[level-membership-for-opthalmology-category]
Chapter 96 Nonrhegmatogenous Retinal Detachment
 For additional online content visit http://ww.expertconsult.com
For additional online content visit http://ww.expertconsult.com
Introduction
A wide variety of diseases may present with sensory retinal detachment without retinal breaks. Nonrhegmatogenous retinal detachment may be exudative in nature or caused by vitreoretinal traction. Some diseases with elevated retina may have both exudative and traction components. In exudative retinal detachment, the subretinal fluid may be confined to a localized area, usually the posterior pole, or may extend to the periphery, even forming bullous retinal detachment. The characteristic feature of a significant exudative retinal detachment is the presence of shifting subretinal fluid.1 The fluid shifts to the most dependent location when patients change body position. The surface of the detached retina is usually smooth; however, retinal folding may occur in some diseases associated with subretinal fibrosis. To reach an accurate diagnosis among many diseases presenting with exudative retinal detachment, careful fundus examination, fluorescein angiography (FA), indocyanine green angiography (ICGA), optical coherence tomography (OCT), ultrasonography, computer tomography (CT), and magnetic resonance imaging (MRI) may be necessary.
Idiopathic
Central serous chorioretinopathy
Bullous retinal detachment
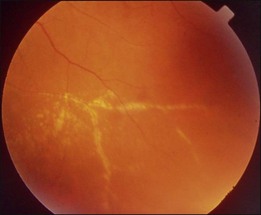
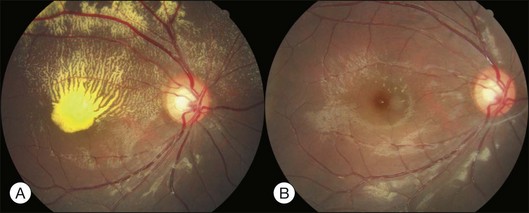
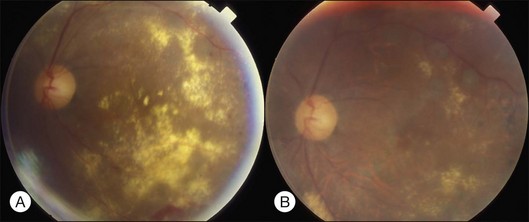
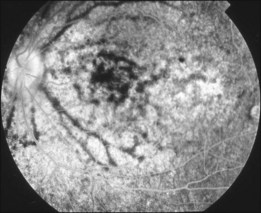
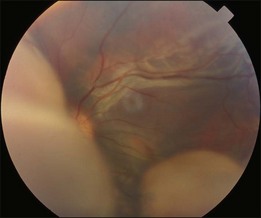
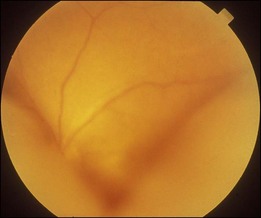
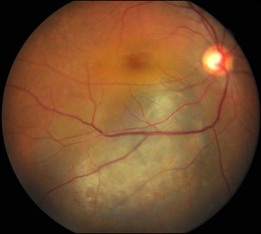
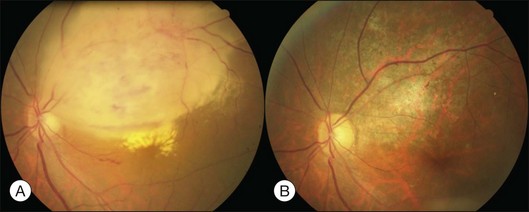
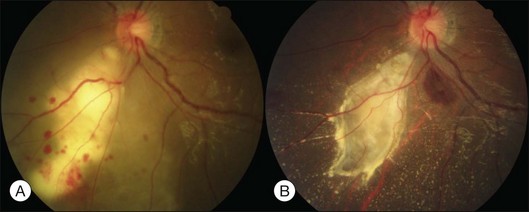
Bullous RD usually has an acute onset with simultaneous or sequential involvement of the two eyes. Fundus examinations reveal multiple areas of serous RD in the posterior retina with lower bullous RD. There may be multiple retinal pigment epithelial detachment (RPED) and one or more grayish or yellow patches of subretinal exudates mimicking focal chorioretinitis (Fig. 96.1). In some cases, retinal folds may form by the contraction of fibrinous patches or fibrotic membrane or bands on the outer surface of the detached retina. Small scattered yellowish granular subretinal deposits may have a tendency to settle along the retinal vessels (Fig. 96.2). The vitreous is usually clear, but may have 1–2 plus cells. The disc is not hyperemic.
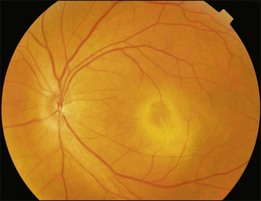
Fig. 96.1 Subretinal fibrin-like deposition mimicking chorioretinitis in a case of acute bullous detachment.
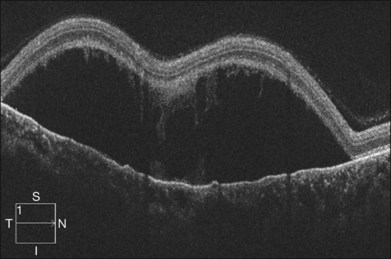
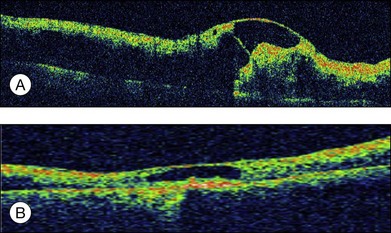
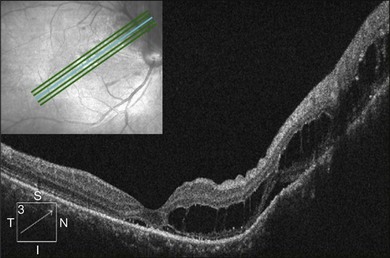
Optical coherence tomography may show retinal pigment epithelial detachment with or without sensory detachment adjacent to or overlying it; the subretinal fluid may be clear or slightly turbid with multiple granular deposits above RPE and on the outer surface of the detached retina, sometimes forming incomplete septa within the subretinal space (Fig. 96.3); subretinal fibrinous mount with surrounding sensory detachment may be seen.2
Complications of bullous RD include: large RPE tear; broad retinal folding; submacular plaques or fibrotic bands; peripheral paravascular exudates; peripheral retinal telangiectasia, occlusion, or even fibrovascular proliferation.2,3 Peripheral vascular changes may be secondary to long-standing sensory detachment.
In treating bullous RD, systemic steroids should be discontinued; patients should keep the head elevated during sleep to prevent fluid shifting to the macular area; FA-guided laser to the leaking points may decrease the subretinal fluid. Once the fluid level recedes, FA should be repeated to identify persistent leaking points and other leaking sites previously hidden by the detached retina. Multiple sessions of laser treatment are usually needed for complete fluid reabsorption. If subretinal fluid (SRF) persists after the above measurements, external drainage of SRF may be undertaken.4 Care should be taken to make sure that the surgical drainage site is posterior enough to access the subretinal space, which is in the dependent area. Alternatively, pars plana vitrectomy with perfluorocarbon liquid injection and simultaneous external drainage through anterior sclerotomy may be performed, followed by focal laser to the exposed leaking sites and air–fluid exchange. The effect of vitrectomy with internal drainage and silicone oil tamponade is controversial. Recently, bevacizumab injection has been shown to rapidly reduce active fluid leakage into the subretinal space as well as decrease the deposition of fibrinous or proteinaceous substances.5 Photodynamic therapy (PDT) with reduced fluence may also reduce choroidal hyperpermeability and facilitate subretinal fluid reabsorption with RPE tear being the major complication.6 Prognosis of bullous RD is variable and is affected by the duration of macular detachment, the presence of submacular fibrosis, the development of submacular RPE tears, and occurrence of fibrovascular proliferation under the macula or in the periphery.
Chronic CSCR
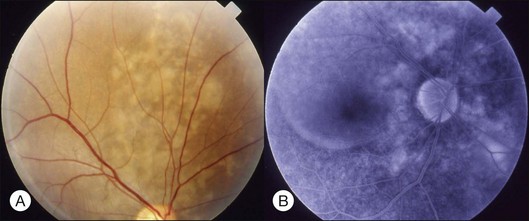
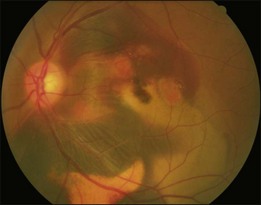
Typical clinical manifestation is the multiple poorly defined areas of chronic persistent or recurrent retinal detachment in the posterior pole. Subtle or obvious areas of RPE changes are noted in the posterior pole and in the juxtapapillary regions; gravity tract forming vertical band or reverse funnel-shaped depigmentation, along with pigment migration or bone-spicule pattern of pigmentary changes within the tract and in the inferior part of the retina are usually found (Fig. 96.4); shallow or bullous detachment in the inferior retina is a frequent finding.
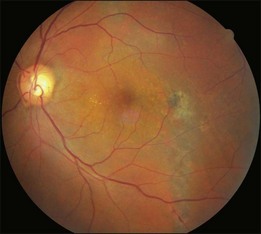
Photocoagulation remains the main treatment method. Conventional laser or the more recently developed MicroPulse laser to leaking points and areas of RPE changes with late fluorescein staining and leakage may stop the leakage.7 PDT with reduced fluence has been advocated to treat leaking points and areas with late oozing shown in FA with good effect. Intravitreal bevacizumab has also been shown to have therapeutic effect.8 However, the prognosis is guarded because of multiple recurrence and permanent macular RPE disturbance.
Uveal effusion syndrome
Histopathological examination shows accumulation of protein-rich extracellular materials in the suprachoroidal and subretinal spaces. Choroidal vessels are dilated without inflammatory cell infiltration. Subarachnoid space around the optic disc is enlarged. The sclera shows deranged fibers with deposition of glycosaminoglycans within.9 Cell culture of the scleral cells reveals intracellular deposition of a glycogen-like substance.10
The pathogenesis is unclear, possibly related to congenital anomaly of the sclera and vortex veins hypoplasia. Excessive glycosaminoglycans accumulate within the sclera combined with defective vortex veins resulting in decreased drainage of extravasated protein through scleral emissary channels of the transscleral outflow pathway; fluid drainage is also compromised from the decreased function of uveoscleral outflow pathways, leading to excessive protein and fluid accumulation in the suprachoroidal space. Later on, the protein and fluid enter the subretinal space when the extracellular protein concentration becomes equal to that within the vessels. Protein in the suprachoroidal space around the disc may gain access to the subarachnoid space and subdural space resulting in an increase in the CSF protein content even without pleocytosis in 50% of the patients.11 Forrester and associates believe that IUES is a kind of ocular mucopolysaccharidosis, with the initial defect resting in the proteodermatan synthesis and/or degradation of the fibroblast of the sclera.12 Other evidence also shows that abnormal mucopolysaccharides of the sclera play an important role in the pathogenesis of IUES. IOP is usually within normal limits because the IOP rising tendency from uveoscleral outflow obstruction is neutralized by decreased aqueous production from the ciliochoroidal detachment.
Best treatment methods have been debated. Vortex vein decompression with scleral resection was initially advocated to treat uveal effusion associated with nanophthalmos. Gass believed that the treatment effect had less to do with vortex vein decompression than with scleral resection to facilitate protein and fluid drainage through the sclera.13 He suggested partial-thickness sclerectomies or full-thickness sclerectomies. After treatment, exudation may gradually disappear within a few months. However, chorioretinal degeneration may continue to develop from chronic mild recurrence of exudation or abnormal metabolism of mucopolysaccharide.
