[level-membership-for-neurosurgery-category]
CHAPTER 175 Normal and Abnormal Embryology of the Brain
It is increasingly becoming apparent that although congenital brain and spinal cord malformations share some common clinical features, the morphology, epidemiology, and natural history of these disorders suggest a diverse and heterogeneous embryologic origin.1,2 In our view, a classification of congenital craniospinal malformations based on reputed embryonic mechanisms is much more helpful. We review the normal embryology of the nervous system, as well as the presumed embryonic mechanisms that have been proposed to give rise to various neural tube malformations, with the intent of providing a mechanistic embryonic framework on which to hang these various disorders.
At the outset, it is important to understand that most of the embryologic mechanisms proposed for congenital craniospinal malformations are putative; that is, there is no absolute proof that they are the cause of a particular human malformation, and there are few adequate animal models to test our hypotheses about the origin of human malformations. However, we can gain considerable insight into the embryopathology of congenital malformations through the study of normal neural development. Throughout this chapter we refer to human development in terms of both postovulatory days (PODs) and the embryonic staging system of O’Rahilly and Müller3; the timing and stages for all major developmental events are listed in Table 175-1. Because this chapter reviews only cranial malformations, it does not include discussions specific to spinal cord development or to congenital spinal cord malformations; interested readers are referred elsewhere for this information.4,5
TABLE 175-1 Embryologic Classification of Congenital Craniospinal Malformations
| DISORDERED MIDLINE AXIAL INTEGRATION DURING GASTRULATION |
| LOCALIZED FAILURE OF NEURULATION |
| PREMATURE ECTODERMAL DYSJUNCTION |
| Some spinal lipomyelomeningoceles (cranial to S2) |
| INCOMPLETE ECTODERMAL DYSJUNCTION |
| Dermal sinus tracts, dermoid/epidermoid tumors, ?encephaloceles |
| DISORDERED TELENCEPHALIC CLEAVAGE |
| Holoprosencephaly |
| DISORDERED COMMISSURAL DEVELOPMENT |
| Callosal agenesis, dysgenesis |
| DISORDERED NEURONAL MIGRATION |
| Schizencephaly, lissencephaly, agyria, polymicrogyria, heterotopias |
| DISORDERED RHOMBIC LIP DEVELOPMENT |
| Dandy-Walker malformation, rhombencephalosynapsis |
| POSTNEURULATION DISORDERS |
Normal Early Human Neural Development
Blastogenesis and Gastrulation (PODs 1 to 13)
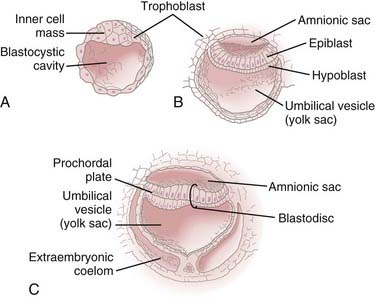
During the first 2 weeks of embryogenesis the human embryo undergoes a number of cell divisions and cellular rearrangements (blastogenesis) that ultimately result in the formation of a blastocyst situated eccentrically within a hollow sphere of trophoblast cells (Fig. 175-1A). The blastocyst later forms a two-layered structure with a dorsal layer, the epiblast, adjacent to the amnionic cavity and a ventral layer, the hypoblast, adjacent to the yolk sac.3 A thickening, the prochordal plate, identifies the cranial end of the embryo (Fig. 175-1B).
Gastrulation begins with the appearance of a midline structure, the primitive streak (PS), at the caudal end of the embryo; the cranial end of the primitive streak is called Hensen’s node. A midline trough, the primitive groove, runs in the midline within the PS; the cranial end of the primitive groove is the primitive pit. The PS elongates cranially over the next 3 days and forms a midline structure in the caudal half of the embryo (Fig. 175-2A). The PS then begins to regress (shorten) back toward the caudal end of the embryo.3 As gastrulation proceeds, cells of the epiblast migrate toward the PS and invaginate through the primitive groove (Fig. 175-2A). The first cells to ingress (while the PS is still elongating) are endodermal cells, which displace the hypoblast cells laterally and form prospective endoderm (the displaced hypoblast cells form extraembryonic tissues).6–10 As the PS regresses, mesodermal cells ingress through the PS between the epiblast and newly formed endoderm and become the mesoderm.9,10 The remaining epiblast cells spread out to replace the cells that have ingressed through the primitive groove and thereby form both the neuroectoderm (the neural tube) and the cutaneous ectoderm (skin). All three germ layers, ectoderm, mesoderm, and endoderm, are derived from the epiblast. Gastrulation transforms the embryo from a two-layered structure to a three-layered structure.11
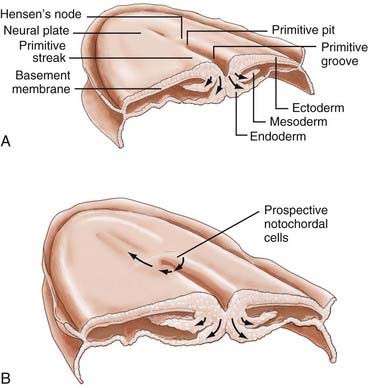
Hensen’s node serves a special role as the “organizer” of the embryo. As the PS elongates, prospective endodermal cells within Hensen’s node migrate through the primitive pit. As the PS regresses, specialized mesodermal cells, prospective notochordal cells, migrate through the primitive pit (Fig. 175-2B) and form the notochordal process in the midline between the neuroectoderm and endoderm.10,11 As we shall see, the notochord plays an important role in directing subsequent neurulation.
To what extent does the notochordal process extend cranially from Hensen’s node, and to what extent does it elongate caudally by the addition of cells to its caudal end from the regressing Hensen’s node? In the chick, the notochordal process grows largely by the latter mechanism.12 In mammals, the available evidence suggests that the situation is much more complex and may involve both mechanisms.13,14 The mechanism of notochord elongation in humans is unknown.
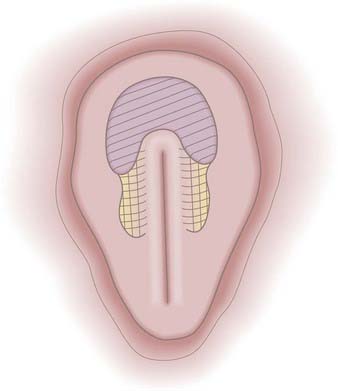
The prospective neuroectoderm is localized to an area of the epiblast that surrounds and flanks Hensen’s node and the cranial half of the PS (Fig. 175-3).15 Fate-mapping studies in chick embryos suggest that each region of the neuroepithelium contributes to multiple neuraxial levels, with the most cranial neuroectoderm contributing to the forebrain and all caudal levels of the neuraxis and more caudal levels of the neuroectoderm contributing to successively more caudal levels of the neuraxis. The prospective brain is derived specifically from neuroectoderm located cranial and lateral to Hensen’s node.15
Formation and Development of the Notochord (PODs 16 to 25)
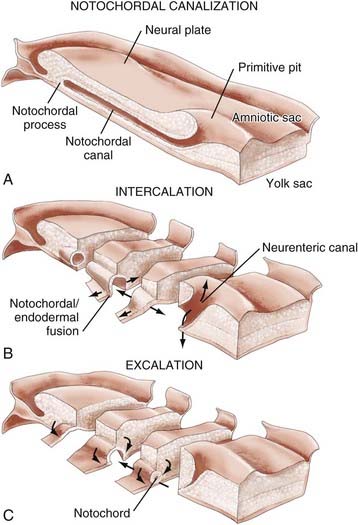
In both human3 and nonhuman primates,16 the notochordal process in cross section consists of cells arranged radially about a central lumen called the notochordal canal (Fig. 175-4A).3 The notochordal canal is continuous dorsally with the amnionic cavity through the primitive pit.17 Between PODs 18 and 20, the notochordal process fuses (intercalates) with the underlying endoderm to form the notochordal plate (Fig. 175-4B). The notochordal plate is therefore incorporated into the roof of the yolk sac, with the notochordal canal becoming continuous with the yolk sac. Intercalation results in a direct communication, the primitive neurenteric canal, that connects the amnionic and yolk sacs at the level of Hensen’s node.3 The neurenteric canal persists for about 3 days, at the end of which the notochordal plate folds dorsoventrally and separates (excalates) from the endoderm (Fig. 175-4C) and the neurenteric canal is obliterated.3,18,19 Thereafter, the true notochord exists as a solid rod of notochordal cells.18 The function or functions of the neurenteric canal are unknown.
Primary Neurulation (PODs 16 to 25)
Human neuroectoderm is first visible at POD 16 as pseudostratified columnar epithelium overlying the midline notochord (Fig. 175-5A) and contiguous with the surrounding squamous epithelium of the cutaneous ectoderm.20 By POD 17 to 19, a midline neural groove (or median hinge point) develops in the midline of the neural plate immediately above the notochord (Fig. 175-5C).21 The neural groove deepens (Fig. 175-5B), and paired neural folds begin to elevate bilaterally.22 Paired dorsolateral grooves (or dorsolateral hinge points) result in further dorsal elevation and medial convergence of the neural folds (Fig. 175-5C). The neural folds separate from the cutaneous ectoderm and fuse (Fig. 175-5D) to form a closed neural tube between POD 21 and 23. Closure generally involves apposition and fusion of first the cutaneous ectoderm and then the neuroectoderm.3,19 The first part of the human neural tube to close is the region of the caudal rhombencephalon or cranial spinal cord, usually when five pairs of somites are present.19
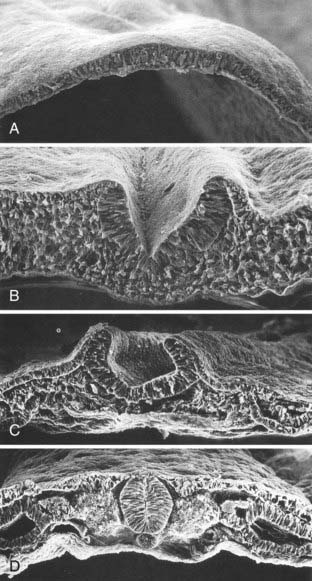
Closure of the neural tube takes place over a period of 4 to 6 days. Although previously thought to close in linear fashion like a zipper extending cranially and caudally from the point of initial closure, mammalian neurulation instead appears to extend from several initiation sites along the craniocaudal neuraxis.19,23–25 Cranial neural tube closure may involve the coordinated interaction of as many as four waves of discontinuous neural tube closure.19,23–26 The spinal cord closes craniocaudally in a linear manner from the point of initial closure to the caudal neuropore.24,25 The last two closure sites are the cranial and caudal neuropores. The cranial neuropore closes between POD 23 and 25, whereas the caudal neuropore closes between POD 25 and 27.27
Neural crest cells are specialized, multipotential migratory cells that arise from the junction between the neural folds and adjacent surface ectoderm18,28–30; in humans, a simultaneous origin for some neural crest cells from the surface ectoderm cannot be excluded.19 Formation of the cranial neural crest begins during elevation of the neural folds before they fuse and continues from the closed neural tube well after the neural folds have fused.18,19,31 The spinal neural crest arises only after closure of the neural tube.3,27
Cranial neural crest cells contribute to the branchial arches (Table 175-2) and the arachnoid and pia mater of the cranium (the dura mater appears to derive from the mesoderm). Spinal neural crest cells undergo terminal differentiation into melanocytes of the body wall and limbs, Schwann cells investing the peripheral nerves, spinal cord meninges, dorsal root and autonomic ganglion cells of the spinal nerves, and adrenal medulla.32 Further details regarding control of migration and terminal differentiation of neural crest cells can be found in several reviews.33–35
TABLE 175-2 Contributions of the Neural Crest to Branchial Clefts
Secondary Neurulation (POD 25)
After caudal neuropore closure at POD 25 to 27, the entire nervous system is covered with cutaneous ectoderm, and more caudal neural development takes place by secondary neurulation.27 This process has been covered in other sources and has no relevance to cranial development; interested readers are referred elsewhere for this information.
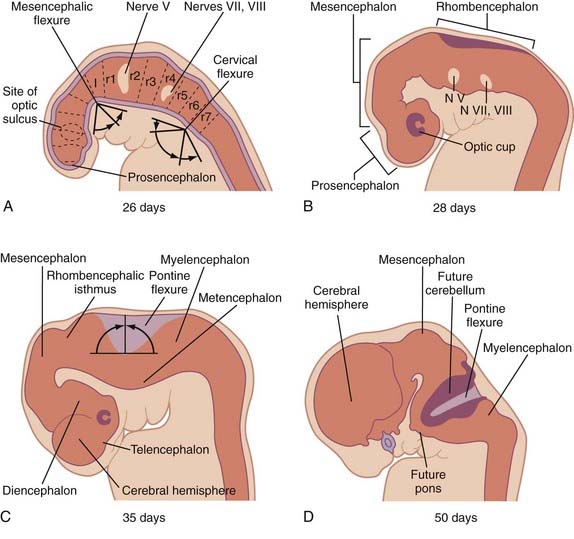
Development of Brain Vesicles, Neural Tube Bending, and Formation of Alar and Basal Plate Derivatives (PODs 19 to 40)
Three neural tube flexures develop during this time as well (Fig. 175-6). The earliest to appear, at POD 19 to 21 during primary neurulation, is the dorsally directed mesencephalic flexure. Later, two additional flexures develop—the dorsally directed cervical flexure (POD 28) and the ventrally directed pontine flexure (POD 32). The pontine flexure continues to bend the future brainstem such that the metencephalon (including the cerebellum) comes to lie dorsal to the myelencephalon by the eighth embryonic week.
