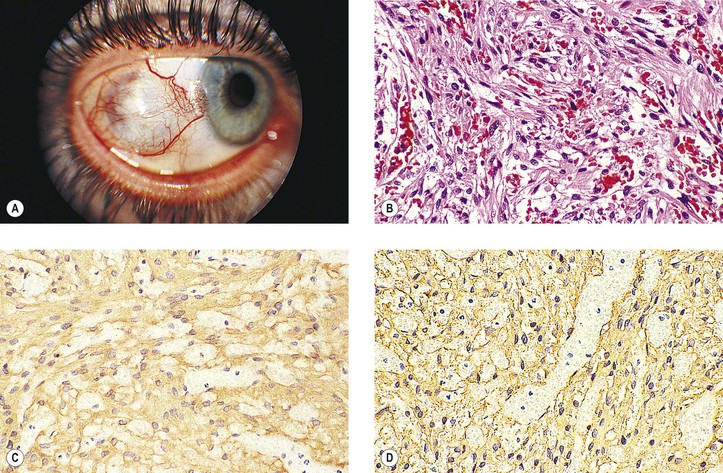
1. Leiomyoma‡ and leiomyosarcoma‡ are very rare orbital tumors.
Histologically, leiomyoma (Fig. 14.26) consists of interlacing fascicles of smooth muscle cells.
c. Leiomyosarcoma, in addition, shows atypical nuclei and mitotic figures.
2. Mesectodermal leiomyoma‡ (see Chapter 9) and leiomyosarcoma‡ are very rare orbital tumors whose origin seems to be from tissue derived from neural crest.
a. Malignant rhabdoid tumor is a rare, highly aggressive renal tumor of infants.
b. Histologically, the tumor is composed of dyscohesive, globoid, and eosinophilic cells, often containing cytoplasmic inclusions.
4. Rhabdomyoma‡ is a very rare, benign orbital tumor.
Histologically, well-differentiated rhabdomyoblasts are present.
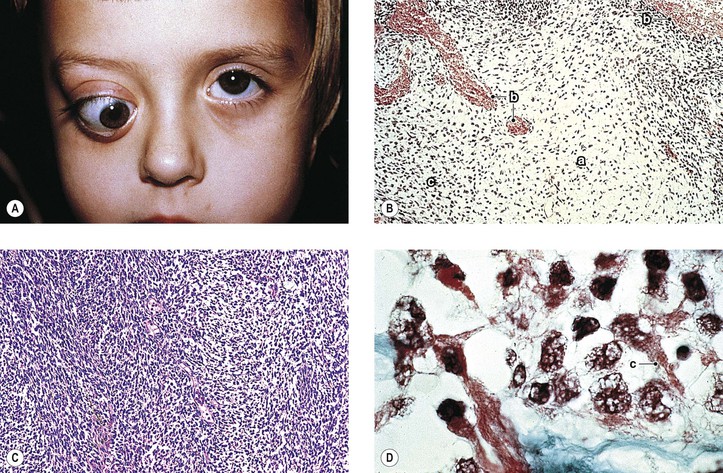
d. The tumor is characterized clinically by a very rapid onset, often simulating an orbital cellulitis.
e. Three types exist: embryonal, differentiated, and alveolar.
f. Embryonal type (Fig. 14.27) is the most common type.
2) Histologically, it is composed of malignant embryonal cells, rhabdomyoblasts, in a loose syncytial arrangement of fascicles of spindle cells running in a haphazard arrangement, usually showing frequent mitotic figures.
2) Cross-striations are easily found in the differentiated type.
1) The alveolar form of rhabdomyosarcoma seems to have the worst prognosis.
b) Molecular confirmation of alveolar rhabdomyosarcoma is important in the treatment of this tumor.
4) It is the most difficult tumor in which to find cross-striations.
i. Prognosis
3) Most deaths occur within the first three years so that a five-year cure probably is a valid one.
