Chapter 49 Neuropsychopharmacology
Introduction
Historically, very few safe and effective medications were available to treat pediatric neuropsychiatric disorders. Children received either no treatment, or treatment that emphasized psychological or behavioral interventions. With a growing evidence base of tolerable and effective psychotropic medications, psychopharmacology has become standard practice for many neuropsychiatric disorders, resulting in increased use of medications in children [Zito, 2007; Zito et al., 2003] and broadening of the practitioner base for prescribing [Olfson et al., 2002].
The database for the safety and efficacy of psychotropic medications in children has increased dramatically in the past decade, although much remains to be done. There is support for the short-term efficacy of many commonly prescribed psychotropics, but there is limited data on their long-term efficacy and safety, the comparative efficacy of psychological and pharmacologic treatments, and the best practices for combining psychological and pharmacologic interventions. For example, there are excellent efficacy and safety data for the short-term treatment of attention-deficit hyperactivity disorder (ADHD) with stimulants [Greenhill et al., 2006; Jensen et al., 2001], of anxiety disorders with selective serotonin reuptake inhibitors [Birmaher et al., 2003; RUPP, 2001; Rynn et al., 2001; Walkup et al., 2008], of obsessive-compulsive disorder (OCD) with clomipramine and selective serotonin reuptake inhibitors [DeVeaugh-Geiss et al., 1992; Geller et al., 2001, 2004; March et al., 1998; POTS, 2004; Riddle et al., 2001], of depression with selective serotonin reuptake inhibitors [Emslie et al., 1997, 2002, 2009; TADS, 2004], and of schizophrenia [Findling et al., 2008b; Haas et al., 2009b; Kryzhanovskaya et al., 2009; Kumra et al., 1996, 2008], bipolar disorder [Delbello et al., 2002, 2006, 2008; Findling et al., 2005, 2009a; Haas et al., 2009a; Tohen et al., 2007], and irritability and behavioral dysregulation in different pediatric disorders with atypical antipsychotics [Aman et al., 2002; Hollander et al., 2006; McCracken et al., 2002; Owen et al., 2009]. In contrast, there remain very little data from placebo-controlled efficacy studies on the short-term benefit of mood stabilizers (e.g., lithium, anticonvulsants) for neuropsychiatric disorders [Wagner et al., 2006, 2009]. There is little information on the long-term usefulness of any psychotropic medication and also increasing concern about the long-term effects of psychotropic drugs on growth and development [MTA, 2004; Nilsson et al., 2004; Swanson et al., 2006; Weintrob et al., 2002]. Although medication combinations are commonly used in children [Martin et al., 2003], studies of medication combinations are uncommon [Abikoff et al., 2005; Delbello et al., 2002].
The expansion of the evidence base in pediatric neuropharmacology has been due, in large part, to the National Institute of Mental Health (NIMH)’s support of partnership between government, academic institutions, and drug companies [Satcher, 2001], and to legislation supporting medications studies in children. In 1997, the passage of the Food and Drug Administration (FDA) Modernization Act mandated the study of medication in children and offered 6-month patent extensions to pharmaceutical companies who studied their products in pediatric populations. As a result, a number of efficacy and safety studies of newer medications targeting a variety of disorders in children and adolescents emerged and contributed to a growing evidence base of medications that have FDA pediatric labeling. The list of psychotropic medications that have FDA approval for use in the treatment of psychiatric conditions in children has grown (Table 49-1). However, a number of studies supporting the safety and efficacy of psychotropic medication (e.g., stimulants for ADHD in children with tics [TSSG, 2002]; selective serotonin reuptake inhibitors for childhood anxiety [Ipser et al., 2009]) are not reflected in current labeling, leaving clinicians in an awkward position as to whether to prescribe medications with evidence for efficacy but without an FDA indication or appropriate labeling. Additionally, older medications that have gone off patent have no labeling, have FDA labeling but have not been subject to the rigorous study, or have out-of-date labeling. As a result, many psychotropic medications prescribed for children are prescribed off-label. Off-label use of medications increased in the past two decades for a variety of indications, including stimulants for children younger than 5 years, selective serotonin reuptake inhibitors for OCD and anxiety, and atypical antipsychotics for aggression. Increased off-label use resulted from downward extension of a current indication to a younger age group (e.g., stimulants in children younger than 5 years, antidepressants in teens and children) and the availability of newer medications with a reduced potential for serious side effects. Sometimes, off-label use has been extremely helpful (e.g., selective serotonin reuptake inhibitors for anxiety disorders), but some off-label prescribing may not be helpful. For example, based on its low side-effect profile, a few positive case reports and open trials, and an aggressive marketing campaign, gabapentin was prescribed for the treatment of bipolar affective disorder; however, subsequent controlled trials have not supported its efficacy [Frye et al., 2000; Pande et al., 2000].
Table 49-1 Labeled and Off-label Use of Neuropsychopharmacologic Agents in Children and Adolescents
| Drug | Labeled Use in Children and Adolescents | Off-label Use/Clinical Practice in Children and Adolescents |
|---|---|---|
| Stimulants Amphetamine, mixed salts Dextroamphetamine |
ADHD (≥3 yrs – IR, ≥6 yr – ER, Narcolepsy (≥6 yrs, IR only) ADHD (≥6 yrs), Narcolepsy (≥6 yrs) ADHD (≥6 yrs), Narcolepsy (≥6 yrs) |
|
| Pemoline | None (withdrawn 2005) | ADHD |
| Atomoxetine | ADHD (≥6 yrs) | |
| Clonidine | ADHD (6–17 yrs – ER) | Aggression, Tic disorders |
| Guanfacine | ADHD (6–17 yrs – ER) | Aggression, Tic disorders |
| Benzodiazepines Clonazepam Lorazepam |
None Anxiety (≥12 yrs), Insomnia (≥12 yrs) |
As a group – agitation, catatonia |
| Tricyclic Antidepressants Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline |
Depression (≥12 yrs) OCD (>10 yrs) None Enuresis Depression |
As a group – depression, anxiety disorders, ADHD and pain syndromes |
| Buproprion | None | Depression, ADHD |
| Serotonin Norepinephrine Re-uptake Inhibitors Desvenlavaxine Duloxetine Venlafaxine |
None |
Depression, anxiety disorders |
| Mirtazapine | None | Depression, Anxiety disorders |
| Serotonin Re-uptake Inhibitors Citalopram Escitalopram Fluoxetine Fluvoxamine Paroxetine Sertraline |
None Depression (≥12 yrs) OCD (≥7 yrs), Depression (≥8 yrs) OCD (≥8 yrs) None OCD (≥6 yrs) |
As a group – depression, anxiety disorders |
| Lithium | Bipolar Disorder (≥12 yrs) | Aggression |
| Anticonvulsants Carbamazepine Gabapentin Lamotrigine Topiramate Valproate |
Seizure disorders Seizure disorders Seizure disorders Seizure disorders Seizure disorders |
As a group – bipolar disorder, aggression, mood instability |
| Typical Neuroleptics Chlorpromazine Haloperidol Piimozide |
Severe behavioral disturbances Psychosis, tics, severe behavioral disturbance Tics |
As a group – psychotic disorders |
| Atypical Neuroleptics Aripiprazole Clozapine Quetiapine Risperidone Ziprasidone |
Irritability in Autistic Disorder (6-17 yrs) Bipolar I Disorder (≥10 yrs), Schizophrenia (≥13 yrs) None Bipolar I Disorder (≥13 yrs), Schizophrenia (≥13 yrs) Bipolar I Disorder (≥10 yrs), Schizophrenia (≥13 yrs) Irritability in Autistic Disorder (6-17 yrs), Bipolar I Disorder (≥10 yrs), Schizophrenia (≥13 yrs) None |
As a group – psychotic disorders, Tourette’s syndrome, aggression |
IR = immediate release, ER = extended release
ADHD, Attention Deficit Hyperactivity Disorder, OCD, Obsessive –Compulsive Disorder
There has also been an increased interest in the perceived safety of commonly prescribed psychotropic agents in children. Although short-term efficacy studies have often demonstrated safety, post-marketing studies have raised some concerns: for example, data suggesting growth suppression with stimulants [MTA, 2004; Swanson et al., 2006] and antidepressants [Nilsson et al., 2004; Weintrob et al., 2002]; increased risk of suicidal ideation in youth [Hammad et al., 2006] and young adults [Stone et al., 2009] with antidepressants; and weight gain and metabolic syndrome with antipsychotics [Fraguas et al., 2010; Correll, 2008]. These adverse events and public concern about the use of medications, particularly in children, have reinforced the importance of on-going research to elucidate the efficacy and safety of psychotropic medications.
The decision about whether to use pharmacologic interventions mandates careful consideration of the benefits and the risks of treatment and of the ability to monitor treatment safely [AACAP, 2009]. To justify the use of medications, the child must first have a disorder or target symptoms with the potential for pharmacologic responsiveness. Second, the child’s level of impairment must cross a threshold of severity such that failure to treat with medication, given all the potential risks, would cause more harm. Third, the child’s symptoms must be unresponsive or insufficiently responsive to nonpharmacologic interventions, or these interventions are not readily available in the community. Finally, the clinician must have the time available to monitor patients adequately – not only for common, and also for rare and important adverse events (e.g., suicidal ideation or behavior). Safe prescribing of medications for children also requires detailed documentation of the decision-making process and an active monitoring plan for outcome and adverse events.
The likelihood of a child with a neuropsychiatric disorder or symptoms presenting to a pediatric neurology practice is high. To treat these children effectively, the pediatric neurologist must have the ability to collect and integrate information from multiple sources (i.e., child, family, school, and other agencies) and to make a diagnostic formulation and treatment plan that addresses the neuropsychiatric symptoms and psychosocial factors that affect the delivery of care and the assessment of outcome. Given the rapidly changing nature and increasing complexity of modern clinical care, pediatric neurologists need to define their comfort level in assessing and treating neuropsychiatric disorders and to determine how this may influence the scope of their practice. For neurologists who treat neuropsychiatric disorders, it is critical to keep abreast of this evolving field, especially new safety data and data on the efficacy of nonpharmacological treatment, e.g., behavioral treatment for tics [Piacentini et al., 2010], that influence the standard of care. This may require a team of colleagues, including psychologists and psychiatrists, who can be involved in the assessment and treatment of these children with complex neuropsychiatric needs.
Stimulants
This section briefly describes the use of stimulants and nonstimulants in the treatment of ADHD (Table 49-2; also see Chapter 47).
Table 49-2 Stimulant and Nonstimulant Medications for Attention-Deficit-Hyperactivity Disorder: Preparations, Pharmacology, and Dosing
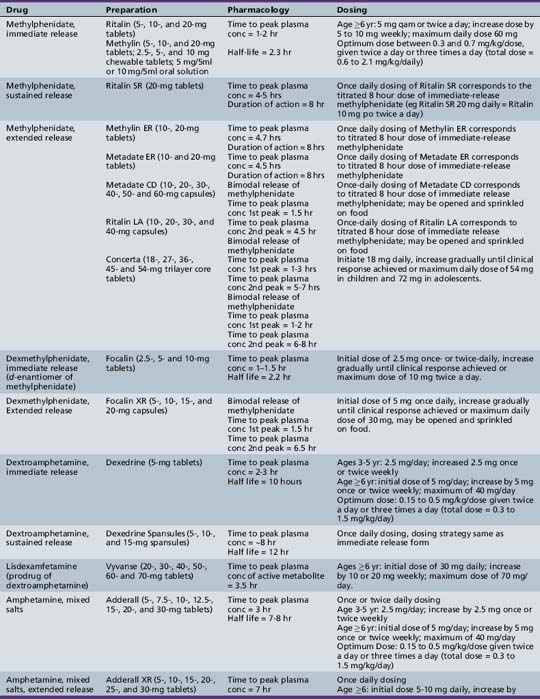
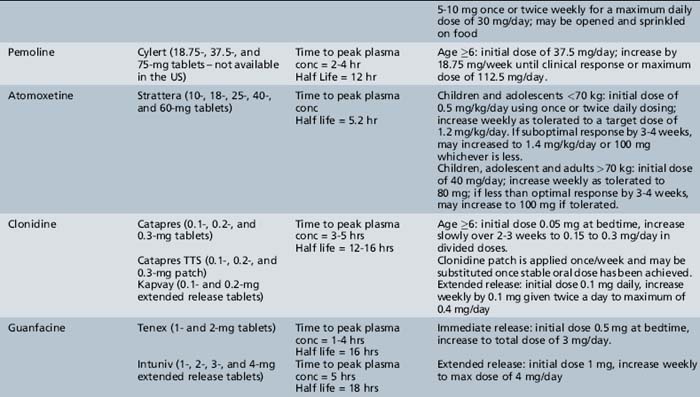
Clinical Applications
Stimulants have been in clinical use since 1937, when it was observed that a group of children in residential treatment showed marked improvement in their behavior with benzedrine (d– and l-amphetamine) [Bradley, 1937]. Since then, the criteria for ADHD have been refined, and other stimulant medications have been evaluated and consistently found efficacious in numerous placebo-controlled studies [Jensen et al., 2001]. Approximately 70–80 percent of school-age patients with ADHD have a positive response to stimulant medication. Although behavioral treatment may be considered, in head-to-head studies medication alone is more beneficial than behavioral therapy alone in children 5 years of age and older. The combination of medication and behavioral therapy is specifically helpful for oppositional behavior and children with ADHD and anxiety [Jensen et al., 2001].
Based on clear evidence of efficacy, the stimulant medications have been used to address attention, hyperactivity, and impulsivity in those with pervasive developmental disorders and in younger children. For many years, stimulant medications were viewed as poorly effective or contraindicated for hyperactivity in children with autism and pervasive developmental disorders. However, in the first large, randomized, controlled trial in this population, methylphenidate demonstrated efficacy. The response rate was less robust than in typically developing children, and rates of adverse events were higher, with nearly 1 in 5 youngsters having to stop treatment [RUPP, 2005]. A single test dose of stimulant may be useful to identify children for whom a longer stimulant trial is contraindicated [Di Martino et al., 2004]. Children who experience a significant worsening of ADHD (e.g., hyperactivity and irritability) or other symptoms (i.e., tics or stereotypies) with a single dose may be excluded from further stimulant treatment [Di Martino et al., 2004].
Preschoolers with ADHD represent another specialized population for whom the use of stimulants may be considered. The Preschoolers with ADHD Treatment Study (PATS), a large randomized, placebo-controlled trial of methylphenidate in children of ages 3–5 years, demonstrated that methylphenidate at doses greater than 2.5 mg three times a day were effective in reducing ADHD in youngsters unresponsive to psychosocial intervention. Again, response rates were less robust than that observed in school-age children, and adverse effects were more common [Greenhill et al., 2006]. Furthermore, methylphenidate may reduce growth rates, even in short-term treatment, and requires careful monitoring of weight and height [Swanson et al., 2006].
Despite substantial data supporting the efficacy of stimulants, their use in children remains controversial. The lay public and media have expressed concerns about the potential for overdiagnosis of ADHD and overuse of stimulant medications in children. However, despite the dramatic increases in stimulant use in the past 10 years [Zito et al., 2003], there is little evidence to support this claim. According to the U.S. Surgeon General, most children with psychiatric disorders are not assessed or treated [Satcher, 2001], and the ratio of stimulant use to prevalence of ADHD is less than 1:1, suggesting that undertreatment is a more prevalent and important issue [Jensen et al., 1999]. Stimulant medications have also come under public scrutiny because of concerns about safety. Concerns about worsening tics, growth suppression, enhancing risk for addiction and, more recently, sudden death, although not substantiated by the evidence, remain important issues for families considering stimulant medications in their child. Some studies actually suggest that children with ADHD who are treated with stimulant early in life have a lower risk of substance abuse than children with ADHD who are not treated [Wilens et al., 2003], but such evidence may not be as reassuring to parents as one would hope. Prudent practice therefore necessitates an evaluation that leads to confidence in the diagnosis, fully informed consent, and use of appropriate doses with close monitoring, combined with effective and available psychosocial treatments. Detailed discussion, in the assessment and treatment phases, with the family and teachers, backed up by full documentation in the medical record, is essential. During the dose adjustment phase, children often are monitored for side effects, including blood pressure, pulse, height, and weight. After a maintenance dose is achieved, visit intervals can vary from 1–4 months.
