Chapter 21 Neurology and the Neuromuscular System
Acetaminophen


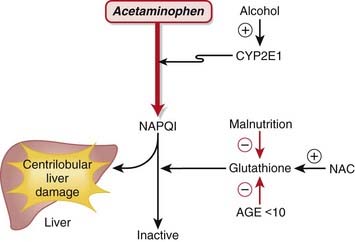
MOA (Mechanism of Action) (Figure 21-1)
 Acetaminophen is neither a narcotic nor a nonsteroidal antiinflammatory drug (NSAID). It is in a drug class of its own called aniline analgesics.
Acetaminophen is neither a narcotic nor a nonsteroidal antiinflammatory drug (NSAID). It is in a drug class of its own called aniline analgesics.

 In contrast to NSAIDs and aspirin, acetaminophen is not an antiplatelet agent, nor does it possess antiinflammatory properties; therefore there are differences in the mechanism of action compared with aspirin or NSAIDs:
In contrast to NSAIDs and aspirin, acetaminophen is not an antiplatelet agent, nor does it possess antiinflammatory properties; therefore there are differences in the mechanism of action compared with aspirin or NSAIDs:
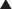





Important Notes
 Therapeutic doses of acetaminophen have no effect on the cardiovascular and respiratory systems or platelet function and do not produce gastric irritation, erosion, or bleeding.
Therapeutic doses of acetaminophen have no effect on the cardiovascular and respiratory systems or platelet function and do not produce gastric irritation, erosion, or bleeding.Overdose
 Acetaminophen exposure is the most commonly reported drug exposure reported to U.S. poison control centers.
Acetaminophen exposure is the most commonly reported drug exposure reported to U.S. poison control centers.


 The mechanism of injury is as follows:
The mechanism of injury is as follows:





Evidence
Analgesia
Acetaminophen versus Placebo for Treatment of Osteoarthritis
 A Cochrane review in 2005 (seven studies) demonstrated that acetaminophen was superior to placebo in five of the seven randomized controlled trials (RCTs). A pooled analysis demonstrated a statistically significant but minimal difference that is of questionable clinical significance. The relative percent improvement in pain score from baseline was 5%, with an absolute change of 4 points on a 0-to-100 scale.
A Cochrane review in 2005 (seven studies) demonstrated that acetaminophen was superior to placebo in five of the seven randomized controlled trials (RCTs). A pooled analysis demonstrated a statistically significant but minimal difference that is of questionable clinical significance. The relative percent improvement in pain score from baseline was 5%, with an absolute change of 4 points on a 0-to-100 scale.Acetaminophen versus Nonsteroidal Antiinflammatory Drugs for Treatment of Osteoarthritis
 The same Cochrane review in 2005 (10 studies) demonstrated that acetaminophen was less effective overall than NSAIDs in terms of pain reduction, global assessments, and improvements in functional status. Patients taking traditional NSAIDS were more likely to experience an adverse GI event (relative risk [RR] 1.47). However, the median trial duration was only 6 weeks, which is too short to adequately assess adverse outcomes.
The same Cochrane review in 2005 (10 studies) demonstrated that acetaminophen was less effective overall than NSAIDs in terms of pain reduction, global assessments, and improvements in functional status. Patients taking traditional NSAIDS were more likely to experience an adverse GI event (relative risk [RR] 1.47). However, the median trial duration was only 6 weeks, which is too short to adequately assess adverse outcomes.Acetaminophen plus Codeine versus Placebo
 A Cochrane review in 2008 (26 studies, N = 2295 patients) of postoperative patients demonstrated significant differences for obtaining at least 50% pain relief over 4 to 6 hours, with a number needed to treat (NNT) of 2.2 for high doses (800 to 1000 mg acetaminophen plus 60 mg codeine) and smaller effect sizes for medium and smaller doses (as low as 325 mg acetaminophen with 30 mg codeine).
A Cochrane review in 2008 (26 studies, N = 2295 patients) of postoperative patients demonstrated significant differences for obtaining at least 50% pain relief over 4 to 6 hours, with a number needed to treat (NNT) of 2.2 for high doses (800 to 1000 mg acetaminophen plus 60 mg codeine) and smaller effect sizes for medium and smaller doses (as low as 325 mg acetaminophen with 30 mg codeine).Acetaminophen Plus Codeine versus Acetaminophen Alone
 A Cochrane review in 2008 (14 studies, N = 926 patients) of postoperative patients demonstrated that addition of codeine increased the proportion of participants achieving at least 50% pain relief over 4 to 6 hours by 10% to 15% and reduced the proportion of patients needing rescue medication by about 15%.
A Cochrane review in 2008 (14 studies, N = 926 patients) of postoperative patients demonstrated that addition of codeine increased the proportion of participants achieving at least 50% pain relief over 4 to 6 hours by 10% to 15% and reduced the proportion of patients needing rescue medication by about 15%.



Opioids






MOA (Mechanism of Action)
 The pain pathways in the body are very complex and only briefly summarized here. In short, opioids reduce the signaling and processing of pain pathways through a variety of receptor types, receptor locations, and complex interactions.
The pain pathways in the body are very complex and only briefly summarized here. In short, opioids reduce the signaling and processing of pain pathways through a variety of receptor types, receptor locations, and complex interactions.
 Exogenous opioids also bind multiple opioid receptors (Table 21-1). Opioid receptors are present in both the brain and the spinal cord, specifically:
Exogenous opioids also bind multiple opioid receptors (Table 21-1). Opioid receptors are present in both the brain and the spinal cord, specifically:




| Receptor | Action |
|---|---|
| Mu (µ) |
Pharmacokinetics (Table 21-2)
 For equivalency, note that:
For equivalency, note that:

 Important metabolites:
Important metabolites:
 Most opioids are metabolized by CYP3A4 and renally eliminated (except where the following information states otherwise).
Most opioids are metabolized by CYP3A4 and renally eliminated (except where the following information states otherwise). Morphine → morphine-3-glucuronide (90%), morphine-6-glucuronide (10%).
Morphine → morphine-3-glucuronide (90%), morphine-6-glucuronide (10%).
• The glucuronides (being water soluble) are eliminated by the kidneys; renal disease can prolong and increase the effects of morphine.
• The glucuronides are water soluble; thus they do not readily cross the blood-brain barrier, but with high concentrations of drug, brain levels will increase.







Side Effects









Important Notes

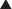












Evidence
Opioids versus Nonsteroidal Antiinflammatory Drugs for Treatment of Renal Colic
 A systematic review in 2004 (20 trials, 1613 participants) found that both NSAIDs and opioids led to clinically important reductions in patient-reported pain scores. Pooled analysis of six trials showed a greater reduction in pain scores for patients treated with NSAIDs than with opioids. Patients treated with NSAIDs were significantly less likely to require rescue analgesia (RR 0.75). Most trials showed a higher incidence of adverse events in patients treated with opioids. Compared with patients treated with opioids, those treated with NSAIDs had significantly less vomiting (0.35).
A systematic review in 2004 (20 trials, 1613 participants) found that both NSAIDs and opioids led to clinically important reductions in patient-reported pain scores. Pooled analysis of six trials showed a greater reduction in pain scores for patients treated with NSAIDs than with opioids. Patients treated with NSAIDs were significantly less likely to require rescue analgesia (RR 0.75). Most trials showed a higher incidence of adverse events in patients treated with opioids. Compared with patients treated with opioids, those treated with NSAIDs had significantly less vomiting (0.35).Opioids for Treatment of Chronic Back Pain
 A systematic review in 2007 examined multiple questions pertaining to opioid treatment of chronic low back pain. 11 studies showed that there was significant variation in how opioids are prescribed for chronic low back pain. A meta-analysis of the four studies assessing the efficacy of opioids compared with placebo or a nonopioid control did not show reduced pain with opioids. A meta-analysis of the five studies directly comparing the efficacy of different opioids demonstrated a nonsignificant reduction in pain from baseline.
A systematic review in 2007 examined multiple questions pertaining to opioid treatment of chronic low back pain. 11 studies showed that there was significant variation in how opioids are prescribed for chronic low back pain. A meta-analysis of the four studies assessing the efficacy of opioids compared with placebo or a nonopioid control did not show reduced pain with opioids. A meta-analysis of the five studies directly comparing the efficacy of different opioids demonstrated a nonsignificant reduction in pain from baseline.
 With respect to risk of addiction, the prevalence of lifetime substance use disorders ranged from 36% to 56%; the prevalence of current substance use disorders was as high as 43%; and aberrant medication-taking behaviors (“drug seeking”) ranged from 5% to 24%. The authors found that the study was limited by retrieval and publication biases and poor study quality. No trial evaluating the efficacy of opioids was longer than 16 weeks.
With respect to risk of addiction, the prevalence of lifetime substance use disorders ranged from 36% to 56%; the prevalence of current substance use disorders was as high as 43%; and aberrant medication-taking behaviors (“drug seeking”) ranged from 5% to 24%. The authors found that the study was limited by retrieval and publication biases and poor study quality. No trial evaluating the efficacy of opioids was longer than 16 weeks.

