[level-membership-for-pathology-category]6
Neurodegenerative disorders of gray matter in childhood
CEREBRAL CORTEX
ALPERS–HUTTENLOCHER SYNDROME OR PROGRESSIVE NEURONAL DEGENERATION OF CHILDHOOD (PNDC)
MACROSCOPIC APPEARANCES
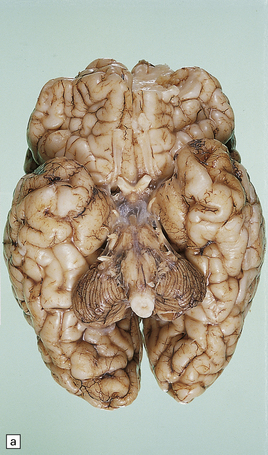
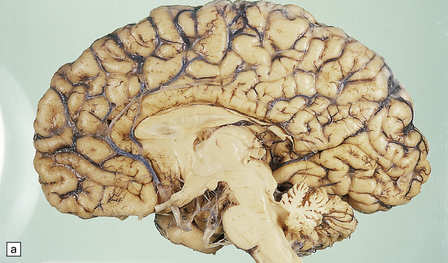
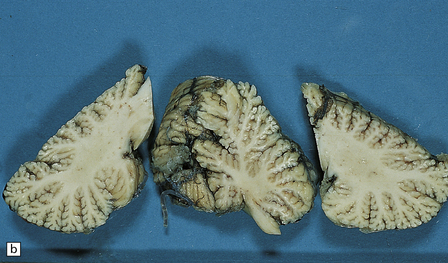
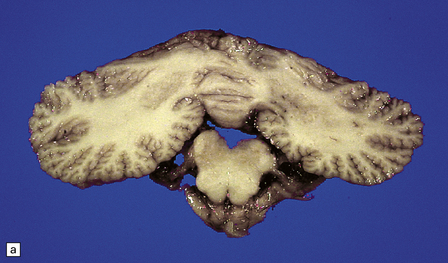
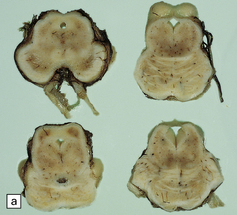
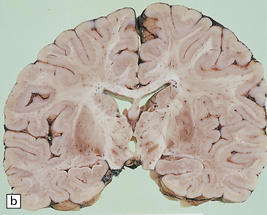
Lesions may be minimal, patchy, or extensive (Fig. 6.1). In affected regions the cortical ribbon is thin, granular, and brown, and even dehiscent in some poorly fixed brains. The calcarine cortex is often picked out in a remarkably selective and characteristic way. Rarely, there is softening of the occipital white matter.
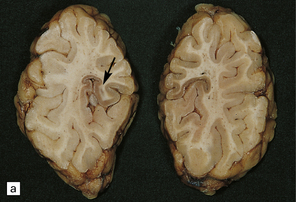
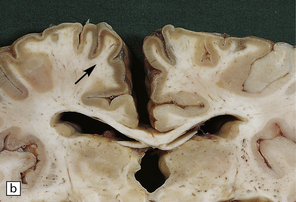
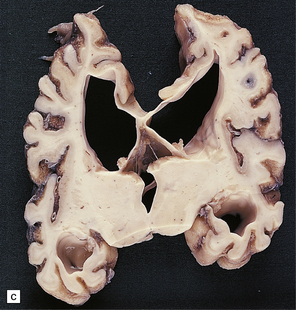
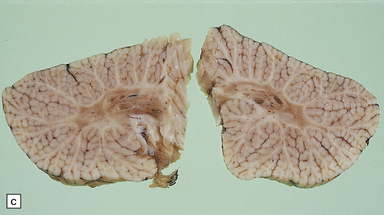
6.1 Alpers–Huttenlocher syndrome.
Selective involvement of the calcarine cortex is a helpful diagnostic pointer. (a) In this child, coronal sections of occipital lobe show the primary visual cortex in both hemispheres delineated by granularity and brown discoloration (arrow). (b) Similar discrete areas of cortical thinning and discoloration are present in the posterior frontal cortex at midthalamic level (arrow). (c) Although cortical lesions are usually patchy, in extreme cases the whole cortical ribbon may be diffusely and uniformly shrunken. In addition, there is ventricular dilatation, some thalamic atrophy, and marked shrinkage of Ammon’s horns.