Pharmacology
Stimulants are sympathomimetic drugs that directly stimulate α- and β-adrenergic receptors. They also stimulate the release of dopamine from presynaptic nerve terminals and inhibit dopamine reuptake. The exact mechanism for efficacy on attention and hyperactivity in ADHD is unknown. Stimulant medications are available in immediate-release and sustained-release preparations (see Table 49-2).
Clinical Management
Assessment
A neuropsychiatric assessment of inattention, impulsivity, and hyperactivity involves gathering information from multiple sources, including the child, parents, teacher, therapist, or other individuals involved with the child (e.g., day-care providers and coaches). Information can be gathered through clinical interview; patient, parent, and teacher rating scales; neuropsychological testing; and medical evaluation, including laboratory screening [AACAP, 1997]. The diagnostic criteria for ADHD are detailed in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR [APA, 2000]). A thorough neuropsychiatric assessment is necessary because inattention, impulsivity, and hyperactivity can be complications of medical conditions, and they are commonly caused by other neuropsychiatric disorders. Children with ADHD often have co-occurring neuropsychiatric disorders, which can make it difficult to determine accurately whether ADHD symptoms are attributable to ADHD or to the co-occurring conditions. Medical and developmental problems in the differential include vision or hearing deficits, seizures, chronic medical illnesses, sleep deprivation, and poor nutrition [Pliszka, 2007]. Children who have unidentified intellectual deficits, learning disabilities, speech and language problems, or substance abuse may appear to exhibit ADHD symptoms. Neuropsychiatric conditions, such as anxiety disorders, depressive disorders, and bipolar disorder, have disturbances in attention, impulse control, and activity level that can be especially difficult to distinguish from ADHD.
Identification of co-occurring conditions and risk factors for adverse effects is essential because some medications may be contraindicated, or medication management may require dosing modification or closer monitoring. For example, a patient with co-occurring ADHD and anxiety or depression may prompt the clinician to initiate treatment with an antidepressant first rather than a stimulant, depending on which of the two conditions is more impairing. A child with a pervasive developmental disorder may have limited response to stimulants or may have an adverse behavioral reaction that can be severe. Similarly, despite evidence that stimulants are unlikely to cause tic worsening relative to placebo [Kurlan, 2003], a patient with a personal or family history of tic disorders may develop tics de novo or experience a worsening of existing tics during an initial stimulant trial.
A medical evaluation to “clear” a child before initiating most psychotropic medications, including stimulants, is prudent. The evaluation should include a recent medical history and a physical examination completed by the primary care provider. Any significant change in health during treatment requires repeat evaluation. Choice of laboratory screening procedures and imaging studies is guided by the medical history and findings on physical or neurologic examination. These may include lead level, thyroid function tests, genetic screening, metabolic studies, magnetic resonance imaging, sleep studies, and electroencephalography (EEG). Other assessments may be indicated, including occupational therapy, physical therapy, speech and language evaluation, and neuropsychology [AACAP, 1997]. For stimulant medications, baseline height, weight, family or personal history of a tic disorder, and family or personal history of cardiac disease, including nonvasovagal syncope and sudden death, are also important. Although baseline electrocardiograph (EKG) screening for all children has been advocated by some, at present the American Academy of Pediatrics and the American Heart Association do not recommend EKG screening of all patients, but rather that the need for an EKG be considered by the treating clinician based on physical exam and history [Vetter et al., 2008].
Initiating medication and dose titration
The starting dose of methylphenidate is 5 mg, given twice daily and dispensed after breakfast and lunch to minimize appetite-suppressant effects. The dose is increased at weekly intervals by increments of 5–10 mg until the therapeutic effect is achieved or side effects are encountered. The manufacturer recommends a maximum daily dose of 60 mg [Ritalin, 2004]; however, some clinicians exceed this figure if higher doses are necessary to control symptoms and the patient is not experiencing adverse effects. Detailed informed consent and adequate monitoring and documentation are essential for safe prescribing. Severity of symptoms, after-school activities, or homework demands may warrant a late-afternoon dose. An alternative method is to calculate the dose by weight [Dulcan, 1990]. The target dose falls between 0.3 and 0.7 mg/kg, administered 2–3 times each day (total dose of 0.6–2.1 mg/kg/day).
With d-amphetamine and mixed salts preparations, the starting dose for children between the ages of 3 and 5 years is 2.5 mg/day, with incremental increases of 2.5 mg weekly until a therapeutic response is achieved. For children 6 years or older, the starting dose is 5 mg given once or twice each day, with weekly increases of 5 mg until a therapeutic response is achieved. The manufacturer does not recommend exceeding 40 mg/day [Adderall, 2004; Green, 2007]. As with methylphenidate, some clinicians use higher doses if clinically indicated. Dosing by weight, the optimum dose may fall between 0.15 and 0.5 mg/kg, administered twice or three times daily, with a maximum daily dose of 0.3–1.5 mg/kg/day [Dulcan, 1990].
Sustained-release preparations have been around for a long time but the options were limited until the mid 1990s. Earlier sustained-release preparations of methylphenidate and d-amphetamine were viewed clinically as less reliable than their immediate-release counterparts [Birmaher et al., 1989; Fitzpatrick et al., 1992; Pelham et al., 1987], and head-to-head comparisons of their efficacy compared to their immediate-release counterparts were lacking. Over the past decade, new and novel delivery systems have developed, which has significantly increased the range of available sustained-release preparations. In clinical practice, many patients experience comparable or greater benefit from sustained-release preparations because of the convenience of once-daily dosing, avoiding dosing at school, increasing compliance, and reducing rebound effects.
Monitoring stimulants
Some clinicians consider the use of “drug holidays” on weekends or during the summer to limit children’s exposure to stimulants and minimize possible long-term adverse effects, such as low weight and growth suppression. However, the practice of drug holidays is controversial because it may result in the rapid return of symptoms and accompanying impairment [Martins et al., 2004; Spencer et al., 2006]. To identify patients who may do well with drug holidays, clinicians can review symptoms and impairment from times of the day when children are not benefiting from stimulants, such as early morning or later in the evening. If patients do well during these times, it may be reasonable to consider more extended time off stimulants. Drug holidays should be planned for times when there are no important school or social activities and they should be reserved for patients whose families can provide adequate structure, behavior management, and supervision during these periods.
Although hyperactive symptoms tend to improve as children mature, inattention and impulsivity often persist. Long-term stimulant treatment may be necessary, as up to 80 percent of ADHD children continue to have symptoms into adolescence and 65 percent into adulthood [Barkley, 1996; Weiss and Hechtmann, 1993].
Adverse Effects
Although most children tolerate stimulant medications well, some do not. The most common side effects of stimulants are insomnia and nervousness [Ritalin, 2004]. Some children with ADHD experience sleep disturbance before exposure to stimulants. The child’s pre-existing sleep history should be obtained, including other factors that may contribute to sleep disturbance (e.g., poor sleep hygiene, caffeine use, oppositional behavior, separation anxiety). Ironically, some children experience difficulty falling asleep related to hyperactivity, and they may actually benefit from a small dose of stimulant to help them stay in bed and fall asleep. In group comparisons, dosing three times daily with short-acting stimulants versus dosing two times daily was not found to increase sleep disturbance [Kent et al., 1995; Stein et al., 1996]; however, new-onset sleep delay after starting stimulants may be associated with a stimulant dose that is too high overall or with dosing stimulants too late in the day. Difficulty with falling asleep because of stimulants can be addressed with a combination of dose adjustment, improved sleep hygiene (i.e., straightforward ritual for falling asleep, and removal of distractions, such as televisions, video games, homework, and night lights, from the room), cooler room temperatures, room-darkening shades, avoidance of late-afternoon or evening doses of stimulants, switching to a different stimulant, or addition of a sedating medication with ADHD treatment effects, such as clonidine [Brown and Gammon, 1992; Prince et al., 1996] or guanfacine.
Short-term reductions in expected weight gain and height have been reported in many studies [MTA, 2004; Swanson et al., 2006], although this has not been universally reported [Spencer et al., 2006; Zeiner, 1995]. Overall, the risk of growth suppression appears mild in school-age children but is moderate in preschool-age children. The Preschool ADHD Treatment Study (PATS) reported an annual reduction in growth rate of 20 percent for height and 50 percent for weight in preschoolers treated with methylphenidate compared to those that were not on stimulants [Swanson et al., 2006]. Because the impact on weight is likely mediated by stimulant-induced appetite suppression, administering stimulant doses immediately after meals [Swanson et al., 1983], allowing the child to eat later in the evening after the appetite suppressant effect of the stimulant has abated, or allowing drug holidays on weekends or evenings may mitigate these effects.
The possibility of growth suppression with long-term use of stimulants remains a controversial topic. Longitudinal studies of children with ADHD treated with stimulants followed into adulthood have not revealed any reduction in height or weight compared to adults who were never exposed to stimulants [Hechtman et al., 1984; Klein and Mannuzza, 1988]. The apparent lack of impact on adult height was thought to result from the discontinuation of stimulants in adolescence. An alternative theory proposes that this pattern of growth is inherent in children with ADHD and not related to stimulant treatment. The dysregulation of several neurotransmitter systems associated with ADHD may alter neuroendocrine function, including those involving growth [Spencer et al., 1998]. It is not clear whether growth suppression identified in the NIMH Multimodal Treatment Study of ADHD (MTA) or PATS studies will continue and have a long-term impact, resolve with stimulant discontinuation, or resolve on its own [Spencer et al., 1998].
Historically, stimulant treatment has been associated with the emergence of tics [Borcherding et al., 1990] or the exacerbation of pre-existing tics [Law and Schachar, 1999]. Children at risk appeared to be those with a personal history or family history of tic disorders. However, the effect on tics is not absolute. In placebo-controlled trials, low to moderate doses of methylphenidate improve attention and behavior in children with chronic tic disorders without significantly worsening tics [Castellanos et al., 1997; Gadow et al., 1995, 1999]. In the Treatment of ADHD in Children with Tics (TACT) study, tic increases categorized as an adverse event occurred in nearly equal numbers of subjects on placebo (22 percent), clonidine (26 percent), and methylphenidate (20 percent). These data suggest that 20–26 percent of youngsters with tics will experience tic worsening shortly after initiation of any medication treatment, including placebo, lending support to the hypothesis that tic increases observed after starting stimulants reflect the natural waxing and waning of tic severity.
Although the product information for the stimulants warns of possible decreases in seizure threshold, no increase in seizure frequency was observed in children treated with stimulants who had co-occurring ADHD and seizure disorder [Feldman et al., 1989; Wroblewski et al., 1992]. There is no increased risk of addiction from appropriate treatment with stimulants for ADHD. Adolescents with ADHD treated with stimulants were less likely to abuse stimulants [Faraone and Wilens, 2003; Hechtman, 1985].
Concerns that stimulants may increase the risk of sudden unexplained death in children followed the publication of case reports and small case series in the early 1990s [Nissen, 2006]. A recently published case-control study comparing 564 cases of sudden death in children of 7–19 years with matched controls who died as passengers in motor vehicle traffic accidents over an 11-year period in the U.S. revealed an association between unexplained sudden death and use of stimulants [Gould et al., 2009]. This finding has enhanced concern that sudden death may be a rare side effect of stimulant use in children. However, the study design does not permit the establishment of causality. The possibility that having ADHD itself confers additional risks for sudden death must be considered, as should the co-occurring increased incidence of risk-taking behavior and substance abuse [Vitiello and Towbin, 2009]. Strategies for managing the cardiac risk of stimulants have been published [Vetter et al., 2008].
Despite the impact on a broad range of ADHD symptoms, including irritability, stimulant medications can be associated with undesirable changes in mood and behavior [Gadow et al., 1992]. Some of these changes occur when the stimulant is reaching peak serum concentrations, or they may occur when serum concentrations are waning – so-called rebound effects. Effects associated with peak concentrations include dysphoria, anxiety, agitation, or the “zombie” effect (i.e., over-focused and passive). Rebound effects include overactivity, talkativeness, excitability, irritability, and insomnia [Rapoport et al., 1978; Zahn et al., 1980]. It is often difficult to determine whether such rebound symptoms are the re-emergence of ADHD symptoms once the patient is off stimulants or true withdrawal or rebound symptoms. These adverse behavioral effects can be managed in a variety of ways, including dose reduction or medication discontinuation, and by improving late-afternoon coverage by adding a low dose of immediate-release stimulant or by switching to a long-acting stimulant [Pliszka, 2007]. Youngsters who experience these side effects on one of the stimulants do not necessarily have the same behavioral effects on another stimulant, and switching to another stimulant or alternative medication can be useful. Clinicians should be comfortable using all stimulant preparations to optimize treatment effects and minimize side effects.
Pemoline (Cylert©), a long acting stimulant with a novel structures has been associated with rare cases of acute hepatic failure [Shevell, 1997]. In 2005, the FDA withdrew labeling of pemoline for use in the treatment of ADHD, and subsequently the pharmaceutical companies voluntarily agreed to stop sales and manufacture of pemoline [Hogan, 2000].
Drug Interactions
The safety of combining stimulants, especially methylphenidate, with clonidine has been controversial. Clonidine has been helpful in managing sleep disturbance related to ADHD or stimulant treatment [Wilens et al., 1994]. The combination appears to help children with ADHD and co-occurring oppositional and conduct disorder [Hunt et al., 1990]. The combination also appears useful in the treatment of ADHD in children with tic disorders [TSSG, 2002; Wilens et al., 2003]. Concerns about the interaction developed after the report of four deaths of children who apparently received clonidine and methylphenidate, as well as other medications in some cases [Fenichel, 1995]. Although the cases did not have a clear pattern of causality [Sallee et al., 2000a; Wilens et al., 1999], clinicians are more cautious about prescribing this combination. When combining clonidine and stimulants, careful screening is recommended for a personal and family history of cardiac abnormalities, arrhythmias, and nonvasovagal syncope, and a baseline and follow-up EKG may be warranted.
Nonstimulant Medications
Atomoxetine
Clinical Applications
Atomoxetine potentially offers several advantages over stimulant medications: longer duration of action, lower misuse or abuse potential, lower risk of rebound effects, lower risk of precipitating tics or psychosis, and ease of prescribing because duplicate or paper prescriptions are not required and multiple refills are possible. Effectiveness of atomoxetine in children has been supported by placebo-controlled trials [Kelsey et al., 2004; Spencer et al., 2002]. Atomoxetine doses of 1.2 and 1.8 mg/kg/day administered in divided doses were superior to placebo and atomoxetine in a dose of 0.5 mg/kg/day; there was no clear superiority of 1.8 versus 1.2 mg/kg/day. Early clinical trials used twice-daily dosing, but once-daily dosing also appears effective [Michelson et al., 2004]. Longer-term treatment (9 months) with atomoxetine appears to be safe and well tolerated [Michelson et al., 2004]. Children maintained therapeutic effect and demonstrated superior psychosocial functioning compared to children who received placebo [Kratochvil et al., 2002]. When compared with methylphenidate treatment, atomoxetine was associated with comparable therapeutic effects [Michelson et al., 2003]. Efficacy of atomoxetine in adults with ADHD was demonstrated at doses of 60–120 mg/day [Strattera, 2010].
Pharmacology
Atomoxetine is well absorbed through the gastrointestinal tract and is metabolized by the liver, predominantly by cytochrome P-450 2D6 (CYP2D6). A small percentage of the population (approximately 7 percent of whites and 2 percent of African Americans) has reduced activity of the cytochrome P-450 isoenzyme system. These individuals are considered to be “poor metabolizers” of medications whose predominant method of metabolism is this isoenzyme. Poor metabolizers can be expected to achieve higher than expected plasma concentrations on a given dose and a prolonged half-life compared with those with normal CYP2D6 activity. The half-life of atomoxetine in poor metabolizers is 21.6 hours, compared with 5.2 hours in normal metabolizers. Atomoxetine is 98 percent bound to plasma proteins, primarily serum albumin [Strattera, 2010].