FYI







α2 Agonists


MOA (Mechanism of Action)
 Glaucoma is characterized by increased intraocular pressure (IOP). Strategies to reduce intraocular pressure include reducing the production and secretion of aqueous humor and facilitating its drainage.
Glaucoma is characterized by increased intraocular pressure (IOP). Strategies to reduce intraocular pressure include reducing the production and secretion of aqueous humor and facilitating its drainage. The exact mechanism by which α2 agonists reduce IOP has not been established, but is likely multifactorial, employing several strategies:
The exact mechanism by which α2 agonists reduce IOP has not been established, but is likely multifactorial, employing several strategies:

 The antihypertensive effect of α2 agonists is a result of inhibition of presynaptic release of vasoconstrictors such as norepinephrine. Recall that the α2 receptor is an autoreceptor (see Chapter 3). An autoreceptor is a receptor that when stimulated by an agonist, reduces release of transmitter into the synaptic cleft.
The antihypertensive effect of α2 agonists is a result of inhibition of presynaptic release of vasoconstrictors such as norepinephrine. Recall that the α2 receptor is an autoreceptor (see Chapter 3). An autoreceptor is a receptor that when stimulated by an agonist, reduces release of transmitter into the synaptic cleft.
Pharmacokinetics






Contraindications









Important Notes




Evidence
In Primary Open Angle Glaucoma and Ocular Hypertension
 A 2007 Cochrane review of all medical interventions for glaucoma and ocular hypertension found three trials comparing brimonidine to timolol. There were no differences in visual field progression (glaucoma) or visual field defects (ocular hypertension) within 1 year. Timolol was better tolerated than brimonidine, as measured by the incidence of dropouts from drug-related adverse events (OR 0.21). There were no trials comparing brimonidine with placebo.
A 2007 Cochrane review of all medical interventions for glaucoma and ocular hypertension found three trials comparing brimonidine to timolol. There were no differences in visual field progression (glaucoma) or visual field defects (ocular hypertension) within 1 year. Timolol was better tolerated than brimonidine, as measured by the incidence of dropouts from drug-related adverse events (OR 0.21). There were no trials comparing brimonidine with placebo.
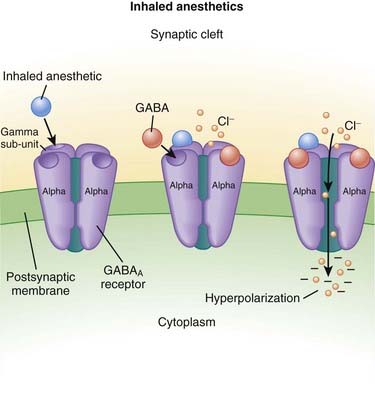
Inhaled Anesthetics


MOA (Mechanism of Action) (Figure 21-2)


Pharmacokinetics
 As their name suggests, these drugs are administered only via inhalation. They are supplied in liquid form and then vaporized using very precise vaporizers that are part of the anesthetic machine, the anesthetic oxygen and air mixture is combined with calculated doses of the inhaled anesthetic.
As their name suggests, these drugs are administered only via inhalation. They are supplied in liquid form and then vaporized using very precise vaporizers that are part of the anesthetic machine, the anesthetic oxygen and air mixture is combined with calculated doses of the inhaled anesthetic.

 Inhaled anesthetics move through the body by dissolving in the blood and distributing into tissues; the important tissue interfaces include the following:
Inhaled anesthetics move through the body by dissolving in the blood and distributing into tissues; the important tissue interfaces include the following:
 Blood ∂ fat (and other vessel-poor tissues)
Blood ∂ fat (and other vessel-poor tissues)
• When inhaled anesthetics are administered, the drug must enter the lungs, then the blood, then the brain.
• For inhaled anesthetics to be eliminated, drug must exit the brain and other tissues, be carried to the lungs, and then exhaled.
• Vessel-poor tissues are slow to take up and release drug. Therefore vessel-poor tissues can act as a sink and slowly absorb drug at the early parts of an anesthetic procedure (lowering the drug levels) but then at the end of a long anesthetic procedure can release drug, prolonging elimination of the drug.
 The solubility of an inhaled anesthetic affects the speed at which it is taken up by the body and exerts its action (speed of onset). The key point is that the partial pressure is the measure of the drug’s active form. If a drug has a high solubility, then a lot of drug needs to be absorbed into blood and tissues before the partial pressure starts to rise; this impedes the onset of action of the drug. If the solubility is low, then the partial pressure will rise quickly. Conversely, eliminating the drug follows the same rules, and high solubility correlates with slower elimination because more drug had to be dissolved into the body initially to achieve the desired partial pressure.
The solubility of an inhaled anesthetic affects the speed at which it is taken up by the body and exerts its action (speed of onset). The key point is that the partial pressure is the measure of the drug’s active form. If a drug has a high solubility, then a lot of drug needs to be absorbed into blood and tissues before the partial pressure starts to rise; this impedes the onset of action of the drug. If the solubility is low, then the partial pressure will rise quickly. Conversely, eliminating the drug follows the same rules, and high solubility correlates with slower elimination because more drug had to be dissolved into the body initially to achieve the desired partial pressure.
• Solubility is described for the blood and is called the blood/gas coefficient. The smaller the number, the less soluble the drug is in blood and therefore the faster it can change its partial pressure (because only a small amount of drug actually needs to be dissolved into the blood). The agents are ranked from fastest to slowest (lowest to highest solubility coefficient) as follows:








Important Notes
 The potency of inhaled anesthetics is measured by the minimum anesthetic concentration (MAC), which is strictly defined as the dose of inhaled anesthetic required to prevent movement in 50% of the population in response to a surgical stimulus when no other drugs are administered. For example, the MAC of desflurane is 6%. Note that this definition is not the same as the dose required to keep someone asleep. Drugs such as opioids decrease the MAC requirements of a drug and are called MAC-sparing agents.
The potency of inhaled anesthetics is measured by the minimum anesthetic concentration (MAC), which is strictly defined as the dose of inhaled anesthetic required to prevent movement in 50% of the population in response to a surgical stimulus when no other drugs are administered. For example, the MAC of desflurane is 6%. Note that this definition is not the same as the dose required to keep someone asleep. Drugs such as opioids decrease the MAC requirements of a drug and are called MAC-sparing agents.



Advanced
 Nitrous oxide has a MAC value of 104%. Therefore, it is not potent enough to be a solo anesthetic agent, because delivering a mixture of 100% nitrous oxide would mean that no oxygen could be delivered and the patient would die of asphyxiation. The greatest concentration that is administered is about 60% to 70%—a MAC value of 0.6 or 0.7. For this reason, nitrous is used only as an adjuvant drug for general anesthesia and is added to one of the other volatile anesthetics.
Nitrous oxide has a MAC value of 104%. Therefore, it is not potent enough to be a solo anesthetic agent, because delivering a mixture of 100% nitrous oxide would mean that no oxygen could be delivered and the patient would die of asphyxiation. The greatest concentration that is administered is about 60% to 70%—a MAC value of 0.6 or 0.7. For this reason, nitrous is used only as an adjuvant drug for general anesthesia and is added to one of the other volatile anesthetics.




Intravenous Anesthetics



MOA (Mechanism of Action)
γ-Aminobutyric Acid (GABA) (Figure 21-3)
 GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
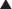

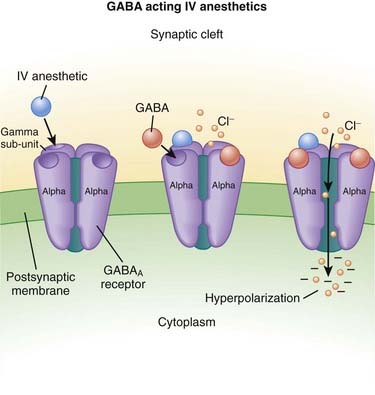



Pharmacokinetics






Side Effects
 Respiratory depression: The respiratory center is depressed with GABA agonists. It is typical for a patient who is administered a dose large enough to induce unconsciousness to develop apnea (to completely stop breathing). For this reason, administration of intravenous anesthetics (regardless of dose) must always be performed in the presence of healthcare workers who are skilled in respiratory resuscitation and who have respiratory resuscitation equipment immediately available.
Respiratory depression: The respiratory center is depressed with GABA agonists. It is typical for a patient who is administered a dose large enough to induce unconsciousness to develop apnea (to completely stop breathing). For this reason, administration of intravenous anesthetics (regardless of dose) must always be performed in the presence of healthcare workers who are skilled in respiratory resuscitation and who have respiratory resuscitation equipment immediately available.



Important Notes

 Dosage: Some general concepts should be applied when choosing the dose of intravenous anesthetic. Administering a larger-than-required dose virtually guarantees significant hypotension:
Dosage: Some general concepts should be applied when choosing the dose of intravenous anesthetic. Administering a larger-than-required dose virtually guarantees significant hypotension:
 Elderly patients require lower doses compared with nonelderly adults, and children require higher doses (on a milligram-per-kilogram scale) compared with adults.
Elderly patients require lower doses compared with nonelderly adults, and children require higher doses (on a milligram-per-kilogram scale) compared with adults.