Vascular
Coats disease
This is a non-familial developmental retinal vasculopathy. The disease is more common in males, is usually unilaterally affected and may occur in infants. Symptoms often develop in children or young adults; one-third had symptoms onset over 30 years of age. All vessels, arteries and veins alike, would be affected, showing telangiectasis combined with a large amount of hard exudates; hemorrhagic retinopathy is occasionally seen. On the other hand, a minor form of the disease mainly involving juxtafoveolar areas may occur; decreased vision will not occur until adulthood when hard exudates and edema develop in the macula. The prognosis is directly influenced by the size of the involved area. Occasionally, other vascular anomalies may appear in the lesion eye or the fellow eye, such as macular macrovessels or arterial tortuosity (Fig. 96.5). The condition may be occasionally associated with other abnormalities such as progressive facial hemiatrophy, facial scapulohumeral muscular dystrophy and deafness, or Alport syndrome.14 It may rarely accompany systemic vascular anomalies.
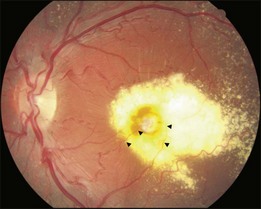
Fundus examinations reveal changes of various severities. The mild form presents with focal telangiectasia and microaneurysms usually at the temporal side of the macula, with or without mild hard exudates. The moderate form ranges from cystoid macular edema with significant hard exudates surrounding the area containing telangiectatic vessels or microaneurysms, to the more extensive vascular abnormalities with massive exudates which may gain access to the subretinal space. The severe form shows wide and scattered vascular lesions, with hard exudates accumulating around the disc and in the posterior pole, causing exudative detachment. The macula may be detached with massive intra- and subretinal exudates, which later may transform to organized subretinal disciform mass or atrophic scar. These changes are likely to be found in infants and children, who visit the ophthalmic clinic because of manifest strabismus secondary to unilateral poor vision or abnormal red reflex from massive exudates in the posterior pole. The accumulation of exudates in the macula may be due to gravity-induced migration of subretinal exudates toward the central area during sleep. The deposition of lipid-rich substance along with macrophage evolves into fibrous tissue. Retinal or choroidal vessels may grow into the lesion to form a disciform scar. The most advanced form presents with bullous detachment with the retina coming in direct contact with the crystalline lens; cholesterol crystals accumulate in the subretinal space.15 There may be dilated abnormal vessels, hard exudates or hemorrhage on the surface or in the retina. However, the abnormal tortuous vessels do not dip into the subretinal space, a sign typical of exophytic retinoblastoma.
Treatment usually involves laser or cryo aiming at the lesions to decrease exudates and preserve vision (Fig. 96.6). For severe exudative detachment, external drainage should be performed first, followed by cryo to the abnormal vessels. Scleral buckling may facilitate retina reattachment, enhance cryo effect and promote regression of abnormal vessels. However, new lesions may develop in nearby or remote areas. Follow-up is crucial to detect and treat new lesions. Sector panretinal photocoagulation (PRP) may be used for a nonperfusion area. For severe cases, vitrectomy to release vitreous traction with external subretinal fluid drainage, laser or cryo may be considered. Recently, repeated intravitreal injection of bevacizumab has been reported to reduce subretinal fluid, facilitating subsequent laser or cryo.16
Accelerated hypertension and pregnancy-induced hypertension
While chronic moderate hypertension is rarely associated with choroidopathy, choroidal ischemia is more frequently associated with accelerated hypertension. Unlike retinal circulation, choroidal vessels do not possess autoregulation; the blood flow during fluctuation of systemic blood pressure is mainly regulated by sympathetic tone. When blood pressure is high, raised sympathetic tone can prevent direct pressure damage to the choriocapillaries; however, if there is a rapid rise in blood pressure, excessively increased sympathetic tone may prompt severe constriction of choroidal arteries and arterioles, leading to ischemic changes of the choriocapillaries. Choroidopathy may be separated into three stages: (1) acute ischemic phase; (2) chronic occlusive phase; (3) chronic reparative phase.17 In the first two phases, fundus examination may observe white or reddish patches in the outer retina, possibly caused by RPE necrosis; exudative detachment is often present. FA may show a large confluent area or scattered areas of choroidal filling delay; in the mid and late phases, multiple dots or a mosaic pattern of hyperfluorescence from RPE leakage may be seen. In the reparative phase, large areas of irregular REP atrophy, Elschnig spots (central hyperpigmented and peripheral hypopigmented lesion of RPE changes), or Siegrist spots (spots of pigmentary changes similar to Elschnig spots arranged linearly along choroidal vessels in the equatorial region) may be seen; exudative detachment disappears, but choroidal delayed filling remains.
Pregnancy-induced hypertension
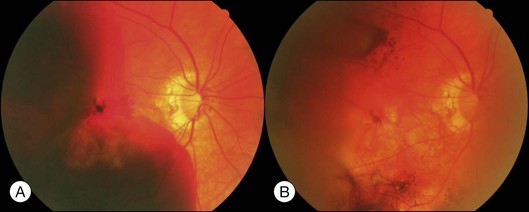
About 1–2% of pregnant women develop exudative retinal detachment pre- or immediately post-delivery, causing visual impairment. Exudative detachment may be limited to the macular area or appear as bullous detachment. The retina may or may not show cotton-wool patches or other changes secondary to hypertension retinopathy (Fig. 96.7). Yellowish-white patches of RPE necrosis may be seen. FA shows delayed choroidal filling and multiple leaking points where RPE has been damaged. After delivery, with control of hypertension, exudative detachment rapidly subsides. Most patients have good visual recovery. The posterior pole may show RPE changes forming hyperpigmented lines or patches mixed with yellow spots of RPE atrophy. The bilateral changes may be mistaken for macular dystrophy. Severe cases may have extensive exudative detachment. Widespread RPE changes similar to tapetoretinal dystrophy and severely compromised vision may result. The cause of chorioretinal changes in pregnancy-induced hypertension is not clear. Blood pressure may not be very high before the onset of exudative detachment. Affected patients may have other symptoms and signs related to disseminated intravascular coagulation, such as hemolysis, low platelet count, and elevation of liver enzymes. It is possible that mechanisms capable of inducing disseminated intravascular coagulation (see below) are also functional in the choroid, causing choroidal ischemia, RPE damage and exudative detachment.
Diabetic retinopathy
Severe diabetic macular edema may sometimes accompany localized macular detachment. Fluid leaking out from the vessels first accumulates within the retina; beyond a certain critical point, fluid may gain access into the subretinal space causing sensory detachment. Severe macular edema not only leads to detachment but may be associated with massive hard exudates (Fig. 96.8). Hard exudates are one of the independent risk factors for vision decrease.18 In addition to vascular hyperpermeability related to diabetic vasculopathy, taut posterior hyaloid membrane may also contribute to macular edema and localized detachment either from the mechanical traction force or from traction-induced increased vascular permeability.
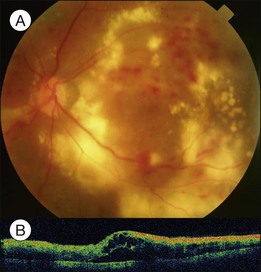
Clinically, early fundus changes leading to severe macular edema may present as a central retinal vein occlusion-like picture with flame-shaped hemorrhage around the disc and scattered perivascular exudates but without fluorescein angiographic evidence of disc leakage and venous delayed filling. In other cases, multiple clusters of microaneurysms may distribute in the posterior pole, accompanied by significant capillary nonperfusion. Sensory detachment and massive exudates may later develop. Exudative detachment combined with macular edema represents severe break down of the inner retinal barrier. Laser alone has little effect in such cases. Multiple sessions of intravitreal anti-VEGF (vascular endothelial growth factors) alone or in combination with subtenon or intravitreal steroid administration may effectively flatten down the retina in most cases. The effect has not been confirmed if subsequent focal laser to the leaking vascular segments or microaneurysms may obtain a more lasting effect. In some cases, exudative detachment is present without significant cystoid macular edema. It may be because the edema is in a resolving phase; thus the response to treatment may be quicker. In severe cases, exudates may consolidate and deposit within or below the macula. If the condition does not improve after several sessions of medical treatment, pars plana vitrectomy combined with hyaloid membrane removal may be performed to reduce edema and hard exudates (Fig. 96.9).19 Because the development of massive hard exudates indicates that the retina is in a relatively hypoxic state or has already gone into the early proliferative stage, panretinal photocoagulation during the operation is required to inhibit production of angiogenic factors, which may induce further macular edema or neovascularization, leading to postoperative vitreous hemorrhage or even neovascular glaucoma.19 In the case of pre-existing posterior vitreous detachment, epiretinal membrane peeling and internal limiting membrane peeling may be considered.20 Massive exudates usually form submacular plaques, affecting vision severely. After surgery, the plaque may reduce in size but does not disappear completely, leaving residual fibrosis or crystal-like deposition, causing permanent decrease of vision. Surgical removal of subretinal exudates through iatrogenic retinotomy has been reported21; the effect has not been firmly established. Patients with severe edema should have a systemic check-up, including blood pressure, blood lipid, and renal function; any abnormalities should be treated, as these may interfere with local response to the treatment.
Vascular occlusive diseases
Severe retinal vein occlusion occasionally is accompanied by serous retinal detachment. Exudative detachment has been described in branch, hemispherical, and central retinal vein occlusion (CRVO). Vascular leakage from congested retinal veins outside the macular area is the major source of subretinal fluid at the fovea. In addition, ischemic retina produces angiogenic factors, such as VEGF, which in turn increase vascular permeability. Both increased intravascular pressure and vascular permeability cause leakage of fluid and blood components into the subretinal space. In eyes with retinal vein occlusion, serous RD is typically located beneath the fovea, and the height of the RD was greatest in the fovea (Fig. 96.10).
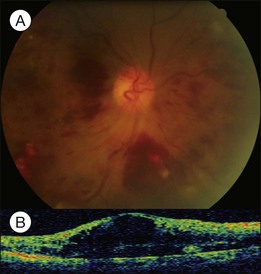
Recent studies with OCT revealed that macular serous retinal detachment is a common complication of retinal vein occlusion. Serous RD secondary to branch retinal vein occlusion was first described by Spaide et al.22 Of the 14 eyes included in that study, 10 (71.4%) had serous RD. Yamaguchi et al. studied 109 eyes with branch retinal vein occlusion (BRVO) by OCT examination, and found that the incidence of serous RD is higher in the group with major BRVO (63%) than in the group with macular BRVO (21%).23 Ozdemir et al. found a high incidence (81.8%) of serous macular detachment in CRVO.24 In a series of 91 patients with retinal vein occlusion examined by OCT, Tsujikawa et al. reported that 76 eyes (83.5%) had serous RD involving the fovea.25 They suggested from their observations that in eyes with retinal vein occlusion, a small pointed RD developed initially just beneath the fovea, but subsequently changed into a dome-shaped RD; the foveal architecture, especially that of the Müller cell cone might be involved in the formation of serous RD.
Application of laser photocoagulation to the affected area of branch retinal vein occlusion has been reported to treat serous RD and can lead to resolution of subretinal fluid. The beneficial effect of laser treatment may be due to the ablation of ischemic retina, decrease of the production of VEGF, closure of incompetent vessels, and stimulation of RPE to enhance the reabsorption of fluid. Intravitreal injection of triamcinolone or bevacizumab has been reported to treat serous RD due to retinal vein occlusion26,27; repeated injection may be necessary for frequent recurrence of macular edema.
