Chapter 86 Neurologic Manifestations of Rheumatic Disorders of Childhood
The rheumatic disorders of childhood include a wide variety of conditions ranging from simple arthritis to complex multisystem autoimmune diseases. The presence and the degree of nervous system impairment vary widely, depending on the diagnosis and course of the disorder. Manifestations of neurologic disease may precede the onset of any other symptoms or occur much later. The current classification of the chronic rheumatic disorders is shown in Box 86-1 [Petty et al., 2004; Dannecker and Quartier, 2009; Horneff and Burgos-Vargas, 2009]. Examples of autoantibodies found in the various rheumatic diseases are given in Table 86-1.
Box 86-1 Classification of the Rheumatic Disorders of Childhood



































(Modified from Cassidy JT, Petty RE. Textbook of pediatric rheumatology, 4th edn. Philadelphia, 2001, WB Saunders.)
Table 86-1 Autoantibodies in Pediatric Rheumatic Diseases
| Antibody | Clinical Finding |
|---|---|
| ANA | 97% SLE, but also positive in MCTD, SSc, 10–85% JDMS, 20–88% JRA, SS, and 2–5% of controls |
| Anti-ds-DNA | 30–70% SLE, rarely in other CTDs |
| Anti-Sm | 30% SLE |
| Anti-RNP | MCTD, also in SLE |
| Anti-SSA/Ro | 25% SLE, 75% SS |
| Anti-SSB/La | 10% SLE, 40% SS |
| Anti-histone | 50% SLE, >90% DILS |
| Anti-centromere | 44–98% CREST syndrome |
| Anti-Scl-70 | 27% SSc |
| Anti-c-ANCA | >90% Wegener’s granulomatosis |
| Anti-p-ANCA | 10% Wegener’s granulomatosis, 70% CSS, 75% UC, 20% Crohn’s disease, 30% SLE |
| Anti-NR2 NMDAR | 10% SLE |
| RF A | 20% polyarticular JRA, 50% SS; 10–30% SLE, MCTD |
| LAC | Correlates with thromboembolic risk in SLE |
| aCL | Correlates with thromboembolic risk in SLE, malignancy; variable in many other diseases |
aCL, anticardiolipin antibody; ANA, antinuclear antibody; c-ANCA, cytoplasmic staining antineutrophil cytoplasmic antibody; CREST, calcinosis, Raynaud’s, esophageal dysmotility, sclerodactyly, telangiectasia; CSS, Churg–Strauss syndrome; CTD, connective tissue disease; DILS, drug-induced lupus syndrome; ds-DNA, double-stranded (native) DNA; JDMS, juvenile dermatomyositis; JRA, juvenile rheumatoid arthritis; LAC, lupus anticoagulant; MCTD, mixed connective tissue disease; NMDAR, N-methyl-d-aspartate receptor; p-ANCA, perinuclear staining antineutrophil cytoplasmic antibody; RF A, rheumatoid factor A; RNP, ribonucleoprotein; SLE, systemic lupus erythematosus; Sm, Smith; SS, Sjögren’s syndrome; SSc, systemic scleroderma; UC, ulcerative colitis.
(Adapted from Okano Y. Antinuclear antibody in systemic sclerosis [scleroderma]. Rheum Dis Clin North Am 22:709, 1996; Moder KG. Use and interpretation of rheumatologic tests: A guide for clinicians. Mayo Clin Proc 71:391, 1996; Bylund DJ, McCallum RM. Vasculitis. In: Henry JB, editor: Clinical diagnosis and management by laboratory methods, Philadelphia, 1996, WB Saunders.)
Neurologic manifestations of rheumatic disorders can arise in both primary and secondary fashion [Benseler and Schneider, 2004]. That is, the antibodies or cellular immune elements responsible for the underlying disease can attack and injure directly or can cause the malfunction of nerves, muscle, brain, spinal cord, and sensory organs. On the other hand, innocent bystander effects of such rheumatic disease accompaniments as the hypercoagulable state, inflammation of the blood vessel wall, and immune complex deposition and side effects of medications used in the treatment of rheumatic disease also take their toll on the nervous system. These neurologic signs and symptoms are often multifactorial in origin, and their treatment may involve approaches to the proximate underlying disease and the more distal symptomatic manifestations.
Rheumatic disease underlies a small but tangible fraction of neurologic syndromes of childhood. For example, vasculitis of infectious or rheumatic origin accounts for approximately 4 percent of childhood stroke [Williams et al., 1997], and more than 50 percent of children with arterial ischemic stroke exhibit focal or multifocal cerebral arteriopathy [Dlamini and Kirkham, 2009]. Rheumatic disease should be considered as a possible etiologic factor in neurologic syndromes of childhood when these syndromes are accompanied by persistent fever, weight loss, myalgias, arthralgias, meningeal signs, or multiple nonanatomically contiguous neurologic deficits [Carvalho and Garg, 2002].
Juvenile Idiopathic Arthritis (Chronic Arthropathies)
The key neurologic and laboratory findings in chronic arthropathies are summarized in Table 86-2 [Dannecker and Quartier, 2009].
Table 86-2 Key Neurologic and Laboratory Findings in the Chronic and Reactive Arthropathies
| Disease | Neurologic Findings | Laboratory Findings |
|---|---|---|
| Systemic juvenile idiopathic arthritis | Encephalopathy, seizures, macrophage activation syndrome (Reye-like syndrome), neuropathies | Elevated WBC and ESR, anemia, DIC, elevated CSF protein and cell count, marked increase in ferritin and LDH |
| Inflammatory bowel disease | Myasthenia gravis, myopathy, neuropathy, seizures, cognitive changes | Elevated ESR, microcytic anemia, melena |
| Acute rheumatic fever | Chorea, personality changes, seizures | Positive ASO titers, elevated ESR and CRP, abnormal EKG |
| Lyme disease | Early infection: aseptic meningitis, headache, chorea, cranial nerve palsies, late neuroborreliosis myelitis, MS-like symptoms, subtle encephalopathy, radiculopathy, mononeuritis multiplex | Positive IgG Lyme titer by ELISA, protein by Western blot in serum, positive PCR in CSF |
ASO, antistreptolysin O; CRP, C-reactive protein; CSF, cerebrospinal fluid; DIC, disseminated intravascular coagulation; EKG, electrocardiogram; ELISA, enzyme-linked immunosorbent assay; ESR, erythrocyte sedimentation rate; IgG, immunoglobulin G; LDH, lactate dehydrogenase; MS, multiple sclerosis; PCR, polymerase chain reaction; WBC, white blood cell count.
The neurologic manifestations and sequelae of juvenile idiopathic arthritis (previously described as juvenile rheumatoid arthritis) and its treatment vary greatly with the subtype of arthritis. Children with pauciarticular or polyarticular disease have only rarely been diagnosed with central nervous system (CNS) disease, but approximately 20 percent of those with pauciarticular and 5 percent of those with polyarticular disease develop uveitis [Duzova and Bakkaloglu, 2008]. In contrast, only 1 percent of children with systemic-onset disease develop uveitis [Rosenberg, 2002]. Approximately 6 percent of children with systemic arthritis develop nervous system symptoms that most often involve the CNS.
Neurologic Manifestations
Systemic Juvenile Idiopathic Arthritis
Acute encephalopathy
The most common form of acute encephalopathy in children with systemic juvenile idiopathic arthritis is macrophage activation syndrome. Symptoms include unremitting fever, rheumatoid rash, seizures, encephalopathy, hepatosplenomegaly, and lymphadenopathy, as well as cardiac, pulmonary, and renal failure. Laboratory studies demonstrate varying degrees of cytopenia, low albumin, elevated D-dimer, and elevated ferritin and lactate dehydrogenase. Elevation of liver enzymes and serum triglycerides is common. Acutely, the sedimentation rate may fall [Ravelli, 2002]. Initial reports of this catastrophic complication implicated the use of acetylsalicylic acid, indomethacin, and gold, but the macrophage activation syndrome has occurred after ingestion of many nonsteroidal anti-inflammatory drugs, including sulfasalazine, and in the context of no drug intake [Bray and Singleton, 1994; Avcin et al., 2006]. Immediate treatment with steroids is associated with resolution in most cases. In steroid-resistant cases, cyclosporine A and etanercept have been reported to be effective [Cortis and Insalaco, 2006; Stabile et al., 2006; Makay et al., 2008].
In one series of patients, acute hepatic dysfunction, metabolic alterations (including hyponatremia), intracranial hemorrhage, and acute encephalopathy have been described with and without disseminated intravascular coagulation [Hadchouel et al., 1985]. Clinical features are suggestive of a Reye-like syndrome, although cerebral edema, hyperammonemia, and hepatic microvascular fatty infiltration have not been observed. Elevated cerebrospinal fluid protein and cell count were common. These patients demonstrated generalized electroencephalographic (EEG) slowing, and the neurologic symptoms in 5 of 7 children were attributable to a metabolic encephalopathy associated with hyponatremia. These five children responded well to high-dose corticosteroids. The remaining two children died from disseminated intravascular coagulation [Hadchouel et al., 1985]. In another series of children with systemic juvenile idiopathic arthritis without disseminated intravascular coagulation, an acute encephalopathy associated with generalized and focal seizures, altered states of consciousness, abnormal ictal and interictal EEG, and moderate elevation of cerebrospinal fluid protein and cells has been reported [Lang et al., 1974]. Perivascular infiltrates of inflammatory cells within the brain parenchyma have been documented in several children with acute encephalopathy. In one patient, cerebrospinal fluid immune complexes were associated with parenchymal perivascular mononuclear cell infiltrates, suggesting an autoimmune basis of this complication [O’Connor et al., 1980]. A more recent case report and review [Ueno et al., 2002] points out that acute necrotizing encephalopathy is characterized by multifocal gray-matter lesions in the thalamus, brainstem tegmentum, and cerebellar dentate nucleus during the acute phase, while Reye’s syndrome presents primarily with cerebral edema.
Two reports have been published in which there was an association of true Reye’s syndrome with chronic administration of acetylsalicylic acid in several patients with juvenile idiopathic arthritis, when acetylsalicylic acid was the drug of choice for the treatment of chronic childhood arthritis [Silverman et al., 1983; Young et al., 1984]. No reports of Reye’s syndrome in children with juvenile idiopathic arthritis have emerged since the use of acetylsalicylic acid in children has declined in the United States; furthermore, this complication has not been reported with the use of newer nonsteroidal anti-inflammatory drugs, such as naproxen.
Neuropathies
Motor and sensory neuropathies have been reported in children but are far more common in adults. As such, mononeuritis multiplex, the most common neuropathy in adults with rheumatoid arthritis, is seldom, if ever, present in children. Neuropathologic studies of adult rheumatoid arthritis patients with peripheral neuropathy have demonstrated vasculitis involving both the leptomeninges and the underlying parenchyma, with infarction of adjacent neuronal tissue. The resulting axonal neuropathy may be associated with demyelination caused by vascular occlusion of the vasa nervorum [Peyronnard et al., 1982].
Mood disturbances
Reports of psychologic studies of children with juvenile idiopathic arthritis remain uncertain about both the relative frequency of psychologic disorders and the relation of age and severity of arthritis to psychologic symptoms [Baildam et al., 1995; Ungerer et al., 1988]. Premorbid family dynamics – specifically, maternal personality – may contribute to subsequent psychologic disorders in the child with juvenile idiopathic arthritis [Vandvik and Eckblad, 1991]. Depression and difficulty with concentration may be secondary to drug interventions, including nonsteroidal anti-inflammatory drugs and sulfasalazine.
Myositis
Approximately 33 percent of children with juvenile idiopathic arthritis have mild elevations of creatine kinase without weakness [Rachelefsky et al., 1976]. Proximal muscle weakness and biopsy-confirmed myositis are extremely rare, although intermittent myalgias are common, especially in systemic juvenile idiopathic arthritis. Myositis has been localized with magnetic resonance imaging (MRI) in a patient with myalgias and elevated muscle enzymes in systemic juvenile idiopathic arthritis [Miller et al., 1995]. Of note, children with Duchenne’s muscular dystrophy, paraplegia, poliomyelitis, and cerebral palsy may have skeletal changes, such as apparent overgrowth of the epiphysis, periarticular osteoporosis, and joint-space narrowing, similar to juvenile idiopathic arthritis [Richardson et al., 1984].
Pauciarticular Juvenile Idiopathic Arthritis (Oligoarthritis)
Iridocyclitis and uveitis
Although prior literature suggests that iridocyclitis and uveitis are more common in children with oligoarthritis than in children with polyarthritis, newer studies found equivalent incidence in the two groups [Ravelli et al., 2005; Saurenmann et al., 2010]. Patients with a positive antinuclear antibody have a higher incidence than those with a negative antinuclear antibody. In girls, but not in boys, incidence increases with age and with duration of clinical disease. In antinuclear antibody-positive girls with juvenile idiopathic arthritis diagnosed before age 2 years, the incidence after a mean follow-up time of 6.9 years was 46 percent; the analogous figure for the entire cohort of 1047 children with juvenile idiopathic arthritis was 11.7 percent [Saurenmann et al., 2010].
Psoriatic, Enthesitis-Related, and Undifferentiated Syndromes
Although primarily an intestinal disorder, inflammatory bowel disease may present with arthritis and neurologic symptoms. Published studies indicate that approximately 3 percent of children with inflammatory bowel disease had neurologic involvement during the course of the disease, including one child with myasthenia gravis and others with myopathy, peripheral neuropathy, venous sinus thrombosis, recurrent strokes, myelopathy, cranial neuropathy, seizures, headache, confusional states, meningitis, and syncope [Bridger et al., 1997; del Rosario et al., 1994; Lossos et al., 1995]. A syndrome has been described that includes ileocolonic lymphoid nodular hyperplasia, mild enterocolitis, and developmental delay with autistic features [Wakefield et al., 1998]. Although initial reports and reviews suggested that this disorder is associated with detection of measles virus in the intestinal mucosa of these children [Martin et al., 2002], causality has not been demonstrated [Murch et al., 2004] and subsequent studies make this association highly unlikely [Hornig et al., 2008]. Other than inflammatory bowel disease, the enthesitis-related syndromes have not been described with neurologic findings.
Between 5 and 6 percent of patients with inflammatory bowel disease, psoriatic arthritis, and other enthesitis-related arthritides develop inflammation of the uveal tract [Girardin et al., 2007]. These patients may experience acute episodes of uveitis with eye redness, pain, photophobia, and blurred vision. Prompt attention is required, and treatment with topical ophthalmic corticosteroids usually clears the inflammation. More recent studies and speculation have focused on the inverse situation: that is, the modulation of gastrointestinal inflammation by the nervous system and its chemical messengers [Anton and Shanahan, 1998; Murch, 1998]. It has become clear that both macrophages and lymphocytes bear receptors for various neuropeptides and CNS-relevant cytokines, growth factors, and hormone-regulating factors. Furthermore, substance P has emerged as an important mediator, not only of sensory signaling but also of mucosal inflammation. Finally, the absence of significant pain in patients with severe inflammatory bowel disease and consequent mucosal erosion has been linked to altered cortical localization of pain perception, as determined by positron emission tomography [Anton and Shanahan, 1998].
Neuropathology
Neuropathologic studies of children with systemic-onset juvenile idiopathic arthritis who have had neurologic complications are rare. Investigations of adults with classic rheumatoid arthritis complicated by CNS disease document the presence of rheumatoid nodules in the cranial and spinal dura, falces, leptomeninges, parenchyma, and choroid plexus [Kim, 1980]. Because adult-type (i.e., polyarticular) rheumatoid arthritis is relatively uncommon in children, these manifestations have not been reported but may occur in older children with seropositive (rheumatoid factor-positive) juvenile idiopathic arthritis who have the same disease as their adult counterparts.
Management
The treatment of juvenile idiopathic arthritis is dictated by the subtype of the disease. Pauciarticular juvenile idiopathic arthritis is usually managed with either nonsteroidal anti-inflammatory drugs, such as naproxen and tolmetin sodium, or steroid intra-articular joint injection. Iridocyclitis usually responds successfully to topical ophthalmic corticosteroids. Polyarticular juvenile idiopathic arthritis is initially treated with nonsteroidal anti-inflammatory drugs; however, the risk of chronic inflammation of multiple joints may require the use of second-line agents, such as methotrexate. If methotrexate fails, a drug blocking tumor necrosis factor-α, such as etanercept or infliximab, is used. Systemic juvenile idiopathic arthritis may be managed with nonsteroidal anti-inflammatory drugs alone but often requires steroids for control of systemic symptoms. Second-line drugs, such as methotrexate, are often needed to control arthritis [Cassidy and Petty, 2001]. Once again, etanercept or infliximab is used for disease poorly responsive to prednisone and methotrexate [Quartier et al., 2003]. High-dose methylprednisolone, intravenous immunoglobulin, and cyclophosphamide have been used in recalcitrant disease [Shaikov et al., 1992; Silverman et al., 1994; Uziel et al., 1996]. Anti-tumor necrosis factor-α therapies, paradoxically, have been associated with worsening or even emergence of new cases of multiple sclerosis [Sicotte and Voskuhl, 2001].
Because of the chronic nature of juvenile idiopathic arthritis, some children have psychological problems, including depression, anger, adjustment disorders, and troubles with peer and family relations. Counseling of the patient and family is beneficial [Baildam et al., 1995; Quirk and Young, 1990]. In addition, the importance of physical and occupational therapy cannot be overemphasized [Rhodes, 1991].
Periodic Fever Syndromes
Neonatal-Onset Multisystem Inflammatory Disease or Chronic Infantile Neurologic Cutaneous and Articular Syndrome
Neonatal-onset multisystem inflammatory disease (NOMID) or chronic infantile neurologic cutaneous and articular (CINCA) Syndrome is an unusual disorder that mimics systemic juvenile idiopathic arthritis. It has its onset during the first year of life, whereas systemic-onset juvenile idiopathic arthritis is a disease of toddlers and children. Clinical manifestations include hectic fever, intermittent rash, lymphadenopathy, hepatosplenomegaly, uveitis, cognitive and developmental delay, chronic meningitis, hydrocephalus, seizures, papilledema, and deforming arthropathy with periosteal changes and bony overgrowth [De Cunto et al., 1997]. Almost all patients have chronic meningitis from infancy and hydrocephalus, and ventriculomegaly can present in utero. Seventy-five percent of patients with CINCA develop progressive sensorineural hearing loss and many progress to deafness; ocular complications occur at an average age of 4 years and include optic nerve changes, uveitis, and corneal stromal keratopathy [Montealegre Sanchez and Hashkes, 2009]. Other neurological sequelae include seizures, abnormal interictal EEG, transient hemiplegia, cerebral atrophy, and an open fontanel. Long-term prognosis is poor [Prieur, 2000].
Familial Mediterranean Fever
Familial Mediterranean fever is inherited as an autosomal-recessive trait as a result of mutations in the MEFV gene. Over 180 individual mutations have been described. Non-neurological symptoms include fever, abdominal pain, peritonitis, pleuritis, and arthritis [Ozdemir et al., 2010]. Myalgias after exertion are common. Headache, febrile seizures, aseptic meningitis, and posterior reversible leukoencephalopathy have been described, as has progressive sensorineural deafness [Montealegre Sanchez and Hashkes, 2009; Ulasli et al., 2010].
Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis Syndrome
Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome has an unclear etiology; it is characterized by febrile episodes persisting for 4 –6 days, separated by afebrile periods lasting 4 weeks to 4 months. Headache can occur during febrile episodes. Recurrent aseptic meningitis accompanied by a generalized tonic clonic seizure has been described in a patient with PFAPA syndrome [Frye, 2006].
Hyper-IgG (Autoimmune Lymphoproliferative) Syndrome
This genetic syndrome is thought to result from abnormal regulation of apoptosis of mature lymphocytes. Proliferation of lymphocytes can present with splenomegaly, skin rashes, enlarged lymph nodes, and autoimmune hemolytic anemia. Neurological symptoms connected with this syndrome are the result of its association with other autoimmune phenomena, including Guillain–Barré syndrome [Sneller et al., 2003].
Arthritis Associated with Infectious Agents
Acute Rheumatic Fever
Acute rheumatic fever is an inflammatory illness that follows group A beta-hemolytic streptococcal pharyngitis. The syndrome affects the heart valves, joints, CNS, skin, and subcutaneous tissues. Common clinical manifestations include migratory polyarthritis, fever, carditis, and, less frequently, Sydenham’s chorea, subcutaneous nodules, and erythema marginatum. The modified Jones criteria, which were last revised in 1992, are used to confirm the diagnosis of acute rheumatic fever and call for the presence of a combination of two major and one minor criteria, or one major and two minor criteria, as well as antibody evidence of preceding streptococcal infection (Box 86-2). In the case of isolated Sydenham’s chorea, demonstration of preceding streptococcal infection may not always be possible, and in these cases is not a requirement for the diagnosis of acute rheumatic fever [Dajani et al., 1992].
Box 86-2 Jones Criteria for Diagnosis of Acute Rheumatic Fever (Revised 1992)









* Prior episodes of acute rheumatic fever are not criteria; if a patient has had a prior attack of acute rheumatic fever, a new attack may be difficult to diagnose on the basis of changing carditis. In this setting, proof of recent streptococcal infection and either one major or one minor criterion may allow a presumptive diagnosis. Proof of recent streptococcal infection is necessary, except for isolated chorea.
(Adapted from Dajani, et al. and The Special Writing Group. Guidelines for the diagnosis of rheumatic fever. Jones Criteria, 1992 update, JAMA 268:2069, 1992.)
Neurological Manifestations
Sydenham’s chorea
Clinical Manifestations
Involuntary, distal, purposeless, rapid movements; hypotonia; weakness; and emotional lability characterize Sydenham’s chorea. It may be associated with other manifestations of rheumatic fever, or “pure” chorea may appear as the sole manifestation of the disease. Isolated chorea represents 20–30 percent of acute rheumatic fever cases and occurs long after the pharyngitis has resolved, which makes the association with streptococcal infection difficult to demonstrate. Indeed, laboratory evidence of preceding streptococcal infection could not be demonstrated in 35 percent of children with Sydenham’s chorea [Ayoub and Wannamaker, 1966]. Although the onset may be explosive, Sydenham’s chorea may develop slowly and insidiously. Chorea may be misdiagnosed as an emotional disorder or as tics with irritability and decreased attention span. Chorea may also be confused with a CNS degenerative process, but this is more common in adults than in young children.
In adolescents, chorea occurs almost exclusively in females and may, on rare occasions, be associated with hemichorea or hemiparesis. The hemiparesis, which may be the initial manifestation of the disorder, has an unusual form of flaccidity combined with hypotonia and slow relaxation of deep tendon reflexes. Choreiform movements are abrupt and erratic without being rhythmic or repetitive, and usually subside during sleep. Face, hands, and feet are most commonly affected; facial movements include grimacing, frowning, grinning, and pouting. Children commonly are unable to sustain prolonged hand contraction, resulting in the “milkmaid” sign. In other patients, emotional lability, personality changes, restlessness, hyperactivity, irritability, and episodes of anger and tearfulness may herald the onset of chorea. Occasionally, typical “spooning” is observed with hyperextension of the hands [Stollerman, 1985]. Choreiform movements usually subside in 2–4 months but may persist for 1 year or more. Chorea and arthritis do not usually accompany each other in acute rheumatic fever; however, carditis frequently develops as the chorea is improving.