 Ketamine:
Ketamine:
 Propofol:
Propofol:









Local Anesthetics




MOA (Mechanism of Action)
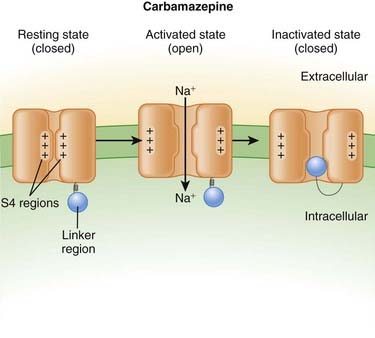
 Neuronal transmission requires that an action potential be propagated from one end of a neuron to the other. Voltage-gated sodium channels open up as the wave of depolarization travels from one end of the neuron to the other. The opening of these ion channels permits sodium to enter the cell, causing depolarization, and is the primary method by which the wave of depolarization occurs.
Neuronal transmission requires that an action potential be propagated from one end of a neuron to the other. Voltage-gated sodium channels open up as the wave of depolarization travels from one end of the neuron to the other. The opening of these ion channels permits sodium to enter the cell, causing depolarization, and is the primary method by which the wave of depolarization occurs. Local anesthetics bind these voltage-gated sodium channels. The ion channels can exist in three states: resting, activated (open), and inactivated. The local anesthetic binds them in the inactivated state and prevents them from transitioning to the open state; thus the ion channel remains closed and unresponsive to incoming depolarizing currents (Figure 21-4).
Local anesthetics bind these voltage-gated sodium channels. The ion channels can exist in three states: resting, activated (open), and inactivated. The local anesthetic binds them in the inactivated state and prevents them from transitioning to the open state; thus the ion channel remains closed and unresponsive to incoming depolarizing currents (Figure 21-4).




 Voltage-gated sodium channels are the primary ion channels in phase 0 of the cardiac action potential. Therefore, local anesthetics are also classified as antiarrhythmics, specifically type 1b. This mechanism of action is discussed in more detail in the discussion of Na+ channel blockers in Chapter 11 and is also responsible for cardiac toxicity of local anesthetics.
Voltage-gated sodium channels are the primary ion channels in phase 0 of the cardiac action potential. Therefore, local anesthetics are also classified as antiarrhythmics, specifically type 1b. This mechanism of action is discussed in more detail in the discussion of Na+ channel blockers in Chapter 11 and is also responsible for cardiac toxicity of local anesthetics.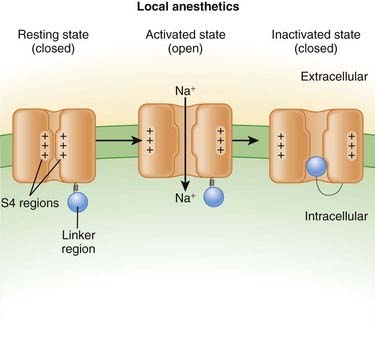
Pharmacokinetics
 Local anesthetics are most commonly injected, but other routes of administration include topical application: oral sprays, creams, and vaporized forms (for airways).
Local anesthetics are most commonly injected, but other routes of administration include topical application: oral sprays, creams, and vaporized forms (for airways). There are important factors that dictate the potency, speed of onset, and duration of a local anesthetic:
There are important factors that dictate the potency, speed of onset, and duration of a local anesthetic:


| Agent | Duration Plain (minutes) | Duration with Epinephrine (minutes) |
|---|---|---|
| 2-Chloroprocaine | 20-30 | 30-45 |
| Procaine | 15-30 | 30 |
| Lidocaine | 30-60 | 120 |
| Mepivacaine | 45-90 | 120 |
| Prilocaine | 30-90 | 120 |
| Bupivacaine | 120-240 | 180-240 |
| Ropivacaine | 120-240 | 180-240 |



Side Effects
 Side effects more commonly occur when the toxic dose is approached or exceeded or if any dose is accidently injected into a blood vessel.
Side effects more commonly occur when the toxic dose is approached or exceeded or if any dose is accidently injected into a blood vessel.



Important Notes
 Onset time and duration are both dose dependent. Higher doses and higher concentrations of solution of local anesthetic will result in faster onset times and longer durations of action. The addition of low-dose epinephrine to the local anesthetic also prolongs the duration of action because it causes vasoconstriction and reduces blood flow, which slows the washout of the drug from the site of action.
Onset time and duration are both dose dependent. Higher doses and higher concentrations of solution of local anesthetic will result in faster onset times and longer durations of action. The addition of low-dose epinephrine to the local anesthetic also prolongs the duration of action because it causes vasoconstriction and reduces blood flow, which slows the washout of the drug from the site of action. The location of injection strongly determines the effect of the local anesthetic. Only a segment of a neuron needs to be bound with local anesthetic to completely block its function (compared with blocking the entire length of the neuron). Therefore if local anesthetic is administered upstream to a large nerve, a very large downstream distribution of sensation can be blocked. This is the principle behind spinal anesthetics, in which a small dose (usually 1 to 3 mL) of a local anesthetic is administered to the cerebral spinal fluid in the lumbar spine and the result is often complete loss of all sensation and motor activity from the chest down to the toes!
The location of injection strongly determines the effect of the local anesthetic. Only a segment of a neuron needs to be bound with local anesthetic to completely block its function (compared with blocking the entire length of the neuron). Therefore if local anesthetic is administered upstream to a large nerve, a very large downstream distribution of sensation can be blocked. This is the principle behind spinal anesthetics, in which a small dose (usually 1 to 3 mL) of a local anesthetic is administered to the cerebral spinal fluid in the lumbar spine and the result is often complete loss of all sensation and motor activity from the chest down to the toes!
 Small doses of epinephrine are often added to local anesthetic to prolong the duration of anesthesia. The epinephrine acts as a vasoconstrictor and thus reduces blood flow to the site of injection and reduces metabolism of the local anesthetic.
Small doses of epinephrine are often added to local anesthetic to prolong the duration of anesthesia. The epinephrine acts as a vasoconstrictor and thus reduces blood flow to the site of injection and reduces metabolism of the local anesthetic.




FYI
 Amide local anesthetics have an –i– in the first part of the generic drug name (lidocaine, prilocaine, bupivacaine), whereas esters (tetracaine, procaine, cocaine) do not.
Amide local anesthetics have an –i– in the first part of the generic drug name (lidocaine, prilocaine, bupivacaine), whereas esters (tetracaine, procaine, cocaine) do not.
Baclofen

MOA (Mechanism of Action)
γ-Aminobutyric Acid (GABA)
 GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.


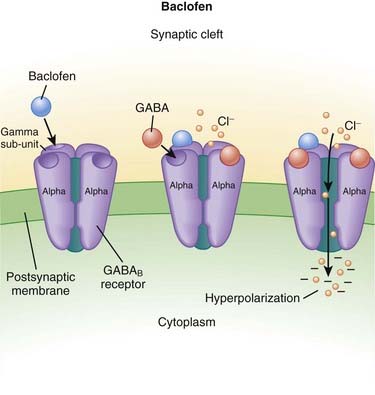



Side Effects









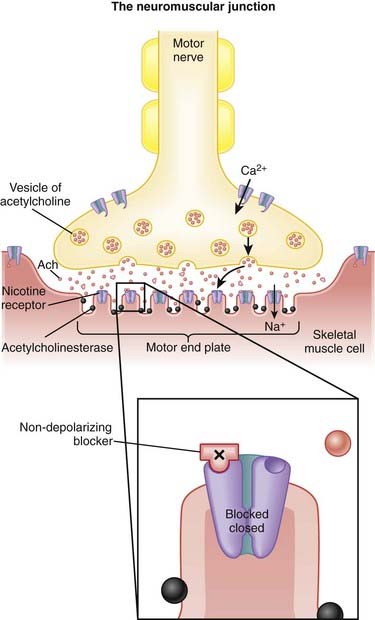
Nondepolarizing Neuromuscular Blockers



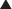
MOA (Mechanism of Action)
 Voluntary skeletal muscle contraction occurs when a motor neuron is depolarized. The distal end of the motor neuron is part of the NMJ, which is the anatomic connection between the neuron and muscle. The presynaptic membrane is the motor nerve, and the postsynaptic membrane is the motor end plate of the muscle cell.
Voluntary skeletal muscle contraction occurs when a motor neuron is depolarized. The distal end of the motor neuron is part of the NMJ, which is the anatomic connection between the neuron and muscle. The presynaptic membrane is the motor nerve, and the postsynaptic membrane is the motor end plate of the muscle cell. The depolarizing motor neuron releases acetylcholine, and the ACh crosses the synapse and binds to nicotinic ACh receptors on the muscle cell. The binding of ACh to the motor end plate induces small mini-depolarizations. When enough mini-depolarizations occur, a full action potential is created in the muscle cell, which results in an increase in intracellular calcium levels and subsequent actin-myosin interactions, resulting in contraction (Figure 21-6).
The depolarizing motor neuron releases acetylcholine, and the ACh crosses the synapse and binds to nicotinic ACh receptors on the muscle cell. The binding of ACh to the motor end plate induces small mini-depolarizations. When enough mini-depolarizations occur, a full action potential is created in the muscle cell, which results in an increase in intracellular calcium levels and subsequent actin-myosin interactions, resulting in contraction (Figure 21-6).



