Neuroepithelial neoplasms displaying neuronal features



Neuroblast-like cells are present in embryonal neoplasms such as primitive neuroectodermal tumors (PNETs), and are admixed with ganglion cells in ganglioneuroblastomas, which are classified with embryonal neoplasms (see Chapter 38). Ganglion cells and/or neurocytes, but not neuroblasts, form a significant component of neoplasms grouped as neuronal and mixed neuronal-glial tumors in the WHO classification (2007):
 ganglion cell neoplasms (i.e. gangliocytoma, ganglioglioma, anaplastic ganglioglioma)
ganglion cell neoplasms (i.e. gangliocytoma, ganglioglioma, anaplastic ganglioglioma)
 desmoplastic infantile ganglioglioma (DIGG) and its variant desmoplastic infantile astrocytoma
desmoplastic infantile ganglioglioma (DIGG) and its variant desmoplastic infantile astrocytoma
 neurocytomas (i.e. central and extraventricular neurocytoma, and cerebellar liponeurocytoma)
neurocytomas (i.e. central and extraventricular neurocytoma, and cerebellar liponeurocytoma)

 rosette-forming glioneuronal tumor of the fourth ventricle
rosette-forming glioneuronal tumor of the fourth ventricle
 paraganglioma (of the filum terminale)
paraganglioma (of the filum terminale)
 dysembryoplastic neuroepithelial tumor (DNET)
dysembryoplastic neuroepithelial tumor (DNET)
 dysplastic gangliocytoma of the cerebellum (Lhermitte–Duclos disease).
dysplastic gangliocytoma of the cerebellum (Lhermitte–Duclos disease).
GANGLIOCYTOMA AND GANGLIOGLIOMA
 Ganglion Cell Tumors
Ganglion Cell Tumors Are rare, accounting for <1% of CNS neoplasms.
Are rare, accounting for <1% of CNS neoplasms.

 Have a predilection for the temporal lobe.
Have a predilection for the temporal lobe.
 Usually present in the first three decades with complex partial seizures.
Usually present in the first three decades with complex partial seizures.
 Are the commonest neoplasms associated with refractory temporal lobe epilepsy.
Are the commonest neoplasms associated with refractory temporal lobe epilepsy.
 Are associated with congenital abnormalities in 5% of cases.
Are associated with congenital abnormalities in 5% of cases.
 TUMOR-RELATED TEMPORAL LOBE EPILEPSY
TUMOR-RELATED TEMPORAL LOBE EPILEPSY


MACROSCOPIC APPEARANCES
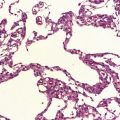
Ganglion cell neoplasms are generally circumscribed; only rare examples have a diffuse glial element (Fig. 37.1). They are usually firm and gray, and frequently contain flecks of calcification and small cysts. They may appear as a mural nodule in a large cyst.
MICROSCOPIC APPEARANCES
Gangliocytomas consist of disorganized ganglion cells set against a neuropil-like background or a lacy, fibrillary background containing a few reactive astrocytes (Fig. 37.2). Groups of ganglion cells may appear in nests enclosed by thin fibrovascular septa. In some cases, extensive desmoplasia may surround elongated cells that have a neuronal immunophenotype. Neoplastic ganglion cells possess abnormal, thickened processes and some have double nuclei. Small round cells with a relatively high nuclear:cytoplasmic ratio, but without nuclear hyperchromasia, represent a further neuronal phenotype encountered in these neoplasms (Fig. 37.2).
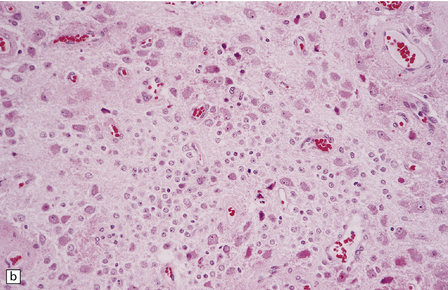
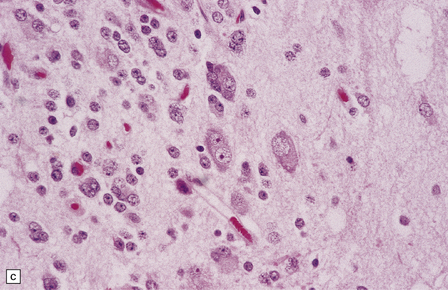
37.2 Gangliocytoma.
(a) and (b) This neoplasm is composed of large and small cells with a neuronal morphophenotype. Many cells have a prominent nucleolus, and some contain basophilic Nissl substance in the periphery of the cytoplasm. (c) Some ganglion cells have double nuclei.
Gangliogliomas also contain neoplastic glial cells, which may greatly outnumber the ganglion cells. Neoplastic glia in gangliogliomas may have an obvious astrocytic morphology, but some ganglion cell neoplasms contain cells of an indeterminate nature (Figs 37.3, 37.4). While the glial component of some gangliogliomas appears as a fibrillary astrocytoma, other gangliogliomas manifest as a pilocytic astrocytoma or pleomorphic xanthoastrocytoma with a ganglion cell component. Eosinophilic granular bodies and neovascularization, of the type seen in pilocytic astrocytomas, may be evident. Degenerative cytologic changes take the form of nuclear pleomorphism and hyperchromasia, in the absence of mitoses. Some gangliogliomas are focally desmoplastic, or may spread into the subarachnoid space, provoking desmoplasia.