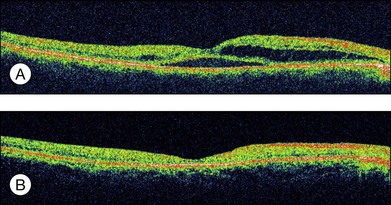
The visual prognosis of serous RD in retinal vein occlusion is variable. In an eye with serous RD associated with retinal vein occlusion, the outer retinal discontinuity does not necessarily lead to poor vision. If the surrounding outer segment of the foveal photoreceptors is preserved, good visual acuity will retain after macular edema and serous RD resolve. However, even after complete resolution of the macular edema and serous RD, diffuse disorganization of the outer photoreceptor layer beneath the fovea often results in poor visual acuity (see Fig. 96.11, online). In addition, a dome-shaped RD sometimes accompanies a focal defect of the outer segment of the photoreceptors above the serous RD. When the defect involves the fovea, visual prognosis is usually poor.
Inflammatory and infectious
Vogt–Koyanagi–Harada syndrome
These associations are high in many populations, including Japanese, Hispanic, Korean, Indian, Italian, Mexican, and Chinese.28
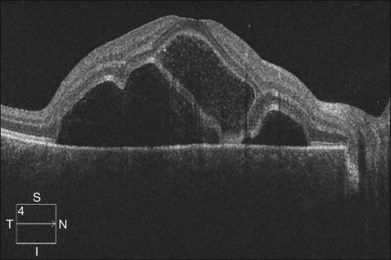
OCT has unique features. In addition to the usual pattern of subretinal fluid accumulation, the subretinal space may develop cystic spaces external to the external limiting membrane (Fig. 96.12); the floors of the cystoid spaces consist mainly of a membranous structure, continuous with the line representing the junction of the photoreceptor inner and outer segments in attached areas. It has been suggested the membranous structures are composed not only of inflammatory products, but also of retinal tissue, probably the outer segment.
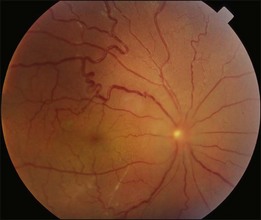
In the convalescent stage, there may be skin and hair changes, including hair loss, alopecia, and vitiligo 2–3 months after the disease onset. Sunset glow appearance of the fundus, which indicates diffuse loss of melanin pigment in the RPE and the choroid, may develop. Pigmented lines radiating from the disc after the subsidence of choroidal thickening indicate previous acute inflammation (Fig. 96.13). Scattered punched out whitish lesions in the peripheral retina (corresponding to the histological diagnosis of Dalen–Fuchs nodules) are often visible. Recurrence after the convalescent stage usually takes the form of chronic iritis instead of exudative detachment. Subfoveal choroidal neovascularization may occur in around 2% of patients with VKH and may require intravitreal injection of an anti-VEGF agent.
Sympathetic ophthalmia (SO)
SO is a bilateral granulomatous uveitis that occurs after ocular trauma or intraocular surgery to an eye. The disease incidence is about 0.3% of eyes with nonsurgical ocular wounds; it is 0.01% for surgical wounds. The inflammation may develop in the contralateral sympathizing eye as short as 2 weeks after trauma to the initial exciting eye. About half of the cases develop this disorder within 1 year after injury. In surgically induced SO, although this disorder is more likely to occur in eyes suffering from multiple intraocular surgeries for complicated vitreoretinal lesions, simple transscleral subretinal fluid drainage procedure has been reported to be associated with SO.29
Posterior scleritis
The presenting symptoms of posterior scleritis are blurred vision and ocular pain associated with eye movement. Clinical signs include serous retinal detachment, choroidal folding on fundus examination, multiple pinpoint leakage in FA and the pathognomonic T-sign by ultrasonography. When there is additional disc swelling and proptosis, CT scan or MRI should be performed to rule out pseudotumor. In rare conditions, posterior scleritis may present with solitary mass instead of diffuse scleritis.30
Posterior scleritis is a subgroup of scleritis, which also includes anterior scleritis. The etiology and treatment of posterior scleritis are similar to those of anterior scleritis, except that the anterior necrotizing type is very rare in posterior scleritis. Reports from most university or tertiary referral centers found that about half of the scleritis cases were associated with systemic diseases. Rheumatoid arthritis is the most commonly associated systemic disease, followed by Wegener granulomatosis and relapsing polychondritis. In community-based referral practice, one-third of the scleritis cases are associated with systemic diseases; most develop after the diagnosis of the systemic disease. Rheumatoid arthritis is the leading cause, with spondyloarthropathy and infectious origin being the second and the third most common etiologies.31 Other systemic diseases associated with scleritis include: Cogan syndrome, herpes simplex and zoster, aspergillosis, inflammatory bowel disease and sarcoidosis.
Topical corticosteroid only is successful in controlling scleritis in less than 10% of cases. Noninfectious, non-necrotizing scleritis should be initially treated with topical corticosteroids and oral nonsteroidal anti-inflammatory drugs (NSAIDs). Patients with necrotizing scleritis or those with non-necrotizing scleritis recalcitrant to NSAIDs are often started on oral prednisone. If the patient does not respond to the treatment within a month, immunomodulatory therapy (IMT) may be introduced. Steroid-sparing IMT may include: antimetabolites (i.e., methotrexate azathioprine, and mycophenolate mofetil); alkylating agents (i.e., chlorambucil and cyclophosphamide); T-cell inhibitors (i.e., cyclosporine and tacrolimus); TNF-α inhibitors (i.e., infliximab or adalimumab), and rituximab – a chimeric monoclonal antibody against CD-20 found on B cells. Subconjunctival and subtenon’s triamcinolone injections are another therapeutic alternative.32
Infections associated with exudative detachment
Bacterial infection
Ocular syphilis may sometimes be concurrent with human immunodeficiency virus (HIV) infection, leading to a more violent and destructive course.33 Ocular syphilis with or without concurrent HIV infection may be associated with exudative retinal detachment.
Peripapillary serous retinal detachment and central serous chorioretinopathy-like manifestations have been reported in patients with cat scratch syndrome.34 The exudates may be absorbed spontaneously, with or without antibiotic treatment, but some severe neuroretinitis cases may be left with optic disk pallor, abnormal color sensation and a relative afferent papillary defect.35
Brucellosis invading the eye is rare, but every structure of the eye could be involved by the disease. The clinical presentations are visual loss, optic disc edema, and serous retinal detachment.36
Fungal infection
Ocular fungal infection usually comes from systemic fungemia, which causes multifocal choroiditis. In ocular fungal infection, serous and hemorrhagic retinal detachments have been found. In some diabetic or immunocompromised patients, mucormycosis may be a fatal fungal infection. Severe rhino-orbital mucormycosis complicated by serous retinal detachment and retinal necrosis has been reported.37
Viral infection
The most common viruses capable of causing serous retinal detachment belong to the herpesvirus family. The Herpesviridae induce acute retinal necrosis, vitritis, retinal arteritis, retinal hemorrhage, exudative retinal detachment and optic neuropathy. Cytomegalovirus (CMV) retinitis usually occurs in HIV-infected patients. CMV retinitis may also have simultaneous rhegmatogenous and exudative retinal detachment.38
Degenerative
Age-related macular degeneration and polypoidal choroidal vasculopathy
In age-related macular degeneration (AMD) with choroidal neovascularization, serous and hemorrhagic detachment of the retina occurs frequently. The neovascular membrane causes leakage of serous exudates and red blood cells into the sub-RPE space and subsequently into the sub-sensory retinal space. Fundus examination typically reveals a light-grayish elevated mass corresponding to a serous and hemorrhagic detachment of the RPE and sensory retina. This mass-like lesion should be differentiated from choroidal melanoma. FA is helpful in the differential diagnosis. In AMD with choroidal neovascularization, the new vessels leak and present as a hyperfluorescent area, and the hemorrhagic lesion shows hypofluorescence because it is blocked by the subretinal blood. A melanoma shows mottled hyperfluorescence in the early phase and increased staining in the late phase. In addition, a mass-like lesion induced by choroidal neovascularization tends to change configurations within a short period of time. A disciform scar eventually evolves from choroidal neovascularization with massive exudates. Extensive hemorrhagic macular detachment may lead to a breakthrough vitreous hemorrhage; the color of the vitreous opacity tends to be yellow instead of red, indicating the presence of old blood or blood degeneration products in the vitreous. In rare cases, massive hemorrhage from active lesion or disciform scar may cause severe hemorrhagic choroidal and retinal detachment (Fig. 96.14).
Polypoidal choroidal vasculopathy has been recognized recently as a distinct exudative macular disorder.39 It is generally thought to be a primary choroidal vascular abnormality characterized by two distinct components: a complex of branching vascular networks and multiple, terminal, reddish-orange, aneurysms, or polypoidal lesions. Clinically, PCV shows multiple, recurrent serosanguineous detachments of the RPE and neurosensory retina secondary to leakage and bleeding from choroidal vascular lesions (Fig. 96.15).40 It may be a cause of severe hemorrhagic choroidal and retinal detachment. Whether PCV is a variant of exudative AMD has not been definitively determined. However, there are significant differences between PCV and exudative AMD in the demographic profile, fundus pictures, natural history, visual outcomes, and response to different treatment modalities. ICGA to identify the characteristic polypoidal vascular dilations is the main method for definite diagnosis of PCV. Recently, OCT has been used extensively to study exudative macular lesions. OCT images can also distinguish the exudative changes associated with PCV from that with exudative AMD. In the study reported by Ozawa et al., serous RD were observed in 78% of the eyes with PCV and in 53% of the eyes with exudative AMD.41 In addition, eyes with a PCV had a greater height of serous RD, and a higher incidence of large sensory detachment than eyes with exudative AMD. The active polypoidal lesions tend to leak more severely and the fluid from polypoidal lesions may leak into the subretinal space through the RPE and cause large serous RD although polypoidal lesions are situated beneath the RPE.
In some cases of PCV, a serous neurosensory detachment at the macula without hemorrhage is the major clinical feature associated with RPE atrophy. FA only shows multifocal areas of granular hyperfluorescence. This type of PCV can masquerade as CSCR. ICGA might help to establish a more definitive diagnosis. Yannuzzi et al. reported a series of 13 patients who were previously diagnosed with CSC, but with further evaluation and follow-up, were diagnosed with PCV.42 These eyes showed the characteristics of exudative macular detachments with a small-caliber, polypoidal vascular abnormality revealed by ICGA. Therefore, the authors recommended that ICGA should be done in the following situations: (1) patients not at risk of CSC, based on age, sex, or race; (2) eyes with persistent serous detachment at the macula associated with lipid accumulation; and (3) recurrent serous detachments with subretinal blood.
The serosanguineous retinal detachments in AMD or PCV can be managed by surgical intervention. Pneumatic displacement of submacular hemorrhage from the macula by intravitreal injection of expansile gas with or without tPA is now the first treatment of choice in most cases (see Fig. 96.16, online).43 Major complications include vitreous hemorrhage and rhegmatogenous retinal detachment. Vitrectomy with subretinal injection of tissue plasminogen activator (tPA) and use of perfluorocarbon liquid to evacuate the liquefied clot from the submacular space has been reported.44 Oshima et al. described the surgical results of patients with massive subretinal hemorrhage extending to the periphery and involving two or more quadrants with hemorrhagic and bullous retinal detachment.45 TPA was injected intravitreally 12–24 hours preoperatively; vitrectomy was performed with peripheral retinotomy, drainage of the subretinal hemorrhage through the retinotomy using perfluorocarbon liquid, and finally gas tamponade with postoperative prone positioning. However, the final visual outcome is limited by the underlying macular pathology.
Tumor and malignancy
Choroidal hemangioma
Patients with focal choroidal hemangioma are mostly middle-aged men or women, complaining of unilateral vision decrease or distortion. Fundus examinations may find localized elevated orange-colored lesions ranging in size from 2 disc diameters (DD) to 10 DD in the juxtapapillary, macular, or extramacular area in the posterior pole. The surface is usually smooth, but retinal thickening with cystic space may be seen above the lesion. Yellow specks or yellowish-white plaque of fibrous metaplasia sometimes exist between the tumor and the overlying retina. The lesion usually does not have pigmentary proliferation within but may have a mild hyperpigmented ring around it. Exudative detachment above and surrounding the tumor or in the inferior peripheral retina, may develop. Cystoid macular edema (CME) may be present when the macula is involved. Gravity tract of pigmentary disturbance may be seen between the tumor and the lower detached retina. Although the detachment is usually limited, severe bullous detachment may occur (see Fig. 96.17, online). Vision is affected by the presence of submacular tumor, submacular fluid or CME.