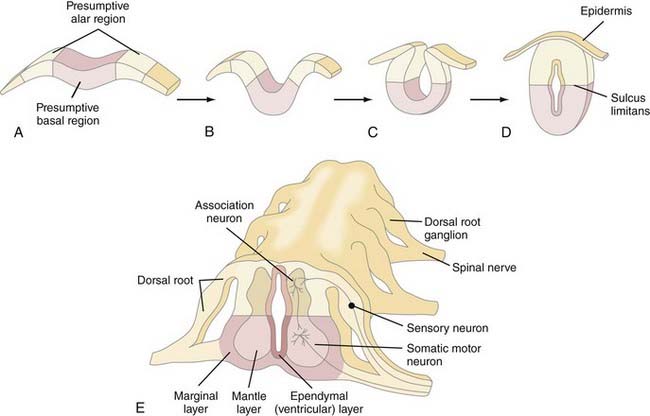
The fundamental cytoarchitectural organization of the spinal cord and brainstem (myelencephalon, metencephalon, and mesencephalon) consists of two components: a basal and an alar plate (Fig. 175-7). The basal plate contains paired ventral neuromotor columns that contribute to the primary motor pathways, whereas the alar plate contains paired dorsal neurosensory columns that contribute to the sensory and integrative pathways. As discussed previously, spinal cord neural crest cells form dorsal root ganglia and dorsal sensory nerves, the central processes of which enter the spinal cord and either ascend within the posterior columns to the nucleus gracilis and cuneatus or synapse locally with dorsal sensory neurons within the alar column (Rexed’s laminae I to VII). Alar plate neurons send projections both cranially in the spinothalamic and spinocerebellar tracts and locally that perform integrative functions with other alar or basal plate neurons within the spinal cord. The basal plate motoneurons receive afferent synapses from alar plate neurons (as well as descending projections from cranial portions of the neuraxis) and, in turn, send efferent axonal projections to the periphery via the ventral root.
The brainstem nuclei are arranged into seven columns, three motor and four sensory. The three motor columns produce somatic efferent, visceral efferent, and special (branchial) efferent nuclei. The four sensory columns produce somatic afferent, visceral afferent, and two special afferent nuclei—special somatic afferent and special visceral afferent nuclei. Each cranial nerve receives contributions from one or more of these seven columns (Table 175-3).
| Olfactory (I) | SVA (olfaction) |
| Optic (II) | SSA (vision) |
| Oculomotor (III) | GSE (oculomotor muscles) |
| GVE (ciliary muscle) | |
| Trochlear (IV) | GSE (superior oblique muscle) |
| Trigeminal (V) | GSA (skin, pharynx sensation) |
| GVA (proprioception) | |
| SVE (branchial muscles) | |
| Abducens (VI) | GSE (lateral rectus) |
| Facial (VII) | SVA (taste in the anterior two thirds of the tongue) |
| GSA (skin of the external auditory meatus) | |
| GVA (taste in the anterior two thirds of the tongue) | |
| SVE (muscles of facial expression) | |
| GVE (salivary, lacrimal glands) | |
| Vestibulocochlear (VIII) | SSA (auditory, vestibular) |
| Glossopharyngeal (IX) | SVA (taste in the posterior third of the tongue) |
| GVA (parotid gland, carotid body and sinus, middle ear) | |
| GSA (external ear) | |
| SVE (stylopharyngeus muscle, branchial arch) | |
| GVE (parotid gland) | |
| Vagus (X) | SVA (taste for the palate, epiglottis) |
| GVA (afferents for the trachea, heart, esophagus, stomach, intestines) | |
| GSA (external auditory meatus) | |
| SVE (intrinsic muscles of the pharynx, superior two thirds of the esophagus) | |
| GVE (efferents for the trachea, digestive tract, heart) | |
| Accessory (XI) | SVE (sternomastoid, trapezius) |
| GVE (soft palate, pharynx) | |
| Hypoglossal (XII) | GSE (tongue) |
GSA, general somatic afferent; GSE, general somatic efferent; GVA, general visceral afferent; GVE, general visceral efferent; SSA, special somatic afferent; SVA, special visceral afferent; SVE, special visceral efferent.
Neuronal Proliferation, Neuronal Migration, and Formation of Commissures (PODs 24 to 115)
The cellular architecture of the brain and spinal cord is based on a similar unifying theme—embryonic neuroblasts proliferate within a ventricular zone and their postmitotic progeny migrate centripetally through the neural tube to take up locations peripherally to form the mantle layer. Axonal processes from mantle layer neurons extend further to form the marginal zone immediately under the pia (Fig. 175-8). Neuronal proliferation begins on POD 24; neuroblasts are later replaced by glioblasts at the ventricular zone, which then give rise to glial cells (astrocytes and oligodendroglia). The remaining cells of the ventricular zone become ependymal cells.
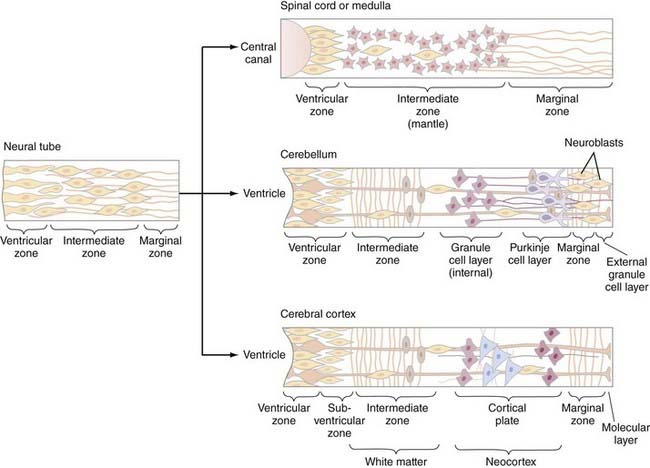
Spinal cord development follows this basic theme and gives rise to the central location of the spinal cord gray matter with surrounding white matter tracts. However, proliferation and migration at other levels of the neuraxis are altered depending on the specific location—two locations that deserve special discussion are the cerebellum and cerebral cortex. In the cerebellum, the postmitotic neuroblasts migrate centripetally to form two zones: the deep cerebellar nuclear layer (which will form the deep cerebellar nuclei) and the Purkinje cell layer (to which the Purkinje, basket, stellate, and Golgi cells migrate) (Fig. 175-8, middle). The granule cells, in contrast, originate in the rhombic lip, migrate superficially around the surface of the cerebellum, and exist superficial to the Purkinje cell layer as the external granule layer. These cells are still mitotically active and produce numerous granule cell progeny, even as long as 2 years after birth.36 Postmitotic granule cells migrate back toward the ventricular zone, through the Purkinje cell layer, to produce the internal granule layer. This second migration is controlled by specialized glial cells, called Bergman glia, the processes of which serve as guideposts for the migration of granule cells through the Purkinje cell layer.
Neurogenesis and neuronal migration are also modified in the cerebral cortex (Fig. 175-8, bottom). The first neuroblasts send axon that form the marginal zone immediately subjacent to the pia. Postmitotic neurons then migrate away from the ventricular zone to form the intermediate zone between the ventricular and marginal zones. Cells from both the ventricular and intermediate zones migrate further centripetally to form the cortical mantle zone—cells that arrive at the mantle zone first form the deeper cortical layers, whereas those that arrive later form more superficial cortical layers that migrate through the previously established layers. As ventricular zone proliferation ceases, proliferation begins in the subventricular zone, located between the ventricular and intermediate zones. Cells from the subventricular zone migrate through the intermediate zone to form a subplate just beneath the cortical mantle zone. Cells from the subplate zone migrate through the mantle zone to form the most superficial cortical layers. The cortex is therefore derived from both the cortical mantle zone and the subplate region and is generated in an inside-out fashion, with superficial layers forming later than deeper layers. The intermediate layer subsequently forms the white matter of the cerebrum. Neuronal migration in the cerebrum also appears to involve specialized radial glial fibers that span from the ventricular to the marginal zones and along which neurons migrate to their final destinations. Neuronal migration involves the interplay of many different molecular species, including reelin, astrotactin, integrins, neurogulins, and others.36
The majority of the commissural fibers of the hemispheres develop from the commissural zone of the lamina terminalis. The lamina terminalis is the site of the cranial neuropore and is located immediately dorsal to the optic chiasm anatomically. Commissural fibers of the anterior commissure, corpus callosum, and hippocampal commissure derive from the dorsal end of the lamina terminalis (the posterior and habenular commissures are formed from the epithalamus of the diencephalon). The earliest commissural fibers, those of the anterior commissure, arise at 54 days, followed by those of the hippocampal commissure during the 11th week and those of the corpus callosum between 84 and 115 days.37,38 The corpus callosum generally develops in an anterior-to-posterior fashion (genu first, followed by the body and then the splenium); because of the later growth of the frontal lobes, the rostrum is the last portion to form. As a result, incomplete abnormalities of the corpus callosum involve the later developing, more posterior portions (and rostrum) with sparing of the earlier developing anterior portions and lead to selective enlargement of the atrium and occipital horns of the ventricles (colpocephaly). An exception to this rule is holoprosencephaly, in which there is selective agenesis of the posterior portions with sparing of the anterior portions.38,39
Abnormal Early Neural Development
Failure of Neural Tube Closure—Anencephaly and Myelomeningocele
Anencephaly and myelomeningocele are neural tube defects (NTDs), or localized failure of primary neurulation, that can arise through one of two mechanisms. The “nonclosure theory” proposes that NTDs represent primary failure of neural tube closure.40 The “overdistention theory,” introduced in 1769 by Morgagni41 and popularized by Gardner,42–45 proposes that NTDs arise through overdistention and rupture of a previously closed neural tube. The nonclosure theory is widely accepted for most cases. However, overdistention may contribute to some experimental NTD models, particularly those caused by vitamin A46 and the T-curtailed mouse mutant47; whether this causes human malformations is unknown. The result is an open, unneurulated segment of neural tube, the nature and severity of which are determined by the location and length of the unneurulated segment. Failure of caudal neurulation produces a myelomeningocele, whereas more cranial failure results in anencephaly.
Because neural tube closure requires the complex interaction of multiple cellular processes, it is not surprising that NTDs may result from a number of embryonic insults. NTDs have been produced experimentally with a number of teratogens (reviewed by Campbell and colleagues2), genetic mutations (reviewed by Copp and associates47,48), and experimental manipulations (reviewed by Schoenwolf and Smith49). Although these processes all suggest a number of potential mechanisms whereby NTDs might arise, the cause of human malformations remains unknown. NTDs are most likely etiologically heterogeneous1,2,47,48 and represent the end result of a variety of embryonic disorders.
Genetic models of NTDs provide a means of identifying candidate gene or genes involved in neural tube closure47,48; three such examples illustrate the variety of ways in which genetic mutations can result in an NTD. The splotch (Sp) mouse mutant is identified by a peculiar patch of white fur and exhibits both exencephaly and myelomeningocele. The genetic locus for the Sp mutation is within the Pax-3 gene locus. Although the Pax-3 gene product appears to be involved in apposition and fusion of the neural folds, neither the mechanisms underlying the normal Pax-3 gene product nor the mechanisms whereby the Sp mutation alters Pax-3 function are known with certainty.
A second mouse mutant, curly tail (ct), exhibits posterior NTDs and tail deformities associated with delay in closure of the caudal neuropore.48 However, the delay is not the result of faulty neuroepithelial development inasmuch as isolated neuroepithelium from ct mutants undergoes normal neurulation.50 Rather, a delay in cell proliferation in the underlying notochord and hindgut endoderm causes an abnormal ventral body axis curvature and impedes posterior neuropore closure.47,48 If the ventral curvature is corrected by splinting the caudal ct embryo with an eyelash51 or by retarding neuroepithelial proliferation with retinoic acid,52 the posterior neuropore closes normally and the incidence of NTDs is reduced.
A third mouse mutant, T-curtailed (T[c]) produces a lumbosacral myelomeningocele with dorsoventral forking of the caudal neural tube. However, rather than delayed or failed neural tube closure, the T(c) mutant causes rupture of the roof plate and reopening of a previously closed neural tube.47
A large number of environmental causes of NTDs have been identified and their underlying mechanisms studied. Recent attention has focused on the role of folate in the embryogenesis of NTDs. Maternal administration of folate antagonists such as aminopterin has long been known to produce NTDs.53 Periconceptional administration of supplemental folate in randomized, placebo-controlled studies reduced both the recurrence rate of NTDs in women with a previously affected pregnancy and the incidence in women who had never had an affected pregnancy.54,55 However, studies of maternal serum and red blood cell folate levels in mothers of infants with myelomeningocele have produced inconsistent results (reviewed by Wald56 and Seller57), and folate deficiency does not cause NTDs in mouse or rat embryos (reviewed by Fleming and Copp58). These observations suggest that NTDs are not usually the result of an absolute folate deficiency.
