Chapter 51 Neurophysiology of Seizures and Epilepsy
A seizure is defined as abnormal neuronal firing leading to a clinical alteration of neurologic function (motor, sensory, autonomic, or psychological). Electrical activity underlying a seizure is the net product of biochemical processes at the cellular level occurring in the context of large neuronal networks and likely involves several key cortical and subcortical structures. The output of this activity is reflected on the surface electroencephalogram (EEG), which is the primary clinical tool for measuring normal and abnormal brain electrical activity. Epilepsy is the condition of recurrent spontaneous seizures arising from aberrant electrical activity within the brain. Epilepsy is not a singular disease, but rather is heterogeneous in terms of clinical expression, underlying causes, and pathophysiology (Table 51-1). An epilepsy syndrome refers to a group of signs and symptoms that usually occur together, such as seizure type, age of seizure onset, responsiveness to a particular AED, and characteristic EEG findings, genetics, and natural history. Epileptogenesis is the process by which neural circuits undergo structural or physiological changes, resulting in an enduring epileptic state.
Table 51-1 Examples of Pathophysiologic Defects Leading to Epilepsy
| Level of Brain Function | Condition | Pathophysiologic Mechanism |
|---|---|---|
| Neuronal network | Cerebral dysgenesis, post-traumatic scar, mesial temporal sclerosis (in TLE) | Altered neuronal circuits; formation of aberrant excitatory connections (i.e., sprouting) |
| Neuron structure | Down syndrome and other syndromes with mental retardation and seizures | Abnormal structure of dendrites and dendritic spines; altered current flow in neuron |
| Neurotransmitter synthesis | Pyridoxine (vitamin B6) dependency | Decreased GABA synthesis; B6, a co-factor for GAD |
| Neurotransmitter receptors, inhibitory | Angelman syndrome, juvenile myoclonic epilepsy | Abnormal GABA receptor subunits |
| Neurotransmitter receptors, excitatory | Nonketotic hyperglycinemia | Excess glycine leads to activation of NMDA receptors |
| Synapse development | Neonatal seizures | Many possible mechanisms, including the depolarizing action of GABA early in development |
| Ion channels (channelopathies) | Benign familial neonatal convulsions Dravet syndrome | Potassium channel mutations Sodium channel mutations |
GABA, gamma-aminobutyric acid; GAD, glutamic acid decarboxylase; NMDA, N-methyl-d-aspartate; TLE, temporal lobe epilepsy.
Classification of Seizures
Seizures are broadly divided into two groups, depending on their site of origin and pattern of spread (Figure 51-1). Partial seizures arise from a localized region of the brain, and the associated clinical manifestations are related to the function ordinarily subserved by that area. Focal discharges can spread locally through synaptic and nonsynaptic mechanisms, distally to subcortical structures, and through commissural pathways that may eventually involve the entire cortex; this evolution is believed to occur when focal seizures secondarily generalize. For example, a seizure arising from the left motor cortex may cause jerking movements of the right upper extremity. If epileptiform discharges subsequently spread to adjacent areas and eventually encompass the entire brain, it is described as a secondarily generalized tonic-clonic seizure.
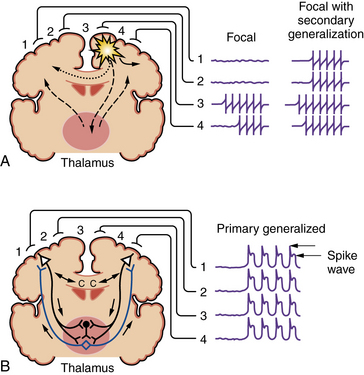
Fig. 51-1 Coronal brain sections depicting seizure types and potential routes of seizure spread.
(From Stafstrom CE. An introduction to seizures and epilepsy: Cellular mechanisms underlying classification and treatment. In: Stafstrom CE, Rho JM, eds. Epilepsy and the ketogenic diet. Totowa, NJ: Humana Press, 2004;3–29. Reprinted with kind permission of Springer Science+Business Media.)
Primary generalized seizures begin with abnormal electrical discharges in both hemispheres simultaneously and involve reciprocal thalamocortical connections (see Figure 51-1). The EEG signature of a primary generalized seizure is bilateral synchronous spike-wave discharges seen across all scalp electrodes. The manifestations of such widespread epileptiform activity can range from brief impairment of consciousness (e.g., an absence seizure) to rhythmic jerking movements of all extremities accompanied by loss of posture and consciousness.
Epilepsies or epilepsy syndromes have likewise been categorized into those in which seizures begin focally (partially) or throughout the brain (generalized), with further division into those that have a known etiology (symptomatic) and those that do not (idiopathic; many of these have a genetic basis) [Commission, 1989]. The classification of epilepsy syndromes is undergoing revision as new knowledge of epilepsy genetics and pathophysiology emerges [Engel, 2006].
Cellular Electrophysiology
Excitation/Inhibition Balance
Although there are differences in the mechanisms that underlie partial and generalized seizures, it is useful to view any seizure activity as a perturbation in the normal balance between inhibition and excitation. This imbalance can involve a localized region, multiple brain areas, or the entire brain, and often reflects a combination of increased excitation and decreased inhibition. However, the traditional concept of a seizure arising when the excitation/inhibition balance is altered is useful but oversimplified. It is now appreciated that factors once thought to be solely inhibitory (e.g., gamma-aminobutyric acid (GABA) synaptic transmission) can actually be excitatory in some circumstances (see Increased Seizure Susceptibility of the Immature Brain, below). Furthermore, synaptic inhibition is critical for certain normal brain rhythms and probably also plays a role in abnormal rhythmic activity, such as spike-wave discharges [Fritschy, 2008].
Structural Correlates of Epilepsy: Hippocampus and Neocortex
Brain regions differ in their propensity to generate seizures, based on intrinsic membrane properties, synaptic organization, cell density, and pattern of cellular inteconnectivity. Even within the same brain region, cell types differ with regard to their excitability [Steriade, 2004]. The hippocampus and neocortex are particularly prone to seizure generation. The hippocampus, with its orderly laminar organization and trisynaptic excitatory circuitry, has been used extensively in electrophysiologic studies of seizure mechanisms [Schwartzkroin and Mueller, 1987].
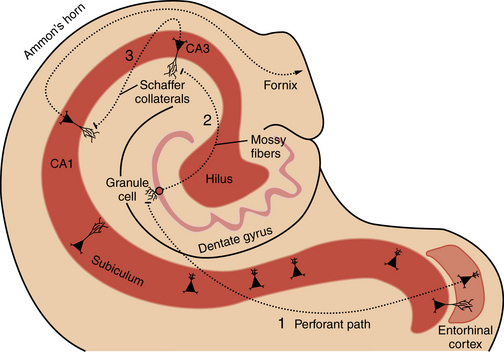
Familiarity with hippocampal anatomy will facilitate understanding of the physiological concepts discussed below (Figure 51-2). The hippocampal formation consists of the dentate gyrus, hippocampus proper (i.e., Ammon’s horn, with subregions CA1, CA2, and CA3), subiculum, and entorhinal cortex. These four regions are linked by prominent excitatory, largely unidirectional, feed-forward connections. The forward-projecting trisynaptic circuit begins with neurons in layer II of the entorhinal cortex that project axons to the dentate gyrus along the perforant pathway, where they synapse on granule cell and interneuron dendrites. Granule cells, the principal cell type of the dentate gyrus, send their axons, called mossy fibers, to synapse on cells in the hilus and in the CA3 field of Ammon’s horn. Several classes of inhibitory interneurons within the dentate hilus modulate on-going excitatory neuronal activity [Lawrence and McBain, 2003]. CA3 pyramidal cells project to other CA3 pyramidal cells through local collaterals, to the CA1 field of Ammon’s horn through Schaffer collaterals, and to the contralateral hippocampus. CA1 pyramidal cells send their axons into the subicular complex. Neurons of the subicular complex project to the entorhinal cortex and other cortical and subcortical targets.
Overview of Ion Channels
The key channels and receptors involved in normal and epileptic firing are summarized in Figure 51-3 and Table 51-2. Two major types of ion channels – voltage-gated and ligand-gated – are responsible for inhibitory and excitatory activity. Voltage-gated channels include sodium and calcium channels that function to depolarize the cell membrane toward the action potential threshold, while voltage-gated potassium channels largely dampen neural excitation. Voltage-gated channels are activated by membrane potential changes that alter the conformational state of the channel and allow selective passage of charged ions through a pore. Ligand-gated channels constitute the second type. In ligand-gated channels, a neurotransmitter (e.g., glutamate, GABA) is released from a presynaptic terminal (after presynaptic calcium influx) into the synaptic cleft and then binds with selective affinity to a membrane-bound receptor on the postsynaptic membrane. This binding activates a cascade of events, including a conformational shift to reveal an ion-permeant pore. Passage of ions across these channels results in depolarization (i.e., inward flux of cations) or hyperpolarization (i.e., inward flux of anions or outward flux of cations). Numerous ion channel mutations underlie epilepsy syndromes, giving rise to the concept of “epilepsy channelopathies” [Reid et al., 2009].