FA shows the characteristic features of perfusion of large vessels within the tumor in the pre-arterial phase followed by irregular hyperfluorescence on the tumor surface; a multiloculated pattern of cystoid macular edema may appear in the late phase.46 A ring of hypofluorescence may be seen surrounding the tumor. In small tumors without significant retinal or pigmentary changes, the lesion shows only mild hyperfluorescence, sometimes difficult to distinguish from the surrounding normal choroid. ICGA examination can demonstrate tumor vessels in the early phase more clearly.
OCT demonstrates choroidal elevation, RPE disturbance, cystic change in the overlying retina, and subretinal fluid (Fig. 96.18). Schisis-like change may be seen in the outer retina over or adjacent to the tumor, sometimes during treatment sessions of PDT or transpupillary thermotherapy (TTT). Ultrasonography shows high internal reflectivity.47
Tumors not causing macular changes do not need treatment. Conventionally, an extramacular tumor with submacular fluid accumulation is treated with a laser, aiming at the leaking tumor surface. Multiple sessions of treatment may be necessary to obtain submacular fluid reabsorption. The tumor size may or may not be reduced with this treatment modality. ICGA-enhanced diode laser is another treatment option to facilitate fluid reabsorption. Recently, TTT and PDT have been advocated to treat the tumor.48,49 With one or a few sessions of treatment at an interval of 2–3 months, fluid reabsorption and tumor reduction, and in some instances, complete flattening down of the tumor may occur (see Fig. 96.19, online). Large tumors may be treated with transscleral cryotherapy, thermotherapy, external beam irradiation, or episcleral plaque. In severe cases, the tumor may be hidden under the detached retina, thus inaccessible to laser or other treatment. In such cases, surgical drainage of the subretinal fluid may be performed to re-expose the tumor for a better treatment effect. Alternatively, repeated injection of bevacizumab may be used to facilitate fluid reabsorption.50 The effect of this treatment has not been well established.
The second type is Sturge–Weber syndrome with diffuse choroidal hemangioma. Compared with the fellow eye, the lesion eye has a more reddish fundus background color; normal choroidal markings are not visible (Fig. 96.20). This may be the only significant fundus finding in some cases. Most cases had mild tortuosity of retinal vessels or scattered pigmentary changes of various degrees. Ultrasonography may detect a thickened choroid. Other more severely affected cases may present with bullous RD, associated with glaucoma secondary to increase resistance of aqueous outflow or abnormalities of the trabecular meshwork. There may be focal choroidal thickening in addition to diffuse thickening. FA may show minimal abnormality or only patchy hypofluorescent and hyperfluorescent areas from pigmentary disturbance. Late phase leakage and cystoid space may be seen in cases with exudative retinal detachment.
Glaucoma in Sturge–Weber syndrome is difficult to control with medication and usually requires operation. Filtering operation is frequently complicated by severe choroidal detachment, with spontaneous recovery in most cases. For exudative detachment with potentially useful vision, Argon laser photocoagulation may be performed to the leaking area to facilitate subretinal fluid reabsorption. Other treatment modalities include low-dose external beam irradiation (1200–2000 cGy) divided into several sessions.51 Recently, PDT applied with multiple spots and sessions has been used to treat diffuse choroidal hemangioma aiming to reduce exudative detachment as well as choroidal thickness.52 Further studies may be needed to confirm its usage.
Choroidal melanoma
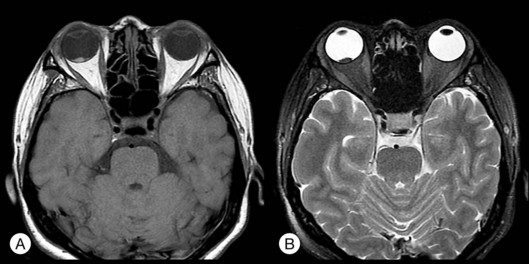
Melanoma often presents as a wide-based or dome-shaped pigmented mass under the retina (see Fig. 96.21, online). A small tumor may contain orange pigment on the surface and part or the entire lesion may be amelanotic. Exudative detachment may develop above and surrounding the tumor or in the lower dependent part away from the tumor location. During tumor growth, choroidal melanoma may invade choroidal capillaries, leading to subretinal hemorrhage. It may later break through the Bruch’s membrane to form a pigmented or amelanotic mushroom-like lesion. Tumor vessels may be visible, especially if the part above the Bruch’s membrane is amelanotic. When the tumor breaks through the Bruch’s membrane, it may cause choroidal, subretinal, or vitreous hemorrhage. Melanoma may invade the optic disc causing disc edema. When invading the vitreous, it may cause pigment dispersion within the vitreous and pigment deposition on the surface of the retina. When invading the retina, the drainage vein may become tortuous and engorged. Rarely, the tumor may assume a diffuse growing pattern causing diffuse choroidal thickening.53 Clinically the diffuse lesion is a more aggressive type than the dome-shaped lesion, and more likely to invade the optic disc and spread outside the sclera, thus having a much worse prognosis.
Histopathologic studies have shown that different tumors may contain different types of cells: spindle A, B, epithelioid cells, or mixed cell types. Epithelioid cell type has the worse visual prognosis. On FA, a typical lesion shows early pinpoint hyperfluorescence with late leakage. If there is a Bruch’s membrane breakthrough, both tumor vessels and retinal vessels can be seen, presenting the so-called double-circulation sign.54 ICGA may show tumor vessels in the late stage. Ultrasonography is valuable in showing the size, height and nodular extraocular extension. Typical lesions present as dome- or mushroom-shaped choroidal elevation with acoustic hollowness and choroidal excavation. A scan usually shows an initial spike with rapid attenuation in tumors with compact spindle A or B cell types.55 The epithelioid cell type is more prone to have a moderate high and low spike within the tumor. MRI inevitably shows the paramagnetic effect of melanin with T1, T2 reverse sign: the lesion shows a hyperdense image in T1 and hypodense in T2, in reverse to the vitreous density (see Fig. 96.22, online).56 In uncertain cases, fine-needle biopsy with 25-gauge needle through the pars plana into the tumor to obtain cells may be used for cytological examinations.
Treatment choice should consider growth potential, size, location, and age. For a small tumor, laser may be used to treat only the leaking points to promote reabsorption of subretinal fluid if exudative detachment involves the macula.57 For definite small-sized malignant melanoma, consider laser or transpupillary thermotherapy.58,59 Photodynamic therapy has been used with mixed results. For medium-sized or large posterior tumors, radiation with plaque or charged particles may be considered. Co-60 plaques or I-125 episcleral plaques are commonly used.60 Ruthenium plaques have also been widely used in Europe.61 Other treatment options include photoirradiation similar to PDT for AMD, local resection, ultrasonic hyperthermia and irradiation, and radiation plaque with photocoagulation. Teletherapy with γ-knife or cyberknife may be used. Severe inflammatory reaction is the most common complication.
The Collaborative Ocular Melanoma Study (COMS) found that preoperative radiation for large choroidal melanomas (height >8 mm, basal diameter >16 mm) does not improve 5-year survival; enucleation and brachytherapy have a similar rate of survival for medium-sized tumors (height 3–8 mm, basal diameter ≤16 mm).62 However, there is evidence from a clinical series that failure to achieve local control after radiation therapy, even when treated by subsequent enucleation, is associated with an increased risk of metastasis.63
Metastatic tumors
Metastatic tumors are the most common malignant neoplasms of the eye,64,65 with the choroid being the most common site for tumor growth. The breast was the first and the lung the second most common primary site for both choroidal and orbital metastases.65 Approximately one-third of patients have no history of primary cancer at the time of ocular diagnosis.65 About 50% of these patients fail to have a primary site detected, despite systemic evaluation by medical oncologists. The most common presenting symptom of intraocular metastasis was blurred vision. When visual acuity was affected, it usually decreased to the range of 20/200 to count fingers. Other presenting symptoms included flashes, floaters and pain. The symptom of pain is rarely found in patients with other primary uveal malignancies, such as malignant melanoma.
Ultrasonography shows that metastatic tumors tend to be flat and extend in surface area rather than thickness. Most choroidal metastases have a plateau- or dome-shaped contour and measure approximately 3–4 mm in thickness. Thicker tumors generally were observed with metastases from the gastrointestinal tract, kidney, lung, and prostate. Shield and associates reported that the mean thickness was 4 mm for metastatic gastrointestinal cancers.65
Workup for a choroidal metastasis includes an MRI of the head to rule out brain metastases and to determine the extent of the tumor. The incidence of central nervous system (CNS) metastases increases from 6% to 28% after development of ocular metastasis.66 Patients with an unknown primary site of origin should obtain a thorough physical examination and have a chest radiography and a mammogram. In women without breast cancer, and in men, the evaluation should be directed initially towards the detection of a primary tumor in the lung, alimentary tract, kidney, thyroid, pancreas, and other organs. Kole et al. suggested that PET-CT might be helpful for detecting a malignancy of unknown origin.67
Common treatment options include chemotherapy, external beam radiation, plaque radiation, hormone therapy, resection, observation, and combination therapy (Fig. 96.23). TTT has been proposed to enhance reabsorption of subretinal fluid.68
Prognosis is poor for patients diagnosed with choroidal metastasis. The average survival time is 8–9 months after diagnosis.69 Cutaneous tumors have the worst prognosis (of 1–2 months survival time), and breast tumors have the best prognosis (7–31 months). Data of 36 patients with choroidal metastases were collected in 11 years in a teaching hospital in Taiwan. The mean age was 53.9 ± 12.8 years. The primary sites of tumors were: lung in 18 (50%); breast in eight (22.2%); gastrointestinal tract in three (8.3%); pancreas in two (5.6%); ovary in two (5.6%); kidney in one (2.8%); liver in one (2.8%), and unknown in one (2.8%).70
Lymphoma
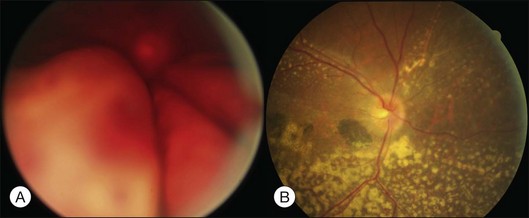
Primary intraocular lymphoma (PIOL) is a rare disease, and can rarely induce serous retinal detachment. It belongs to a subset of primary central nervous system lymphoma (PCNSL). Up to 80% of PIOL patients have CNS involvement at the time of, or following, PIOL diagnosis. Around one-quarter of PCNSL patients develop intraocular involvement.71 It is known as the most important cause of the uveitis masquerade syndrome (see Chapter 79, Intermediate uveitis-pars planitis, and Chapter 123, Diagnostic and therapeutic vitrectomy for uveitis). The clinical signs include cellular infiltration of the vitreous, which mimics vitritis, and multiple creamy subretinal infiltrative mounds with surface pigmented clumps (Fig. 96.24). Some cases may show localized subretinal yellowish infiltration with exudative detachment, or intraretinal infiltration resembling acute retinal necrosis. The diagnosis of PIOL requires a high index of suspicion. When elderly patients develop vitreous opacity with vitreous cells, PIOL should be kept in the list of differential diagnosis, especially when the so-called vitritis is refractory to corticosteroid treatment. When there is a suspicion of PIOL, pars plana vitrectomy and lumbar puncture are the two important diagnostic procedures. Cytological examination of the vitreous sample leads to a definite diagnosis. A negative result is likely to occur when patients are receiving treatment with corticosteroid. Some found a higher IL-10 or IL-10 to IL-6 ratio in the vitreous of patients with PIOL. Although the cytokine levels are not diagnostic of PIOL, they are useful adjuncts to the cytological examination. When the clinical suspicion of PIOL is high, and IL-10 level or IL-10:IL-6 ratio is high, even if the initial vitrectomy and lumbar puncture are negative, more aggressive diagnostic procedures are warranted. In some cases with only scanty vitreous infiltration, cytological examination of samples from a subretinal biopsy may be required.72
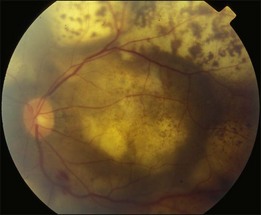
Fig. 96.24 Intraocular lymphoma showing multiple yellowish subretinal mounts with overlying pigmented clumps.