More recent attention has focused on the possibility that NTDs are caused by abnormalities involving metabolic pathways (in either the mother or fetus) that require folate59; such abnormalities predispose an individual to NTDs and might therefore be overcome by folate supplementation. Folate and its metabolites tetrahydrofolate and 5-methyltetrahydrofolate are important in a variety of mammalian metabolic reactions, including purine and pyrimidine (and therefore DNA) synthesis, and in the transfer of methyl groups during the metabolism of methionine and homocysteine. In particular, the role of folate in methionine and homocysteine metabolism has generated considerable interest.60–68
One hypothesis is that maternal or fetal mutations in either methionine synthase or 5,10-methylenetetrahydrofolate reductase may slow this “methylation cycle” down, drive the conversion of methionine to homocysteine, and lead to methionine deficiency or homocysteine excess, or both. Ingestion of higher doses of folate may overcome this relative deficiency by restoring more normal homocysteine and methionine levels.59,67,69 A number of observations support this view. For example, elevated maternal serum68 and amnionic fluid67 homocysteine levels have been identified in women carrying a fetus with an NTD. Disordered methionine metabolism has been demonstrated in nonpregnant women who had previously given birth to a child with an NTD.67,69 Finally, mutations of the 5,10-methylenetetrahydrofolate reductase gene have been demonstrated in 18% of individuals with NTDs, in 13% of parents of children with NTDs, and in just 6% in controls.65,70 Abnormalities of cystathionine synthase have also been identified.67
A number of teratogens are known to act through a variety of cellular mechanisms to cause NTDs in animals and humans (see Copp and coworkers47 for review). One important teratogen, valproic acid (VPA), produces NTDs in both animal models and humans, probably by inhibiting neural fold fusion.47 Although the exact mechanism is not known, VPA appears to disrupt folate metabolic pathways,71 perhaps by interfering with the conversion of tetrahydrofolate to 5-formyltetrahydrofolate.67 Maternal folate administration has reduced the incidence of VPA-associated NTDs in some, but not all studies,71 and mouse strains that are susceptible to valproate-induced NTDs have significantly lower levels of 5,10-methylenetetrahydrofolate after valproate administration than do resistant strains.72 In addition, a number of developmental regulatory genes (such as transcription factors and cell cycle checkpoint genes) may be altered in these susceptible strains.73 One hypothesis is that VPA may act by changing folate-dependent methylation of regulatory proteins such as transcription factors.
Incomplete Dysjunction—Dermal Sinus Tracts, Dermoid and Epidermoid Tumors, Meningocele Manqué and Meningoceles
The most widely accepted theory of DST embryogenesis proposes that they arise through faulty separation (dysjunction) of neuroectoderm from the overlying cutaneous ectoderm with a tract of cutaneous ectoderm left sequestered between the skin and neural tube.74 The histology of this tract may represent a number of cutaneous ectodermal abnormalities, including epithelial-lined sinuses (DSTs), epidermoid tumors (containing only ectodermal tissue as pseudostratified squamous epithelium), and dermoid tumors (containing both ectodermal and mesodermal tissues such as sebaceous glands and hair follicles) located anywhere between the skin and neural tube. Hoving and colleagues have provided evidence that cranial dysjunction at the anterior neuropore involves apoptosis; misexpression or failure of apoptosis may underlie NTDs, particularly those involving the nasofrontal region.75 Another way in which abnormal dysjunction could potentially arise might be through misexpression of cell adhesion molecules or other molecular markers located on cutaneous ectoderm or neuroectoderm, or both.
Dermal sinuses may involve any level of the neuraxis, although there is a predilection for neuropores in both the lumbosacral and frontonasal regions. Frontonasal dermal sinuses arise through incomplete dysjunction at the site of the anterior neuropore and commonly involve the frontobasal skull. The embryogenesis and surgical anatomy of frontonasal dermal sinuses are predicted by the normal development of the anterior cranial base (Fig. 175-9). The prosencephalic primordium, olfactory epithelium, adenohypophysis, and facial cutaneous ectoderm all share a common embryonic origin (at least in avian embryos) from the region of the anterior neuropore.76,77 The frontal bones develop later from bilaterally paired mesenchymal anlagen and are separated by the metopic suture. Simultaneously, nasal bones develop along the nasal spine and are separated from the frontal bones by a fibrous capsule, the fonticulus nasofrontalis. The nasal spine is separated from the deeper nasal cartilaginous capsule by the prenasal space. During normal embryogenesis, a tongue of dura extends ventrally from the inferior aspect of the anterior cranial fossa and is interposed anteriorly between the frontal and nasal bones at the fonticulus nasofrontalis and inferiorly between the prenasal cartilage and the nasal bones within the prenasal space. During normal development, this dural reflection becomes surrounded by ossifying bone and regresses; remnants of the tract persist as the foramen cecum along the floor of the anterior cranial fossa, between the insertion of the falx cerebri anteriorly and the crista galli posteriorly.78
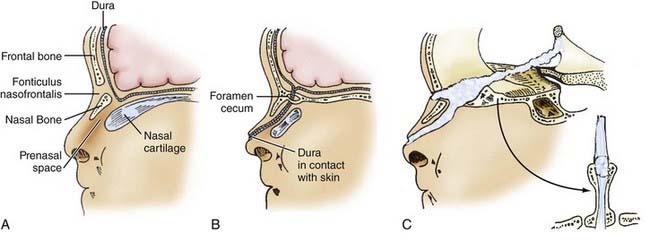
Nasal DSTs typically arise along the nasal dorsum or the glabella; 90% end extracranially. When they extend intracranially, they pass through the foramen cecum and typically end within the falx; on rare occasion the tract extends entirely through the falx and travels within the subarachnoid space to end at the lamina terminalis (the site of the anterior neuropore) as predicted embryologically (Fig. 175-9). Associated dermoid tumors may occur anywhere along the DST and involve extracranial or intracranial sites.78–83 Finally, sequestered neuroepithelium within the prenasal space may develop independently into a mass of neuroglial tissue, the nasal cerebral heterotopia or “nasal glioma.”84–86
Finally, dermal sinuses are also common in the occipital region,87,88 where they arise from either the roof of the fourth ventricle, the cerebellar vermis, or the subdural space posterior to the cerebellum. The tract may extend either beneath the tentorium into the posterior fossa or above the tentorium toward the occipital lobe or branch to involve both compartments. Although some authors have suggested that occipital dermal sinuses arise from the region of the anterior neuropore,87 this is inconsistent with the association of occipital dermal sinuses with structures derived from the dorsal aspect of the rhombencephalon (cerebellum and fourth ventricle) well distant from the lamina terminalis. Occipital dermal sinuses more likely arise from the rhombencephalic neural tube during neural tube closure. Why dermal sinuses have a predilection for this region of the neuraxis is unclear. The rhombencephalon in humans is among the first regions of the neural tube to undergo neural fold fusion and may therefore be particularly vulnerable to disorders of neurulation; in addition, the presence of the pontine flexure may lend additional physical stress to the neural tube at this site. Finally, the frequent occurrence of dermal sinuses may simply reflect the relatively large size of this region of the neural tube at the time of neurulation.
Disorders of Gastrulation—Combined Spina Bifida, Split Cord Malformations, and Neurenteric Cysts
A number of seemingly unrelated disorders are thought to share a common embryogenesis, including combined (anterior and posterior) spina bifida (also called the split notochord syndrome); split cord malformations (SCMs); neurenteric cysts; intestinal malrotations, duplications, and fistulas; some cases of the Klippel-Feil malformation sequence; some cases of sacral agenesis and Currarino’s triad; and a number of other unclassified complex congenital craniospinal malformations, all of which exhibit disorders of all three primary germ layers. Variably described as the “split notochord syndrome,”89 “endodermal-ectodermal adhesion syndrome,”90 “accessory neurenteric canal syndrome,”91 or “disordered midline integration during gastrulation” syndrome,1 the embryogenesis of these disorders is fully discussed by Dias and Walker.1
All these malformations share some type of SCM. All have in common a splitting of the neuroectoderm into two parts over a portion of its length. The two “hemicords” thus formed are typically separated by either a bony or cartilaginous midline extradural spur (type I SCM) or a thinner, fibrous intradural midline band of tissue (SCM type II).92 In both cases, atretic and nonfunctional midline dorsal (and in rare instances ventral) rootlets arise from one or both “hemicords” and end on the midline tissue.93–97 The presence of both lateral and median sets of nerve roots arising from each “hemicord” supports the presence of at least a partial spinal cord duplication in both malformations. Finally, pathologic specimens of both malformations demonstrate neither absolute splitting nor complete duplication of the cord but rather incomplete duplications with relatively well preserved lateral halves and dystrophic medial halves.93,97–100
SCMs are seen in association with a variety of anomalies, including combined (anterior and posterior) spina bifida; hemimyelomeningoceles; myelomeningoceles (occurring in up to a third of cases); cervical myelomeningoceles; neurenteric cysts; some examples of the Klippel-Feil anomaly, iniencephaly, and caudal agenesis; certain intestinal duplications and diverticula (reviewed by Dias and Walker1); and Currarino’s triad. All these complex congenital craniospinal malformations have in common both SCMs and anomalies of all three primary germ layers and therefore probably share a common embryonic origin.1,45 Four theories have attempted to explain the underlying embryopathy.
Beardmore and Wigglesworth101 proposed an adhesion between the epiblast and hypoblast that interferes with notochordal outgrowth. On encountering this “endodermal-ectodermal adhesion,” the notochord might split around the adhesion and produce two “heminotochords,” each inducing a neural “hemicord.” Associated remnants of the adhesion could give rise to endodermal remnants located anywhere between the gut and cutaneous ectoderm. This works only if the notochord extends cranially from Hensen’s node; a modification of this theory90 to account for the possibility of notochordal elongation as Hensen’s node regresses postulates an adhesion within the PS caudal to Hensen’s node around which the notochord might be split during node regression.
Bremer noted the similarity between patients with combined anterior and posterior malformations, in which both the involved vertebrae and spinal cord are split midsagittally to form two laterally displaced “hemivertebral” columns and an SCM surrounding a central cleft, and the primitive neurenteric canal of embryogenesis, both seeming to bisect the embryo and connect the amnionic and yolk sacs.91 This led to the proposal that a more cranial “accessory neurenteric canal” might split the notochord and neuroepithelium and result in a split cord and related malformations.
McLetchie and associates102 and Saunders103 proposed a primary duplication of the notochord that results in a secondary endodermal-ectodermal interaction between the duplicated notochords. Dodds suggested that during normal embryogenesis, bilaterally paired prospective notochordal cell anlagen might be integrated into a single midline structure during regression of the PS.104 Feller and Sternberg proposed that abnormal rests of undifferentiated cells in Hensen’s node might interfere with proper midline integration and result in paired notochords; subsequent differentiation of these cell rests could give rise to a variety of midline anomalies composed of tissues derived from any of the three primary germ cell layers.105
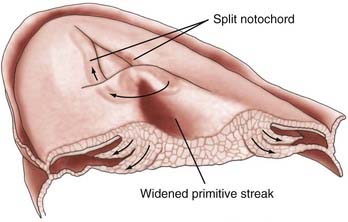
Finally, Dias and Walker proposed a disorder involving midline axial integration during gastrulation (Fig. 175-10).1 During normal development, paired notochordal anlagen within Hensen’s node are integrated to form a single notochordal process; bilaterally paired prospective neuroepithelial cells flanking both sides of Hensen’s node and the PS are similarly integrated to form a single neural plate. According to this theory, complex congenital craniospinal malformations arise when both the paired notochordal anlagen and adjacent prospective neuroepithelial regions remain separate (perhaps because of an abnormally wide Hensen node or PS) during gastrulation. The intervening space between the paired “hemicords” is composed of pluripotent PS cells and could give rise to a variety of tissue types from any of the three primary germ layers (all having been described in association with SCMs).106–108
Although most SCMs involve the thoracic or lumbar spinal cord, rare cranial lesions have been reported.109,110 Because the cranial notochord reaches only to the clivus (adjacent to the midbrain), one would expect cranial SCMs to involve only the rhombencephalon (medulla and pons) or mesencephalon; SCMs involving the diencephalon or telencephalon have not been described, nor would they be expected based on this embryologic mechanism.
Intracranial Lipomas
Lipomas may arise at any level of the neuraxis, although spinal lipomas are the most common and most frequently involve the lumbosacral spinal cord, conus medullaris, and filum terminale. Spinal lipomas (particularly those that develop cranial to S2) reputedly arise from premature dysjunction during primary neurulation.111,112 The cutaneous ectoderm, prematurely separated from the neuroepithelium before neural fold fusion, allows surrounding mesenchymal cells access to the central canal of the developing neural tube. These mesenchymal cells, which are normally induced to form meninx when exposed to the outer surface of the neural tube, are instead induced to form fat on exposure to the central canal of the spinal cord.112 More caudal spinal lipomas (those arising caudal to S2) probably involve some (not yet defined) abnormality of secondary neurulation.