F. Cartilage—chondroma‡ and chondrosarcoma‡
1. Chondroma and chondrosarcoma are extremely rare orbital tumors.
G. Bone (all are extremely rare orbital tumors)
Nonneoplastic and neoplastic diseases of bone usually cause exophthalmos by decreasing orbital volume.
2. Fibrous dysplasia† (Fig. 14.29)
4. Giant cell reparative granuloma‡
5. Juvenile fibromatosis‡ (psammomatoid ossifying fibroma)
6. Cholesterol granuloma‡ and cholesteatoma‡ (see previously in this chapter)
8. Osteitis fibrosa cystica (brown tumor)†
10. Paget’s disease†
11. Osteoma,‡ osteoblastoma,‡ and osteogenic sarcoma (osteosarcoma)‡
13. Ameloblastoma‡
IV. Neural tumors
1. An amputation neuroma is rare in the orbit.
B. Neurofibromas† (see Figs. 2.3–2.5)
C. Neurilemmoma (schwannoma†; Figs. 14.30 and 14.31)
1. A neurilemmoma is a rare orbital tumor composed of neoplastic Schwann cells.
2. Histologically, nuclei of spindle-shaped Schwann cells show a tendency toward palisading.
a. When the texture is compact and composed of interwoven bundles of long bipolar spindle cells, often with ribbons of palisading cells alternating with relatively acellular areas, the Antoni type A pattern is present.
1) Areas of the tumor may mimic tactile corpuscles and are called Verocay bodies.
b. The tumor is usually encapsulated in the perineurium of the originating nerve.
d. Electron microscopy may show Luse bodies (see Fig. 14.31C; i.e., aggregates of long-spaced collagen).
3. Malignant peripheral nerve sheath tumor (malignant schwannoma, malignant neurilemmoma, neurofibrosarcoma, perineural fibrosarcoma, and neurogenic sarcoma)‡ is extremely rare, but when present it is associated with neurofibromatosis in 50% of cases.
4. Juvenile pilocytic astrocytoma (glioma) of optic nerve† (see Chapter 13)
D. Peripheral primitive neuroectodermal tumors (PNETs‡; Fig. 14.32)
1. PNETs are a group of soft-tissue tumors of presumed neural crest origin arising outside the central and sympathetic nervous system.
b. All share in a chromosomal aberration translocation (11;22)(q24]2).
2. Adult neuroblastoma (one of the PNETs)
a. Adult neuroblastoma most rarely involves the orbit as a primary tumor.
b. The two-mutation model of tumorigenesis applies to neuroblastoma as well as to retinoblastoma.
c. More commonly, it is a childhood metastatic disease (see previously in this chapter).
A. Meningioma† (see Chapter 13)
B. Nonchromaffin paraganglioma (carotid body tumor)‡
2. Histologically, it is composed of clusters of relatively clear (epithelioid) or dark (chief) cells surrounded by a vascularized connective tissue stroma.
C. Granular cell tumor (granular cell myoblastoma‡; Fig. 14.33)
1. Granular cell tumor is a rare, benign orbital tumor.
2. Histologically, it is composed of round to polygonal cells in solid groups and cords and occasionally in alveolated collections.
b. Silver stains frequently show reticulin surrounding individual cells.
c. Electron microscopy reveals oval and round membrane-bound cytoplasmic bodies.
D. Alveolar soft-part sarcoma‡ (Fig. 14.34)
1. An alveolar soft-part sarcoma is a rare, malignant orbital tumor.
2. Histologically, it is composed of alveolated groups of round and polygonal cells circumscribed by bands of connective tissue, some of which contain delicate vascular channels in a distinct organoid pattern.
E. Malignant melanoma‡ (see Chapter 17)
F. Endodermal sinus tumor (parietal yolk sac carcinoma)‡
1. Myxoma may arise in orbital bones as a benign solitary lesion or be part of Carney’s syndrome (see Chapter 7).
VI. Epithelial cysts and neoplasms of lacrimal gland
A. Lacrimal ductal cysts (dacryops)†
2. Histologically, the cysts are lined by a double layer of epithelium.
B. Localized amyloidosis of the lacrimal gland‡
1. Localized amyloidosis of the lacrimal gland can occur unilaterally or bilaterally.
2. Characteristic amyloid (see Chapter 7) is found by light and electron microscopy and by immunohistochemistry (monoclonal λ light chains).
