Nongranulomatous Inflammation
Uveitis, Endophthalmitis, Panophthalmitis, and Sequelae
Classification
Terminology
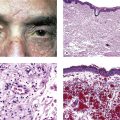
II. Endophthalmitis (Fig. 3.1) is an inflammation of one or more coats of the eye and adjacent cavities.
III. Panophthalmitis (Fig. 3.2) is an inflammation of all three coats of the eye (and adjacent cavities); it often starts as an endophthalmitis that then involves the sclera and spreads to orbital structures.
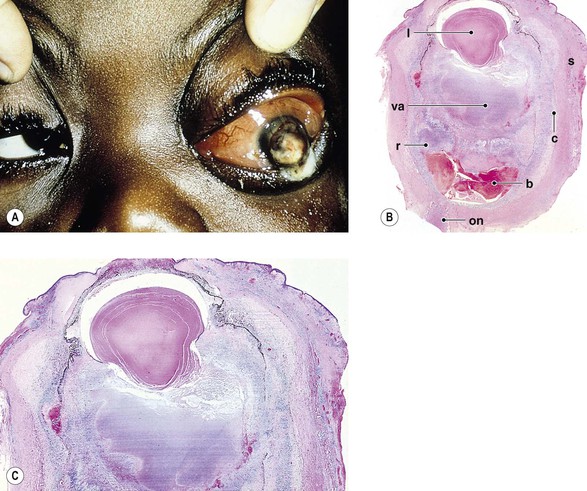
Sources of Inflammation
Suppurative Endophthalmitis and Panophthalmitis
Clinical Features
Classification
I. Exogenous
C. Postoperative suppurative inflammation in the first day or two after surgery is usually purulent, fulminating, and caused by bacteria.
2. A bacterial infection is also a possible cause of delayed endophthalmitis, especially with less virulent bacteria such as Staphylococcus epidermidis and Propionibacterium acnes (see Chapter 5).
II. Endogenous
Histology
Suppurative inflammation is characterized by a polymorphonuclear leukocytic infiltrate (Figs. 3.3 and 3.4). Tissue necrosis causes a suppurative or purulent exudate (pus).
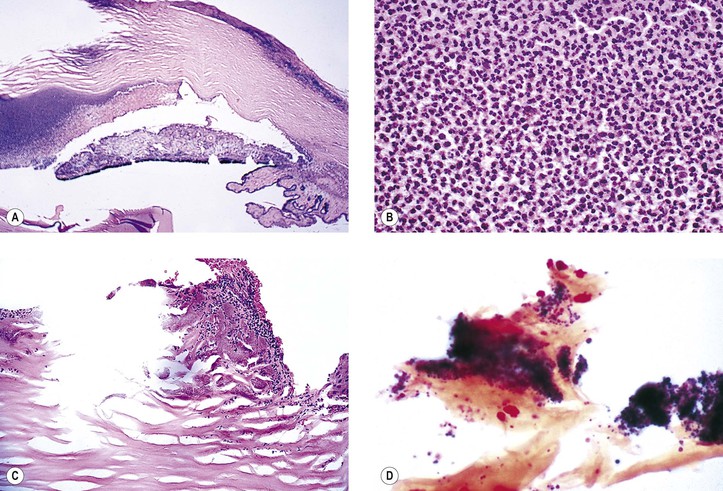
Examples
I. Behçet’s disease (see Fig. 3.3) is a chronic endogenous endophthalmitis.
D. Plasminogen activator levels may be decreased.
E. A hypercoagulable or general vascular endothelial dysfunction is usually found.
H. The ocular inflammation is characterized by recurrent iridocyclitis and hypopyon (often motile), usually involving both eyes but not necessarily simultaneously.
2. Small patches of retinal whitening are characteristic.
4. Rarely, a bilateral immune corneal ring (Wessely ring) may occur.
I. Biopsy of mucocutaneous lesions shows vasculitis.
L. Histologically, the main process appears to be a small or moderate-sized blood vessel obliterative and necrotizing vasculitis.