Clinical Management
Children should undergo standard psychiatric and medical assessment for ADHD (see “Clinical Management of Stimulant Medication”). No laboratory screening is required, although obtaining baseline and follow-up liver function studies should be considered in view of reports of severe acute liver dysfunction [Lim et al., 2006]. Baseline values for weight, heart rate, and blood pressure should be obtained. Concomitant use of medications with cardiovascular effects and medications that inhibit cytochrome P-450 may necessitate dosage adjustment. Although the determination of cytochrome P-450 metabolizer status is available, it appears to be unnecessary because regular dosing parameters provide the opportunity for assessment of outcome and adverse events that may be experienced by poor metabolizers.
In children, the usual starting dose of atomoxetine is 0.5 mg/kg/day, given in the morning or divided into morning and late-afternoon doses. Starting with a split dosing regimen may decrease gastrointestinal side effects and irritability. Decreasing side effects on initiation will ultimately lead to better compliance. The dose can be consolidated after a target dose is achieved. The dose may be increased every 3 days to a target dose of 1.2 mg/kg/day. For adults or for children and adolescents weighing more than 70 kg, atomoxetine may be initiated at 40 mg/day. The dose may be increased gradually every 3 days to a maximum dose of 80 mg. If there is an inadequate response after a 2- to 4-week trial, the dose may be increased to 100 mg/day. Patients taking atomoxetine concomitantly with potent inhibitors of CYP2D6 should be maintained at the initial dose for at least 4 weeks before a dosage increase is considered. Dosage reduction is required in patients with hepatic impairment [Strattera, 2010].
In clinical practice, combination therapy may be used during initiation and titration of atomoxetine, especially in children with severe ADHD symptoms. The effectiveness and tolerability of combining atomoxetine with methylphenidate in children who have not responded to monotherapy have been reported in a few patients [Brown, 2004]. Discontinuation of atomoxetine does not require dose tapering. Abrupt discontinuation of atomoxetine has not been associated with an acute discontinuation syndrome [Wernicke et al., 2003].
Adverse Effects
The most common side effects of atomoxetine in children and adolescents are upset stomach, decreased appetite, nausea, vomiting, dizziness, tiredness, and mood swings. Atomoxetine was associated with increases in heart rate of 6 beats per minute, and increases in systolic and diastolic blood pressure of 1.5 mmHg compared with placebo. In an analysis of short-term and long-term treatment with atomoxetine, increased systolic blood pressure was observed in adults, and increased diastolic blood pressure occurred in children and adolescents. Heart rate increased in both groups, and no prolongation of the QTc interval was observed [Strattera, 2010]. Seizures and prolonged QTc intervals were reported after an overdose with atomoxetine [Sawant and Daviss, 2004]. During post-marketing experience, several cases of significant liver injury in children treated with atomoxetine have been reported [Lim et al., 2006; Stojanovski et al., 2007]. Lilly Research Laboratories subsequently reviewed all hepatobiliary events during clinical trials and through post-marketing voluntary adverse events reporting, and identified three cases of probable severe, reversible liver injury related to atomoxetine use [Bangs et al., 2008]. Strattera labeling now warns that severe liver injury is possible and states that atomoxetine should be discontinued and not restarted when laboratory tests show liver injury or when there is clinical evidence of jaundice. Use in patients with liver disease probably should be avoided.
Drug Interactions
Inhibitors of CYP2D6 may increase serum concentrations of atomoxetine. Atomoxetine does not have significant effects on the cytochrome P-450 system. Combination with monoamine oxidase inhibitors may precipitate a hypertensive crisis. Atomoxetine has potential interactions with cardiovascular agents and adrenergic agonists. Albuterol’s tendency to increase heart rate and blood pressure may be potentiated by atomoxetine. No increase in the cardiovascular effects of methylphenidate was observed when atomoxetine was added, and no interactions between atomoxetine and other protein-bound medications have been observed [Strattera, 2010].
Alpha2-Agonists
Clinical Applications
Clonidine is available in immediate-release and extended-release (ER) formulations. Immediate-release clonidine has been shown to improve behavior of children with ADHD; however, the degree of its effect in a meta-analysis was less than that of stimulants, and it was associated with many side effects [Connor et al., 1999]. The largest randomized controlled study to compare clonidine, methylphenidate, and the combination directly confirmed this earlier analysis [Palumbo et al., 2008]. Greatest improvement is seen with hyperactivity and impulsivity [Hunt, 1987; Hunt et al., 1982] and with frustration tolerance [Hunt, 1987] than with distractibility [Palumbo et al., 2008]. Children have been maintained on immediate-release clonidine for up to 5 years with continued benefit [Hunt et al., 1990, 1991]. Recently, clonidine ER was found effective in 2 randomized controlled trials (RCTs) in patient age 6-17 years (Kapvay, 2010, Kollins et al, 2011). One RCT used clonidine ER monotherapy and one RCT used clonidine ER as adjunctive therapy to a stimulant. To date, only the RCT with adjunctive therapy has been published in a peer reviewed journal (Kollins et al, 2011).
The combination of clonidine and methylphenidate has been helpful in adolescents with co-occurring ADHD and oppositional or conduct disorder [Hunt et al., 1990]. This combination allowed the dose of methylphenidate to be reduced by 40 percent [Hunt et al., 1990]. In children with co-occurring ADHD and tic disorders, those treated with clonidine experienced improvements in both conditions [Steingard et al., 1993]. A randomized controlled trial of clonidine, alone and in combination with methylphenidate, in children with ADHD and tics (TACT Trial) showed that clonidine was equally effective as methylphenidate but that the combination was most effective for treating ADHD symptoms [TSSG, 2002]. Clonidine has been used to treat sleep disturbances in children with ADHD [Wilens et al., 1994], and it appears to be helpful in managing severe aggression [Kemph et al., 1993]. Studies of clonidine in youngsters with Tourette’s syndrome show mixed results [Leckman et al., 1982], but significant improvements in tics and behavior have been reported [Cohen et al., 1980; Comings et al., 1990; Leckman et al., 1991].
Guanfacine is available in immediate-release and extended-release (ER) formulations through different manufacturing companies. Limited data exist on immediate-release guanfacine in children. One open trial demonstrated its effectiveness in ADHD [Hunt et al., 1995]. Findings for patients with ADHD and Tourette’s syndrome have been mixed [Chappell et al., 1995; Horrigan and Barnhill, 1995]. Results of more recent studies have been more promising [Scahill et al., 2001]. More data are available on the extended-release formulation (Intuniv®). Randomized, double-blind, placebo-controlled studies have demonstrated efficacy of guanfacine ER for treatment of symptoms of ADHD in short-term treatment [Biederman et al., 2008; Sallee et al., 2009b]. Continued efficacy is seen in longer-term treatment of up to 24 months [Sallee et al., 2009a]. Review of data suggests that guanfacine ER may be most effective in younger children and children with ADHD, combined type [Biederman et al., 2008].
Pharmacology
Clonidine and guanfacine exert agonist effects on presynaptic α2-adrenergic receptors in the sympathetic nuclei of the brain, resulting in decreased release of norepinephrine from presynaptic nerve terminals. In the central nervous system (CNS), α2-adrenergic agonists are thought to regulate noradrenergic activity in the locus ceruleus [Arnsten et al., 1988].
Clinical Management
Clonidine is initiated at a 0.05-mg dose taken at bedtime, gradually titrating the dose over several weeks to 0.15–0.3 mg/day in divided doses. Clonidine is also available in a transdermal patch, which may have the advantage of increased medication compliance and more stable blood levels. After a therapeutic dose of oral clonidine is achieved, the equivalent patch can be substituted [Catapres, 2010]. Clonidine ER is initiated at 0.1 mg at bedtime and increased weekly by 0.1 mg to a maximum dose of 0.4 mg divided twice daily (Kapvay, 2010).
Guanfacine immediate release offers the advantage over clonidine of a longer half-life and therefore less frequent dosing, and it may cause less sedation than clonidine [Hunt et al., 1995]. It is initiated at 0.5 mg at bedtime and is titrated up to a maximum of 3 mg/day. Guanfacine ER has the greatest ease of administration and allows once-daily dosing. It is initiated at 1 mg daily and increased weekly to a maximum daily dose of 4 mg/day [Intuniv, 2011].
Adverse Effects
The most common side effects of clonidine and guanfacine include sedation, dizziness, fatigue, dry mouth and eyes, nausea, hypotension, and constipation [Catapres, 2010; Iintuniv, 2011]. Syncope has been reported in 1 percent of children on guanfacine ER [Intuniv, 2011]. Clonidine in patch form may be associated with contact dermatitis, which may be reduced by changing its location on the body. Because of the risk of rebound hypertension with abrupt discontinuation, these medications should be prescribed only to patients with reliable medication compliance. Likewise, the dose should be tapered gradually when discontinuing the medication [Catapres, 2010; Intuniv, 2011].
Case reports of sudden death in children treated with clonidine has raised concerns about the cardiovascular safety of clonidine by itself or in combination with methylphenidate [Fenichel, 1995]. The most commonly reported cardiovascular side effect is bradycardia in patients taking clonidine either alone or in combination with methylphenidate [Chandran, 1994; Connor et al., 2000; Daviss et al., 2008], although this has not been universally reported [Kofoed et al., 1999; Leckman et al., 1991; Wilens et al., 2003]. One study reported prolongation of the PR interval that was not clinically significant [Connor et al., 2000], and one study reported bradycardia accompanied by a variety of EKG changes, which was confounded by concomitant treatment with other medications with potential for cardiovascular side effects [Chandran, 1994]. Some studies have suggested that the risk for bradycardia is higher when clonidine is given concomitantly with methylphenidate [Connor et al., 2000], but others have shown no additional risk [Daviss et al., 2008; Leckman et al., 1991]. To date, studies of guanfacine ER have not reported any EKG abnormalities considered to be serious adverse events, although a few subjects have been noted with clinically asymptomatic bradycardia [Biederman et al., 2008; Sallee et al., 2009a]. Practitioners should monitor bradycardia in patients taking clonidine.
Drug Interactions
Clonidine may potentiate the CNS-depressant effects of alcohol and sedating medications. The manufacturer advises caution in combining clonidine with medications that affect cardiac conduction and sinus node function because of potential additive effects, such as bradycardia and atrioventricular block [Catapres, 2010].
Antidepressants
Tricyclic Antidepressants
Clinical Applications
Tricyclic antidepressants are approved for the treatment of depression in adults and adolescents (Table 49-3). Placebo-controlled studies have demonstrated the efficacy of tricyclic antidepressants in the treatment of depression in adults, but studies enrolling children and adolescents have yielded inconsistent results. Some open studies show no significant response to tricyclic antidepressants [Kashani et al., 1984; Kramer and Feiguine, 1981]. Others suggest efficacy of desipramine, imipramine, and nortriptyline [Boulos et al., 1991; Geller et al., 1986; Ryan et al., 1986]. Placebo-controlled studies, however, do not support efficacy [Geller et al., 1989, 1990; Puig-Antich et al., 1987]. The lack of efficacy in placebo-controlled trials may be related to methodological problems, such as a small number of subjects, types of subjects enrolled (with depression that is either too mild or too severe), and the high placebo response rates in many studies. The use of tricyclic antidepressants for the treatment of depression in children and adolescents has declined because of their equivocal efficacy and safety in this population and the availability of antidepressants with a more favorable side-effect profile (i.e., selective serotonin reuptake inhibitors). Clomipramine has been approved for use in children 10 years of age or older for OCD, following publication of several randomized, controlled trials [DeVeaugh-Geiss et al., 1992; Leonard et al., 1989]. However, clomipramine is used less commonly for OCD than selective serotonin reuptake inhibitors and is considered second-line treatment in clinical practice, in part because of a less favorable side-effect profile.
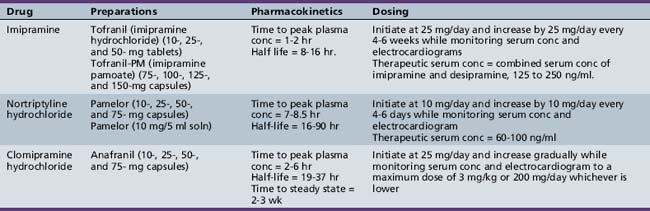
Tricyclic antidepressants have been prescribed as third-line treatments for patients with ADHD who have failed adequate trials of stimulants, who could not tolerate the adverse effects of stimulants, and who had co-occurring conditions such as depression, anxiety, tic disorders [Wilens et al., 1993], or enuresis. Studies have demonstrated significant improvement in ADHD symptoms with imipramine, nortriptyline [Saul, 1985; Wilens et al., 1993], and desipramine [Biederman et al., 1986, 1989; Gastfriend et al., 1984]. However, tricyclic antidepressants appear to be less effective than methylphenidate [Rapoport et al., 1974], and are associated with a large dropout rate over time due to loss of efficacy or side effects [Quinn and Rapoport, 1975].
Studies of imipramine in the treatment of separation anxiety disorder and school refusal have yielded inconsistent findings [Bernstein et al., 2000; Gittelman-Klein and Klein, 1971; Klein et al., 1992]. The largest double-blind, placebo-controlled study of adolescents with school refusal and comorbid depression and anxiety found greatest improvement in school attendance in youngsters who received a combination of imipramine and cognitive behavioral therapy when compared to youngsters receiving cognitive behavioral therapy alone [Bernstein et al., 2000]. Case reports describe the usefulness of tricyclic antidepressants in the treatment of panic disorder [Ballenger et al., 1989; Black and Robbins, 1990], but no placebo-controlled trials have been carried out.
Imipramine appears to be effective in treating enuresis as an initial agent.[Fritz et al., 2004], and in children who have failed nonpharmacological interventions and desmopressin [Gepertz and Neveus, 2004]. Efficacy appears to be correlated with serum concentration [Fritz, 1994].
Clomipramine is approved for the treatment of OCD in adults and children older than 10 years. In short-term clinical trials, clomipramine has demonstrated superiority over placebo [DeVeaugh-Geiss et al., 1992; Leonard et al., 1989] in reducing symptoms of OCD and preventing relapse [Leonard et al., 1991]. Improvement in clomipramine-treated subjects was sustained during 1-year open treatment [DeVeaugh-Geiss et al., 1992].
Clinical Management
Assessment
As with ADHD, assessment for anxiety disorders and depression requires the gathering of information from multiple sources, including the child, parents, school, therapist, and others involved with the child. Information can be gathered through clinical interview, other informants, self-reports, and medical evaluation, including laboratory screening. Diagnostic criteria for anxiety disorders and depressive disorders are detailed in the DSM-IV TR [APA, 2000].
A medical evaluation is required before initiating tricyclic antidepressants. A physical examination should have been completed within the past 1 year. A baseline EKG, orthostatic blood pressure and heart rate, complete blood cell count (CBC), electrolyte determination, renal and hepatic function tests, thyroid function tests, and urinalysis should be performed [Green, 2007]. Other laboratory screening measures may be indicated, based on findings in the history or physical examination.
Initiation and titration of tricyclic antidepressants
Dosing strategies, as well as the utility of serum blood levels of tricyclic antidepressants, are not as well established in children and adolescents as for adults, but the literature does provide some guidance. Imipramine may be initiated at 25 mg and increased by 25 mg every 4–6 days. Because there is large variability in plasma drug concentration with a given dose of tricyclic antidepressants among individuals, monitoring of serum concentration has been recommended [Biederman et al., 1989; Geller et al., 1986]. A combined serum concentration of imipramine and its primary metabolite, desipramine, in the range of 125–250 ng/mL is considered therapeutic for the treatment of depression. Higher serum concentrations within this range have correlated with higher response rates compared with placebo in the treatment of pediatric depression [Puig-Antich et al., 1987]. Serum concentrations greater than 250 ng/mL have not improved response rates and were associated with increased side effects [Preskorn et al., 1989]. Low doses of imipramine are recommended for treatment of enuresis, not to exceed 15 mg in children and 75 mg in adolescents [Fritz et al., 2004].