Laboratory Findings
Laboratory studies include serologic documentation of antecedent streptococcal infection with an antistreptolysin-O titer, demonstration of a prolonged PR interval on electrocardiogram, elevated C-reactive protein or erythrocyte sedimentation rate, and leukocytosis. Although throat culture may show group A beta-hemolytic streptococci, an elevated or increasing antistreptolysin-O titer is required. An elevated anti-DNase-B increases the sensitivity of an antistreptolysin-O titer alone from 80 to 95 percent and may be necessary, especially in isolated chorea, in which sensitivity may only be 65 percent. To exclude other conditions manifesting with chorea, patients may need additional diagnostic studies, including serum ceruloplasmin, thyroxine, calcium, and antinuclear antibody titers. In patients with Sydenham’s chorea, EEGs may demonstrate diffuse paroxysmal features and generalized or posterior slowing [Ganji et al., 1988]. Cerebrospinal fluid examinations and neuroimaging are rarely necessary. Of interest is the finding of antineuronal antibodies in the cerebrospinal fluid of patients with chorea [Swedo, 1994]. The specificity of these antibodies is not well known, and they are frequently documented in patients with CNS lupus [Bluestein, 1997].
A 3-year-old child with chorea was reported as having the first example of a persistently abnormal MRI, showing a cystic abnormality in the caudate and putamen [Emery and Vieco, 1997]. Subsequent longitudinal radiologic studies of patients with Sydenham’s chorea demonstrated that, although the majority of patients with this disorder have a normal brain MRI, those with abnormalities during the symptomatic period often continue to demonstrate these same abnormalities when the disease is clinically quiescent or resolved [Faustino et al., 2003]. Most patients with MRI abnormalities demonstrate abnormal signal intensity or cystic changes in the caudate nuclei; subcortical foci and multiple peripheral white matter foci of abnormal signal have also been reported [Emery and Vieco, 1997; Faustino et al., 2003; Robertson and Smith, 2002].
Neuropathology
Neuropathologic findings in acute rheumatic fever are rare. Rheumatic proliferative endarteritis is limited to the small cortical and meningeal vessels, with spotty patches of gray-matter degeneration [Halbreich et al., 1976].
Treatment
All patients with acute rheumatic fever, including those whose only manifestation is chorea, should receive a 10-day course of penicillin or erythromycin. Prophylaxis with penicillin or sulfadiazine should be started immediately and continued at least until adulthood because of frequent reinfection and the risk of rheumatic heart disease with subsequent streptococcal pharyngitis. More specifically, when residual valvular disease exists, prophylaxis should continue for at least 10 years after the last episode and at least until age 40. If there is no residual valvular disease, the duration of treatment beyond 10 years or into adulthood is not clearly defined. When Sydenham’s chorea is diagnosed and there is no valvular disease, the duration of prophylaxis should be at least 5 years or until age 21, whichever is longer [Dajani et al., 1995].
Children who develop chorea as the sole manifestation of acute rheumatic fever during the initial episode of illness have an approximate 50 percent risk of developing rheumatic heart disease with subsequent infection [Aron et al., 1965]. In addition, studies have suggested that, in certain chorea-prone patients, recurrence of Sydenham’s chorea may follow either undetectable streptococcal infection or another infectious trigger [Berrios et al., 1985].
Sydenham’s chorea has been treated successfully with chlorpromazine, haloperidol [Shenker et al., 1973], phenobarbital, diazepam, valproic acid [Daoud et al., 1990], and corticosteroids [Green, 1978]. In mild cases, cyproheptadine may be effective. Other agents, such as clonidine, pimozide [Shannon and Fenichel, 1990], and corticosteroids [Barash et al., 2005], may be beneficial in refractory cases. Complete recovery can be expected within 2–6 months, although some children may have residual motor, visuomotor, or cognitive dysfunction and a variety of neuropsychiatric manifestations [Bird et al., 1976; Faustino et al., 2003; Leonard et al., 1993; Stehbens and MacQueen, 1972; Swedo et al., 1989].
In some children with Sydenham’s chorea, atlantoaxial subluxation may occur during the acute episode of chorea, with symptoms of neck stiffness, decreased mobility, and pain [Coster and Cole, 1990]. In such patients, differentiating this complication from juvenile rheumatoid arthritis may require further clinical and laboratory evaluation and specific orthopedic intervention. In rare instances, recurrences of chorea may occur in patients without evidence of rheumatic cardiac involvement decades after the childhood onset of symptoms [Gibb and Lees, 1989].
Post-Infectious Tourette’s Syndrome and PANDAS
Chorea, obsessive-compulsive disorder, tic disorder, and Tourette’s syndrome may all have a common autoimmune pathway. It has been reported that some children with chorea have obsessive-compulsive disorder, and that the obsessive-compulsive disorder resolves before or simultaneously with the resolution of the chorea [Swedo et al., 1993, 1998]. Additionally, an increased prevalence of obsessive-compulsive disorder has been noted in children with tics and Tourette’s syndrome. An increased prevalence of antineuronal antibodies, as well as increased levels of antistreptococcal antibodies, has been reported in all four of these diseases. It has been shown that, in patients with chorea, obsessive-compulsive disorder, or tic disorder, there is an increased incidence of the histocompatability locus antigen marker D8/17, which has been reported more frequently in rheumatic fever patients [Allen et al., 1995; Murphy et al., 1997; Swedo, 1994; Swedo et al., 1997]. The acronym PANDAS (pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections) has been suggested for some of these conditions in which there is a combination of behavioral problems, obsessive-compulsive behavior, and tics when associated with an antecedent group A beta-hemolytic streptococcal infection [Garvey et al., 1998]. Some studies refute the connection between group A beta-hemolytic streptococcal infection and obsessive-compulsive disorder with tics, citing evidence that, although children with Sydenham’s chorea have behavioral difficulties, they do not have an increased incidence of obsessive-compulsive disorder [Faustino et al., 2003]. In addition, a more recent series of studies compared antibody profiles in patients with obsessive-compulsive disorder alone, obsessive-compulsive disorders with PANDAS, or obsessive-compulsive disorder with chronic tic disorder, respectively, and PANDAS, Tourette’s syndrome, and controls, respectively, and found no difference among them in either anti-brain antibodies or anti-streptolysin O antibody titers [Morris et al., 2009; Gause et al., 2009].
Other Central Nervous System Manifestations
Rarely, acute rheumatic fever may be accompanied by other neurologic problems, such as meningoencephalitis, encephalitis [Benda, 1948], seizures [Goldenberg et al., 1992], pseudotumor cerebri [Mitkov, 1961], papilledema [Chun et al., 1961], diplopia [Schieken et al., 1973], central retinal occlusion [Ling et al., 1969], transient intellectual loss [Gatti and Rosenheim, 1969], and acute psychosis [Wertheimer, 1961]. Combined, these complications occur in 3–5 percent of all patients.
Lyme Disease
Lyme disease is an important cause of neurologic symptoms in children. The illness follows a tick bite and occurs in endemic areas during the summer months. The clinical course of Lyme disease is marked by stages similar to the course of syphilis, another spirochetal infection. The early stage begins with the tick bite and includes a flulike illness and the appearance of an oval, expanding rash at the site of the tick bite. At this stage, systemic infection with Borrelia burgdorferi may be documented by culture. Within several weeks, the patient may develop early neurologic manifestations, which most commonly include facial nerve palsy and aseptic meningitis, but can also include other cranial neuropathies or transverse myelitis and can represent direct invasion of the organism into cerebrospinal fluid [Huisman et al., 1999]. Acute sinovenous thrombosis with consequent pseudotumor cerebri has been reported, as well [Ansari et al., 2002]. The illness resolves spontaneously, but the resolution may be hastened by antibiotic treatment (amoxicillin or erythromycin in children younger than 9 years old and tetracycline in children age 9 years or older for 10–30 days).
Weeks to months later, the patient who was not treated with antibiotics may develop episodic arthritis of the large joints, primarily the knee. Characteristically, the knee becomes acutely effused, but not particularly tender or hot. The often dramatic joint swelling lasts for several days and resolves but returns multiple times if treatment with antibiotics is not started. The diagnosis may be confirmed after the first few weeks with a positive serum IgG titer against B. burgdorferi. Western blotting may be used to confirm the diagnosis if the titer is equivocal [Steere, 1989]. Polymerase chain reaction amplification of the B. burgdorferi genome in the cerebrospinal fluid is available, as well [Ansari et al., 2002].
Years after the acute infection, some patients develop late neuroborreliosis. In children, this rare complication may manifest as a subtle encephalopathy with stuttering and memory disturbances. Adults may develop a multiple sclerosis-like illness, optic neuritis, seizures, and chronic meningitis. The diagnosis of late neuroborreliosis is confirmed by the demonstration of an elevated intrathecal IgG titer compared with serum titer. Treatment with intravenous ceftriaxone for 1 month to penetrate the blood–brain barrier is indicated, but the response is variable. There is no evidence that treatment lasting longer than 4 weeks is of additional benefit [Bingham et al., 1995; Logigian et al., 1990; Szer et al., 1991].
Reiter’s Syndrome
Reiter’s syndrome is one of several spondyloarthopathies. It is characterized by arthritis, uveitis, and urethritis. Progressive myelopathy [Kim et al., 2007], cerebral vasculitis [Niederwieser et al., 2001], axonal polyneuropathy [Cuchacovich et al., 1991], and seizures [Fishel et al., 1995] have been reported as rare neurological complications in adults with Reiter’s syndrome.
Connective Tissue Disorders
The key neurologic and laboratory findings in connective tissue disorders are summarized in Table 86-3 [Tan, 1986].
Table 86-3 Key Neurologic and Laboratory Findings in Connective Tissue Diseases
| Disease | Neurologic Findings | Laboratory Findings |
|---|---|---|
| SLE | Encephalopathy, chorea, seizures, aseptic meningitis, psychosis, behavioral or cognitive dysfunction, headaches, strokes, neuropathy, myelitis | Elevated ANA, low C3 and C4, pancytopenia, hematuria, proteinuria, autoantibodies, LAC, elevated aCL |
| Scleroderma: coup-de-sabre deformity | Seizures, blurred vision, bulbar palsy, optic neuritis, trigeminal neuropathy | Elevated ANA and rheumatoid factor |
| Mixed connective tissue disease | Same as SLE | Same as SLE plus elevated anti-RNP, elevated CK |
| Sjögren’s syndrome | Encephalopathy, optic neuritis, aseptic meningitis, recurrent paresis, myelopathy, neuropathy, autonomic dysfunction | Positive ANA, rheumatoid factor, antibodies to SSA/Ro and SSB/La |
aCL, anticardiolipin antibody; ANA, antinuclear antibody; C3, third component of complement; C4, fourth component of complement; CK, creatine kinase; LAC, lupus anticoagulant; RNP, ribonucleoprotein; SLE, systemic lupus erythematosus.
Systemic Lupus Erythematosus
Revised criteria for the classification of SLE, developed by the American College of Rheumatology (Box 86-3), call for the presence of at least 4 of 11 specific criteria, including a positive test for antinuclear antibody. The antinuclear antibody alone is not sufficient to establish the diagnosis. It must be associated with multiorgan involvement. Arthritis, arthralgia, fever, and photosensitive rash are the most common initial complaints, with renal, cardiac, and neurologic involvement responsible for chronic disability. Lymphadenopathy, hepatosplenomegaly, pleural and pericardial effusions, pulmonary infiltrates, pericarditis, abdominal pain, and even peritonitis may be present at the time of initial evaluation [Cassidy and Petty, 2001; Szer and Jacobs, 1992].
Box 86-3 Revised Criteria for the Classification of Systemic Lupus Erythematosus (1982)











aCL, anticardiolipin antibody; ds-DNA, double-stranded DNA; LAC, lupus anticoagulant; Sm, Smith.
(Adapted from Tan EM, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271, 1982; Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725, 1997.)
The prognosis for children with SLE has improved dramatically, with an estimated 10-year survival of 85 percent [Cassidy and Petty, 2001; Szer and Jacobs, 1992; Takei et al., 1997].
Neurologic Manifestations
Nervous system involvement in SLE accounts for some of the most frequent manifestations of the disease during childhood [Hiraki et al., 2008; Muscal and Brey, 2010]. Even in SLE patients without overt neuropsychological symptoms, abnormalities have been reported in quantitative EEG studies [Ritchlin et al., 1992]. Early reports suggested that CNS involvement was the second most common cause of death in children with SLE. More recently, the outcome of children with CNS lupus appears favorable, with most showing recovery [Hiraki et al., 2008].
Seizures
Between 20 and 50 percent of children with SLE develop generalized and occasionally focal motor seizures [Parikh et al., 1995; Steinlin et al., 1995; Vieira-Karuta et al., 2008]. Seizures usually occur during the first year of illness and may be the initial manifestation of SLE, but they may occur at any stage. Interictal EEG may show multifocal paroxysmal sharp-wave or slow-wave activity, particularly in the temporal lobes and particularly in patients with recurrent seizures. Patients who present with a single seizure may have a normal interictal EEG [Appenzeller et al., 2004].
Neuropsychiatric lupus
Although the diagnostic criteria for SLE (see Box 86-3; reviewed in Ruiz-Irastorza et al. [2001]) include only two neuropsychiatric syndromes (seizure and psychosis), numerous neuropsychiatric manifestations have been associated with SLE. The prevalence of these complications in children with SLE ranges from 22 to 95 percent, depending on the report [Muscal and Brey, 2010]. This wide range in reported prevalence rates primarily reflects the use of different diagnostic criteria. Nineteen neuropsychiatric SLE (NPSLE) syndromes based on specific case definitions have been defined for adults [American College of Rheumatology Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature, 1999; summarized in Muscal and Brey, 2010]. Initial studies that extended these definitions to children and adolescents indicated that neurological manifestations include headache in 72 percent, mood disorder in 57 percent, cognitive dysfunction in 55 percent, seizures in 51 percent, acute confusional state in 35 percent, peripheral nervous system dysfunction in 15 percent, psychosis in 12 percent, and stroke in 12 percent [Sibbitt et al., 2002]. Neuropsychiatric manifestations of SLE are generally classified as either primary – related to direct involvement of the neuropsychiatric system; or secondary – related to complications of the disease and its treatment. The latter includes infections, metabolic repercussions of organ failure (e.g., uremia), and drug-induced toxicity, such as hypertension associated with glucocorticoid use. A more recent study of 256 children with SLE confirmed the higher frequency with which they develop neuropsychiatric symptoms relative to adults with SLE, the association of renal disease in SLE with secondary neurological complications, and, despite this, the excellent (97 percent) overall survival of children with NPSLE [Hiraki et al., 2008].
In about half of the SLE patients with CNS involvement, the neuropsychiatric pathologies are already present at disease onset. Studies have shown that, in about 70 percent of affected children, the CNS manifestations occur within the first year of diagnosis; in the remaining 30 percent, CNS manifestations may not develop for up to 13 years [Harel et al., 2006]. Others have reported a median disease duration of 11 months [Yu et al., 2006] and a mean of 2.5 ± 5.2 years [Fragoso-Loyo and Sanchez-Guerrero, 2007] prior to the development of neuropsychiatric symptoms. About 30–70 percent of affected children have more than one neuropsychiatric symptom [Steinlin et al., 1995]. Onset may be acute or indolent. In some children, neuropsychiatric manifestations appear to be the only presenting symptom of SLE. However, it remains controversial whether neuropsychiatric manifestations can truly appear as an isolated event [Toubi et al., 1995; Harel et al., 2006], or whether there is always some involvement of other organs [Weiner and Allen, 1991]. Numerous autoantibodies have been seen in association with NPSLE, including antineuronal antibodies, antiphospholipid antibodies, anti-NR2 N-methyl-d-aspartate (NMDA) receptor antibody, and antiribosomal P antibody [Levy et al., 2009]. Anti-NR2 subunit NMDA receptor antibodies are found more frequently in SLE patients and their healthy first-degree relatives than in healthy unrelated controls, but there is no difference between SLE patients with and without NPSLE, respectively, or between SLE patients and their healthy first-degree relatives, respectively. This suggests that, while the familial propensity to have antibodies to the NMDA receptor NR2 subunit is associated with the propensity for autoimmune diseases like SLE, there is no association between these antibodies and the neuropsychiatric pathology seen in SLE [Steup-Beekman et al., 2007]. Diagnostic testing for NPSLE is controversial, as traditional imaging techniques, such as MRI, detect only a subset of abnormalities in these patients. Use of neuropsychiatric testing to diagnose neurocognitive impairment is increasingly being used in this population, but remains a challenging area.
Headache
Headaches are common in children and adolescents with SLE [Benseler and Silverman, 2007; Muscal and Brey, 2010]. Headache usually occurs during exacerbation of systemic symptoms and frequently is associated with other neurologic symptoms. Unless objective neurologic symptoms are present, the diagnostic work-up may yield few abnormalities, although evaluation for a hypertensive encephalopathy should be considered [Parikh et al., 1995; Steinlin et al., 1995]. Most patients will respond to increased dosages of corticosteroids.
In two prospective studies of adult patients with SLE, it was found that vascular headache developed in 26 percent of adult patients, and muscle contraction headache in 40 percent [Vazquez-Cruz et al., 1990; Bicakci et al., 2008]. Although a relationship between migraine and SLE has been hypothesized to be the result of cerebrovascular endothelial dysfunction in vasculitis, a recent study failed to demonstrate evidence of such dysfunction [Davey et al., 2010].
Chorea
Chorea occurs in perhaps 5 percent of children with SLE and is the initial symptom in 25–30 percent of patients who present with neurologic symptoms [Herd et al., 1978]. Systemic lupus manifestations often occur within 1 year of onset; rarely, a prolonged latent interval may ensue after the onset of chorea [Parikh et al., 1995; Steinlin et al., 1995]. Approximately 50 percent of children with lupus chorea develop other CNS manifestations, including seizures and neuropsychiatric disturbances. Chorea has also been associated with thromboembolic disease and elevated anticardiolipin antibody [Besbas et al., 1994].
Reye-like syndrome
A Reye-like syndrome associated with acetylsalicylic acid treatment of SLE has been recognized [Hansen et al., 1985]. Salicylate hepatocellular injury appears to be relatively common in patients with rheumatic disorders, but its relation to Reye’s syndrome has raised the possibility of some other etiologic relation between salicylate therapy, various rheumatic disorders, and idiopathic Reye’s syndrome. This syndrome has not been seen with other nonsteroidal anti-inflammatory agents.
Cerebrovascular disease
Although it is unusual for a cerebrovascular syndrome to be the initial manifestation of SLE, approximately 3 percent of children develop cerebrovascular occlusive disease that results in hemiplegia, aphasia, or sensory and visual impairments [Parikh et al., 1995; Steinlin et al., 1995]. Most of these patients have serious renal, cardiac, pulmonary, or hematologic disease with hypertension or thrombocytopenia. Even if there is recurrent hemiplegia, significant recovery may occur. Antiphospholipid antibody has been associated with thrombosis in SLE, especially when lupus anticoagulant is present [Levy et al., 2003]. Microembolic signals in cerebral vasculature have been observed by transcranial Doppler in patients with lupus; patients with antiphospholipid antibodies are more likely to exhibit this phenomenon [Baizabal-Carvallo and Samson, 2009]. Alternatively, thrombotic or thromboembolic disease may be due to a vasculopathy with or without autoimmunity, atheromatous disease, valvular disease, or vasculitis [Bruyn, 1995; West, 1994]. In adults, it has been reported that elevated homocysteine levels increase the risk of atherothrombotic events [Petri et al., 1996].
One case report indicates that perivenous inflammation with secondary calcification may occur in SLE. This postmortem finding was preceded by computed tomography (CT) and MRI evidence of foci of breakdown of the blood–brain barrier, calcifications, and a clinical picture consistent with focal vascular dysfunction and diffuse encephalopathy [Matsumoto et al., 1998].
Multifocal cerebral dysfunction with patchy areas of altered signal intensity on MRI has been interpreted as acute vascular lesions associated with pulse steroid therapy in patients with SLE. This syndrome responds well to substitution of cyclophosphamide for pulse methylprednisolone, and, as such, it is imperative that it be promptly recognized [Tabata et al., 2002].
Hypertensive encephalopathy
Headache, seizures, coma, and focal ischemic CNS injury have been reported as manifestations of hypertensive encephalopathy associated with lupus nephritis [Cassidy et al., 1977]. Recent reports demonstrate in adolescent and adult lupus patients the classic posterior reversible leukoencephalopathy associated with hypertension of a variety of etiologies. Controlling blood pressure and treatment with corticosteroids frequently result in substantial neurologic improvement [Muscal et al., 2010; Bag et al., 2010].
Cranial nerve, brainstem, and spinal cord dysfunction
Ophthalmoplegia, ptosis, diplopia, facial numbness, vertigo, sensorineural hearing loss [Hisashi et al., 1993], vocal cord paralysis [Teitel et al., 1992], and ataxia have been described in children with SLE. Brainstem involvement is usually observed in conjunction with other nervous system symptoms, such as chorea and seizures [Gold and Yahr, 1960].
Approximately 6 percent of children with SLE manifest visual symptoms, which include blurred vision, sudden blindness, and field loss [Brandt et al., 1975]. Retinal hemorrhages, cotton wool exudates, papilledema, optic neuritis, and cytoid bodies have all been reported [Cassidy et al., 1977; Hackett et al., 1974]. Retinal artery occlusion may also occur, with resulting transient or permanent visual loss. Papilledema associated with pseudotumor cerebri caused by either the disease or corticosteroid therapy also has been reported [Green et al., 1995]. If papilledema is present, the possible presence of increased pressure or a mass lesion should be assessed before lumbar puncture is performed [Brandt et al., 1975; Carlow and Glaser, 1974]. Patients with either retinal artery occlusion or papilledema should be treated with high doses of corticosteroids after elimination of structural or occlusive cerebrovascular disease.
Transverse myelopathy that causes both paraplegia and sensory loss as the initial or late neurologic manifestations of SLE has been reported [Andrianakos et al., 1975; Meislin and Rothfield, 1968]. Other studies in adults have shown an association with antiphospholipid antibodies [Kovacs et al., 1993]. In addition, Devic’s disease with neuromyelitis optica (NMO) antibody seropositivity has been reported in patients with lupus [Karim and Majithia, 2009; Nasir et al., 2009]. Early treatment with high doses of corticosteroids, alone or in combination with cyclophosphamide or rituximab, has been used, but improvement was variable [Propper and Bucknall, 1989; Boumpas et al., 1990; Berlanga et al., 1992; Chan and Boey, 1996; Karim and Majithia, 2009; Nasir et al., 2009].
Central nervous system infections
Infection of the CNS in children with SLE is relatively rare [Cassidy and Petty, 2001; Fish et al., 1977; Walravens and Chase, 1976]. Bacterial meningitis, opportunistic bacterial infection, and fungal meningitis (aspergillosis, nocardiosis, and cryptococcosis) have been reported. Brain abscess may be difficult to differentiate from a multifocal vasculitis, but differentiation may be facilitated by using serial CT scans and angiography. Multiple abscesses of the CNS may be relatively silent; if they are suspected, broad-spectrum antimicrobial or antifungal therapy should be instituted after appropriate cultures are obtained.
Lupus aseptic meningitis
The syndrome of lupus aseptic meningitis, accompanied by a sterile cerebrospinal fluid lymphocytic pleocytosis, may be manifested by nuchal rigidity, fever, and headache, and may occur early in childhood lupus. This syndrome has been reported in association with nonsteroidal anti-inflammatory drugs and trimethoprim-sulfamethoxazole use [Escalante and Stimmler, 1992]. Clinical manifestations and cerebrospinal fluid abnormalities may persist for several weeks before resolving spontaneously. Therapy with corticosteroids may improve this condition, but the data are inconclusive because of the self-limited course [Canoso and Cohen, 1975; Keefe et al., 1974].