Intracranial lipomas, in contrast, most likely represent postneurulation abnormalities arising from an abnormality of the meninx primitiva (the anlagen of the pia and arachnoid derived from the cranial neural crest).113 A unifying theory by Truwit and Barkovich (based on previous work by Verga, Krainer, and Osaka as referenced in their study) proposes that abnormal or persistent rests of neural crest cells that form the meninx primitiva form intracranial lipomas at predictable sites—the cistern of the lamina terminalis, pericallosal cistern, velum interpositum, and the suprasellar, quadrigeminal, and cerebellopontine angle cisterns. According to this theory, the meninx primitiva forms initially in what is to become the intracranial cisterns. Between approximately PODs 44 and 57 the cisterns of the subarachnoid space develop coincident with regression of the meninx; the cisterns ventral to the brainstem develop earliest and extend lateral and dorsal to the brainstem (41 days), to the telencephalic vesicles and dorsal midbrain (44 days), and finally to the cistern of the lamina terminalis (56 to 57 days). Maldevelopment of the meninx would therefore least frequently affect those cisterns that develop earlier and most commonly affect those that develop later. Both the preferred anatomic locations, the relationship to (and lipomatous extension into) the choroidal fissure, the passage of cranial nerves through the lipomas, and the maldevelopment of surrounding structures such as the corpus callosum are all predicted by this unifying theory.113
Postneurulation Disorders—Encephaloceles
The embryology of encephaloceles was reviewed by Chapman and colleagues.114 Although thought by von Recklinghausen to arise from failure of closure of the cranial neuropore,115 these malformations are now thought to represent a postneurulation disorder of mesenchyme through which neural tissue herniates.114,116–118 Unlike anencephaly (failure of cranial neural tube closure), in which the involved neural tissue is exposed and completely disorganized, encephaloceles are skin-covered malformations containing well-developed neural (unilateral or bilateral basifrontal or occipital cortex, cerebellum, or brainstem) and mesenchymal (choroid plexus, pia/arachnoid) tissues that have undergone considerable histogenesis.119,120 Marin-Padilla has suggested that mesodermal insufficiency may result in growth impairment of the chondrocranium and a delay in closure of the membranous neurocranium.116,118 The subsequent explosive growth of the telencephalon may eventually surpass the accommodative capacity of the neurocranium, and neural tissues may then herniate through the mesenchyme to end extracranially. Microcephaly is a common concomitant of encephaloceles; however, it is uncertain whether this is a primary or secondary event. Alternatively, all that may be necessary is a focal area of mesenchymal insufficiency or weakness to allow the rapidly growing telencephalon to herniate; occipital encephaloceles may be produced in chickens simply by incising the occipital mesenchyme overlying the cranial neural tube after neurulation is complete.121 Chapman and coworkers have suggested that associated hydrocephalus may contribute to the genesis of encephaloceles.114 However, encephaloceles may occur in the absence of hydrocephalus; moreover, associated hydrocephalus may be merely a consequence rather than the cause of the encephalocele.
Although there is greater agreement on the genesis of occipital and parietal encephaloceles as postneurulation disorders, the embryology of nasofrontal encephaloceles and whether they share a common embryogenesis with frontonasal DSTs and nasal gliomas are unresolved issues. Some have described all three as part of a continuum of anomalies sharing a common embryologic mechanism.78,85 Hoving and colleagues suggest that all three entities arise from abnormal dysjunction involving the cranial neuropore, perhaps through abnormal apoptosis.75,122 This theory is based on (1) the constant anatomic relationship between these encephaloceles and the foramen cecum and (2) a sustained connection between neuroectoderm and cutaneous ectoderm within the wall of encephaloceles. However, other evidence suggests that nasal encephaloceles do not share a common embryogenetic mechanism with either nasofrontal DSTs or nasal gliomas and arise through a postneurulation disorder. First, nasofrontal encephaloceles take origin from the frontobasal cortex, whereas nasofrontal DSTs and nasal gliomas with intracranial extension take origin from the lamina terminalis and commissural plate, the site of the anterior neuropore.82,123,124 Second, nasal gliomas contain only disorganized and poorly differentiated neuroglial tissue with almost no cytoarchitectural features,85 whereas frontal encephaloceles often contain well-differentiated basifrontal cortex,119,120 in some cases arising from only one hemisphere, thus suggesting a disorder that follows telencephalic cleavage (at least after POD 32). Finally, frontobasal encephaloceles are common in Southeast Asian populations120 and are associated with a number of genetic syndromes,125 whereas frontobasal dermal sinuses, dermoids, and nasal gliomas are not.
Disordered Telencephalic Cleavage—Holoprosencephaly
Formation and cleavage of the telencephalon into the two cerebral hemispheres are controlled by the underlying prochordal plate, a mesodermal region anterior to the notochordal process that expresses the gene product sonic hedgehog. Sonic hedgehog performs a number of important functions during development, including induction of the neural groove from the median hinge point cells, as well as directed axonal outgrowth from ventral horn motoneurons. Mouse mutants that misexpress sonic hedgehog resemble humans with holoprosencephaly, and many families with holoprosencephaly have mutations involving the sonic hedgehog gene (reviewed by Gilbert126), but the mechanism whereby misexpression results in holoprosencephaly is unknown. Although the molecular cause of holoprosencephaly is probably manifested during the preneurulation stage, holoprosencephaly ultimately represents a postneurulation disorder caused by improper telencephalic cleavage. The three forms of holoprosencephaly in descending order of severity are alobar, semilobar, and lobar. The alobar form is represented by a monoventricle with no apparent telencephalic cleavage; a dorsal interhemispheric cyst represents a dorsal outpouching of the third ventricle. Diencephalic involvement produces midline fusion of the pallidum and thalami with an absent third ventricle, as well as hypotelorism (remember that the neural retina is an outpouching of the diencephalon) or even cyclopia. The semilobar form is characterized by more advanced posterior hemispheric cleavage but abnormal anterior hemisphere cleavage and incomplete formation of the falx cerebri; the thalami are partially fused, the third ventricle is small, and the hypotelorism is less pronounced. The lobar form is characterized by incomplete formation of the anterior falx with unseparated frontal horns and absence of the septum pellucidum. Perhaps considered the mildest form of disordered telencephalic cleavage, septo-optic dysplasia is characterized by partial or complete absence of the septum pellucidum associated variably with optic nerve hypoplasia and abnormal pituitary-hypothalamic function.
Disorders of Commissural Connections—Callosal Agenesis
Abnormalities of corpus callosum development frequently accompany a variety of other disorders. As discussed previously, development of the corpus callosum proceeds in a rostral-to-caudal sequence, with the genu developing first, then the body, and subsequently the splenium. The rostrum, the last portion of the corpus callosum to develop, is the sole exception to this rostrocaudal sequence. Partial disorders of the corpus callosum therefore characteristically involve the more posterior callosum with sparing of the anterior portions. A significant exception to this rule is holoprosencephaly, in which the anterior corpus callosum is absent but the posterior portions are present.38
Predictably, agenesis of the corpus callosum is often accompanied by agenesis of both the anterior and hippocampal commissures because all three are derived from the commissural plate of the lamina terminalis. It is important to understand that commissural axons form but never cross the midline. Instead, these axons are redirected and run along the medial hemispheres as the bundles of Probst.38 Failure of corpus callosum fibers to cross the midline results in eversion of the cingulate gyrus and failure of formation of the cingulate sulcus.38
The prevailing theory explaining the origin of callosal agenesis is failure of apoptosis involving midline glial cells in the commissural plate. These glial cells serve as a passive bed for the passage of axons from the two hemispheres. Subependymal pioneering glial cells from the medial walls of the lateral ventricles migrate toward the commissural plate and serve as a “glial sling” below the interhemispheric fissure; crossing axons use this cellular “bridge” to cross the interhemispheric fissure. Mouse mutants in which the midline glial sling is disrupted exhibit callosal agenesis.127–129
Disorders of Neuronal Migration—Schizencephaly, Lissencephaly, Polymicrogyria, and Heterotopias
Disorders of neuronal migration occur between the 5th and 16th gestational week, during which neurons migrate from the ventricular zone to their final locations within the cerebrum. These disorders can take several forms. Schizencephaly represents a focal disorder of migration from the periventricular zone during the 5th to 7th embryonic week, perhaps the result of abnormalities of or prenatal injury to radial glial units. Unilateral and bilateral forms exist and are characterized by gray matter–lined clefts (either open or closed lipped) extending from the ventricular ependyma to the pial surface. The gray matter lining the cleft is often polymicrogyric. The origin of the cleft within the ventricle is marked by a small outpouching (or “nipple”). Other neuronal migrational disorders such as cortical heterotopias, lissencephaly/pachygyria, and septal agenesis often coexist (in 80% to 90%).38
Agyria refers to complete absence of sulcation, whereas lissencephaly, or pachygyria, refers to incomplete sulcation, usually with a few broad and flat gyri and shallow sulci. Lissencephaly is thought to represent abnormal migration of neurons during the 12th to 16th week.38 Neurons apparently begin migration but are unable to complete it.130 Posterior callosal agenesis and colpocephaly (enlargement of the atria and occipital horns) are common. Two types of lissencephaly have been described: type I largely consists of a four-layered cortex with a molecular layer, a disorganized outer cellular layer, a cell-sparse layer, and an inner cell layer. Histologically, the cortex resembles that of a 12-week-old fetal brain. Type II lissencephaly is characterized by a thickened, disorganized, and unlayered cortex. Lissencephaly has been described in association with chromosomal and genetic conditions; for example, lissencephaly type I is associated with Miller-Dieker syndrome (deletion of 17p13.3), whereas lissencephaly type II is associated with Walker-Warburg syndrome.131 Lissencephaly has been associated with mutations in six genes, although mutations in the LIS1 and DCS genes, both involved in regulation of microtubule and cytoplasmic dynein function, account for nearly 80%.130
Polymicrogyria is a condition characterized by too numerous and abnormally small gyri. Both focal and diffuse forms exist. The cortex appears to be thicker with closely packed sulcal folds. Most commonly, the cortex is four layered, although variations can occur, with different areas or even the same areas containing unlayered cortex or poorly laminated or parallel four-layered cortex coexisting with four-layered cortex.131 The distinguishing histologic feature of polymicrogyria is cortical laminar necrosis involving cortical layer V. Polymicrogyria appears to represent a postmigrational abnormality, probably ischemic in origin, that occurs later than the 20th gestational week because migration of neurons to the older, outer cortical layers has already taken place before that time.38 However, polymicrogyria is a heterogeneous disorder, and genetic forms have been described as well.130
Finally, localized areas of cortical neuronal migration may result in focal heterotopias in which neurons find themselves buried within white matter. A focal abnormality, either misguided or arrested neuronal migration, is the likely culprit and may involve disorders related to the generation or expression of glial precursors or the glial-neuronal interaction that ordinarily results in focused neuronal migration. Nodular and laminar forms are identified. Laminar forms are characterized by the formation of an intermediate, irregular (and often nodular) band of gray matter between the ventricular and cortical zones; in some cases a “double cortex” occurs.131 Focal nodular heterotopia is characterized by localized nodular heterotopia adjacent to the ventricular zone. Heterotopias are associated with a number of chromosomal and other congenital disorders, including Aicardi’s syndrome, Chiari II malformation (see later), aplasia cutis congenita, and polymicrogyria. Nodular heterotopias may be associated with a defect in filamin-1 located on the long arm of the X chromosome.130,132
Disordered Rhombic Lip Development—Dandy-Walker Malformation, Rhombencephalosynapsis, and Other Cerebellar Malformations
Dandy-Walker malformation is characterized by the triad of (1) dysgenesis or agenesis of the cerebellar vermis, (2) cystic enlargement of the fourth ventricle, and (3) an enlarged posterior fossa with upward displacement of the tentorium.133 Hydrocephalus is common but is not usually obvious at birth, instead developing over the first 3 months of age.133 Histologically, the membrane of the fourth ventricular cyst is composed of ependymal, neuroglial, and pial layers, thus suggesting agenesis of the cerebellar vermis between the ependymal and pial layers. The fourth ventricular outlets are usually patent, and communication between the ventricular and subarachnoid spaces occurs in more than 80% of cases.134 Although the vermis is most severely affected, some degree of hypoplasia involving the cerebellar hemispheres occurs in most cases.135 Associated supratentorial abnormalities are present in 68% of cases and include callosal dysgenesis or agenesis (in up to 50%),135 schizencephaly, lissencephaly, and cortical heterotopias.134 The Dandy-Walker variant describes a similar, although less severe form with a hypoplastic vermis, a less distended fourth ventricle, a less diminutive posterior fossa, and more normal tentorial position. Dandy-Walker malformation appears to arise as a result of an abnormality involving the rhombic lip. Recent studies have identified chromosome 3 deletions in the region of the zinc finger in cerebellum family of transcription factors. Mice mutations involving homologous regions similarly have a Dandy-Walker phenotype.136
Rhombencephalosynapsis is a rare example of focal cerebellar dysplasia.135,137 Its cardinal features include absence of the midline vermis with fusion of the cerebellar hemispheres and, frequently, dentate nuclei.135 Abnormalities also involve other midline structures, such as fusion of the colliculi and absence of the septum pellucidum.135 Its embryogenesis is unknown; although “excessive midline fusion” of the cerebellar primordia has been proposed,138 this fails to account for abnormalities involving other midline structures. The condition is sporadic and not genetically inherited.137
Acquired Central Nervous System Anomalies—Chiari II Malformation
The Chiari II malformation, associated almost universally with a myelomeningocele, is an example of a secondarily acquired malformation that to a variable extent encompasses anomalies of virtually the entire neuraxis. Most prominent among these anomalies are caudal displacement of the cerebellar vermis and tonsils into the cervical canal; elongation, kinking, and caudal displacement of the lower brainstem below the foramen magnum; and upward displacement of the superior cerebellum through a dysplastic, low-lying tentorial incisura. A small posterior fossa and lückenschädel of the skull, as well as many associated telencephalic and diencephalic anomalies (Table 175-4), suggest a pancerebral disorder involving much of the cranial neuraxis and chondrocranium.139
TABLE 175-4 Chiari-Associated Central Nervous System Malformations
| DISORDERS OF THE SKULL |
Theories regarding the embryology of Chiari malformations can generally be grouped into four types.139 The first group, regarded as the dysgenesis/developmental arrest theories, presume the Chiari malformation to be the result of primary dysgenesis of the neuraxis140; others suggest that failure of the pontine flexure might lead to herniation of the hindbrain contents.141,142 Neither of these theories can account for the frequent associated cerebral malformations.