The treatment of choice for PCNSL, with or without PIOL, is high-dose methotrexate intravenously, which can pass the blood–brain barrier and blood–ocular barrier and reach the therapeutic level in the vitreous. Some advocate additional intravitreal methotrexate (0.4 mg/0.1 mL) injections in patients with PCNSL and PIOL (Fig. 96.25). The side-effects of intravitreal methotrexate injection include corneal epithelial toxicity and cataract. In relapsing cases with intraocular involvement only, treatment success has been reported with intravitreal injection of methotrexate only. In refractory cases, radiotherapy on the brain and the involved eye may be an option. However, recurrence is the rule with radiotherapy only. Intravitreal rituximab is a promising alternative for treatment of PIOL, with no significant side-effect.73
Leukemia
About 40–70% of the patients with leukemia have eye involvement.74 Pathological changes from anterior segment to posterior segment may occur, including corneal ring ulcer, iris infiltration, glaucoma, retinopathy, choroidopathy, and optic neuropathy.74 Structural alteration may be caused by direct leukemic cells infiltration or by accompanied hematological abnormalities, such as anemia, hyperviscosity, or both.74 Posterior segment manifestations are usually associated with retinal vascular changes, retinal hemorrhage or even retinal infiltrations. Exudative retinal detachment may occasionally be seen secondary to leukemic choroidopathy.74
Previous study has shown about half of leukemia cases have uveal infiltration.75 Most do not have clinical symptoms or fundus changes. Ultrasonography may show mild choroidal thickening. Some patients may present with localized or diffuse leopard-spot changes secondary to retinal pigment epithelium damage by either extensive leukemic infiltration of the choroid capillaries or chemotherapy. Focal or diffuse choroidal elevation may occasionally be seen from choroidal infiltration of leukemia, especially acute lymphocytic lymphoma. Exudative detachment may occur as well as RPE detachment (Fig. 96.26). When localized, it may be similar to CSCR; in more extensive detachment, FA may show pinpoint leakage secondary to RPE damage, similar to Harada disease, posterior scleritis or other infiltrative diseases of the choroid. Systemic chemotherapy may prompt reabsorption of subretinal fluid.76 During this process, numerous yellow exudates-like patches may be seen beneath the retina (Fig. 96.27).
Disc anomalies
Optic nerve pit
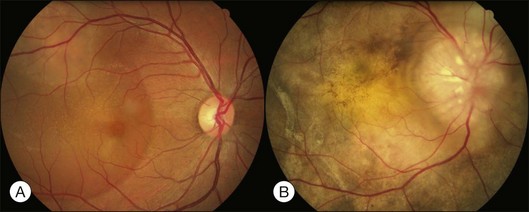
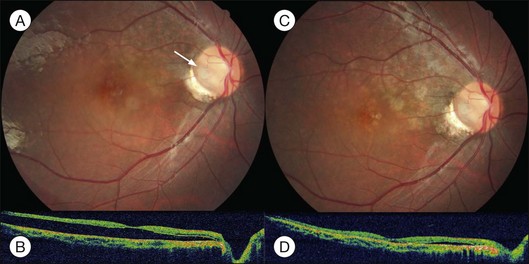
Optic pit is a congenital defect within the optic nerve head and appears as a gray dimple. It varies in size but on average, is less than one-third of the DD in width. Clinical examination shows a localized excavation of the disc that typically measures less than one half of the DD; 50% of the pits locate on the temporal side, and one-third locate centrally without association with retinal detachment. The incidence of the disease is 1/11 00077; 25–75% (40%) are associated with retinal detachment.78 The condition is unilateral in 90% of cases. In 85% of cases, the abnormal optic disc is larger than the contralateral one. The color of the pit may be gray, yellow, or less frequently, black. Various juxtapapillary changes including peripapillary retinal pigment epithelial change or choroidal atrophy or both are present in 95%. Visual field defect is caused by retinal elevation in 40% and enlarged optic pit in 60%. The patterns of visual field defect are nasal and temporal steps, altitudinal defects, paracentral scotomas, arcuate scotomas, or generalized or localized constriction.77 The macula may show the following changes: serous macular elevation, macular cystic degeneration and schisis formation, or macular mottling without evidence of RD. Schisis and RD may be present simultaneously (see Fig. 96.28, online). There is communication between the schisis cavity or subretinal space and the optic disc pit. Retinal detachment including schisis occurs in 40% of cases, usually extending into the macular region or slightly beyond.79 Larger pit, temporal location pit, and macular hole may be predisposing factors for RD. Long-standing serous retinal detachment can eventually lead to cystic degeneration of the macula and loss of pigment in the underlying retinal pigment epithelium. Lamellar and, rarely, a full-thickness macular hole may develop. Spontaneous resolution of the macular detachment is reported in 25% of cases.
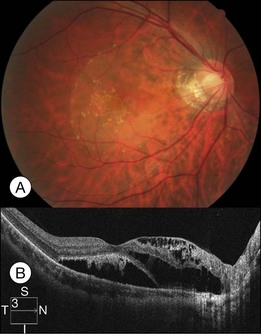
The origin of the schisis fluid or subretinal fluid remains controversial. A collie dog model suggested a connection between the vitreous cavity and the subretinal space (India ink).80 OCT may show a thin fenestrated sheet of tissue covering the pit. In addition, some patients treated with vitrectomy and tamponade have gas extending into or under the retina postoperatively. Regenbogen et al. proposed the idea that subretinal fluid could be derived from cerebrospinal fluid.77,81 Lincoff and Kreissig suggested fluid emanating from an optic disc pit was creating a schisis-like separation of the inner layers of the retina. A detachment of the outer layers from pigment epithelium was a secondary process that began in the center of the macula and did not connect with the optic disc pit.82
Persistent macular elevation may require surgery (Fig. 96.29). The greater the separation between the peripapillary RPE and the retina, the less the chance of a successful treatment. Laser photocoagulation (along the disc margin adjacent to RD), pneumatic displacement, pars plana vitrectomy, pars plana vitrectomy with autologous platelets, and macular buckling (vertically at posterior pole) have been reported.82–84 Cox and associates compared various surgical modalities. They concluded that combined surgery of vitrectomy, gas injection and laser photocoagulation to the temporal margin of the disc is the most effective therapy.84
Morning glory syndrome
RD is noted in about one-third of the cases.85 It may be confined to the peripapillary area or involve a large part of the entire retina (Fig. 96.30). The detachment may be rhegmatogenous or nonrhegmatogenous; the cause is difficult to determine from clinical presentation alone, although bullous detachment is more likely to occur in rhegmatogenous detachment. Definite diagnosis relies on the identification of retinal breaks, which are usually small and slit-like on the surface or margin of the abnormal disc. Because of poor contrast, retinal breaks are difficult to find preoperatively. During operation, SRF may be seen coming out from the hidden break by active or passive suction.
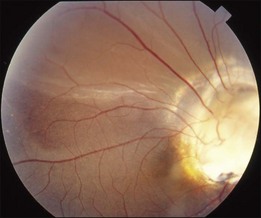
For nonrhegmatogenous detachment, the condition may undergo spontaneous improvement and recurrence. The source of fluid is debated. There may be communication between the subretinal space and subarachnoid space or the vitreous cavity, as in the optic pit. The fact that certain cases had a successful reattachment after an optic nerve fenestration operation suggests that CSF may be an important source of the subretinal fluid.86,87
Other conditions
Postsurgical exudative retinal detachment
Exudative detachment may occur after scleral buckling and cryopexy to treat rhegmatogenous retinal detachment.88 Excessive high circumferential buckle placement or multiple vortex veins compression may induce choroidal detachment with or without exudative retinal detachment. Old age and a medical history of cardiovascular diseases are risk factors.88 Excessive cryopexy may induce exudative retinal detachment causing delayed reabsorption or transient increase of subretinal fluid. As long as retinal breaks sit on the buckle, no specific treatment needs to be done. Choroidal detachment may be treated with systemic steroid especially if the anterior chamber depth becomes shallow or intraocular pressure is high.
External drainage may induce choroidal hemorrhage entering into the subretinal space in the detached area. If subretinal blood deposits under the macula, visual acuity may be greatly compromised. The complication rate is high if drainage is done after cryotherapy because of the engorgement of choroidal vessels. To avoid this complication, one should perform external drainage as infrequently as possible, or shift to vitrectomy when drainage is judged necessary with scleral buckling procedure. Once subretinal hemorrhage and choroidal hemorrhage occur, gas injection with head down position may help to push the blood away from the macula.89,90 Simultaneous use of intravitreal TPA to better mobilize the blood clot has been advocated.91
Prolonged hypotony during or after intraocular surgery may induce serous or hemorrhagic choroidal detachment with or without exudative or hemorrhagic retinal detachment. This complication may occur in cataract operations, either phacoemulsification or extracapsular cataract extraction; filtering operation for glaucoma; penetrating keratoplasty; secondary intraocular lens implantation, especially sutured lens; or vitrectomy. The popularization of small gauze vitrectomy may result in more cases experiencing postoperative hypotony secondary to sclerotomy wound leakage, leading to choroidal detachment, particularly in high myopic eyes.92 Indications and timing for surgical intervention depend on the extent of the detachment, the degree of blood clot lysis within the suprachoroidal space, the level of intraocular pressure, severity of the symptoms, and whether or not rhegmatogenous retinal detachment exists. Additional information available online.

Post-organ transplantation or hemodialysis exudative detachment
There are two types of fundus changes: type 1 has orange-colored RPE proliferation mixed with RPE atrophic changes, forming a leopard-spot pattern, associated with exudative retinal detachment. FA shows multiple RPE leaking points. Subretinal fluid usually disappears within weeks or months; recurrence is common. The onset of the lesions may not be related to graft rejection. Most patients are under steroid, or other antimetabolites treatment such as cyclosporine or azathioprine. The lesions are compatible with acute RPE damage. The underlying cause may be due to choroidal ischemia secondary to localized intravascular coagulopathy. The other type shows multifocal RPE detachment associated with subretinal serofibrinous substance and bullous detachment, similar to atypical CSCR.93,94 The condition tends to develop in patients under hemodialysis or after kidney transplantation. Most reported hemodialysis cases were taking systemic steroids. The pathogenesis may be similar to those inducing atypical CSCR.
Conclusion
 Bonus images for this chapter can be found online at http://www.expertconsult.com
Bonus images for this chapter can be found online at http://www.expertconsult.com
Fig. 96.11 (A) Optical coherence tomography image showing severe macular edema in a case of nonischemic central retinal vein occlusion. (B) Resolution of macular edema after intravitreal bevacizumab 1.25 mg and posterior subtenon triamcinolone acetonide 40 mg.
Fig. 96.16 (A) Massive subretinal hemorrhage in a case of polypoidal choroidal vasculopathy. (B) Reabsorption of the blood after pneumatic displacement with 0.2 mL C3F8 and intravitreal injection of bevacizumab 1.25 mg.