Peripheral nervous system involvement
Involvement of the peripheral nervous system occurs in approximately 5 percent of children with SLE. Peripheral neuropathies, with symptoms consisting of paresthesias, numbness, and distal weakness, are usually relatively mild in severity and course, although severe forms of acute lumbosacral plexopathies have been reported [Bailey et al., 1956; Jacob, 1963]. Neuropathy manifesting as either mononeuritis multiplex or acute demyelinating polyneuropathy may occur at any time during SLE, may recur, and generally worsens when CNS involvement is greater. Polyradiculoneuropathy may mimic Guillain–Barré syndrome in children; however, this pattern is extremely rare [Norris et al., 1977; Robson et al., 1994].
Myopathy
Myositis is rare in SLE, although myalgias and generalized weakness are common. Myositis can be distinguished from corticosteroid-related myopathy by demonstrating elevated levels of muscle enzymes, electromyography consistent with myopathic and fibrillation activity, and lack of vacuolization in muscle biopsies. One case report demonstrated pyridostigmine-responsive myasthenia gravis complicating childhood SLE [Nishimura et al., 1997]. Although children with myasthenia gravis may have circulating serum antinuclear antibodies, clinical SLE is highly unusual.
Drug-induced lupus syndrome
Many drugs have been reported to induce a lupus-like syndrome in children and adults [Rubin, 1997]. In general, this disorder is milder than spontaneous SLE and occurs with equal frequency in males and females [Totoritis and Rubin, 1985]. Arthritis, pneumonitis, and pericarditis are common, whereas rashes and alopecia are less frequent. Hepatosplenomegaly, lymphadenopathy, and acute pancreatitis may occur in some patients; renal disease appears less often [Rubin, 1997]. Procainamide, hydralazine, and isoniazid are the three drugs that have most commonly induced this syndrome. These drugs share a primary amine or hydrazine portion that is acetylated by the N-acetyl transferase system of the liver in two different phenotypic expressions [Rubin, 1997]. The risk of a drug-induced syndrome appears to be much greater in the “slow” acetylators than in the “fast” acetylators.
Antiepileptic drugs and phenothiazines have also been associated with a drug-induced lupus syndrome. Antiepileptic drugs reported to cause this disorder in children include phenytoin, ethosuximide, carbamazepine, and trimethadione [Rubin, 1997; Singsen et al., 1976]. Although children receiving these drugs are usually asymptomatic, approximately 20 percent of them produce antinuclear antibodies; these children commonly have normal immunoglobulins and serum complement levels, and remain free of a clinical lupus-like syndrome. Antiepileptic medication therefore may be continued [Singsen et al., 1976]. Among patients in whom antiepileptic drugs have been discontinued, the presence of antinuclear antibodies may persist for several years.
Drug-induced SLE has been associated with etanercept therapy in a child with juvenile idiopathic arthritis [Lepore et al., 2003].
Laboratory Findings
Laboratory features of SLE commonly include a positive antinuclear antibody titer, low C3 and C4 levels, leukopenia, direct Coombs-positive hemolytic anemia, hematuria, and proteinuria. Abnormal autoantibodies may include antibody to double-stranded DNA, Sm (Smith), RNP (ribonucleoprotein), Ro (or SSA), La (or SSB), and anticardiolipin. In addition, there may be a paradoxical prolongation of the partial thromboplastin time because of antiphospholipid antibodies. Antibodies to antiphospholipid may also produce a biological false-positive rapid plasma reagin (RPR) or Venereal Disease Research Laboratories (VDRL) test. In adults, a strong correlation between MRI changes and the presence of a positive lupus anticoagulant or anticardiolipin antibody exists, but there is no clear correlation with neuropsychiatric disease [Ishikawa et al., 1994; Manco-Johnson and Nuss, 1995; Molad et al., 1992; Toubi et al., 1995]. Pediatric case reports have also been published showing some correlation between disease and antiphospholipid antibodies, as demonstrated by one of these tests [Steinlin et al., 1995; von Scheven et al., 1996]. Elevated lupus anticoagulant (LAC) appears to confer a significant increased risk of arterial or venous thrombotic events [Galli et al., 2003]. Anticardiolipin IgG fraction and anti-β2-glycoprotein I antibodies may also correlate, but their association is less certain [Galli et al., 2003].
Several mechanisms explaining the pathogenesis of CNS lupus have been reported [Bruyn, 1995]. In most patients with documented CNS lupus, evidence of immune-mediated abnormalities cannot be found, suggesting multifactorial causes of CNS disease. Interestingly, one study found that approximately 75 percent of observed neurologic events were attributed to metabolic, hematologic, or infectious factors rather than to the primary disease process [Kaell et al., 1986]. Serum complement and autoantibody levels may remain normal. The cerebrospinal fluid is often benign, and imaging modalities are of little value, unless there is an ischemic event [Hirohata et al., 1985; Szer and Jacobs, 1992]. However, cerebrospinal fluid examination is often necessary to rule out infectious causes.
In support of a diagnosis of immune disease, studies of cerebrospinal fluid immunoglobulin production have documented elevations of IgG, IgG/albumin ratio, and the cerebrospinal fluid IgG index and the presence of oligoclonal IgG, suggesting accelerated CNS IgG synthesis. Further support for immune-mediated disease may be found in the relatively high incidence of antineuronal and antiribosomal P antibody in some children with SLE [Reichlin, 2003; Silverman, 1996; West et al., 1995]; however, antiribosomal P was not found to be sensitive for neuropsychiatric manifestations of SLE in another series [Press et al., 1996]. In addition, endothelial or vascular injury may be a complement-mediated immunologic insult, because the choroid plexus has been reported as the deposition site of complement and immune complexes; cerebrospinal fluid C4 also appears to be reduced [Hadler et al., 1973; Sher and Pertschuk, 1974]. In addition, cerebrospinal fluid anti-double-stranded DNA complexes and lymphocytotoxic antibodies have been documented in CNS lupus [Bluestein, 1997; Carr et al., 1975]. Serial cerebrospinal fluid C4 complement levels may help to distinguish between neuropsychiatric symptoms caused by corticosteroids and those caused by SLE. A serial decrease in C4 complement level suggests increased disease activity rather than a drug-induced phenomenon. More recent studies demonstrate moderate levels of interleukin-6 in neuropsychiatric SLE. The importance of this finding must be interpreted with caution because significantly high levels of interleukin-6 may be found in CNS infection [Tsai et al., 1994]. At present, cerebrospinal fluid findings cannot reliably confirm the diagnosis of neuropsychiatric symptoms associated with CNS lupus.
False-positive elevation of antistreptococcal antibody titers can occur in lupus chorea and may incorrectly result in the diagnosis of Sydenham’s chorea, unless antinuclear antibody titers are obtained. Serum complement is decreased in lupus chorea, but cerebrospinal fluid complement, anti-double-stranded DNA antibody titers, and immunoglobulin synthesis have not been studied [Kukla et al., 1978]. In adults, a significant correlation exists between chorea and the presence of antiphospholipid antibodies [Asherson et al., 1987]. This association has also been reported in children [Besbas et al., 1994].
In patients with transverse myelopathy, cerebrospinal fluid analysis may demonstrate increased protein concentration, decreased glucose, and a monocytic pleocytosis [Al-Husaini and Jamal, 1985].
Neurodiagnostic Testing
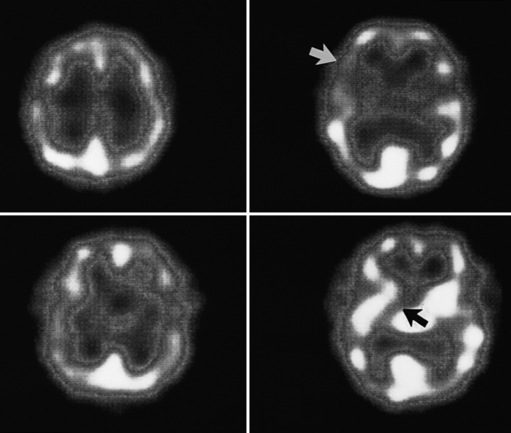
Neuroimaging with either CT or MRI scans is essential for evaluating the patient with SLE who is suspected of having intracranial disease [Carette et al., 1982; Provenzale et al., 1994]. Such studies may show cortical or cerebellar atrophy (Figure 86-1), infarction (Figure 86-2), low-density lesions in the cerebral white matter (Figure 86-3) [Isshi et al., 1994; Muscal et al., 2010], or hemorrhage [Aisen et al., 1985; Kovacs et al., 1993]. MR angiography or venography may detect sinovenous thrombosis [Steinlin et al., 1995]. Cerebral angiography may be helpful in further differentiating arterial thrombotic from embolic disease, but may be normal in children because small vessel arterial changes may not be demonstrable [Jones et al., 1975]. MRI has been used to detect myelopathy in a child as young as 5 years of age [Vieira et al., 2002]. In a meta-analysis of the literature on lupus patients (age 12–53 years) with transverse myelitis affecting four or more spinal segments, MRI demonstrated increased spinal cord T2 signal, most frequently in the cervical to mid-lower thoracic spinal segments [Espinosa et al., 2010]. Single-photon emission computed tomography (SPECT) is a sensitive tool for demonstrating diffuse and multiple perfusion abnormalities in children and adults with CNS events (Figure 86-4). Unfortunately, the usefulness of SPECT is limited. It is not specific for CNS lupus, and longitudinal assessment with this modality correlates poorly with clinical status [Szer et al., 1993]. There is some suggestion that combining SPECT and MRI findings may have some value in identifying and monitoring lupus patients with neuropsychiatric involvement [Castellino et al., 2008].
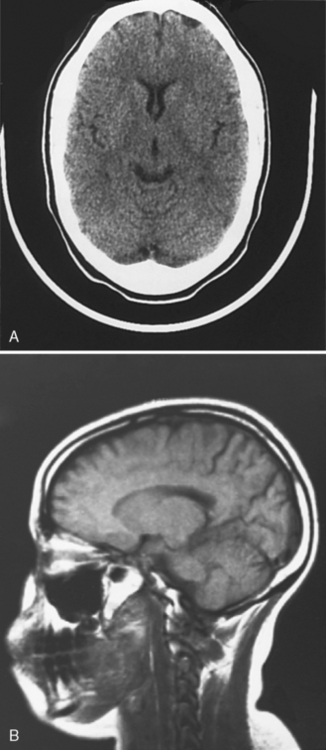
Fig. 86-1 Brain imaging in systemic lupus erythematosus.
(A, Courtesy of Dr. Joseph Thompson, Department of Neuroradiology, Loma Linda University Children’s Hospital. B, Courtesy of Dr. David B Hinshaw, Jr., Department of Neuroradiology, Loma Linda University Children’s Hospital.)
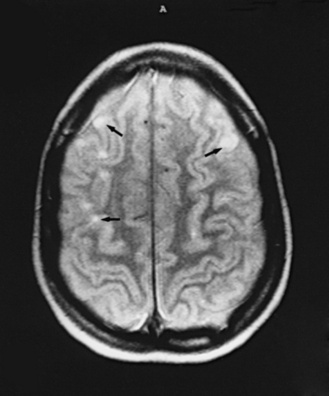
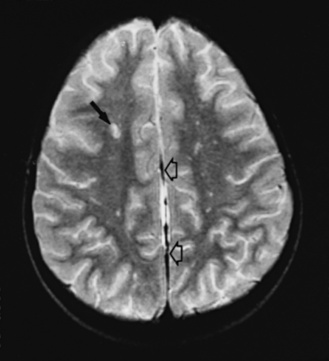
Treatment of Neurologic Manifestations
Treatment of the neurologic complications seen in children with SLE can be categorized as follows.
Treatment of generalized symptoms, where the nervous system is involved as a bystander, requires controlling the underlying inflammatory disorder, correcting metabolic or systemic abnormalities such as hypertension, and administering specific symptom-directed medications, such as antiepileptic drugs for the treatment of seizures; analgesic medications for headache; antidepressants, sedatives or tranquilizers, or antipsychotic agents for specific psychiatric symptoms; and dopamine-blocking agents for the treatment of chorea. Commonly used antiepileptic drugs in children with seizures associated with SLE include phenobarbital, phenytoin, diazepam, lorazepam, valproate, and carbamazepine. Antiepileptic drug use should not be prolonged unnecessarily; discontinuation should be considered when the primary disease is well controlled. In moderate doses, corticosteroids do seem to have beneficial effects in studies of adults with mood or cognitive disorders, suggesting that these are often secondary to neuropsychiatric disease [Carbotte et al., 1995]. However, corticosteroids have also been associated with a variety of behavioral and mood disorders and with psychosis, although the mechanism of this toxic response is unknown. Treatment of lupus chorea with haloperidol, chlorpromazine, valproate, or corticosteroids is usually successful in conjunction with treatment of the underlying disorder.
Anticoagulation may be considered in patients with ischemic cerebrovascular insults who have antiphospholipid antibodies, because there is a significant risk of recurrent thrombotic episodes [Bruyn, 1995; Khamashta et al., 1995]. Agents used in adults have included acetylsalicylic acid, heparin, and warfarin. In adults, prevention of recurrent venous thrombosis may require doses of warfarin with an international normalized ratio of 2 to 3 [Meroni et al., 2003]. It is unclear, even in studies of adults with arterial thromboembolic disease, if anticoagulation is effective, even with a higher international normalized ratio [Brey et al., 2003]. Because of the small number of children with SLE who have such events, it is unlikely that controlled clinical studies will be completed. Given the nature of children’s activities and the reports of hemorrhage in children without SLE but with a lupus-type anticoagulant [Becton and Stine, 1997], it might be suspected that the risk of serious or life-threatening hemorrhage would be greater than in adults. Various alternative regimens have been recommended in children [Ravelli and Martini, 1997; Silverman, 1996]. Steroids have not been found to alter the pathology of antiphospholipid manifestations. It is imperative that patients with hypertension and thrombocytopenia do not receive anticoagulants because of the potential risk of CNS hemorrhage. Patients on long-term steroid therapy for SLE who also require anticoagulation must be monitored for the increased risk of gastrointestinal bleeding. Although strategies to prevent thrombosis occurrence should be part of the management of patients with SLE, to date, no laboratory test can predict recurrent thrombotic events and no pharmacogenomics studies that could help with anticoagulation monitoring have been carried out in SLE patients [Burgos and Alarcon, 2009].
Immunosuppressive therapy with corticosteroids has proved to be the mainstay of treatment for patients with neuropsychiatric symptoms or symptoms associated with vasculitis [Hanmer and Saltissi, 1986; Sanna et al., 2003]. High-dose steroids have also been used for the treatment of coma, seizures, chorea, and transverse myelitis [Chan and Boey, 1996; Eyanson et al., 1980; Harisdangkul et al., 1995; West, 1994]. Adverse neuropsychiatric effects, such as psychosis or vacuolar myopathy, seen with high doses or chronic administration of corticosteroids, are unusual [Wysenbeek et al., 1990].
Cytotoxic agents, such as mycophenolate [Buratti et al., 2001; Contreras et al., 2004; Kapitsinou et al., 2004] or cyclophosphamide, are used in patients with serious renal or neurologic disease. Candidates for such treatment include those who have evidence of diffuse proliferative glomerulonephritis, and those with CNS lupus or myelopathy who are refractory to corticosteroids. As there are no prospective controlled trials comparing different immunosuppressive and cytotoxic modalities in the various CNS lupus disorders, it is difficult to feel confident about a specific therapeutic regimen [Berlanga et al., 1992; Neuwelt et al., 1995; Propper and Bucknall, 1989]. Whereas mycophenolate is being used increasingly in steroid-resistant major organ disease and diffuse proliferative lupus nephritis, there are no data about the effectiveness of azathioprine or mycophenolate in CNS lupus.
Neuropathology
Neuropathologic studies of childhood and adult SLE are rare, and available reports document a variety of abnormalities. Foci of acute cortical and cerebellar encephalomalacia with neuronal loss and demyelination have been described [Gold and Yahr, 1960; Smith et al., 1994]. Postmortem examination reports of adults who have died from CNS lupus have documented diffuse microthrombi and demyelination [Hanly et al., 1992]. Vascular changes have also included vasculitis. Proliferative intimal changes, fibrinoid degeneration, and perivascular inflammation in cerebral arterial vessels have been reported in older studies, but have rarely been observed in recent investigations, suggesting that therapy may have altered the pathologic findings. These vascular changes are seen less frequently in children than in adults [Walravens and Chase, 1976].
Segmental small artery involvement with leptomeningeal and parenchymal thrombosis may occur, as well as venous sinus thrombosis [Falko et al., 1979; Steinlin et al., 1995]. These individuals have both occlusion and recanalization of vessels without changes in the media and adventitia, suggesting that the cerebrovascular lesions in SLE may result from processes acting at the endothelial-blood interface [Smith et al., 1994]. Intravascular coagulation and occlusion may be the primary mechanisms responsible for microinfarction [Falko et al., 1979]. More recent neuropathologic studies in adults implicate platelet thrombi, possibly mediated by antiphospholipid antibodies [Ellison et al., 1993]. Other studies, which do not document vascular involvement, suggest a role for antineuronal antibody-mediated damage in adults with SLE [Kuroe et al., 1994; Bluestein, 1997].
A review of the histopathologic studies of lupus patients with chorea rarely indicated abnormalities of the basal ganglia [Kuroe et al., 1994], but there were other CNS changes in these patients, such as microinfarction [Penn and Rowan, 1968; Kovacs et al., 1993; Scolding and Joseph, 2002].
Some neuropathologic studies of patients with peripheral neuropathies have revealed, although rarely, focal areas of necrosis in small arteries supplying nerve bundles, as well as perivascular inflammatory changes, fibrinous exudates, and thrombus formation. Adult patients with myelopathy exhibited large spinal cord infarcts, spinal cord subdural hematoma, and subpial leukomyelopathy [Provenzale and Bouldin, 1992].
Scleroderma
Scleroderma in children occurs in two clinically distinct forms: localized and systemic. Localized scleroderma is further subdivided into morphea, generalized morphea, linear scleroderma, and coup-de-sabre lesions. Coup-de-sabre lesions present as linear sclerodermatous changes of the head or oral cavity. Since there may be underlying CNS changes, this form of scleroderma is an important neurologic finding in children. Systemic scleroderma is subdivided into progressive systemic sclerosis and a generally milder syndrome termed CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) [Lehman, 1996].
Progressive systemic sclerosis in childhood is a multisystem disease manifesting with progressive hardening of the skin and subcutaneous tissues, with involvement of the gastrointestinal tract, joints, heart, lungs, and kidneys [Uziel et al., 1995]. Children tend to have fewer signs and laboratory parameters of vascular disease compared with adults [Vancheeswaran et al., 1996], but generally the disease is the same [Kornreich et al., 1977; Martinez-Cordero et al., 1993]. Raynaud’s phenomenon, severe cardiac and pulmonary disease with congestive heart failure, pulmonary interstitial fibrosis, pulmonary vascular sclerosis, and renal sclerosis with acute renal failure contribute significantly to the mortality associated with this disorder. Although the two disorders may be difficult to differentiate clinically, the CREST syndrome is generally thought to be milder than progressive systemic sclerosis and carries a more favorable prognosis. Systemic sclerosis may have all of the features of the CREST syndrome but is characterized by more severe internal organ involvement [Cassidy and Petty, 2001]. Skin biopsy is diagnostic, demonstrating increased thickness and density of the dermal collagen beneath the epidermis, with scattered foci of perivascular mononuclear cell infiltrate and no evidence of immune complex deposition in any affected organs, including the kidney. Radiographic assessment often demonstrates subcutaneous calcinosis, joint effusions, and diminished esophageal peristalsis. Pulmonary function studies reveal decreased lung diffusion capacity, and echocardiography may document pericardial effusion or pulmonary hypertension.
Neurologic Manifestations
Children with the coup-de-sabre form of localized scleroderma are at risk for CNS involvement [Appenzeller et al., 2004]. Coup-de-sabre lesions have been associated with intractable partial seizures, although the causality of the association has been questioned [Rigante et al., 2008; Chiang et al., 2009]. Other associated neurological abnormalities have included pseudopapilledema, strabismus, hemiparesis contralateral to the facial lesions, and developmental regression [Zannin et al., 2007; Verhelst et al., 2008]. Vasculitis of small and medium-sized cerebral vessels ipsilateral to the skin lesions has been documented by angiography. A case study with neuropathology has suggested that this may not be a form of CNS vasculitis but rather a neurocutaneous syndrome with vascular dysgenesis [Chung et al., 1995].
Periarticular muscle atrophy, common in children with systemic scleroderma, may be attributed to both disuse and subtle myopathic involvement. Muscle biopsy demonstrates findings of a mixed neuromyopathy with group atrophy, suggesting vascular neuropathic involvement as one of the mechanisms associated with the primary disease [Clements et al., 1978]. Despite elevation of creatine kinase activity in 33 percent of patients with scleroderma, muscle atrophy is rare [Dabich et al., 1974], but clinical weakness is frequent, particularly during the early inflammatory phase of the illness. MRI may be useful in the localization of suspected muscle disease in patients with scleroderma [Olsen et al., 1996].
Children with progressive systemic sclerosis generally do not have primary CNS involvement, although cerebral hemorrhage secondary to thrombocytopenia has been reported [Gordon and Silverstein, 1970; Kornreich et al., 1977].
Although neurologic manifestations have been reported rarely in adults with scleroderma, some reviewers [Cerinic et al., 1996; Hietaharju et al., 1993] have argued that these manifestations may be more common than originally believed and include neuropsychiatric symptoms, bulbar palsy, optic neuritis, trigeminal neuropathy, mononeuritis multiplex, and polyneuropathy. There is no consensus regarding treatment of these complications. Of interest is a recent study that demonstrated cerebral hypoperfusion on SPECT scans in half of the neurologically asymptomatic adults with progressive systemic sclerosis [Nobili et al., 2002]. This hypoperfusion is hypothesized to be the result of a noninflammatory microangiopathy that has its origins in endothelial cell damage and dysfunction.
Laboratory Findings
Laboratory evaluation should include determination of antinuclear antibody titers, immunoglobulins, erythrocyte sedimentation rate, rheumatoid factor, and creatine kinase activity. A mild elevation of serum immunoglobulins may be present, and there may be markers of inflammation. Patients with progressive systemic sclerosis may have antibodies to DNA topoisomerase 1 (Scl-70) or RNA polymerase I, II, or III (RNAPs). Patients with the CREST syndrome may have an anticentromere antibody. Approximately 27 percent of children with progressive systemic sclerosis have Scl-70 antibodies [Vancheeswaran et al., 1996], compared with 14–77 percent of adults, whereas an uncertain number of children with the CREST syndrome have anticentromere antibody, compared with 44–98 percent of adults [Okano, 1996].
Treatment
Treatment of progressive systemic scleroderma is largely supportive, and management of the underlying disease is often not successful. Initial management includes a course of corticosteroids to improve weakness associated with myopathy. d-Penicillamine was initially reported as beneficial for skin fibrosis but generally has not been associated with improvement [Murray and Laxer, 2002]. There has been anecdotal support for the use of steroids and methotrexate in combination for the treatment of linear scleroderma [Uziel et al., 2000]. Raynaud’s phenomenon should be treated with calcium channel blockers, such as nifedipine or amlodipine. Gastrointestinal symptoms may be relieved by bethanechol chloride, metoclopramide, or cisapride [Pope, 1996]. Occupational and physical therapy is the mainstay of treatment, and is aimed at maintaining and improving joint mobility that is impaired secondary to scarring.
Recent efforts have shown promising results with mechanistic therapeutic strategies aimed at newly identified targets, including signal transduction effectors, effector T cells, and vascular endothelial cells [Ong and Denton, 2010].
Mixed Connective Tissue Disease
Mixed connective tissue disease is characterized by a combination of signs and symptoms of SLE, scleroderma, and dermatomyositis/polymyositis, with specific serologic association of antibodies reactive with the ribonuclease-sensitive component of extractable nuclear antigen (anti-RNP) and speckled pattern antinuclear antibody [Oetgen et al., 1981]. Some children progress to a systemic sclerosis pattern [Kotajima et al., 1996], whereas others follow a course typical of mild SLE.