Reversal of Blockade
 If the patient remains paralyzed for longer than desired, then reversal medication can be administered to facilitate return of muscle strength.
If the patient remains paralyzed for longer than desired, then reversal medication can be administered to facilitate return of muscle strength.
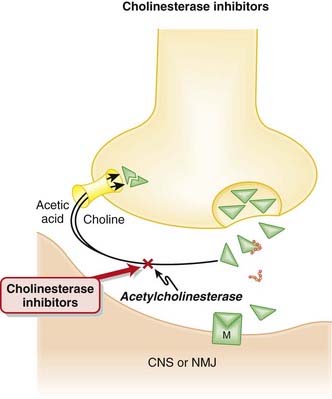
 Cholinesterase is the enzyme that breaks down ACh. Anticholinesterases are the drugs that inhibit cholinesterase, resulting in increased levels of ACh everywhere in the body.
Cholinesterase is the enzyme that breaks down ACh. Anticholinesterases are the drugs that inhibit cholinesterase, resulting in increased levels of ACh everywhere in the body.
Pharmacokinetics
 There are important differences in duration of action and method of metabolism among the different drugs in this class. See Table 21-4.
There are important differences in duration of action and method of metabolism among the different drugs in this class. See Table 21-4.





Important Notes
 Potency and time to onset: Onset time is determined by the dose administered. For a given drug, giving a larger dose will result in a faster onset of paralysis. For drugs that are more potent (e.g., pancuronium is the most potent), a smaller dose is required for paralysis, but because a smaller dose is required, more potent drugs have a slower onset of action.
Potency and time to onset: Onset time is determined by the dose administered. For a given drug, giving a larger dose will result in a faster onset of paralysis. For drugs that are more potent (e.g., pancuronium is the most potent), a smaller dose is required for paralysis, but because a smaller dose is required, more potent drugs have a slower onset of action.
 The degree of paralysis can be measured with a small battery-powered device called a nerve stimulator. It essentially delivers a small electric shock. Electrodes (usually just electrocardiographic patches) are applied on top of the motor nerve of interest (usually the ulnar nerve at the wrist or the facial nerve at the temple). When the electrical shock is applied, a muscle that is not paralyzed will vigorously contract; a partially paralyzed muscle will demonstrate a small twitch, and a fully paralyzed muscle will not contract at all.
The degree of paralysis can be measured with a small battery-powered device called a nerve stimulator. It essentially delivers a small electric shock. Electrodes (usually just electrocardiographic patches) are applied on top of the motor nerve of interest (usually the ulnar nerve at the wrist or the facial nerve at the temple). When the electrical shock is applied, a muscle that is not paralyzed will vigorously contract; a partially paralyzed muscle will demonstrate a small twitch, and a fully paralyzed muscle will not contract at all.
 If four electrical shocks are applied in succession, the test is called a train of four. This is important because each successive shock will release a slightly smaller amount of ACh from the presynaptic nerve; because the drugs are competitive antagonists, the partially blocked muscle will demonstrate progressively smaller contractions, a phenomenon called fade. A patient can have zero fade when as many as 50% of the nicotinic receptors are still occupied; therefore four full contractions with the nerve stimulator TOF test does not guarantee that the patient will have 100% strength.
If four electrical shocks are applied in succession, the test is called a train of four. This is important because each successive shock will release a slightly smaller amount of ACh from the presynaptic nerve; because the drugs are competitive antagonists, the partially blocked muscle will demonstrate progressively smaller contractions, a phenomenon called fade. A patient can have zero fade when as many as 50% of the nicotinic receptors are still occupied; therefore four full contractions with the nerve stimulator TOF test does not guarantee that the patient will have 100% strength.




FYI
 Curare is the historical prototype of nondepolarization neuromuscular blockers, but it is no longer used clinically. Curare (also called D-tubocurare) was the first paralytic used in anesthesia, but it has been replaced by newer agents. It was introduced to anesthesia around 1940. It was discovered in South America and was first used in poison arrows for hunting. It is harvested from the plant Strychnos toxifera. The toxin strychnine is also from this genus of plant (but from a different species).
Curare is the historical prototype of nondepolarization neuromuscular blockers, but it is no longer used clinically. Curare (also called D-tubocurare) was the first paralytic used in anesthesia, but it has been replaced by newer agents. It was introduced to anesthesia around 1940. It was discovered in South America and was first used in poison arrows for hunting. It is harvested from the plant Strychnos toxifera. The toxin strychnine is also from this genus of plant (but from a different species).



Depolarizing Neuromuscular Blockers
Description
Depolarizing neuromuscular blockers are paralyzing drugs also known as muscle relaxants.

MOA (Mechanism of Action)
 Voluntary skeletal muscle contraction occurs when a motor neuron is depolarized. The distal end of the motor neuron is part of the neuromuscular junction (NMJ), which is the anatomic connection between the neuron and muscle. The presynaptic membrane is the motor nerve, and the postsynaptic membrane is the motor end plate of the muscle cell.
Voluntary skeletal muscle contraction occurs when a motor neuron is depolarized. The distal end of the motor neuron is part of the neuromuscular junction (NMJ), which is the anatomic connection between the neuron and muscle. The presynaptic membrane is the motor nerve, and the postsynaptic membrane is the motor end plate of the muscle cell. The depolarizing motor neuron releases acetylcholine (ACh), which crosses the synapse and binds to nicotinic ACh receptors on the muscle cell. The binding of ACh to the motor end plate induces small mini-depolarizations via opening sodium channels. When enough mini-depolarizations occur, a full action potential is created in the muscle cell, which results in an increase in intracellular calcium levels and subsequent actin-myosin interactions, resulting in contraction (Figure 21-7).
The depolarizing motor neuron releases acetylcholine (ACh), which crosses the synapse and binds to nicotinic ACh receptors on the muscle cell. The binding of ACh to the motor end plate induces small mini-depolarizations via opening sodium channels. When enough mini-depolarizations occur, a full action potential is created in the muscle cell, which results in an increase in intracellular calcium levels and subsequent actin-myosin interactions, resulting in contraction (Figure 21-7).



Pharmacokinetics

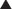



Contraindications


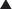
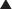
 Risk of hyperkalemia:
Risk of hyperkalemia:
 Hyperresponders: In some patients there are extrajunctional (outside the NMJ) nicotinic receptors on the muscle that ACh can bind to; this binding can cause a greater-than-normal release of potassium.
Hyperresponders: In some patients there are extrajunctional (outside the NMJ) nicotinic receptors on the muscle that ACh can bind to; this binding can cause a greater-than-normal release of potassium.


Side Effects



Important Notes
 Because succinylcholine binds irreversibly, there are important differences between it and the nondepolarizing drugs, which bind competitively:
Because succinylcholine binds irreversibly, there are important differences between it and the nondepolarizing drugs, which bind competitively:
 Increasing the ACh concentration in the synaptic cleft via administration of anticholinesterase drugs is not effective in reversing the block.
Increasing the ACh concentration in the synaptic cleft via administration of anticholinesterase drugs is not effective in reversing the block. The train-of-four (TOF) test (see the discussion of nondepolarizing NMJ blockers) exhibits different results with succinylcholine versus nondepolarizers:
The train-of-four (TOF) test (see the discussion of nondepolarizing NMJ blockers) exhibits different results with succinylcholine versus nondepolarizers:
• With each stimulation, the amount of ACh released is slightly less than with the previous stimulation; because succinylcholine is not competitively bound, the concentration of ACh in the synaptic cleft does not influence the strength of contraction. The strength of contraction is solely dependent on nicotinic receptor occupancy. Therefore there is no fade when there is partial blockade by succinylcholine, which is in contrast to nondepolarizers.



Advanced
 In attempts to reduce muscle pain after succinylcholine-induced fasciculations, a defasciculating dose of a nondepolarizing paralytic (such as rocuronium or vecuronium) is given 5 minutes beforehand. This is only a very small dose so as not to produce paralysis in the awake patient; usually about 5% to 10% of the ED95 is used (
In attempts to reduce muscle pain after succinylcholine-induced fasciculations, a defasciculating dose of a nondepolarizing paralytic (such as rocuronium or vecuronium) is given 5 minutes beforehand. This is only a very small dose so as not to produce paralysis in the awake patient; usually about 5% to 10% of the ED95 is used (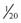 to
to 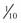 of the intubating dose). Lidocaine has also been used to reduce postsuccinylcholine muscle pain.
of the intubating dose). Lidocaine has also been used to reduce postsuccinylcholine muscle pain.