Fig. 96.17 Bullous retinal detachment in a case of solitary choroidal hemangioma.
Fig. 96.19 (A) Optical coherence tomography image of the macular area in a case with upper part choroidal hemangioma showing submacular fluid and intraretinal cysts. (B) Resolution of the macular changes after photodynamic therapy to the choroidal hemangioma.
Fig. 96.21 Malignant choroidal melanoma inferior to the macula.
Fig. 96.22 Magnetic resonance imaging study of the above-mentioned case showing typical hyperintense T1 (A) and hypointense T2 (B) images with respect to the vitreous.
Fig. 96.28 Color fundus picture (A) and optical coherence tomography image (B) showing optic pit with macular sensory detachment with subretinal yellow deposit and retinoschisis nasal to the fovea.
1 Schepens CL. Retinal detachment and allied diseases, 1st edn. Philadelphia: Saunders; 1983.
2 Wang M, Sander B, la Cour M, et al. Clinical characteristics of subretinal deposits in central serous chorioretinopathy. Acta Ophthalmol Scand. 2005;83:691–696.
3 Schatz H, McDonald HR, Johnson RN, et al. Subretinal fibrosis in central serous chorioretinopathy. Ophthalmology. 1995;102:1077–1088.
4 Yang CM, Lin CP. Bullous retinal detachment in a patient with central serous chorioretinopathy. J Formos Med Assoc. 1998;97:711–714.
5 Seong HK, Bae JH, Kim ES, et al. Intravitreal bevacizumab to treat acute central serous chorioretinopathy: short-term effect. Ophthalmologica. 2009;223:343–347.
6 Canakis C, Conway MD, Livir-Rallatos C, et al. Ocular photodynamic therapy in choroidal neovascularization complicating idiopathic central serous chorioretinopathy. Ophthalmic Surg Lasers Imaging. 2004;35:168–171.
7 Chen SN, Hwang JF, Tseng LF, et al. Subthreshold diode micropulse photocoagulation for the treatment of chronic central serous chorioretinopathy with juxtafoveal leakage. Ophthalmology. 2008;115:2229–2234.
8 Schaal KB, Hoeh AE, Scheuerle A, et al. Intravitreal bevacizumab for treatment of chronic central serous chorioretinopathy. Eur J Ophthalmol. 2009;19:613–617.
9 Uyama M, Takahashi K, Kozaki J, et al. Uveal effusion syndrome: clinical features, surgical treatment, histologic examination of the sclera, and pathophysiology. Ophthalmology. 2000;107:441–449.
10 Shiono T, Shoji A, Mutoh T, et al. Abnormal sclerocytes in nanophthalmos. Graefes Arch Clin Exp Ophthalmol. 1992;230:348–351.
11 Schepens CL, Brockhurst RJ. Uveal effusion. 1. Clinical picture. Arch Ophthalmol. 1963;70:189–201.
12 Forrester JV, Lee WR, Kerr PR, et al. The uveal effusion syndrome and trans-scleral flow. Eye (Lond). 1990;4:354–365.
13 Gass JD. Uveal effusion syndrome. A new hypothesis concerning pathogenesis and technique of surgical treatment. Retina. 1983;3:159–163.
14 Desai UR, Sabates FN. Long-term follow-up of facioscapulohumeral muscular dystrophy and Coats’ disease. Am J Ophthalmol. 1990;110:568–569.
15 Coats’ disease. In: Eagle RCJ, ed. Eye pathology. 1st edn. Philadelphia: WB Saunders; 1999:214–215.
16 Stergiou PK, Symeonidis C, Dimitrakos SA. Coats’ disease: treatment with intravitreal bevacizumab and laser photocoagulation. Acta Ophthalmol. 2009;87:687–688.
17 Kishi S, Tso MO, Hayreh SS. Fundus lesions in malignant hypertension. I. A pathologic study of experimental hypertensive choroidopathy. Arch Ophthalmol. 1985;103:1189–1197.
18 Chew EY, Klein ML, Ferris FL, 3rd., et al. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch Ophthalmol. 1996;114:1079–1084.
19 Yang CM. Surgical treatment for severe diabetic macular edema with massive hard exudates. Retina. 2000;20:121–125.
20 Gandorfer A, Messmer EM, Ulbig MW, et al. Resolution of diabetic macular edema after surgical removal of the posterior hyaloid and the inner limiting membrane. Retina. 2000;20:126–133.
21 Takaya K, Suzuki Y, Mizutani H, et al. Long-term results of vitrectomy for removal of submacular hard exudates in patients with diabetic maculopathy. Retina. 2004;24:23–29.
22 Spaide RF, Lee JK, Klancnik JK, Jr., et al. Optical coherence tomography of branch retinal vein occlusion. Retina. 2003;23:343–347.
23 Yamaguchi Y, Otani T, Kishi S. Serous macular detachment in branch retinal vein occlusion. Retina. 2006;26:1029–1033.
24 Ozdemir H, Karacorlu M, Karacorlu S. Serous macular detachment in central retinal vein occlusion. Retina. 2005;25:561–563.
25 Tsujikawa A, Sakamoto A, Ota M, et al. Serous retinal detachment associated with retinal vein occlusion. Am J Ophthalmol. 2010;149:291–301.
26 Karacorlu M, Karacorlu SA, Ozdemir H, et al. Intravitreal triamcinolone acetonide for treatment of serous macular detachment in central retinal vein occlusion. Retina. 2007;27:1026–1030.
27 Ferencz JR, Rosen E, Tam G, et al. Treatment of total exudative retinal detachment due to central retinal vein occlusion by intravitreal bevacizumab in a patient with p-ANCA vasculitis. Clin Ophthalmol. 2007;1:347–351.
28 Damico FM, Kiss S, Young LH. Vogt-Koyanagi-Harada disease. Semin Ophthalmol. 2005;20:183–190.
29 Gupta V, Gupta A, Dogra MR. Posterior sympathetic ophthalmia: a single centre long-term study of 40 patients from North India. Eye (Lond). 2008;22:1459–1464.
30 Sainz de la Maza M. Foster CS, Jabbur NS. Scleritis associated with systemic vasculitic diseases. Ophthalmology. 1995;102:687–692.
31 Raiji VR, Palestine AG, Parver DL. Scleritis and systemic disease association in a community-based referral practice. Am J Ophthalmol. 2009;148:946–950.
32 Rachitskaya A, Mandelcorn ED, Albini TA. An update on the cause and treatment of scleritis. Curr Opin Ophthalmol. 2010;21:463–467.
33 Passo MS, Rosenbaum JT. Ocular syphilis in patients with human immunodeficiency virus infection. Am J Ophthalmol. 1988;106:1–6.
34 Matsuo T, Kato M. Submacular exudates with serous retinal detachment caused by cat scratch disease. Ocul Immunol Inflamm. 2002;10:147–150.
35 Wade NK, Levi L, Jones MR, et al. Optic disk edema associated with peripapillary serous retinal detachment: an early sign of systemic Bartonella henselae infection. Am J Ophthalmol. 2000;130:327–334.
36 Tunc M, Durukan H. Bilateral severe visual loss in brucellosis. Ocul Immunol Inflamm. 2004;12:233–236.
37 Kim IT, Shim JY, Jung BY. Serous retinal detachment in a patient with rhino-orbital mucormycosis. Jpn J Ophthalmol. 2001;45:301–304.
38 Sandy CJ, Bloom PA, Graham EM, et al. Retinal detachment in AIDS-related cytomegalovirus retinitis. Eye (Lond). 1995;9:277–281.
39 Yannuzzi LA, Sorenson J, Spaide RF, et al. Idiopathic polypoidal choroidal vasculopathy (IPCV). Retina. 1990;10:1–8.
40 Yannuzzi LA, Wong DW, Sforzolini BS, et al. Polypoidal choroidal vasculopathy and neovascularized age-related macular degeneration. Arch Ophthalmol. 1999;117:1503–1510.
41 Ozawa S, Ishikawa K, Ito Y, et al. Differences in macular morphology between polypoidal choroidal vasculopathy and exudative age-related macular degeneration detected by optical coherence tomography. Retina. 2009;29:793–802.
42 Yannuzzi LA, Freund KB, Goldbaum M, et al. Polypoidal choroidal vasculopathy masquerading as central serous chorioretinopathy. Ophthalmology. 2000;107:767–777.
43 Fang IM, Lin YC, Yang CH, et al. Effects of intravitreal gas with or without tissue plasminogen activator on submacular haemorrhage in age-related macular degeneration. Eye (Lond). 2009;23:397–406.
44 Lewis H. Intraoperative fibrinolysis of submacular hemorrhage with tissue plasminogen activator and surgical drainage. Am J Ophthalmol. 1994;118:559–568.
45 Oshima Y, Ohji M, Tano Y. Pars plana vitrectomy with peripheral retinotomy after injection of preoperative intravitreal tissue plasminogen activator: a modified procedure to drain massive subretinal haemorrhage. Br J Ophthalmol. 2007;91:193–198.
46 Shields CL, Honavar SG, Shields JA, et al. Circumscribed choroidal hemangioma: clinical manifestations and factors predictive of visual outcome in 200 consecutive cases. Ophthalmology. 2001;108:2237–2248.
47 Liu W, Zhang Y, Xu G, et al. Optical coherence tomography for evaluation of photodynamic therapy in symptomatic circumscribed choroidal hemangioma. Retina. 2011;31:336–343.
48 Othmane IS, Shields CL, Shields JA, et al. Circumscribed choroidal hemangioma managed by transpupillary thermotherapy. Arch Ophthalmol. 1999;117:136–137.
49 Boixadera A, Garcia-Arumi J, Martinez-Castillo V, et al. Prospective clinical trial evaluating the efficacy of photodynamic therapy for symptomatic circumscribed choroidal hemangioma. Ophthalmology. 2009;116:100–105.
50 Sagong M, Lee J, Chang W. Application of intravitreal bevacizumab for circumscribed choroidal hemangioma. Korean J Ophthalmol. 2009;23:127–131.
51 Gottlieb JL, Murray TG, Gass JD. Low-dose external beam irradiation for bilateral diffuse choroidal hemangioma. Arch Ophthalmol. 1998;116:815–817.
52 Anand R. Photodynamic therapy for diffuse choroidal hemangioma associated with Sturge Weber syndrome. Am J Ophthalmol. 2003;136:758–760.
53 Shields CL, Shields JA, De Potter P, et al. Diffuse choroidal melanoma. Clinical features predictive of metastasis. Arch Ophthalmol. 1996;114:956–963.
54 Augsburger JJ, Golden MI, Shields JA. Fluorescein angiography of choroidal malignant melanomas with retinal invasion. Retina. 1984;4:232–241.
55 Farah ME, Byrne SF, Hughes JR. Standardized echography in uveal melanomas with scleral or extraocular extension. Arch Ophthalmol. 1984;102:1482–1485.
56 Bond JB, Haik BG, Mihara F, et al. Magnetic resonance imaging of choroidal melanoma with and without gadolinium contrast enhancement. Ophthalmology. 1991;98:459–466.
57 Char DH, Bove R, Phillips TL. Laser and proton radiation to reduce uveal melanoma-associated exudative retinal detachments. Am J Ophthalmol. 2003;136:180–182.
58 Shields JA, Glazer LC, Mieler WF, et al. Comparison of xenon arc and argon laser photocoagulation in the treatment of choroidal melanomas. Am J Ophthalmol. 1990;109:647–655.
59 Shields CL, Shields JA, Perez N, et al. Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology. 2002;109:225–234.
60 Diener-West M, Earle JD, Fine SL, et al. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol. 2001;119:969–982.
61 Kleineidam M, Guthoff R, Bentzen SM. Rates of local control, metastasis, and overall survival in patients with posterior uveal melanomas treated with ruthenium-106 plaques. Radiother Oncol. 1993;28:148–156.