Neurologic Manifestations
Proximal muscle weakness, increased creatine kinase activity, myopathic electromyograms, and muscle biopsy are consistent with inflammatory myositis. In one study, asymptomatic children with mixed connective tissue disease had abnormalities detectable on electromyography and biopsy of proximal muscles [Singsen et al., 1980]. A more recent study revealed myositis in 58 percent of patients [Rayes et al., 2002].
Seizures, headache, increased cerebrospinal fluid protein content, and aseptic meningitis have been reported in children with mixed connective tissue disease [Oetgen et al., 1981]. In the original 1972 description of 25 patients, of whom at least 4 were children, none had neurologic symptoms [Sharp et al., 1972]. A follow-up study of 14 of the original cohort indicated myositis as the only neurologic finding in 5 adults. The only surviving child was asymptomatic; the other three died of non-neurologic causes [Nimelstein et al., 1980]. Two additional children have been described with neurologic manifestations [Graf et al., 1993]. The first had an internal carotid artery occlusion and stroke, eventually recovered without evidence of antiphospholipid antibody. The second child had a large intracerebral hematoma and died. Postmortem examination found fibrinoid necrosis of intracerebral capillaries. Trigeminal neuralgia has been observed in adults but has not been reported in children [Sharp, 1975]. Studies of adults have indicated a significant incidence of neuropsychiatric symptoms [Bennett and O’Connell, 1980]. Additionally, adults with recurrent optic neuropathy and/or transverse myelopathy have been reported [Flechtner and Baum, 1994; Mok and Lau, 1995; Bhinder et al., 2007].
Treatment
Treatment of mixed connective tissue disease depends on disease manifestations. For disease with mild organ involvement, hydroxychloroquine may be adequate. For more severe organ system involvement or significant myositis, corticosteroids may be necessary. As there are few long-term studies that clarify the prognosis and best therapy, treatment is empirical [Mier et al., 1996; Tiddens et al., 1993]. Methotrexate, azathioprine, and cyclophosphamide have been used, similar to therapy for SLE.
Sjögren’s Syndrome
Sjögren’s syndrome is a chronic autoimmune disorder in which lymphocytic infiltration of the salivary, lacrimal, and other exocrine glands leads to keratoconjunctivitis sicca, xerostomia, and recurrent inflammation of the salivary glands. Primary Sjögren’s syndrome is extremely rare in children, but the secondary form – preceding, accompanying, or following SLE, juvenile idiopathic arthritis, mixed connective tissue disease, juvenile dermatomyositis, or scleroderma – is more frequent [Nikitakis et al., 2003].
Neurologic Manifestations
Several children with primary Sjögren’s syndrome and neurologic involvement have been reported. A 10-year-old female with optic neuritis and severe CNS disease has been described [Berman et al., 1990], as well as an 18-year-old female with aseptic meningitis and small infarctions in both temporal regions and in the right posterior parietal region [Gerraty et al., 1993]. In addition, a 17-year-old female has been described with bilateral carotid and vertebral artery occlusions, who was treated with surgical bypass [Nagahiro et al., 1996]. A 9-year-old female who was eventually diagnosed with Sjögren’s syndrome developed recurrent paresis with associated nonenhancing lucencies of the left internal capsule and subcortical white matter in the right temporo-occipital regions on CT. MRI confirmed the CT findings and exhibited multiple regions of increased signal intensity on T2-weighted images in the internal capsule and subcortical and periventricular white matter [Ohtsuka et al., 1995]. She developed a rapidly increasing paresis that resulted in quadriplegia but responded fairly well to corticosteroids. T2-weighted images on MRI indicated diffuse swelling of the cervical cord and increased signal intensity from the first cervical to the seventh thoracic vertebrae [Ohtsuka et al., 1995]. In adult patients with Sjögren’s syndrome and biopsy-documented inflammatory vascular disease, both CNS and peripheral nervous system involvement have been reported in 25–66 percent of patients [Alexander, 1992]. Neurologic manifestations in these patients consisted of focal and diffuse brain disease, including seizures, cognitive and behavioral disorders, acute encephalopathy, aseptic meningitis, and forms of progressive myelopathies. Peripheral nervous system involvement included motor and sensory neuropathies and carpal tunnel syndrome [Malinow et al., 1985; Molina et al., 1985]. Further study of these patients revealed one or more cerebrospinal fluid abnormalities in 16 of 21 patients, including increased protein content, pleocytosis, elevated IgG levels, elevated IgG index, increased oligoclonal bands, and abnormal cerebrospinal fluid/serum glucose ratios [Malinow et al., 1985]. Because classification criteria for Sjögren’s syndrome vary, it is difficult to interpret the frequency of neurologic symptoms in adults [Alexander, 1992]. MRI may indicate subcortical or periventricular lesions in the white matter in up to 80 percent of adult patients [Alexander, 1992]. Although the pathogenesis of Sjögren’s syndrome is unknown, it has been suggested that the anti-Ro (anti-SSA) antibody may play an immunopathologic role in CNS disease [Alexander, 1992].
Laboratory Findings
Characteristic laboratory findings include hypergammaglobulinemia, positive antinuclear antibody, anti-SSA, anti-SSB, and classic rheumatoid factor [Anaya et al., 1995]. Confirmation of the diagnosis is obtained by labial salivary gland biopsy, demonstrating periductal lymphocytic infiltration [Cassidy and Petty, 2001]. The Schirmer test, sialogram, and salivary scintiphotography support the diagnosis [Chudwin et al., 1981].
Treatment
Hydroxychloroquine and supportive care are the mainstays of treatment of non-neurologic manifestations of Sjögren’s syndrome [Fox, 1992]. Immunosuppressive management with corticosteroids is needed for significant neurologic manifestations [Anaya et al., 1995; Ostuni et al., 1996]. If there is progressive neurologic disease, cyclophosphamide in addition to corticosteroids has been used successfully [Alexander, 1992]. Long-term prognosis in children with and without neurologic disease is unclear [Anaya et al., 1995; Ostuni et al., 1996].
Primary Vasculitic Diseases
Vasculitis may be either primary or secondary to a multisystem disorder. As the causes of vasculitic disorders remain unknown, classification of the vasculitides is difficult. (In Box 86-1, the vasculitides are organized by pathologic type.) Size and location of the affected vessels may aid in identifying specific disorders. Alternatively, because many of these disorders have distinct clinical patterns of involvement, the diagnosis is often established on clinical grounds with the aid of specific laboratory tests, which help rule out other disorders and confirm the diagnosis.
Although multiple etiologies have been proposed to explain the development of vasculitis, the formation of circulating immune complexes and the production of antiendothelial antibodies are the most commonly accepted theories [Conn, 1990; Kissel, 1989]. In the immune complex model, deposition of circulating antigen-antibody complexes in the vascular wall causes activation of the complement system and release of chemotactic substances that attract circulating polymorphonuclear leukocytes. Leukocytic infiltration of the vascular wall and phagocytosis of immune complexes result in the release of intracellular enzymes that cause localized vascular injury. Activation of the coagulation and kallikrein-kinin systems further contributes to the development of this type of vascular tissue damage. In the antiendothelial cell antibody model, viral infections or the underlying inflammatory disease process may alter the endothelial cell so that a specific antigen site precipitates antibody formation [Cines, 1989; Conn, 1990]. However, the presence of other factors, such as tumor necrosis factor, interleukin-1, and interferon-gamma, appears necessary to sensitize the endothelial cells so that lysis can occur when these cells are exposed to antiendothelial cell antibodies. Once endothelial injury occurs, altered vascular tone causes localized contraction and vasospasm, which, presumably, happens in conjunction with the release of vasoconstrictor substances, such as prostaglandins and calcium, or possibly as the result of inhibition of the release of endothelium-derived relaxing factor [Miller and Burnett, 1990]. The key neurologic findings in the vasculitides are listed in Table 86-4.
Table 86-4 Key Neurologic and Laboratory Findings in Childhood Vasculitides
| Disease | Neurologic Findings | Laboratory Findings |
|---|---|---|
| Polyarteritis nodosa | Headache, encephalopathy, stroke, seizures, neuropathies | Elevated WBC, ESR, positive HBsAg and c-ANCA |
| Kawasaki’s disease | Aseptic meningitis, focal neurologic findings | Coronary aneurysms, thrombocytosis |
| Cogan’s syndrome | Neurosensory hearing loss | None |
| Henoch–Schönlein purpura | Encephalopathy | Elevated IgA in 50%, hematuria, melena |
| Churg–Strauss syndrome | Headache, encephalopathy, stroke, seizures, various peripheral neuropathies, coma, intracranial hemorrhage | Eosinophilia, eosinophils on skin biopsy, p-ANCA |
| Wegener’s granulomatosis | Encephalopathy, intracranial hemorrhage, meningitis | c-ANCA |
| Primary angiitis of the CNS | Headache, encephalopathy, seizures, stroke, myelopathy | Elevated ESR |
| Sarcoidosis | Obstructive hydrocephalus, seventh nerve palsy, meningitis, seizures, peripheral neuropathies | Noncaseating granuloma |
| Temporal arteritis | Blindness, encephalopathy, headache | Elevated ESR |
| Takayasu’s arteritis | Headache, stroke, syncope, visual loss | Elevated ESR and factor VIII-related antigen |
| Behçet’s disease | Headache, meningitis, psychiatric disorders, encephalopathy, pseudotumor cerebri, brainstem signs | Elevated ESR |
c-ANCA, cytoplasmic staining antineutrophil cytoplasmic antibody; CNS, central nervous system; ESR, erythrocyte sedimentation rate; HBsAg, hepatitis B surface antigen; IgA, immunoglobulin A; p-ANCA, perinuclear staining antineutrophil cytoplasmic antibody; WBC, white blood cell count.
Necrotizing Vasculitis
Polyarteritis Nodosa
Polyarteritis nodosa occurs principally in older children and adolescents. Classic polyarteritis nodosa is characterized by unexplained fever, arthralgias, calf discomfort, abdominal pain, recurrent pulmonary infection, renal disease with hypertension, fatigability, weight loss, malar rash, and purpura. Severe kidney impairment and marked hypertension occur early in the course of the disease. Diffuse glomerulonephritis or necrotizing arteritis is found in renal biopsy material. Mesenteric arteritis and resultant bowel wall infarction may lead to gastrointestinal hemorrhage [Bakkaloglu et al., 2001; Maeda et al., 1997].
Before corticosteroid therapy, progressive neurologic, renal, and cardiovascular involvement led to a mortality rate of 80–90 percent. Currently, the use of corticosteroids and cytotoxic agents has dramatically reduced the mortality rate to as low as 5 percent [Bakkaloglu et al., 2001; Maeda et al., 1997]. Unfortunately, polyarteritis nodosa often follows a chronic course, characterized by periods of exacerbations despite aggressive immune suppression [Maeda et al., 1997].
Neurologic manifestations
Approximately one-quarter of patients with polyarteritis nodosa demonstrate neurologic manifestations during their lifetimes [Dillon, 1997]. Seizures frequently accompany polyarteritis nodosa and generally appear early in the illness, become more difficult to control as the disease becomes chronic, and may culminate in episodes of status epilepticus in terminally ill patients. Headache, disturbances of higher cortical function, and affective disorders commonly precede the onset of seizures. This finding suggests CNS vasculitis in addition to vasculitis of the internal organs and skin. Cerebrospinal fluid is usually normal, and the presence of blood strongly suggests a ruptured cerebral microaneurysm [Peal et al., 1946]. Commonly reported EEG abnormalities include diffuse and focal paroxysmal activity and background slowing. Cerebral angiography may indicate segmental narrowing of small and medium-sized vessels, and occasional microaneurysmal dilatation. Biopsy of the superficial temporal artery is not recommended because only 11 percent of patients have demonstrable arteritis in the external carotid circulation [Goder, 1956].
Polyarteritis nodosa may manifest with sudden onset of visual field defects, hemiparesis, or increased intracranial pressure. These symptoms tend to occur only in patients with a protracted course. Visual loss, associated with proliferative retinitis and fundal hemorrhages, may occur independently or accompanying increased intracranial pressure [Fager et al., 1951; Magilavy et al., 1977]. Nystagmus, ophthalmoplegia, diminished corneal reflexes, and ataxia are reported rarely [Ford and Siekert, 1965].
The sudden onset of stroke in a patient with polyarteritis nodosa requires complete assessment of the cerebral circulation, initially with MRI and MR angiography, followed by cerebral angiography in selected patients. Although angiography may initially be normal, evidence of segmental arterial narrowing may be demonstrated within several weeks of the CNS event (Figure 86-5B). Pathology indicates necrotizing angiitis, ruptured microaneurysms of the superficial and deep arteries, subarachnoid hemorrhage, and encephalomalacia caused by ischemia or infarction.
Fig. 86-5 Angiography in polyarteritis nodosa.
(Courtesy of Dr. Joseph Thompson, Department of Neuroradiology, Loma Linda University Children’s Hospital.)
Neuropsychiatric symptoms are uncommon in polyarteritis nodosa, despite the diffuse cerebral arteritis [Ford and Siekert, 1965]. Extremely ill patients may develop organic psychosis, a progressive confusional state, and variable states of consciousness that are associated with renal insufficiency. Depression is unusual in the stable patient.
Children with polyarteritis nodosa often experience diffuse myalgia; however, myositis is rarely diagnosed clinically, although it is demonstrated in approximately 11 percent of postmortem studies. There may be an accompanying increase of creatine kinase activity or evidence of myopathy indicated by electromyography [Blau et al., 1977; Magilavy et al., 1977]. Muscle biopsy may reveal necrotizing arteritis with fibrinoid necrosis and perivascular inflammation. A myopathic pattern is demonstrated in approximately 50 percent of children with polyarteritis nodosa who undergo electromyography.
Peripheral neuropathy responds well to corticosteroids. Mononeuritis multiplex is the most common and typically early neurologic feature of adult polyarteritis nodosa [Nadeau, 2002]. Sensory and motor nerve conduction velocities are decreased in childhood polyarteritis nodosa. Pathologic studies of the peripheral nervous system reveal fulminant arteritis with thrombotic occlusion of small and medium-sized arteries, and accompanying profound nerve infarction and demyelination. Myelopathy occurs rarely and usually only in patients who suffer from chronic disease [Carr and Bryer, 1993].
Microscopic polyangiitis is a subgroup of polyarteritis nodosa and is seen rarely in children [Ozen, 2004]. There is frequent pulmonary hemorrhage, and all patients have glomerulonephritis. They are frequently positive for perinuclear staining antineutrophil cytoplasmic antibody (p-ANCA) due to myeloperoxidase antibody. Peripheral neuropathy is less common than in polyarteritis nodosa.
Laboratory findings
Laboratory evaluation reveals leukocytosis, anemia, increased sedimentation rate, abnormal urinalysis, and evidence of nephrosis and nephritis, in the absence of complement consumption, antinuclear antibodies, and rheumatoid factor [Ozen et al., 1992]. Patients with polyarteritis nodosa may have circulating antineutrophil cytoplasmic antibodies [Bakkaloglu et al., 2001], elevated factor VIII-related antigen [Ates et al., 1994], and positive serology for hepatitis B surface antigen [Guillevin et al., 1995]. In addition, an association with preceding streptococcal infection has been noted [David et al., 1993; Hoyne and Steiner, 1940]. Angiography may demonstrate aneurysmal dilatation of the medium-sized mesenteric, celiac, or renal arteries (Figure 86-5A).
Neuropathology
Postmortem studies of pediatric patients with arteritis demonstrate necrotizing angiitis accompanied by polymorphonuclear infiltration of small and medium-sized arteries [Ford and Siekert, 1965].
Treatment
Corticosteroids are the mainstay of treatment. Children whose disease follows a chronic course or who develop catastrophic events should be treated with cyclophosphamide, both to avoid side effects associated with the use of chronic steroids and to provide greater immune suppression [Lhote and Guillevin, 1995]. Poorer prognosis has been associated with significant CNS disease, renal insufficiency, gastrointestinal disease, and cardiomyopathy. Aggressive treatment with high-dose intravenous corticosteroids and cyclophosphamide should be considered for such patients. Plasmapheresis has not been found to be of benefit [Lhote and Guillevin, 1995]. Overall 5-year survival has improved from 10 percent before corticosteroid use to about 80 percent in adults, and perhaps even higher in children [Lhote and Guillevin, 1995].
Kawasaki’s Disease
Kawasaki’s disease is characterized by aneurysms that are largely limited to the coronary arteries, although aneurysms have been reported in multiple vessels as well [Burns and Glodé, 2004]. Kawasaki’s disease is diagnosed by the presence of fever of at least 5 days’ duration and by 4 of the following 5 criteria:
Prognosis has been excellent since the use of intravenous immunoglobulin and acetylsalicylic acid, with immediate clinical improvement in the majority of children and a decreased rate of coronary artery aneurysmal formation from 20–25 percent to less than 8 percent. However, myocardial infarction, dysrhythmias, and sudden death were reported in 1 percent of patients [Melish, 1996].
Neurologic manifestations
Although pronounced irritability, lethargy, and aseptic meningitis are quite common, other neurologic manifestations are rare [Melish, 1981]. With the exception of a single case report [Engel et al., 1995], cerebral aneurysms have not been demonstrated in infants with Kawasaki’s disease. This contrasts with polyarteritis nodosa, in which cerebral aneurysms have been reported. In patients who present with an out-of-hospital cardiac arrest secondary to coronary artery involvement, an anoxic encephalopathy may occur.
Treatment
Treatment of Kawasaki’s disease consists of acetylsalicylic acid, 80–100 mg/kg per day in four divided doses, until the patient has been afebrile for about 3–7 days, followed by 3–5 mg/kg per day in a single dose and continued until inflammatory markers have returned to normal, the thrombocytosis has resolved, and no coronary artery disease exists on follow-up echocardiogram at 4–6 weeks. During the acute stage, intravenous immunoglobulin, 2 g/kg as a single dose over 12 hours, is recommended early in the course and preferably before the 10th day [Melish, 1996; Shulman et al., 1995]. The use of corticosteroids in Kawasaki’s disease is controversial because of one early report of an associated increased frequency of coronary artery aneurysms. This report was not confirmed by others, and children with Kawasaki’s disease who have failed intravenous immunoglobulin therapy have successfully received corticosteroids [Wright et al., 1996].
Cogan’s Syndrome
Cogan’s syndrome has been reported rarely in children [Ndiaye et al., 2002]. Although vasculitis may be diffuse, the characteristic features are vertigo, deafness, photophobia, and interstitial keratitis. Eye and ear involvement may precede other systemic symptoms by years. Aortitis and aortic valve insufficiency may also be present [Cassidy and Petty, 2001], along with arthralgia, myalgia, anorexia, episcleritis and/or uveitis, and fever. The noncardiac features generally respond well to corticosteroid therapy, but the aortic valve disease may require surgical replacement of the valve [Olfat and Al-Mayouf, 2001]. Neurologic symptoms have been described in adults [Bicknell and Holland, 1978].
Leukocytoclastic Vasculitis
Henoch–Schönlein Purpura
Henoch–Schönlein purpura is characterized by palpable purpura, petechiae, or ecchymotic rash, typically found over the buttocks and the lower extremities. It is often associated with large-joint arthritis, cramping abdominal pain, fever, peripheral edema, and renal involvement, with a median age of onset of about 6 years [Cassidy and Petty, 2001; Szer, 1996]. Henoch–Schönlein purpura is a common, self-limited illness lasting from about 1 to 3 months. Recurrences may occur over 1–2 years. Laboratory tests are usually normal, except for microscopic hematuria and guaiac-positive stools. Approximately 50 percent of patients have elevated IgA levels. Skin, renal, and gastrointestinal biopsies demonstrate leukocytoclastic vasculitis with polymorphonuclear cells within the vessel walls and deposition of IgA, complement, and properdin.
Neurologic characteristics
CNS complications have been reported in 1–8 percent of children and are secondary to hypertension, renal failure, and vasculitis. Headache, seizures, altered states of consciousness, cerebral vasculitis, intracranial hemorrhage, spastic paralysis, and chorea occur in some patients [Belman et al., 1985; Chen et al., 2000; Østergaard and Storm, 1991; Ritter et al., 1983]. A diffuse encephalopathy in association with or without hypertension has been reported [Belman et al., 1985]. Although the pathogenesis of CNS symptoms has not been clearly defined, it has been suggested that IgA immune-complex deposition initiates arteriolar inflammation [Østergaard and Storm, 1991]. Peripheral nervous system complications previously reported include lesions of the femoral, sciatic, and facial nerves; Guillain–Barré syndrome; and isolated mononeuropathies, including mononeuritis multiplex [Bulun et al., 2001; Ritter et al., 1983].
Treatment
There is consensus regarding the management of arthritis with nonsteroidal anti-inflammatory drugs and painful cutaneous edema with steroids. Supportive care is required for children who develop massive gastrointestinal bleeding or who become dehydrated or hypertensive [Szer, 1996]. High-dose steroids and plasmapheresis have been used anecdotally for severe CNS disease with success [Bulun et al., 2001; Chen et al., 2000; Eun et al., 2003].
Hypersensitivity Angiitis
Hypersensitivity, or allergic, angiitis is an acute necrotizing inflammation of blood vessels similar to Henoch–Schönlein purpura. Unlike in the latter, however, recurrent attacks are uncommon. The disorder may be acute and rapidly fatal, and may be caused by a hypersensitivity reaction to various drugs, infections, or serums [Cassidy and Petty, 2001; Farooki et al., 1974]. Among adults manifesting similar clinical symptoms, decreased serum complement and cryoglobulinemia have been reported [Cassidy and Petty, 2001]. The occurrence of neurologic complications in the various forms of hypersensitivity angiitis appears to be rare because the cerebral vessels tend to be spared.
Granulomatous Angiitis
Granulomatous angiitis represents a category of systemic necrotizing arteritis manifested by extravascular granulomatous nodules, which consist of a central area of fibrinoid degeneration with surrounding eosinophils and epithelioid and giant cells [Farooki et al., 1974]. Although Churg–Strauss syndrome (allergic granulomatosis), Wegener’s granulomatosis, and primary angiitis of the CNS are the primary forms of this type of vasculitis, necrotizing sarcoid granulomatosis and sarcoidosis also are discussed in this section because of their granulomatous pathology [Cassidy and Petty, 2001].
Churg–Strauss Syndrome
Churg–Strauss syndrome, also known as allergic granulomatosis, is associated clinically with asthma, fever, eosinophilia, cardiac failure, renal damage, and peripheral neuropathy [Sehgal et al., 1995]. Criteria established by the American College of Rheumatology require the presence of four of the following seven findings:
Approximately 21 percent of patients are children [Farooki et al., 1974]. The vasculitis is manifested by fibrinoid necrosis of the media of small arteries and veins, with arterial and capillary involvement.
Neurologic manifestations
Neurologic involvement appears to be limited to the peripheral nervous system; no reported CNS involvement has been observed in children, except for a case report of a 13-year-old female who presented with chorea and associated MRI changes [Kok et al., 1993]. Mononeuritis multiplex or symmetric polyneuropathy is found in 65–80 percent of adult patients. Symptoms are due to a vasculitis of the vasa nervorum. CNS involvement in adults tends to occur late in the course of the disease. Intracranial and subarachnoid hemorrhage, convulsions, and psychosis have been reported. Cranial nerve involvement and optic neuritis also have been described [Acheson et al., 1993; Liou et al., 1994; Sehgal et al., 1995; Tervaert and Kallenberg, 1993].