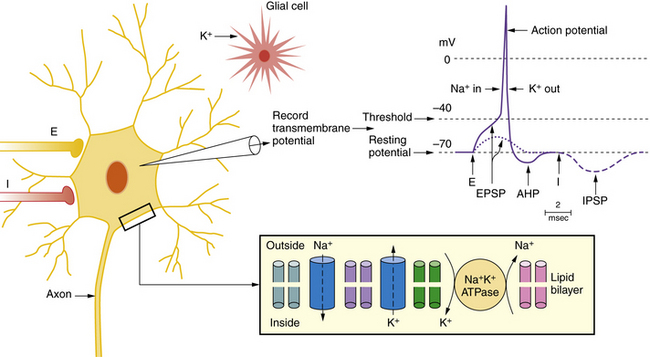
Table 51-2 Roles of Channels and Receptors in Normal and Epileptic Firing
| Channel or Receptor | Role in Normal Neuronal Function | Possible Role in Epilepsy |
|---|---|---|
| Voltage-gated Na+ channel | Subthreshold EPSP; action potential upstroke | Repetitive action potential firing |
| Voltage-gated K+ channel | Action potential downstroke | Abnormal action potential repolarization |
| Ca2+-dependent K+ channel | AHP after action potential; sets refractory period | Limits repetitive firing |
| Voltage-gated Ca2+ channel | Transmitter release; carries depolarizing charge from dendrites to soma | Excess transmitter release; activates pathophysiologic intracellular processes |
| Non-NMDA receptor (i.e., AMPA) | Fast EPSP | Initiates PDS |
| NMDA receptor | Prolonged, slow EPSP | Maintains PDS; Ca2+ activates pathophysiologic intracellular processes |
| GABAA receptor | IPSP | Limits excitation |
| GABAB receptor | Prolonged IPSP | Limits excitation |
| Electrical synapses | Ultrafast excitatory transmission | Synchronization of neuronal firing |
| Na+-K+ pump | Restores ionic balance | Prevents K+-induced depolarization |
AHP, after-hyperpolarization; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; EPSP, excitatory postsynaptic potential; GABA, γ-aminobutyric acid; IPSP, inhibitory postsynaptic potential; NMDA, N-methyl-d-aspartate; PDS, paroxysmal depolarization shift.
Voltage-Dependent Membrane Conductances
Depolarizing Conductances
A rapidly inactivating inward sodium conductance underlies the depolarizing (excitatory) phase of the action potential, and a non-inactivating, persistent sodium current can augment cell depolarization (e.g., produced by excitatory synaptic input) in the subthreshold voltage range; both are critical for regulation of neuronal firing and play a role in epilepsy [Stafstrom, 2007b; Ragsdale, 2008]. Many AEDs act in part through interactions with voltage-dependent sodium channels [Rogawski and Loscher, 2004]. Each sodium channel is composed of a complex of three polypeptide subunits: a major α subunit and two smaller β subunits that influence the kinetic properties of the α subunit. The shape of action potentials is determined by the types of α and β subunits present in an individual neuron [Catterall et al., 2005].
Neurons also display voltage-gated inward calcium conductances. Calcium currents underlie burst discharges in hippocampal CA3 neurons. Activation of voltage-dependent calcium channels contributes to the depolarizing phase of the action potential, and can affect neurotransmitter release, gene expression, and neuronal firing patterns. Several distinct subtypes of calcium channels are distinguished on the basis of electrophysiologic properties, pharmacologic profile, molecular structure, and cellular localization [Catterall et al., 2003]. The molecular structure of voltage-gated calcium channels is similar to that of sodium channels. Voltage-dependent calcium channels are hetero-oligomeric complexes containing a principal pore-forming α-1 subunit and one or more smaller subunits (α2, β, γ, δ) that are not obligatory for normal activity but modulate the kinetic properties of the channel.
Hyperpolarizing Conductances
Depolarizing sodium and calcium currents are counterbalanced by an array of voltage-dependent hyperpolarizing (inhibitory) currents, primarily mediated by potassium channels. Potassium channels represent the largest and most diverse family of voltage-gated ion channels, and they function to decrease neuronal excitation [Gutman et al., 2005]. The prototypic voltage-gated potassium channel is composed of four membrane-spanning α subunits and four regulatory β subunits that are assembled in an octameric complex to form an ion-selective pore. Potassium conductances include a leak conductance, which is a major determinant of the resting membrane potential; an inward rectifier (involving the flux of other ions), which is activated by hyperpolarization; a large set of delayed rectifiers, which are involved in the termination of action potentials and repolarization of the neuron’s membrane potential; an A-current, which helps to determine interspike interval and affects the rate of cell firing; an M-current, which is activated by cholinergic muscarinic agonists and affects resting membrane potential and cell firing rate; and a set of calcium-activated potassium conductances, which are sensitive to intracellular calcium concentration and affect cell firing rate and interburst interval. (Rectification refers to differences in conductance depending on the direction of ion flow through the channel; rectification can also result from blocking of the pore by other ions.)
Modulation or facilitation of hyperpolarizing conductances can be viewed as potentially antiepileptic, and some newer AEDs act directly on voltage-gated potassium channels [Wickenden, 2002]. For example, topiramate induces a steady membrane hyperpolarization mediated by a potassium conductance, and levetiracetam blocks sustained repetitive firing by paradoxically decreasing voltage-gated potassium currents. Retigabine, an opener of Kv7 subtype potassium channels, has broad efficacy in animal seizure models and enhances activation of KCNQ2 and KCNQ3 potassium channels [Miceli et al., 2008]. This finding is particularly intriguing, given that mutations in genes encoding these proteins have been linked to a rare form of inherited epilepsy, benign familial neonatal seizures [Singh et al., 2003].
Synaptic Physiology
Inhibitory Synaptic Transmission
GABA, the main inhibitory neurotransmitter in the mature mammalian central nervous system, is a neutral amino acid synthesized from glutamic acid by the rate-limiting enzyme glutamic acid decarboxylase. GABA released from axon terminals binds to at least two classes of receptors, GABAA and GABAB, which are found on almost all cortical neurons [Sieghart, 2006].
The GABAA receptor is a macromolecular receptor complex consisting of an ion pore and binding sites for agonists and a variety of allosteric modulators, such as benzodiazepines and barbiturates, each differentially affecting the kinetic properties of the receptor. The GABAA receptor is a heteropentameric complex composed of combinations of several polypeptide subunits arranged in topographic fashion to form an ion channel. This channel is selectively permeable to chloride and bicarbonate ions. Seven types of subunits (α, β, γ, δ, ε, π, ρ) have been described, each with one or more subtypes [Wafford et al., 2004]. Although several thousand receptor isoforms are possible from differential expression and assembly of various subunits, only a limited number of functional combinations is likely to exist in the brain. Most functional GABAA receptors follow the general motif of containing either α and β or α, β, and γ subunits with variable stoichiometry. Because individual subunits may be differentially sensitive to pharmacologic agents, GABAA receptor subunits represent potentially useful molecular targets for new AEDs [Macdonald and Kang, 2009].
Activation of GABAA receptors on the somata of mature neurons generally results in the influx of chloride ions and consequent membrane hyperpolarization, inhibiting cell firing. In neurons of the immature brain, however, GABAA receptor activation causes depolarization of the postsynaptic membrane [Staley, 2006a]. This reversal of the conventional GABAA effect reflects a reversed chloride electrochemical gradient, a consequence of the evolving expression of cation chloride co-transporters during development (see Development of Neurotransmitters, Receptors, and Transporters, below).
In addition to GABAA receptors, metabotropic GABAB receptors are located on postsynaptic membrane and presynaptic terminals [Bettler and Tiao, 2006]. GABAB receptors act through guanosine triphosphate (GTP)-binding proteins to control calcium or potassium conductances. Whereas GABAA receptors generate fast, high-conductance, inhibitory postsynaptic potentials close to the cell body, GABAB receptors on the postsynaptic membrane mediate slow, long-lasting, low-conductance inhibitory postsynaptic potentials, primarily in dendrites. Perhaps of greater functional significance, activation of GABAB receptors on axon terminals blocks neurotransmitter release. It is thought that GABAB receptors are associated with terminals that release GABA on to postsynaptic GABAA receptors. In such cases, activation of GABAB receptors reduces the amount of GABA released, resulting in disinhibition [Simeone et al., 2003]. Abnormalities of GABAergic function, including synthesis, synaptic release, receptor composition, trafficking or binding, and metabolism, can each lead to a hyperexcitable, epileptic state [Cossart et al., 2005; Macdonald and Kang, 2009].