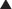
Nicotine



MOA (Mechanism of Action)
 There are several nicotine receptors, composed of alpha (α) and beta (β) subunits. The various α subunits and β subunits all appear in different combinations throughout the body. The most common subunits are α4β2, α3β4, and α7.
There are several nicotine receptors, composed of alpha (α) and beta (β) subunits. The various α subunits and β subunits all appear in different combinations throughout the body. The most common subunits are α4β2, α3β4, and α7.
 Nicotine binds to presynaptic nicotinic α4β2 receptors, which in turn leads to the release of dopamine into the nucleus accumbens, the “reward” pathway of the brain (Figure 21-8).
Nicotine binds to presynaptic nicotinic α4β2 receptors, which in turn leads to the release of dopamine into the nucleus accumbens, the “reward” pathway of the brain (Figure 21-8).



Pharmacokinetics






Side Effects
Full Agonists
Local Effects



Partial Agonists
 Nausea: The mechanism has not been described; however, other agents that elevate dopamine levels in the CNS also promote nausea.
Nausea: The mechanism has not been described; however, other agents that elevate dopamine levels in the CNS also promote nausea.Serious
 Neuropsychiatric: Events such as depression, agitation, hostility, and suicidality have been observed in the postmarketing period (see Important Notes for further details). The mechanism is unknown; however, agents that elevate dopamine levels in the CNS have been known to produce some of the same side effects.
Neuropsychiatric: Events such as depression, agitation, hostility, and suicidality have been observed in the postmarketing period (see Important Notes for further details). The mechanism is unknown; however, agents that elevate dopamine levels in the CNS have been known to produce some of the same side effects.Important Notes
 Despite the acute effects of nicotine on the cardiovascular system (see Mechanism of Action), there is no clear evidence that nicotine itself increases risk of cardiovascular events. Recent concerns have arisen about an increased risk of mortality after coronary artery bypass surgery; however, these findings need to be confirmed by well-designed prospective studies. Currently the risk-to-benefit ratio of nicotine replacement in patients with cardiovascular disease is unknown.
Despite the acute effects of nicotine on the cardiovascular system (see Mechanism of Action), there is no clear evidence that nicotine itself increases risk of cardiovascular events. Recent concerns have arisen about an increased risk of mortality after coronary artery bypass surgery; however, these findings need to be confirmed by well-designed prospective studies. Currently the risk-to-benefit ratio of nicotine replacement in patients with cardiovascular disease is unknown.




Advanced
Pharmacogenetics
 The rate of nicotine metabolism is determined in part by polymorphisms in the genes for CYP2A6 as well as uridine 5′-diphosphate (UDP)–glucuronosyltransferases (UGTs), a secondary pathway of elimination. These differences are believed to explain why women metabolize nicotine more rapidly than men and why patients of Caucasian and Hispanic descent metabolize nicotine more rapidly than patients of Asian and Black descent.
The rate of nicotine metabolism is determined in part by polymorphisms in the genes for CYP2A6 as well as uridine 5′-diphosphate (UDP)–glucuronosyltransferases (UGTs), a secondary pathway of elimination. These differences are believed to explain why women metabolize nicotine more rapidly than men and why patients of Caucasian and Hispanic descent metabolize nicotine more rapidly than patients of Asian and Black descent.Evidence
Nicotine Replacement Therapies for Smoking Cessation
 A 2008 Cochrane review (111 trials, N > 40,000 participants) compared nicotine replacement therapies to other interventions and placebo as an aid to smoking cessation. Overall, nicotine replacement therapies performed better than control for improving abstinence rates (relative risk [RR] 1.58). Abstinence rates for individual interventions varied slightly: gum, RR 1.43; patch, RR 1.66; inhaler, RR 1.90; nasal spray, RR 2.02; and tablets or lozenges, RR 2.00. Only one study compared quit rates between nicotine replacement and other active comparators (bupropion), and quit rates were lower for the nicotine patch in this study, compared with bupropion.
A 2008 Cochrane review (111 trials, N > 40,000 participants) compared nicotine replacement therapies to other interventions and placebo as an aid to smoking cessation. Overall, nicotine replacement therapies performed better than control for improving abstinence rates (relative risk [RR] 1.58). Abstinence rates for individual interventions varied slightly: gum, RR 1.43; patch, RR 1.66; inhaler, RR 1.90; nasal spray, RR 2.02; and tablets or lozenges, RR 2.00. Only one study compared quit rates between nicotine replacement and other active comparators (bupropion), and quit rates were lower for the nicotine patch in this study, compared with bupropion.Partial Agonists versus Placebo or Active Comparators for Smoking Cessation and Relapse Prevention
 A 2008 Cochrane review (nine trials, 7267 participants) compared varenicline with either placebo or bupropion for smoking cessation and relapse prevention. The pooled RR for continuous abstinence after 6 months versus placebo was 2.3. Varenicline also performed better than bupropion (RR 1.5) and nicotine replacement therapy (RR 1.3) for continuous abstinence at 12 months.
A 2008 Cochrane review (nine trials, 7267 participants) compared varenicline with either placebo or bupropion for smoking cessation and relapse prevention. The pooled RR for continuous abstinence after 6 months versus placebo was 2.3. Varenicline also performed better than bupropion (RR 1.5) and nicotine replacement therapy (RR 1.3) for continuous abstinence at 12 months.
FYI
 Nicotine vaccines are currently in development. The vaccines would stimulate antibodies to nicotine, which would then form complexes with nicotine and prevent absorption of nicotine across the blood-brain barrier. Early results are promising, although positive responses are typically seen in those with sufficient antibody production.
Nicotine vaccines are currently in development. The vaccines would stimulate antibodies to nicotine, which would then form complexes with nicotine and prevent absorption of nicotine across the blood-brain barrier. Early results are promising, although positive responses are typically seen in those with sufficient antibody production.

Dopamine and Dopamine Agonists


MOA (Mechanism of Action)
 Parkinson’s disease is characterized by the progressive loss of dopaminergic neurons in the substantia nigra. Dopamine is a catecholamine that is synthesized in these dopaminergic neurons and is released onto two pathways in the striatum that regulate coordinated movement.
Parkinson’s disease is characterized by the progressive loss of dopaminergic neurons in the substantia nigra. Dopamine is a catecholamine that is synthesized in these dopaminergic neurons and is released onto two pathways in the striatum that regulate coordinated movement.







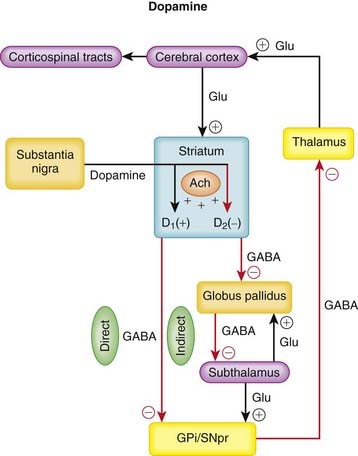
Pharmacokinetics
 Levodopa is the precursor of dopamine and is readily and completely converted to dopamine by the enzyme dopa decarboxylase (DDC). Levodopa is rapidly absorbed when administered orally, with a short plasma half-life of 1 to 3 hours. Dietary amino acids may compete for absorption sites in the small intestine; therefore administration with meals may delay absorption and reduce peak plasma concentrations.
Levodopa is the precursor of dopamine and is readily and completely converted to dopamine by the enzyme dopa decarboxylase (DDC). Levodopa is rapidly absorbed when administered orally, with a short plasma half-life of 1 to 3 hours. Dietary amino acids may compete for absorption sites in the small intestine; therefore administration with meals may delay absorption and reduce peak plasma concentrations. Once converted, dopamine is then metabolized and inactivated by catechol-O-methyltransferase (COMT) and monoamine oxidase (MAO). Another approach to enhancing dopaminergic transmission is to inhibit these enzymes, thereby increasing dopamine levels. These agents, COMT inhibitors and MAO inhibitors, are discussed in Chapters 21 and 23.
Once converted, dopamine is then metabolized and inactivated by catechol-O-methyltransferase (COMT) and monoamine oxidase (MAO). Another approach to enhancing dopaminergic transmission is to inhibit these enzymes, thereby increasing dopamine levels. These agents, COMT inhibitors and MAO inhibitors, are discussed in Chapters 21 and 23.







Important Notes
 l-Dopa is almost always administered with a peripheral decarboxylase inhibitor. The reason for this is that l-dopa is readily converted to dopamine by decarboxylases before it even reaches the CNS. This dopamine is then converted to inactive metabolites and eliminated, or it acts on peripheral dopamine receptors, resulting in a number of side effects.
l-Dopa is almost always administered with a peripheral decarboxylase inhibitor. The reason for this is that l-dopa is readily converted to dopamine by decarboxylases before it even reaches the CNS. This dopamine is then converted to inactive metabolites and eliminated, or it acts on peripheral dopamine receptors, resulting in a number of side effects.