MICROSCOPIC APPEARANCES
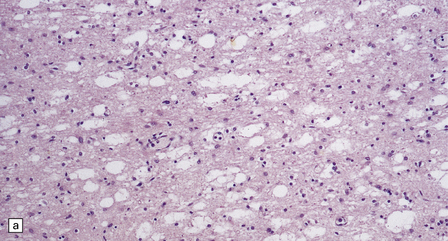
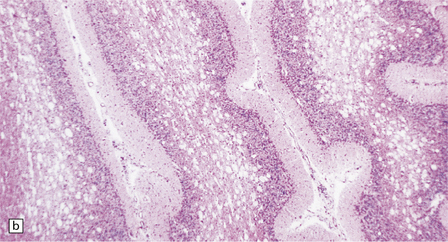
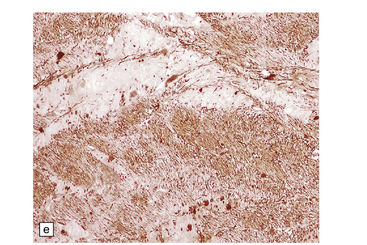
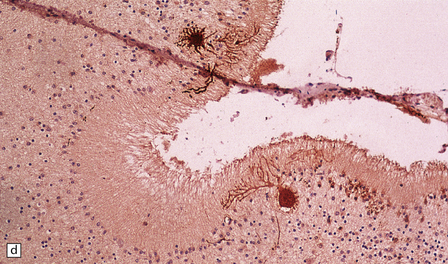
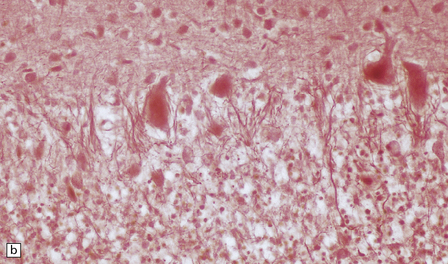
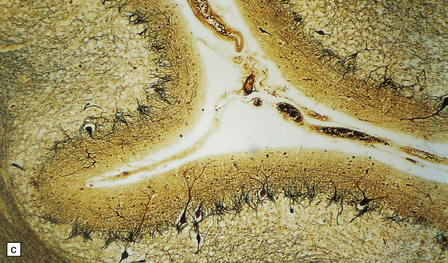
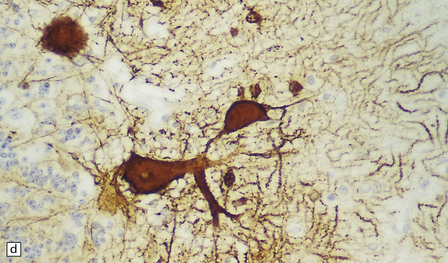
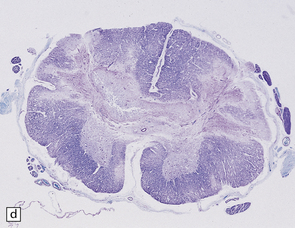
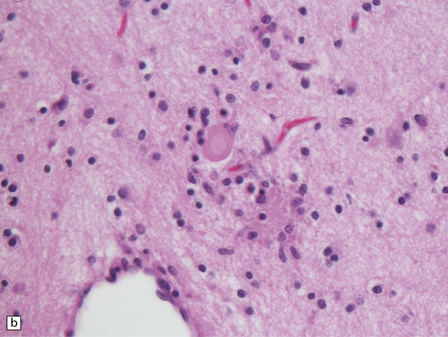
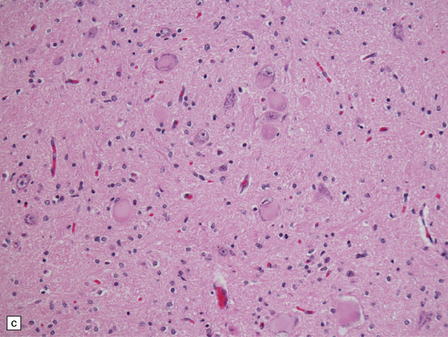
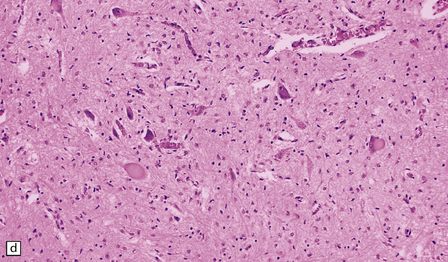
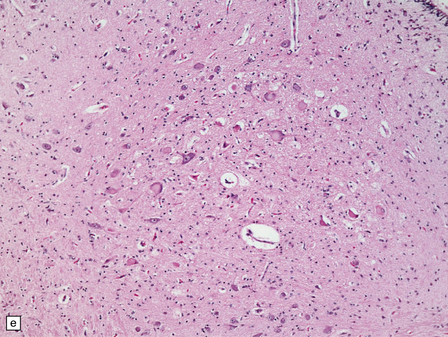
Histologic abnormalities are more widespread than expected from the macroscopic appearances. The patchy lesions do not conform to vascular territories or watershed zones and show a graded intensification and extension of the degenerative process through the depth of the cortical gray matter. Mild superficial spongiosis gives way to increasing sponginess, neuronal loss, and gliosis extending down through the cortex. In severe lesions, the whole ribbon is replaced by a narrow remnant of hypertrophic astrocytes devoid of nerve cells (Fig. 6.2). Neutral fat may be deposited in considerable amounts. Lesions may be symmetric or asymmetric, but there is a striking predilection for the striate cortex. Secondary changes are found in the white matter. Other variable findings include hippocampal sclerosis, cerebellar cortical infarcts, spinal cord tract degeneration, and spongiosis and gliosis in the thalamus (Fig. 6.3), amygdala, substantia nigra, and dentate nuclei.