The second group, referred to as the hydrocephalus/hydrodynamic theories, relies on the presence of “fetal hydrocephalus,” which because of a presumed imbalance (either static or dynamic) between the supratentorial and infratentorial compartments, displaces the posterior fossa contents caudally. However, these theories ignore the small cranial volumes and absence of hydrocephalus in all early human fetuses with dysraphism and Chiari malformations.143 Moreover, they fail to explain the small size of the posterior fossa, upward herniation of the superior cerebellar vermis, and associated cerebral anomalies.
The third group, referred to as the traction theories, suggests that traction on the caudal spinal cord by a myelomeningocele may pull the hindbrain caudally.144,145 However, experimental evidence suggests that the forces generated by spinal cord traction are dissipated within four spinal segments.146 Moreover, these theories do not explain the upwardly herniated superior cerebellar vermis, the medullary kink and vermian peg, or associated cerebral anomalies.
The fourth group, referred to as the small posterior fossa/overgrowth theories, suggests that the Chiari malformation is caused by a primary disorder of the paraxial mesoderm that results in a small posterior fossa that cannot adequately accommodate the burgeoning cerebellum and brainstem.147 Padget and Lindenberg suggested that leakage of cerebrospinal fluid (CSF) from an open neural placode might result in an acquired microcephaly with a small posterior fossa that leads to premature fusion of the cerebellar primordia and, with subsequent cerebellar growth, a Chiari malformation.148,149
McLone and Knepper proposed a unifying theory of embryogenesis for Chiari malformations that incorporates elements of each of the preceding theories.139,143 According to this model, an open neural placode allows CSF to escape from the central canal of the caudal neural tube. In addition, spinal occlusion (which in normal animals and humans precedes and is responsible for rapid brain enlargement) is incomplete in animal models of dysraphism and allows further leakage of CSF from the ventricles through the central canal. Finally, CSF leakage continues from the still open placode after spinal occlusion ends at stage 14 (POD 32). This persistent venting of CSF interferes with proper ventricular enlargement and eventually results in multiple central nervous system anomalies. For example, incomplete dilation of the telencephalic ventricles results in disorganized migration of neurons from the ventricular zone, thereby producing cortical heterotopias, gyral anomalies (stenogyria), and callosal dysgenesis. Inadequate distention of the third ventricle results in prolonged contact between the two thalami and an enlarged massa intermedia. Finally, inadequate enlargement of the rhombencephalic ventricle may similarly influence brainstem development and produce abnormalities of cranial nerve nuclei and their afferent and efferent connections.139
Most importantly, impaired ventricular enlargement affects development of the chondrocranium, especially the posterior fossa. Growth and development of the chondrocranium are normally dependent on cues provided by expansion of the underlying neural mass (the developing brain and ventricular system) during early embryogenesis.150,151 Incomplete distention of the rhombencephalic ventricle leaves the posterior fossa chondrocranium without an adequate inductive force; the posterior fossa is therefore smaller than normal, and the tentorium is low set and deficient.139,143 This small posterior fossa is incapable of accommodating the later growth of the cerebellum, and as a consequence the posterior fossa contents are displaced both cranially through the tentorial incisura and caudally through the foramen magnum. Impaction of neural tissues at these levels impairs CSF flow through the foramina of Luschka and Magendie, as well as through the subarachnoid space at the foramen magnum and tentorial incisura, and results in hydrocephalus.
Finally, although the secondary neuronal migration and other malformations are fixed once they occur, the hindbrain abnormalities appear to be anatomically reversible if fetal surgery is peformed,152 thus confirming that this is a prenatally acquired anomaly rather than a true developmental malformation.
Barkovich AJ, Lyon G, Evrard P. Formation, maturation, and disorders of white matter. AJNR Am J Neuroradiol. 1992;13:447-461.
Campbell LR, Dayton DH, Sohal GS. Neural tube defects: a review of human and animal studies on the etiology of neural tube defects. Teratology. 1986;34:171-187.
Chapman PH, Swearingen B, Caviness VS. Subtorcular occipital encephaloceles. Anatomical considerations relevant to operative management. J Neurosurg. 1989;71:375-381.
Copp AJ, Brook FA, Estibeiro P, et al. The embryonic development of mammalian neural tube defects. Prog Neurobiol. 1990;35:363-403.
Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229-253.
Golden JA, Chernoff GF. Multiple sites of anterior neural tube closure in humans: evidence from anterior neural tube defects (anencephaly). Pediatrics. 1995;95:506-510.
Guerrini R, Dobyns WB, Brakovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences, and treatment options. Trends Neurosci. 2007;31:154-162.
Hoving EW, Vermeij-Keers C. Frontoethmoidal encephaloceles: a study of their pathogenesis. Pediatr Neurosurg. 1997;27:246-256.
McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosurg. 1989;15:1-12.
Millen KJ, Gleeson JG. Cerebellar development and disease. Curr Opin Neurobiol. 2008;18:12-19.
Müller F, O’Rahilly R. The first appearance of the future cerebral hemispheres in the human embryo at stage 14. Anat Embryol. 1988;177:495-511.
Müller F, O’Rahilly R. The development of the human brain, the closure of the caudal neuropore, and the beginning of secondary neurulation at stage 12. Anat Embryol. 1987;176:413-430.
Müller F, O’Rahilly R. The development of the human brain and the closure of the rostral neuropore at stage 11. Anat Embryol. 1986;175:205-222.
Müller F, O’Rahilly R. The first appearance of the neural tube and optic primordium in the human embryo at stage 10. Anat Embryol. 1985;172:157-169.
Müller F, O’Rahilly R. The first appearance of the major subdivisions of the human brain at stage 9. Anat Embryol. 1983;168:419-432.
Naidich TP, McLone DG, Mutluer S. A new understanding of dorsal dysraphism with lipoma (lipomyeloschisis): radiologic evaluation and surgical correction. AJR Am J Roentgenol. 1983;140:1065-1078.
Norman MG, McGillivray BC, Kalousek DK, et al, editors. Congenital Malformations of the Brain: Pathological, Embryological, Clinical, Radiological, and Genetic Aspects. New York: Oxford University Press, 1995.
O’Rahilly R, Müller F. Developmental Stages in Human Embryos. Washington, DC: Carnegie Institution of Washington; 1987.
O’Rahilly R, Müller F. The first appearance of the human nervous system at stage 8. Anat Embryol. 1981;163:1-13.
Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I. A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23:1074-1087.
Sarnat HB, editor. Cerebral Dysgenesis: Embryology and Clinical Expression. New York: Oxford University Press, 1992.
Schoenwolf GC, Smith JL. Mechanisms of neurulation: traditional viewpoint and recent advances. Development. 1990;109:243-270.
Suwanwela C, Suwanwela N. A morphological classification of sincipital encephalomeningoceles. J Neurosurg. 1972;36:201.
Van Allen MI, Kalousek DK, Chernoff GF, et al. Evidence for multi-site closure of the neural tube in humans. Am J Med Genet. 1993;47:723-743.
1 Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229-253.
2 Campbell LR, Dayton DH, Sohal GS. Neural tube defects: a review of human and animal studies on the etiology of neural tube defects. Teratology. 1986;34:171-187.
3 O’Rahilly R, Müller F. Developmental Stages in Human Embryos. Washington, DC: Carnegie Institution of Washington; 1987.
4 Dias MS, McLone DG, Partington M. Normal and abnormal embryology of the spinal cord and spine. In: Winn HR, editor. Youmans Neurological Surgery, Vol 4. Philadelphia: WB Saunders; 2004:4239-4288.
5 Dias MS, McLone DG. Normal and abnormal early development of the nervous system. In: McLone DG, editor. Pediatric Neurosurgery: Surgery of the Developing Nervous System. Philadelphia: WB Saunders; 2001:31-71.
6 Vakaet L. Some new data concerning the formation of the definitive endoblast in the chick embryo. J Embryol Exp Morphol. 1962;10:38-57.
7 Modak SP. Experimental analysis of the origin of the embryonic endoblast in birds. Rev Suisse Zool. 1966;73:877-908.
8 Fontaine J, Le Douarin NM. Analysis of endoderm formation in the avian blastoderm by the use of quail-chick chimaeras. The problem of the neurectodermal origin of the cells of the APUD system. J Embryol Exp Morphol. 1977;41:209-222.
9 Rosenquist GC. A radioautographic study of labeled grafts in the chick blastoderm. Development from primitive streak stages to stage 12. Contrib Embryol. 1966;38(262):73-110.
10 Nicolet G. Analyse autoradiographique de la localisation des différentes ébauches présomptives dans la ligne primitive de l’embryon de Poulet. J Embryol Exp Morphol. 1970;23:70-108.
11 Nicolet G. Avian gastrulation. Adv Morphol. 1971;9:231-262.
12 Spratt NT. Regression and shortening of the primitive streak in the explanted chick blastoderm. J Exp Zool. 1947;104:69-100.
13 Schoenwolf GC, Smith JL. Gastrulation and early mesodermal patterning in vertebrates. In: Tuan RS, Lo CW, editors. Methods in Molecular Biology. Developmental Biology Protocols, Vol 1. Totowa, NJ: Human Press; 1998.
14 Tam PPL, Behringer RR. Mouse gastrulation: the formation of a mammalian body plan. Mech Dev. 1997;68:3-25.
15 Schoenwolf GC, Sheard P. Fate mapping the avian epiblast with focal injections of a fluorescent-histochemical marker: ectodermal derivatives. J Exp Zool. 1990;255:323-339.
16 Hendrickx AG. Description of stages IX, X, and XI. In: Hendrickx AG, editor. Embryology of the Baboon. London: University of Chicago Press; 1971:69-85.
17 Shaw W. Observations on two specimens of early human ova. Br Med J. 1932;1:411-415.
18 Müller F, O’Rahilly R. The development of the human brain and the closure of the rostral neuropore at stage 11. Anat Embryol. 1986;175:205-222.
19 Müller F, O’Rahilly R. The first appearance of the neural tube and optic primordium in the human embryo at stage 10. Anat Embryol. 1985;172:157-169.
20 O’Rahilly R. Developmental stages in human embryos, including a survey of the Carnegie collection. Part A: embryos of the first three weeks (stages 1 to 9). Washington, DC: Carnegie Inst Wash Publ 631; 1973.
21 O’Rahilly R, Müller F. The first appearance of the human nervous system at stage 8. Anat Embryol. 1981;163:1-13.
22 Müller F, O’Rahilly R. The first appearance of the major subdivisions of the human brain at stage 9. Anat Embryol. 1983;168:419-432.
23 Golden JA, Chernoff GF. Multiple sites of anterior neural tube closure in humans: evidence from anterior neural tube defects (anencephaly). Pediatrics. 1995;95:506-510.
24 Golden JA, Chernoff GF. Intermittent pattern of neural tube closure in two strains of mice. Teratology. 1993;47:73-80.
25 Van Allen MI, Kalousek DK, Chernoff GF, et al. Evidence for multi-site closure of the neural tube in humans. Am J Med Genet. 1993;47:723-743.
26 Urioste M, Rosa A. Anencephaly and faciocranioschisis: evidence of complete failure of closure 3 of the neural tube in humans. Am J Med Genet. 1998;75:4-6.
27 Müller F, O’Rahilly R. The development of the human brain, the closure of the caudal neuropore, and the beginning of secondary neurulation at stage 12. Anat Embryol. 1987;176:413-430.
28 Streeter GL. Developmental horizons in human embryos. Description of age group XI, 13 to 20 somites, and age group XII, 21 to 29 somites. Contrib Embryol. 1942;30:211-245.