C. General information on neoplasms
1. Characteristically, lacrimal gland tumors cause a “down and in” type of exophthalmos.
6. Simplified classification of tumors of the lacrimal gland
a. Lymphoid tumors and inflammatory pseudotumors: 75% (most benign)
b. Epithelial and cystic lesions: 25%
7. Prognosis
b. All malignant tumors: the mortality rate is 50% or more.
D. Pleomorphic adenoma (benign mixed tumor)† (Fig. 14.35)
1. Pleomorphic adenoma occurs in young adults, with a median age of 35 years. Males predominate 2 : 1.
2. It is a locally invasive tumor and may infiltrate its own pseudocapsule to involve adjacent periosteum.
a. Acute pain and progression are rare.
3. Histologically, the tumor shows marked structural variation from patient to patient and within the same tumor.
E. Other types of benign tumors (all are rare)
1. Hemangioma‡
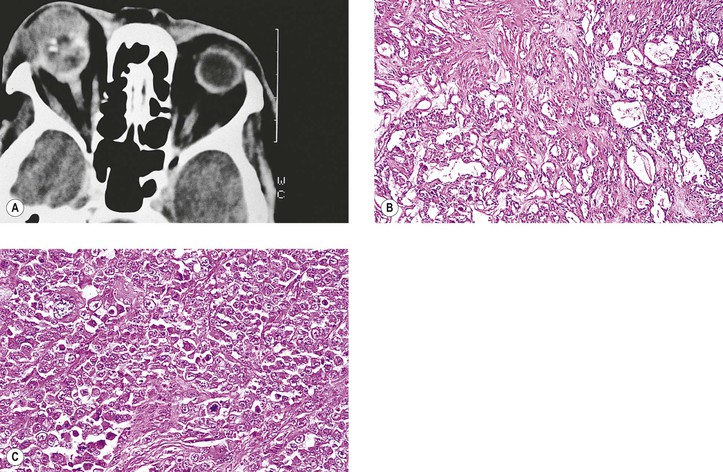
F. Malignant mixed tumor† (Fig. 14.36)
2. It arises from a pleomorphic adenoma.
3. Histologically, areas resembling a pleomorphic adenoma are seen along with adenocarcinomatous areas.
G. Adenoid cystic carcinoma† (malignant cylindroma; Fig. 14.37)
2. The tumor soon invades perineural lymphatics and has an extremely poor prognosis.
3. Acute pain and progression are common.
4. Histologically, under lower power it has a characteristic “Swiss-cheese” pattern.
a. Aggregates or islands of poorly differentiated, small, tightly packed epithelial cells are sharply outlined against the surrounding typical, hyaline-like stroma.
1) Aggregates may be very small, moderate, or quite large, but they are always sharply outlined.
2) Aggregates contain mucin-filled cystic spaces of different sizes, hence the Swiss-cheese pattern.
H. Other types of carcinomas (all are rare and all have a very poor prognosis)‡
2. Median age group is approximately 53 years, with a 3 : 1 male predominance.
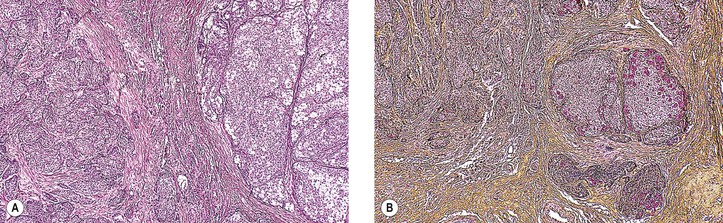
3. Mucoepidermoid carcinoma (Fig. 14.38) is the most common primary carcinoma of the major salivary glands, but it is rare in the lacrimal gland. The tumor contains both epidermoid (squamous) and mucin-producing cells.