Nortriptyline is not FDA-approved for any indication in children and adolescents, but the limited studies to date have not reported serious adverse effects [Geller et al., 1989, 1992]. Although studies of nortriptyline in depressed adults reveal correlation between response and serum blood level with a therapeutic window of 50–150 ng/mL, correlations between therapeutic efficacy and serum nortriptyline levels have not been consistently established in pediatric populations [Geller et al., 1989, 1992]. A useful algorithm has been published for nortriptyline dosing, based on steady state plasma levels determined 24 hours after a single test dose of 25 mg in 5–9-year-olds and 50 mg in 10–17-year-olds for treatment of depression [Geller et al., 1985]. As the half-life is significantly shorter in children and adolescents, twice-daily dosing is recommended and there may be value in monitoring serum concentrations [Geller et al., 1986]. The therapeutic range for nortriptyline is postulated to be 60–100 ng/mL [Geller and Carr, 1988; Geller et al., 1987]. EKGs should be obtained at baseline and after steady-state plasma levels are achieved [Geller et al., 1987].
Because of the risk of adverse cardiac effects, serial EKGs are recommended with the use of all tricyclic antidepressants. They should be obtained at baseline, at the middle point in titration, and at the final dose [Elliot and Popper, 1990/1991]. The following parameters have been suggested as safety parameters for cardiovascular monitoring:
Adverse Effects
Eight sudden deaths have been reported in children and adolescents taking tricyclic antidepressant medications [Popper and Zimnitzky, 1995; Riddle et al., 1993; Varley and McClellan, 1997]. Six were taking desipramine and two were taking imipramine. Although the role of the tricyclic antidepressants in these tragic deaths is unclear, it has been speculated that the cause of death was cardiac in nature, most likely due to malignant arrhythmias. After the reports, child psychiatrists have become more cautious about prescribing tricyclics in general, carefully screening patients at risk for adverse effects and monitoring cardiac status with serial EKGs [Elliot and Popper, 1990/1991]. It is not clear whether the vulnerability to such medication effects can be determined from routine monitoring of cardiac function. Those who experience increased QTc above that which is considered safe while on tricyclic antidepressants should be monitored closely, and the dose should be adjusted downward until resolution of QTc prolongation.
Behavioral side effects include early and acute increases in anxiety or depression. Tricyclic antidepressants may also induce manic episodes or rapid cycling in patients with a history of bipolar disorder. A family history of bipolar disorder may be a risk factor for this effect, but such a history is not a contraindication for the use of antidepressants. A discontinuation syndrome (i.e. nausea, headache, and malaise), hypomania, or mania may result from abrupt cessation of antidepressants [Haddad, 2001] and may be associated with manic reactions [Narayan and Haddad, 2010]. The FDA mandated a black box warning regarding increases in suicidal ideation for all antidepressants (see “Selective Serotonin Reuptake Inhibitors”).
Selective Serotonin Reuptake Inhibitors
Clinical Applications
The selective serotonin reuptake inhibitors, including citalopram, escitalopram, fluvoxamine, fluoxetine, paroxetine, and sertraline, have FDA approval for a number of mood, anxiety, and eating disorders in adults (Table 49-4). In children and adolescents, fluoxetine, fluvoxamine, and sertraline have FDA approval for OCD; fluoxetine also has FDA approval for major depression. Escitalopram has FDA approval for major depression in adolescents (12 years and older). These indications are based on large-scale, randomized, controlled trials for OCD [Geller et al., 2001; March et al., 1998; Riddle et al., 2001] and major depression [Emslie et al., 1997, 2002, 2009; TADS, 2004]. Other large, randomized, controlled trials of the agents have been completed, but not all have been published. Published trials demonstrating efficacy include citalopram for depression [Wagner et al., 2004b]; fluoxetine [Birmaher et al., 2003], fluvoxamine [RUPP, 2001], and sertraline [Walkup et al., 2008] for separation, social, and generalized anxiety; sertraline for depression [Wagner et al., 2003] and generalized anxiety [Rynn et al., 2001]; paroxetine [Wagner et al., 2004a] and venlafaxine [March et al., 2007] for social phobia, and fluoxetine for stereotypies in children with autistic spectrum disorder [Hollander et al., 2001].
The use of the selective serotonin reuptake inhibitors for depression and anxiety disorder increased significantly in the 1990s, with upwards of 1–2 percent of teens being prescribed these medications for any indication [Olfson et al., 2002a]. Despite the marked increase in use, the prevalence of disorders in children and adolescents that are potentially responsive to the selective serotonin reuptake inhibitors (e.g., depression and anxiety disorders) is much greater (minimum prevalence of 3–5 percent) than the number of children who are taking these medications (1–2 percent), suggesting overall under-utilization of these evidenced-based treatments.
In 2004, the FDA issued a black box warning for increased risk of suicidal ideation and behavior in children and adolescents using all antidepressants. This warning was based on the analysis of data from 24 short-term, placebo-controlled trials of nine antidepressant drugs, involving a total of over 4400 children and adolescents being treated for major depressive disorder, OCD, and other psychiatric disorders. Results of this analysis showed an increased risk of suicidal thoughts and behavior in the treatment groups compared to placebo (approximately 4 percent versus 2 percent). Since this time, prescriptions of antidepressants in pediatric populations have fallen off and recent reports suggest that rates of youth suicide increased after many years of decline [Gibbons et al., 2007]. There is no evidence that the prevalence of mood disorders in children has diminished over this same time period, or that patients who are identified as being depressed are being offered alternative treatments to medication. This is especially ironic, as the current evidence base suggests the limited value of nonpharmacological approaches to the treatment of depression [TADS, 2004].
Pharmacology
Pharmacokinetics
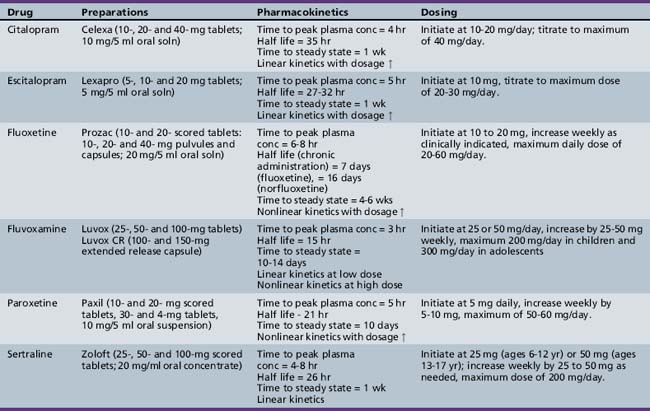
Citalopram
Peak blood levels of citalopram (Celexa®) occur at about 4 hours, with a half-life of about 35 hours. Single- and multiple-dose pharmacokinetics of citalopram are linear throughout the standard dose range (10-40 mg/day). Steady-state plasma concentrations are reached in approximately 1 week. Citalopram is metabolized in the liver to a number of mostly inactive metabolites. Citalopram appears to have limited ability to inhibit cytochrome P-450 isoenzymes; inhibition of these isoenzymes is primarily responsible for the demethylation of citalopram. CYP3A4 and CYP2C19 do not appear to affect citalopram blood levels or half-life significantly. The (S)-enantiomer of citalopram is mostly responsible for the serotonin reuptake [Dietz and Robinson, 2005].
Escitalopram
Escitalopram (Lexapro®) is the (S)-enantiomer of a 50:50 racemic mixture of (R)- and (S)-enantiomers of citalopram. The (S)-enantiomer in escitalopram is approximately 100 times more potent at serotonin reuptake inhibition than the (R)-enantiomer. Evidence suggests that adolescents metabolize escitalopram more quickly than adults but once-daily dosing is adequate [Lexapro, 2009]. Steady state is achieved in about 1 week. The pharmacokinetic profiles of escitalopram for single and multiple doses are linear in the range of doses used clinically (10–30 mg/day). Escitalopram is metabolized primarily in the liver by CYP3A4, CYP2C19, and other enzymes. The primary metabolites are found in lesser concentrations in the plasma than escitalopram and are not thought to be clinically active. In a similar way to citalopram, it is not anticipated that escitalopram will inhibit cytochrome P-450 or be inhibited by medication that inhibits the cytochrome P-450 system [Lexapro, 2009].
Fluoxetine
Fluoxetine (Prozac®) reaches peak plasma levels approximately 6–8 hours after oral administration and is almost completely protein-bound. Fluoxetine is extensively metabolized in the liver to norfluoxetine (active metabolite) through a variety of isoenzymes of the cytochrome P-450 system. The kinetic profiles are generally nonlinear, with chronic plasma levels not directly linked to dose. With chronic administration, the half-life of fluoxetine and norfluoxetine increases to 7 days and up to 16 days, respectively. Steady-state concentrations are therefore delayed for up to 4–6 weeks. In pediatric populations, the steady-state concentrations of fluoxetine in children were 2 times higher than in adolescents at the same dose, mostly attributable to weight, although the overall levels were still within the range seen in studies of adult populations [Prozac, 2009]. The long duration of activity can have potential benefits (e.g., on-going medication exposure with occasional missed doses, and single-dose weekly administration for the sustained-release fluoxetine) and some risks – side effects may emerge as the plasma level increases 4–6 weeks after initiation of treatment, and newly prescribed medications may interact with fluoxetine or norfluoxetine for up to 4 weeks after fluoxetine discontinuation. The prolonged half-life may be problematic for patients who do not tolerate the medication, develop manic reactions, or have other problematic behavioral side effects [Reinblatt et al., 2009; Reinblatt and Riddle, 2006], when the need to discontinue and clear the medication from the body is critical.
Fluvoxamine
Fluvoxamine (Luvox®) reaches peak plasma levels in up to 8 hours after oral administration and up to 12 hours with the extended-release preparation. Pharmacokinetic profiles are nearly linear in the lower range of treatment doses, with a loss of linearity with higher doses. The half-life of fluvoxamine increases slightly from 15 hours to between 17 and 22 hours after single and multiple doses, respectively. Steady-state plasma levels are usually achieved within 10–14 days. Fluvoxamine undergoes extensive metabolism in the liver to a number of clinically inactive metabolites, but the specific cytochrome P-450 isoenzymes involved are unknown. Fluvoxamine does inhibit CYP1A2 and, to a lesser extent, CYP3A4 and CYP2D6, resulting in potentially significant drug interactions, including tertiary amine tricyclic antidepressants (e.g., imipramine, clomipramine), some benzodiazepines, propranolol, warfarin, and theophylline [van Harten, 1995]. Pharmacokinetic studies suggest that children, especially young girls, metabolize fluvoxamine more slowly and that they therefore may require lower treatment doses and have steady-state plasma levels that are 2–3 times that seen in adolescents [Labellarte et al., 2004].
Paroxetine
Paroxetine (Paxil®) reaches peak plasma concentrations at approximately 5 hours, and the average terminal half-life is about 21 hours after chronic administration of 30 mg/day. Steady-state paroxetine plasma levels are reached after about 10 days. Pharmacokinetics were nonlinear for multiple dosing and approximately eight times what would be expected based on single-dose kinetics. A nonlinear kinetic profile results from the saturation of CYP2D6, which is primarily responsible for paroxetine metabolism. Paroxetine undergoes extensive first-pass metabolism to a number of metabolites, none of which appears to have any significant clinical activity. Paroxetine appears to be a strong inhibitor of cytochrome P-450, which suggests the potential for clinically meaningful drug–drug interactions. Pediatric data suggest that children metabolize paroxetine more quickly than adults but that once-daily dosing is likely adequate [Findling et al., 1999].
Sertraline
Sertraline (Zoloft®) reaches peak plasma concentrations at approximately 4–8 hours after a 2-week period of oral administration, with an average terminal half-life of about 26 hours. Steady-state sertraline plasma levels are reached after about 1 week. Pharmacokinetic profiles were linear for single doses in the treatment range of 50–200 mg/day. Sertraline undergoes extensive first-pass metabolism by multiple cytochrome P-450 isoenzymes to N-desmethylsertraline, which is substantially less clinically active than sertraline. Sertraline does not appear to be a strong inducer or inhibitor of cytochrome P-450; coupled with the fact that sertraline is metabolized by multiple isoenzymes, this suggests that few clinically meaningful drug–drug interactions should occur. Pediatric data suggest that, although children metabolize sertraline more quickly than adults, they may require lower doses because of their lower body weight to avoid excessive plasma levels [Zoloft, 2009].
Clinical Management
Assessment
The assessment of mood and anxiety disorders before initiation of a selective serotonin reuptake inhibitor is similar to that described earlier for the tricyclic antidepressants. Assessment requires gathering information from multiple sources, including the child, parents, school, therapist, or others involved with the child. Information can be gathered through clinical interview, other informants, self-reports, and medical evaluation, including laboratory screening. Diagnostic criteria for anxiety disorders and depressive disorders are detailed in the DSM-IV TR [APA, 2000].
Initiation and titration of dose
Escitalopram
Escitalopram comes in 5-, 10-, and 20-mg tablets. The 10- and 20-mg tablets are scored. The one positive double-blind, placebo-controlled study in adolescents used standard adult dosing of 10–20 mg/day [Emslie et al., 2009].
Fluoxetine
Fluoxetine comes in 10-, 20-, and 40-mg pulvules; 10-mg scored tablets; and Prozac Weekly, which contains approximately 90 mg of fluoxetine in a time-released preparation. Fluoxetine is also available in a liquid (20 mg/5 mL). In most clinical trials of fluoxetine in children and adolescents, dosing began at 10 mg/day for 1–2 weeks, with a subsequent increase to 20 mg. The dose is often held at this level up to 4–6 weeks before going to 30 or 40 mg/day [TADS, 2004]. Doses as high as 60 mg/day have been used in clinical trials, but maximum doses are used only if this dose is reached after a patient has demonstrated partial benefit after 10–12 weeks of treatment [TADS, 2004].
Fluvoxamine
Fluvoxamine comes in 25-, 50-, and 100-mg scored tablets. In clinical trials of fluvoxamine in children, dosing began at 25 or 50 mg/day and increased by 50 mg on a weekly basis to a maximum dose of up to 200 mg/day. Average therapeutic doses at the end of treatment were between 150 and 200 mg/day [Riddle et al., 2001]. In clinical trials, the method of titration often leads to the highest safe dose rather than least effective dose. With slower dosage adjustment, it may be possible to establish benefit at lower doses. This is particularly true for prepubertal children (especially girls), who may metabolize fluvoxamine more slowly and have higher blood levels than teens and adults [Labellarte et al., 2004].
Sertraline
Sertraline comes in 25-, 50-, and 100-mg scored pills or tablets and in a concentrate of 20 mg/mL. Generally, 50 mg/day given as a single dose appears to be as effective as other doses for most conditions. Dosing can begin at 25 or 50 mg/day. For those who do not respond or only partially respond, higher doses may be necessary. Clinical trials enrolling adults and children using sertraline have usually used doses up to 200 mg/day as tolerated. One clinical trial of sertraline in children demonstrated efficacy on a fixed dose of 50 mg/day [Rynn et al., 2001]; however, higher doses up to 140 mg/day are well tolerated [Walkup et al., 2008].
Adverse Effects
Common effects
Although clinicians usually think of adverse effects as physical changes for selective serotonin reuptake inhibitors, the behavioral and psychiatric adverse effects and their impact on growth and development are a greater concern and require review with the patient and parent before starting treatment. (For review, see Walkup and Labellarte [2001].) Perhaps the most important of these effects is activation, which occurs in about 10–20 percent of patients [March et al., 1998; Reinblatt et al., 2009; RUPP, 2001]. This activation syndrome (distinct from the mood improvement effects of the antidepressants) can result in a number of complaints, including anxiety, mental restlessness, increased activity level or akathisia, increased impulsivity, and disinhibition. This activation is very similar to but milder than what some children experience when they take diphenhydramine. Activation effects usually appear very early in treatment or after a dose change. Patients do not appear to develop tolerance to these symptoms, but they resolve with dose reduction or discontinuation, consistent with the pharmacokinetics of the medication.