Evidence
Parkinson’s Disease
Ropinirole versus Bromocriptine
 A 2001 Cochrane review (three trials, N = 482 patients) found that ropinirole and bromocriptine had similar effects in improving off-time and reducing l-dopa dose, without increasing adverse events such as dyskinesia. The authors noted that the three included studies might not have enough power to distinguish between agents. Off-time refers to wearing off of the effects of the drug, severely limiting the mobility of the patient.
A 2001 Cochrane review (three trials, N = 482 patients) found that ropinirole and bromocriptine had similar effects in improving off-time and reducing l-dopa dose, without increasing adverse events such as dyskinesia. The authors noted that the three included studies might not have enough power to distinguish between agents. Off-time refers to wearing off of the effects of the drug, severely limiting the mobility of the patient.Pramipexole versus Placebo
 A 2000 Cochrane review (four trials, N = 669 patients) compared pramipexole with placebo in patients with Parkinson’s disease and long-term complications from l-dopa. Pramipexole reduced off-time, improved motor impairments and disability, and reduced l-dopa dose requirements, but it also increased dyskinetic adverse events.
A 2000 Cochrane review (four trials, N = 669 patients) compared pramipexole with placebo in patients with Parkinson’s disease and long-term complications from l-dopa. Pramipexole reduced off-time, improved motor impairments and disability, and reduced l-dopa dose requirements, but it also increased dyskinetic adverse events.Early Parkinson’s Disease: L-Dopa versus Bromocriptine
 A 2007 Cochrane review examined six trials (N = 850 patients), but the studies were too heterogeneous for a meta-analysis to be conducted. The authors arrived at a qualitative conclusion that bromocriptine may be beneficial in delaying motor complications and dyskinesias, with comparable effects on impairment and disability.
A 2007 Cochrane review examined six trials (N = 850 patients), but the studies were too heterogeneous for a meta-analysis to be conducted. The authors arrived at a qualitative conclusion that bromocriptine may be beneficial in delaying motor complications and dyskinesias, with comparable effects on impairment and disability.FYI
 In the future we will likely discover that the pathophysiology of Parkinson’s disease is much more complicated than described in the Mechanism of Action section and that several other neurotransmitters may be involved, including substance P and dynorphin. The significance of these transmitters in the pathophysiology of Parkinson’s disease is an active area of research.
In the future we will likely discover that the pathophysiology of Parkinson’s disease is much more complicated than described in the Mechanism of Action section and that several other neurotransmitters may be involved, including substance P and dynorphin. The significance of these transmitters in the pathophysiology of Parkinson’s disease is an active area of research.
Catechol-O-Methyl Transferase (COMT) Inhibitors


MOA (Mechanism of Action)
 Parkinson’s disease is characterized by the progressive loss of dopaminergic neurons in the substantia nigra. Dopamine is a catecholamine that is synthesized in these dopaminergic neurons and is released onto two pathways in the striatum that regulate coordinated movement.
Parkinson’s disease is characterized by the progressive loss of dopaminergic neurons in the substantia nigra. Dopamine is a catecholamine that is synthesized in these dopaminergic neurons and is released onto two pathways in the striatum that regulate coordinated movement.

 COMT inhibitors increase the amount of dopamine available to the CNS. COMT is one of the two major enzymes involved in the metabolism of catecholamines (epinephrine, norepinephrine, and dopamine). Thus one of the ways COMT inhibitors increase dopamine is by inhibiting its breakdown (Figure 21-10).
COMT inhibitors increase the amount of dopamine available to the CNS. COMT is one of the two major enzymes involved in the metabolism of catecholamines (epinephrine, norepinephrine, and dopamine). Thus one of the ways COMT inhibitors increase dopamine is by inhibiting its breakdown (Figure 21-10). The COMT inhibitors also free up transporters for levodopa:
The COMT inhibitors also free up transporters for levodopa:

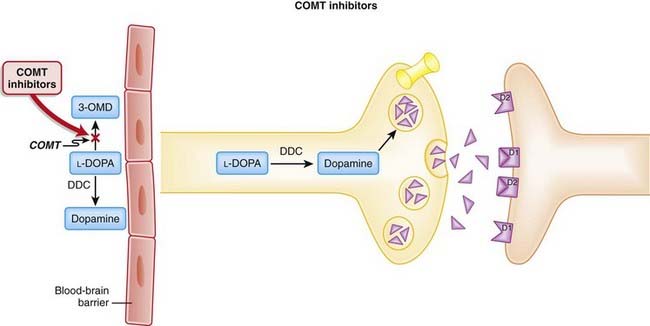
Pharmacokinetics



Contraindications
 Concomitant use with nonselective MAOIs: This includes the use of an MAO-A and an MAO-B inhibitor in combination. The MAO pathway becomes the key metabolic route for epinephrine and norepinephrine in the presence of COMT blockade. There should be at least a 2-week washout before initiation of treatment with a COMT inhibitor. Caution should also be exercised in patients on MAO-B selective inhibitors, as these become nonselective at higher doses.
Concomitant use with nonselective MAOIs: This includes the use of an MAO-A and an MAO-B inhibitor in combination. The MAO pathway becomes the key metabolic route for epinephrine and norepinephrine in the presence of COMT blockade. There should be at least a 2-week washout before initiation of treatment with a COMT inhibitor. Caution should also be exercised in patients on MAO-B selective inhibitors, as these become nonselective at higher doses.


Side Effects






Evidence
COMT Inhibitors versus Dopamine Therapy in Parkinson’s Disease Patients with Motor Complications on L-Dopa
 A 2004 Cochrane review compared COMT inhibitors (tolcapone) with pergolide (one trial, N = 203 over 12 weeks) and bromocriptine (one trial, N = 146 over 8 weeks). Tolcapone allowed for a greater reduction in l-dopa dose than bromocriptine and was similar to pergolide. Tolcapone produced similar benefits in motor impairment and disability ratings versus both bromocriptine and pergolide. The studies were underpowered to detect statistical differences for these efficacy outcomes, and there were no studies involving entacapone.
A 2004 Cochrane review compared COMT inhibitors (tolcapone) with pergolide (one trial, N = 203 over 12 weeks) and bromocriptine (one trial, N = 146 over 8 weeks). Tolcapone allowed for a greater reduction in l-dopa dose than bromocriptine and was similar to pergolide. Tolcapone produced similar benefits in motor impairment and disability ratings versus both bromocriptine and pergolide. The studies were underpowered to detect statistical differences for these efficacy outcomes, and there were no studies involving entacapone.

Cholinesterase Inhibitors
Description
Cholinesterase inhibitors are a collection of agents that increase acetylcholine levels.


MOA (Mechanism of Action)
 Acetylcholine is the main neurotransmitter in the parasympathetic arm of the ANS and a major neurotransmitter in the CNS.
Acetylcholine is the main neurotransmitter in the parasympathetic arm of the ANS and a major neurotransmitter in the CNS.







Pharmacokinetics







Contraindications



Important Notes
 The cholinesterase inhibitors used to treat Alzheimer’s disease are reversible inhibitors. Irreversible cholinesterase inhibitors lead to a profound increase in cholinergic neurotransmission, which in turn leads to convulsions, thick secretions that obstruct the airways, cardiac arrhythmias, and death. Irreversible cholinesterase inhibitors have been used as nerve gases in warfare (sarin gas) for decades and are also used in many pesticides.
The cholinesterase inhibitors used to treat Alzheimer’s disease are reversible inhibitors. Irreversible cholinesterase inhibitors lead to a profound increase in cholinergic neurotransmission, which in turn leads to convulsions, thick secretions that obstruct the airways, cardiac arrhythmias, and death. Irreversible cholinesterase inhibitors have been used as nerve gases in warfare (sarin gas) for decades and are also used in many pesticides.Evidence
Donepezil for Treatment of Alzheimer’s Disease
 A 2006 Cochrane review (24 trials, N = 5796 participants) assessed whether donepezil improves the well-being of patients with dementia from Alzheimer’s disease. Most of the participants had mild to moderate disease, and most studies were less than 6 months in duration. Donepezil improved cognition versus placebo. Some improvement was seen in global clinical scale, and improvements were also seen in activities of daily living and behavior but not in quality of life. Withdrawals were more frequent with donepezil 10 mg versus placebo. Results were similar for all severities of disease. Health resource use was reported in only two studies, and there were no differences versus placebo.
A 2006 Cochrane review (24 trials, N = 5796 participants) assessed whether donepezil improves the well-being of patients with dementia from Alzheimer’s disease. Most of the participants had mild to moderate disease, and most studies were less than 6 months in duration. Donepezil improved cognition versus placebo. Some improvement was seen in global clinical scale, and improvements were also seen in activities of daily living and behavior but not in quality of life. Withdrawals were more frequent with donepezil 10 mg versus placebo. Results were similar for all severities of disease. Health resource use was reported in only two studies, and there were no differences versus placebo.
Galantamine for Mild Cognitive Impairment or Alzheimer’s Disease
 A 2006 Cochrane review (10 trials, N = 6805 participants) assessed the clinical effects of galantamine in patients with mild cognitive impairment or probable or possible Alzheimer’s disease. In Alzheimer’s disease the authors found that galantamine was more effective than placebo in improving cognitive function, as well as some evidence of improvement on measures of activity of daily living and behavioral symptoms. In mild cognitive impairment, data from two trials suggest marginal clinical benefit but an increased incidence of death with galantamine.
A 2006 Cochrane review (10 trials, N = 6805 participants) assessed the clinical effects of galantamine in patients with mild cognitive impairment or probable or possible Alzheimer’s disease. In Alzheimer’s disease the authors found that galantamine was more effective than placebo in improving cognitive function, as well as some evidence of improvement on measures of activity of daily living and behavioral symptoms. In mild cognitive impairment, data from two trials suggest marginal clinical benefit but an increased incidence of death with galantamine.Rivastigmine for Treatment of Alzheimer’s Disease
 A 2009 Cochrane review (9 trials, N = 4775 participants) determined the efficacy and safety of rivastigmine for patients with dementia of the Alzheimer’s type. High-dose rivastigmine (6 to 12 mg daily) improved measures of cognition and activities of daily living versus placebo. These differences were statistically significant for cognition only when lower doses of rivastigmine were used. The incidence of side effects such as nausea, vomiting, diarrhea, anorexia, headache, syncope, abdominal pain, and dizziness was higher than with placebo. One trial compared patches with capsules, finding no differences in efficacy but a lower incidence of side effects with the lower dose patch versus capsules.
A 2009 Cochrane review (9 trials, N = 4775 participants) determined the efficacy and safety of rivastigmine for patients with dementia of the Alzheimer’s type. High-dose rivastigmine (6 to 12 mg daily) improved measures of cognition and activities of daily living versus placebo. These differences were statistically significant for cognition only when lower doses of rivastigmine were used. The incidence of side effects such as nausea, vomiting, diarrhea, anorexia, headache, syncope, abdominal pain, and dizziness was higher than with placebo. One trial compared patches with capsules, finding no differences in efficacy but a lower incidence of side effects with the lower dose patch versus capsules.