29 Nichols DH. Formation and distribution of neural crest mesenchyme to the first pharyngeal arch region of the mouse embryo. Am J Anat. 1986;176:221-231.
30 Tan SS, Morris-Kay G. The development and distribution of the cranial neural crest in the rat embryo. Cell Tissue Res. 1985;240:403-416.
31 Müller F, O’Rahilly R. The first appearance of the future cerebral hemispheres in the human embryo at stage 14. Anat Embryol. 1988;177:495-511.
32 Bronner-Fraser M. The neural crest: what can it tell us about cell migration and determination? Curr Top Dev Biol.. 1980;15 (Part 1):1-25.
33 Perris R, Bronner-Fraser M. Recent advances in defining the role of the extracellular matrix in neural crest development. Comments in Developmental Neurobiology, Vol 1. London: Gordon and Breach Science Publishers S.A.. 1989. 61-83
34 Erickson CA. Morphogenesis of the neural crest. In: Browder LW, editor. Developmental Biology, Vol 2. New York: Plenum Publishing Corp.; 1986:481-543.
35 Anderson DJ. The neural crest cell lineage problem: neuropoiesis? Neuron. 1989;3:1-12.
36 Sanes DH, Reh TA, Harris WA, editors. Development of the Nervous System, 2nd ed, Amsterdam: Elsevier, 2006.
37 Sarnat HB, editor. Cerebral Dysgenesis: Embryology and Clinical Expression. New York: Oxford University Press, 1992.
38 Barkovich AJ, Lyon G, Evrard P. Formation, maturation, and disorders of white matter. AJNR Am J Neuroradiol. 1992;13:447-461.
39 Barkovich AJ, Norman D. Anomalies of the corpus callosum: correlation with further anomalies of the brain. AJNR Am J Neuroradiol. 1988;9:493-501.
40 von Recklinghausen E. Untersuchungen über die Spina bifida. Arch Path Anat. 1886;105:243-373.
41 Morgagni JB. The Seats and Causes of Disease Investigated by Anatomy. London: A. Millar and T. Cadell; 1769.
42 Gardner WJ. Diastematomyelia and the Klippel-Feil syndrome. Relationship to hydrocephalus, syringomyelia, meningocele, meningomyelocele, and iniencephalus. Clev Clin Q. 1964;31:19-44.
43 Gardner WJ. Embryologic origin of spinal malformations. Acta Radiol Diagn. 1966;5:1013-1023.
44 Gardner WJ. Hypothesis: overdistention of the neural tube may cause anomalies of non-neural organs. Teratology. 1980;22:229-238.
45 Gardner WJ. The Dysraphic States from Syringomyelia to Anencephaly. Amsterdam: Excerpta Medica; 1973.
46 Caldarelli M, McLone DG, Collins JA, et al. Vitamin A induced neural tube defects in a mouse. Concepts Pediatr Neurosurg. 1985;6:161-171.
47 Copp AJ, Brook FA, Estibeiro P, et al. The embryonic development of mammalian neural tube defects. Prog Neurobiol. 1990;35:363-403.
48 Copp AJ. Genetic models of mammalian neural tube defects. In: Bock G, Marsh J, editors. Neural Tube Defects. CIBA Foundation Symposium, Number 181. Chichester, England: John Wiley & Sons; 1994:118-143.
49 Schoenwolf GC, Smith JL. Mechanisms of neurulation: traditional viewpoint and recent advances. Development. 1990;109:243-270.
50 van Straaten HWM, Hekking JWM, Consten C, et al. Intrinsic and extrinsic factors in the mechanisms of neurulation: effect of curvature of the body axis on closure of the posterior neuropore. Development. 1993;117:1163-1172.
51 Brooke FA, Shum ASW, van Straaten HWM, et al. Curvature of the caudal region is responsible for failure of neural tube closure in the curly tail (ct) mouse embryo. Development. 1991;113:671-678.
52 Seller MJ, Embury S, Polani PE, et al. Neural tube defects in curly-tail mice. II. Effect of maternal administration of vitamin A. Proc R Soc Lond Ser B Biol Sci. 1979;206:95-107.
53 Seller MJ. Maternal nutrition factors and neural tube defects in experimental animals. In: Dobbing J, editor. Prevention of Spina Bifida and Other Neural Tube Defects. New York: Academic Press; 1983:1-22.
54 Czeizel AE, Dudás I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;26:1832-1835.
55 MRC Vitamin Research Study. Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131-137.
56 Wald NJ. Folic acid and neural tube defects: the current evidence and implications for prevention. In: Bock G, Marsh J, editors. Neural Tube Defects. CIBA Foundation Symposium Number 181. Chichester, England: John Wiley & Sons; 1994:192-211.
57 Seller MJ. Vitamins, folic acid and the cause and prevention of neural tube defects. In: Bock GMJ, editor. Neural Tube Defects. CIBA Foundation Symposium Number 181. Chichester, England: John Wiley & Sons; 1994:161-179.
58 Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280:2107-2109.
59 Scott JM, Wier DG, Molloy A, et al. Folic acid metabolism and mechanisms of neural tube defects. In: Bock GMJ, editor. Neural Tube Defects. CIBA Foundation Symposium Number 181. Chichester, England: John Wiley & Sons; 1994:180-191.
60 Gordon N. Folate metabolism and neural tube defects. Brain Dev. 1995;17:307-311.
61 Buehler JW, Mulinare J. Preventing neural tube defects. Pediatr Ann. 1997;26:535-539.
62 Ubbink JB. Is an elevated circulating maternal homocysteine concentration a risk factor for neural tube defects? Nutr Rev. 1995;53:173-175.
63 Bower C. Folate and neural tube defects. Nutr Rev. 1995;53:S33-S38.
64 Rosenquist TH, Ratashak SA, Selhub J. Homocysteine induces congenital defects of the heart and neural tube: effect of folic acid. Proc Nat Acad Sci U S A. 1996;93:15227-15232.
65 Whitehead AS, Gallagher P, Mills JL, et al. A genetic defect in 5,10 methylenetetrahydrofolate reductase in neural tube defects. Q J Med. 1995;88:763-766.
66 Steegers-Theunissen RPM, Boers GHJ, Trijbels FJM, et al. Neural-tube defects and derangement of homocysteine metabolism. N Engl J Med. 1991;324:199-200.
67 Steegers-Theunissen RP. Folate metabolism and neural tube defects: a review. Eur J Obstet Gynecol Reprod Biol. 1995;61:39-48.
68 Minns RA. Folic acid and neural tube defects. Spinal Cord. 1996;34:460-465.
69 Steegers-Theunissen RPM, Boers GHJ, Trijbels JMF, et al. Maternal hyperhomocysteinemia: a risk factor for neural tube defects? Metabolism. 1994;43:1475-1480.
70 Goyette P, Sumner JS, Milos R, et al. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat Genet. 1994;7:195-200.
71 Nau H. Valproic acid–induced neural tube defects. In: Bock GMJ, editor. Neural Tube Defects. CIBA Foundation Symposium Number 181. Chichester, England: John Wiley & Sons; 1994:144-152.
72 Finnell RH, Wlodarczyk BC, Craig JC, et al. Strain-dependent alterations in the expression of folate pathway genes following teratogenic exposure to valproic acid in a mouse model. Am J Med Genet. 1997;70:303-311.
73 Wlodarczyk BC, Craig J, Bennett GD, et al. Valproic acid–induced changes in gene expression during neurulation in a mouse model. Teratology. 1996;54:284-297.
74 Walker AE, Bucy PC. Congenital dermal sinuses; a source of spinal meningeal infection and subdural abscesses. Brain. 1934;57:401-421.
75 Hoving EW, Vermeij-Keers C, Mommaas-Kienhuis AM, et al. Separation of neural and surface ectoderm after closure of the rostral neuropore. Anat Embryol. 1990;182:455-463.
76 Couly G, Le Douarin NM. The fate map of the cephalic neural primordium at the presomitic to the 3-somite stage in the avian embryo. Development. 1988;103(suppl):101-113.
77 Couly G, Le Douarin NM. Mapping of the early neural primordium in quail-chick chimeras II. The prosencephalic neural plate and neural folds: implications for the genesis of cephalic human congenital malformations. Dev Biol. 1987;120:198-214.
78 Sessions RB. Nasal dermal sinuses: new concepts and explanations. Laryngoscope. 1982;92(suppl 29):1-28.
79 Pensler JM, Bauer BS, Naidich TP. Craniofacial dermoids. Plastic Reconstr Surg. 1988;82:953-958.
80 Okuda Y, Shizuo O. Nasal dermal sinus and dermoid cyst with intrafalcial extension. Case report and review of literature. Child’s Nerv Syst. 1987;3:40-43.
81 McQuown SA, Smith JD, Gallo AE. Intracranial extension of nasal dermoids. Neurosurgery. 1983;12:531-535.
82 Wardinsky TD, Pagon RA, Kropp RJ, et al. Nasal dermoid sinus cysts: association with intracranial extension and multiple malformations. Cleft Palate Craniofac J. 1991;28:87-95.
83 Barkovich AJ, Vandermarck P, Edwards MSB, et al. Congenital nasal masses: CT and MR imaging features in 16 cases. AJNR Am J Neuroradiol. 1991;12:105-116.
84 Hirsh LF, Stool SE, Langfitt TW, et al. Nasal glioma. J Neurosurg. 1977;46:85-91.
85 Yeoh GPS, Bale PMB, de Silva M. Nasal cerebral heterotopia: the so-called nasal glioma or sequestered encephalocele and its variants. Pediatr Pathol. 1989;9:531-549.
86 Gorenstein A, Kern EB, Facer GW, et al. Nasal gliomas. Arch Otolaryngol. 1980;106:536-540.
87 Schijman E, Monges J, Cragnaz R. Congenital dermal sinuses, dermoid and epidermoid cysts of the posterior fossa. Childs Nerv Syst. 1986;2:83-89.
88 Cheek WR, Laurent JP. Dermal sinus tracts. Concepts Pediatr Neurosurg. 1985;6:63-75.
89 Bentley JFR, Smith JR. Developmental posterior enteric remnants and spinal malformations. The split notochord syndrome. Am J Dis Child. 1960;35:76-86.
90 Prop N, Frensdorf EL, van de Stadt FR. A postvertebral entodermal cyst associated with axial deformities: a case showing the “entodermal-ectodermal adhesion syndrome.”. Pediatrics. 1967;39:555-562.
91 Bremer JL. Dorsal intestinal fistula; accessory neurenteric canal; diastematomyelia. Arch Pathol. 1952;54:132-138.
92 Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I. A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
93 Herren RY, Edwards JE. Diplomyelia (duplication of the spinal cord). Arch Pathol. 1940;30:1203-1214.
94 James CCM, Lassman JP. Diastematomyelia. A critical survey of 24 cases submitted to laminectomy. Arch Dis Child. 1964;39:125-130.
95 Pang D. Tethered Cord Syndrome. Neurosurgery: State of the Art Reviews, Vol 1. Philadelphia: Hanley & Belfus. 1986. 45-79
96 Pang D. Split cord malformation: Part II. The clinical syndrome. Neurosurgery. 1992;31:481-500.
97 Ross GW, Swanson SA, Perentes E, et al. Ectopic midline spinal ganglion in diastematomyelia: a study of its connections. J Neurol Neurosurg Psychiatry. 1988;51:1231-1234.
98 Cohen J, Sledge CB. Diastematomyelia. An embryological interpretation with report of a case. Am J Dis Child. 1960;100:127-133.
99 Lichtenstein BW. “Spinal dysraphism.” Spina bifida and myelodysplasia. Arch Neurol. 1940;44:792-810.
100 Rokos J. Pathogenesis of diastematomyelia and spina bifida. J Pathol. 1975;117:155-161.
101 Beardmore HE, Wigglesworth FW. Vertebral anomalies and alimentary duplications. Pediatr Clin North Am. 1958;5:457-474.
102 McLetchie NGB, Purves JK, Saunders RL. The genesis of gastric and certain intestinal diverticula and enterogenous cysts. Surg Gynecol Obstet. 1954;99:135-141.
103 Saunders RL. Combined anterior and posterior spina bifida in a living neonatal human female. Anat Rec. 1943;87:255-278.
104 Dodds GS. Anterior and posterior rhachischisis. Am J Pathol. 1941;17:861-872.
105 Feller A, Sternberg H. Zur Kenntnis der Fehlbildungen der Wirbelsäule. I. Die Wirbelkörperspalte und ihre formale Genese. Virchows Arch Pathol Anat. 1929;272:613-640.
106 Fernbach SK, Naidich TP, McLone DG, et al. Computed tomography of primary intrathecal Wilms tumor with diastematomyelia. J Comput Assist Tomogr. 1984;8:523-528.
107 Cameron AH. Malformations of the neuro-spinal axis, urogenital tract and foregut in spina bifida attributable to disturbances of the blastopore. J Pathol Bacteriol. 1957;73:213-221.
108 Ugarte N, Gonzalez-Crussi F, Sotelo-Avila C. Diastematomyelia associated with teratoma. J Neurosurg. 1980;53:720-725.