The selective serotonin reuptake inhibitors have also been reported to induce an apathy syndrome. Although it may manifest early in treatment, those with anxiety and depression may not be sensitive to this subtle medication effect until after a period of recovery. Awareness of the potential for apathy improves the likelihood that it will be recognized. Even though patients who have experienced apathy can readily differentiate it from depression or sedation, careful interviewing may be required to make these differentiations. Apathy responds to dose reduction; however, dose reductions required to eliminate apathy may lead to loss of symptom control. Patients who report loss of response, not feeling like themselves, or feeling better but numb, or who appear to have lost interest without significant depressive symptoms should be interviewed closely to assess for apathy [Hoehn-Saric et al., 1990; Reinblatt and Riddle, 2006].
Sexual side effects are not commonly documented in clinical trials with children and adolescents, but in clinical practice, it is not uncommon for physicians to find that teens begin to complain spontaneously about sexual side effects when they explore their sexual responsiveness when alone or with another person. Awareness of sexual side effects can make it more likely that these effects are identified and managed, and that that they will not lead to poor adherence, discontinuation, and loss of treatment response. Dose reduction, changing to antidepressants with a lower risk for sexual side effects, or the addition of medication that may improve sexual function (i.e., bupropion or stimulants) have all been reported to be effective [Woodrum and Brown, 1998].
Although epistaxis is common in children and adolescents, some children experience increased rate or severity of epistaxis and increased bruising. Such changes appear to be an effect of the selective serotonin reuptake inhibitors on platelet functions, not on coagulation factors [Lake et al., 2000] and may be reflected in prolonged bleeding times.
Selective serotonin reuptake inhibitors, specifically fluoxetine, have been associated with change in the rate of linear growth. This was first reported in a small case series [Weintrob et al., 2002] and in a review of the large fluoxetine database [Nilsson et al., 2004]. It appears that the deceleration of growth stopped with medication discontinuation and recurred in one case when the medication was restarted [Weintrob et al., 2002]. It is not clear whether this is an effect of all the selective serotonin reuptake inhibitors or specific to fluoxetine. It is possible that this effect may be mediated by selective serotonin reuptake inhibitor-induced sleep disruption and resulting impact on growth hormone, and is not mediated by reduction in appetite, as can occur with the stimulants. It does not appear to affect all children uniformly. Because the follow-up period in these reports is relatively short, it is unclear whether children can catch up while on medication or whether medication discontinuation is required for children to obtain their optimal height. The assessment of changes in growth can be complicated by the potential effects of psychiatric disorders on growth [Pine et al., 1996].
Although there are no data to suggest that selective serotonin reuptake inhibitor treatment leads to drug-seeking behavior (i.e., is addictive), some individuals do appear to have a discontinuation syndrome with abrupt discontinuation [Ditto, 2003]. There are three complications to abrupt discontinuation. The first complication is a withdrawal reaction that includes flu-like symptoms, such as malaise and gastrointestinal symptoms, but it may also include unusual symptoms, such as sensory or psychological disturbances. Some patients with abrupt discontinuation also experience an abrupt return of symptoms, including suicidal risk. Ironically, case reports suggest that abrupt discontinuation of antidepressants can be associated with hypomanic reactions [Narayan and Haddad, 2010]. Antidepressants with shorter half-lives appear to be more commonly associated with discontinuation syndromes than selective serotonin reuptake inhibitors with longer half-lives.
Antidepressants and risk for suicidal behavior
The data on which these new warnings are based include more than 24 randomized, controlled clinical trials (>4000 subjects) of the newer nontricyclic antidepressants [Hammad et al., 2006], completed for the purpose of extended exclusivity under the FDA Modernization Act [FDA, 1998]. Although a number of methodological limitations plague the analysis of these studies, it appears that there is a small but significant increased risk for more suicidality adverse events on active medication (about 4 events per 100 patients) versus placebo (about 2 events per 100 patients). There does not appear to be a groupwise risk for emergence or worsening of suicidal behavior for the medications, but the data do support an association with activation syndrome. Although the risk appears to be small (number needed to harm is about 50), the fact that, in these same studies, there is marginal antidepressant efficacy suggests that more caution is required when considering the use of these medications [Hammad et al., 2006]. In contrast to the industry-sponsored studies completed for exclusivity, National Institutes of Health (NIH)-funded studies of the treatment of depression and anxiety in children suggest significant benefit for the selective serotonin reuptake inhibitors [Birmaher et al., 2003; Emslie et al., 1997, 2009; RUPP, 2001; TADS, 2004; Walkup et al., 2008]. The big difference between industry-sponsored and NIH-sponsored clinical trials is the large placebo response rate in industry-sponsored trials, with resulting smaller effect sizes than those observed in NIH-funded trials. The increased risk for suicidality previously observed was principally seen in depressed patients, and its use for other indications, such as anxiety disorders, may not share this increased risk [Bridge et al., 2007]. This may be an important consideration for child neurologists because they are more likely to use selective serotonin reuptake inhibitors for such purposes. The risk for suicide is higher overall among teenagers and less so in younger children.
Serotonin-Norepinephrine Reuptake Inhibitors
Clinical Applications
The serotonin-norepinephrine reuptake inhibitors, which include duloxetine, venlafaxine, and desvenlafaxine, have FDA approval for treatment of depression and anxiety disorders in adults. Duloxetine also has FDA approval for treatment of fibromyalgia and diabetic neuropathic pain in adults. Venlafaxine has also shown promise in the treatment of ADHD in adults [Findling et al., 1996], and in co-occurring major depressive disorder and ADHD in adults [Hornig-Rohan and Amsterdam, 2002]. The use of serotonin-norepinephrine reuptake inhibitors in children and adolescents is off-label. At present, the only published randomized controlled studies of serotonin-norepinephrine reuptake inhibitors in pediatric populations include a positive trial of venlafaxine ER for social phobia [March et al., 2007], and equivocal trials for generalized anxiety disorder [Rynn et al., 2007] and depression [Emslie et al., 2007]. Two small open-label studies of venlafaxine in children and adolescents with ADHD suggest potential benefit for impulsivity and hyperactivity [Findling et al., 2007; Olvera et al., 1996] but were complicated by a high dropout rate due to adverse effects, including behavioral activation [Olvera et al., 1996]. A few case reports have been published on the successful use of duloxetine in youngsters with chronic pain and comorbid major depressive disorder [Meighen, 2007]; essentially, no data are available on use of desvenlafaxine. This review will limit its discussion to venlafaxine use in children and adolescents, given the limited data for duloxetine and desvenlafaxine.
Pharmacology
Venlafaxine
Venlafaxine is available in immediate-release tablets (Effexor®) and extended-release capsules (Effexor XR®). In adults, venlafaxine at doses of less than 150 mg/day selectively inhibit serotonin reuptake, and at higher doses, inhibit both serotonin and norepinephrine reuptake. Venlafaxine does not have significant activity at muscarinic, histaminic, or α-adrenergic receptors. It is well absorbed through the gastrointestinal tract, metabolized by the liver, and excreted in the urine. The serum half-lives of venlafaxine and its active metabolite, O-desmethylvenlafaxine, are about 5 and 11 hours, respectively (Table 49-5) [Effexor, 2009]. The pharmacodynamic behavior of venlafaxine ER is not well understood in children and adolescents, although some preliminary work suggests that there is no clear relationship between dose and plasma serotonin and norepinephrine [Emslie et al., 2007; Findling et al., 2007]. Various dosing strategies have been employed in clinical trials with higher dosing strategies used in youngsters with mood and anxiety symptoms and lower dosing strategies in youngsters with ADHD. In studies of depression, children under 40 kg were begun at 37.5 mg daily and increased weekly up to 112.5 mg; children of 40–49 kg also began at 37.5 mg and increased weekly up to 150 mg, and children over 50 kg were titrated up to a maximum daily dose of 225 mg daily [Emslie et al., 2007], although most individuals did not require the upper limits of these dosing strategies. Dosing strategies for venlafaxine in studies of ADHD used lower doses, ranging from 0.5 mg to 2.0 mg/kg daily in divided doses.
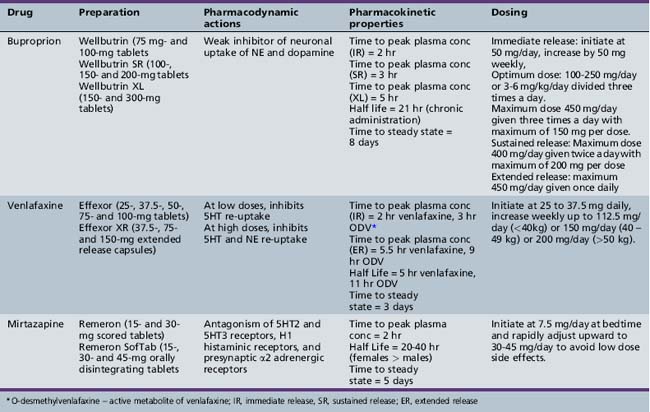
Adverse Effects
In adults, common adverse events associated with venlafaxine include asthenia, sweating, nausea, constipation, anorexia, vomiting, somnolence, dry mouth, dizziness, nervousness, anxiety, tremor, blurred vision, and sexual dysfunction. In addition, venlafaxine has been associated with increases in diastolic blood pressure that appear dose-dependent, especially with doses above 300 mg daily [Effexor, 2009]. In children and adolescents, treatment-emergent side effects include asthenia, anorexia, nausea, weight loss, abdominal pain, dizziness, somnolence, and nervousness [Emslie et al., 2007; March et al., 2007; Rynn et al., 2007]. Small increases in diastolic blood pressure have been reported in randomized clinical trials but were not considered clinically significant. Venlafaxine appears to have one of the highest rates of treatment-emergent suicide-related behaviors (relative risk of 8.84 percent) compared to other antidepressant medication in children and adolescents [Hammad et al, 2006], and has been associated with other treatment-emergent behavioral abnormalities, including hostility, overactivity, and agitation.
Drug Interactions
Venlafaxine or venlafaxine ER is contraindicated to be used in conjunction with a monoamine oxidase inhibitor and requires a 14-day washout period. Additionally, venlafaxine should only be used cautiously in conjunction with triptans due to the increased risk of serotonin syndrome. Although venlafaxine is metabolized by the CYP2D6 isoenzyme, the combined serum concentration of venlafaxine and its active metabolite does not appear to be altered by poor metabolizers [Effexor, 2009].
Other Antidepressants
Bupropion
Clinical applications
Bupropion (Wellbutrin®, Zyban®) is FDA-approved for the treatment of depression, nicotine dependence, and smoking cessation in adults 2009 [Wellbutrin, 2009]. Off-label uses of bupropion include monotherapy in the treatment of depression in children and adolescents, augmentation of selective serotonin reuptake inhibitor therapy in the treatment of depression [Lam et al., 2004], management of selective serotonin reuptake inhibitor-induced sexual side effects [Masand et al., 2001], and as a second-line agent in the treatment of ADHD. Bupropion may improve symptoms of ADHD in prepubertal children [Casat et al., 1989; Simeon et al., 1986]. Effect sizes of bupropion and placebo differences were smaller than for standard stimulant medications [Casat et al., 1989]. However, bupropion appeared to have comparable efficacy compared with methylphenidate in a double-blind, crossover-design study [Barrickman et al., 1995]. Adolescents with depression [Glod et al., 2003] and adolescents with ADHD and co-occurring depression [Daviss et al., 2001] demonstrated improvement with bupropion.
Pharmacology
Bupropion is available in immediate-release tablets (Wellbutrin®, Zyban®) and extended-release tablets (Wellbutrin SR®, Wellbutrin XL®). The mechanism of action in ameliorating depression is unknown. In animals, bupropion selectively inhibits noradrenergic neurons in the locus ceruleus and inhibits the reuptake of dopamine, but it does not have significant activity at α-adrenergic, cholinergic, or histaminic receptors. Bupropion is well absorbed through the gastrointestinal tract, with a peak serum concentration attained in 2–3 hours and a half-life of 14–21 hours. Steady-state concentrations are achieved in 8 days. Steady-state plasma levels achieved with the extended-release form were about 85 percent of the immediate-release preparation. Bupropion is 80 percent bound to serum albumin. It is metabolized by the liver (primarily by CYP2B6) and excreted in urine and feces. Although bupropion has been found to induce its own metabolism in animals, this has not been specifically studied in humans (see Table 49-5) [Wellbutrin, 2009].
Clinical management
In addition to the standard assessment for ADHD or depression, possible risk factors for adverse effects with bupropion should be identified. Bupropion has been associated with seizures in approximately 0.4 percent of patients treated at doses up to 450 mg/day. The risk of seizures appears to be strongly associated with peak serum concentrations and dose. The incidence of seizures appears to increase significantly above daily doses of 450 mg and with rapid dose titration [Wellbutrin, 2009]. The risk of seizures with bupropion increases with conditions that lower the seizure threshold, such as preexisting seizure disorder, metabolically unstable anorectic or bulimic patients, or those with head trauma, CNS tumors, concomitant use of medications that lower seizure threshold, excessive use of alcohol, and abrupt withdrawal of alcohol or sedatives. Patients with hepatic cirrhosis may have impaired metabolism of bupropion, resulting in elevated bupropion levels and increased risk of seizures, even at modest doses. No specific laboratory tests are recommended by the manufacturer before initiation. To minimize the risk of seizures, the manufacturer recommends that clinicians titrate bupropion slowly and divide the dose three times per day (immediate release), with each single dose no greater than 150 mg to avoid large peak serum concentrations. The dose should not exceed 450 mg/day for immediate-release preparations and 400 mg/day for extended-release preparations [Wellbutrin, 2009].
Bupropion has not been FDA-approved for use in children or adolescents. Conservative dosing strategies begin at 50 mg/day and are increased by 50 mg/day at weekly intervals [Simeon et al., 1986]. Optimal doses in studies have ranged from 100 mg to 250 mg/day in prepubertal children [Clay et al., 1988; Simeon et al., 1986] or 3–6 mg/kg/day [Conners et al., 1996]. No incidence of seizure was reported in these studies. If symptoms fail to respond to therapeutic doses of bupropion over this period, the medication should be tapered slowly to avoid adverse effects.
Adverse effects
Common adverse effects include agitation, dry mouth, insomnia, headache, nausea, vomiting, constipation, and tremor [Wellbutrin, 2009]. Neuropsychiatric side effects have been observed in some patients, including psychotic symptoms and confusion. In general, antidepressants carry a risk of inducing manic symptoms in patients with bipolar affective disorder. The risk of inducing mania with bupropion is thought to be less than with tricyclic antidepressants and selective serotonin reuptake inhibitors [Sachs et al., 1994] . Bupropion has been reported to exacerbate tics in children with co-occurring ADHD and Tourette’s syndrome [Spencer et al., 1993].
Drug interactions
Because bupropion is metabolized by CYP2B6, plasma concentrations may increase if it is given concomitantly with inhibitors of this enzyme (e.g., orphenadrine, cyclophosphamide). Bupropion inhibits CYP2D6 and potentially increases the plasma concentrations of other medications metabolized by this isoenzyme (e.g., tricyclic antidepressants and some selective serotonin reuptake inhibitors). The combination of bupropion with monoamine oxidase inhibitors may precipitate a hypertensive crisis. Both Wellbutrin® and Zyban® contain bupropion, and this combination is contraindicated because of an increased risk of seizure. Concurrent use of l-DOPA and bupropion has been associated with an increased rate of adverse effects [Wellbutrin, 2009].
Mirtazapine
Clinical applications
Mirtazapine (Remeron®) is available in 15-, 30-, and 45-mg tablets and orally disintegrating tablets. It is FDA-approved for the treatment of major depressive disorder in adults. In short-term studies, it appears to be more effective than placebo and comparable to amitriptyline in improving depressive symptoms [Bremner, 1995; Claghorn and Lesem, 1995]. Use of mirtazapine is off-label for any indication in children and adolescents. Two unpublished randomized, placebo-controlled studies of mirtazapine in children and adolescents failed to show superiority of mirtazapine to placebo for depression. One open-label study of mirtazapine showed improvement in children and adolescents with social phobia, although tolerability was limited due to weight gain and high discontinuation rate for adverse events [Mrakotsky et al., 2008].