62 The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol. 2006;124:1684–1693.
63 Egan KM, Ryan LM, Gragoudas ES. Survival implications of enucleation after definitive radiotherapy for choroidal melanoma: an example of regression on time-dependent covariates. Arch Ophthalmol. 1998;116:366–370.
64 Shields JA. Metastatic tumors to the uvea. Int Ophthalmol Clin. 1993;33:155–161.
65 Shields CL, Shields JA, Gross NE, et al. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104:1265–1276.
66 Demirci H, Shields CL, Chao AN, et al. Uveal metastasis from breast cancer in 264 patients. Am J Ophthalmol. 2003;136:264–271.
67 Kole AC, Nieweg OE, Pruim J, et al. Detection of unknown occult primary tumors using positron emission tomography. Cancer. 1998;82:1160–1166.
68 Wang TJ, Chen MS, Yang CM, et al. Subthreshold transpupillary thermotherapy for early resolution of foveal subretinal fluid in choroidal metastasis. Retina. 2006;26:391–395.
69 Shields CL. Plaque radiotherapy for the management of uveal metastasis. Curr Opin Ophthalmol. 1998;9:31–37.
70 Wang TJ, Yang CM, Ho TC, et al. Metastatic choroidal tumors in Taiwan: an 11-year experience. Am J Ophthalmol. 2005;140:735–737.
71 Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17:69–72.
72 Coupland SE, Chan CC, Smith J. Pathophysiology of retinal lymphoma. Ocul Immunol Inflamm. 2009;17:227–237.
73 Pe’er J, Hochberg FH, Foster CS. Clinical review: treatment of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2009;17:299–306.
74 Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol. 1983;27:211–232.
75 Robb RM, Ervin LD, Sallan SE. An autopsy study of eye involvement in acute leukemia of childhood. Med Pediatr Oncol. 1979;6:171–177.
76 Miyamoto K, Kashii S, Honda Y. Serous retinal detachment caused by leukemic choroidal infiltration during complete remission. Br J Ophthalmol. 2000;84:1318–1319.
77 Brown GC, Shields JA, Goldberg RE. Congenital pits of the optic nerve head. II. Clinical studies in humans. Ophthalmology. 1980;87:51–65.
78 Krivoy D, Gentile R, Liebmann JM, et al. Imaging congenital optic disc pits and associated maculopathy using optical coherence tomography. Arch Ophthalmol. 1996;114:165–170.
79 Imamura Y, Zweifel SA, Fujiwara T, et al. High-resolution optical coherence tomography findings in optic pit maculopathy. Retina. 2010;30:1104–1112.
80 Brown GC, Shields JA, Patty BE, et al. Congenital pits of the optic nerve head. I. Experimental studies in collie dogs. Arch Ophthalmol. 1979;97:1341–1344.
81 Regenbogen L, Stein R, Lazar M. Macular and Juxtapapillar Serous Retinal Detachment Associated with Pit of Optic Disc. Ophthalmologica. 1964;148:247–251.
82 Lincoff H, Kreissig I. Optical coherence tomography of pneumatic displacement of optic disc pit maculopathy. Br J Ophthalmol. 1998;82:367–372.
83 Garcia-Arumi J, Guraya BC, Espax AB, et al. Optical coherence tomography in optic pit maculopathy managed with vitrectomy-laser-gas. Graefes Arch Clin Exp Ophthalmol. 2004;242:819–826.
84 Cox MS, Witherspoon CD, Morris RE, et al. Evolving techniques in the treatment of macular detachment caused by optic nerve pits. Ophthalmology. 1988;95:889–896.
85 Haik BG, Greenstein SH, Smith ME, et al. Retinal detachment in the morning glory anomaly. Ophthalmology. 1984;91:1638–1647.
86 Coll GE, Chang S, Flynn TE, et al. Communication between the subretinal space and the vitreous cavity in the morning glory syndrome. Graefes Arch Clin Exp Ophthalmol. 1995;233:441–443.
87 Irvine AR, Crawford JB, Sullivan JH. The pathogenesis of retinal detachment with morning glory disc and optic pit. Retina. 1986;6:146–150.
88 Maruyama Y, Yuuki T, Kimura Y, et al. Ciliary detachment after retinal detachment surgery. Retina. 1997;17:7–11.
89 Chen SN, Ho CL, Kuo YH, et al. Intravitreous tissue plasminogen activator injection and pneumatic displacement in the management of submacular hemorrhage complicating scleral buckling procedures. Retina. 2001;21:460–463.
90 Gopalakrishan M, Giridhar A, Bhat S, et al. Pneumatic displacement of submacular hemorrhage: safety, efficacy, and patient selection. Retina. 2007;27:329–334.
91 Hassan AS, Johnson MW, Schneiderman TE, et al. Management of submacular hemorrhage with intravitreous tissue plasminogen activator injection and pneumatic displacement. Ophthalmology. 1999;106:1900–1907.
92 Bourla DH, Bor E, Axer-Siegel R, et al. Outcomes and complications of rhegmatogenous retinal detachment repair with selective sutureless 25-gauge pars plana vitrectomy. Am J Ophthalmol. 2010;149:630–634.
93 Gass JD. Bullous retinal detachment and multiple retinal pigment epithelial detachments in patients receiving hemodialysis. Graefes Arch Clin Exp Ophthalmol. 1992;230:454–458.
94 Fawzi AA, Holland GN, Kreiger AE, et al. Central serous chorioretinopathy after solid organ transplantation. Ophthalmology. 2006;113:805–813.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 96 Nonrhegmatogenous Retinal Detachment
For additional online content visit http://ww.expertconsult.com
Introduction
A wide variety of diseases may present with sensory retinal detachment without retinal breaks. Nonrhegmatogenous retinal detachment may be exudative in nature or caused by vitreoretinal traction. Some diseases with elevated retina may have both exudative and traction components. In exudative retinal detachment, the subretinal fluid may be confined to a localized area, usually the posterior pole, or may extend to the periphery, even forming bullous retinal detachment. The characteristic feature of a significant exudative retinal detachment is the presence of shifting subretinal fluid.1 The fluid shifts to the most dependent location when patients change body position. The surface of the detached retina is usually smooth; however, retinal folding may occur in some diseases associated with subretinal fibrosis. To reach an accurate diagnosis among many diseases presenting with exudative retinal detachment, careful fundus examination, fluorescein angiography (FA), indocyanine green angiography (ICGA), optical coherence tomography (OCT), ultrasonography, computer tomography (CT), and magnetic resonance imaging (MRI) may be necessary.
Idiopathic
Central serous chorioretinopathy
Bullous retinal detachment
Bullous RD usually has an acute onset with simultaneous or sequential involvement of the two eyes. Fundus examinations reveal multiple areas of serous RD in the posterior retina with lower bullous RD. There may be multiple retinal pigment epithelial detachment (RPED) and one or more grayish or yellow patches of subretinal exudates mimicking focal chorioretinitis (Fig. 96.1). In some cases, retinal folds may form by the contraction of fibrinous patches or fibrotic membrane or bands on the outer surface of the detached retina. Small scattered yellowish granular subretinal deposits may have a tendency to settle along the retinal vessels (Fig. 96.2). The vitreous is usually clear, but may have 1–2 plus cells. The disc is not hyperemic.
Fig. 96.1 Subretinal fibrin-like deposition mimicking chorioretinitis in a case of acute bullous detachment.
Optical coherence tomography may show retinal pigment epithelial detachment with or without sensory detachment adjacent to or overlying it; the subretinal fluid may be clear or slightly turbid with multiple granular deposits above RPE and on the outer surface of the detached retina, sometimes forming incomplete septa within the subretinal space (Fig. 96.3); subretinal fibrinous mount with surrounding sensory detachment may be seen.2
Complications of bullous RD include: large RPE tear; broad retinal folding; submacular plaques or fibrotic bands; peripheral paravascular exudates; peripheral retinal telangiectasia, occlusion, or even fibrovascular proliferation.2,3 Peripheral vascular changes may be secondary to long-standing sensory detachment.
In treating bullous RD, systemic steroids should be discontinued; patients should keep the head elevated during sleep to prevent fluid shifting to the macular area; FA-guided laser to the leaking points may decrease the subretinal fluid. Once the fluid level recedes, FA should be repeated to identify persistent leaking points and other leaking sites previously hidden by the detached retina. Multiple sessions of laser treatment are usually needed for complete fluid reabsorption. If subretinal fluid (SRF) persists after the above measurements, external drainage of SRF may be undertaken.4 Care should be taken to make sure that the surgical drainage site is posterior enough to access the subretinal space, which is in the dependent area. Alternatively, pars plana vitrectomy with perfluorocarbon liquid injection and simultaneous external drainage through anterior sclerotomy may be performed, followed by focal laser to the exposed leaking sites and air–fluid exchange. The effect of vitrectomy with internal drainage and silicone oil tamponade is controversial. Recently, bevacizumab injection has been shown to rapidly reduce active fluid leakage into the subretinal space as well as decrease the deposition of fibrinous or proteinaceous substances.5 Photodynamic therapy (PDT) with reduced fluence may also reduce choroidal hyperpermeability and facilitate subretinal fluid reabsorption with RPE tear being the major complication.6 Prognosis of bullous RD is variable and is affected by the duration of macular detachment, the presence of submacular fibrosis, the development of submacular RPE tears, and occurrence of fibrovascular proliferation under the macula or in the periphery.
Chronic CSCR
Typical clinical manifestation is the multiple poorly defined areas of chronic persistent or recurrent retinal detachment in the posterior pole. Subtle or obvious areas of RPE changes are noted in the posterior pole and in the juxtapapillary regions; gravity tract forming vertical band or reverse funnel-shaped depigmentation, along with pigment migration or bone-spicule pattern of pigmentary changes within the tract and in the inferior part of the retina are usually found (Fig. 96.4); shallow or bullous detachment in the inferior retina is a frequent finding.
Photocoagulation remains the main treatment method. Conventional laser or the more recently developed MicroPulse laser to leaking points and areas of RPE changes with late fluorescein staining and leakage may stop the leakage.7 PDT with reduced fluence has been advocated to treat leaking points and areas with late oozing shown in FA with good effect. Intravitreal bevacizumab has also been shown to have therapeutic effect.8 However, the prognosis is guarded because of multiple recurrence and permanent macular RPE disturbance.
Uveal effusion syndrome
Histopathological examination shows accumulation of protein-rich extracellular materials in the suprachoroidal and subretinal spaces. Choroidal vessels are dilated without inflammatory cell infiltration. Subarachnoid space around the optic disc is enlarged. The sclera shows deranged fibers with deposition of glycosaminoglycans within.9 Cell culture of the scleral cells reveals intracellular deposition of a glycogen-like substance.10
The pathogenesis is unclear, possibly related to congenital anomaly of the sclera and vortex veins hypoplasia. Excessive glycosaminoglycans accumulate within the sclera combined with defective vortex veins resulting in decreased drainage of extravasated protein through scleral emissary channels of the transscleral outflow pathway; fluid drainage is also compromised from the decreased function of uveoscleral outflow pathways, leading to excessive protein and fluid accumulation in the suprachoroidal space. Later on, the protein and fluid enter the subretinal space when the extracellular protein concentration becomes equal to that within the vessels. Protein in the suprachoroidal space around the disc may gain access to the subarachnoid space and subdural space resulting in an increase in the CSF protein content even without pleocytosis in 50% of the patients.11 Forrester and associates believe that IUES is a kind of ocular mucopolysaccharidosis, with the initial defect resting in the proteodermatan synthesis and/or degradation of the fibroblast of the sclera.12 Other evidence also shows that abnormal mucopolysaccharides of the sclera play an important role in the pathogenesis of IUES. IOP is usually within normal limits because the IOP rising tendency from uveoscleral outflow obstruction is neutralized by decreased aqueous production from the ciliochoroidal detachment.