109 Sandberg GD, Wong K, Horkayne-Szakaly I, et al. Trimyelia with divergent cord pathways and three foramina magni. Childs Nerv Syst. 2007;23:249-253.
110 Rustamzaheh E, Graupman PC, Lam CH. Basicranial diplomyelia: an extension of the split cord malformation theory. Case report. J Neurosurg. 2006;104(suppl 5):362-365.
111 Naidich TP, McLone DG, Mutluer S. A new understanding of dorsal dysraphism with lipoma (lipomyeloschisis): radiologic evaluation and surgical correction. AJR Am J Roentgenol. 1983;140:1065-1078.
112 McLone DG, Naidich TP. Spinal dysraphism: Experimental and clinical. In: Holtzman RN, Stein BM, editors. The Tethered Spinal Cord. New York: Thieme-Stratton; 1985:14-28.
113 Truwit CL, Barkovich AJ. Pathogenesis of intracranial lipoma: an MR study in 42 patients. AJNR Am J Neuroradiol. 1990;11:665-674.
114 Chapman PH, Swearingen B, Caviness VS. Subtorcular occipital encephaloceles. Anatomical considerations relevant to operative management. J Neurosurg. 1989;71:375-381.
115 von Recklinghausen G. Untersuchungen über die Spina bifida. Part 2 (Über die Art un die Entstenhung der Spina bifida, ihre Bezichung zur Ruckenmarks und Darmspalte). Virchows Arch. 1886;105:296-330.
116 Marin-Padilla M. Notochordal-basichondrocranium relationships: abnormalities in experimental axial skeletal (dysraphic) disorders. J Embryol Exp Morphol. 1979;53:15-38.
117 Marin-Padilla M. Study of the skull in human cranioschisis. Acta Anat. 1965;62:1-20.
118 Marin-Padilla M. Morphogenesis of experimental encephalocele (cranioschisis occulta). J Neurol Sci. 1980;46:83-99.
119 Blackwood W, Corsellis JAN. Greenfield’s Neuropathology. Chicago: Year Book Medical Publishers; 1976.
120 Suwanwela C, Suwanwela N. A morphological classification of sincipital encephalomeningoceles. J Neurosurg. 1972;36:201.
121 Gluckman TJ, George TM, McLone DG. Postneurulation rapid brain growth represents a critical time for encephalocele formation: a chick model. Pediatr Neurosurg. 1996;25:109-164.
122 Hoving EW, Vermeij-Keers C. Frontoethmoidal encephaloceles: a study of their pathogenesis. Pediatr Neurosurg. 1997;27:246-256.
123 Plewes J, Jacobson I. Familial frontonasal dermoid cyst. J Neurosurg. 1971;34:683-686.
124 Kahn MA, Gibb AG. Median dermoid cysts of the nose; familial occurrence. J Laryngol Otol. 1970;84:709-718.
125 Cohen MM, Lemire RM. Syndromes with cephaloceles. Teratology. 1982;25:161-172.
126 Gilbert SF. Developmental Biology. Sunderland, MA: Sinauer Associates; 1997.
127 Shu T, Butz KG, Gronostajski RM, et al. Abnormal development of forebrain midline glia and commissural projections in NFia knock-out mice. J Neurosci. 2008;23:203-212.
128 Silver J. Glia-neuron interactions at the midline of the developing mammalian brain and spinal cord. Perspect Dev Neurobiol. 1993;1:227-236.
129 Silver J, Ogawa MY. Postnatally induced formation of the corpus callosum in acallosal mice on glia-coated cellulose bridges. Science. 1983;220:1067-1069.
130 Guerrini R, Dobyns WB, Brakovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences, and treatment options. Trends Neurosci. 2007;31:154-162.
131 Norman MG, McGillivray BC, Kalousek DK, et al, editors. Congenital Malformations of the Brain: Pathological, Embryological, Clinical, Radiological, and Genetic Aspects. New York: Oxford University Press, 1995.
132 Grossman RI, Yousen DM, editors. Neuroradiology: The Requisites, 2nd ed, Philadelphia: Elsevier, 2003.
133 Altman NR, Naidich TP, Braffman BH. Posterior fossa malformations. AJNR Am J Neuroradiol. 1992;13:691-724.
134 Hirsch JF, Pierre-Kahn A, Renier D, et al. The Dandy-Walker malformation. J Neurosurg. 1984;61:515-522.
135 Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23:1074-1087.
136 Millen KJ, Gleeson JG. Cerebellar development and disease. Curr Opin Neurobiol. 2008;18:12-19.
137 Savolaine ER, Fadell RJ, Patel YP. Isolated rhomboencephalosynapsis diagnosed by magnetic resonance imaging. Clin Imaging. 1991;15:125-129.
138 Keppes JJ, Clough C, Villaneuva A. Congenital fusion of the thalami (atresia of the third ventricle) and associated abnormalities in a 6-month-old infant. Acta Neuropathol (Berlin). 1969;13:97-104.
139 McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosurg. 1989;15:1-12.
140 Cleland J. Contribution to the study of spina bifida, encephalocele, and anencephalus. J Anat Physiol. 1883;17:257-292.
141 Daniel PM, Strich SJ. Some observations on the congenital deformity of the central nervous system known as the Arnold-Chiari malformation. J Neuropathol Exp Neurol. 1958;17:255-266.
142 Peach B. The Arnold-Chiari malformation. Morphogenesis. Arch Neurol. 1965;12:527-535.
143 McLone DG, Nakahara S, Knepper PA. Chiari II malformation: pathogenesis and dynamics. Concepts Pediatr Neurosurg. 1991;11:1-17.
144 Penfield W, Coburn DF. Arnold-Chiari malformation and its operative treatment. Arch Neurol Psychiatry. 1938;40:328-366.
145 Lichtenstein BW. Distant neuroanatomic complications of spina bifida (spinal dysraphism): hydrocephalus, Arnold-Chiari deformity, stenosis of the aqueduct of Sylvius, etc., pathogenesis and pathology. Arch Neurol Psychiatry. 1942;47:195-214.
146 Goldstein F, Kepes JJ. The role of traction in the development of the Arnold-Chiari malformation. An experimental study. J Neuropathol Exp Neurol. 1966;25:654-666.
147 Marin-Padilla M, Marin-Padilla TM. Morphogenesis of experimentally induced Arnold-Chiari malformation. J Neurol Sci. 1981;50:29-55.
148 Padget DH. Development of so-called dysraphism: with embryologic evidence of clinical Arnold-Chiari and Dandy-Walker malformations. Johns Hopkins Med J. 1972;130:127-165.
149 Padget DH, Lindenberg R. Inverse cerebellum morphogenetically related to Dandy-Walker and Arnold-Chiari syndromes: bizarre malformed brain with occipital encephalocele. Johns Hopkins Med J. 1972;131:228-246.
150 Jelínek R, Pexieder T. Pressure of the CSF and the morphogenesis of the CNS I. Chick embryo. Folia Morphol (Warsz). 1970;18:102-110.
151 Coulombre AJ, Coulombre JL. The role of mechanical factors in brain morphogenesis. Anat Rec. 1958;130:289-290.
152 Tulipan N, Hernandez-Schulman M, Lowe LH, et al. Intrauterine myelomeningocele repair reverses preexisting hindbrain herniation. Pediatr Neurosurg. 1999;31:137-142.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 175 Normal and Abnormal Embryology of the Brain
It is increasingly becoming apparent that although congenital brain and spinal cord malformations share some common clinical features, the morphology, epidemiology, and natural history of these disorders suggest a diverse and heterogeneous embryologic origin.1,2 In our view, a classification of congenital craniospinal malformations based on reputed embryonic mechanisms is much more helpful. We review the normal embryology of the nervous system, as well as the presumed embryonic mechanisms that have been proposed to give rise to various neural tube malformations, with the intent of providing a mechanistic embryonic framework on which to hang these various disorders.
At the outset, it is important to understand that most of the embryologic mechanisms proposed for congenital craniospinal malformations are putative; that is, there is no absolute proof that they are the cause of a particular human malformation, and there are few adequate animal models to test our hypotheses about the origin of human malformations. However, we can gain considerable insight into the embryopathology of congenital malformations through the study of normal neural development. Throughout this chapter we refer to human development in terms of both postovulatory days (PODs) and the embryonic staging system of O’Rahilly and Müller3; the timing and stages for all major developmental events are listed in Table 175-1. Because this chapter reviews only cranial malformations, it does not include discussions specific to spinal cord development or to congenital spinal cord malformations; interested readers are referred elsewhere for this information.4,5
TABLE 175-1 Embryologic Classification of Congenital Craniospinal Malformations
| DISORDERED MIDLINE AXIAL INTEGRATION DURING GASTRULATION |
| LOCALIZED FAILURE OF NEURULATION |
| PREMATURE ECTODERMAL DYSJUNCTION |
| Some spinal lipomyelomeningoceles (cranial to S2) |
| INCOMPLETE ECTODERMAL DYSJUNCTION |
| Dermal sinus tracts, dermoid/epidermoid tumors, ?encephaloceles |
| DISORDERED TELENCEPHALIC CLEAVAGE |
| Holoprosencephaly |
| DISORDERED COMMISSURAL DEVELOPMENT |
| Callosal agenesis, dysgenesis |
| DISORDERED NEURONAL MIGRATION |
| Schizencephaly, lissencephaly, agyria, polymicrogyria, heterotopias |
| DISORDERED RHOMBIC LIP DEVELOPMENT |
| Dandy-Walker malformation, rhombencephalosynapsis |
| POSTNEURULATION DISORDERS |
Normal Early Human Neural Development
Blastogenesis and Gastrulation (PODs 1 to 13)
During the first 2 weeks of embryogenesis the human embryo undergoes a number of cell divisions and cellular rearrangements (blastogenesis) that ultimately result in the formation of a blastocyst situated eccentrically within a hollow sphere of trophoblast cells (Fig. 175-1A). The blastocyst later forms a two-layered structure with a dorsal layer, the epiblast, adjacent to the amnionic cavity and a ventral layer, the hypoblast, adjacent to the yolk sac.3 A thickening, the prochordal plate, identifies the cranial end of the embryo (Fig. 175-1B).
Gastrulation begins with the appearance of a midline structure, the primitive streak (PS), at the caudal end of the embryo; the cranial end of the primitive streak is called Hensen’s node. A midline trough, the primitive groove, runs in the midline within the PS; the cranial end of the primitive groove is the primitive pit. The PS elongates cranially over the next 3 days and forms a midline structure in the caudal half of the embryo (Fig. 175-2A). The PS then begins to regress (shorten) back toward the caudal end of the embryo.3 As gastrulation proceeds, cells of the epiblast migrate toward the PS and invaginate through the primitive groove (Fig. 175-2A). The first cells to ingress (while the PS is still elongating) are endodermal cells, which displace the hypoblast cells laterally and form prospective endoderm (the displaced hypoblast cells form extraembryonic tissues).6–10 As the PS regresses, mesodermal cells ingress through the PS between the epiblast and newly formed endoderm and become the mesoderm.9,10 The remaining epiblast cells spread out to replace the cells that have ingressed through the primitive groove and thereby form both the neuroectoderm (the neural tube) and the cutaneous ectoderm (skin). All three germ layers, ectoderm, mesoderm, and endoderm, are derived from the epiblast. Gastrulation transforms the embryo from a two-layered structure to a three-layered structure.11
Hensen’s node serves a special role as the “organizer” of the embryo. As the PS elongates, prospective endodermal cells within Hensen’s node migrate through the primitive pit. As the PS regresses, specialized mesodermal cells, prospective notochordal cells, migrate through the primitive pit (Fig. 175-2B) and form the notochordal process in the midline between the neuroectoderm and endoderm.10,11 As we shall see, the notochord plays an important role in directing subsequent neurulation.
To what extent does the notochordal process extend cranially from Hensen’s node, and to what extent does it elongate caudally by the addition of cells to its caudal end from the regressing Hensen’s node? In the chick, the notochordal process grows largely by the latter mechanism.12 In mammals, the available evidence suggests that the situation is much more complex and may involve both mechanisms.13,14 The mechanism of notochord elongation in humans is unknown.
The prospective neuroectoderm is localized to an area of the epiblast that surrounds and flanks Hensen’s node and the cranial half of the PS (Fig. 175-3).15 Fate-mapping studies in chick embryos suggest that each region of the neuroepithelium contributes to multiple neuraxial levels, with the most cranial neuroectoderm contributing to the forebrain and all caudal levels of the neuraxis and more caudal levels of the neuroectoderm contributing to successively more caudal levels of the neuraxis. The prospective brain is derived specifically from neuroectoderm located cranial and lateral to Hensen’s node.15
Formation and Development of the Notochord (PODs 16 to 25)
In both human3 and nonhuman primates,16 the notochordal process in cross section consists of cells arranged radially about a central lumen called the notochordal canal (Fig. 175-4A).3 The notochordal canal is continuous dorsally with the amnionic cavity through the primitive pit.17 Between PODs 18 and 20, the notochordal process fuses (intercalates) with the underlying endoderm to form the notochordal plate (Fig. 175-4B). The notochordal plate is therefore incorporated into the roof of the yolk sac, with the notochordal canal becoming continuous with the yolk sac. Intercalation results in a direct communication, the primitive neurenteric canal, that connects the amnionic and yolk sacs at the level of Hensen’s node.3 The neurenteric canal persists for about 3 days, at the end of which the notochordal plate folds dorsoventrally and separates (excalates) from the endoderm (Fig. 175-4C) and the neurenteric canal is obliterated.3,18,19 Thereafter, the true notochord exists as a solid rod of notochordal cells.18 The function or functions of the neurenteric canal are unknown.