Pharmacology
Mirtazapine increases synaptic levels of norepinephrine and serotonin through antagonism of α2-adrenergic receptors in the CNS. It is an antagonist at 5HT2 and 5HT3 receptors, increasing relative 5HT1 activity. Mirtazapine has antagonist activity at H1 histaminic, α1-adrenergic, and muscarinic receptors. Mirtazapine is well absorbed from the gastrointestinal tract. It reaches peak concentration in 2 hours, and its elimination half-life is in the range of 20–40 hours. Mirtazapine is metabolized by the liver and excreted in the urine. The isoenzymes CYP2D6, CYP1A2, and CYP3A4 are involved in its metabolism (see Table 49-5) [L’italien, 2003; Remeron, 2007].
Clinical management
In clinical practice, the sedating and appetite-stimulating effects of mirtazapine may be useful for patients who suffer from insomnia, agitation, decreased appetite, or anxiety as part of their depressive syndrome. Patients may be started at 7.5 mg (orally disintegrating tablets cannot be split) to 15 mg at bedtime. The dose may be increased up to a maximum of 45 mg daily at intervals of 1–2 weeks [L’italien, 2003; Remeron, 2007]. More rapid dose escalation and uses of higher doses may actually be associated with fewer side effects, especially sedation and increased appetite [Preskorn, 2000].
Adverse effects
Common side effects of mirtazapine include sedation, dizziness, nausea, increased appetite, weight gain, and increased serum levels of cholesterol. Effects on sedation and appetite decrease at higher doses of mirtazapine [Preskorn, 2000]. Less common side effects include liver enzyme elevations (i.e., aspartate transaminase) greater than three times the upper limit of the normal range and induction of mania or hypomania. Mirtazapine has been associated with rare cases of neutropenia or agranulocytosis in premarketing clinical trials [L’italien, 2003; Remeron, 2007].
Drug interactions
Mirtazapine may have additive effects if combined with alcohol or other CNS depressants. Mirtazapine is not a potent inhibitor of CYP2D6, CYP1A2, or CYP3A4. Combination of mirtazapine and monoamine oxidase inhibitors may induce a hypertensive crisis [L’italien, 2003; Remeron, 2007].
Anxiolytics
Anxiety disorders are one of the most prevalent categories of childhood and adolescent psychopathology. Approximately 8.9 percent of prepubertal children [Costello, 1989] in a general pediatric sample and approximately 8.7 percent of an adolescent sample [Kashani and Orvaschel, 1988] met criteria for at least one anxiety disorder. Anxiety disorders in children tend to have a chronic course with low remission rates [Keller et al., 1992]. Co-occurring conditions, such as other anxiety disorders [Kendall et al., 2010], depression [Bernstein and Borchardt, 1991; Kashani and Orvaschel, 1988; Strauss and Last, 1993], and ADHD [Anderson et al., 1987; Last et al., 1987] are common.
Treatment of anxiety disorders in children and adolescents is multimodal, including psychoeducational and cognitive behavioral therapy, family therapy, school interventions, and pharmacotherapy [Connolly and Bernstein, 2007]. Choices include tricyclic antidepressants, selective serotonin reuptake inhibitors, benzodiazepines, and α2-agonists and β-blockers. Selective serotonin reuptake inhibitors are the pharmacologic treatment of choice. Tricyclic antidepressants may be a reasonable choice for patients with co-occurring depression [Bernstein et al., 1996], ADHD [Pliszka, 2003], or enuresis [Fritz et al., 2004], or who cannot tolerate selective serotonin reuptake inhibitors. The role of benzodiazepines is limited to the short-term management of anxiety symptoms. In cases of severe anxiety, they may be added to tricyclic antidepressants or selective serotonin reuptake inhibitors for acute management of anxiety symptoms until the therapeutic effects of the antidepressants emerge. In clinical practice, the use of benzodiazepines is limited by concerns about possible abuse or dependence, drug diversion, and adverse cognitive effects.
Benzodiazepines
Clinical Applications
Benzodiazepines are classified as sedative-hypnotic medications. They act as sedatives or anxiolytics at low doses and sleep-inducing hypnotics at high doses. Although rapidly effective in relieving symptoms, they carry a risk of tolerance, dependence, and withdrawal with long-term use. These effects may be avoided by using only moderate doses for short-term therapy (1–2 weeks) [Sadock et al., 2007]. Benzodiazepines differ from each other with regard to relative potency and half-life. In contrast to long-half-life benzodiazepines, short-half-life benzodiazepines are associated with less daytime sedation, more frequent dosing, earlier and more severe withdrawal syndromes, rebound insomnia, and anterograde amnesia. Long-half-life benzodiazepines require less frequent dosing and have a less severe withdrawal syndrome, but high plasma concentrations of the drug may accumulate and cause signs of toxicity over time [Sadock et al., 2007].
Case reports and small open trials indicate that clonazepam and alprazolam may be helpful in treating anxiety disorders in children and adolescents [Biederman, 1987; Kutcher and MacKenzie, 1988; Simeon and Ferguson, 1987]. However, placebo-controlled studies have not supported their use [Graae et al., 1994; Simeon et al., 1992].
Pharmacology
Benzodiazepines are well absorbed from the gastrointestinal tract. They are extensively metabolized in the liver and excreted in urine. Benzodiazepine plasma concentrations are sensitive to medications that inhibit or induce cytochrome P-450 isoenzymes (see “Drug Interactions”). Benzodiazepines activate gamma-aminobutyric acid (GABA) benzodiazepine binding sites, which open chloride channels, resulting in a reduced rate of neuronal firing and muscle activity (Table 49-6) [Sadock, 2007].
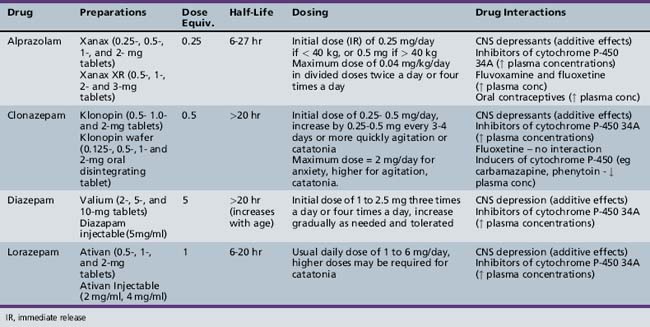
Clinical Management
In considering benzodiazepine therapy for a patient, the clinician should inquire about any possible history of substance abuse, as combined use of benzodiazepines and other CNS depressants can be lethal [Moody, 2004], and benzodiazepines can also be abused or diverted [Longo and Johnson, 2000]. Family history of substance abuse may identify patients who are at risk for drug abuse or dependence, and household members with substance use histories may indicate a risk for drug diversion. Concomitant use of medications and co-occurring medical conditions that may affect the metabolism of benzodiazepines should be identified. The patient and family should be informed about the risks of tolerance, dependence, and withdrawal effects, as well as possible effects on cognitive and motor functioning. During treatment, the clinician must ensure that medications are not overused, abused, or diverted, and should monitor for possible adverse cognitive or behavioral side effects. After the treatment trial is complete, the medication should be tapered slowly because of the potential for withdrawal and rebound anxiety [Coffey, 1993; Kutcher et al., 1992].
Adverse Effects
The most frequent adverse effects in children and adolescents are sedation, drowsiness, and decreased mental acuity [Biederman, 1991]. Benzodiazepines may cause paradoxical behavioral disinhibition, which manifests as increased agitation, aggression, and hyperactivity. This has been described with clonazepam in children [Graae et al., 1994] and adolescents [Reiter and Kutcher, 1991]. Anterograde amnesia also has been associated with benzodiazepines, particularly high-potency benzodiazepines [Uzun et al., 2010].
Benzodiazepine therapy carries a risk of tolerance, dependence, and withdrawal effects. Abrupt discontinuation or rapid dose reduction has been associated with seizures, delirium, and withdrawal symptoms [Uzun et al., 2010].
Drug Interactions
Additive CNS depression may occur when benzodiazepines are administered concomitantly with other CNS depressants, such as alcohol. Disulfiram may decrease the clearance of diazepam and chlordiazepoxide. Because benzodiazepines are metabolized by cytochrome P-450 isoenzymes, inhibitors and inducers of these isoenzymes can significantly alter the plasma concentration of benzodiazepines. Clonazepam and phenytoin decrease each other’s plasma concentrations. Valproate may increase plasma concentrations of lorazepam [Depakote, 2009].
Buspirone
Clinical Applications
Buspirone (BuSpar®) is approved for use in the treatment of anxiety disorders and for short-term relief of symptoms of anxiety [Buspar, 2007]. It is typically used in the treatment of generalized anxiety disorder. Buspirone does not appear to be effective as monotherapy in other anxiety disorders, such as post-traumatic stress disorder, panic disorder [Sheehan et al., 1988], or social phobia [van Vliet et al., 1997]. Although randomized, controlled trials do not support the use of buspirone to augment selective serotonin reuptake inhibitors in partial responders, some patients with depression [Dimitriou and Dimitriou, 1998] and OCD [Jenike et al., 1991] may benefit.
Open trials of buspirone in adolescents with generalized anxiety disorder [Kutcher et al., 1992] and children and adolescents with varied anxiety disorders [Simeon, 1991] demonstrate benefit. Additionally, buspirone appeared to improve anxiety and irritability in children with pervasive developmental disorders [Buitelaar et al., 1998] and may improve anxiety and aggression symptoms in some hospitalized children [Pfeffer et al., 1997]. However, two large unpublished, industry-sponsored, randomized, controlled trials in children and adolescents with generalized anxiety disorder did not demonstrate benefit [Buspar, 2007]. Buspirone offers several potential benefits over benzodiazepines, including lack of euphoric effects, performance impairment, abuse potential, and withdrawal symptoms with discontinuation. Although buspirone appears to be as effective as benzodiazepines in decreasing psychic symptoms of anxiety, it appears to be less effective for somatic symptoms of anxiety, and the onset of therapeutic effect may be delayed 2–4 weeks [Kutcher et al., 1992].
Pharmacology
Buspirone differs from benzodiazepines in that it has no activity at the GABA-benzodiazepine receptor complex. Buspirone acts as a partial agonist or mixed agonist/antagonist at 5HT1A receptors, as an agonist at postsynaptic dopamine receptors, and as an antagonist at presynaptic autoreceptor and postsynaptic sites in the CNS. Buspirone is rapidly absorbed from the gastrointestinal tract. Only 4 percent of the dose reaches systemic circulation because of extensive first-pass metabolism. Buspirone is metabolized in the liver, primarily by the CYP3A4 isoenzyme. Its primary metabolite, 1-pyrimidinylpiperazine (1-PP), has clinical activity. The elimination half-life is about 2–4 hours in all age groups and is prolonged by renal or hepatic impairment. Unlike adolescents or adults, prepubertal children show the highest concentration of the active metabolite, 1-PP, after oral administration of buspirone at doses of 15–60 mg/day, which may account for higher incidence of side effects in this group [Salazar et al., 2001]. The onset of clinical response may appear within the first 2 weeks of therapy, but optimal therapeutic effect is usually delayed [Kutcher et al., 1992].
Clinical Management
Laboratory screening is not required before prescribing buspirone. The typical starting dose is 5 mg taken 2–3 times daily, with gradual increases to 15 mg taken three times daily. Younger children may be started at 2.5–5 mg/day and gradually increased to a total dose of 20–30 mg/day, administered in divided doses 2–3 times daily [Scahill and Martin, 2002].
Adverse Effects
The most common adverse effects of buspirone are dizziness, headache, drowsiness, and lightheadedness [Buspar, 2007]. Side effects in adults appear to be dose-related and to decrease over time [Feighner and Cohn, 1989]. CNS depression is less common than in benzodiazepines. Behavioral activation, including agitation, aggression and euphoric mania, has been described in prepubertal children [Pfeffer et al., 1997].
Drug Interactions
Combination of buspirone and monoamine oxidase inhibitors may result in a hypertensive crisis, requiring emergency medical attention. Because of reports of liver transaminase elevations with the concomitant use of buspirone and trazodone, the manufacturer recommends monitoring liver function when prescribing this combination. Because buspirone is highly protein-bound, it may displace other medications that are protein-bound and increase their free plasma level. Inducers of CYP3A4 may decrease plasma levels of buspirone. Alternatively, inhibitors of CYP3A4 (e.g., fluvoxamine, erythromycin, grapefruit juice) may increase the serum concentration of buspirone. Increased serum concentrations of haloperidol have been reported when combined with buspirone [Kudo and Ishizaki, 1999].
Mood Stabilizers
The diagnosis of bipolar disorder in children is controversial for a number of reasons. Historically, it was thought not to exist in children; the presentation of bipolar disorder in children does not always resemble the presentation in adults; there is the perception of overdiagnosis; and with limited evidence of efficacy, use of medications for bipolar disorder in prepubertal children is rising. Although juvenile bipolar disorder has sometimes been overdiagnosed [Carlson, 1990], it is often under-recognized, or misdiagnosed as schizophrenia or disruptive behavior disorders [Carlson et al., 1994]. The phenotype of juvenile bipolar disorder has not been well defined and different investigators have utilized varying criteria [DelBello and Grcevich, 2004; Leibenluft et al., 2006]. Efforts to operationalize the diagnosis of juvenile bipolar disorder better are on-going [Leibenluft et al., 2003]. Bipolar children can, but are not likely to, exhibit classic symptoms of adult bipolar disorder. Rather, children with bipolar disorder are more likely to exhibit atypical manic or mixed-mood symptoms [Swann et al., 2005]. Adding to the complexity of making the diagnosis is the broad differential diagnosis and co-occurrence of other neuropsychiatric disorders [McClellan et al., 2007].
Evidence supporting the use of mood stabilizers to treat the acute episodes of mood disturbances and for prophylactic use during the maintenance phase of bipolar disorder [APA, 2002] will be reviewed. Mood stabilizers used in clinical practice can be grouped into four categories:
 lithium, which is approved for use in the acute treatment of mania and for mania prophylaxis in adults
lithium, which is approved for use in the acute treatment of mania and for mania prophylaxis in adults


Each mood stabilizer is not equally effective for all phases of the disorder or as prophylaxis. In adults, lithium, valproate, carbamazepine, risperidone, olanzapine, ziprasidone, aripiprazole, and quetiapine are currently approved for treating acute mania. Lamotrigine, seroquel, and olanzapine are labeled for use during the depressive phase of bipolar disorder [Calabrese et al., 1999; Evins, 2003]. Lithium, lamotrigine, quetiapine, olanzapine, and aripiprazole are FDA-approved for use during the maintenance phase of treatment as prophylaxis against recurrent episodes.
Combining medications in the treatment of bipolar affective disorder in adults is common. Typical treatment consists of long-term maintenance with a mood stabilizer as prophylaxis. An antipsychotic or an additional mood stabilizer may be added to stabilize the acute symptoms of a manic episode [Drug treatments for bipolar disorder, 2005; Narayan and Haddad, 2010]. During a depressive episode, an antidepressant, lamotrigine, or atypical antipsychotic may be added while cautiously monitoring for the possible emergence of antidepressant-induced manic or rapid-cycling symptoms [Drug treatments for bipolar disorder, 2005; Narayan and Haddad, 2010].
The pediatric literature is less well developed than that for adults and is complicated by diagnostic variability between studies. However there is an emerging evidence base for the treatment of bipolar I disorder. In the past several years, risperidone, olanzapine, quetiapine, and aripiprazole were FDA-approved for treatment of bipolar I disorder in youngsters 10 years and older (with the exception of olanzapine, which is 13 years and older). Additionally, lithium is FDA-approved for use in youngsters of 12 years and older for bipolar disorder, although the evidence base for this labeling is not robust and results from a downward extension of the adult literature. As with childhood depression, children with bipolar affective disorder are thought to be less responsive to medication compared with adults [McClellan et al., 2007]. Combination of mood stabilizers [Findling et al., 2003], or a mood stabilizer with an antipsychotic [Kafantaris, 1995], is sometimes required to stabilize acute manic symptoms. Studies looking at efficacy of combination therapies in children and adolescents are lacking.