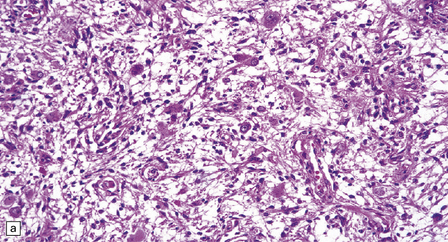
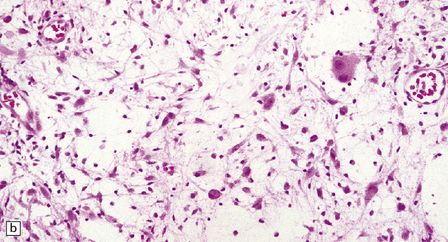
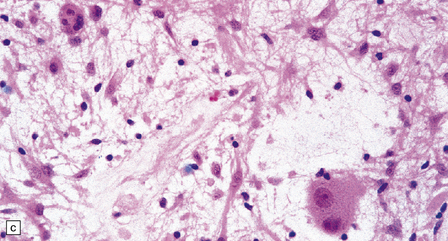
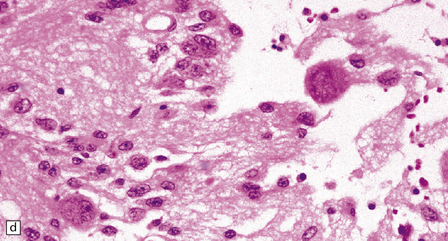
37.3 Ganglioglioma.
(a) Ganglion cells are seen among disorganized neoplastic cells with an astrocytic morphology. (b) A microcystic pattern is found in another part of the same neoplasm. (c) Astrocytic cells with fibrillary processes surround ganglion cells. While the glial component of a ganglioglioma may clearly have an astrocytic phenotype, its features may not be sufficiently typical to allow classification as fibrillary or pilocytic. (d) The histogenesis of some cells is unclear; in standard histologic preparations, some cells are not readily classified as neuronal or glial.
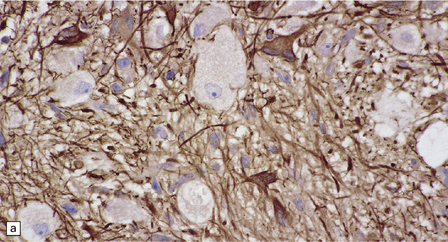
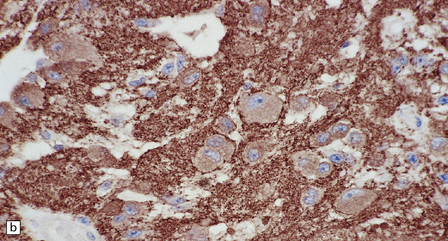
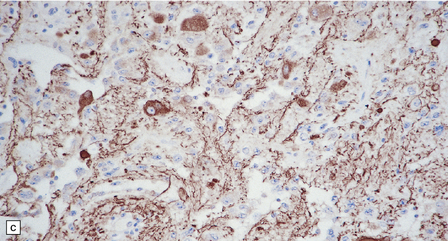
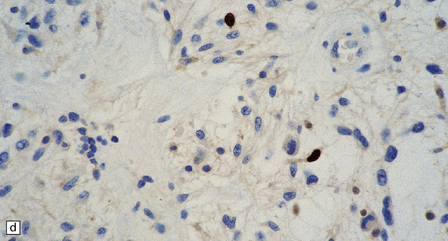
37.4 Ganglioglioma.
(a) Ganglion cells are surrounded by the processes of astrocytic cells that are immunoreactive for GFAP. (b) The cytoplasm of ganglion cells and a meshwork of neuritic processes are immunoreactive for synaptophysin. (c) The cell bodies of ganglion cells and some fine processes are also immunoreactive for neurofilament proteins. (d) The low growth fraction of a ganglioglioma is illustrated by the paucity of Ki-67-labeled nuclei.
Ganglion cells are immunoreactive for NEU-N, synaptophysin, and neurofilament proteins (Fig. 37.4). Electron microscopy reveals dense-core synaptic vesicles.
Small round undifferentiated cells with a high nuclear:cytoplasmic ratio and nuclear hyperchromasia are combined with ganglion cells in the ganglioneuroblastoma, which is regarded as a variant of PNET (Fig. 37.5). Mitoses and apoptotic bodies are usually identifiable among the small cells. Recognition of the embryonal element is important because its presence confers a much poorer prognosis and has implications for therapy. The combination of neoplastic neuronal and glial cells that are both undifferentiated and differentiated (i.e. ganglioneuroblastoma/glioblastoma, or malignant glioma/PNET) is recognized as an entity, but extremely rare. Like the ganglioneuroblastoma, these neoplasms should probably be regarded as variants of PNET.
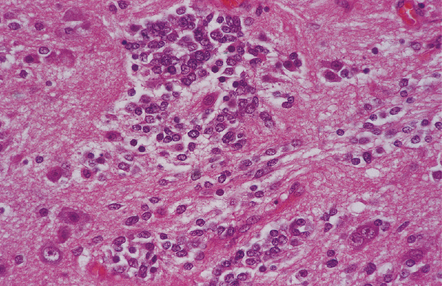
37.5 Ganglioneuroblastoma.
A closely packed group of small undifferentiated cells with hyperchromatic nuclei is seen among ganglion cells.