Best treatment methods have been debated. Vortex vein decompression with scleral resection was initially advocated to treat uveal effusion associated with nanophthalmos. Gass believed that the treatment effect had less to do with vortex vein decompression than with scleral resection to facilitate protein and fluid drainage through the sclera.13 He suggested partial-thickness sclerectomies or full-thickness sclerectomies. After treatment, exudation may gradually disappear within a few months. However, chorioretinal degeneration may continue to develop from chronic mild recurrence of exudation or abnormal metabolism of mucopolysaccharide.
Vascular
Coats disease
This is a non-familial developmental retinal vasculopathy. The disease is more common in males, is usually unilaterally affected and may occur in infants. Symptoms often develop in children or young adults; one-third had symptoms onset over 30 years of age. All vessels, arteries and veins alike, would be affected, showing telangiectasis combined with a large amount of hard exudates; hemorrhagic retinopathy is occasionally seen. On the other hand, a minor form of the disease mainly involving juxtafoveolar areas may occur; decreased vision will not occur until adulthood when hard exudates and edema develop in the macula. The prognosis is directly influenced by the size of the involved area. Occasionally, other vascular anomalies may appear in the lesion eye or the fellow eye, such as macular macrovessels or arterial tortuosity (Fig. 96.5). The condition may be occasionally associated with other abnormalities such as progressive facial hemiatrophy, facial scapulohumeral muscular dystrophy and deafness, or Alport syndrome.14 It may rarely accompany systemic vascular anomalies.
Fundus examinations reveal changes of various severities. The mild form presents with focal telangiectasia and microaneurysms usually at the temporal side of the macula, with or without mild hard exudates. The moderate form ranges from cystoid macular edema with significant hard exudates surrounding the area containing telangiectatic vessels or microaneurysms, to the more extensive vascular abnormalities with massive exudates which may gain access to the subretinal space. The severe form shows wide and scattered vascular lesions, with hard exudates accumulating around the disc and in the posterior pole, causing exudative detachment. The macula may be detached with massive intra- and subretinal exudates, which later may transform to organized subretinal disciform mass or atrophic scar. These changes are likely to be found in infants and children, who visit the ophthalmic clinic because of manifest strabismus secondary to unilateral poor vision or abnormal red reflex from massive exudates in the posterior pole. The accumulation of exudates in the macula may be due to gravity-induced migration of subretinal exudates toward the central area during sleep. The deposition of lipid-rich substance along with macrophage evolves into fibrous tissue. Retinal or choroidal vessels may grow into the lesion to form a disciform scar. The most advanced form presents with bullous detachment with the retina coming in direct contact with the crystalline lens; cholesterol crystals accumulate in the subretinal space.15 There may be dilated abnormal vessels, hard exudates or hemorrhage on the surface or in the retina. However, the abnormal tortuous vessels do not dip into the subretinal space, a sign typical of exophytic retinoblastoma.
Treatment usually involves laser or cryo aiming at the lesions to decrease exudates and preserve vision (Fig. 96.6). For severe exudative detachment, external drainage should be performed first, followed by cryo to the abnormal vessels. Scleral buckling may facilitate retina reattachment, enhance cryo effect and promote regression of abnormal vessels. However, new lesions may develop in nearby or remote areas. Follow-up is crucial to detect and treat new lesions. Sector panretinal photocoagulation (PRP) may be used for a nonperfusion area. For severe cases, vitrectomy to release vitreous traction with external subretinal fluid drainage, laser or cryo may be considered. Recently, repeated intravitreal injection of bevacizumab has been reported to reduce subretinal fluid, facilitating subsequent laser or cryo.16
Accelerated hypertension and pregnancy-induced hypertension
While chronic moderate hypertension is rarely associated with choroidopathy, choroidal ischemia is more frequently associated with accelerated hypertension. Unlike retinal circulation, choroidal vessels do not possess autoregulation; the blood flow during fluctuation of systemic blood pressure is mainly regulated by sympathetic tone. When blood pressure is high, raised sympathetic tone can prevent direct pressure damage to the choriocapillaries; however, if there is a rapid rise in blood pressure, excessively increased sympathetic tone may prompt severe constriction of choroidal arteries and arterioles, leading to ischemic changes of the choriocapillaries. Choroidopathy may be separated into three stages: (1) acute ischemic phase; (2) chronic occlusive phase; (3) chronic reparative phase.17 In the first two phases, fundus examination may observe white or reddish patches in the outer retina, possibly caused by RPE necrosis; exudative detachment is often present. FA may show a large confluent area or scattered areas of choroidal filling delay; in the mid and late phases, multiple dots or a mosaic pattern of hyperfluorescence from RPE leakage may be seen. In the reparative phase, large areas of irregular REP atrophy, Elschnig spots (central hyperpigmented and peripheral hypopigmented lesion of RPE changes), or Siegrist spots (spots of pigmentary changes similar to Elschnig spots arranged linearly along choroidal vessels in the equatorial region) may be seen; exudative detachment disappears, but choroidal delayed filling remains.
Pregnancy-induced hypertension
About 1–2% of pregnant women develop exudative retinal detachment pre- or immediately post-delivery, causing visual impairment. Exudative detachment may be limited to the macular area or appear as bullous detachment. The retina may or may not show cotton-wool patches or other changes secondary to hypertension retinopathy (Fig. 96.7). Yellowish-white patches of RPE necrosis may be seen. FA shows delayed choroidal filling and multiple leaking points where RPE has been damaged. After delivery, with control of hypertension, exudative detachment rapidly subsides. Most patients have good visual recovery. The posterior pole may show RPE changes forming hyperpigmented lines or patches mixed with yellow spots of RPE atrophy. The bilateral changes may be mistaken for macular dystrophy. Severe cases may have extensive exudative detachment. Widespread RPE changes similar to tapetoretinal dystrophy and severely compromised vision may result. The cause of chorioretinal changes in pregnancy-induced hypertension is not clear. Blood pressure may not be very high before the onset of exudative detachment. Affected patients may have other symptoms and signs related to disseminated intravascular coagulation, such as hemolysis, low platelet count, and elevation of liver enzymes. It is possible that mechanisms capable of inducing disseminated intravascular coagulation (see below) are also functional in the choroid, causing choroidal ischemia, RPE damage and exudative detachment.
Diabetic retinopathy
Severe diabetic macular edema may sometimes accompany localized macular detachment. Fluid leaking out from the vessels first accumulates within the retina; beyond a certain critical point, fluid may gain access into the subretinal space causing sensory detachment. Severe macular edema not only leads to detachment but may be associated with massive hard exudates (Fig. 96.8). Hard exudates are one of the independent risk factors for vision decrease.18 In addition to vascular hyperpermeability related to diabetic vasculopathy, taut posterior hyaloid membrane may also contribute to macular edema and localized detachment either from the mechanical traction force or from traction-induced increased vascular permeability.
Clinically, early fundus changes leading to severe macular edema may present as a central retinal vein occlusion-like picture with flame-shaped hemorrhage around the disc and scattered perivascular exudates but without fluorescein angiographic evidence of disc leakage and venous delayed filling. In other cases, multiple clusters of microaneurysms may distribute in the posterior pole, accompanied by significant capillary nonperfusion. Sensory detachment and massive exudates may later develop. Exudative detachment combined with macular edema represents severe break down of the inner retinal barrier. Laser alone has little effect in such cases. Multiple sessions of intravitreal anti-VEGF (vascular endothelial growth factors) alone or in combination with subtenon or intravitreal steroid administration may effectively flatten down the retina in most cases. The effect has not been confirmed if subsequent focal laser to the leaking vascular segments or microaneurysms may obtain a more lasting effect. In some cases, exudative detachment is present without significant cystoid macular edema. It may be because the edema is in a resolving phase; thus the response to treatment may be quicker. In severe cases, exudates may consolidate and deposit within or below the macula. If the condition does not improve after several sessions of medical treatment, pars plana vitrectomy combined with hyaloid membrane removal may be performed to reduce edema and hard exudates (Fig. 96.9).19 Because the development of massive hard exudates indicates that the retina is in a relatively hypoxic state or has already gone into the early proliferative stage, panretinal photocoagulation during the operation is required to inhibit production of angiogenic factors, which may induce further macular edema or neovascularization, leading to postoperative vitreous hemorrhage or even neovascular glaucoma.19 In the case of pre-existing posterior vitreous detachment, epiretinal membrane peeling and internal limiting membrane peeling may be considered.20 Massive exudates usually form submacular plaques, affecting vision severely. After surgery, the plaque may reduce in size but does not disappear completely, leaving residual fibrosis or crystal-like deposition, causing permanent decrease of vision. Surgical removal of subretinal exudates through iatrogenic retinotomy has been reported21; the effect has not been firmly established. Patients with severe edema should have a systemic check-up, including blood pressure, blood lipid, and renal function; any abnormalities should be treated, as these may interfere with local response to the treatment.
Vascular occlusive diseases
Severe retinal vein occlusion occasionally is accompanied by serous retinal detachment. Exudative detachment has been described in branch, hemispherical, and central retinal vein occlusion (CRVO). Vascular leakage from congested retinal veins outside the macular area is the major source of subretinal fluid at the fovea. In addition, ischemic retina produces angiogenic factors, such as VEGF, which in turn increase vascular permeability. Both increased intravascular pressure and vascular permeability cause leakage of fluid and blood components into the subretinal space. In eyes with retinal vein occlusion, serous RD is typically located beneath the fovea, and the height of the RD was greatest in the fovea (Fig. 96.10).
Recent studies with OCT revealed that macular serous retinal detachment is a common complication of retinal vein occlusion. Serous RD secondary to branch retinal vein occlusion was first described by Spaide et al.22 Of the 14 eyes included in that study, 10 (71.4%) had serous RD. Yamaguchi et al. studied 109 eyes with branch retinal vein occlusion (BRVO) by OCT examination, and found that the incidence of serous RD is higher in the group with major BRVO (63%) than in the group with macular BRVO (21%).23 Ozdemir et al. found a high incidence (81.8%) of serous macular detachment in CRVO.24 In a series of 91 patients with retinal vein occlusion examined by OCT, Tsujikawa et al. reported that 76 eyes (83.5%) had serous RD involving the fovea.25 They suggested from their observations that in eyes with retinal vein occlusion, a small pointed RD developed initially just beneath the fovea, but subsequently changed into a dome-shaped RD; the foveal architecture, especially that of the Müller cell cone might be involved in the formation of serous RD.
Application of laser photocoagulation to the affected area of branch retinal vein occlusion has been reported to treat serous RD and can lead to resolution of subretinal fluid. The beneficial effect of laser treatment may be due to the ablation of ischemic retina, decrease of the production of VEGF, closure of incompetent vessels, and stimulation of RPE to enhance the reabsorption of fluid. Intravitreal injection of triamcinolone or bevacizumab has been reported to treat serous RD due to retinal vein occlusion26,27; repeated injection may be necessary for frequent recurrence of macular edema.
The visual prognosis of serous RD in retinal vein occlusion is variable. In an eye with serous RD associated with retinal vein occlusion, the outer retinal discontinuity does not necessarily lead to poor vision. If the surrounding outer segment of the foveal photoreceptors is preserved, good visual acuity will retain after macular edema and serous RD resolve. However, even after complete resolution of the macular edema and serous RD, diffuse disorganization of the outer photoreceptor layer beneath the fovea often results in poor visual acuity (see Fig. 96.11, online). In addition, a dome-shaped RD sometimes accompanies a focal defect of the outer segment of the photoreceptors above the serous RD. When the defect involves the fovea, visual prognosis is usually poor.