Lithium
Clinical Applications
Lithium is FDA-approved for use in the treatment of bipolar affective disorder. It is effective in managing acute manic and depressive episodes, preventing or diminishing the intensity of subsequent episodes in maintenance treatment, and decreasing mood instability between episodes [Goodwin and Jamison, 1990]. Data in children and adolescents with bipolar disorder are limited and offer mixed results. Several earlier studies showed good response rates [DeLong and Aldershof, 1987; Strober et al., 1990; Varanka et al., 1988], although this was not universally demonstrated [Carlson et al., 1992]. These studies were largely uncontrolled and naturalistic, varied in diagnostic criteria, and included mixed diagnostic groups (e.g., bipolar I, bipolar II, bipolar not otherwise specified (NOS)), making interpretation of results difficult. A more recent large open-label study has provided additional support for the use of lithium in adolescents with bipolar disorder [Kafantaris et al., 2003], although the need for larger randomized, controlled studies of children and adolescents in both the acute and maintenance phases is recognized and protocols are currently in development [Findling et al., 2008]. Lithium also has been used with positive results in the treatment of severe aggression in adults [Sheard, 1975] and children [Campbell et al., 1984; DeLong and Aldershof, 1987; Malone et al., 2000].
Pharmacology
Lithium is an alkali metal, similar to sodium and potassium, and it is available in salt form as lithium carbonate or lithium citrate. Lithium has effects at the cellular level (i.e., cell membrane ion channels and cyclic adenosine monophosphate second-messenger cellular processes) and in neurotransmitter systems. However, the specific mechanism of action is unclear [Lenox and Wang, 2003].
Lithium is available in immediate-release and sustained-release preparations. Ninety percent of immediate-release lithium is absorbed by the gastrointestinal tract; 60–90 percent of extended-release lithium is absorbed. Immediate-release lithium reaches peak serum levels in 1–1.5 hours (4–12 hours for the extended-release form), and it is almost completely excreted in urine, with the remainder excreted in the feces and breast milk. Lithium has a half-life of about 20 hours and reaches steady-state levels after 1 week of administration [FDA, 2010].
Clinical Management
Lithium is started at dosages ranging from 300 to 600 mg/day. Formulas [Weller et al., 1986] and nomograms [Alessi et al., 1999; Geller and Fetner, 1989] have been developed to guide lithium dosing in pediatric populations. Trough lithium levels (10–12 hours after last dose) are drawn when steady state is achieved, at least 5 days after initiating lithium or changing the dose. The recommended therapeutic serum levels range from 0.6 to 1.2 mEq/L in adults, although therapeutic level has not been firmly established in children. Lithium levels between 0.8 and 1.0 mEq/L may offer the best prophylaxis against mania, whereas lower levels may be adequate for prophylaxis against depression [Severus et al., 2005, 2009]. Lithium levels above the therapeutic range increase the risk of toxicity. Early therapeutic effects may be detected at 10–14 days after achieving a therapeutic level; however, there is often a significant lag of 4–6 weeks [Kowatch et al., 2000]. Between 4 and 6 weeks of lithium at therapeutic serum concentrations constitutes an adequate trial. Because several days are required to achieve therapeutic plasma concentrations, antipsychotic medications may be necessary to stabilize severe mania acutely, especially if psychotic symptoms are present.
After mood symptoms are stabilized, lithium levels should be routinely checked every 3–6 months. Other indications to obtain a lithium level include symptoms of toxicity, worsening side effects, and breakthrough mood symptoms. Urinalysis, renal evaluation [Jefferson, 2010], and thyroid function tests should be repeated every 3–6 months to monitor for the emergence of subclinical renal and thyroid dysfunction (Table 49-7).
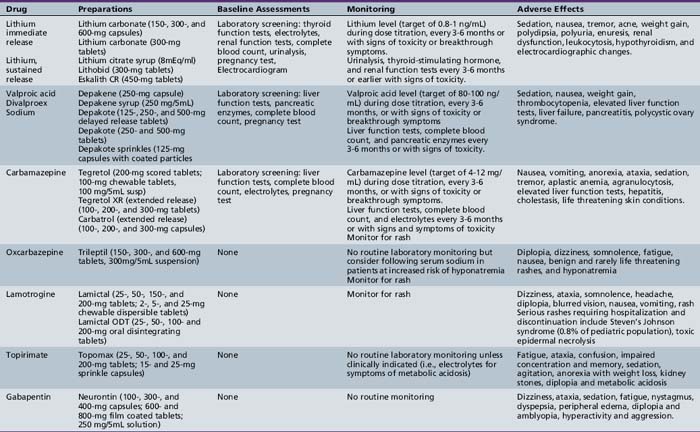
Stabilization of manic symptoms may be prolonged up to several years [Biederman et al., 1998]. Lithium should be continued for at least 18 months after manic symptoms have been stabilized [APA, 2002] because discontinuation of lithium therapy, especially abrupt discontinuation, is associated with increased relapse rates [Strober et al., 1990].
Adverse Effects
Side effects of lithium are polydipsia, polyuria or enuresis, gastric distress (i.e., nausea, vomiting, and diarrhea), weight gain, tremor, fatigue, leukocytosis, acne, and mild cognitive impairment [Alessi et al., 1999; Silva et al., 1992]. Serious renal side effects are rare and include nephrotic syndrome, features of distal renal tubular acidosis, nonspecific interstitial fibrosis, and renal failure [Alessi et al., 1999; Gelenberg and Schoonover, 1991]. Lithium therapy can induce alterations in thyroid function tests and produce hypothyroidism [Alessi et al., 1999]. Lithium can have rare cardiac side effects, including first-degree atrioventricular block, irregular sinus rhythm, and increased premature ventricular contractions [Gelenberg and Schoonover, 1991]. Reversible conduction abnormalities in children have been demonstrated on their EKGs [Campbell et al., 1972]. Studies examining the long-term effects of lithium treatment of children are lacking. Intrauterine exposure to lithium during the first trimester has been associated with teratogenic effects, most commonly Ebstein’s anomaly of the tricuspid valve [Giles and Bannigan, 2006]. Lithium has been associated with pseudotumor cerebri in children [Kelly et al., 2009].
Drug Interactions
Lithium levels are affected by medications or conditions that can alter the body’s fluid and electrolyte status. Dehydration and extremely low salt intake can result in elevated lithium levels. Likewise, an extremely high salt intake can result in lowered lithium levels. Many medications, such as diuretics, nonsteroidal anti-inflammatory drugs, and tetracycline, can cause an increase in lithium levels by influencing renal clearance [Finley et al., 1995].
Lithium is commonly used in combination with other psychotropic medications in the treatment of bipolar disorder. However, a number of case reports have described neurotoxicity occurring rarely when combined with other mood-stabilizing medications, including antipsychotic medications [Finley et al., 1995].
Valproic Acid
Clinical Applications
Valproic acid is approved for the treatment of seizures, migraine prophylaxis, and the manic phase of bipolar affective disorder in adults [Depakote, 2009]. The initial support for use of valproic acid in child psychiatric practice came from the adult literature. The evidence for its use in the treatment of juvenile bipolar disorder, however, is limited. Although some support for the use of valproic acid was derived from earlier case series and open trials [Henry et al., 2003; Kowatch et al., 2000; Wagner et al., 2002], a large, double-blind, randomized, placebo-controlled trial did not demonstrate superiority of divalproex extended release over placebo in the acute phase management of bipolar disorder [Wagner et al., 2009]. Divalproex sodium has also been shown in small studies to benefit children and adolescents with explosive temper and mood lability [Donovan et al., 2000] and for treatment of core symptoms of autism and associated symptoms of affective instability, aggression, and impulsivity [Hollander et al., 2001].
Pharmacology
Valproate rapidly converts to valproic acid in the stomach. Divalproex dissociates into valproic acid in the gastrointestinal tract. Valproic acid is rapidly and almost entirely absorbed from the gastrointestinal tract. Peak plasma concentrations are achieved 1–4 hours, 3–5 hours, and 7–14 hours after oral administration of valproate, divalproex, and extended-release tablets, respectively. Food intake can retard the time to peak plasma concentrations, although this has little clinical consequence once steady-state concentrations have been achieved. Valproic acid binds plasma proteins; therefore, the presence of other protein-binding drugs can increase the unbound or free fraction of valproic acid. Conversely, the addition of valproate with another protein-bound drug can increase the unbound or free fraction of the other drug. Renal and hepatic insufficiency can reduce protein binding, which can increase unbound valproate. Additionally, plasma protein binding of valproic acid is dependent on drug concentration. Concentrations of free valproic acid increase at serum levels above 100 mg/mL. Peak and trough concentrations with delayed-release tablets and capsules containing coated particles may differ from the equivalent dose of valproic acid. Valproic acid is metabolized in the liver and excreted predominantly in the urine [Malone et al., 2000; Depakote, 2009]. No therapeutic serum concentrations for the treatment of behavioral or psychiatric disorders have been determined.
Clinical Management
In addition to the routine psychiatric or neurologic history, patients should be asked about risk factors affecting treatment with valproate, including pregnancy, disorders of metabolism, hepatic or renal disease, concomitant medications, and coagulation disorders. Laboratory screening should include liver function tests and a CBC, and a pregnancy test in females [Danielyan and Kowatch, 2005]. Parents should be educated about potential signs of hepatic or pancreatic toxicity and polycystic ovary disease.
Recommended initial dosing of valproic acid is 10–15 mg/kg/day, given in divided doses two or three times per day. Dosage may be increased by 5–10 mg/kg/day at weekly intervals [Malone et al., 2000; Depakote, 2009]. The manufacturer’s maximum recommended dosage is 60 mg/kg/day. However, starting at lower doses and using slower upward adjustments of dose may be required for tolerability. Although therapeutic levels for treating mania have not been established, achieving a plasma concentration of 85–110 mg/L [Kowatch et al., 2000] is needed to consider a trial adequate for determining outcome in an individual patient. Divalproex extended release allows for once-daily dosing.
In adult psychiatry, an oral loading dose of valproate is sometimes used to achieve a therapeutic plasma concentration and to gain rapid control over mania. The tolerability of an oral loading dose of divalproex sodium was examined in children and adolescents [Good et al., 2001]. A loading dose of 15 mg/kg/day resulted in therapeutic plasma levels by day 5 of treatment and it was well tolerated. Dosage adjustment was required for overweight children, in order to avoid supratherapeutic drug levels [Good et al., 2001]. Total valproic acid levels should be checked when steady state is reached, after any dosage change, after conversion to a different preparation of valproate, for signs of toxicity, or for a change in clinical status.
Maintenance involves monitoring of the therapeutic response and side effects and requires laboratory screening. Liver enzymes, CBC, and serum valproic acid levels should be periodically monitored, although the exact frequency of testing has not been determined. Initially, monitoring may be done every 3–6 months for the first year and semiannually thereafter (see Table 49-7).
Adverse Effects
Adverse effects include sedation, nausea, vomiting, gastrointestinal upset, increased appetite, weight gain, and hair loss. Rare side effects include thrombocytopenia, platelet dysfunction, prolonged bleeding time, elevation in liver transaminase levels, and lactate dehydrogenase, and can occur without clinical symptoms. Children younger than 2 years appear to have an increased risk of hepatotoxicity, and extreme caution should be used in this population. This heightened risk may result from an undiagnosed metabolic or mitochondrial disorder. In particular, children with urea cycle defects, such as ornithine transcarbamylase deficiency, should not receive this medication. Carnitine supplementation should be considered when prescribing this medication, as this may facilitate the processing of valproate in the mitochondrial β-oxidation pathway. Valproate has also been associated with rare cases of hyperammonemic encephalopathy [Barrueto and Hack, 2001; Elgudin et al., 2003], which occurs more commonly with polypharmacy and with associated liver dysfunction [Verrotti et al., 1999]. One case reported in the child psychiatry literature manifested as increased aggression and confusion, an EEG with symmetric 5- to 6-Hz waves, elevated serum ammonia levels, and normal levels of serum liver transaminases [Yehya et al., 2004]; the situation resolved with valproic acid discontinuation. Elevated ammonia levels have also been observed in the absence of encephalopathy, but the clinical relevance of such laboratory abnormalities is unclear. Other reports suggest that oral carnitine can be useful in reducing ammonia levels [Bohan et al., 2001]. Rare cases of pancreatitis have been reported [Malone et al., 2000; Depakote, 2009]. Cases of polycystic ovary disease have been reported in some women treated with valproate for seizure disorder [Isojarvi et al., 1993]. It is unclear whether this is related to the medication or is an endocrine abnormality related to the epilepsy [Davis et al., 2000]. Intrauterine exposure to valproic acid during the first trimester is associated with neural tube defects [Malone et al., 2000; Depakote, 2009].
Drug Interactions
As it is metabolized by the cytochrome-P450 system, valproic acid has many interactions with medications that are also metabolized by this system. Medications that may increase valproic acid concentrations include erythromycin, selective serotonin reuptake inhibitors, guanfacine, and aspirin. Medications that may decrease concentrations of valproate include carbamazepine. Additionally, valproic acid may result in increases in concentration of other anticonvulsants (phenytoin, phenobarbital, primidone, carbamazepine, and lamotrigine), tricyclic antidepressants, diazepam, and warfarin. Great caution should be utilized when valproic acid is used in combination with lamotrigine due to enhanced risk of Stevens–Johnson syndrome. When valproic acid is combined with antipsychotics, CNS depressant effects and extrapyramidal side effects may increase. Valproic acid may increase lithium-associated tremor [Danielyan and Kowatch, 2005].
Carbamazepine
Clinical Applications
Limited data are available on the efficacy of carbamazepine for the treatment of bipolar disorder children and adolescents. Justification for its use in clinical practice has rested on the adult literature, which is difficult to interpret. Carbamazepine was superior to placebo and comparable to lithium [Lerer et al., 1987; Small et al., 1991] in improving manic symptoms. However, it was not as effective as valproate, and patients on carbamazepine required adjunctive medication to achieve stabilization [Vasudev et al., 2000]. Similarly, in children, carbamazepine was as effective as lithium but less so than valproic acid [Kowatch et al., 2000]. Carbamazepine may be helpful in the depressive phase of bipolar disorder [Dilsaver et al., 1996].
Pharmacology
Carbamazepine is structurally related to tricyclic antidepressants. It reduces activity at sodium and calcium channels, resulting in reduced synaptic transmission. It is available in immediate-release forms (e.g., Tegretol® tablets, chewable tablets, suspension) and extended-release capsules (i.e., Tegretol XR®). It is slowly absorbed from the gastrointestinal tract. The plasma half-life gradually decreases over 3–5 weeks from 25–65 hours to approximately 12–17 hours due to autoinduction. Steady-state concentrations are achieved in 2–4 days. Between 75 and 90 percent of the drug is bound to plasma proteins. It is metabolized in the liver and excreted in the urine [Sachs et al., 1994; Tallian et al., 1996].
Clinical Management
Patients should be evaluated for medical conditions that may contraindicate use of carbamazepine, including liver disease, hematologic disorders, immune disorders, cardiac disease, and pregnancy. Laboratory screening should include a urine pregnancy screen, liver function tests, and a CBC. Because laboratory screening can fail to predict impending hepatic and hematologic complications, parents should be aware of signs and symptoms indicating serious adverse effects, such as rash, symptoms of aplastic anemia or agranulocytosis, and hepatitis or cholestasis. Sexually active patients should be informed of its teratogenic effects and the possible decrease in effectiveness of oral contraceptives [Sachs et al., 1994; Tallian et al., 1996].
Dosage is initiated at 100–300 mg/day, increasing as tolerated while monitoring blood levels. Therapeutic blood levels in the treatment of mania have not been established. Recommended levels are the same as those for seizure disorder, 4–12 mg/mL [Rosenberg et al., 1994]. Blood levels should be checked 5 days after initiating or changing doses, and again in 4 weeks because of an anticipated reduction in plasma levels due to autoinduction. Although it is recommended that liver function tests, bilirubin levels, and CBCs be routinely checked every 3 months, the utility of such testing in predicting serious hepatic or hematologic complications is questionable (see Table 49-7). Hyponatremia resulting from carbamazepine use is rare in children, and if it is clinically suspected, sodium levels should be obtained.