Primary Neurulation (PODs 16 to 25)
Human neuroectoderm is first visible at POD 16 as pseudostratified columnar epithelium overlying the midline notochord (Fig. 175-5A) and contiguous with the surrounding squamous epithelium of the cutaneous ectoderm.20 By POD 17 to 19, a midline neural groove (or median hinge point) develops in the midline of the neural plate immediately above the notochord (Fig. 175-5C).21 The neural groove deepens (Fig. 175-5B), and paired neural folds begin to elevate bilaterally.22 Paired dorsolateral grooves (or dorsolateral hinge points) result in further dorsal elevation and medial convergence of the neural folds (Fig. 175-5C). The neural folds separate from the cutaneous ectoderm and fuse (Fig. 175-5D) to form a closed neural tube between POD 21 and 23. Closure generally involves apposition and fusion of first the cutaneous ectoderm and then the neuroectoderm.3,19 The first part of the human neural tube to close is the region of the caudal rhombencephalon or cranial spinal cord, usually when five pairs of somites are present.19
Closure of the neural tube takes place over a period of 4 to 6 days. Although previously thought to close in linear fashion like a zipper extending cranially and caudally from the point of initial closure, mammalian neurulation instead appears to extend from several initiation sites along the craniocaudal neuraxis.19,23–25 Cranial neural tube closure may involve the coordinated interaction of as many as four waves of discontinuous neural tube closure.19,23–26 The spinal cord closes craniocaudally in a linear manner from the point of initial closure to the caudal neuropore.24,25 The last two closure sites are the cranial and caudal neuropores. The cranial neuropore closes between POD 23 and 25, whereas the caudal neuropore closes between POD 25 and 27.27
Neural crest cells are specialized, multipotential migratory cells that arise from the junction between the neural folds and adjacent surface ectoderm18,28–30; in humans, a simultaneous origin for some neural crest cells from the surface ectoderm cannot be excluded.19 Formation of the cranial neural crest begins during elevation of the neural folds before they fuse and continues from the closed neural tube well after the neural folds have fused.18,19,31 The spinal neural crest arises only after closure of the neural tube.3,27
Cranial neural crest cells contribute to the branchial arches (Table 175-2) and the arachnoid and pia mater of the cranium (the dura mater appears to derive from the mesoderm). Spinal neural crest cells undergo terminal differentiation into melanocytes of the body wall and limbs, Schwann cells investing the peripheral nerves, spinal cord meninges, dorsal root and autonomic ganglion cells of the spinal nerves, and adrenal medulla.32 Further details regarding control of migration and terminal differentiation of neural crest cells can be found in several reviews.33–35
TABLE 175-2 Contributions of the Neural Crest to Branchial Clefts
Secondary Neurulation (POD 25)
After caudal neuropore closure at POD 25 to 27, the entire nervous system is covered with cutaneous ectoderm, and more caudal neural development takes place by secondary neurulation.27 This process has been covered in other sources and has no relevance to cranial development; interested readers are referred elsewhere for this information.
Development of Brain Vesicles, Neural Tube Bending, and Formation of Alar and Basal Plate Derivatives (PODs 19 to 40)
Three neural tube flexures develop during this time as well (Fig. 175-6). The earliest to appear, at POD 19 to 21 during primary neurulation, is the dorsally directed mesencephalic flexure. Later, two additional flexures develop—the dorsally directed cervical flexure (POD 28) and the ventrally directed pontine flexure (POD 32). The pontine flexure continues to bend the future brainstem such that the metencephalon (including the cerebellum) comes to lie dorsal to the myelencephalon by the eighth embryonic week.
The fundamental cytoarchitectural organization of the spinal cord and brainstem (myelencephalon, metencephalon, and mesencephalon) consists of two components: a basal and an alar plate (Fig. 175-7). The basal plate contains paired ventral neuromotor columns that contribute to the primary motor pathways, whereas the alar plate contains paired dorsal neurosensory columns that contribute to the sensory and integrative pathways. As discussed previously, spinal cord neural crest cells form dorsal root ganglia and dorsal sensory nerves, the central processes of which enter the spinal cord and either ascend within the posterior columns to the nucleus gracilis and cuneatus or synapse locally with dorsal sensory neurons within the alar column (Rexed’s laminae I to VII). Alar plate neurons send projections both cranially in the spinothalamic and spinocerebellar tracts and locally that perform integrative functions with other alar or basal plate neurons within the spinal cord. The basal plate motoneurons receive afferent synapses from alar plate neurons (as well as descending projections from cranial portions of the neuraxis) and, in turn, send efferent axonal projections to the periphery via the ventral root.
The brainstem nuclei are arranged into seven columns, three motor and four sensory. The three motor columns produce somatic efferent, visceral efferent, and special (branchial) efferent nuclei. The four sensory columns produce somatic afferent, visceral afferent, and two special afferent nuclei—special somatic afferent and special visceral afferent nuclei. Each cranial nerve receives contributions from one or more of these seven columns (Table 175-3).
| Olfactory (I) | SVA (olfaction) |
| Optic (II) | SSA (vision) |
| Oculomotor (III) | GSE (oculomotor muscles) |
| GVE (ciliary muscle) | |
| Trochlear (IV) | GSE (superior oblique muscle) |
| Trigeminal (V) | GSA (skin, pharynx sensation) |
| GVA (proprioception) | |
| SVE (branchial muscles) | |
| Abducens (VI) | GSE (lateral rectus) |
| Facial (VII) | SVA (taste in the anterior two thirds of the tongue) |
| GSA (skin of the external auditory meatus) | |
| GVA (taste in the anterior two thirds of the tongue) | |
| SVE (muscles of facial expression) | |
| GVE (salivary, lacrimal glands) | |
| Vestibulocochlear (VIII) | SSA (auditory, vestibular) |
| Glossopharyngeal (IX) | SVA (taste in the posterior third of the tongue) |
| GVA (parotid gland, carotid body and sinus, middle ear) | |
| GSA (external ear) | |
| SVE (stylopharyngeus muscle, branchial arch) | |
| GVE (parotid gland) | |
| Vagus (X) | SVA (taste for the palate, epiglottis) |
| GVA (afferents for the trachea, heart, esophagus, stomach, intestines) | |
| GSA (external auditory meatus) | |
| SVE (intrinsic muscles of the pharynx, superior two thirds of the esophagus) | |
| GVE (efferents for the trachea, digestive tract, heart) | |
| Accessory (XI) | SVE (sternomastoid, trapezius) |
| GVE (soft palate, pharynx) | |
| Hypoglossal (XII) | GSE (tongue) |
GSA, general somatic afferent; GSE, general somatic efferent; GVA, general visceral afferent; GVE, general visceral efferent; SSA, special somatic afferent; SVA, special visceral afferent; SVE, special visceral efferent.
Neuronal Proliferation, Neuronal Migration, and Formation of Commissures (PODs 24 to 115)
The cellular architecture of the brain and spinal cord is based on a similar unifying theme—embryonic neuroblasts proliferate within a ventricular zone and their postmitotic progeny migrate centripetally through the neural tube to take up locations peripherally to form the mantle layer. Axonal processes from mantle layer neurons extend further to form the marginal zone immediately under the pia (Fig. 175-8). Neuronal proliferation begins on POD 24; neuroblasts are later replaced by glioblasts at the ventricular zone, which then give rise to glial cells (astrocytes and oligodendroglia). The remaining cells of the ventricular zone become ependymal cells.
Spinal cord development follows this basic theme and gives rise to the central location of the spinal cord gray matter with surrounding white matter tracts. However, proliferation and migration at other levels of the neuraxis are altered depending on the specific location—two locations that deserve special discussion are the cerebellum and cerebral cortex. In the cerebellum, the postmitotic neuroblasts migrate centripetally to form two zones: the deep cerebellar nuclear layer (which will form the deep cerebellar nuclei) and the Purkinje cell layer (to which the Purkinje, basket, stellate, and Golgi cells migrate) (Fig. 175-8, middle). The granule cells, in contrast, originate in the rhombic lip, migrate superficially around the surface of the cerebellum, and exist superficial to the Purkinje cell layer as the external granule layer. These cells are still mitotically active and produce numerous granule cell progeny, even as long as 2 years after birth.36 Postmitotic granule cells migrate back toward the ventricular zone, through the Purkinje cell layer, to produce the internal granule layer. This second migration is controlled by specialized glial cells, called Bergman glia, the processes of which serve as guideposts for the migration of granule cells through the Purkinje cell layer.
Neurogenesis and neuronal migration are also modified in the cerebral cortex (Fig. 175-8, bottom). The first neuroblasts send axon that form the marginal zone immediately subjacent to the pia. Postmitotic neurons then migrate away from the ventricular zone to form the intermediate zone between the ventricular and marginal zones. Cells from both the ventricular and intermediate zones migrate further centripetally to form the cortical mantle zone—cells that arrive at the mantle zone first form the deeper cortical layers, whereas those that arrive later form more superficial cortical layers that migrate through the previously established layers. As ventricular zone proliferation ceases, proliferation begins in the subventricular zone, located between the ventricular and intermediate zones. Cells from the subventricular zone migrate through the intermediate zone to form a subplate just beneath the cortical mantle zone. Cells from the subplate zone migrate through the mantle zone to form the most superficial cortical layers. The cortex is therefore derived from both the cortical mantle zone and the subplate region and is generated in an inside-out fashion, with superficial layers forming later than deeper layers. The intermediate layer subsequently forms the white matter of the cerebrum. Neuronal migration in the cerebrum also appears to involve specialized radial glial fibers that span from the ventricular to the marginal zones and along which neurons migrate to their final destinations. Neuronal migration involves the interplay of many different molecular species, including reelin, astrotactin, integrins, neurogulins, and others.36
The majority of the commissural fibers of the hemispheres develop from the commissural zone of the lamina terminalis. The lamina terminalis is the site of the cranial neuropore and is located immediately dorsal to the optic chiasm anatomically. Commissural fibers of the anterior commissure, corpus callosum, and hippocampal commissure derive from the dorsal end of the lamina terminalis (the posterior and habenular commissures are formed from the epithalamus of the diencephalon). The earliest commissural fibers, those of the anterior commissure, arise at 54 days, followed by those of the hippocampal commissure during the 11th week and those of the corpus callosum between 84 and 115 days.37,38 The corpus callosum generally develops in an anterior-to-posterior fashion (genu first, followed by the body and then the splenium); because of the later growth of the frontal lobes, the rostrum is the last portion to form. As a result, incomplete abnormalities of the corpus callosum involve the later developing, more posterior portions (and rostrum) with sparing of the earlier developing anterior portions and lead to selective enlargement of the atrium and occipital horns of the ventricles (colpocephaly). An exception to this rule is holoprosencephaly, in which there is selective agenesis of the posterior portions with sparing of the anterior portions.38,39
Abnormal Early Neural Development
Failure of Neural Tube Closure—Anencephaly and Myelomeningocele
Anencephaly and myelomeningocele are neural tube defects (NTDs), or localized failure of primary neurulation, that can arise through one of two mechanisms. The “nonclosure theory” proposes that NTDs represent primary failure of neural tube closure.40 The “overdistention theory,” introduced in 1769 by Morgagni41 and popularized by Gardner,42–45 proposes that NTDs arise through overdistention and rupture of a previously closed neural tube. The nonclosure theory is widely accepted for most cases. However, overdistention may contribute to some experimental NTD models, particularly those caused by vitamin A46 and the T-curtailed mouse mutant47; whether this causes human malformations is unknown. The result is an open, unneurulated segment of neural tube, the nature and severity of which are determined by the location and length of the unneurulated segment. Failure of caudal neurulation produces a myelomeningocele, whereas more cranial failure results in anencephaly.
Because neural tube closure requires the complex interaction of multiple cellular processes, it is not surprising that NTDs may result from a number of embryonic insults. NTDs have been produced experimentally with a number of teratogens (reviewed by Campbell and colleagues2), genetic mutations (reviewed by Copp and associates47,48), and experimental manipulations (reviewed by Schoenwolf and Smith49). Although these processes all suggest a number of potential mechanisms whereby NTDs might arise, the cause of human malformations remains unknown. NTDs are most likely etiologically heterogeneous1,2,47,48 and represent the end result of a variety of embryonic disorders.
Genetic models of NTDs provide a means of identifying candidate gene or genes involved in neural tube closure47,48; three such examples illustrate the variety of ways in which genetic mutations can result in an NTD. The splotch (Sp
[/not-level-membership-for-neurosurgery-category]