Wegener’s Granulomatosis
A necrotizing granulomatous vasculitis of the small vessels in the upper and lower respiratory tract and the kidneys characterizes Wegener’s granulomatosis. Symptoms frequently include fever, malaise, rhinorrhea, epistaxis, chronic sinusitis, nasal obstruction, pharyngeal ulcers, parenchymal pulmonary lesions, and chronic glomerulonephritis. The most common laboratory abnormality is a cytoplasmic staining antineutrophil cytoplasmic antibody due to anti-PR3 antibodies found in about 90 percent of patients [Cassidy and Petty, 2001]. Children are rarely affected. The cause of this disorder is unknown.
Neurologic manifestations
Of 17 children with Wegener’s granulomatosis, 3 had neurologic complications manifested by peripheral neuropathy [Orlowski et al., 1978]. In a more recent review of 23 children, the course was similar to that described in adults, except that there was more frequent subglottic stenosis and nasal septal deformity. CNS involvement occurred in 17 percent of patients and peripheral nervous system involvement in 9 percent [Rottem et al., 1993]. Two patients had multiple cranial nerve palsies and two had seizures. Cerebral vasculitis has been reported in individual case reports [Haas et al., 2002; von Scheven et al., 1998]. Keratitis, optic nerve granuloma, proptosis, orbital pseudotumor [Wardyn et al., 2003], laryngitis with accompanying aphonia, spasticity, proximal muscle weakness, and diminished deep tendon reflexes have been described [Drachman, 1963; Moorthy et al., 1977]. In contrast to children, approximately 10 percent of adults with Wegener’s granulomatosis had CNS involvement, and about 44 percent had peripheral nervous system involvement [Cruz and Segal, 1997].
Treatment
Prednisone and daily oral cyclophosphamide are often considered the treatments of choice and have lowered the previous mortality rate of up to 100 percent to the current survival rate of greater than 90 percent [Langford, 2003]. Pulse cyclophosphamide, intravenous immunoglobulin, trimethoprim-sulfamethoxazole, cyclosporine A, and methotrexate also have been reported to have variable success in inducing remission [Calabrese et al., 1995; Georganas et al., 1996; Jayne and Lockwood, 1996; Reinhold-Keller et al., 1996; Sneller et al., 1995; Stegeman et al., 1996].
Primary Angiitis of the Central Nervous System
Primary angiitis of the CNS, also called granulomatous angiitis, is characterized by granulomas in 80 percent of cases. Numerous case reports have been published in the last decade. Clinical criteria require a history or finding of an acquired neurologic deficit associated with angiographic or histopathologic demonstration of vasculitic changes, exclusion of other etiologies, and no evidence of systemic disease [Calabrese, 1995]. It is increasingly recognized as the cause of otherwise unexplained cerebral ischemic episodes in the young [Dillon, 1997] and has been mistaken on occasion for Rasmussen’s encephalitis [Derry et al., 2002]. Clinical features include headache, confusion, nausea, altered mental status, focal neurologic deficits, and progressive intellectual deterioration. Primary angiitis of the CNS may occur in children as young as 2 years of age and has an equal sex distribution [Barron et al., 1993; Matsell et al., 1990].
Neurologic manifestations
Clinical manifestations of primary CNS angiitis correlate with the size of the affected vessels. Patients with small-vessel disease tend to present with headache, focal seizures, or progressive behavioral or multifocal neurologic impairment. In contrast, patients with medium-sized or large-vessel disease tend to present with acute ischemic stroke or transient ischemic attack [Benseler and Schneider, 2004; Lanthier et al., 2001]. In both groups, cranial nerve abnormalities have been described, including diplopia, blurred vision, nystagmus, dysarthria, and pupillary abnormalities. Hemiparesis and language disorders have been observed in approximately 50 percent of patients. Seizures, spinal cord abnormalities, cerebral hemorrhage, fever, and weight loss also have been reported [Calabrese, 1995].
The course of this disorder fluctuates, and, although initial studies reported that 90 percent of patients died within a year of onset, it is clear from more recent studies that this disease has a broad spectrum and that the prognosis is much better [Benseler and Schneider, 2004; Calabrese, 1995]. In general, the prognosis for patients with small-vessel disease tends to be better than that for patients with stroke due to large-vessel disease [Lanthier et al., 2001].
The disease in a subgroup of patients seems to follow a benign course termed benign angiopathy of the CNS. These patients tend to be young women who have a sudden onset of symptoms and near-normal cerebrospinal fluid studies, and whose disease often remits after a monophasic course. Unfortunately, some of these patients progress years later to serious and even lethal disease [Calabrese, 1995]. Although most of these patients have been adults, two pediatric patients who experienced complete recovery have been described [Calabrese and Mallek, 1988].
Laboratory findings
Laboratory studies reveal elevated erythrocyte sedimentation rate, elevated cerebrospinal fluid protein concentration, and moderately increased monocytosis with increased pressure [Calabrese et al., 1992; Sabharwal et al., 1982]. However, normal acute phase reactants and spinal fluid have been reported in children [Benseler and Schneider, 2004]. Cerebral angiography documents small and medium-sized vessel occlusion, or segmental narrowing with saccular aneurysms and areas of infarction. Neuropathologic studies indicate segmental necrotizing granulomatous vasculitis diffusely affecting the parenchymal and leptomeningeal vessels of the cortex but not the subcortical areas of the brain [Hankey, 1991; Sabharwal et al., 1982]. Patients with small-vessel disease are more likely to demonstrate multifocal, hyperintense lesions on T2-weighted MRI, with normal cerebrospinal fluid, erythrocyte sedimentation rate, and cerebral angiograms. Those with medium-sized or large-vessel disease often demonstrate infarcts on CT and MRI, and are more likely than those with small-vessel disease to have abnormal cerebrospinal fluid studies, erythrocyte sedimentation rates, and angiograms (Figure 86-6) [Lanthier et al., 2001].
Necrotizing Sarcoid Granulomatosis
Necrotizing sarcoid granulomatosis, first described in 1973, is clinically and pathologically intermediate between sarcoidosis and Wegener’s granulomatosis [Kwong et al., 1994]. It consists of a necrotizing granulomatosis and vasculitis similar to that found in Wegener’s granulomatosis, with noncaseating granulomas resembling those of sarcoidosis. The condition has been reported in children with multifocal neurologic disease and retinal angiitis. In one child, progressive asymmetric lower limb weakness associated with urinary retention, and diagnosed as an acute myelitis and polyneuritis, responded to treatment with corticosteroids [Beach et al., 1980]. A more recent review discussed the previous reported cases, defined the vasculitic features, and differentiated childhood sarcoidosis from familial granulomatous arteritis with arthritis, uveitis, and fever [Kwong et al., 1994]. Findings reported in this disease are similar to those in sarcoidosis and include disc edema and retinal periphlebitis responsive to corticosteroid therapy. Necrotizing sarcoid granulomatosis usually responds to corticosteroids and does not require cytotoxic therapy. No significant reports of neurologic involvement have been made [Heinrich et al., 2003]. The extent of this disorder in children and the degree of neurologic involvement are unknown.
Sarcoidosis
Sarcoidosis is a chronic, multisystem granulomatous disease of unknown etiology that is rare in children. Interestingly, sarcoidosis in older children is indistinguishable from adult sarcoidosis, but younger children (younger than 4 years) tend to have different clinical manifestations. In the early-onset form, the children tend to present in infancy to preschool years [Fink and Cimaz, 1997]. This form is characterized by relatively painless, boggy polyarthritis with well-preserved range of motion. Uveitis and cutaneous sarcoid lesions are usually present, but pulmonary involvement is rare. Older children tend to present with pulmonary symptoms, hilar adenopathy, fatigue, weight loss, anorexia, headache, fever, and parotid enlargement [Kwong et al., 1994; Pattishall and Kendig, 1996]. This form of sarcoidosis is characterized by hilar and paratracheal adenopathy with interstitial infiltrates, hypercalciuria, leukopenia, and eosinophilia. An elevated erythrocyte sedimentation rate and the presence of elevated serum immunoglobulins further support the diagnosis. Pulmonary function study results are abnormal in most older patients. Noncaseating granulomas obtained from mediastinal or peripheral lymph node biopsies are found in 90 percent of patients.
Neurologic manifestations
Neurologic findings include obstructive hydrocephalus with noncaseating granulomas throughout the CNS, transient cranial nerve VII palsies, and myelopathy. There are reports of optic nerve and orbital involvement, pituitary and hypothalamic lesions, meningitis, and seizures [Cohen-Gadol et al., 2003; Lee et al., 1998; Leiba et al., 1996; Monfort-Gourand et al., 1996; Pattishall and Kendig, 1996].
In adults, about 5 percent of patients with sarcoidosis have neurologic involvement. CNS findings include meningeal, parameningeal, hypothalamic, and pituitary sarcoid infiltration. Adults have presented with symptoms of an apparent intracranial or intramedullary spinal cord tumor. Furthermore, peripheral neuropathies, including postnuclear cranial neuropathies, are associated with sarcoidosis [Scott, 1993]. It appears that the neurologic symptoms are different in children than in adults. Seizures are the most common CNS manifestation. Cranial nerve palsy and hypothalamic dysfunction are also frequent. About 25 percent of patients present with a CNS mass lesion [Baumann and Robertson, 2003]. Uveitis has been reported in both children and adults with sarcoid [Gallagher et al., 2007; Tugal-Tutkun et al., 2007].
Two reports of intramedullary spinal cord lesions studied by MRI and subsequent biopsy indicate that patchy contrast enhancement and increased T2-weighted signal intensity almost always occurred in sarcoid lesions of the spinal cord. These two relatively small cohorts of patients with non-neoplastic spinal cord lesions differed from one another with regard to the question of whether there was significant expansion of the spinal cord in the region of sarcoid involvement [Cohen-Gadol et al., 2003; Lee et al., 1998].
Treatment
Treatment of the early-onset form of sarcoidosis has been anecdotal and has consisted of corticosteroids, azathioprine, methotrexate, or, occasionally, cyclophosphamide [Fink and Cimaz, 1997]. Therapy of late-onset or adult-type sarcoidosis depends on whether the arthritis is acute, chronic, or relapsing. Nonsteroidal anti-inflammatory drugs have been used for indolent disease, as well as for acute attacks. More painful episodes have been successfully managed with colchicine. Attacks unresponsive to colchicine have often responded to corticosteroids. Chronic disease unresponsive to nonsteroidal anti-inflammatory drugs has responded to low-dose, alternate-day steroids or methotrexate [Sequeira, 1997]. Steroid-refractory uveitis of childhood has been reported to respond to therapy with biological response modifiers [Gallagher et al., 2007].
Giant Cell Arteritis
Temporal Arteritis
Classic giant cell arteritis (temporal arteritis) most often affects men older than 50 years of age [Nordborg et al., 1995]. A recent report of temporal arteritis in a 9-year-old Haitian female with a past medical history remarkable for sensorineural hearing loss; an initial presentation with monocular blindness and a tortuous superficial temporal artery, with a temporal artery biopsy study consistent with giant cell arteritis; and a disease course that included a systemic illness with encephalopathy most consistent with polyarteritis nodosa make it clear that this disorder can occur in childhood [Bert et al., 1999]. Headache, scalp tenderness, visual disturbances, and jaw claudication appearing in association with an elevated sedimentation rate suggest the diagnosis. Biopsy of the superficial temporal artery reveals giant cell perivascular inflammatory infiltrates [Small, 1984]. Hemiparesis, vertigo, hearing loss, brainstem strokes, seizures, oculomotor disorders, and peripheral neuropathies have been reported. Optic neuritis is the most common problem. The superficial temporal, vertebral, ophthalmic, and posterior ciliary arteries are frequently involved. The internal and external carotid and central retinal arteries are less commonly affected, but occlusion of the ophthalmic or central retinal artery can lead to blindness. Intracranial arterial involvement has been documented, although it is rare [Small, 1984]. Myelopathy, transient ischemic attacks, and cerebral infarctions have been reported in adults with temporal arteritis. It is unclear whether the latter two disorders are due to the underlying condition or the patient’s age [Caselli and Hunder, 1993].
Before the case report of Bert et al. [1999], juvenile temporal arteritis was regarded as a different and even uncommon clinical entity than adult-type temporal arteritis. Two children, 7 and 8 years of age, initially developed unilateral nodules of the temporal arteries without systemic or neurologic symptoms [Lie et al., 1975]. Studies of these lesions failed to demonstrate the classic giant cells but instead revealed perivascular lymphoid hyperplasia with eosinophilia. Occlusion of the temporal artery lumen with thrombus formation and diffuse intimal thickening and focal disruption of the internal elastic lamina and media were observed. This disorder demonstrates a form of non-giant cell granulomatous inflammation of the temporal artery; it is distinct from classic temporal arteritis of adults [Lie et al., 1975]. A more recent review included an additional case report of this syndrome [Lie, 1995].
Takayasu’s Arteritis
Takayasu’s arteritis, a form of arteritis involving the aorta and its branches, has been reported in children as young as 5 months of age. Onset occurs before 16 years of age in 25–30 percent of patients [Morales et al., 1991], and a recent postmortem study from India demonstrated a mean age at death of 22.6 ± 10.2 years [Sharma et al., 1998]. In children, the abdominal aorta and its branches are more commonly affected than the aortic arch. The clinical course is variable. Most patients come to medical attention in the pre-pulseless stage with nonspecific symptoms of fever, fatigue, dyspnea, anorexia, and arthralgia. The obstructive phase of the illness is characterized by aneurysmal dilatation and stenosis of the aorta or pulmonary arteries. As the abdominal aorta is involved in young children, initial symptoms may include abdominal pain or mass, congestive heart failure, and occlusive vascular disease of the abdominal organs or lower extremities [Gronemeyer and Demello, 1982].
Symptoms of claudication, pulselessness, carotid hypersensitivity, and cerebrovascular involvement have been reported in children [Pantell and Goodman, 1981]. Hypertension caused by renal artery stenosis occurs in approximately 70 percent of patients. Hypertensive encephalopathy has been reported in two children with renal artery stenosis [Millar et al., 1996]. One review noted that neurologic symptoms in childhood are quite rare, but that adults may develop carotid thromboembolic disease, cerebral hypertension, subclavian steal syndrome, or hypersensitivity of the carotid sinus [Tervaert and Kallenberg, 1993]. A review of 26 children emphasized that hypertension, constitutional symptoms, gastrointestinal pain, and heart failure were fairly common. In addition, 50 percent of patients had significant headache, 12 percent had convulsions, and 8 percent developed hemiplegia [Morales et al., 1991]. Another review of 24 patients found similar findings, except for a lack of neurologic symptoms [Jain et al., 2000]. In one small study of patients who succumbed to Takayasu’s arteritis, 20 percent had seizures and 10 percent had hemiplegia [Sharma et al., 1998]. All of these patients had hypertension as the presenting feature of their disease.
Treatment with corticosteroids and other immunosuppressive agents improves the long-term prognosis, but most immunosuppressive drugs provide little improvement over corticosteroids [Hoffman, 1996]. Significant improvement has been made with the use of bypass grafts and angioplasty [Hoffman, 1995; Kohrman and Huttenlocher, 1986].
Miscellaneous Vasculitic Disorders
Not all vasculitic disorders can be classified in the previously noted categories of polyarteritis – leukocytoclastic, granulomatous, and giant cell vasculitides. As many as one-third of all primary vasculitic diseases of childhood are categorized as idiopathic or “polyangiitis overlap syndrome” [Cassidy and Petty, 2001]. Other diseases, such as Behçet’s syndrome, do not fit any of these categories.
Behçet’s Disease
Behçet’s disease is rare in children [Cassidy and Petty, 2001; Rakover et al., 1989], usually having its onset in the second decade of life. This rarity has led some authors to postulate that juvenile Behçet’s disease may differ from adult Behçet’s disease in pathogenesis [Koné-Paut et al., 1998]. One study of juvenile Behçet’s disease placed the onset of this disorder at or around age 7 [Krause et al., 1999]. The classic clinical triad of Behçet’s disease consists of recurrent oral and genital ulcerations and recurrent uveitis [Schor, 2000]. The uveitis tends to be a panuveitis with retinal vasculitis [Tugal-Tutkun and Urgancioglu, 2003]. The following criteria have been proposed for the diagnosis of Behçet’s disease: the presence of recurrent aphthous stomatitis and two of the following:
Neurologic manifestations
CNS manifestations are reported in as many as 66 percent of adult patients during the course of the illness. One study found that the prevalence of neurologic manifestations other than headache was much higher in their cohort of patients with childhood Behçet’s disease (26.3 percent) than in their adult counterparts (3.8 percent) [Krause et al., 1999]. Three forms of CNS involvement have been reported in adults and children:
The CNS symptoms occur as isolated recurrent events that evolve slowly [Bahabri et al., 1996; Devlin et al., 1995; Fujikawa and Suemitsu, 1997; Rosenberger et al., 1982].
Laboratory findings
MRI with or without gadolinium enhancement and SPECT have demonstrated CNS involvement in adult patients [Erdem et al., 1993; Mizukami et al., 1992; Wechsler et al., 1993], although both techniques underestimate the frequency of brain lesions in adults and children. In a study of seven children and one adult with Behçet’s disease, all patients demonstrated hypoperfusion, especially in the basal ganglia and frontal and temporal cortex, on 99mTc-HMPAO SPECT scanning [Nobili et al., 2002].
Neuropathology
Neuropathologic studies demonstrate lymphocytic meningitis, vasculitis with perivascular cuffing, and thrombosis of arterioles, venules, veins, and dural venous sinuses. Although the etiopathogenesis of Behçet’s disease is unknown, the intraocular fluid of patients with Behçet’s disease-associated uveitis, but not that of other uveitis patients, harbors T cells specific for nonpeptide prenyl pyrophosphate antigens. This finding suggests that perhaps an autoimmune attack on a specific antigen underlies susceptibility to Behçet’s disease [Verjans et al., 2002].
Treatment
Treatment remains controversial, although early treatment with high doses of corticosteroids appears beneficial [Cassidy and Petty, 2001; O’Duffy and Goldstein, 1976]. Ten children were reviewed recently in whom thalidomide was used successfully in some resistant cases, with good improvement in oral and genital ulcerations [Kari et al., 2001]. In patients with intracranial venous or dural sinus thrombosis, heparinization may be beneficial [Wechsler et al., 1992]. Immunosuppressive agents have been used for the CNS manifestations of the disease [Zelenski et al., 1989].
Miscellaneous Disorders
Thrombotic Thrombocytopenic Purpura
Rapid onset of thrombocytopenia, severe microangiopathic hemolytic anemia, renal disease, neurologic symptoms, and fever characterizes thrombotic thrombocytopenic purpura [Amorosi and Ultmann, 1966].
Neurologic manifestations
Thrombotic thrombocytopenia purpura results in widespread microvascular thrombosis of numerous organs. Neurologic involvement can present as headache, confusion, seizure, and occasionally focal deficits [Lawlor et al., 1997]. CNS symptoms usually persist, unlike the intermittent fluctuating course observed in adults [Thompson et al., 1992]. Retinal hemorrhage is frequent. There have been rare reports of thrombotic thrombocytopenic purpura occurring with SLE [Chak et al., 2003]. Thrombotic occlusion may occur in any tissue and may involve the CNS, peripheral nervous system, or skeletal muscles. Cerebrospinal fluid examination results are normal in most patients, and EEG changes are nonspecific. MRI, MR angiography, cerebral angiography [Rinkel et al., 1991; Wijdicks, 1994], and CT scans may reveal evidence of arterial occlusion. A recent study of 12 adult patients with thrombotic thrombocytopenic purpura [Bakshi et al., 1999] demonstrated acute MRI abnormalities in 75 percent, most commonly reversible posterior leukoencephalopathy and edema associated with hypertension. Ischemic strokes, sometimes multiple in the same patient, were seen in 25 percent of the patients in this study, and many were complicated by secondary hemorrhage into the area of infarction. Half of the strokes were in the posterior cerebral artery territory, and, in general, patients with stroke or primary hematoma (1 patient in this series) had an unfavorable long-term outcome.
Laboratory findings
Decreased levels of protease activity with the presence of von Willebrand factor-protease inhibitors contribute to the bleeding diathesis in this condition [Horton et al., 2003]. Recent studies have demonstrated a deficiency of ADAMTS13 in affected individuals. Although often resulting from genetic mutations in the ADAMTS13 gene, individuals may also develop this syndrome as the result of autoimmune inhibitors against ADAMTS13. ADAMTS13 is a von Willebrand factor-cleaving metalloprotease resulting in the release of multimers.
Neuropathology
Pathologically, amorphous hyaline material occluding small cerebral arteries usually is seen and is indistinguishable from that found in the heart and kidneys. Cerebral gray matter contains many vascular lesions [Silverstein, 1968; Tsai, 2010].
Treatment
Previously, only 10 percent of children with thrombotic thrombocytopenic purpura survived for more than 1 year, despite aggressive intervention with heparin, intravenous corticosteroids, whole-blood transfusion, dextran infusion, and splenectomy. Plasma exchange transfusion has been beneficial for some children, decreasing the mortality to very low levels [Byrnes and Khurana, 1977; Lawlor et al., 1997]. It has been suggested that plasma exchange removes pathogenic substances and supplies other factors that may be deficient in patients with thrombotic thrombocytopenic purpura [Lawlor et al., 1997]. Inhibitors of ADAMTS13 are usually IgG, are short-lived, and are readily cleared by treatment with plasma exchange; however, relapses may occur and thus these patients often require long-term immunosuppressive therapy.
Erythromelalgia and Erythermalgia
Erythromelalgia, a rare disorder, consists of intense, asymmetric burning sensations with associated erythema and elevated temperature in the extremities [Mandell et al., 1977]. The transient episodes may last for minutes, hours, or even days. Due to the intense pain and discomfort, patients often do not walk or use their hands. Patients find relief by immersing their hands or feet in ice water. One form of the disorder is associated with thrombocythemia (a fixed increase in the number of platelets). Blood pressure and peripheral pulses are normal during the symptomatic phase. In older patients, this condition has been associated with hypertension, SLE, diabetes mellitus, and myeloproliferative diseases. A defect in prostaglandin metabolism was postulated after many patients reported relief of symptoms after using acetylsalicylic acid [Jorgensen and Sondergaard, 1978]. This so-called secondary erythromelalgia has no parallel in childhood [Drenth et al., 1994].
Primary erythromelalgia or “erythermalgia” is a more rare, chronic, symmetric, burning sensation that occurs in children and adults. It is not reliably responsive to medication [Davis et al., 2000], although some patients have responded well to amitriptyline, sodium nitroprusside, regional anesthetic blockade, and gabapentin [Chan et al., 2002; Harrison et al., 2003; McGraw and Kosek, 1997; Rauck et al., 1996]. Regional nerve block has also been used to control pain [Harrison et al., 2003]. Histopathology reveals no consistent vasculitic changes, and erythermalgia is not associated with thrombocythemia [Drenth et al., 1994]. Some studies have demonstrated autonomic and small sensory fiber axonal neuropathy in patients with this disorder [Chan et al., 2002; Davis et al., 2000; Harrison et al., 2003].
The differential for this disorder includes Raynaud’s phenomenon and reflex sympathetic dystrophy, but it can usually be distinguished from these conditions by its unique clinical appearance and the relief provided by ice water immersion. In one report, a child with a syndrome resembling erythermalgia was found to have growth hormone deficiency and remission of symptoms with growth hormone replacement therapy; this has raised the question of whether a disorder involving the insulin-like growth factor family of trophic factors and receptors is involved [Cimaz et al., 2001]. Familial instances of erythermalgia have facilitated localization of the gene for susceptibility to this condition to chromosome 2q31–32 [Drenth et al., 2001].