Excitatory Synaptic Transmission
Glutamate, an excitatory amino acid, is the principal excitatory neurotransmitter of the mammalian central nervous system. Glutamatergic pathways are widespread throughout the brain, and excitatory amino acid activity is critical to normal brain development and activity-dependent synaptic plasticity [Simeone et al., 2004]. There are two broad classes of glutamate receptors – ionotropic and metabotropic. Ionotropic glutamate receptors are divided into N-methyl-d-aspartate (NMDA) and non-NMDA receptors, based on biophysical properties and pharmacologic profiles. Each subtype of glutamate receptor consists of a multimeric assembly of subunits that determine its distinct functional properties. An NMDA receptor consists of an NR1 subunit plus NR2A, NR2B, NR2C, NR2D, and/or NR3A.
The NMDA receptor contains a binding site for glutamate (or NMDA) and a recognition site for a variety of modulators (e.g., glycine, polyamines, MK-801, zinc). NMDA receptors also demonstrate voltage-dependent block by magnesium ions. When the membrane is depolarized and the magnesium block of the NMDA receptor is alleviated, activation of the NMDA receptor results in an influx of calcium and sodium ions. Calcium entry is central to the initiation of a number of second messenger pathways, such as stimulation of a variety of kinases that subsequently activate signal transduction cascades, leading to changes in transcriptional regulation. Activation of the NMDA receptor leads to generation of relatively slow and long-lasting excitatory postsynaptic potentials. These synaptic events contribute to epileptiform burst discharges, and NMDA receptor blockade results in the attenuation of bursting activity in many models of epileptiform activity [Kalia et al., 2008].
Non-NMDA ionotropic receptors are divided into α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors [Vincent and Mulle, 2009]. AMPA receptors are composed of combinations of GluR1, GluR2, GluR3, and/or GluR4 subunits, while kainate receptors are composed of combinations of GluR5, GluR6, GluR7, KA1, and/or KA2 subunits. AMPA receptors are responsible for the major part of the excitatory postsynaptic potential – fast-rising and brief in duration – generated by release of glutamate on to postsynaptic neurons. The depolarization generated by AMPA receptors is necessary for effective activation of NMDA receptors. Consequently, AMPA receptor antagonists block most excitatory synaptic activity. Non-NMDA receptors typically pass sodium current, but certain subunit combinations, such as a relative deficit of GluR2, GluR5, or GluR6, endow the receptor with increased calcium permeability, a finding that has implications for development, as well as for epilepsy (see Development of Neurotransmitters, Receptors, and Transporters below) [Rakhade and Jensen, 2009; Santos et al., 2009].
Metabotropic glutamate receptors represent a large, heterogeneous family of G-protein-coupled receptors that subsequently activate various transduction pathways – phosphoinositide hydrolysis and activation of adenylate cyclase and phospholipases C and D. Metabotropic receptors are important modulators of voltage-dependent potassium and calcium channels, nonselective cation currents, and ligand-gated receptors (i.e., GABA and glutamate receptors), and they can regulate glutamate release. Different metabotropic glutamate receptor subtypes are specific for different intracellular processes. Although ubiquitous within the central nervous system, subtypes of metabotropic receptors appear to be differentially localized. Metabotropic glutamate receptors have been implicated in a wide variety of normal neurologic processes (e.g., long-term potentiation) [Anwyl, 2009] and disease states (e.g., epilepsy) [Ure et al., 2006].
Abnormal Neuronal Firing
Specific pathophysiological mechanisms mediate each stage of seizure evolution, including transitions from a normal neuronal firing pattern to interictal epileptiform bursts, from interictal firing to seizure activity, and from the seizure to the postictal state [Stafstrom, 2004; Lado and Moshe, 2008]. Figure 51-4 depicts EEG and intracellular changes that can be seen in normal, interictal, and ictal states. In the normal situation, action potentials are generated in neuron 1 when the membrane potential reaches threshold for firing. These discharges may influence the activity of an adjacent neuron (neuron 2) synaptically, resulting in an excitatory postsynaptic potential. An adjacent interneuron (neuron 3, which is inhibitory) may also be activated by a discharge from neuron 1 after a brief delay, giving rise to an inhibitory postsynaptic potential. The activity recorded in neuron 2 reflects the temporal and spatial summation of excitatory and inhibitory postsynaptic potentials.
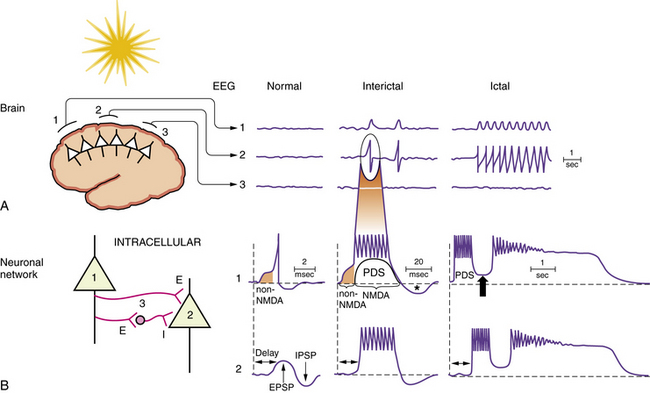
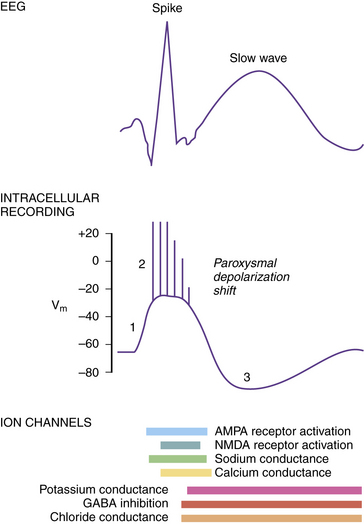
Paroxysmal Depolarization Shift
The intracellular correlate of the focal interictal epileptiform discharge on the EEG is known as the paroxysmal depolarization shift (PDS) [Gorji and Speckmann, 2009]. Initially, there is a rapid shift in the membrane potential in a depolarizing direction, followed by a burst of repetitive action potentials lasting several hundred milliseconds (Figure 51-5). The initial depolarization is mediated by AMPA receptors, whereas the sustained depolarization is a consequence of NMDA receptor activation. Afterward, the PDS terminates with a repolarization phase, primarily a consequence of inhibitory potassium and chloride conductances carried by voltage-gated potassium channels and GABA receptors. The prolonged period of hyperpolarization after the PDS is mediated by inhibitory conductances and constitutes a refractory period. PDS activity in several adjacent neurons would be expected to facilitate synchronous firing [LeDuigou et al., 2009].
Synchronizing Mechanisms
The hippocampal formation (see Figure 51-2) normally displays robust neuronal synchronization. Sharp waves, dentate spikes, theta activity, 40-Hz oscillations, and 200-Hz oscillations are all forms of neuronal synchronization that can be recorded in various regions of the hippocampal formation [Buzsaki and Draguhn, 2004]. Synchronization of neuronal activity appears to be intrinsic to the mechanism by which the hippocampus performs its normal functions. However, neuronal synchronization is also a hallmark of epilepsy and exaggerated synchrony among hippocampal neurons may directly generate seizures [Engel et al., 2009; Hughes, 2008]. Forms of synchronized activity that do not trigger seizures in a normal hippocampus may trigger epileptiform discharges in a hippocampus that has undergone selective neuronal loss, synaptic reorganization, or changes in receptor expression.
In the hippocampus, synchronizing mechanisms include inputs from subcortical nuclei and intrinsic interneuron-mediated synchronization. For example, high-amplitude theta activity is a salient feature of the normal hippocampus. The theta rhythm represents synchronized activity of hippocampal neurons and largely depends on input from the septum [Buzsaki and Draguhn, 2004]. Subcortical nuclei, such as the septum, have divergent inputs that target hippocampal interneurons. The divergent axon projections of interneurons and the powerful effect of the GABAA receptor-mediated conductances that they produce enable interneurons to entrain the activity of large populations of principal neurons. These characteristics make interneurons a very effective target for subcortical modulation of hippocampal principal cell activity.
Recurrent excitatory circuits are another substrate for neuronal synchronization. Recurrent excitatory collaterals are a normal feature of the hippocampal CA3 region. CA3 pyramidal cells form direct, monosynaptic connections with other CA3 pyramidal cells, contributing to the synchronized bursts that characterize this region [Traub et al., 2004a]. In the epileptic temporal lobe, synaptic reorganization and axonal sprouting may lead to aberrant recurrent excitation, providing a synchronizing mechanism in other parts of the hippocampal formation, including CA1, subiculum, entorhinal cortex, and dentate gyrus. In the dentate gyrus of the normal hippocampus, for example, granule cells form few or no monosynaptic contacts with neighboring granule cells. However, in the epileptic hippocampus, mossy fiber sprouting results in direct excitatory interactions among granule cells [Nadler, 2003] (Figure 51-6).