VII. Reticuloendothelial system
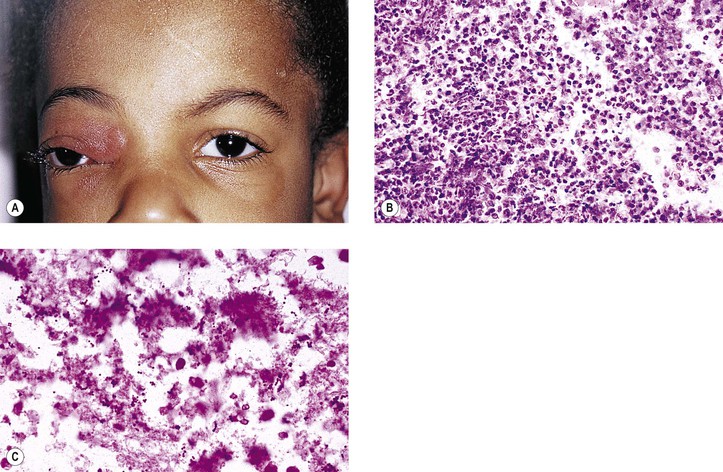
A. Langerhans’ cell histiocytosis (LCH; Langerhans’ granulomatosis, histiocytosis X)† (Tables 14.1 and 14.2)
1. LCH occurs primarily in children, adolescents, and young adults. It is characterized by a proliferation of Langerhans’ cells in an inflammatory background, often containing many eosinophils.
b. 80% of patients with orbital involvement are male, have unifocal, unisystem disease, and present with periorbital swelling and/or mass.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Orbit
Normal Anatomy
I. The orbital volume is approximately 30 ml, and the orbital depth (anterior to posterior) is approximately 4.5 cm (Figs. 14.1–14.5).
A. The medial orbital wall is quite thin (<0.5 mm) and transparent.
II. In addition to the bony walls, the eye, and the optic nerve, the orbit contains many soft-tissue structures such as fat, muscle (striated and nonstriated), cartilage, bone, fibrous tissue, nerves, and blood vessels.
B. All orbital structures may be involved in disease processes.
Exophthalmos
I. The main clinical manifestation of orbital disease is exophthalmos (ocular proptosis), the extent and direction of which depend on a number of factors1: (1) size of lesion, (2) character of lesion (expansile vs. infiltrative growth, rapid vs. slow growth), (3) location of the lesion in the orbit (small lesion in muscle cone causes more exophthalmos than lesion of same size outside the muscle cone; lesions anterior to septum orbitale do not produce exophthalmos unless they also grow posteriorly), and (4) lesion’s effect on the extraocular muscles (complete paralysis of all muscles by itself can cause 2 mm of exophthalmos).
Developmental Abnormalities
Developmental Abnormalities of Bony Orbit
Developmental abnormalities are usually associated with abnormalities of the cranial and facial bones such as tower skull or hypertelorism.