3. A secondary chronic nongranulomatous inflammatory infiltrate is frequently noted in adjacent tissues (see Fig. 3.4).
Nonsuppurative, Chronic Nongranulomatous Uveitis and Endophthalmitis
Clinical Features
Classification
I. Exogenous: The inflammation is usually secondary to trauma.
II. Endogenous (Fig. 3.5)
B. The inflammation may be associated with viral infections such as rubella and subacute sclerosing panencephalitis (SSPE); bacterial infections such as syphilis; local ocular (nonsystemic) entities such as pars planitis, Fuchs’ heterochromic iridocyclitis, uveal effusion (see Chapter 9), and glaucomatocyclitic crisis (Posner–Schlossman syndrome; see Chapter 16); and systemic diseases such as Reiter’s syndrome, Behçet’s disease (see earlier), Kawasaki’s disease (mucocutaneous lymph node syndrome), phacoanaphylactic endophthalmitis (the uvea usually shows a chronic, nongranulomatous uveitis; see Chapter 4), collagen vascular disease (including rheumatoid arthritis), Crohn’s disease (regional enteritis; see later in this chapter), ulcerative colitis, and Whipple’s disease (see Chapter 12); and atopy.
C. A history of cigarette smoking is a significant risk factor.
Examples
I. Viral infections such as herpes simplex and zoster, Epstein–Barr virus (EBV), SSPE, rubella (see Chapter 2), and rubeola may cause an endogenous nonsuppurative, chronic nongranulomatous uveitis.
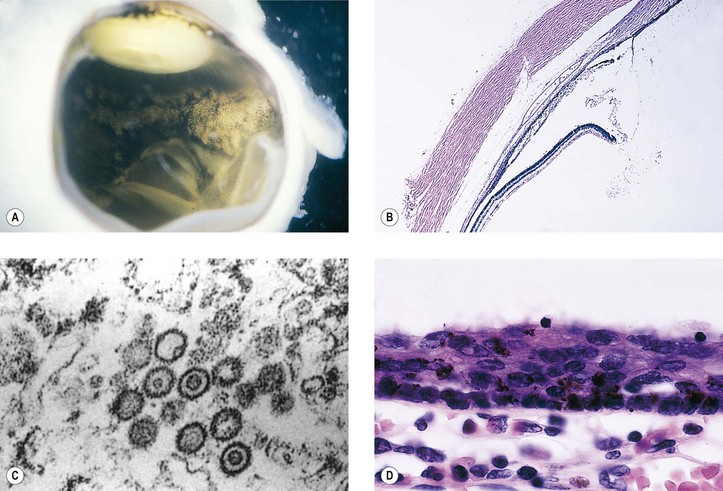
A. Herpes simplex virus (HSV; Fig. 3.6)
1. HSV consists of a linear, double-stranded DNA packaged in an icosahedral capsid and covered by a lipid-containing membrane.
2. Neonatal HSV most commonly causes a nonfollicular conjunctivitis followed by keratitis.
3. Acquired HSV in children and adults is similar to that in neonates.
a. A mucocutaneous eruption is common.
c. The most common ocular manifestation is keratitis (see Chapter 8).
4. Histologically, the infected area reveals both acute and chronic nongranulomatous inflammation.
a. Intranuclear inclusions (Cowdry type A) may be seen.
1. The EBV, a B-lymphotrophic virus that accounts for most cases of infectious mononucleosis, is associated with Burkitt’s lymphoma (see Chapter 14), and it is detected in up to 50% of B-cell malignancies encountered in immunosuppressed patients.
4. Histologically, a chronic nongranulomatous inflammation is seen.
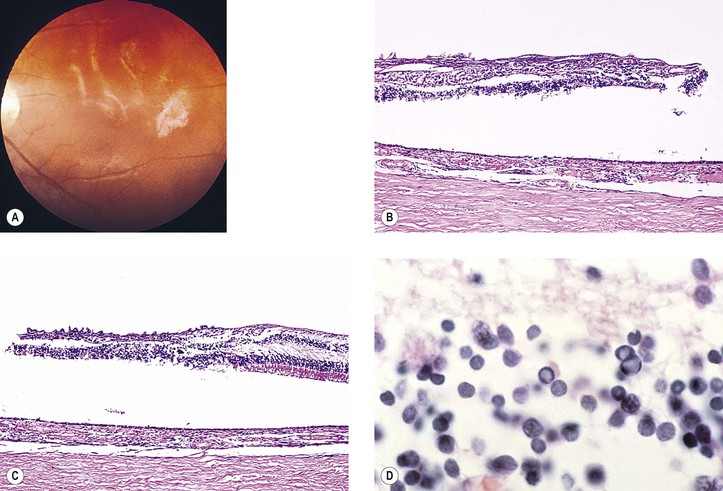
C. Subacute sclerosing panencephalitis (SSPE, Fig. 3.7)
2. The disease usually emerges 5–7 years after the child has had an uneventful measles (rubeola) infection. The ocular signs and symptoms may antedate those of the central nervous system by as long as two years.