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome is a multisystem, autoimmune disorder characterized by the clinical features of recurrent thrombosis, pregnancy loss, or thrombocytopenia, and the laboratory feature of antiphospholipid antibodies (aPL) directed against negatively charged phospholipids [Wilson et al., 1999]. Numerous aPL have been described, including lupus anticoagulants (LAC), anticardiolipin antibodies (aCL), antibodies to β2 glycoprotein-I (β2GPI), and antibodies directed against prothrombin, annexin V, phosphatidylserine, and phosphatidylinositol [Passam and Krillis, 2004]. LAC are a group of antibodies directed against plasma proteins such as β2GPI, prothrombin, and annexin V, which are bound to anionic phospholipids. They are detected by functional clotting tests, such as the activated partial thromboplastin time, dilute Russell viper venom time, kaolin clotting time, and, infrequently, prothrombin time. In clotting factor deficiencies such as hemophilia, diluting a patient’s plasma 1:1 with normal plasma can correct the observed prolonged clotting, but this does not occur in the presence of a LAC. Criteria for the diagnosis of a LAC include both the absence of correction in a 1:1 mixing reaction with a healthy person’s plasma, and confirmation of phospholipid dependence with the addition of a phospholipid-containing substrate [Brandt et al., 1995]. aCL can be detected in enzyme-linked immunosorbent assays, or indirectly in the VDRL assay. Although initially developed as a serologic test for syphilis, this latter assay detects agglutination of lipid particles that contain cholesterol and cardiolipin, thus resulting in a false-positive result in patients with antibodies against cardiolipin. Although the significance of the isotype is controversial, elevated levels of IgG aCL likely have a greater association with thrombotic risk than IgM and IgA isotypes. In addition to cardiolipin, aPL may react with other phospholipids, such as phosphatidyl serine and phosphatidyl inositol; however, the clinical significance of these antibodies is less clear. Beta2GPI is the most common target of aPL; it becomes antigenic upon binding to a negatively charged surface, thereby inhibiting clotting. Antibodies to β2GPI are likely critical to the pathogenesis of clinical manifestations of antiphospholipid antibody syndrome, particularly stroke, although many patients with antiphospholipid antibody syndrome do not test positive on these assays.
The diagnosis of antiphospholipid antibody syndrome requires specific clinical manifestations in addition to detection of antibodies. Antiphospholipid antibody syndrome is a heterogeneous syndrome, and thus strict diagnostic criteria for “definite” antiphospholipid antibody syndrome have been developed. The most recent international Sapporo criteria for antiphospholipid antibody syndrome require that individuals meet at least one of the clinical criteria and one of the laboratory criteria [Miyakis et al., 2006]. Clinical criteria include either:
One of the following three laboratory criteria must be met:
Patients with antiphospholipid antibody syndrome are usually classified as having “primary antiphospholipid antibody syndrome” if there is no evidence for another underlying autoimmune disorder, or “secondary antiphospholipid antibody syndrome” if another autoimmune disorder, such as SLE, Wegener’s granulomatosus, scleroderma, or psoriatic arthritis, is present [Falcini et al., 1991]. When seen in the setting of a lupus-like disorder that does not meet the strict American College of Rheumatology criteria for SLE, the disease is referred to as SLE-like antiphospholipid antibody syndrome [Weber et al., 1999]. These syndromes are seen in both children and adults; in some children, primary antiphospholipid antibody syndrome is a precursor of secondary antiphospholipid antibody syndrome.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Acheson J.F., Cockerell O.C., Bentley C.R., et al. Churg-Strauss vasculitis presenting with severe visual loss due to bilateral sequential optic neuropathy. Br J Ophthalmol. 1993;77:118.
Aisen A.M., Gabrielsen T.O., McCune W.J. MR imaging of systemic lupus erythematosus involving the brain. Am J Roentgenol. 1985;144:1027.
Alexander E. Central nervous system disease in Sjögren’s syndrome. New insights into immunopathogenesis. Rheum Dis Clin North Am. 1992;18:637.
Al-Husaini A., Jamal G.A. Myelopathy as the main presenting feature of systemic lupus erythematosus. Eur Neurol. 1985;24:94.
Allen A.J., Leonard H.L., Swedo S.E. Case study: A new infection-triggered, autoimmune subtype of pediatric OCD and Tourette’s syndrome. J Am Acad Child Adolesc Psychiatry. 1995;34:307.
American College of Rheumatology Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599-608.
Amorosi E.L., Ultmann J.E. Thrombotic thrombocytopenic purpura: Report of 16 cases and review of the literature. Medicine. 1966;45:139.
Anaya J.M., Ogawa N., Talal N. Sjögren’s syndrome in childhood. J Rheumatol. 1995;22:1152.
Andrianakos A.A., Duffy J., Suzuki M., et al. Transverse myelopathy in systemic lupus erythematosus. Ann Intern Med. 1975;83:616.
Ansari I., Crichlow B., Gunton K.B., et al. A child with venous sinus thrombosis with initial examination findings of pseudotumor syndrome. Arch Ophthalmol. 2002;120:867.
Anton P.A., Shanahan F. Neuroimmunomodulation in inflammatory bowel disease. How far from “bench” to “bedside”? Ann N Y Acad Sci. 1998;840:723.
Appenzeller S., Montenegro M.A., Dertkigil S.S., et al. Neuroimaging findings in scleroderma en coup de sabre. Neurology. 2004;62:1585.
Aron A.M., Freeman J.M., Carter S. The natural history of Sydenham’s chorea. Am J Med. 1965;38:83.
Asherson R.A., Derksen R., Harris E.N., et al. Chorea in systemic lupus erythematosus and “lupus-like”disease: association with antiphospholipid antibodies. Semin Arthritis Rheum. 1987;16:253.
Ates E., Bakkaloglu A., Saatci U., et al. Von Willebrand factor antigen compared with other factors in vasculitic syndromes. Arch Dis Child. 1994;70:40.
Avcin T., Tse S.M., Schneider R., et al. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686.
Ayoub E.M., Wannamaker L.W. Streptococcal antibody titers in Sydenham’s chorea. Pediatrics. 1966;38:946.
Bag A.K., Curé J.K., Sullivan J.C., et al. Central variant of posterior reversible encephalopathy syndrome in systemic lupus erythematosus: new associations? Lupus. 2010;19(2):225-226.
Bahabri S.A., al-Mazyed A., al-Balaa S., et al. Juvenile Behçet’s disease in Arab children. Clin Exp Rheumatol. 1996;14:331.
Baildam E.M., Holt P.J.L., Conway S.C., et al. The association between physical function and psychological problems in children with juvenile chronic arthritis. Br J Rheum. 1995;34:470.
Bailey A.A., Sayre G.P., Clark E.C. Neuritis associated with systemic lupus erythematosus. AMA Arch Neurol Psychiatry. 1956;75:251.
Baizabal-Carvallo J.F., Samson Y. Microembolic signals in systemic lupus erythematosus and other cerebral small vessel diseases. J Neurol. 2009. Epub ahead of print
Bakkaloglu A., Ozen S., Baskin E., et al. The significance of antineutrophil cytoplasmic antibody in microscopic polyangiitis and classic polyarteritis nodosa. Arch Dis Child. 2001;85:427.
Bakshi R., Shaikh Z.A., Bates V.E., et al. Thrombotic thrombocytopenic purpura: Brain CT and MRI findings in 12 patients. Neurology. 1999;52:1285.
Barash J., Margalith D., Matitiau A. Corticosteroid treatment in patients with Sydenham’s chorea. Pediatr Neurol. 2005;32(3):205-207.
Barron T.F., Ostrov B.E., Zimmerman R.A., et al. Isolated angiitis of the CNS: Treatment with pulse cyclophosphamide. Pediatr Neurol. 1993;9:73.
Baumann R.J., Robertson W.C.Jr. Neurosarcoid presents differently in children than in adults. Pediatrics. 2003;112:e480.
Beach R.C., Corrin B., Scopes J.W., et al. Necrotizing sarcoid granulomatosis with neurological lesions in a child. J Pediatr. 1980;97:950.
Becton D.L., Stine K.C. Transient lupus anticoagulants associated with hemorrhage rather than thrombosis: The hemorrhagic lupus anticoagulant syndrome. J Pediatr. 1997;130:998.
Belman A.L., Leicher C.R., Moshé S.L., et al. Neurologic manifestations of Schönlein-Henoch purpura: Report of three cases and review of the literature. Pediatrics. 1985;75:687.
Benda C.E. Chronic rheumatic encephalitis. Arch Neurol Psychiatry. 1948;59:262.
Bennett R.M., O’Connell D.J. Mixed connective tissue disease: A clinico-pathologic study of 20 cases. Semin Arthritis Rheum. 1980;10:25.
Benseler S.M., Silverman E.D. Neuropsychiatric involvement in pediatric systemic lupus erythematosus. Lupus. 2007;16(8):564-571.
Benseler S., Schneider R. Central nervous system vasculitis in children. Curr Opin Rheumatol. 2004;16:43.
Berlanga B., Rubio F.R., Moga I., et al. Response to intravenous cyclophosphamide treatment in lupus myelopathy. J Rheumatol. 1992;19:829.
Berman J.L., Kashii S., Trachtman M.S., et al. Optic neuropathy and central nervous system disease secondary to Sjögren’s syndrome in a child. Ophthalmology. 1990;97:1606.
Berrios X., Quensey S., Morales A., et al. Are all recurrences of “pure” Sydenham chorea true recurrences of acute rheumatic fever? J Pediatr. 1985;107:867.
Bert R.J., Antonacci V.P., Berman L., et al. Polyarteritis nodosa presenting as temporal arteritis in a 9-year-old child. Am J Neuroradiol. 1999;20:167.
Besbas N., Damarguc I., Ozen S., et al. Association of antiphospholipid antibodies with systemic lupus erythematosus in a child presenting with chorea: A case report. Eur J Pediatr. 1994;153:891.
Bhinder S., Harbour K., Majithia V. Transverse myelitis, a rare neurological manifestation of mixed connective tissue disease – a case report and a review of literature. Clin Rheumatol. 2007;26(3):445-447.
Bicakci S., Ozbek S., Bicakci K., et al. Recurrent headache and MRI findings in systemic lupus erythematosus. J Natl Med Assoc. 2008;100(3):323-326.
Bicknell J.M., Holland J.V. Neurologic manifestations of Cogan syndrome. Neurology. 1978;28:278.
Bingham P.M., Galetta S.L., Arthreya B., et al. Neurologic manifestation in children with Lyme disease. Pediatrics. 1995;96:1053.
Bird M.T., Palkes H., Prensky A.L. A follow-up study of Sydenham’s chorea. Neurology. 1976;26:601.
Blau E.B., Morris R.F., Yunis E.J. Polyarteritis nodosa in older children. Pediatrics. 1977;60:227.
Bluestein H.G. Antibodies to brain. In: Wallace D.J., Hahn B.H., editors. Dubois’ Lupus Erythematosus. Baltimore: Williams & Wilkins, 1997.
Boumpas D.T., Patronas N.J., Dalakas M.R., et al. Acute transverse myelitis in systemic lupus erythematosus: Magnetic resonance imaging and review of the literature. J Rheumatol. 1990;17:89.
Brandt J.T., Triplett D.A., Alving B., et al. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost. 1995;74:1185-1190.
Brandt K.D., Lessell S., Cohen A.S. Cerebral disorders of vision in systemic lupus erythematosus. Ann Intern Med. 1975;83:163.
Bray V.J., Singleton J.D. Disseminated intravascular coagulation in Still’s disease. Semin Arthritis Rheum. 1994;24:222.
Brey R.L., Chapman J., Levine S.R., et al. Stroke and the antiphospholipid syndrome: Consensus meeting Taormina 2002. Lupus. 2003;12:508.
Bridger S., Evans N., Parker A., et al. Multiple cerebral venous thromboses in a child with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1997;25:533.
Bruyn G.A. Controversies in lupus: Nervous system involvement. Ann Rheum Dis. 1995;54:159.
Bulun A., Topaloglu R., Duzova A., et al. Ataxia and peripheral neuropathy: Rare manifestations in Henoch-Schonlein purpura. Pediatr Nephrol. 2001;16:1139.
Buratti S., Szer I.S., Spencer C.H., et al. Mycophenolate mofetil treatment of severe renal disease in pediatric onset systemic lupus erythematosus. J Rheumatol. 2001;28:2103.
Burgos P.I., Alarcón G.S. Thrombosis in systemic lupus erythematosus: risk and protection. Expert Rev Cardiovasc Ther. 2009;7(12):1541-1549.
Burns J.C., Glodé M.P. Kawasaki syndrome. Lancet. 2004;364:533.
Byrnes J.J., Khurana M. Treatment of thrombotic thrombocytopenic purpura with plasma. N Engl J Med. 1977;297:1386.
Calabrese L.H. Vasculitis of the central nervous system. Rheum Dis Clin North Am. 1995;21:1059.
Calabrese L.H., Furlan A.J., Gragg L.A., et al. Primary angiitis of the central nervous system: Diagnostic criteria and clinical approach. Cleve Clin J Med. 1992;59:293.
Calabrese L.H., Hoffman G.S., Guillevin L. Therapy of resistant systemic necrotizing vasculitis. Polyarteritis, Churg-Strauss syndrome, Wegener’s granulomatosis, and hypersensitivity vasculitis group disorders. Rheum Dis Clin North Am. 1995;21:41.
Calabrese L.H., Mallek J.A. Primary angiitis of the central nervous system: Report of eight new cases, review of the literature and proposal for diagnostic criteria. Medicine. 1988;67:20.
Canoso J.J., Cohen A.S. Aseptic meningitis in systemic lupus erythematosus: Report of three cases. Arthritis Rheum. 1975;18:369.
Carbotte R.M., Denburg S.D., Denburg J.A. Cognitive dysfunction in systemic lupus erythematosus is independent of active disease. J Rheumatol. 1995;22:863.
Carette S., Urowitz M.B., Grosman H., et al. Cranial computerized tomography in systemic lupus erythematosus. J Rheumatol. 1982;9:855.
Carlow T.J., Glaser J.S. Pseudotumor cerebri syndrome in systemic lupus erythematosus. JAMA. 1974;228:197.
Carr J., Bryer A. An isolated myelopathy as a presentation of polyarteritis nodosa. Br J Rheumatol. 1993;32:644.
Carr R.I., Harbeck R.J., Hoffman A.A., et al. Clinical studies on the significance of DNA anti-DNA complexes in the systemic circulation and cerebrospinal fluid (CSF) of patients with systemic lupus erythematosus. J Rheumatol. 1975;2:184.
Carvalho K.S., Garg B.P. Arterial strokes in children. Neurol Clin. 2002;20:1079.
Caselli R.J., Hunder G.G. Neurologic aspects of giant cell (temporal) arteritis. Rheum Dis Clin North Am. 1993;19:941.
Cassidy J.T., Petty R.E. Textbook of pediatric rheumatology, ed 4. Philadelphia: WB Saunders; 2001.
Cassidy J.T., Sullivan D.B., Petty R.E., et al. Lupus nephritis and encephalopathy: Prognosis in 58 children. Arthritis Rheum. 1977;20(Suppl 2):315.
Castellino G., Padovan M., Bortoluzzi A., et al. Single photon emission computed tomography and magnetic resonance imaging evaluation in SLE patients with and without neuropsychiatric involvement. Rheumatology (Oxford). 2008;47(3):319-323.
Cerinic M.M., Generini S., Pignone A., et al. The nervous system in systemic sclerosis (scleroderma). Clinical features and pathogenetic mechanisms. Rheum Dis Clin North Am. 1996;22:879.
Chak W.K., Lam D.S., Lo W.H., et al. Thrombotic thrombocytopenic purpura as a rare complication in childhood systemic lupus erythematosus: Case report and literature review. Hong Kong Med J. 2003;9:363.
Chan K.F., Boey M.L. Transverse myelopathy in SLE: Clinical features and functional outcomes. Lupus. 1996;5:294.
Chan M.K.H., Tucker A.T., Madden S., et al. Erythromelalgia: An endothelial disorder responsive to sodium nitroprusside. Arch Dis Child. 2002;87:229.
Chen C.L., Chiou Y.H., Wu C.Y., et al. Cerebral vasculitis in Henoch-Schonlein purpura: A case report with sequential magnetic resonance imaging changes and treated with plasmapheresis alone. Pediatr Nephrol. 2000;15:276.
Chiang K.L., Chang K.P., Wong T.T., et al. Linear scleroderma “en coup de sabre”: initial presentation as intractable partial seizures in a child. Pediatr Neonatol. 2009;50(6):294-298.
Chudwin D.S., Daniels T.E., Wara D.W., et al. Spectrum of Sjögren syndrome in children. J Pediatr. 1981;98:213.
Chun R.W.M., Smith N.J., Forster F.M. Papilledema in Sydenham’s chorea. Am J Dis Child. 1961;101:641.
Chung M.H., Sum J., Morrell M.J., et al. Intracerebral involvement in scleroderma en coup de sabre: Report of a case with neuropathologic findings. Ann Neurol. 1995;37:679.
Cimaz R., Rusconi R., Fossali E., et al. Unexpected healing of cutaneous ulcers in a short child. Lancet. 2001;358:211.
Cines D.B. Disorders associated with autoantibodies to endothelial cells. Rev Infect Dis. 1989;2(Suppl 4):S705.
Clements P.J., Furst D.E., Campion D.E., et al. Muscle disease in progressive systemic sclerosis. Diagnostic and therapeutic considerations. Arthritis Rheum. 1978;21:62.
Cohen-Gadol A., Zikel O.M., Miller G.M., et al. Spinal cord biopsy: A review of 38 cases. Neurosurgery. 2003;52:806.
Conn D.L. Polyarteritis. Rheum Dis Clin North Am. 1990;16:341.
Contreras G., Pardo V., Leclercq B., et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med. 2004;350:971.
Cortis E., Insalaco A. Macrophage activation syndrome in juvenile idiopathic arthritis. Acta Paediatr Suppl. 2006;95(452):38-41.
Coster T.A., Cole H.C. Atlanto-axial dislocation in association with rheumatic fever: A case report. Spine. 1990;15:591.
Cruz D.N., Segal A.S. A patient with Wegener’s granulomatosis presenting with a subarachnoid hemorrhage: Case report and review of CNS disease associated with Wegener’s granulomatosis. Am J Nephrol. 1997;17:181.
Cuchacovich M., Gatica H., Contreras L. [Neurologic involvement in Reiter’s disease. Report of 2 cases]. Rev Med Chil. 1991;119(6):687-690. Spanish
Dabich L., Sullivan D.B., Cassidy J.T. Scleroderma in the child. J Pediatr. 1974;85:770.
Dajani A.S., Ayoub E., Bierman F.Z., et al. Guidelines for the diagnosis of rheumatic fever. Jones Criteria, 1992 update. Special Writing Group of the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young of the American Heart Association. JAMA. 1992;268:2069.
Dajani A., Taubert K., Ferrieri P., et al. Treatment of acute streptococcal pharyngitis and prevention of rheumatic fever: A statement for health professions. Pediatrics. 1995;96:758.
Dannecker G.E., Quartier P. Juvenile idiopathic arthritis: classification, clinical presentation and current treatments. Horm Res. 2009;72(Suppl 1):4-12.
Daoud A.S., Zaki M., Shakir R., et al. Effectiveness of sodium valproate in the treatment of Sydenham’s chorea. Neurology. 1990;40:1140.
Davey R., Bamford J., Emery P. The role of endothelial dysfunction in the pathogenesis of neuropsychiatric systemic lupus erythematosus. Lupus. 2010. Epub ahead of print
David J., Ansell B.M., Woo P. Polyarteritis nodosa associated with streptococcus. Arch Dis Child. 1993;69:685.
Davis L.E., Hodgin U.G., Kornfield M. Recurrent meningoencephalitis with recovery from Behçet’s disease. West J Med. 1986;145:238.
Davis M.D.P., O’Fallon W.M., Rogers R.S., et al. Natural history of erythromelalgia: Presentation and outcome in 168 patients. Arch Dermatol. 2000;136:330.
De Cunto C.L., Liberatore D.I., San Roman J.L., et al. Infantile-onset multisystem inflammatory disease: A differential diagnosis of systemic juvenile rheumatoid arthritis. J Pediatr. 1997;130:551.
del Rosario J.F., Orenstein S.R., Bhargava S., et al. Thrombotic complications in pediatric inflammatory bowel disease. Clin Pediatr (Phila). 1994;33:159.
Derry C., Dale R.C., Thom M., et al. Unihemispheric cerebral vasculitis mimicking Rasmussen’s encephalitis. Neurology. 2002;58:327.
Devlin T., Gray L., Allen N.B., et al. Neuro-Behçet’s disease: Factors hampering proper diagnosis. Neurology. 1995;45:1754.
Dillon M.J. Rare vasculitic syndromes. Ann Med. 1997;29:175.
Dlamini N., Kirkham F.J. Stroke and cerebrovascular disorders. Curr Opin Pediatr. 2009;21(6):751-761.
Drachman D.A. Neurologic complications of Wegener’s granulomatosis. Arch Neurol. 1963;8:145.
Drenth J.P., Finley W.H., Breedveld G.J., et al. The primary erythermalgia-susceptibility gene is located on chromosome 2q31-32. Am J Hum Genet. 2001;68:1277.
Drenth J.P., van Genderen P.J.J., Michiels J.J. Thrombocythemic erythromelalgia, primary erythermalgia, and secondary erythermalgia: Three distinct clinicopathologic entities. Angiology. 1994;45:451.
Duzova A., Bakkaloglu A. Central nervous system involvement in pediatric rheumatic diseases: current concepts in treatment. Curr Pharm Des. 2008;14(13):1295-1301.
Ellison D., Gatter K., Heryet A., et al. Intramural platelet deposition in cerebral vasculopathy of systemic lupus erythematosus. J Clin Pathol. 1993;46:37.
Emery E.S., Vieco P.T. Sydenham chorea: Magnetic resonance imaging reveals permanent basal ganglia injury. Neurology. 1997;48:531.
Engel D.G., Gospe S.M.Jr, Tracy K.A., et al. Fatal infantile polyarteritis nodosa with predominant central nervous system involvement. Stroke. 1995;26:699.
Erdem E., Carlier R., Idir A.B., et al. Gadolinium-enhanced MRI in central nervous system Behçet’s disease. Neuroradiology. 1993;35:142.
Escalante A., Stimmler M.M. Trimethoprim-sulfamethoxazole induced meningitis in systemic lupus erythematosus. J Rheumatol. 1992;19:800.
Espinosa G., Mendizábal A., Mínguez S., et al. Transverse myelitis affecting more than 4 spinal segments associated with systemic lupus erythematosus: clinical, immunological, and radiological characteristics of 22 patients. Semin Arthritis Rheum. 2010;39(4):246-256.
Eun S.H., Kim S.J., Cho D.S., et al. Cerebral vasculitis in Henoch-Schonlein purpura: MRI and MRA findings, treated with plasmapheresis alone. Pediatr Int. 2003;45:484.
Eyanson S., Passo M.H., Aldo-Benson M.A., et al. Methylprednisolone pulse therapy for nonrenal lupus erythematosus. Ann Rheum Dis. 1980;39:377.
Fager D.B., Bigler J.A., Simonds J.P. Polyarteritis nodosa in infancy and childhood. J Pediatr. 1951;39:65.
Falcini F., Taccetti G., Trapani S., et al. Primary antiphospholipid syndrome: a report of two pediatric cases. J Rheumatol. 1991;18:1085-1087.
Falko J.M., Williams J.C., Harvey D.G., et al. Hyperlipoproteinemia and multifocal neurologic dysfunction in systemic lupus erythematosus. J Pediatr. 1979;95:523.
Farooki Z.Q., Brough A.J., Green E.W. Necrotizing arteritis. Am J Dis Child. 1974;128:837.
Faustino P.C., Terreri M.T.R.A., da Rocha A.J., et al. Clinical, laboratory, psychiatric and magnetic resonance findings in patients with Sydenham chorea. Neuroradiology. 2003;45:456.