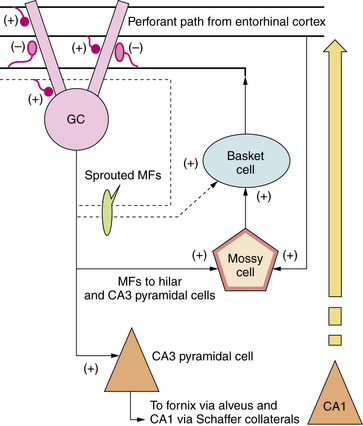
Fig. 51-6 Hippocampal circuitry and seizure-induced circuit reorganization.
(From Sankar R et al. Paroxysmal disorders. In: Menkes JH et al., eds. Child Neurology, 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2006;857–942.)
Mechanisms independent of chemical synaptic transmission may synchronize neuronal firing under some circumstances. Such nonsynaptic mechanisms include gap junctions, electrical field (i.e., ephaptic) effects, and changes in extracellular ion concentrations. Gap junctions allow electrical signals to pass directly between cells. Gap junctions are upregulated in epileptic brain tissue and blockade of gap junctions significantly affects the duration of seizure activity [Traub et al., 2004b]. Ephaptic effects, generated by current flow through the extracellular space, can enhance epileptogenic neuronal synchronization [Dudek et al., 1998]. Increases in extracellular potassium concentration have long been known to affect epileptic excitability and synchronization, and experiments have demonstrated clear epileptogenic effects of blocking potassium regulation (e.g., through inwardly rectifying potassium channels) [Traynelis and Dingledine, 1988; McBain et al., 1993].
Glial Mechanisms for Modulating Epileptogenicity
Glial contributions to epileptic mechanisms have been relatively neglected until recently [Wetherington et al., 2008]. Because the ionic balance between the intracellular and extracellular compartments is altered after neuronal activity (especially after repetitive discharges seen with seizures), there must exist mechanisms to restore ion homeostasis. Astrocytes are perhaps most closely associated with regulation of extracellular potassium levels (i.e., potassium buffering) because glial cell membranes are preferentially permeable to potassium. A variety of inwardly rectifying potassium channels mediate potassium uptake, and the association of glial endfeet with brain microvasculature provides a convenient “sink” for potassium release. Glial cell membrane potential changes are directly correlated with changes in extracellular potassium concentration, and blockade of potassium channels selective for glial cells results in neuronal hyperexcitability. Therefore, certain glial cells can modulate neuronal discharge by regulating extracellular potassium.
Another important role of glia is transporting synaptically released glutamate out of the extracellular space. Glia cell membranes are uniquely equipped for this role, possessing powerful glutamate transporters [Shigeri et al., 2004]. Rapid and efficient removal of extracellular glutamate characterizes normal, healthy brain tissue, and this is essential because residual glutamate would continue to excite surrounding neurons. Blockade of glutamate transporters (or knockout of the genes for these transport proteins) results in epilepsy or excitotoxicity [Meldrum et al., 1999].
Glial cells modulate excitability in other ways, as well. First, glia play a critical role in regulating extracellular pH by a proton exchanger and by bicarbonate transporter mechanisms. Even low levels of neuronal activity create significant pH transients. NMDA receptor function is very sensitive to pH. Second, glial cells are thought to release powerful neuroactive agents into the extracellular space. Glial glutamate release can excite neighboring neurons, and some investigations have suggested that other glia-related factors – such as the cytokine interleukin-1 – can have profound proconvulsant effects [Vezzani et al., 2008].
Physiology of Absence Epilepsy
The generalized spike-wave discharges accompanying absence seizures reflect a widespread, phase-locked oscillation between excitation (i.e., spike) and inhibition (i.e., slow wave) in mutually connected thalamocortical networks [McCormick and Contreras, 2001; Huguenard and McCormick, 2007]. Pyramidal neurons from layer VI of the neocortex send excitatory projections to thalamic relay neurons and inhibitory GABAergic neurons comprising the nucleus reticularis thalami (Figure 51-7). Excitatory outputs of the thalamic relay neurons impinge on the apical dendrites of layer VI pyramidal neurons in the neocortex. This thalamocortical relay is a critical substrate for the generation of cortical rhythms and is influenced by sensory inputs (e.g., from the retina) and by several brainstem nuclei that constitute the origin of cholinergic, noradrenergic, serotonergic, and other projections. This reciprocal circuitry, which is responsible in large part for normal EEG oscillations during wake and sleep states, can become hyperactive and generate generalized spike-wave discharges or can be dampened to reduce spontaneous cortical rhythms. The anatomy implies that spike-wave discharges are interrupted at cortical or thalamic levels.
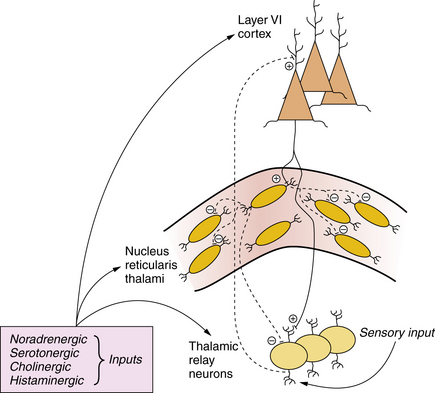
Although multiple ionic conductances are involved in rhythmic pacemaking activity, two channels are believed to play a key role in regulating thalamocortical activity. The first is a subtype of voltage-gated calcium channel known as the low-threshold (T-type) calcium channel. These channels are activated by small depolarizations of the plasma membrane. In many neurons, calcium influx through these channels triggers low-threshold spikes and activates a burst of action potentials [McCormick and Contreras, 2001; Perez-Reyes, 2003]. Such an excitatory burst is thought to underlie the spike portion of a generalized spike-wave oscillation. AEDs known to be clinically effective against absence seizures (e.g., ethosuximide, valproic acid) block T-type calcium currents.
The second important ion channel involved in the regulation of thalamocortical rhythmicity is the hyperpolarization-activated cation channel, responsible for the so-called h-current. These channels, densely expressed in the thalamus and hippocampus, are activated by hyperpolarization and produce a depolarizing current carried by an inward flux of sodium and potassium ions. This depolarization helps to bring the resting membrane potential toward threshold for activation of T-type calcium channels, which produces a calcium spike and a burst of action potentials. Hyperpolarization-activated cation channels are highly expressed in dendrites and are expressed to a lesser extent in the soma. They are also critically involved in developmental plasticity [Bender and Baram, 2008].
Unlike other voltage-gated conductances that can be labeled either inhibitory or excitatory, h-currents are both inhibitory and excitatory [Poolos, 2004; Dyhrfjeld-Johnsen et al., 2009]. Hyperpolarization-activated cation channels possess an inherent negative-feedback property; hyperpolarization activates certain cation channels, which then leads to depolarization that then deactivates these channels. The net effect of hyperpolarization-activated cation channel activation is a decrease in the input resistance of the membrane, which is the voltage change produced by a given synaptic current. H-currents tend to stabilize membrane potential around the resting potential, countering hyperpolarizing and depolarizing inputs. The relevance of hyperpolarization-activated cation channels in the pathogenesis of absence seizures was underscored by the demonstration that lamotrigine, an anti-absence agent, enhances dendritic h-currents in hippocampal pyramidal neurons and by the experimental finding that deletion of a specific hyperpolarization-activated cation isoform results in absence epilepsy [Ludwig et al., 2003].
With respect to antiepileptic action, it is uncertain whether known T-type calcium channel blockers, such as ethosuximide, in fact block absence seizures through interactions with this type of calcium channel [Huc et al., 2009]. Antagonists of GABAB receptors and dopaminergic agonists can also interrupt abnormal thalamocortical discharges in experimental absence epilepsy models [Snead, 1996]. GABAB receptors are involved in mediating long-lasting thalamic inhibitory postsynaptic potentials involved in the generation of normal thalamocortical rhythms, whereas brainstem monoaminergic projections disrupt these rhythms. Although it is appealing to think of absence seizures as a byproduct of dysfunction of the T-type calcium channel or h-channel, or both, the actual pathophysiologic mechanisms are probably much more complex [Hughes, 2009].