a. Patients have high titers of measles antibody in their serum and cerebrospinal fluid.
b. Measles antigen can be demonstrated in brain tissue by immunofluorescence.
II. Local ocular (nonsystemic) syndromes such as pars planitis and Fuchs’ heterochromic iridocyclitis may cause a nonsuppurative, chronic nongranulomatous uveitis.
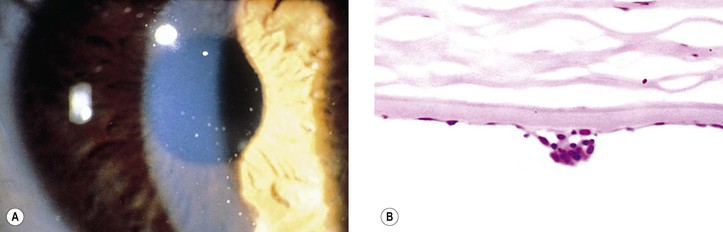
A. Pars planitis (intermediate uveitis, peripheral uveitis, chronic cyclitis)
2. Histologically, a chronic nongranulomatous inflammation of the vitreous base, retinal perivasculitis, and microcystoid degeneration of the macular retina are seen.
b. The layer appears continuous with similar preretinal fibroglial membranes.
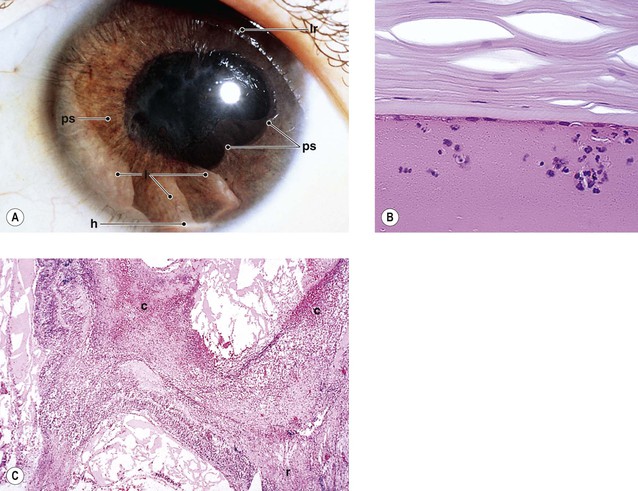
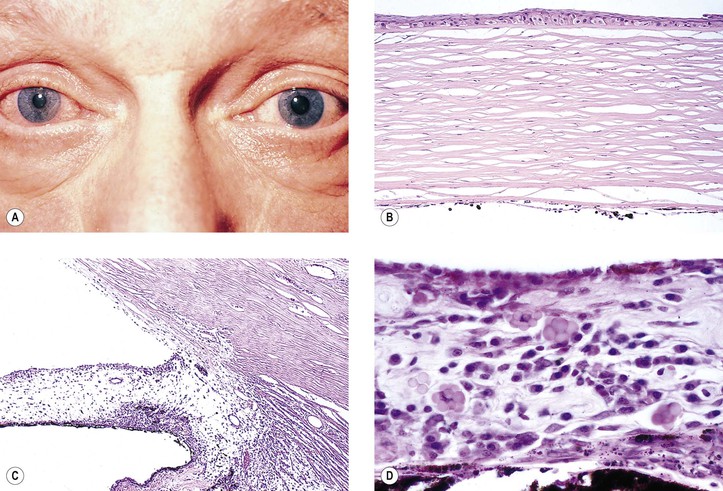
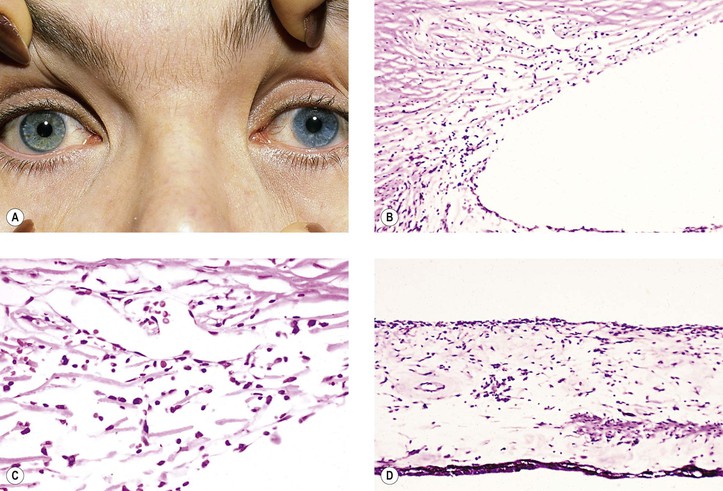
B. Fuchs’ heterochromic iridocyclitis (FHI; also called Fuchs uveitic syndrome) (Figs 3.8 and 3.9)
3. White, opalescent iris nodules may develop in black patients.
III. Systemic syndromes such as Reiter’s syndrome, rheumatoid arthritis, and Crohn’s disease
2. HLA-B27 is positive in a high percentage of patients.
2. Juvenile rheumatoid arthritis (JR) is the most common specific childhood entity associated with uveitis in children.
b. Approximately 12% of patients with JR in whom uveitis develops eventually become blind.
Sequelae of Uveitis, Endophthalmitis, and Panophthalmitis
Cornea
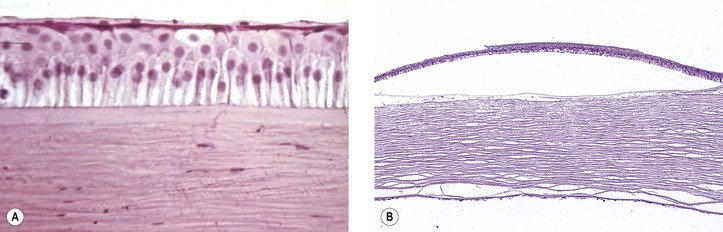