 DIFFERENTIAL DIAGNOSIS OF GANGLION CELL NEOPLASMS
DIFFERENTIAL DIAGNOSIS OF GANGLION CELL NEOPLASMS
 Are neoplastic ganglion cells present? Disorganized neoplastic ganglion cells do not show the uniform orientation and orderly arrangement of neurons in the cerebral cortex, may be identified away from gray matter in the deep white matter or subarachnoid space (though a few scattered neurons are normally found in the subcortical white matter – especially of the temporal lobe), and have abnormal cytologic characteristics, including double nuclei.
Are neoplastic ganglion cells present? Disorganized neoplastic ganglion cells do not show the uniform orientation and orderly arrangement of neurons in the cerebral cortex, may be identified away from gray matter in the deep white matter or subarachnoid space (though a few scattered neurons are normally found in the subcortical white matter – especially of the temporal lobe), and have abnormal cytologic characteristics, including double nuclei.




DESMOPLASTIC INFANTILE GANGLIOGLIOMA (DIGG)
MACROSCOPIC APPEARANCES
 DIFFERENTIAL DIAGNOSIS OF DIGG
DIFFERENTIAL DIAGNOSIS OF DIGG


MICROSCOPIC APPEARANCES
In areas of prominent desmoplasia, neoplastic glial cells are spindle-shaped with elongated hyperchromatic nuclei (Fig. 37.6). In some areas, the fascicular architecture and desmoplasia give way to foci of plump astrocytic cells or even limited numbers of poorly differentiated neuroepithelial cells. Ganglion cells with large nuclei are found within DIGGs (Fig. 37.6), but not DIAs, and can be numerous, mimicking a ganglioglioma. A small number of mitoses with a normal configuration may be present among the poorly differentiated cells.
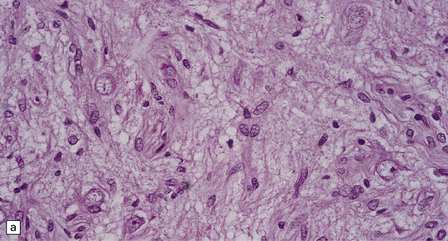
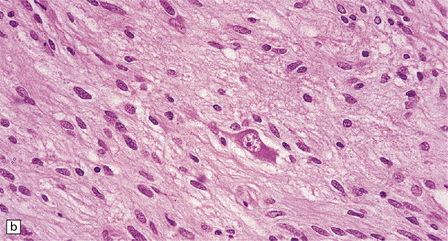
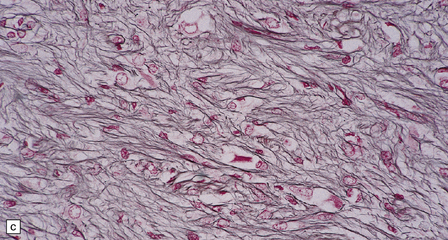
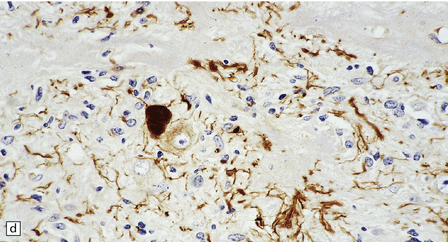
37.6 Desmoplastic infantile ganglioglioma.
Ganglion cells are scattered among cells with fibrillary cytoplasm and oval nuclei that have either (a) a random or (b) a fascicular arrangement. (c) A dense pericellular reticulin pattern is characteristic. (d) Ganglion cells and some fine processes show immunoreactivity for neurofilament protein.
CENTRAL AND EXTRAVENTRICULAR NEUROCYTOMA
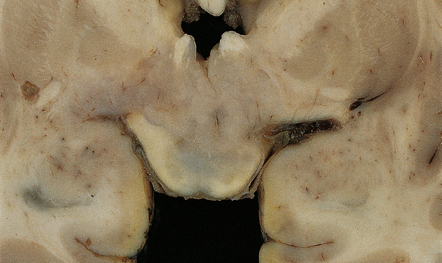
The central neurocytoma is a distinctive intraventicular neoplasm usually located in the region of the septum pellucidum/foramen of Monro (Fig. 37.7). Since the neuronal nature of its isomorphous cells was demonstrated, it has become the archetypal neurocytic neoplasm, despite its idiosyncratic clinicopathologic features. Rare circumscribed intraparenchymal tumors with a similar phenotype have been reported to occur throughout the neuraxis, and the term extraventricular neurocytoma has been coined for these. The cerebellar liponeurocytoma (below) also belongs to this category.
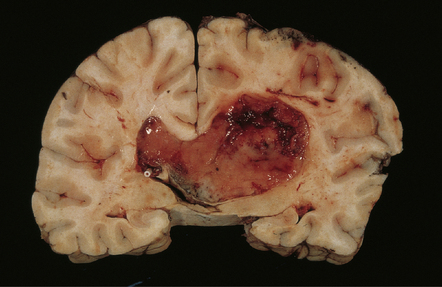
37.7 Central neurocytoma.
A coronal section through the cerebrum at the level of the hippocampus reveals a circumscribed, fleshy tumor in the ventricles.