Fink C.W., Cimaz R. Early onset sarcoidosis: Not a benign disease. J Rheumatol. 1997;24:174.
Fish A.J., Blau E.B., Westberg N.G., et al. Systemic lupus erythematosus within the first two decades of life. Am J Med. 1977;62:99.
Fishel B., Wollschlaeger B., Tishler M. Epileptic seizures as a neurological complication of Reiter’s syndrome. Clin Exp Rheumatol. 1995;13(4):533.
Flechtner K.M., Baum K. Mixed connective tissue disease: Recurrent episodes of optic neuropathy and transverse myelopathy. Successful treatment with plasmapheresis. J Neurol Sci. 1994;126:146.
Ford R.G., Siekert R.G. Central nervous system manifestations of periarteritis nodosa. Neurology. 1965;15:114.
Fox R.I. Treatment of the patient with Sjögren’s syndrome. Rheum Dis Clin North Am. 1992;18:699.
Fragoso-Loyo H.E., Sanchez-Guerrero J. Effect of severe neuropsychiatric manifestations on short-term damage in systemic lupus erythematosus. J Rheumatol. 2007;34:76-80.
Frye R.E. Recurrent aseptic encephalitis in periodic fever, aphthous stomatitis, pharyngitis and adenopathy (PFAPA) syndrome. Pediatr Infect Dis J. 2006;25(5):463-465.
Fujikawa S., Suemitsu T. Behçet’s disease in children: A nationwide retrospective survey in Japan. Acta Paediatr Jpn. 1997;39:285.
Gallagher M., Quinones K., Cervantes-Castañeda R.A., et al. Biological response modifier therapy for refractory childhood uveitis. Br J Ophthalmol. 2007;91(10):1341-1344.
Galli M., Luciani D., Bertolini G., et al. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome. A systematic review of the literature. Blood. 2003;101:1827.
Ganji S., Duncan M.C., Frazier E. Sydenham’s chorea: Clinical, EEG, CT scan, and evoked potential studies. Clin Electroencephalogr. 1988;19:114.
Garvey M.A., Giedd J., Swedo S.E. PANDAS: The search for environmental triggers of pediatric neuropsychiatric disorders. Lessons from rheumatic fever. J Child Neurol. 1998;13:413.
Gatti F.M., Rosenheim E. Sydenham’s chorea associated with transient intellectual impairment. Am J Dis Child. 1969;118:915.
Gause C., Morris C., Vernekar S., et al. Antineuronal antibodies in OCD: comparisons in children with OCD-only, OCD+chronic tics and OCD+PANDAS. J Neuroimmunol. 2009;214(1–2):118-124.
Georganas C., Ioakimidis D., Iatrou C., et al. Relapsing Wegener’s granulomatosis: Successful treatment with cyclosporin A. Clin Rheumatol. 1996;15:189.
Gerraty R.P., McKelvie P.A., Byrne E. Aseptic meningoencephalitis in primary Sjögren’s syndrome. Response to plasmapheresis and absence of CNS vasculitis at autopsy. Acta Neurol Scand. 1993;88:309.
Gibb W.R., Lees A.J. Tendency to late recurrence following rheumatic chorea. Neurology. 1989;39:999.
Girardin M., Waschke K.A., Seidman E.G. A case of acute loss of vision as the presenting symptom of Crohn’s disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4:695-698.
Goder G. Periarteritis nodosa in childhood. Z Gesamte Exp Med. 1956;11:652.
Gold A.P., Yahr M.D. Childhood lupus erythematosus: A clinical and pathological study of the neurological manifestations. Trans Am Neurol Assoc. 1960;85:96.
Goldenberg J., Ferraz M.B., Fonseca A.S., et al. Sydenham chorea: Clinical and laboratory findings. Analysis of 187 cases. Rev Paul Med. 1992;110:152.
Gordon R.M., Silverstein A. Neurologic manifestations in progressive systemic sclerosis. Arch Neurol. 1970;22:126.
Graf W.D., Milstein J.M., Sherry D.D. Stroke and mixed connective tissue disease. J Child Neurol. 1993;8:256.
Green L.N. Corticosteroids in the treatment of Sydenham’s chorea. Arch Neurol. 1978;35:53.
Green L., Vinker S., Amital H., et al. Pseudotumor cerebri in systemic lupus erythematosus. Semin Arthritis Rheum. 1995;25:103.
Gronemeyer P.S., Demello D.E. Takayasu’s disease with aneurysms of right common iliac artery in iliocaval fistula in a young infant. Case report and review of the literature. Pediatrics. 1982;69:626.
Guillevin L., Lhote F., Cohen P., et al. Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine (Baltimore). 1995;74:238.
Haas J.P., Metzler M., Ruder H., et al. An unusual manifestation of Wegener’s granulomatosis in a 4-year-old girl. Pediatr Neurol. 2002;27:71.
Hackett E.R., Martinez R.D., Larson P.F., et al. Optic neuritis in systemic lupus erythematosus. Arch Neurol. 1974;31:9.
Hadchouel M., Prieur A., Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis. Possible relationship to drugs or infection. J Pediatr. 1985;106:561.
Hadler N.M., Gerwin R.D., Frank M.M., et al. The fourth component of complement in the cerebrospinal fluid in systemic lupus erythematosus. Arthritis Rheum. 1973;16:507.
Halbreich U., Assael M., Kauly N., et al. Rheumatic brain disease: A disease in its own right. J Nerv Ment Dis. 1976;163:24.
Hankey G.J. Isolated angiitis/angiopathy of the central nervous system. Cerebrovasc Dis. 1991;1:2.
Hanly J.G., Walsh N.M., Sangalang V. Brain pathology in systemic lupus erythematosus. J Rheumatol. 1992;19:732.
Hanmer O., Saltissi D. Response of acute cerebral lupus in childhood to pulse methylprednisolone in reduced dosage. Ann Rheum Dis. 1986;45:606.
Hansen J.R., McCray P.B., Bale J.F., et al. Reye syndrome associated with aspirin therapy for systemic lupus erythematosus. Pediatrics. 1985;76:202.
Harel L., Sandborg C., Lee T., et al. Neuropsychiatric manifestations in pediatric systemic lupus erythematosus and association with antiphospholipid antibodies. J Rheumatol. 2006;33:1873-1877.
Harisdangkul V., Doorenbos D., Subramony S.H. Lupus transverse myelopathy: Better outcome with early recognition and aggressive high-dose intravenous corticosteroid pulse treatment. J Neurol. 1995;242:326.
Harrison C.M., Goddard J.M., Rittey C.D. The use of regional anaesthetic blockade in a child with recurrent erythromelalgia. Arch Dis Child. 2003;88:65.
Heinrich D., Gordjani N., Trusen A., et al. Necrotizing sarcoid granulomatosis: A rarity in childhood. Pediatr Pulmonol. 2003;35:407.
Herd J.K., Medhi M., Uzendoski U.M., et al. Chorea associated with SLE: Report of 2 cases and review of literature. Pediatrics. 1978;61:308.
Hietaharju A., Jantti V., Korpela M., et al. Central nervous system involvement and psychiatric manifestations in systemic sclerosis (scleroderma): Clinical and neurophysiological evaluation. Acta Neurol Scand. 1993;87:382.
Hiraki L.T., Benseler S.M., Tyrrell P.N., et al. Clinical and laboratory characteristics and long-term outcome of pediatric systemic lupus erythematosus: a longitudinal study. J Pediatr. 2008;152(4):550-556.
Hirohata S., Hirose S., Miyamoto T. Cerebrospinal fluid IgA, IgM and IgG indexes in systemic lupus erythematosus. Arch Intern Med. 1985;145:1843.
Hisashi K., Komune S., Taira T., et al. Anticardiolipin antibody-induced sudden profound sensorineural hearing loss. Am J Otolaryngol. 1993;14:275.
Hoffman G.S. Takayasu arteritis: Lessons from the American National Institutes of Health experience. Int J Cardiol. 1996;54:S99.
Hoffman G.S. Treatment of resistant Takayasu’s arteritis. Rheum Dis Clin North Am. 1995;21:73.
Horneff G., Burgos-Vargas R. Juvenile idiopathic arthritis. Subgroup characteristics and comparisons between rheumatoid arthritis-like subgroups and ankylosing spondylitis-like subgroups. Clin Exp Rheumatol. 2009;27(4 Suppl 55):S131-S138.
Hornig M., Briese T., Buie T., et al. Lack of association between measles virus vaccine and autism with enteropathy: a case-control study. PLoS ONE. 2008;3(9):e3140.
Horton T.M., Stone J.D., Yee D., et al. Case series of thrombotic thrombocytopenic purpura in children and adolescents. J Pediatr Hematol Oncol. 2003;25:336.
Hoyne A.L., Steiner M.D. Periarteritis nodosa complicating scarlet fever. Am J Dis Child. 1940;59:1271.
Huisman T.A., Wohlrab G., Nadal D., et al. Unusual presentations of neuroborreliosis (Lyme disease) in childhood. J Comput Assist Tomog. 1999;23:39.
International Study Group for Behçet’s Disease. Evaluation of diagnostic (“classification”) criteria in Behçet’s Disease: Towards internationally agreed criteria. Br J Rheumatol. 1992;31:299.
Ishikawa O., Ohnishi K., Miyachi Y., et al. Cerebral lesions in systemic lupus erythematosus detected by magnetic resonance imaging. Relationship to anticardiolipin antibody. J Rheumatol. 1994;21:87.
Isshi K., Hirohata S., Hashimoto T., et al. Systemic lupus erythematosus presenting with diffuse low density lesions in the cerebral white matter on computed axial tomography scans: Its implication in the pathogenesis of diffuse central nervous system lupus. J Rheumatol. 1994;21:1758.
Jacob J.C. Systemic lupus erythematosus in childhood: Report of 35 cases with discussion of 7 apparently induced by anticonvulsant medication, and of prognosis and treatment. Pediatrics. 1963;32:257.
Jain S., Sharma N., Singh S., et al. Takayasu arteritis in children and young Indians. Int J Cardiol. 2000;75(Suppl 1):S153.
Jayne D.R., Lockwood C.M. Intravenous immunoglobulin as sole therapy for systemic vasculitis. Br J Rheumatol. 1996;35:1150.
Jones J.M., Martinez A.J., Joshi V.V., et al. Systemic lupus erythematosus. Arch Pathol. 1975;99:152.
Jorgensen H.P., Sondergaard J. Pathogenesis of erythromelalgia. Arch Dermatol. 1978;114:112.
Kaell A.T., Shetty M., Lee B.C., et al. The diversity of neurologic events in systemic lupus erythematosus. Arch Neurol. 1986;43:273.
Kapitsinou P.P., Boletis J.N., Skopouli F.N., et al. Lupus nephritis: Treatment with mycophenolate mofetil. Rheumatol (Oxford). 2004;43:377.
Kari J.A., Shah V., Dillon M.J. Behcet’s disease in UK children: Clinical features and treatment including thalidomide. Rheumatology. 2001;40:933.
Karim S., Majithia V. Devic’s syndrome as initial presentation of systemic lupus erythematosus. Am J Med Sci. 2009;338(3):245-247.
Keefe E.B., Bardana E.J.Jr, Harbeck R.J., et al. Lupus meningitis antibody to deoxyribonucleic acid (DNA) and DNA/anti-DNA complexes in cerebrospinal fluid. Ann Intern Med. 1974;80:58.
Khamashta M.A., Cuadrado M.J., Mujic F., et al. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993.
Kim R.C. Rheumatoid disease with encephalopathy. Ann Neurol. 1980;7:86.
Kim S.K., An J.Y., Park M.S., et al. A Case Report of Reiter’s Syndrome with Progressive Myelopathy. J Clin Neurol. 2007;3(4):215-218.
Kissel J.T. Neurologic manifestations of vasculitis. Neurol Clin. 1989;7:655.
Kohrman M.H., Huttenlocher P.R. Takayasu arteritis. A treatable cause of stroke in infancy. Pediatr Neurol. 1986;2:154.
Kok J., Bosseray A., Brion J.P., et al. Chorea in a child with Churg-Strauss syndrome. Stroke. 1993;24:1263.
Koné-Paut I., Yurdakul S., Bahabri S.A., et al. Clinical features of Behçet’s disease in children: An international collaborative study of 86 cases. J Pediatr. 1998;132:721.
Kornreich H.K., King K.K., Bernstein B.H., et al. Scleroderma in childhood. Arthritis Rheum. 1977;20:343.
Kotajima L., Aotsuka S., Sumiya M., et al. Clinical features of patients with juvenile onset mixed connective tissue disease: Analysis of data collected in a nationwide collaborative study in Japan. J Rheumatol. 1996;23:1088.
Kovacs J.A., Urowitz M.B., Gladman D.D. Dilemmas in neuropsychiatric lupus. Rheum Dis Clin North Am. 1993;19:795.
Krause I., Uziel Y., Guedj D., et al. Childhood Behçet’s disease: Clinical features and comparison with adult-onset disease. Rheumatology. 1999;38:457.
Kukla L.F., Reddy C., Silkalns G., et al. Systemic lupus erythematosus presenting as chorea. Arch Dis Child. 1978;53:345.
Kuroe K., Kurahashi K., Nakano I., et al. A neuropathological study of a case of lupus erythematosus with chorea. J Neurol Sci. 1994;123:59.
Kwong T., Valderrama E., Paley C., et al. Systemic necrotizing vasculitis associated with childhood sarcoidosis. Semin Arthritis Rheum. 1994;23:388.
Langford C.A. Wegener’s granulomatosis: Current and upcoming therapies. Arthritis Res Ther. 2003;5:180.
Lang H., Anttila R., Svekus A., et al. EEG findings in juvenile rheumatoid arthritis and other connective tissue diseases in children. Acta Paediatr Scand. 1974;63:373.
Lanthier S., Lortie A., Michaud J., et al. Isolated angiitis of the CNS in children. Neurology. 2001;56:837.
Lawlor E.R., Webb D.W., Hill A., et al. Thrombotic thrombocytopenic purpura: A treatable cause of childhood encephalopathy. J Pediatr. 1997;130:313.
Lee M., Epstein F.J., Rezai A., et al. Nonneoplastic intramedullary spinal cord lesions mimicking tumors. Neurosurgery. 1998;43:788.
Lehman T.J. Systemic and localized scleroderma in children. Curr Opin Rheumatol. 1996;8:576.
Leiba H., Siatkowski R.M., Culbertson W.W., et al. Neurosarcoidosis presenting as an intracranial mass in childhood. J Neuroophthalmol. 1996;16:269.
Leonard H.L., Swedo S.E., Lenane M.C., et al. A 2- to 7-year follow-up study of 54 obsessive-compulsive children and adolescents. Arch Gen Psychiatry. 1993;50:429.
Lepore L., Marchetti F., Facchini S., et al. Drug-induced systemic lupus erythematosus associated with etanercept therapy in a child with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2003;21:276.
Levy D.M., Ardoin S.P., Schanberg L.E. Neurocognitive impairment in children and adolescents with systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2009;5(2):106-114.
Levy D.M., Massicotte M.P., Harvey E., et al. Thromboembolism in paediatric lupus patients. Lupus. 2003;12:741.
Lhote F., Guillevin L. Polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome. Rheum Dis Clin North Am. 1995;21:911.
Lie J.T. Bilateral juvenile temporal arteritis. J Rheumatol. 1995;22:774.
Lie J.T., Gordon L.P., Titus J.L. Juvenile temporal arteritis: Biopsy study of 4 cases. JAMA. 1975;234:496.
Ling W., Oftedal G., Simon T. Central retinal artery occlusion in Sydenham’s chorea. Am J Dis Child. 1969;118:525.
Liou H.H., Yip P.K., Chang Y.C., et al. Allergic granulomatosis and angiitis (Churg-Strauss syndrome) presenting as prominent neurologic lesions and optic neuritis. J Rheumatol. 1994;21:2380.
Logigian E.L., Kaplan R.F., Steere A.C. Chronic neurologic manifestations of Lyme disease. N Engl J Med. 1990;323:1438.
Lossos A., River Y., Eliakim A., et al. Neurologic aspects of inflammatory bowel disease. Neurology. 1995;45:416.
Maeda M., Kobayashi M., Okamoto S., et al. Clinical observation of 14 cases of childhood polyarteritis nodosa in Japan. Acta Paediatr Jpn. 1997;39:277.
Magilavy D.B., Petty R.E., Cassidy J.T., et al. A syndrome of childhood polyarteritis. J Pediatr. 1977;91:25.
Makay B., Yilmaz S., Türkyilmaz Z., et al. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer. 2008;50(2):419-421.
Malinow K.L., Molina R., Gordon B., et al. Neuropsychiatric dysfunction in primary Sjögren’s syndrome. Ann Intern Med. 1985;103:344.
Manco-Johnson M.J., Nuss R. Lupus anticoagulant in children with thrombosis. Am J Hematol. 1995;48:240.
Mandell F., Folkman J., Matsumoto S. Erythromelalgia. Pediatrics. 1977;59:45.
Martin C.M., Uhlmann V., Killalea A., et al. Detection of measles virus in children with ileo-colonic lymphoid nodular hyperplasia, enterocolitis and developmental disorder. Mol Psychiatry. 2002;7:S47.
Martinez-Cordero E., Fonseca M.C., Aquilar-Leon D.E., et al. Juvenile systemic sclerosis. J Rheumatol. 1993;20:405.
Masi A.T., Hunder G.G., Lie J.T., et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33:1094.
Matsell D.G., Keene D.L., Jimenez C., et al. Isolated angiitis of the central nervous system in childhood. Can J Neurol Sci. 1990;17:151.
Matsumoto R., Shintaku M., Suzuki S., et al. Cerebral perivenous calcification in neuropsychiatric lupus erythematosus: A case report. Neuroradiology. 1998;40:583.
McGraw T., Kosek P. Erythromelalgia pain managed with gabapentin. Anesthesiology. 1997;86:988.
Meislin A.G., Rothfield N. Systemic lupus erythematosus in childhood: Analysis of 42 cases, with comparative data on 200 adult cases followed concurrently. Pediatrics. 1968;42:37.
Melish M.E. Kawasaki syndrome. Pediatr Rev. 1996;17:153.
Melish M.E. Kawasaki syndrome. A new infectious disease. J Infect Dis. 1981;143:317.
Meroni P.L., Moia M., Derksen R.H., et al. Venous thromboembolism in the antiphospholipid syndrome: Management guidelines for secondary prophylaxis. Lupus. 2003;12:504.
Mier R., Ansell B., Hall M.A., et al. Long term follow-up of children with mixed connective tissue disease. Lupus. 1996;5:221.
Millar A.J., Gilbert R.D., Brown R.A., et al. Abdominal aortic aneurysms in children. J Pediatr Surg. 1996;31:1624.
Miller M.L., Levinson L., Pachman L.M., et al. Abnormal muscle MRI in a patient with systemic juvenile arthritis. Pediatr Radiol. 1995;25(Suppl 1):S107.
Miller W.L., Burnett J.C.Jr. Blood vessel physiology and pathophysiology. Rheum Dis Clin North Am. 1990;16:251.
Mitkov V. Cerebral manifestations of rheumatic fever. World Neurol. 1961;2:920.
Miyakis S., Lockshin M.D., Atsumi T., et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
Mizukami K., Shiraishi H., Tanaka Y., et al. CNS changes in neuro-Behçet’s disease: CT, MR, and SPECT findings. Comput Med Imaging Graph. 1992;16:401.
Mok C.C., Lau C.S. Transverse myelopathy complicating mixed connective tissue disease. Clin Neurol Neurosurg. 1995;97:259.
Molad Y., Sidi Y., Gornish M., et al. Lupus anticoagulant: Correlation with magnetic resonance imaging of brain lesions. J Rheumatol. 1992;19:556.
Molina R., Provost T.T., Alexander E.L. Peripheral inflammatory vascular disease in Sjögren’s syndrome: Association with nervous system complications. Arthritis Rheum. 1985;28:1341.
Monfort-Gourand M., Chokre R., Dubiez M., et al. Inflammatory pseudotumor of the orbit and suspected sarcoidosis. Arch Pediatr. 1996;3:697.
Montealegre Sanchez G.A., Hashkes P.J. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev Med Child Neurol. 2009;51(6):420-428.
Moorthy A.V., Chesney R.W., Segar W.E., et al. Wegener’s granulomatosis in childhood: Prolonged survival following cytotoxic therapy. J Pediatr. 1977;91:616.
Morales E., Pineda C., Martínez-Lavín M. Takayasu’s arteritis in children. J Rheumatol. 1991;18:1081.
Morris C.M., Pardo-Villamizar C., Gause C.D., et al. Serum autoantibodies measured by immunofluorescence confirm a failure to differentiate PANDAS and Tourette syndrome from controls. J Neurol Sci. 2009;276(1–2):45-48.
Murch S.H. Local and systemic effects of macrophage cytokines in intestinal inflammation. Nutrition. 1998;14:780.
Murch S.H., Anthony A., Casson D.H., et al. Retraction of an interpretation. Lancet. 2004;363:750.
Murphy T.K., Goodman W.K., Fudge M.W., et al. B lymphocyte antigen D8/17: A peripheral marker for childhood-onset obsessive-compulsive disorder and Tourette’s syndrome. Am J Psychiatr. 1997;154:402.
Murray K.J., Laxer R.M. Scleroderma in children and adolescents. Rheum Dis Clin North Am. 2002;28:603.
Muscal E., Brey R.L. Neurologic manifestations of systemic lupus erythematosus in children and adults. Neurol Clin. 2010;28(1):61-73.
Muscal E., Traipe E., de Guzman M.M., et al. MR imaging findings suggestive of posterior reversible encephalopathy syndrome in adolescents with systemic lupus erythematosus. Pediatr Radiol. 2010. Epub ahead of print
Nadeau S.E. Neurologic manifestations of systemic vasculitis. Neurol Clin. 2002;20:123.
Nagahiro S., Mantani A., Yamada K., et al. Multiple cerebral arterial occlusions in a young patient with Sjögren’s syndrome: Case report. Neurosurgery. 1996;38:592.
Nasir S., Kerr D.A., Birnbaum J. Nineteen episodes of recurrent myelitis in a woman with neuromyelitis optica and systemic lupus erythematosus. Arch Neurol. 2009;66(9):1160-1163.
Ndiaye I.C., Rassi S.J., Wiener-Vacher S.R. Cochleovestibular impairment in pediatric Cogan’s syndrome. Pediatrics. 2002;109:E38.
Neuwelt C.M., Lacks S., Kaye B.R., et al. Role of intravenous cyclophosphamide in the treatment of severe neuropsychiatric systemic lupus erythematosus. Am J Med. 1995;98:32.
Niederwieser G., Bonelli R.M., Pongratz R., et al. Cerebral vasculitis associated with Reiter’s syndrome. J Neurol. 2001;248(11):988-989.
Nikitakis N.G., Rivera H., Lariccia C., et al. Primary Sjögren syndrome in childhood: Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:42.
Nimelstein S.H., Brody S., McShane D., et al. Mixed connective tissue disease: A subsequent evaluation of the original 25 patients. Medicine. 1980;59:239.
Nishimura A., Yamazaki H., Fuchigami T., et al. [A childhood case of systemic lupus erythematosus associated with myasthenia gravis]. No To Hattatsu (in Japanese). 1997;29:390.
Nobili F., Cutolo M., Sulli A., et al. Brain functional involvement by perfusion SPECT in systemic sclerosis and Behçet’s disease. N Y Acad Sci. 2002;966:409.
Nordborg E., Nordborg C., Malmvall B.E., et al. Giant cell arteritis. Rheum Dis Clin North Am. 1995;21:1013.
Norris D.G., Colon A.R., Stickler G.B. Systemic lupus erythematosus in children: The complex problems of diagnosis and treatment encountered in 101 such patients at the Mayo Clinic. Clin Pediatr. 1977;16:774.