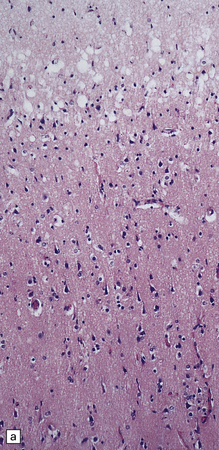
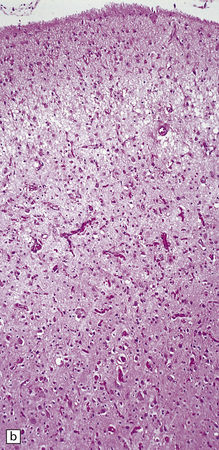
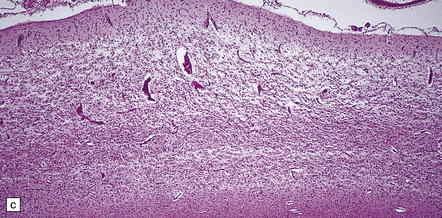
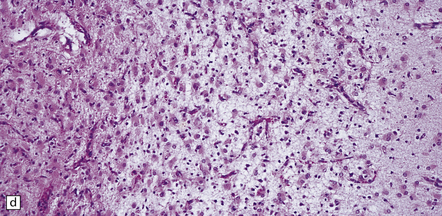
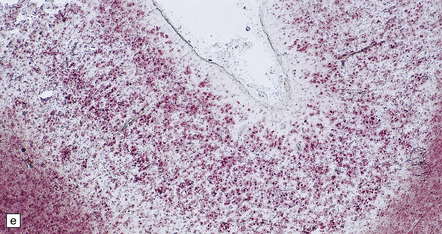
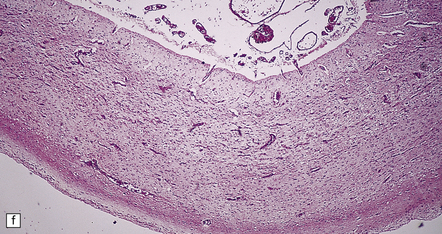
6.2 Alpers–Huttenlocher syndrome.
(a) The mildest histologic changes consist of a fine spongiosis in the superficial cortical layers. (b) Neuronal loss and gliosis gradually extend more deeply through the cortical ribbon. (c) Eventually, the whole cortex is replaced by hypertrophic astrocytes. This shows the striate cortex, which is especially prone to such severe destruction. (d) The neurons and neuropil of the striate cortex are entirely replaced by a cystic meshwork of glial processes emanating from plump hypertrophic astrocytes. (e) Massive amounts of neutral fat can be deposited in the degenerating cortex. (f) The chronic end stage of this process is a thin, poorly cellular gliotic remnant. This shows striate cortex from the same case as in Fig. 6.1c.
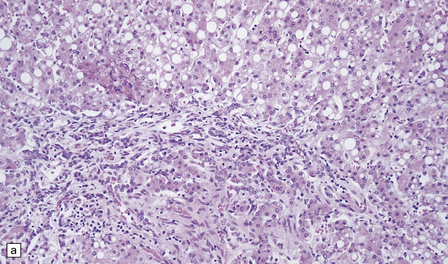
HEPATIC PATHOLOGY
Nearly all patients show characteristic changes in the liver: the hepatocytes undergo severe microvesicular fatty or oncocytic change (Fig. 6.4). There are hepatocyte necrosis, diffuse haphazard bile duct proliferation, and bridging fibrosis, with disorganization and regeneration that amount to cirrhosis at one end of the histologic spectrum (Fig. 6.5), or end-stage collapse and fibrosis at the other.
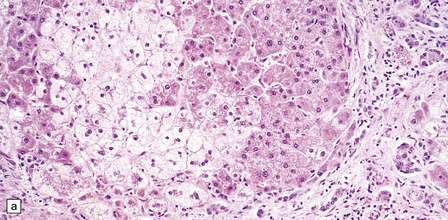
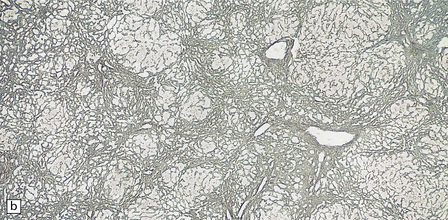
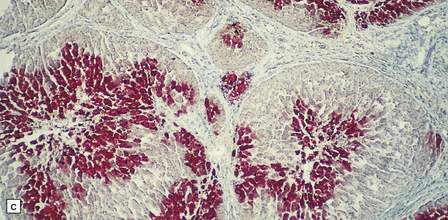
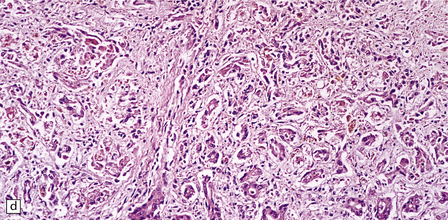
6.4 Alpers–Huttenlocher syndrome: liver pathology.
Histologic changes in the liver are characteristic and essential for a confident diagnosis. (a) The principal features are hepatic steatosis and oncocytic change along with hepatocyte necrosis and a profuse and haphazard proliferation of bile ductules. (b) Fibrosis and nodular regeneration often give rise to a cirrhotic pattern. (c) Fat may be heavily deposited within the regenerative nodules. (d) In some patients, regeneration does not occur and the liver is replaced by dense fibrous tissue and irregular bile ductules. Note the bile retention.