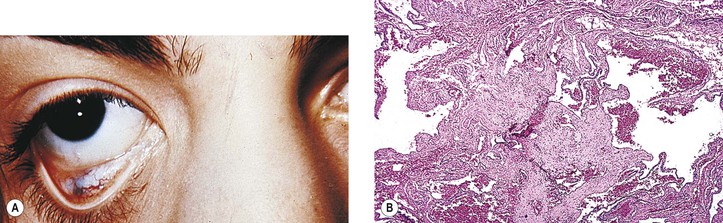
Microphthalmos with Cyst
I. Microphthalmos with cyst (see Fig. 2.10) is usually a unilateral condition, but it may be bilateral.
II. The cyst may be so large as to obscure the microphthalmic eye.
IV. The condition is caused by incomplete closure of the fetal cleft.
Orbital Inflammation
Acute
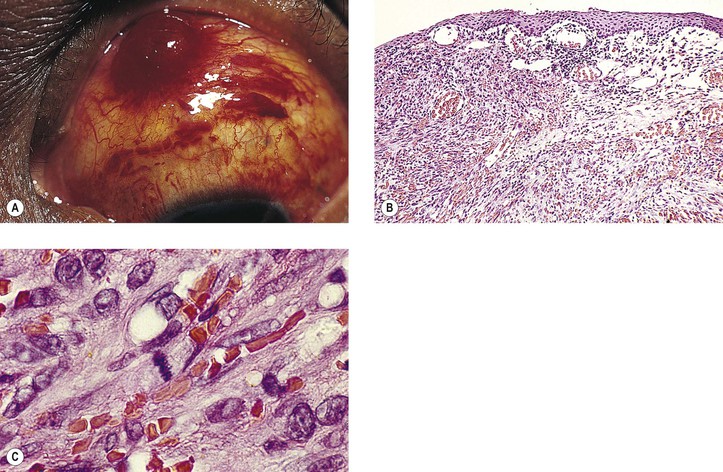
I. Nonsuppurative (see section Nonsuppurative, Chronic Nongranulomatous Uveitis and Endophthalmitis in Chapter 3)—orbital cellulitis is most commonly caused by extension of an inflammation from the paranasal sinuses (Fig. 14.6).
II. Suppurative (see section Suppurative Endophthalmitis and Panophthalmitis in Chapter 3)
A. Purulent infection (e.g., with Staphylococcus) occurs commonly after trauma.
B. Phycomycosis (mucormycosis) is a devastating cause of suppurative orbital inflammation (Fig. 14.7; see Chapter 4).
Chronic
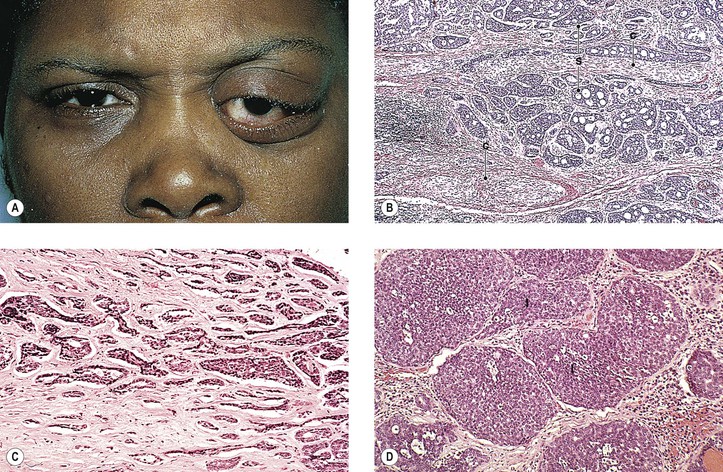
B. A rare cause is the benign lymphoepithelial lesion of Godwin2 (Fig. 14.8).
2. It may be part of Sjögren’s syndrome (see later).
3. Rarely, it may become malignant.
C. Sjögren’s syndrome (see Fig. 14.8)
2. The tissue destruction results in the symptoms of keratoconjunctivitis sicca and xerostomia.
3. Indirect support exists for a putative role of the Epstein–Barr virus (see Chapter 3) in the pathogenesis of the disease.
D. Inflammatory pseudotumor (see later in this chapter)
II. Granulomatous
A. Granulomatous inflammations rarely involve the orbit.
4. Histologically, a granulomatous reaction surrounds cholesterol crystals and altered blood.
Injuries
Penetrating Wounds
I. Direct effect on whatever tissue may be wounded by the injury
II. Complications (indirect effect)
A. Infection
1. Organisms may be introduced at the time of injury.
a. Bacteria cause an acute purulent inflammation.
b. Fungi cause a delayed, chronic, granulomatous inflammation.