Adverse Effects
The most common adverse effects are nausea, vomiting, sedation, and ataxia [Sachs et al., 1994; Tallian et al., 1996]. Carbamazepine can cause benign elevations in liver transaminases; hepatitis, indicated by elevated liver transaminase levels greater than three times normal; and cholestasis, indicated by elevations in bilirubin and alkaline phosphatase. Carbamazepine therapy has been associated with leukopenia in 1–2 percent of patients, and agranulocytosis or aplastic anemia in 1 of 250,000 patients. Although blood dyscrasias are not always predicted by leukopenia, CBCs are recommended at 3- to 6-month intervals. Other adverse effects observed with carbamazepine include cognitive or behavioral disturbance [Carpenter and Vining, 1993], CNS toxicity, altered cardiac conduction, and life-threatening dermatologic conditions (i.e., exfoliative dermatitis, toxic epidermal necrolysis, and Stevens–Johnson syndrome). Intrauterine exposure has been associated with neural tube defects and other fetal malformations [Rosa, 1991]. Case reports of manic reactions to carbamazepine may be related to its antidepressant structure [Myers and Carrera, 1989].
Drug Interactions
Carbamazepine is an inducer of CYP3A4, leading to decreased plasma concentrations of medications metabolized by this isoenzyme. Several medications that are commonly combined with carbamazepine in psychiatric practice may be affected, including anticonvulsants (e.g., lamotrigine, clonazepam, valproic acid), antidepressants (e.g., bupropion, clomipramine, desipramine, imipramine, nefazodone), and antipsychotics (e.g., clozapine, fluphenazine, haloperidol, olanzapine, ziprasidone). Carbamazepine levels can be increased by fluoxetine, lamotrigine, valproate, cimetidine, and erythromycin. Coadministration of carbamazepine with nefazodone results in inadequate levels of nefazodone and its active metabolite, and is therefore contraindicated. Combination with lithium or antipsychotics may increase the risk of adverse neurologic effects (e.g., drowsiness, dizziness, ataxia) [Sachs et al., 1994; Tallian et al., 1996].
Other Mood Stabilizers
Although lithium and valproate are first-line pharmacologic treatments for bipolar disorder in adults [APA, 2002], many children and adolescents do not respond adequately to these medications and cannot tolerate the side effects or the requirement of laboratory monitoring. In clinical practice, newer anticonvulsants are beginning to be used for the treatment of bipolar disorder. These medications include oxcarbazepine, lamotrigine, gabapentin, and topiramate. Many of these newer anticonvulsants have a more favorable adverse effect profile and do not require blood tests. Support for their use in clinical practice, however, has been primarily based on case reports and open trials. Use of these medications is more appropriate for patients with bipolar disorder who are unresponsive to traditional therapies or cannot tolerate traditional agents.
Lamotrigine
Clinical management
Lamotrigine (Lamictal®) is approved for use in the treatment of epilepsy and for prophylaxis of mania during maintenance therapy for type 1 bipolar disorder [Lamictal, 2009]. In adults, lamotrigine appears to be helpful in the treatment of patients with bipolar depression [Calabrese et al., 1999; Evins, 2003], some patients with rapid-cycling bipolar disorder [Bowden et al., 1999; Calabrese et al., 2000; Frye et al., 2000], and as a prophylactic agent to prevent or attenuate recurrences of bipolar episodes [Bowden et al., 2003; Calabrese et al., 2000, 2003; McElroy et al., 2004]. Data on lamotrigine in pediatric bipolar disorder are limited to case series and open-label trials but suggest that, in adolescents, lamotrigine may be effective for the treatment of bipolar depression [Chang et al., 2006; Kusumakar and Yatham, 1997] and for the maintenance treatment of bipolar disorder [Pavuluri et al., 2009].
Pharmacology
Lamotrigine (Lamictal®) is a member of the phenyltriazine class of antiepileptic drugs and is chemically unrelated to other antiepileptic drugs. The mechanism of action of lamotrigine is unknown in both epilepsy and bipolar disorder. Lamotrigine may exert its effect on sodium channels and modulate release of excitatory amino acids, such as glutamate and aspartate [Lamictal, 2009]. Lamotrigine is available in scored tablets (Lamictal®), chewable dispersible tablets (which can be chewed or dissolved in juice), and orally disintegrating tablets (Lamictal ODT®). Lamotrigine is rapidly absorbed through the gastrointestinal tract, metabolized in the liver, and excreted in urine. Pharmacokinetic studies reveals that lamotrigine clearance is influenced by total body weight and by co-administration of other antiepileptic drug therapy. Specifically, children tend to have a higher lamotrigine clearance than adults, especially those under 30 kg, and may require a higher weight-corrected dose. As with adults, the clearance of lamotrigine is affected by concurrent antiepileptic drug treatment [Lamictal, 2009].
Clinical management
Before initiating treatment, a urine pregnancy test should be obtained. The patient and family should be educated about the risk of serious rash or hypersensitivity reaction (i.e., Stevens–Johnson syndrome), and instructed on how to recognize the signs and symptoms requiring immediate medical attention and the need for a slow titration [Joe et al., 2009] and slow retitration after discontinuation to avoid the rash.
Dosing for treatment of pediatric bipolar disorder is not established, but existing published reports in adolescent populations used gradual titration of lamotrigine over 6–8 weeks, starting at 12.5–25 mg with weekly or biweekly increments of 12.5–25 mg up, reaching doses between 100 and 200 mg daily based on clinical response [Chang et al., 2006; Pavuluri et al., 2009]. A painstaking approach to dose adjustment decreases the risk for rash, including Stevens–Johnson syndrome [Joe et al., 2009]. Doses were adjusted for adolescents on valproate with initial dosing of 12.5 mg, with weekly increments of 12.5 mg for a maximum daily dose between 50 and 100 mg daily. There are published dosing recommendations for pediatric patients who are being treated for epilepsy with and without concomitant antiepileptic drug treatment [Lamictal, 2009]. For children between the ages of 2 and 12 years who are not on other antiepileptic medications, 0.3 mg/kg/day of lamotrigine is administered in one or two divided doses (rounded down to the nearest 5 mg) for the first 2 weeks. During the third and fourth week, 0.6 mg/kg/day is administered in two divided doses (rounded down to the nearest 5 mg). The dose may be titrated up in increments of 0.6 mg/kg/day (rounded down to the nearest tab) every 2 weeks. The usual maintenance dose of seizures ranges from 4.5 to 7.5 mg/kg/day. For pediatric patients older than 12 years, 25 mg of lamotrigine is administered daily during the first 2 weeks of treatment. The dose is increased to 50 mg/day for the third and fourth week and given in two divided doses. The dose may be increased by increments of 50 mg/day every 1–2 weeks (see Table 49-7). Maintenance dosage ranges from 225 to 375 mg daily, given in two divided doses. Dosing must be adjusted upward for children taking concomitant carbamazepine, phenytoin, phenobarbital, or primidone, which increases lamotrigine clearance, and dosing must be reduced for children taking concomitant valproate, which decreases lamotrigine clearance; physicians are referred to the product prescribing information for detailed recommendations regarding dosing with valproate and other antiepileptic medications [Lamictal, 2009].
Adverse effects
Common adverse effects observed in clinical studies include dizziness, ataxia, somnolence, headache, diplopia, blurred vision, nausea, vomiting, and rash [Lamictal, 2009; Salee et al., 2009]. Lamotrigine has been associated with serious rashes requiring hospitalization and discontinuation of treatment. The manufacturer reports the incidence of Stevens–Johnson syndrome as 0.8 percent in the pediatric population, which is higher than that observed in adult populations. Rare cases of toxic epidermal necrolysis and rash-related death have been reported. Potential risk factors for serious rash are concomitant use of valproate, rapid dose escalation, and higher than recommended starting doses. Most rashes emerge during the first 6–8 weeks of treatment [Lamictal, 2009; Salee et al., 2009]. Rashes have been reported in 10 percent of patients in clinical trials. As it is difficult to distinguish a benign rash from the early stages of a more serious rash, the manufacturer recommends discontinuation of treatment at the first sign of a rash, unless it is clearly not medication-related. Rare but potentially life-threatening hypersensitivity reactions have occurred with lamotrigine. Symptoms such as fever and lymphadenopathy, with or without a rash, should prompt immediate evaluation and possible discontinuation of treatment. Lamotrigine accumulates in melanin-rich tissues, such as skin and the eye. Clinical trials did not detect ophthalmologic adverse effects; however, no long-term data are available. Lamotrigine inhibits dihydrofolate reductase; administration of lamotrigine to rats reduced serum folate levels. Low folate levels have been associated with teratogenic effects [Lamictal, 2009; Salee et al., 2009].
Drug interactions
Any drug interaction that might lead to higher blood levels or longer duration of action may increase the risk for rash. Valproate, for example, decreases clearance of lamotrigine and may increase the risk of potentially life-threatening rashes [Lamictal, 2009; Salee et al., 2009]. In contrast, carbamazepine, phenytoin, phenobarbital, and primidone increase clearance of lamotrigine and may require dosing adjustments.
Oxcarbazepine
Clinical applications
Oxcarbazepine is a 10-keto analog of carbamazepine and is currently approved for both monotherapy and adjunctive therapy for partial seizures in children and adolescents. Data in adults suggest that oxcarbazepine is efficacious for treatment of acute mania [Hirschfeld and Kasper, 2004]. Data are limited for treatment of pediatric bipolar disorder, but oxcarbazepine was not superior to placebo in the one published randomized, placebo-controlled trial in children and adolescents with bipolar I disorder [Wagner et al., 2006].
Pharmacology
The mechanism of action of oxcarbazepine (Trileptal®) in the treatment of seizures and bipolar disorder is unknown. Oxcarbazepine is well absorbed after oral administration and is rapidly reduced in the liver to the active 10-monohydroxy metabolite (MHD), which is responsible for its pharmacologic activity. The MHD is conjugated with glucuronic acid and almost entirely excreted in the urine. The half-life of the oxcarbazepine is 2 hours and the half-life of the MHD is 9 hours. Dose adjustments are not required with mild to moderate hepatic insufficiency, but are required with renal disease [Trileptil, 2009].
Drug interactions
Oxcarbazepine has little activity on most of the cytochrome P-450 enzymes, with the exception of CYP2C19 and CYPCA4/5. Due to its activity on the CYPCA4/5 isoenzyme, it can reduce plasma concentrations of oral contraceptives and cyclosporine. Additionally, plasma levels of oxcarbazepine and its active metabolite are lowered in the presence of inducers of the P450 enzyme system, including carbamazepine, phenytoin, and phenobarbital. Unlike carbamazepine, oxcarbazepine does not induce its own metabolism and is not highly protein-bound: therefore, its co-administration with other medications is less complex [Trileptil, 2009].
Clinical management
As efficacy has not been established, dosing requirements for treatment of pediatric bipolar disorder are unknown. Dosing for pediatric epilepsy is weight-based. It is recommended that oxcarbazepine should be started at 8–10 mg/kg daily in divided doses, not to exceed a total dose of 600 mg. The dose may be increased over a period of 2 weeks to a target dose (900 mg if <30 kg, 1200 mg if <40 kg, and 1800 mg if >40 kg) [Trileptil, 2009]. Although rapid dose escalation is possible, there is suggestion that slower titration of oxcarbazepine improves tolerability [Wagner et al., 2006].
Gabapentin
Clinical applications
Gabapentin (Neurontin®) is approved as a treatment for postherpetic neuralgia in adults and as an adjunctive therapy for treatment of partial seizures in children and adults [Neurontin, 2009]. Case reports and open studies of gabapentin as adjuvant therapy in adult bipolar patients were promising; however, double-blind trials have not confirmed efficacy in mania as monotherapy [Frye et al., 2000] or adjunctive treatment [Pande et al., 2000], or in treatment of resistant rapid-cycling bipolar disorder [Evins, 2003; Yatham et al., 2002]. Data are very sparse on mood or behavioral symptoms in pediatric populations. One small case series suggested that gabapentin may reduce irritability in children with severe neurologic impairment [Hauer et al., 2007], but there are no safety or efficacy data in pediatric bipolar disorder.
Pharmacology
Gabapentin (Neurontin®) is available in capsules, tablets, and solution. Plasma half-life is 5–7 hours. It is not metabolized by the liver and is excreted unchanged in the urine. It is not protein-bound and has no clinically significant drug interactions [Neurontin, 2009].
Clinical management
Pediatric dosing for partial seizures is established, and for adolescents typically ranges from 900 mg to 1800 mg daily [Neurontin, 2009]. Clinical trial data do not indicate any routine monitoring of laboratory screens. No therapeutic plasma concentrations have been established (see Table 49-7).
Adverse effects
Gabapentin is generally well tolerated. Common side effects include dizziness, ataxia, sedation, fatigue, and nystagmus. Less common side effects include dyspepsia, peripheral edema, diplopia, and amblyopia [Neurontin, 2009]. Behavioral side effects are possible and include aggression and hyperactivity [Lee et al., 1996; Tallian et al., 1996].
Topiramate
Clinical applications
Topiramate (Topamax®) is approved for migraine prophylaxis in adults, monotherapy of partial and primary generalized tonic-clonic seizures for ages 12 years and up, adjunctive therapy for partial seizures and primary generalized tonic-clonic seizures for ages 2 years and up, and for seizures associated with Lennox–Gastaut syndrome for ages 2 years and up [Sheard, 1975; Tohen et al., 2007]. In adults, despite early promise that topiramate may be effective as adjunctive therapy in treatment-resistant bipolar disorder [Ghaemi et al., 2001; Vieta et al., 2002] and for management of acute mania or rapid-cycling bipolar disorder [Chengappa et al., 1999; McElroy et al., 2000], placebo-controlled trials did not demonstrate superiority over placebo in the acute management of mania [Kushner et al., 2006]. Data are quite limited in pediatric populations. Retrospective chart reviews of outpatient and inpatient children and adolescents with bipolar disorder treated with topiramate as an adjuvant mood stabilizer indicated response rates of 73 percent for mania [Barzman et al., 2005; DelBello et al., 2002], but the only double-blind, placebo-controlled trial was terminated prematurely when the adult trials failed to show efficacy [Delbello et al., 2005]. Potential advantages of topiramate over other mood stabilizers are its weight-neutral and appetite-suppressive effects [Chengappa et al., 1999; McElroy et al., 2000]. Topiramate augmentation of olanzapine for the treatment of pediatric bipolar disorder helped to limit weight gain compared to youngsters treated with olanzapine alone, although did not confer any benefit on mood stabilization [Wozniak et al., 2009].
Pharmacology
Topiramate (Topomax®) is available in tablets or sprinkle capsules, which are bioequivalent. Its absorption is rapid, reaching peak plasma concentrations at 2 hours. Antacids can decrease absorption of topiramate. Plasma elimination half-life is 21 hours, and steady state is reached in about 4 days. About 70 percent of topiramate is excreted unchanged in the urine. The remainder is metabolized in the liver [Topomax, 2009].
Clinical management
Topiramate should be initiated at 25–50 mg daily (1–3 mg/kg/day), with small weekly increases of 1–3 mg/kg/day to minimize adverse effects. The recommended total daily dose in the treatment of seizures is 5–9 mg/kg/day or 400 mg in divided doses [Weiner et al., 2004]. No guidelines are available for dosing in pediatric bipolar disorder. Lower doses, 50–100 mg daily, of topiramate given adjunctively with olanzapine were effective in limiting weight gain [Wozniak et al., 2009]. Monitoring the plasma concentration is unnecessary, unless topiramate is used concomitantly with phenytoin or carbamazepine (see Table 49-7).
Adverse effects and drug interactions
Adverse effects include fatigue, ataxia, confusion, impaired concentration and memory, sedation, agitation, anorexia with weight loss, kidney stones, and diplopia. Topiramate may decrease systemic levels of bicarbonate, leading to a metabolic acidosis. Consequently, slow titration, clinical observation, and laboratory monitoring are advised. Carbamazepine, valproic acid, and phenytoin can lower the plasma concentration of topiramate. Topiramate may increase the concentration of phenytoin and decrease that of valproic acid [Sheard, 1975; Tohen et al., 2007].