 CENTRAL NEUROCYTOMAS
CENTRAL NEUROCYTOMAS



MACROSCOPIC AND MICROSCOPIC APPEARANCES
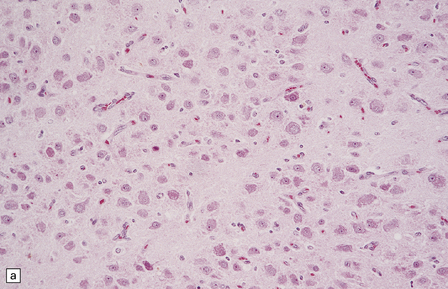
The central neurocytoma is a soft, well-circumscribed neoplasm that often contains flecks of calcification. It consists of monotonous round to oval cells, which have fibrillary cytoplasm and round nuclei with finely granular chromatin (Fig. 37.8). A fixation artifact giving a ‘fried egg’ appearance to the cells is sometimes seen. The sheet of neoplastic cells is divided by anuclear fibrillary zones, which contain fine capillaries. Mitotic figures are seldom seen. Rarely, ganglion cells are present, evincing the neuronal lineage of the neoplasm.
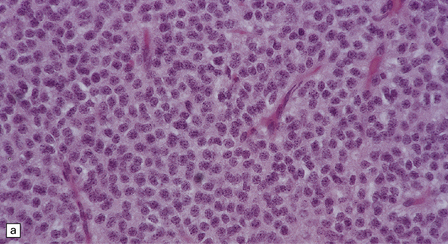
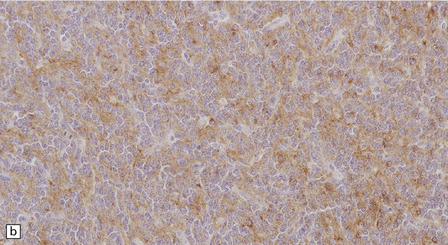
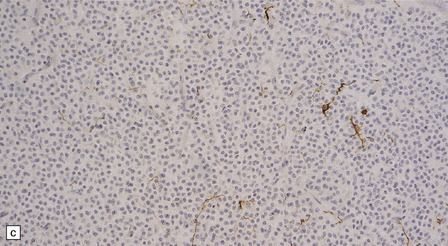
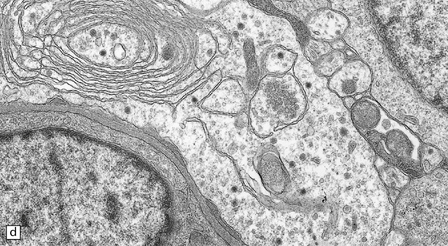
37.8 Central neurocytoma.
(a) A sheet of cells with uniform, round nuclei. This is typical of the central neurocytoma. (b) The cells show immunoreactivity for synaptophysin. (c) A few reactive astrocytes express GFAP. The neoplastic cells are rarely GFAP-positive. (d) Ultrastructural view of a central neurocytoma revealing dense-core synaptic vesicles.
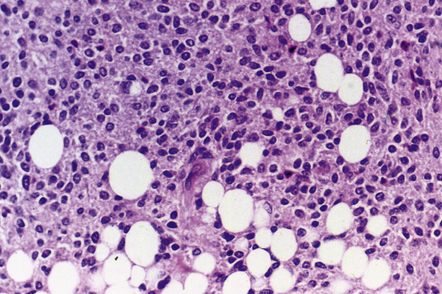
CEREBELLAR LIPONEUROCYTOMA
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The liponeurocytoma tends to be well circumscribed and soft, not unlike a central neurocytoma. It is composed of sheets of monomorphic neurocytic cells with round or oval nuclei, which are admixed with groups of lipidized tumor cells (Fig. 37.9). Immunohistochemistry reveals a neuronal phenotype in most cells, but the reactivities for synaptophysin and MAP-2 are usually supplemented by labeling of a few tumor cells for GFAP. The Ki-67 labeling index is characteristically low.
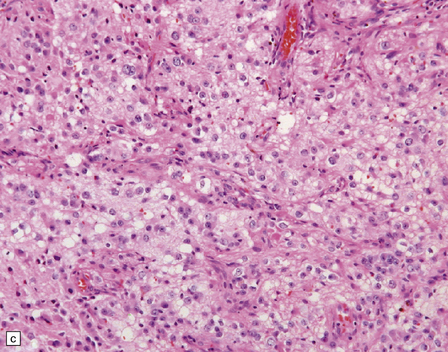
PAPILLARY GLIONEURONAL TUMOR
MACROSCOPIC AND MICROSCOPIC APPEARANCES
PGNTs can be solid or cystic and often show focal calcification. At the microscopic level, there is a pseudo-papillary architecture characterized by discohesive elements centered on hyalinized blood vessels. Small, poorly differentiated, GFAP-immunopositive cells with round nuclei and a high nuclear:cytoplasmic ratio have a perivascular distribution, but give way to interpapillary groups of cells with a (mainly neurocytic) neuronal morphology and immunophenotype. Mitoses are rare, and necrosis and microvascular proliferation are not features of the neoplastic process (Fig. 37.10).