Increased Seizure Susceptibility of the Immature Brain
Seizure incidence is highest during the first decade and especially during the first year of life. Multiple physiologic factors (Table 51-3 and Figure 51-8) contribute to the increased susceptibility of the developing brain to seizures [Sanchez and Jensen, 2001; Velísková et al., 2004; Wong, 2005; Ben-Ari and Holmes, 2006; Silverstein and Jensen, 2007; Stafstrom, 2007a]. This information is mainly derived from experimental epilepsy models in rodents, in which a window of heightened excitability in the second postnatal week gives rise to a lowered seizure threshold. This period of hyperexcitability in the rodent is approximately analogous to the first year of life in the human infant [Avishai-Eliner et al., 2002].
Table 51-3 Factors Promoting Increased Seizure Susceptibility in the Developing Brain
| Factor | Consequence | References |
|---|---|---|
| Input resistance and time constant: increased in immature neurons | Small inputs result in relatively large voltage changes | McCormick and Prince, 1987 |
| Voltage-gated ion channels: earlier maturation of sodium and calcium channels, delayed development of potassium channels | Longer action potentials, shorter refractory periods, increased neuron firing | Spitzer, 2006 |
| Synapse development: excitatory synapses appear before inhibitory synapses | Relative predominance of excitation over inhibition early in development | Swann et al., 1999 |
| Synapse development: overexpression of excitatory synapses during critical period | Corresponds to window of heightened seizure susceptibility | Swann et al., 1999 |
| Developmental changes in glutamate receptor subunits: NR2B/NR2A ratio favors prolonged depolarizing responses; NR2D relative overexpression reduces Mg2+ block | Favor relative hyperexcitability | Rakhade and Jensen, 2009 |
| Late appearance of functional inhibitory synapses | Along with other factors favoring excitation, contributes to neuronal excitatory drive and lack of functional inhibition | Brooks-Kayal, 2005 |
| Developmental changes in GABAA receptor function and Cl− gradient | GABA is depolarizing early in life, enhancing excitability | Ben-Ari, 2002 |
| Developmental changes in GABAA receptor subunits | Partially accounts for developmental differences in inhibitory effectiveness and benzodiazepine responsiveness | Brooks-Kayal, 2005 |
| Developmental sensitivity to glutamate toxicity | Less glutamate-induced excitotoxicity early in development | Rakhade and Jensen, 2009 |
| Immature GABAA binding pattern in substantia nigra | Proconvulsant effect | Velisek et al., 1995 |
| Electrical synapses; more common early in development | Mechanism for enhanced synchrony of neuronal networks | Talhouka et al., 2007) |
| Immature homeostatic mechanisms: Na+,K+-ATPase, glial K+ regulation, K+-Cl− cotransporters | Prolonged exposure to elevated extracellular K+ leads to further neuronal depolarization | Haglund and Schwartzkroin, 1990 |
GABA, gamma-aminobutyric acid; NR, N-methyl-d-aspartate (NMDA) receptor.
(Adapted from Stafstrom CE et al. Consequences of epilepsy in the developing brain: Implications for surgical management. Semin Pediatr Neurol 2000;7:147.)
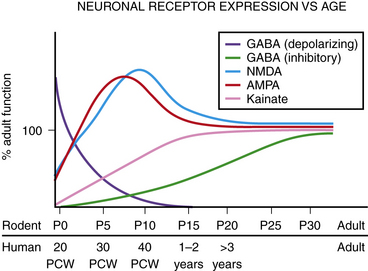
Fig. 51-8 Schematic depiction of changes in receptor expression in the developing brain.
(From Silverstein FS, Jensen FE. Neonatal seizures. Ann Neurol 2007;62:112–120. Wiley–Liss, Inc.)
Each seizure susceptibility factor alters the net brain excitatory–inhibitory balance in favor of enhanced excitation, and each involves a multiplicity of substrates and mediators, including ion channels, neurotransmitters and their receptors, structural changes in the maturing brain, and ionic gradients. Seizure propensity in the very young involves a complex interplay between the timing of these cellular and molecular changes [Rakhade and Jensen, 2009]. Although there are disadvantages to the physiologic adaptations that make the immature brain especially vulnerable to hyperexcitability and seizure generation, such idiosyncrasies of early brain development also provide the opportunity to develop novel, age-specific therapies.
Development of Ionic Channels and Membrane Properties
The relative timing of ion channel development plays a major role in the enhanced excitability of the immature brain [Spitzer, 2006]. Sodium and calcium ion channels, which mediate neuronal excitation, develop relatively early. Action potentials persist longer in early development, and this prolonged excitation mediates a greater calcium current, particularly at the presynaptic terminal; these factors increase excitability by enhancing neurotransmitter release. However, the brain must achieve a critical balance; activity-driven calcium channel activation is necessary for normal developmental processes such as cellular differentiation, migration, and synaptogenesis, but excessive inward calcium current can cause seizures and neuronal damage. Mutations of the α-1 subunit of the sodium channel are responsible for a spectrum of developmental epilepsies, including generalized epilepsy with febrile seizures plus and Dravet syndrome (also known as severe myoclonic epilepsy of infancy) [Meisler and Kearney, 2005; Harkin et al., 2007; Stafstrom, 2009].
Through a large variety of potassium channels outward currents counter depolarizing influences and tend to stabilize membrane excitability. Potassium channels develop slightly later than sodium and calcium channels. During a window of development, the net balance of ion fluxes favors excitation [Rakhade and Jensen, 2009].
Intrinsic membrane properties, such as membrane resistance and time constant, contribute to the neuron’s response to a synaptic input. During development, neurons have relatively higher membrane resistances and shorter time constants, and a depolarizing stimulus of a given amplitude and duration can have a greater effect in an immature neuron than in a mature one [McCormick and Prince, 1987]. Such electrical factors act in concert with ionic conductances to determine the magnitude of the membrane’s response to a synaptic input.
Development of Neurotransmitters, Receptors, and Transporters
Excitatory synapses tend to form before inhibitory ones. Each glutamate receptor type, and each receptor subunit within the receptor type, possesses a distinct ontogenetic profile. For example, excitatory NMDA receptors are transiently overexpressed early in postnatal development, when they are needed for critical developmental processes [Simeone et al., 2004]. The early developmental stoichiometry of NMDA receptor subunits favors prolonged depolarizing responses. Early in development, NMDA receptor subunits NR2A, NR2B, and NR3A are elevated, favoring longer depolarizations, decreased senstivity to blockade by magnesium ions, and enhanced calcium influx. All of these physiological adaptations favor depolarization, seizures, and excitotoxicity. Similarly, the developmental profile of AMPA receptors favors subunit stoichiometries (fewer combinations with GluR2 subunits) with enhanced calcium influx. Metabotropic glutamate receptors may also be expressed developmentally to favor seizure generation [Avallone et al., 2006].
Electrical synapses appear to be more prevalent in the developing brain than in the mature brain. Electrical synapses have been reported in several brain regions, including neonatal neocortex and hippocampus. Fast-acting electrical transmission can facilitate rapid synchrony of the neuronal network and precipitate seizures and excitotoxicity [Velazquez and Carlen, 2000; Talhouka et al., 2007].
GABA receptors also exhibit a developmental profile, with different expression of GABAA receptor subunits at different stages of development [Brooks-Kayal, 2005; Huang, 2009]. GABA’s physiologic action also varies during development. Early in development, GABA exerts an excitatory action, rather than the inhibitory effect seen later. Emerging evidence suggests that GABA is depolarizing early in development because of the particular distribution of chloride ions (Figure 51-9) [Staley, 2006b]. In the developing brain (up to about the second week of life in rats, which corresponds roughly to a few months of age in humans), the intracellular chloride concentration is much higher than in mature neurons. This distribution is due to the presence of a membrane pump, called sodium, potassium, chloride co-transporter 1 (NKCC1), that actively influxes chloride ions, utilizing the energy stored in the transmembrane sodium gradient to import a potassium ion, along with one sodium ion and two chloride ions. NKCC1’s activity results in an intracellular chloride concentration about 3–4 times greater than that found in mature neurons. Thus, when GABAA receptors are activated by GABA release from interneurons, the postsynaptic chloride-permeable channels open and chloride effluxes out of the neuron down its concentration gradient. Chloride efflux results in a loss of intracellular negativity and a depolarizing current; the membrane potential deviates toward the chloride equilibrium potential, which is several tens of millivolts less negative than resting potential (i.e., depolarizing). The resulting depolarizing current can lead to action potential generation. This membrane depolarization is sufficient to activate NMDA receptors and allow entry of calcium ions into the cell.