I. Corneal endothelial degeneration or glaucoma, or both, may result in chronic stromal and epithelial edema and ultimately in bullous keratopathy (Fig. 3.10).
A. Pannus degenerativus may follow bullous keratopathy.
B. Keratic precipitates of mononuclear cells (mainly lymphocytes and plasma cells) along with pigment (see Figs. 3.5B and 3.9B) may be found on the endothelium.
II. Ruptured corneal bullae may become infected secondarily, leading to a corneal ulcer.
III. Band keratopathy (i.e., calcium deposition; see Figs. 8.23–8.25) is common beneath the corneal epithelium in chronically inflamed eyes, especially in children who have Still’s disease.
Iris
II. Chronic anterior uveitis may induce peripheral anterior synechiae formation (see Fig. 3.5C).
III. Neovascularization of the anterior surface of the iris (rubeosis iridis or red iris, as seen clinically) may cause secondary anterior chamber angle synechiae and secondary angle closure.
Shrinkage of the fibrovascular membrane on the anterior iris surface may evert the pupillary border of the iris, termed an ectropion uveae (Fig. 3.11; see Fig. 15.5).
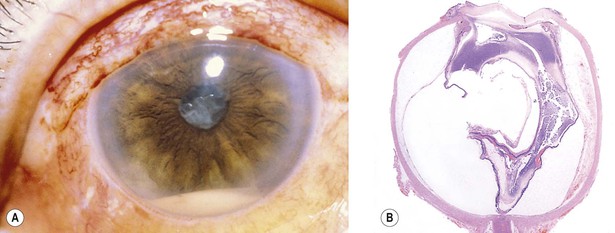
IV. Inflammatory and fibrous iris membranes may attach the pupillary margin of the iris to an underlying lens, to a lens implant or lens capsule in pseudophakic eyes, or to the anterior surface of the vitreous in aphakic eyes, resulting in an immobile pupil, seclusio pupillae (Fig. 3.12).