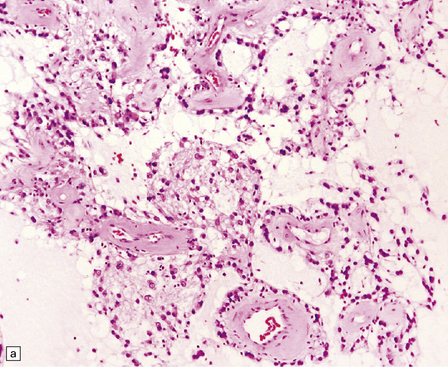
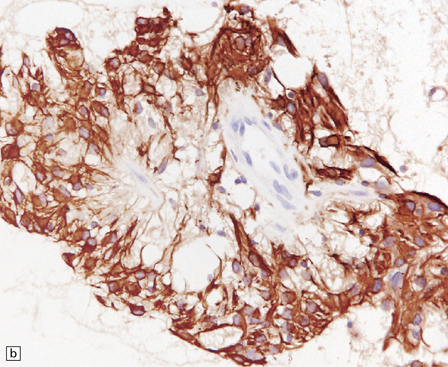
ROSETTE-FORMING GLIONEURONAL TUMOR OF THE FOURTH VENTRICLE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The RGNT is a circumscribed tumor, with restricted infiltration on microscopy. Two histological elements are present: neuronal and glial. Small uniform neurocytic cells form ring-like rosettes, usually with a core of dense synaptophysin-positive neurofibrillary material, or cluster around capillaries. Microcystic degeneration of the focally myxoid matrix may occur. The glial element resembles a pilocytic astrocytoma (Fig. 37.11).
PARAGANGLIOMA OF THE FILUM TERMINALE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
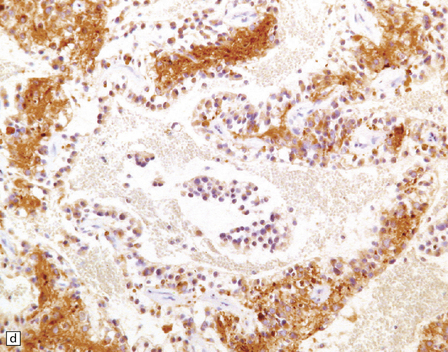
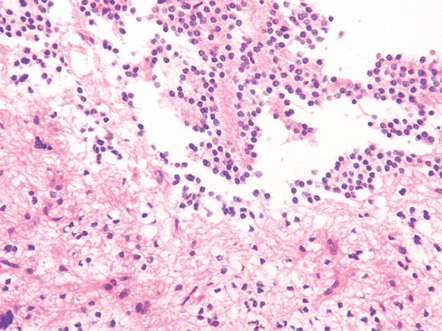
Microscopically, nests of oval or polyhedral cells with granular cytoplasm are surrounded by a delicate fibrovascular network (Fig. 37.12). Some cytologic pleomorphism may be apparent, but is of no biologic significance. Nuclei contain stippled chromatin. Ganglion cells may make up a large proportion of the cells (gangliocytic paraganglioma).
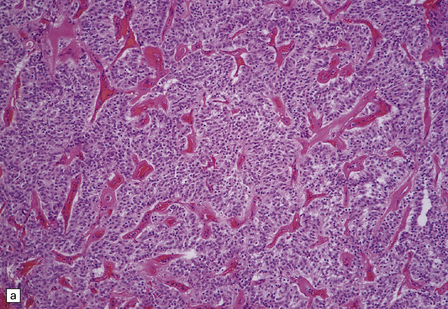
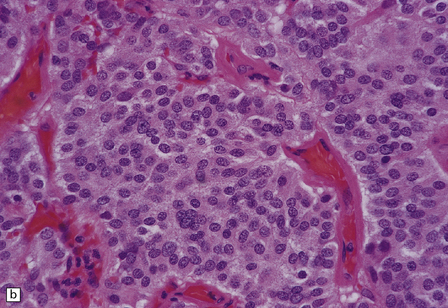
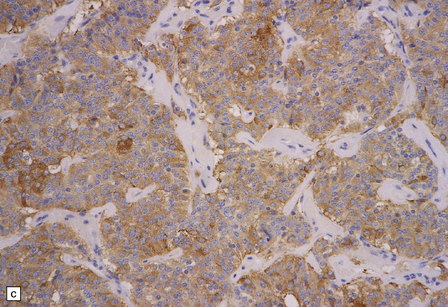
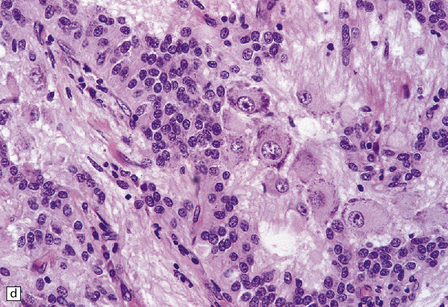
37.12 Paraganglioma of the filum terminale.
(a) A network of capillaries surrounds nests of uniform cells with stippled chromatin. (b) The same neoplasm at higher magnification. (c) Strong synaptophysin immunoreactivity of the tumor cells. (d) Paraganglioma containing numerous ganglion cells.
 PARAGANGLIOMA AND EPENDYMOMA
PARAGANGLIOMA AND EPENDYMOMA

DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR (DNT)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
DNTs are currently divided into two forms: simple and complex (Figs 37.13, 37.14). Both have the characteristic ‘specific glioneuronal element’, which has a nodular architecture and contains masses of oligodendrocyte-like cells (OLCs). The OLCs are set against a mucinous matrix, in which microcyst formation occurs. Focally, the OLCs have a columnar arrangement and seem to be suspended from neuritic processes or capillaries. Small cysts sometimes contain a mature neuron, which appears suspended (sometime described as ‘floating’) in a mucopolysaccharide-rich medium. The complex form also includes groups of glial cells, often with a nodular architecture. In isolation, the glial cells have the features of an astrocytoma, oligodendroglioma, or mixed glioma. Focal calcification is seen in some DNTs.