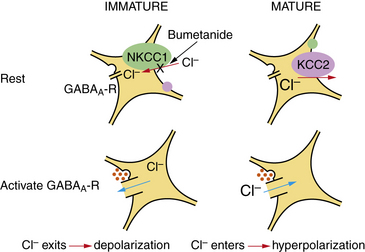
Even when GABA is depolarizing, another mechanism – shunting – can effectively produce inhibition by increasing membrane conductance. Shunting dissipates depolarizing synaptic currents without causing membrane potential change [Isaev et al., 2007]. That is, when chloride is passively distributed across the membrane, increasing chloride conductance will not generate transmembrane chloride currents. Therefore, the neonatal brain retains some inhibition, even in the face of depolarizing GABA action.
As development proceeds, the expression and activity of the NKCC1 transporter declines and another membrane pump is expressed, potassium chloride co-transporter 2 (KCC2) [Rivera et al., 1999]. KCC2 has the opposite action – it actively extrudes chloride to the outside of the neuron, leaving the intracellular compartment with a lower basal chloride concentration (see Figure 51-9). Binding of GABA to the GABAA receptor of a mature neuron that expresses KCC2 will cause channel opening and influx of chloride into the neuron, down its concentration gradient. Increased intracellular negativity will hyperpolarize the neuron, moving its membrane potential closer to the chloride reversal potential, which is more negative than resting potential. These actions will keep the neuron further from the threshold for action potential firing, comprising its inhibitory action.
The transition from GABAergic excitation to inhibition is an important physiological milestone. As glutamatergic synapses develop and begin to mediate their characteristic excitatory action, GABA switches to its mature inhibitory action (via change in expression of the chloride pumps described above; also see Figure 51-8). This time point also corresponds to other developmental milestones: namely, the appearance of GABAB inhibitory responses and disappearance of large oscillatory currents known as giant depolarizing potentials (GDPs) [Mohajerani and Cherubini, 2006]. GDPs are seen only at early ages, are dependent upon both NMDA and depolarizing GABA action, and function to facilitate the formation of synapses and neuronal circuits. The peak point of seizure susceptibility resides during this transition, when GABA is still excitatory, excitation via glutamate receptors has not yet matured, and GABAB inhibition is not yet complete. This maximum susceptibility occurs around postnatal (P) day 10–14 in rats, corresponding to approximately term in humans, an observation of major relevance for neonatal seizures and their treatment.
Phenobarbital, which blocks GABAA receptors, is typically used to suppress seizures in the neonatal period, but its efficacy against neonatal seizures is imperfect [Painter et al., 1999]. GABAA receptor ontogeny is also associated with varying sensitivity to benzodiazepines, an observation relevant to seizure treatment. Therefore, novel approaches to the management of neonatal seizures are needed. In this regard, bumetanide, a diuretic similar to furosemide that blocks NKCC1 (see Figure 51-9), has been shown to suppress epileptic activity in brain slices and animal models [Dzhala et al., 2005], an effect that is enhanced with concomitant phenobarbital use [Dzhala et al., 2008].
Structural Maturation of the Brain and Seizure Susceptibility
In the developing brain, synaptic connections are formed and pruned as a function of neuronal activity. Neuronal activity also determines which connections remain stable and which are lost. Such changes are referred to as plasticity, a general phenomenon that likely influences the brain’s capacity to learn and respond to the environment. The same properties that govern plasticity may also contribute to increased susceptibility for the epileptic network to synchronize in association with brain pathology or injury. For example, during the second week of life in the rat, the hippocampal CA3 region is characterized by an exuberance of excitatory connections between pyramidal cells that endow the region with heightened excitability and epileptiform activity [Swann, 2005]. These connections are later pruned, with stabilization of the excessive excitation seen during this developmental window.
Regulation of the Ionic Environment
The development of several ion transport mechanisms favors excitability early in life [Shigeri et al., 2004]. Expression of the chloride gradient depends on the development of the KCC2 transporter, which determines whether GABA mediates a depolarizing or hyperpolarizing effect. Expression of the sodium-potassium ion pump (Na+,K+-ATPase) also follows a developmental pattern [Haglund and Schwartzkroin, 1990]. Extracellular accumulation of potassium after neuronal activity (e.g., during a seizure) can further depolarize the neuronal membrane, exacerbating or prolonging ictal activity. The ability of glial cells to clear extracellular potassium improves with age.
Epileptogenesis in the Developing Brain
Numerous factors likely play a role in epileptogenesis, and these are best considered according to a temporal sequence [Bender and Baram, 2007; Pitkanen and Lukasiuk, 2009; Rakhade and Jensen, 2009]. In the first hours to days following a precipitating brain injury or severe seizure, immediate early genes are activated that lead to transcription of proteins that alter subsequent excitability. In addition, after a seizure, many receptors and other proteins involved in excitability are phosphorylated, including potassium channels and GABAA, AMPA, and NMDA receptors. Later, in the time frame of days to weeks, inflammation occurs, with elevations of cytokines and other inflammatory compounds, neurotrophic factors, ion channels, and neurotransmitter receptors. These changes presage chronic epileptogenic events that are expressed weeks to months later, including altered neurogenesis and gliosis. Therefore, the epileptogenic cascade progresses from changes at the gene level to changes at the structural level; hopefully, these multifactorial processes will provide biomarkers to identify which individuals are at most risk for the development of epilepsy and optimal ways to intervene in the process (Sankar and Rho, 2007).
Antiepileptic Drug Mechanisms
Procedures for Antiepileptic Drug Testing
Since the introduction of phenobarbital in 1912, dozens of AEDs have been approved for medical use by the U.S. Food and Drug Administration after extensive testing in a variety of animal seizure models and in human clinical trials. Until recently, the paradigms of drug discovery have been biased toward the development of drugs sharing properties exhibited by older agents, such as phenytoin and phenobarbital. Now, AED screening programs incorporate advances in molecular and cellular neurobiology, resulting in more rational screening programs, with new molecular targets being discovered beyond neuronal membrane-bound ion channels and enzymes involved in neurotransmitter metabolism and reuptake [Rogawski, 2006].
The two most commonly used models for the routine screening and identification of new AEDs are the maximal electroshock (MES) and subcutaneous pentylenetetrazol (PTZ) tests conducted with mice or rats [White et al., 2007]. MES tests the ability of a compound to eliminate the tonic extensor component of an electrically evoked seizure, whereas PTZ (a GABAA receptor antagonist) tests the ability of a drug to inhibit a generalized clonic seizure. MES seizures can be blocked by agents such as phenytoin and carbamazepine, which are effective in treating seizures with partial onset. In contrast, drugs effective in treating generalized absence seizures (e.g., ethosuximide) effectively inhibit PTZ-induced seizures. Valproic acid, a broad-spectrum agent, is effective in MES and PTZ tests, and is clinically effective against most seizure types. Alternative screeing tests are currently being used to assess AED effects on limbic seizures (e.g., 6-Hz test, kindling) and other seizure types [Barton et al., 2001; Bertram, 2007].
Antiepileptic Drug Mechanisms of Action
Most AEDs exert their principal effects on the following molecular targets: GABAA receptors, glutamate receptors (both NMDA and non-NMDA), voltage-dependent sodium channels, and voltage-dependent calcium channels [Rogawski and Loscher, 2004]. Each AED is likely to have multiple mechanisms of action because there is no single action of an AED that can completely account for all of its clinical effects. Several AEDs block voltage-gated sodium channels, but the interactions with other molecular targets help to define a drug’s clinical spectrum. Although this discussion focuses primarily on studies of hippocampal neurons and circuits, insights into antiepileptic mechanisms have emerged from studies of many neural tissues (e.g., spinal cord, peripheral nerve, neocortex) and experimental preparations [Rho and Sankar, 1999]. Putative molecular targets of clinically approved antiepileptic drugs are shown in Figure 51-10 and Figure 51-11, which depict key elements of central nervous system excitatory and inhibitory synapses, respectively.
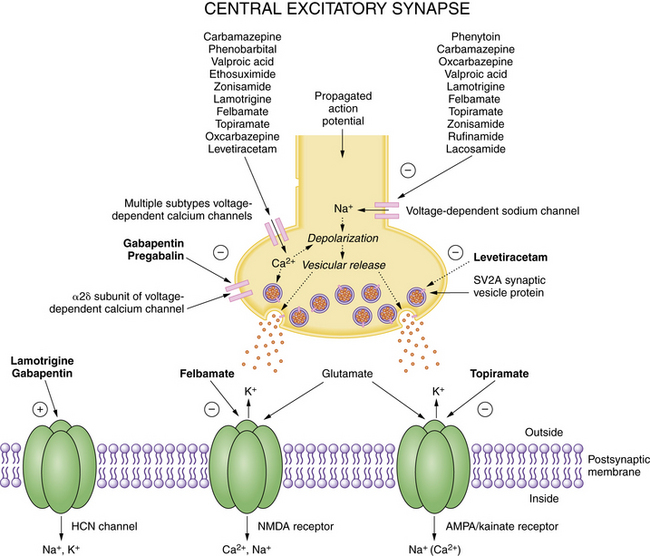
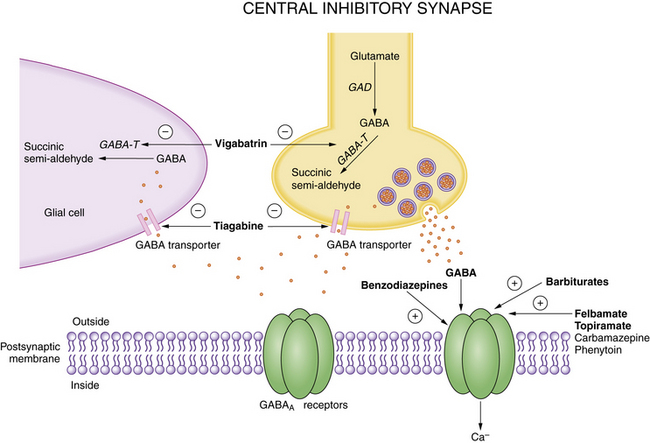
GABA, gamma-aminobutyric acid; GABA-T, GABA transaminase; GAD, glutamic acid decarboxylase.