B. Inflammation
1. The inflammation may be secondary to toxic products of tissue destruction.
2. Orbital thrombophlebitis may result.
3. A retained intraorbital foreign body may induce inflammation.
Nonpenetrating Wounds
The effects of nonpenetrating wounds are those secondary to contusion and concussion, mainly hemorrhage, secondary muscle palsies, and infraorbital nerve involvement.
Vascular Disease
Primary
I. Primary orbital vascular disease is rare.
II. Causes include varices, arteriovenular aneurysm, thrombophlebitis, and cavernous sinus thrombosis.
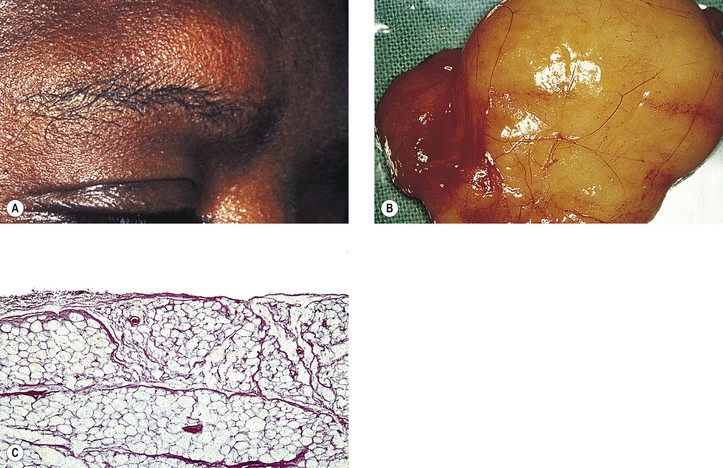
Orbital varix, also called distensible venous malformation (Fig. 14.9), may occur anterior to the septum orbitale and not cause exophthalmos, or it may occur posterior to the septum orbitale, causing exophthalmos. The exophthalmos may be acute if the varix undergoes thrombosis.
Part of Systemic Disease
Ocular Muscle Involvement in Systemic Disease
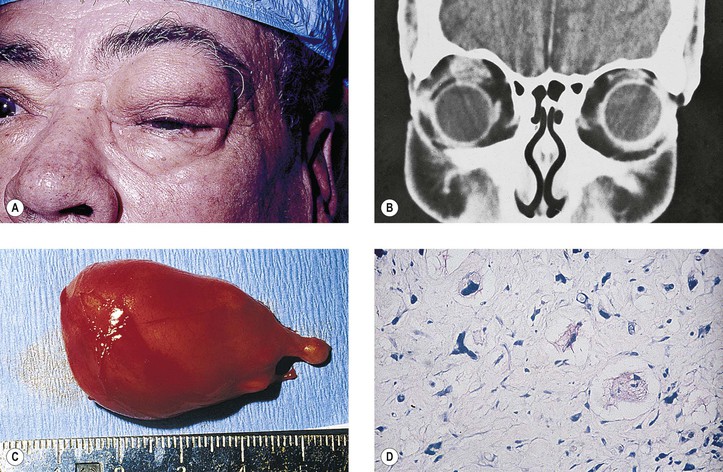
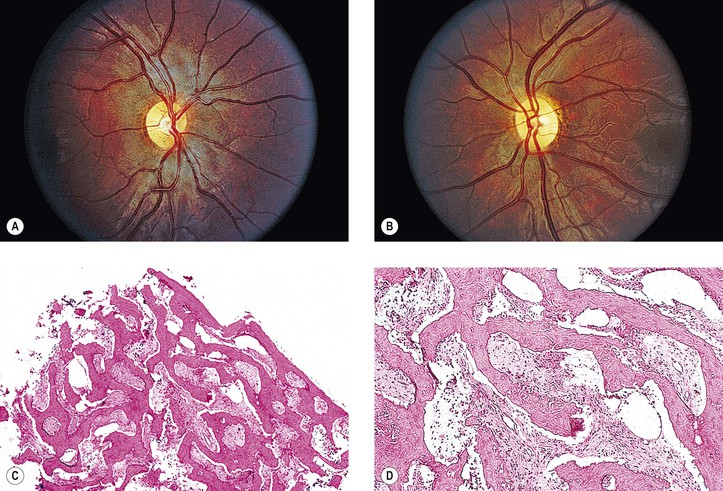
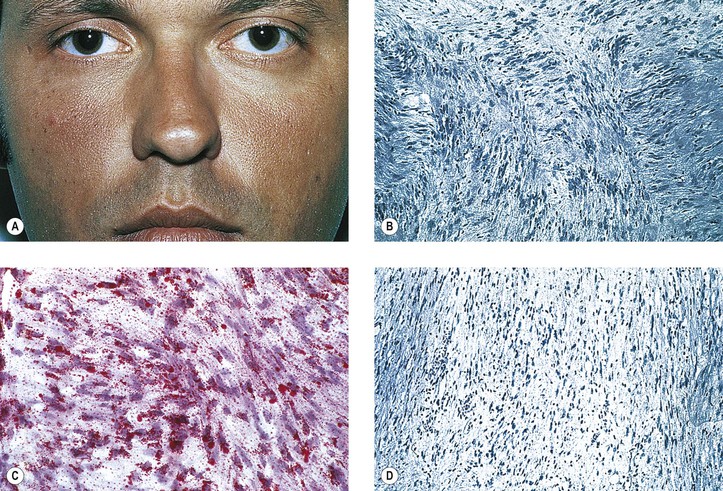
Graves’ Disease (Fig. 14.10)
I. Classification of eye changes of Graves’ disease (Box 14.1)
II. Mild form (“thyrotoxic” exophthalmos)
B. It may present initially with unilateral involvement but usually becomes bilateral.
C. Clinically and chemically, the patient is hyperthyroid.
D. Lid retraction, the most common clinical sign, may simulate exophthalmos.
E. Occasionally exophthalmos is present.
F. Prognosis for vision is good.
III. Severe form (“thyrotropic” or “malignant” exophthalmos; thyroid ophthalmopathy; thyroid orbitopathy)
A. The severe form is an autoimmune disease that affects people in middle age (average age, 50 years).