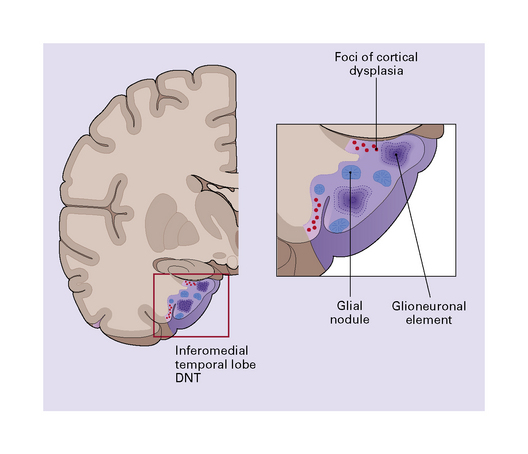
37.13 Dysembryoplastic neuroepithelial tumor.
This neoplasm sometimes contains glial nodules in addition to the specific glioneuronal elements and focal areas of cortical dysplasia. Nodules may contain cells with an astrocytic or oligodendroglial morphophenotype. Astrocytomatous nodules may resemble pilocytic or fibrillary astrocytomas. Glial nodules may occur in normal cortex away from the glioneuronal element.
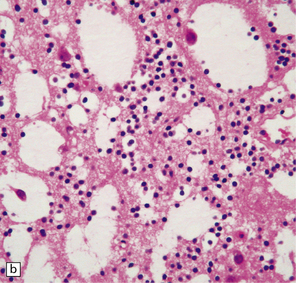
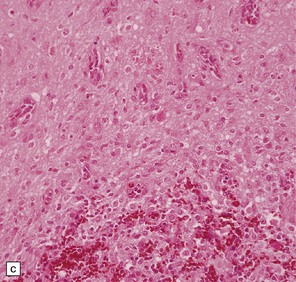
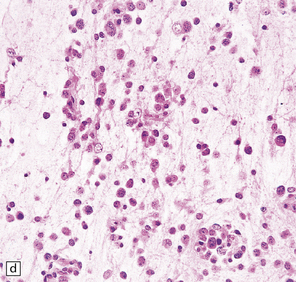
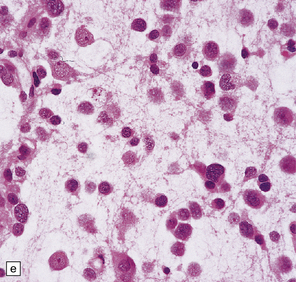
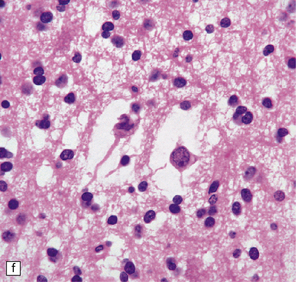
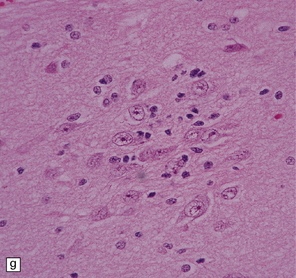
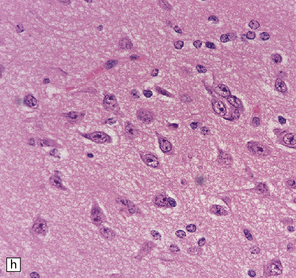
37.14 Dysembryoplastic neuroepithelial tumor.
(a) Nodular cluster of cells in a DNT. (b) OLCs are typical of DNTs, but cells with an astrocytic morphology may also be present (c). (d) In the specific glioneuronal element, OLCs appear in festoons against a mucinous background. (e) OLCs are admixed with larger cells that show neuronal features and may be suspended within microcysts (f). (g) A cluster of neurons and small OLCs is seen away from the main tumor mass. (h) Cortical dysplasia in association with a DNT.
 DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR
DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR
 Accounts for 1–2% of CNS neoplasms.
Accounts for 1–2% of CNS neoplasms.
 Occurs mainly in the temporal lobe.
Occurs mainly in the temporal lobe.
 Usually presents in children or young adults.
Usually presents in children or young adults.

 Is not associated with neurologic deficit or significant mass effect.
Is not associated with neurologic deficit or significant mass effect.
 Has characteristic radiologic features, such as nodularity.
Has characteristic radiologic features, such as nodularity.
 Is the diagnosis in about 20% of patients undergoing temporal lobe surgery for epilepsy.
Is the diagnosis in about 20% of patients undergoing temporal lobe surgery for epilepsy.
 DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR
DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR

DYSPLASTIC GANGLIOCYTOMA OF THE CEREBELLUM
 DYSPLASTIC GANGLIOCYTOMA OF THE CEREBELLUM
DYSPLASTIC GANGLIOCYTOMA OF THE CEREBELLUM


MACROSCOPIC AND MICROSCOPIC APPEARANCES
The folia within a dysplastic gangliocytoma of the cerebellum appear abnormally thickened (Fig. 37.15a). Enlarged abnormal neurons replace internal granule cells, distorting the normal cerebellar architecture (Fig. 37.15b). The cells are immunoreactive for synaptophysin and neurofilament proteins and their axons in the molecular layer are often myelinated. There is a loss of Purkinje cells and rarefaction or cavitation of white matter in the core of affected folia.