(Adapted from Rho JM, Sankar R. The pharmacological basis of antiepileptic drug action. Epilepsia 1999;40:1471–1483. Reprinted with the permission of Blackwell Publishing Ltd.)
Although structurally unrelated to classic agents such as phenytoin and carbamazepine, lamotrigine blocks sodium channels in a voltage-, frequency-, and use-dependent manner. However, lamotrigine has a much broader spectrum of activity than phenytoin or carbamazepine, demonstrating efficacy against several forms of generalized seizures in addition to partial seizures. The mechanistic basis for this difference may be related to the drug’s enhancement of dendritic hyperpolarization-activated cation channels [Rogawski and Loscher, 2004].
Contrary to the prediction that gabapentin, a GABA analog, may act on GABAergic systems, gabapentin does not interact with GABAA or GABAB receptors, and does not affect GABA reuptake, synthesis, or metabolism. However, the existence of a specific binding site for gabapentin has been reported. Unlike other pharmacologic agents (e.g., flunarizine, dihydropyridines) that interact with the α1 subunit of voltage-dependent calcium channels, gabapentin binds to the α2δ subunit of the L-type calcium channel [Rogawski and Bazil, 2008]. Gabapentin (much like lamotrigine) enhances h-currents in hippocampal neurons, but the clinical significance of this remains unclear. Recent data confirms an interaction of gabapentin with GABAB receptor coupling to G-proteins and modulation of an inwardly rectifying potassium channel and N-type voltage-gated calcium channels. Whatever the relevant mechanisms of antiepileptic action, elevated brain GABA levels are found in patients taking gabapentin.
Felbamate is the first pharmacologic agent to potentiate GABAA receptor-mediated responses and inhibit NMDA receptor-mediated responses within the same drug concentration range [Rho and Sankar, 1999]. These dual actions may result in synergism with respect to seizure protection. Similarly, topiramate inhibits the AMPA or kainate subtype of glutamate receptor (specifically the GluR5 kainate receptor) and augments GABAA receptor-mediated chloride currents [Rogawski and Loscher, 2004].
Zonisamide is a broad-spectrum agent that has a unique mechanistic profile [Rogawski and Loscher, 2004]. In cultured spinal cord neurons, zonisamide reduces sustained repetitive firing of action potentials, consistent with actions on voltage-gated sodium channels; in cultured neurons from rat cerebral cortex, zonisamide blocks low-threshold T-type calcium currents, which predicts efficacy against generalized spike-wave epilepsies.
Levetiracetam has challenged our conceptual understanding of relevant mechanisms of action. It failed the MES and PTZ tests in animals, but had a profound effect in retarding amygdala kindling in rats, and has proven clinical efficacy against partial and perhaps some generalized seizures. The strongest clue to identifying levetiracetam’s principal molecular target is the demonstration of a specific high-affinity neuronal binding site [Kaminski et al., 2008]. Levetiracetam binds to a specific synaptic vesicle protein, SV2A, which is involved in neurotransmitter release. The exact mechanism by which levetiracetam is dependent on SV2A for its antiepileptic action remains unknown [van Vliet et al., 2009].
The most recently approved AEDs in the United States are pregabalin, lacosamide, and rufinamide. Pregabalin, like gabapentin, seems to work by binding to the α2δ subunit of voltage-activated calcium channels, which reduces synaptic release of glutamate [Rogawski and Bazil, 2008]. Lacosamide reportedly possesses a novel mechanism of action – enhancing slow inactivation of sodium channels (older AEDs that block sodium channels do so by enhancing fast inactivation, which is mechanistically distinct) [Errington et al., 2008]. Whether this molecular mechanism will lead to enhanced seizure suppression is currently unkown, but it does represent an attempt to explore novel therapeutic approaches using physiologically relevant targets. Further preliminary evidence links lacosamide to modulation of collapsin-response mediator protein-2, which is involved in neuronal growth and plasticity; whether this action affects seizure suppression is unknown [Beydoun et al., 2009]. Rufinamide’s mechanism of action is currently unknown, but preliminary results point to an effect on fast sodium channel inactivation [Arroyo, 2007].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735-740.
Arroyo S. Rufinamide. Neurotherapeutics. 2007;4:155-162.
Avallone J., Gashi E., Magrys B., et al. Distinct regulation of metabotropic glutamate receptor (mGluR1 alpha) in the developing limbic system following multiple early-life seizures. Exp Neurol. 2006;202:100-111.
Avishai-Eliner S., Brunson K.L., Sandman C.A., et al. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518-524.
Barton M.E., Klein B.D., Wolf H.H., et al. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217-227.
Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728-739.
Ben-Ari Y., Holmes G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055-1063.
Bender R.A., Baram T.Z. Epileptogenesis in the developing brain: what can we learn from animal models? Epilepsia. 2007;48(Suppl 5):2-6.
Bender R.A., Baram T.Z. Hyperpolarization activated cyclic-nucleotide gated HCN channels in developing neuronal networks. Prog Neurobiol. 2008;86:129-140.
Bertram E. The relevance of kindling for human epilepsy. Epilepsia. 2007;48(Suppl 2):65-74.
Bettler B., Tiao T.Y. Molecular diversity, trafficking and subcellular localization of GABAB receptors. Pharmacol Ther. 2006;1103:533-543.
Beydoun A., D’Souza J., Hebert D., et al. Lacosamide: pharmacology, mechanisms of action and pooled efficacy and safety data in partial-onset seizures. Expert Rev Neurother. 2009;91:33-42.
Brooks-Kayal A.R. Rearranging receptors. Epilepsia. 2005;46(Suppl 7):29-38.
Buzsaki G., Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926-1929.
Catterall W.A., Golden A.L., Waxman S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397-409.
Catterall W.A., Striessnig J., Snutch T.P. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev. 2003;55:579.
Commission on Classification and Terminology of the International league Against Epilepsy. Proposal for revised classification of epilepsies and epilepsy syndromes. Epilepsia. 1989;30:389-399.
Cossart R., Bernard C., Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108-115.
Dudek F.E., Yasumura T., Rash J.E. ‘Non-synaptic’ mechanisms in seizures and epileptogenesis. Cell Biol Int. 1998;22:793-805.
Dyhrfjeld-Johnsen J., Morgan R.J., Soltesz I. Double trouble? Potential for hyperexcitability following both channelopathic up- and downregulation of Ih in epilepsy. Front Neurosci. 2009:31.
Dzhala V.I., Brumback A.C., Staley K.J. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222-235.
Dzhala V.I., Talos D.M., Sdrulla D.A., et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205-1213.
Engel J.Jr. Report of the ILAE classification core group. Epilepsia. 2006;47:1558-1568.
Engel J.Jr, Bragin A., Staba R., et al. High-frequency oscillations: what is normal and what is not? Epilepsia. 2009;50:598-604.
Errington A.C., Stohr T., Heers C., et al. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol. 2008;73:157-169.
Fritschy J.-M. Epilepsy, E/I balance and GABA-A receptor plasticity. Front Mol Neurosci. 2008;1:5.
Gorji A., Speckmann E.J. Epileptiform EEG spikes and their functional significance. Clin EEG Neurosci. 2009;40:230-233.
Gutman G.A., Chandy K.G., Grissmer S., et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473-508.
Haglund M., Schwartzkroin P.A. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63:225-239.
Harkin L.A., McMahon J.M., et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(Part 3):843-852.
Huang Z.J. Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J Physiol. 2009;587(Pt 9):1881-1888.
Huc S., Monteil A., Bidaud I., et al. Regulation of T-type calcium channels: signalling pathways and functional implications. Biochim Biophys Acta. 2009;1793:947-952.