1. The disease is characterized by an increased percentage of suppressor/cytotoxic T lymphocytes.
2. Circulating T cells are directed against thyroid follicular cell antigens.
C. Clinically and chemically, the patient may be hyperthyroid, hypothyroid, or euthyroid.
E. Prognosis for vision is poor.
F. Histologically, the orbital tissue is characterized predominantly by extraocular and periorbital muscle involvement by edema, lymphocytic infiltration (mainly CD4+ and CD8+ T cells along with some focal aggregates of B cells, plasma cells, and mast cells), endomysial fibrosis, and mucopolysaccharide deposition.
Myotonic Dystrophy (Myotonia Dystrophica; Steinert’s Disease)
A. Its onset is between 20 and 30 years of age, with patients rarely living beyond 40 or 50 years.
B. Frontal baldness and endocrinopathy, especially testicular atrophy, are common.
II. Ocular findings
B. Foveal dystrophy that hardly affects vision and pigmentary retinopathy
III. Histologically, selective atrophy of muscle fibers is seen.
Mitochondrial Myopathies
A. Leber’s hereditary optic atrophy (see Chapter 13)
B. CPEO
2. Ptosis, external ophthalmoplegia, and often a pigmentary retinopathy can be seen.
C. KSS
2. Patients have short stature and increased serum and cerebrospinal fluid lactate levels.
5. Histologically, along with the characteristic ragged-red appearance seen under light microscopy with the modified trichrome stain, electron microscopy shows an increased number of mitochondria containing abnormal cristae.
a. The eyes show abnormalities of the macular photoreceptor–retinal pigment epithelium (RPE)–choriocapillaris complex, namely absent or degenerated outer segments and hyperpigmented and hypopigmented RPE.
1) The cytoplasm of RPE cells show ballooned, structurally abnormal (“giant”) mitochondria.