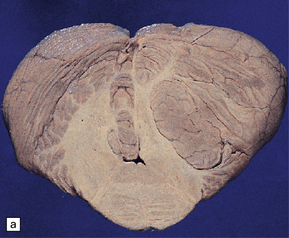
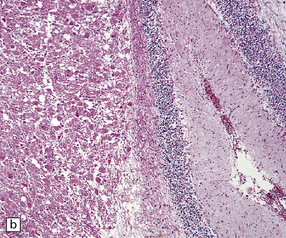
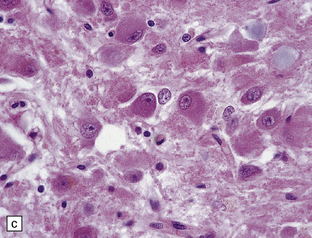
HYPOTHALAMIC NEURONAL HAMARTOMA
This non-neoplastic lesion is evident as a small mass ventral to the hypothalamus, and is composed of ganglion cells arranged in clusters against a neuropil-like background (Fig. 37.16). Generally, the hamartoma is asymptomatic, but it occasionally produces epilepsy (sometimes gelastic seizures) or endocrine syndromes, such as precocious puberty, which result from the capacity of the ganglion cells to secrete hormones in an aberrant fashion.
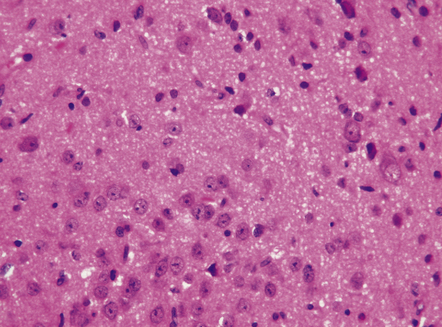
REFERENCES
Blümcke, I., Löbach, M., Wolf, H.K., et al. Evidence for developmental precursor lesions in epilepsy-associated glioneuronal tumors. Microsc Res Tech.. 1999;46:53–58.
Campos, M.G., Zentner, J., Ostertun, B., et al. Anaplastic ganglioglioma: case report and review of the literature. Neurol Res.. 1994;16:317–320.
Celli, P., Scarpinati, M., Nardacci, B., et al. Gangliogliomas of the cerebral hemispheres. Report of 14 cases with long-term follow-up and review of the literature. Acta Neurochir (Wien). 1993;125:52–57.
Dash, R.C., Provenzale, J.M., McComb, R.D., et al. Malignant supratentorial ganglioglioma (ganglion cell-giant cell glioblastoma): a case report and review of the literature. Arch Pathol Lab Med.. 1999;123:342–345.
Felix, I., Bilbao, J.M., Asa, S.L., et al. Cerebral and cerebellar gangliocytomas: a morphological study of nine cases. Acta Neuropathol (Berl).. 1994;88:246–251.
Forshew, T., Tatevossian, R.G., Lawson, A.R., et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol.. 2009;218:172–181.
Haddad, S.F., Moore, S.A., Menezes, A.H., et al. Ganglioglioma: 13 years of experience. Neurosurgery.. 1992;31:171–178.
Hakim, R., Loeffler, J.S., Anthony, D.C., et al. Gangliogliomas in adults. Cancer.. 1997;79:127–131.
Jay, V., Squire, J., Becker, L.E., et al. Malignant transformation in a ganglioglioma with anaplastic neuronal and astrocytic components. Report of a case with flow cytometric and cytogenetic analysis. Cancer.. 1994;73:2862–2868.
Johannsson, J.H., Rekate, H.L., Roessmann, U. Gangliogliomas: pathological and clinical correlation. J Neurosurg.. 1981;54:58–63.
Kleinschmidt-DeMasters, B.K., Birks, D.K., Aisner, D.L., et al. Atypical teratoid/rhabdoid tumor arising in a ganglioglioma: genetic characterization. Am J Surg Pathol.. 2011;35:1894–1901.
Kordek, R., Biernat, W., Sapieja, W., et al. Pleomorphic xanthoastrocytoma with a gangliomatous component: an immunohistochemical and ultrastructural study. Acta Neuropathol (Berl).. 1995;89:194–197.
Luyken, C., Blümcke, I., Fimmers, R., et al. Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer.. 2004;101:146–155.
Schindler, G., Capper, D., Meyer, J., et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol (Berl).. 2011;121:397–405.
Wolf, H.K., Müller, M.B., Spänle, M., et al. Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol (Berl).. 1994;88:166–173.
Zentner, J., Wolf, H.K., Ostertun, B., et al. Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry.. 1994;57:1497–1502.
Ellison, D.W., Zygmunt, S.C., Weller, R.O. Neurocytoma/lipoma (neurolipocytoma) of the cerebellum. Neuropathol Appl Neurobiol.. 1993;19:95–98.
Figarella-Branger, D., Pellissier, J.F., Daumas-Duport, C., et al. Central neurocytomas. Critical evaluation of a small-cell neuronal tumor. Am J Surg Pathol.. 1992;16:97–109.
Hessler, R.B., Lopes, M.B., Frankfurter, A., et al. Cytoskeletal immunohistochemistry of central neurocytomas. Am J Surg Pathol.. 1992;16:1031–1038.
Jackson, T.R., Regine, W.F., Wilson, D., et al. Cerebellar liponeurocytoma. Case report and review of the literature. J Neurosurg. 2001;95:700–703.