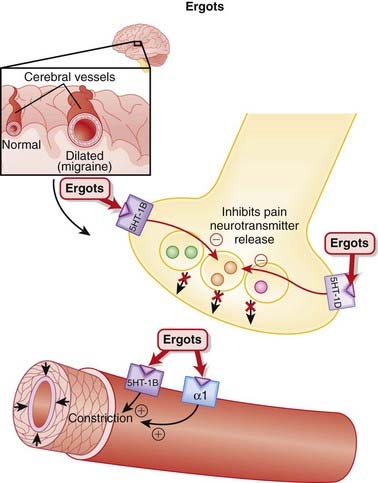
Ergot Alkaloids


MOA (Mechanism of Action)
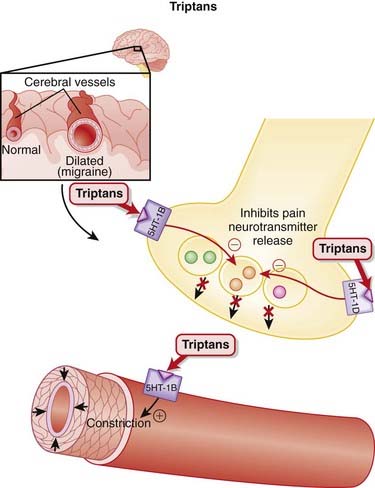
 Migraines are believed to be caused by cerebral vasodilation and subsequent activation of pain fibers.
Migraines are believed to be caused by cerebral vasodilation and subsequent activation of pain fibers.




Pharmacokinetics
 Ergotamine undergoes extensive first-pass metabolism and has a very low oral bioavailability. In contrast, ergonovine and methylergonovine are rapidly absorbed and reach peak plasma levels in under 90 minutes, with plasma levels 10 times those of ergotamine.
Ergotamine undergoes extensive first-pass metabolism and has a very low oral bioavailability. In contrast, ergonovine and methylergonovine are rapidly absorbed and reach peak plasma levels in under 90 minutes, with plasma levels 10 times those of ergotamine.


 Ergotamine derivatives are metabolized via CYP3A4. Given the dangers associated with excessive ergotamine levels (see Side Effects), the concomitant use of strong CYP3A4 inhibitors should be avoided.
Ergotamine derivatives are metabolized via CYP3A4. Given the dangers associated with excessive ergotamine levels (see Side Effects), the concomitant use of strong CYP3A4 inhibitors should be avoided.

Contraindications
 Coronary artery disease: Vasoconstriction or spasm of coronary arteries may exacerbate symptoms and lead to myocardial ischemia.
Coronary artery disease: Vasoconstriction or spasm of coronary arteries may exacerbate symptoms and lead to myocardial ischemia.








Important Notes
 The first use of ergots was to promote uterine contractions (an oxytocic) in childbirth in the 1500s. However, widespread use for this indication in the early 1800s was accompanied by an increase in the number of stillbirths. Excessive uterine contraction reduces blood flow to the fetus, and this is the likely reason for the stillbirths. In an early version of adverse drug reaction surveillance, ergot was no longer recommended for induction of labor but instead for postpartum hemorrhage, an indication that exists to this day.
The first use of ergots was to promote uterine contractions (an oxytocic) in childbirth in the 1500s. However, widespread use for this indication in the early 1800s was accompanied by an increase in the number of stillbirths. Excessive uterine contraction reduces blood flow to the fetus, and this is the likely reason for the stillbirths. In an early version of adverse drug reaction surveillance, ergot was no longer recommended for induction of labor but instead for postpartum hemorrhage, an indication that exists to this day.








Triptans


MOA (Mechanism of Action)




Pharmacokinetics




| Drug | Peak Plasma Concentration | Elimination Half-Life |
|---|---|---|
| Sumatriptan |
PO, Orally; SC, subcutaneously.

Contraindications








Evidence
Sumatriptan versus Other Therapies or Placebo
 A 2003 Cochrane review (25 trials, N = 16,200 patients) found that sumatriptan elicited significantly more pain-free responses at 2 hours at the 25-mg (NNT 5) and 100-mg (NNT 7.5) doses but not the 50-mg dose, compared with placebo. All doses were statistically different from placebo for pain relief. Adverse events were more common with sumatriptan 100 mg versus placebo (number needed to harm [NNH] 7).
A 2003 Cochrane review (25 trials, N = 16,200 patients) found that sumatriptan elicited significantly more pain-free responses at 2 hours at the 25-mg (NNT 5) and 100-mg (NNT 7.5) doses but not the 50-mg dose, compared with placebo. All doses were statistically different from placebo for pain relief. Adverse events were more common with sumatriptan 100 mg versus placebo (number needed to harm [NNH] 7).



Topiramate

MOA (Mechanism of Action)



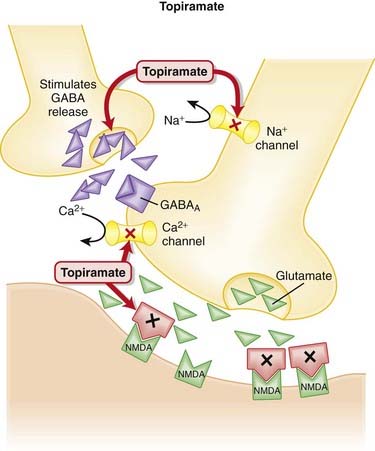






Side Effects
Serious






Important Notes
 Topiramate is being tried in a variety of off-label indications, with varying degrees of success. The diversity of this agent is likely a reflection of its multiple mechanisms of action, although concerns have also been raised about its overuse and misuse. One of the major concerns is the significant cognitive effects associated with its use.
Topiramate is being tried in a variety of off-label indications, with varying degrees of success. The diversity of this agent is likely a reflection of its multiple mechanisms of action, although concerns have also been raised about its overuse and misuse. One of the major concerns is the significant cognitive effects associated with its use.


Phenytoin









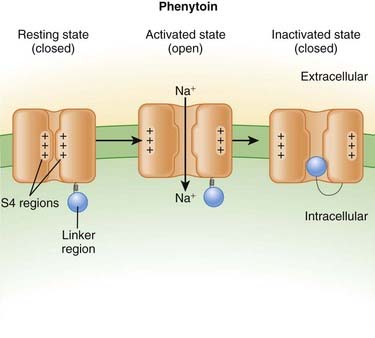









Contraindications
 Cardiac conduction abnormalities (intravenous phenytoin): Intravenous phenytoin, being a Na+ channel blocker, affects ventricular automaticity and is therefore contraindicated in patients at risk for being dependant on a ventricular escape rhythm, including those with sinus bradycardia, sinoatrial block, and second- and third-degree block.
Cardiac conduction abnormalities (intravenous phenytoin): Intravenous phenytoin, being a Na+ channel blocker, affects ventricular automaticity and is therefore contraindicated in patients at risk for being dependant on a ventricular escape rhythm, including those with sinus bradycardia, sinoatrial block, and second- and third-degree block.Side Effects







Evidence

Status Epilepticus (Refractory Seizures)
 A 2005 Cochrane review compared various agents used in status epilepticus. In the only study (198 patients) that had a phenytoin arm, intravenous lorazepam was more effective at terminating seizures than intravenous phenytoin (RR of 0.62 for seizures continuing). There was no difference in adverse events between the two agents.
A 2005 Cochrane review compared various agents used in status epilepticus. In the only study (198 patients) that had a phenytoin arm, intravenous lorazepam was more effective at terminating seizures than intravenous phenytoin (RR of 0.62 for seizures continuing). There was no difference in adverse events between the two agents.FYI
 Phenytoin was first synthesized in 1908, although it was not discovered to have anticonvulsant properties until 1938. Researchers at the time were looking for structural relatives of phenobarbital that lacked sedative properties and that were capable of suppressing electroshock convulsions in laboratory animals.
Phenytoin was first synthesized in 1908, although it was not discovered to have anticonvulsant properties until 1938. Researchers at the time were looking for structural relatives of phenobarbital that lacked sedative properties and that were capable of suppressing electroshock convulsions in laboratory animals.