Hughes J.R. Absence seizures: a review of recent reports with new concepts. Epilepsy Behav. 2009;15:404-412.
Hughes J.R. Gamma, fast, and ultrafast waves of the brain: their relationships with epilepsy and behavior. Epilepsy Behav. 2008;13:25-31.
Huguenard J.R., McCormick D.A. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350-356.
Isaev D., Isaeva E., Khazipov R., et al. Shunting and hyperpolarizing GABAergic inhibition in the high-potassium model of ictogenesis in the developing rat hippocampus. Hippocampus. 2007;17:210-219.
Kalia L.V., Kalia S.K., Salter M.W. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742-755.
Kaminski R.M., Matagne A., Leclercq K., et al. SV2A protein is a broad-spectrum anticonvulsant target: functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology. 2008;54:715-720.
Lado F.A., Moshé S.L. How do seizures stop? Epilepsia. 2008;49:1651-1664.
Lawrence J.J., McBain C.J. Interneuron diversity series: containing the detonation–feedforward inhibition in the CA3 hippocampus. Trends Neurosci. 2003;26:631-640.
LeDuigou C., Huberfeld G., Miles R.. Recurrent synaptic actions and the genesis of epileptiform activity. Schwartzkroin P.A., editor. Encyclopedia of Basic Epilepsy Research. vol 2. Oxford: Elsevier; 2009:836-841.
Ludwig A., Budde T., Stieber J., et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216-224.
Macdonald R.L., Kang J.Q. Molecular pathology of genetic epilepsies associated with GABAA receptor subunit mutations. Epilepsy Curr. 2009;9:18-23.
McBain C.J., Traynelis S.F., Dingledine R. High potassium-induced synchronous bursts and electrographic seizures. In: Schwartzkroin P.A., editor. Epilepsy: models, mechanisms, and concepts. Cambridge, U.K.: Cambridge University Press; 1993:437-461.
McCormick D.A., Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815-846.
McCormick D.A., Prince D.A. Post-natal development of electrophysiological properties of rat cerebral cortical pyramidal neurones. J Physiol. 1987;393:743-762.
Meisler M.H., Kearney J.A. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010-2017.
Meldrum B.S., Akbar M.T., Chapman A.G. Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res. 1999;36:189.
Miceli F., Soldovieri M.V., Martire M., et al. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol. 2008;8:65-74.
Mohajerani M.H., Cherubini E. Role of giant depolarizing potentials in shaping synaptic currents in the developing hippocampus. Crit Rev Neurobiol. 2006;18:12-23.
Nadler J.V. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res. 2003;28:1649-1658.
Painter M.J., Scher M.S., Stein A.D., et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485-489.
Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117-161.
Pitkanen A., Lukasiuk K. Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 2009;14:16-25.
Poolos N.P. The yin and yang of the h-channel and its role in epilepsy. Epilepsy Curr. 2004;4:3-6.
Ragsdale D.S. How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res Rev. 2008;58:149-159.
Rakhade S.N., Jensen F.E. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380-391.
Reid C.A., Berkovic S.F., Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41-57.
Rho J.M., Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40:1471-1483.
Rivera C., Voipio J., Payne J.A., et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251-255.
Rogawski M.A. Molecular targets versus models for new antiepileptic drug discovery. Epilepsy Res. 2006;681:22-28.
Rogawski M.A., Bazil C.W. New molecular targets for antiepileptic drugs: alpha2delta, SV2A, and Kv7/KCNQ/M potassium channels. Curr Neurol Neurosci Rep. 2008;8:345-352.
Rogawski M.A., Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553-564.
Sanchez R.M., Jensen F.E. Maturational aspects of epilepsy mechanisms and consequences for the immature brain. Epilepsia. 2001;42:577-585.
Sankar S., Rho J.M. Do seizures affect the developing brain? Lessons from the laboratory. J Child Neurol. 2007;22:21S-29S.
Santos S.D., Carvalho A.L., Caldeira M.V., et al. Regulation of AMPA receptors and synaptic plasticity. Neuroscience. 2009;158:105-125.
Schwartzkroin P.A., Mueller A.L.. Electrophysiology of hippocampal neurons. Jones E.G., Peters A., editors. Cerebral Cortex: Further Aspects of Cortical Function, Including Hippocampus. vol 6. New York: Plenum Press; 1987:295-343.
Shigeri Y., Seal R.P., Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Rev. 2004;45:250-265.
Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231-263.
Silverstein F.S., Jensen F.E. Neonatal seizures. Ann Neurol. 2007;62:112-120.
Simeone T.A., Donevan S.D., Rho J.M. Molecular biology and ontogeny of gamma-aminobutyric acid (GABA) receptors in the mammalian central nervous system. J Child Neurol. 2003;18:39-48.
Simeone T.A., Sanchez R.M., Rho J.M. Molecular biology and ontogeny of glutamate receptors in the mammalian central nervous system. J Child Neurol. 2004;19:343-360.
Singh N.A., Westenskow P., Charlier C., et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726-2737.
Snead O.C.3rd. Presynaptic GABAB– and gamma-hydroxybutyric acid-mediated mechanisms in generalized absence seizures. Neuropharmacology. 1996;35:359-367.
Spitzer N.C. Electrical activity in early neuronal development. Nature. 2006;444:707-712.
Stafstrom C.E. An introduction to seizures and epilepsy: cellular mechanisms underlying classification and treatment. In: Stafstrom C.E., Rho J.M., editors. Epilepsy and the Ketogenic Diet. Totowa. New Jersey: Humana Press; 2004:3-29.
Stafstrom C.E. Neurobiological mechanisms of developmental epilepsy: translating experimental findings into clinical application. Semin Pediatr Neurol. 2007;14:164-172.
Stafstrom C.E. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;71:15-22.
Stafstrom C.E. Severe epilepsy syndromes of early childhood: the link between genetics and pathophysiology with a focus on SCN1A mutations. J Child Neurol. 2009;248(Suppl):15S-23S.
Staley K.J. Role of the depolarizing GABA response in epilepsy. Adv Exp Med Biol. 2006;548:104-109.
Staley K.J. Wrong-way chloride transport: is it a treatable cause of some intractable seizures? Epilepsy Curr. 2006;6:124-127.
Steriade M. Neocortical cell classes are flexible entities. Nat Rev Neurosci. 2004;5:121-134.
Swann J., Pierson M., Smith K.L., et al. Developmental neuroplasticity: roles in early life seizures and chronic epilepsy. Delgado-Escueta A., Wilson W., Olsen R., Porter R., editors. Jasper’s Basic Mechanisms of the Epilepsies. vol 79. Philadelphia: Lippincott Williams & Wilkins; 1999:203-216.
Swann J.W. The impact of seizures on developing hippocampal networks. Prog Brain Res. 2005;147:347-354.
Talhouka R.S., Zeinieha M.P., Mikati M.A., et al. Gap junctional intercellular communication in hypoxia–ischemia-induced neuronal injury. Prog Neurobiol. 2007;84:57-76.
Traub R.D., Bibbig A., Le Beau F.E., et al. Cellular mechanisms of neuronal population oscillations in the hippocampus in vitro. Annu Rev Neurosci. 2004;27:247-278.
Traub R.D., Michelson-Law H., Bibbig A.E., et al. Gap junctions, fast oscillations and the initiation of seizures. Adv Exp Med Biol. 2004;548:110-122.
Traynelis S., Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259-276.
Ure J., Baudry M., Perassolo M. Metabotropic glutamate receptors and epilepsy. J Neurol Sci. 2006;247:1-9.
van Vliet E.A., Aronica E., Redeker S., et al. Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy. Epilepsia. 2009;503:422-433.
Velazquez J.P., Carlen P. Gap junctions, synchrony and seizures. Trends Neurosci. 2000;23:68-74.
Velisek L., Velisková J., Moshé S.L. Developmental seizure models. Ital J Neurol Sci. 1995;16:127-133.
Velísková J., Claudio O.I., Galanopoulou A.S., et al. Seizures in the developing brain. Epilepsia. 2004;45(Suppl 8):6-12.
Vezzani A., Balosso S., Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797-803.
Vincent P., Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158:309-323.
Wafford K.A., Macaulay J.A., Fradley R., et al. Differentiating the role of gamma-aminobutyric acid type A (GABAA) receptor subtypes. Biochem Soc Trans. 2004;32(Pt. 3):553-556.
Wetherington J., Serrano G., Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168-178.
White H.S., Smith M.D., Wilcox K.S. Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol. 2007;81:85-110.
Wickenden A.D. Potassium channels as anti-epileptic drug targets. Neuropharmacology. 2002;43:1055-1060.
Wong M. Advances in the pathophysiology of developmental epilepsies. Semin Pediatr Neurol. 2005;12:72-87.