Jouvet, A., Lellouch-Tubiana, A., Boddaert, N., et al. Fourth ventricle neurocytoma with lipomatous and ependymal differentiation. Acta Neuropathol (Berl).. 2005;109:346–351.
Mrak, R.E., Yasargil, M.G., Mohapatra, G., et al. Atypical extraventricular neurocytoma with oligodendroglioma-like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol.. 2004;35:1156–1159.
Söylemezoglu, F., Scheithauer, B.W., Esteve, J., et al. Atypical central neurocytoma. J Neuropathol Exp Neurol.. 1997;56:551–556.
von Deimling, A., Janzer, R., Kleihues, P., et al. Patterns of differentiation in central neurocytoma. An immunohistochemical study of eleven biopsies. Acta Neuropathol (Berl). 1990;79:473–479.
Craver, R.D., Nadell, J., Nelson, J.S., et al. Desmoplastic infantile ganglioglioma. Pediatr Dev Pathol.. 1999;2:582–587.
De Munnynck, K., Van Gool, S., Van Calenbergh, F., et al. Desmoplastic infantile ganglioglioma: a potentially malignant tumor? Am J Surg Pathol.. 2002;26:1515–1522.
Komori, T., Scheithauer, B.W., Parisi, J.E., et al. Mixed conventional and desmoplastic infantile ganglioglioma: an autopsied case with 6-year follow-up. Mod Pathol.. 2001;14:720–726.
Louis, D.N., von Deimling, A., Dickersin, G.R., et al. Desmoplastic cerebral astrocytomas of infancy: a histopathologic, immunohistochemical, ultrastructural, and molecular genetic study. Hum Pathol.. 1992;23:1402–1409.
Ng, T.H., Fung, C.F., Ma, L.T. The pathological spectrum of desmoplastic infantile gangliogliomas. Histopathology.. 1990;16:235–241.
Paulus, W., Schlote, W., Perentes, E., et al. Desmoplastic supratentorial neuroepithelial tumours of infancy. Histopathology.. 1992;21:43–49.
VandenBerg, S.R., May, E.E., Rubinstein, L.J., et al. Desmoplastic supratentorial neuroepithelial tumors of infancy with divergent differentiation potential (desmoplastic infantile gangliogliomas). Report on 11 cases of a distinctive embryonal tumor with favorable prognosis. J Neurosurg. 1987;66:58–71.
Dysembryoplastic neuroepithelial tumors
Baisden, B.L., Brat, D.J., Melhem, E.R., et al. Dysembryoplastic neuroepithelial tumor-like neoplasm of the septum pellucidum: a lesion often misdiagnosed as glioma: report of 10 cases. Am J Surg Pathol.. 2001;25:494–499.
Daumas-Duport, C., Scheithauer, B.W., Chodkiewicz, J.P., et al. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery.. 1988;23:545–556.
Hirose, T., Scheithauer, B.W. Mixed dysembryoplastic neuroepithelial tumor and ganglioglioma. Acta Neuropathol (Berl).. 1998;95:649–654.
Hirose, T., Scheithauer, B.W., Lopes, M.B., et al. Dysembryoplastic neuroepithelial tumor (DNT): an immunohistochemical and ultrastructural study. J Neuropathol Exp Neurol.. 1994;53:184–195.
Honavar, M., Janota, I., Polkey, C.E. Histological heterogeneity of dysembryoplastic neuroepithelial tumour: identification and differential diagnosis in a series of 74 cases. Histopathology.. 1999;34:342–356.
Raymond, A.A., Halpin, S.F., Alsanjari, N., et al. Dysembryoplastic neuroepithelial tumor. Features in 16 patients. Brain. 1994;117:461–475.
Barbashina, V., Salazar, P., Ladanyi, M., et al. Glioneuronal tumor with neuropil-like islands (GTNI): a report of 8 cases with chromosome 1p/19q deletion analysis. Am J Surg Pathol.. 2007;31:1196–1202.
Coons, S.W., Rekate, H.L., Prenger, E.C., et al. The histopathology of hypothalamic hamartomas: study of 57 cases. J Neuropathol Exp Neurol.. 2007;66:131–141.
Edgar, M.A., Rosenblum, M.K. Mixed glioneuronal tumors: recently described entities. Arch Pathol Lab Med.. 2007;131:228–233.
Kamalian, N., Abbassioun, K., Amirjamshidi, A., et al. Paraganglioma of the filum terminale internum. Report of a case and review of the literature. J Neurol.. 1987;235:56–59.
Kim, D.H., Suh, Y.L. Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol (Berl).. 1997;94:187–191.
Komori, T., Scheithauer, B.W., Anthony, D.C., et al. Papillary glioneuronal tumor: a new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol.. 1998;22:1171–1183.
McLendon, R.E., Bentley, R.C., Parisi, J.E., et al. Malignant supratentorial glial-neuronal neoplasms: report of two cases and review of the literature. Arch Pathol Lab Med.. 1997;121:485–492.
Prayson, R.A. Papillary glioneuronal tumor. Arch Pathol Lab Med.. 2000;124:1820–1823.
Teo, J.G., Gultekin, S.H., Bilsky, M., et al. A distinctive glioneuronal tumor of the adult cerebrum with neuropil-like (including rosetted) islands: report of 4 cases. Am J Surg Pathol.. 1999;23:502–510.
Wolf, H.K., Campos, M.G., Zentner, J., et al. Surgical pathology of temporal lobe epilepsy. Experience with 216 cases. J Neuropathol Exp Neurol.. 1993;52:499–506.