O’Connor D., Bernstein B., Hanson V., et al. Disease of central nervous system in juvenile rheumatoid arthritis. Arthritis Rheum. 1980;23:727.
O’Duffy J.D., Goldstein N.P. Neurologic involvement in seven patients with Behçet’s disease. Am J Med. 1976;61:170.
Oetgen W.J., Boice J.A., Lawless O.J. Mixed connective tissue disease in children and adolescents. Pediatrics. 1981;67:333.
Ohtsuka T., Saito Y., Hasegawa M., et al. Central nervous system disease in a child with primary Sjögren syndrome. J Pediatr. 1995;127:961.
Okano Y. Antinuclear antibody in systemic sclerosis (scleroderma). Rheum Dis Clin North Am. 1996;22:709.
Olfat M., Al-Mayouf S.M. Cogan’s syndrome in childhood. Rheumatol Int. 2001;20:246.
Olsen N.J., King L.E.Jr, Park J.H. Muscle abnormalities in scleroderma. Rheum Dis Clin North Am. 1996;22:783.
Ong V.H., Denton C.P. Innovative therapies for systemic sclerosis. Curr Opin Rheumatol. 2010. Epub ahead of print
Orlowski J.P., Clough J.D., Dyment P.G. Wegener’s granulomatosis in the pediatric age group. Pediatrics. 1978;61:83.
Østergaard J.R., Storm K. Neurologic manifestations of Schönlein-Henoch purpura. Acta Paediatr Scand. 1991;80:339.
Ostuni P.A., Ianniello A., Sfriso P., et al. Juvenile onset of primary Sjögren’s syndrome: Report of 10 cases. Clin Exp Rheumatol. 1996;14:689.
Ozdemir O., Sezgin I., Kurtulgan H.K., et al. Prevalence of known mutations in the MEFV gene in a population screening with high rate of carriers. Mol Biol Rep. 2010. Epub ahead of print
Ozen S. Juvenile polyarteritis: Is it a different disease? J Rheumatol. 2004;31:831.
Ozen S., Besbas N., Saatci U., et al. Diagnostic criteria for polyarteritis nodosa in childhood. J Pediatr. 1992;120:206.
Pantell R.H., Goodman B.W.Jr. Takayasu’s arteritis. The relationship with tuberculosis. Pediatrics. 1981;67:84.
Parikh S., Swaiman K.F., Kim Y. Neurologic characteristics of childhood lupus erythematosus. Pediatr Neurol. 1995;13:198.
Passam F., Krillis S. Laboratory tests for the antiphospholipid syndrome: current concepts. Pathology. 2004;36:129-138.
Pattishall E.N., Kendig E.L.Jr. Sarcoidosis in children. Pediatr Pulmonol. 1996;22:195.
Peal A.R., Gildersleeve N., Lucchesi P.F. Periarteritis nodosa complicating scarlet fever. Am J Dis Child. 1946;72:310.
Penn A.S., Rowan A.J. Myelopathy in systemic lupus erythematosus. Arch Neurol. 1968;18:337.
Petri M., Roubenoff R., Dallal G.E., et al. Plasma homocysteine as a risk factor for atherothrombotic events in systemic lupus erythematosus. Lancet. 1996;348:1120.
Petty R.E., Southwood T.R., Manners P., et al. International League of Associations for Theumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J Rheumatol. 2004;31:390.
Peyronnard J., Charron L., Beaudet F., et al. Vasculitic neuropathy in rheumatoid disease and Sjögren syndrome. Neurology. 1982;32:839.
Pope J.E. Treatment of systemic sclerosis. Rheum Dis Clin North Am. 1996;22:893.
Press J., Palayew K., Laxer R.M., et al. Antiribosomal P antibodies in pediatric patients with systemic lupus erythematosus and psychosis. Arthritis Rheum. 1996;39:671.
Prieur A.M. A recently recognised chronic inflammatory disease of early onset characterised by the triad of rash, central nervous system involvement and arthropathy. Clin Exp Rheumatol. 2000;19:103.
Propper D.J., Bucknall R.C. Acute transverse myelopathy complicating systemic lupus erythematosus. Ann Rheum Dis. 1989;48:512.
Provenzale J., Bouldin T.W. Lupus-related myelopathy: Report of three cases and review of the literature. J Neurol Neurosurg Psychiatry. 1992;55:830.
Provenzale J.M., Barboriak D.P., Gaensler E.H.L., et al. Lupus-related myelitis: Serial MR findings. Am J Neuroradiol. 1994;15:1911.
Quartier P., Taupin P., Bourdeaut F., et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003;48:1093.
Quirk M.E., Young M.H. The impact of JRA on children, adolescents, and their families: Current research and implication for future studies. Arthritis Care Res. 1990;3:36.
Rachelefsky G.S., Kar N.C., Coulson A., et al. Serum enzyme abnormalities in juvenile rheumatoid arthritis. Pediatrics. 1976;58:730.
Rakover Y., Adar J., Tal I., et al. Behçet disease: Long-term follow-up of three children and review of the literature. Pediatrics. 1989;83:986.
Rauck R.L., Naveira F., Speight K.L., et al. Refractory idiopathic erythromelalgia. Anesth Analg. 1996;82:1097.
Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548.
Ravelli A., Felici E., Magni-Manzoni S., et al. Patients with antinuclear antibody-positive juvenile idiopathic arthritis constitute a homogeneous subgroup irrespective of the course of joint disease. Arthritis Rheum. 2005;52(3):826-832.
Ravelli A., Martini A. Antiphospholipid antibody syndrome in pediatric patients. Rheum Dis Clin North Am. 1997;23:657.
Rayes H.A., Al-Sheikh A., Al Dalaan A., et al. Mixed connective tissue disease: the King Faisal Specialist Hospital experience. Ann Saudi Med. 2002;22(1–2):43-46.
Reichlin M. Ribosomal P antibodies and CNS lupus. Lupus. 2003;12:916.
Reinhold-Keller E., De-Groot K., Rudert H., et al. Response to trimethoprim/sulfamethoxazole in Wegener’s granulomatosis depends on the phase of disease. Q J Med. 1996;89:15.
Rhodes V.J. Physical therapy management of patients with juvenile rheumatoid arthritis. Phys Ther. 1991;71:910.
Richardson M.L., Helms C.A., Vogler J.B., et al. Skeletal changes in neuromuscular disorders mimicking juvenile rheumatoid arthritis and hemophilia. Am J Roentgenol. 1984;143:893.
Rigante D., Battaglia D., Contaldo I., et al. Longstanding epileptic encephalopathy and linear localized scleroderma: two distinct pathologic processes in an adolescent. Rheumatol Int. 2008;28(9):925-929.
Rinkel G.J.E., Wijdicks E.F.M., Hene R.J. Stroke in relapsing thrombotic thrombocytopenic purpura. Stroke. 1991;22:1087.
Ritchlin C.T., Chabot R.J., Alper K., et al. Quantitative electroencephalography: A new approach to the diagnosis of cerebral dysfunction in systemic lupus erythematosus. Arthritis Rheum. 1992;35:1330.
Ritter F.J., Seay A.R., Lahey M.E. Peripheral mononeuropathy complicating anaphylactoid purpura. J Pediatr. 1983;103:77.
Robertson W.C.Jr, Smith C.D. Sydenham’s chorea in the age of MRI: A case report and review. Pediatr Neurol. 2002;27:65.
Robson M.G., Walport M.J., Davies K.A. Systemic lupus erythematosus and acute demyelinating polyneuropathy. Br J Rheumatol. 1994;33:1074.
Rosenberg A.M. Uveitis associated with childhood rheumatic diseases. Curr Opin Rheumatol. 2002;14:542.
Rosenberger A., Adler O.B., Haim S. Radiologic aspects of Behçet’s disease. Radiology. 1982;144:261.
Rottem M., Fauci A.S., Hallahan C.W., et al. Wegener granulomatosis in children and adolescents: Clinical presentation and outcome. J Pediatr. 1993;122:26.
Rubin R.L. Drug-induced lupus. In: Wallace D.J., Hahn B.H., editors. Dubois’ lupus erythematosus. Baltimore: Williams & Wilkins, 1997.
Ruiz-Irastorza G., Khamashta M.A., Castellino G., et al. Systemic lupus erythematosus. Lancet. 2001;357(9261):1027-1032.
Sabharwal U.K., Keogh L.H., Weisman M.H., et al. Granulomatous angiitis of the nervous system. Case report and review of the literature. Arthritis Rheum. 1982;25:342.
Sanna G., Bertolaccini M.L., Mathieu A. Central nervous system lupus: A clinical approach to therapy. Lupus. 2003;12:935.
Saurenmann R.K., Levin A.V., Feldman B.M., et al. Risk factors for development of uveitis differ between girls and boys with juvenile idiopathic arthritis. Arthritis Rheum. 2010. Epub ahead of print
Schieken R., Anderson W.T., Anthony C.L. Diplopia: A rare manifestation of chorea. Am J Dis Child. 1973;125:586.
Schor N.F. Neurology of systemic autoimmune disorders: A pediatric perspective. Semin Pediatr Neurol. 2000;7:108.
Scolding N.J., Joseph F.G. The neuropathology and pathogenesis of systemic lupus erythematosus. Neuropathol Appl Neurobiol. 2002;28(3):173-189.
Scott T.F. Neurosarcoidosis: Progress and clinical aspects. Neurology. 1993;43:8.
Sehgal M., Swanson J.W., DeRemee R.A., et al. Neurologic manifestations of Churg-Strauss syndrome. Mayo Clin Proc. 1995;70:337.
Sequeira W. Rheumatic manifestations of sarcoidosis. In Kelley W.N., Harris E.D.Jr, Ruddy S., et al, editors: Textbook of rheumatology, ed 5, Philadelphia: WB Saunders, 1997.
Shaikov A.V., Maximov A.A., Speransky A.I., et al. Repetitive use of pulse therapy with methylprednisolone and cyclophosphamide in addition to oral methotrexate in children with systemic juvenile rheumatoid arthritis – preliminary results of a long-term study. J Rheumatol. 1992;19:612.
Shannon K.M., Fenichel G.M. Pimozide treatment of Sydenham’s chorea. Neurology. 1990;40:186.
Sharma B.K., Jain S., Radotra B.D. An autopsy study of Takayasu arteritis in India. Int J Cardiol. 1998;66(Suppl 1):S85.
Sharp G.C. Mixed connective tissue disease. Bull Rheum Dis. 1975;25:828.
Sharp G.C., Irvin W.S., Tan E.M., et al. Mixed connective tissue disease: An apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52:148.
Shenker D.M., Grossman H.J., Klawans H.L. Treatment of Sydenham’s chorea with haloperidol. Dev Med Child Neurol. 1973;15:19.
Sher J.H., Pertschuk L.P. Immunoglobulin G deposits in the choroid plexus of a child with systemic lupus erythematosus. J Pediatr. 1974;85:385.
Shulman S.T., De lnocencio J., Hirsch R. Kawasaki disease. Pediatr Clin North Am. 1995;42:1205.
Sibbitt W.L.Jr, Brandt J.R., Johnson C.R., et al. The incidence and prevalence of neuropsychiatric syndromes in pediatric onset systemic lupus erythematosus. J Rheumatol. 2002;29:1536.
Sicotte N.L., Voskuhl R.R. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology. 2001;57:1885.
Silverman E. What’s new in the treatment of pediatric SLE? J Rheumatol. 1996;23:1657.
Silverman E.D., Cawkwell G.D., Lovell D.J., et al. Intravenous immunoglobulin in the treatment of systemic juvenile rheumatoid arthritis: A randomized placebo controlled trial. Pediatric Rheumatology Collaborative Study Group. J Rheumatol. 1994;21:2353.
Silverman E.D., Miller J.J., Bernstein B., et al. Consumption coagulopathy associated with systemic juvenile rheumatoid arthritis. J Pediatr. 1983;103:872.
Silverstein A. Thrombotic thrombocytopenic purpura: The initial neurological manifestations. Arch Neurol. 1968;18:358.
Singsen B.H., Fishman L., Hanson V. Antinuclear antibodies and lupus-like syndromes in children receiving anticonvulsants. Pediatrics. 1976;57:529.
Singsen B.H., Swanson V.L., Bernstein B.H., et al. A histologic evaluation of mixed connective tissue disease in childhood. Am J Med. 1980;68:710.
Small P. Giant cell arteritis presenting as a bilateral stroke. Arthritis Rheum. 1984;27:819.
Smith R.W., Ellison D.W., Jenkins E.A., et al. Cerebellum and brainstem vasculopathy in systemic lupus erythematosus: Two clinico-pathological cases. Ann Rheum Dis. 1994;53:327.
Sneller M.C., Dale J.K., Straus S.E. Autoimmune lymphoproliferative syndrome. Curr Opin Rheumatol. 2003;15(4):417-421.
Sneller M.C., Hoffman G.S., Talar-Williams C., et al. An analysis of forty-two Wegener’s granulomatosis patients treated with methotrexate and prednisone. Arthritis Rheum. 1995;38:608.
Stabile A., Bertoni B., Ansuini V., et al. The clinical spectrum and treatment options of macrophage activation syndrome in the pediatric age. Eur Rev Med Pharmacol Sci. 2006;10(2):53-59.
Steere A.C. Lyme disease. N Engl J Med. 1989;321:586.
Stegeman C.A., Tervaert J.W.C., de Jong P.E., et al. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-trimoxazole Wegener Study Group. N Engl J Med. 1996;335:16.
Stehbens J.A., MacQueen J.C. The psychological adjustment of rheumatic fever patients with and without chorea comparisons ten years later. Clin Pediatr. 1972;11:638.
Steinlin M.I., Blaser S.I., Gilday D.L., et al. Neurologic manifestations of pediatric systemic lupus erythematosus. Pediatr Neurol. 1995;13:191.
Steup-Beekman G., Steens S., van Buchem M., et al. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus. 2007;16(5):329-334.
Stollerman G.H. Rheumatic fever. In: Kelley W.N., Harris E.D.Jr, Ruddy S., et al, editors. Textbook of Rheumatology. Philadelphia: WB Saunders, 1985.
Swedo S.E. Sydenham’s chorea. A model for childhood autoimmune neuropsychiatric disorders. JAMA. 1994;272:1788.
Swedo S.E., Leonard H.L., Garvey M., et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: Clinical description of the first 50 cases. Am J Psychiatry. 1998;155:264.
Swedo S.E., Leonard H.L., Mittleman B.B., et al. Identification of children with pediatric autoimmune neuropsychiatric disorder associated with streptococcal infections by a marker associated with rheumatic fever. Am J Psychiatry. 1997;154:110.
Swedo S.E., Leonard H.L., Schapiro M.B., et al. Sydenham’s chorea: Physical and psychological symptoms of St. Vitus dance. Pediatrics. 1993;91:706.
Swedo S.E., Rapoport J.L., Cheslow D.L., et al. High prevalence of obsessive-compulsive symptoms in patients with Sydenham’s chorea. Am J Psychiatry. 1989;146:246.
Szer I.S. Henoch-Schönlein purpura: When and how to treat. J Rheumatol. 1996;23:1661.
Szer I.S., Jacobs J.C. Pediatric systemic lupus erythematosus. In Lahita R.G., editor: Systemic lupus erythematosus, ed 2, New York: Churchill Livingstone, 1992.
Szer I.S., Miller J.H., Rawlings D., et al. Cerebral perfusion abnormalities in children with central nervous system manifestations of lupus detected by single photon emission computed tomography. J Rheumatol. 1993;20:2143.
Szer I.S., Taylor E., Steere A.C. The long-term course of Lyme arthritis in children. N Engl J Med. 1991;325:159.
Tabata Y., Kobayashi I., Kawamura N., et al. Central nervous system manifestations after steroid pulse therapy in systemic lupus erythematosus. Eur J Pediatr. 2002;161:503.
Takei S., Maeno N., Shigemori M., et al. Clinical features of Japanese children and adolescents with systemic lupus erythematosus: Results of 1980-1994 survey. Acta Paediatr Jpn. 1997;39:250.
Tan E.M. Systemic autoimmunity and antinuclear antibodies. Clin Aspects Autoimmunity. 1986;1:2.
Teitel A.D., MacKenzie C.R., Stern R., et al. Laryngeal involvement in systemic lupus erythematosus. Semin Arthritis Rheum. 1992;22:203.
Tervaert J.W.C., Kallenberg C. Neurologic manifestations of systemic vasculitides. Rheum Dis Clin North Am. 1993;19:913.
Thompson C.E., Damon L.E., Ries C.A., et al. Thrombotic microangiopathies in the 1980’s: Clinical features, response to treatment, and the impact of the human immunodeficiency virus epidemic. Blood. 1992;80:1890.
Tiddens H.A., van der Net J.J., de Graeff-Meeder E.R., et al. Juvenile-onset mixed connective tissue disease: Longitudinal follow-up. J Pediatr. 1993;122:191.
Totoritis M.C., Rubin R.L. Drug-induced lupus: Genetic, clinical, and laboratory features. Postgrad Med. 1985;78:149.
Toubi E., Khamashta M.A., Panarra A., et al. Association of antiphospholipid antibodies with central nervous system disease in systemic lupus erythematosus. Am J Med. 1995;99:397.
Tsai C.Y., Wu T.H., Tsai S.T., et al. Cerebrospinal fluid interleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand J Rheumatol. 1994;23:57.
Tsai H.M. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol. 2010;91:1.
Tugal-Tutkun I., Aydin-Akova Y., Güney-Tefekli E., et al. Referral patterns, demographic and clinical features, and visual prognosis of Turkish patients with sarcoid uveitis. Ocul Immunol Inflamm. 2007;15(4):337.
Tugal-Tutkun I., Urgancioglu M. Childhood-onset uveitis in Behçet’s disease: A descriptive study of 36 cases. Am J Ophthalmol. 2003;136:1114.
Ueno H., Katamura K., Hattori H., et al. Acute lethal encephalopathy in systemic juvenile rheumatoid arthritis. Pediatr Neurol. 2002;26(4):315-317.
Ulaşlı A.M., Kutlu G., Kocatürk O., et al. Posterior reversible encephalopathy during an attack of familial Mediterranean fever. Rheumatol Int. 2010. Epub ahead of print
Ungerer J.A., Horgan B., Chaitow J., et al. Psychosocial functioning in children and young adults with juvenile arthritis. Pediatrics. 1988;81:195.
Uziel Y., Feldman B.M., Krafchik B.R., et al. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136:91.
Uziel Y., Laxer R.M., Schneider R., et al. Intravenous immunoglobulin therapy in systemic juvenile rheumatoid arthritis: A follow-up study. J Rheumatol. 1996;23:910.
Uziel Y., Miller M.L., Laxer R.M. Scleroderma in children. Pediatr Clin North Am. 1995;42:1171.
Vancheeswaran R., Black C.M., David J., et al. Childhood-onset scleroderma. Arthritis Rheum. 1996;39:1041.
Vandvik I.H., Eckblad G. Mothers of children with recent onset of rheumatic disease: Associations between maternal distress psychosocial variables and the disease of the children. J Dev Behav Pediatr. 1991;12:84.
Vazquez-Cruz J., Traboulssi H., Rodriguez-De la Serna A., et al. A prospective study of chronic or recurrent headache in systemic lupus erythematosus. Headache. 1990;30:232.
Verhelst H.E., Beele H., Joos R., et al. Hippocampal atrophy and developmental regression as first sign of linear scleroderma “en coup de sabre”. Eur J Paediatr Neurol. 2008;12(6):508-511.
Verjans G.M., van Hagen P.M., van der Kooi A., et al. Vgamma9Vdelta2 T cells from eyes of patients with Behçet’s disease recognize non-peptide prenyl pyrophosphate antigens. J Neuroimmunol. 2002;130:46.
Vieira J.P., Ortet O., Barata D., et al. Lupus myelopathy in a child. Pediatr Neurol. 2002;27:303.
Vieira-Karuta S.C., Silva I.C., Liberalesso P.B., et al. Epileptic seizures and EEG features in juvenile systemic lupus erythematosus. Arq Neuropsiquiatr. 2008;66(3A):468-470.
von Scheven E., Athreya B.H., Rose C.D., et al. Clinical characteristics of antiphospholipid antibody syndrome in children. J Pediatr. 1996;129:339.
von Scheven E., Lee C., Berg B.O. Pediatric Wegener’s granulomatosis complicated by central nervous system vasculitis. Pediatr Neurol. 1998;19:317.
Wakefield A.J., Murch S.H., Anthony A., et al. Ileal lymphoid-nodular hyperplasia, non-specific colitis, and pervasive developmental disorder in children. Lancet. 1998;351:637.
Walravens P.A., Chase H.P. The prognosis of childhood systemic lupus erythematosus. Am J Dis Child. 1976;130:929.
Wardyn K.A., Ycinska K., Matuszkiewicz-Rowinska J., et al. Pseudotumour orbitae as the initial manifestation in Wegener’s granulomatosis in a 7-year-old girl. Clin Rheumatol. 2003;22:472.
Weber M., Hayem G., De Bandt M., et al. Classification of an intermediate group of patients with antiphospholipid syndrome and lupus-like disease: primary or secondary antiphospholipid syndrome. J Rheumatol. 1999;26:2131-2136.
Wechsler B., Dell’Isola B., Vidailhet M., et al. MRI in 31 patients with Behçet’s disease and neurological involvement: Prospective study with clinical correlation. J Neurol Neurosurg Psychiatry. 1993;56:793.
Wechsler B., Vidaihet M., Piette J.C., et al. Cerebral venous thrombosis in Behçet’s disease: Clinical study and long-term follow-up of 25 cases. Neurology. 1992;42:614.
Weiner D.K., Allen N.B. Large vessel vasculitis of the central nervous system in systemic lupus erythematosus: report and review of the literature. J Rheumatol. 1991;18:748-751.
Wertheimer N.M. Rheumatic’ schizophrenia. Arch Gen Psychiatry. 1961;4:579.
West S.G. Neuropsychiatric lupus. Rheum Dis Clin North Am. 1994;20:129.
West S.G., Emlen W., Wener M.H., et al. Neuropsychiatric lupus erythematosus: A 10-year prospective study on the value of diagnostic tests. Am J Med. 1995;99:153.
Wijdicks E.F. Silent brain infarct in thrombotic thrombocytopenic purpura. Stroke. 1994;25:1297.
Williams L.S., Garg B.P., Cohen M., et al. Subtypes of ischemic stroke in children and young adults. Neurology. 1997;49:1541.
Wilson W.A., Gharavi A.E., Koike T., et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
Wright D.A., Newburger J.W., Baker A., et al. Treatment of immune globulin-resistant Kawasaki disease with pulsed doses of corticosteroids. J Pediatr. 1996;128:146.
Wysenbeek A.J., Leibovici L., Zoldan J. Acute central nervous system complications after pulse steroid therapy in patients with systemic lupus erythematosus. J Rheumatol. 1990;17:1695.
Young R.S., Torretti D., Williams R.H., et al. Reye’s syndrome associated with long-term aspirin therapy. JAMA. 1984;251:754.
Yu H.H., Lee J.H., Wang L.C., et al. Neuropsychiatric manifestations in pediatric systemic lupus erythematosus: a 20-year study. Lupus. 2006;15:651-657.
Zannin M.E., Martini G., Athreya B.H., et alJuvenile Scleroderma Working Group of the Pediatric Rheumatology European Society (PRES). Ocular involvement in children with localised scleroderma: a multi-centre study. Br J Ophthalmol. 2007;91(10):1311-1314.
Zelenski J.D., Holden D., Calabrese L.H. Central nervous system vasculitis in Behçet’s syndrome: Angiographic improvement after therapy with cytotoxic agents. Arthritis Rheum. 1989;32:217.