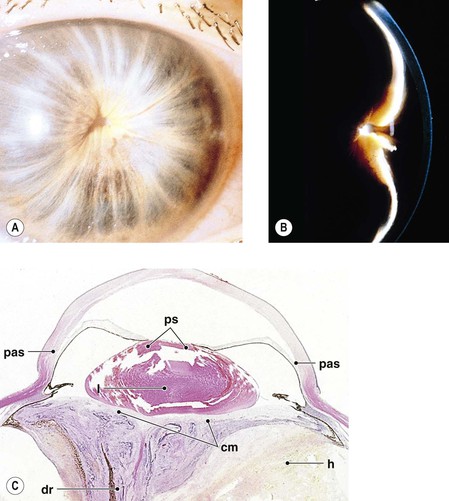
The same membrane may grow over the pupil and cover or occlude the area completely, called occlusio pupillae (see Fig. 3.12).
V. Total (i.e., 360°) posterior synechiae cause a complete pupillary block, preventing aqueous flow into the anterior chamber.
A. The pressure builds up in the posterior chamber, bowing the iris forward (iris bombé; see Fig. 3.12).
Lens
Ciliary Body
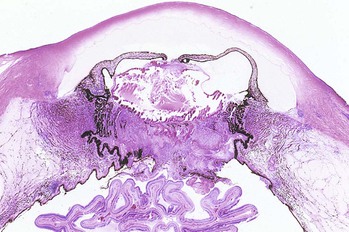
III. Intraocular inflammation may organize and fibrose behind the lens or lens implant (or behind the pupil in an aphakic eye) between portions of the pars plicata of the ciliary body. Such a fibrous membrane spanning the retrolental space and often incorporating proliferated ciliary epithelium and vitreous base is called a cyclitic membrane (see Fig. 3.12C; Fig. 3.13).
Glaucoma
End Stage of Diffuse Ocular Diseases
B. The best example is the diffuse atrophy with long-standing glaucoma.
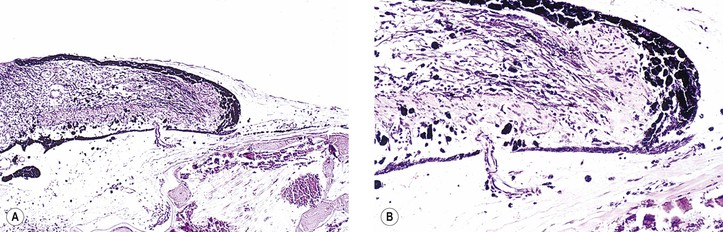
II. Atrophy with shrinkage (atrophia bulbi; Fig. 3.14)
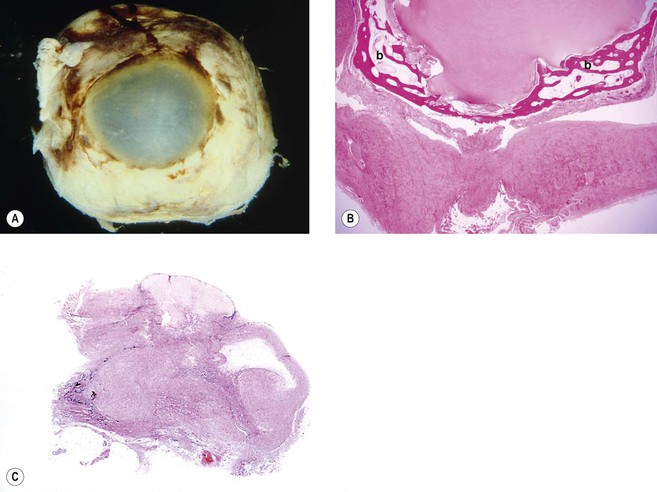
III. Atrophy with shrinkage and disorganization (phthisis bulbi; see Fig. 3.14)
A. This is common in atrophia bulbi (see Figs. 3.14 and Fig. 18.11) and phthisis bulbi.
Access the complete reference list online at