Lamotrigine







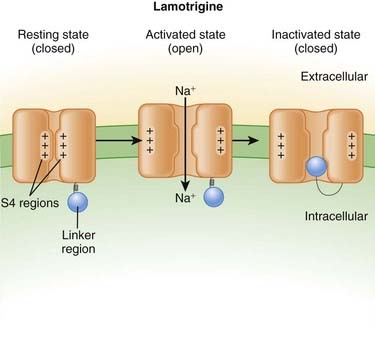




Side Effects
 CNS effects include dizziness, ataxia, blurred vision, and double vision. These types of sensory side effects occur with agents that inhibit sodium channels, although a mechanism has not been established.
CNS effects include dizziness, ataxia, blurred vision, and double vision. These types of sensory side effects occur with agents that inhibit sodium channels, although a mechanism has not been established.
Serious
 Rash: Rashes can range from mild to severe, including Stevens-Johnson syndrome and toxic epidermal necrolysis. These rashes can be fatal in patients who are otherwise compromised, or if treatment is not discontinued and/or if the rash is not treated. The exact mechanism behind these severe reactions is not known.
Rash: Rashes can range from mild to severe, including Stevens-Johnson syndrome and toxic epidermal necrolysis. These rashes can be fatal in patients who are otherwise compromised, or if treatment is not discontinued and/or if the rash is not treated. The exact mechanism behind these severe reactions is not known.Important Notes
 Lamotrigine is increasingly being used for psychiatric indications, including bipolar disorder. The idea to use lamotrigine in bipolar disorder first originated with the observation of its antiglutamatergic effects and the belief that this excitatory neurotransmitter may have a role in the pathogenesis of this illness.
Lamotrigine is increasingly being used for psychiatric indications, including bipolar disorder. The idea to use lamotrigine in bipolar disorder first originated with the observation of its antiglutamatergic effects and the belief that this excitatory neurotransmitter may have a role in the pathogenesis of this illness.Evidence
Lamotrigine versus Carbamazepine Monotherapy for Seizure Disorders
 A 2006 Cochrane review (five trials, N = 1384 participants) compared lamotrigine to carbamazepine as monotherapy in partial-onset or generalized-onset tonic-clonic seizures. The authors found that time to treatment withdrawal was improved with lamotrigine over carbamazepine (hazard ratio 0.55), whereas time to first seizure and freedom from seizure were not statistically different between lamotrigine and carbamazepine.
A 2006 Cochrane review (five trials, N = 1384 participants) compared lamotrigine to carbamazepine as monotherapy in partial-onset or generalized-onset tonic-clonic seizures. The authors found that time to treatment withdrawal was improved with lamotrigine over carbamazepine (hazard ratio 0.55), whereas time to first seizure and freedom from seizure were not statistically different between lamotrigine and carbamazepine.FYI
 Lamotrigine was initially developed as an antifolate agent, at a time when folate was believed to play a role in seizure development. Although it proved to be only a weak inhibitor of dihydrofolate reductase, its other pharmacologic properties as an Na+ channel blocker led to its success as an antiseizure medication.
Lamotrigine was initially developed as an antifolate agent, at a time when folate was believed to play a role in seizure development. Although it proved to be only a weak inhibitor of dihydrofolate reductase, its other pharmacologic properties as an Na+ channel blocker led to its success as an antiseizure medication.γ-Aminobutyric acid (GABA) Analogues
Description
GABA analogues are primarily used in the treatment of seizures and management of neuropathic pain.


MOA (Mechanism of Action)


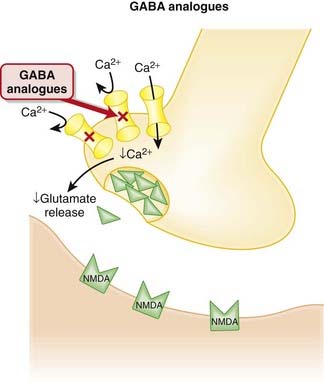
 Gabapentin’s major mechanism in the treatment of seizure is via binding to the α2δ subunit of presynaptic voltage-gated Ca2+ channels in the brain. It is believed that by reducing the influx of Ca2+ at nerve terminals, the release of excitatory neurotransmitters such as glutamate is reduced, although the connection between the reduction of Ca2+ influx and inhibition of glutamate release has not been established (Figure 21-17).
Gabapentin’s major mechanism in the treatment of seizure is via binding to the α2δ subunit of presynaptic voltage-gated Ca2+ channels in the brain. It is believed that by reducing the influx of Ca2+ at nerve terminals, the release of excitatory neurotransmitters such as glutamate is reduced, although the connection between the reduction of Ca2+ influx and inhibition of glutamate release has not been established (Figure 21-17).
Pharmacokinetics





Side Effects




Evidence
Pregabalin as Adjunctive Therapy in Drug-Resistant Partial Seizures
 A 2008 Cochrane review (four trials, N = 1397 participants) compared a range of doses of pregabalin with placebo. The authors found that patients treated with pregabalin were more likely to experience a 50% reduction in seizures. However, pregabalin was not associated with freedom from seizures, and participants were significantly more likely to withdraw from treatment for any reason and for adverse events. Pregabalin elicited ataxia, dizziness, somnolence, and weight gain compared with placebo.
A 2008 Cochrane review (four trials, N = 1397 participants) compared a range of doses of pregabalin with placebo. The authors found that patients treated with pregabalin were more likely to experience a 50% reduction in seizures. However, pregabalin was not associated with freedom from seizures, and participants were significantly more likely to withdraw from treatment for any reason and for adverse events. Pregabalin elicited ataxia, dizziness, somnolence, and weight gain compared with placebo.Gabapentin for Acute and Chronic Pain
 A 2005 Cochrane review (15 studies, N = 1468 participants) examined RCTs assessing the analgesic effectiveness and adverse effects of gabapentin for pain management. Only one small study assessed acute pain, and it found no differences between gabapentin and placebo. In chronic pain, 42% of patients improved versus 19% on placebo.
A 2005 Cochrane review (15 studies, N = 1468 participants) examined RCTs assessing the analgesic effectiveness and adverse effects of gabapentin for pain management. Only one small study assessed acute pain, and it found no differences between gabapentin and placebo. In chronic pain, 42% of patients improved versus 19% on placebo.
Carbamazepine


MOA (Mechanism of Action)







Pharmacokinetics












Side Effects











Evidence
Seizure Disorder
Carbamazepine versus Phenytoin
 A 2002 Cochrane review (three trials, N = 551 participants) compared these two agents in patients with partial-onset or generalized tonic-clonic seizures, using individual patient data from three trials (551 patients). The authors found no differences between carbamazepine and phenytoin for time to 6- or 12-month remission, time to first seizure, or time to withdrawal.
A 2002 Cochrane review (three trials, N = 551 participants) compared these two agents in patients with partial-onset or generalized tonic-clonic seizures, using individual patient data from three trials (551 patients). The authors found no differences between carbamazepine and phenytoin for time to 6- or 12-month remission, time to first seizure, or time to withdrawal.Carbamazepine versus Lamotrigine
 A 2006 Cochrane review (five trials, N = 1384 participants) compared these two agents in patients with partial-onset or generalized-onset tonic-clonic seizures, using individual patient data from five trials (1384 patients). The time to treatment withdrawal was significantly improved with lamotrigine compared with carbamazepine, whereas the time to first seizure and freedom from seizure at 6 months favored carbamazepine, although not by a statistically significant amount.
A 2006 Cochrane review (five trials, N = 1384 participants) compared these two agents in patients with partial-onset or generalized-onset tonic-clonic seizures, using individual patient data from five trials (1384 patients). The time to treatment withdrawal was significantly improved with lamotrigine compared with carbamazepine, whereas the time to first seizure and freedom from seizure at 6 months favored carbamazepine, although not by a statistically significant amount.
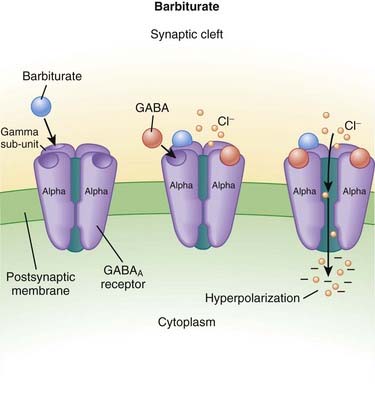
Barbiturates



MOA (Mechanism of Action)
 GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.





Pharmacokinetics
 Barbiturates can be administered orally, intravenously, intramuscularly, and, less commonly, rectally.
Barbiturates can be administered orally, intravenously, intramuscularly, and, less commonly, rectally.