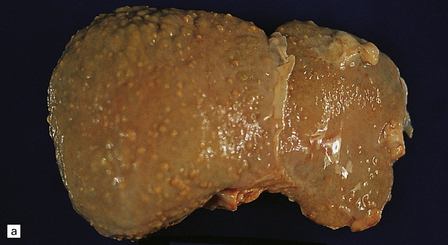
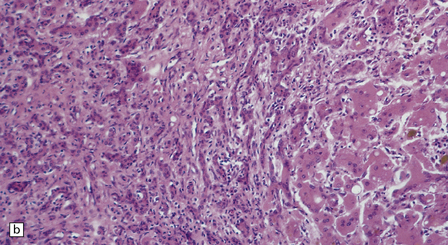
6.5 Alpers–Huttenlocher syndrome.
Liver pathology in two brothers who died from PNDC. One of these brothers received valproate therapy, but both showed typical liver pathology. (a) Macroscopic appearance of the finely nodular cirrhotic liver of one brother. (b) Liver histology shows the typical changes of PNDC in the other brother. The role of drug therapy and in particular valproate toxicity in the pathogenesis of PNDC is controversial.
 ALPERS–HUTTENLOCHER SYNDROME
ALPERS–HUTTENLOCHER SYNDROME














 DIFFERENTIAL DIAGNOSIS OF ALPERS–HUTTENLOCHER SYNDROME WITH EPILEPSY IN THE CONTEXT OF PROGRESSIVE CEREBRAL ATROPHY
DIFFERENTIAL DIAGNOSIS OF ALPERS–HUTTENLOCHER SYNDROME WITH EPILEPSY IN THE CONTEXT OF PROGRESSIVE CEREBRAL ATROPHYThe following discriminating features should be sought:
 Hypoxic–ischemic encephalopathy: predilection for vascular boundary zones, relative preservation of gyral crests; striate cortex spared.
Hypoxic–ischemic encephalopathy: predilection for vascular boundary zones, relative preservation of gyral crests; striate cortex spared.


 Hepatic encephalopathy: involvement of basal ganglia; cavitation of gray–white matter interface.
Hepatic encephalopathy: involvement of basal ganglia; cavitation of gray–white matter interface.
 Neuronal storage disorders: neuronal abnormalities.
Neuronal storage disorders: neuronal abnormalities.


 ETIOLOGY OF ALPERS–HUTTENLOCHER SYNDROME
ETIOLOGY OF ALPERS–HUTTENLOCHER SYNDROME
 Prominent spongiosis prompted speculation of a kinship with Creutzfeldt–Jakob disease but one apparently successful animal transmission experiment has not been duplicated.
Prominent spongiosis prompted speculation of a kinship with Creutzfeldt–Jakob disease but one apparently successful animal transmission experiment has not been duplicated.

 Evidence of mitochondrial dysfunction in some patients includes cytochrome oxidase deficiency.
Evidence of mitochondrial dysfunction in some patients includes cytochrome oxidase deficiency.
 Most patients are compound heterozygotes with two mutations of the mtDNA polymerase gene POLG1.
Most patients are compound heterozygotes with two mutations of the mtDNA polymerase gene POLG1.
BASAL GANGLIA
HOLOTOPISTIC STRIATAL NECROSIS (FAMILIAL STRIATAL DEGENERATION)
 DIFFERENTIAL DIAGNOSIS OF BASAL GANGLIA DISORDERS IN CHILDHOOD
DIFFERENTIAL DIAGNOSIS OF BASAL GANGLIA DISORDERS IN CHILDHOOD
The following discriminating clinicopathological features should be sought.
 Holotopistic striatal necrosis: acute necrosis or slow degeneration; recessive inheritance.
Holotopistic striatal necrosis: acute necrosis or slow degeneration; recessive inheritance.


 Wilson’s disease: striatal, retinal, and hepatic degeneration.
Wilson’s disease: striatal, retinal, and hepatic degeneration.

 Juvenile Parkinson disease: degeneration of substantia nigra and locus ceruleus.
Juvenile Parkinson disease: degeneration of substantia nigra and locus ceruleus.
 Glutaric acidemia: striatal degeneration; specific biochemical abnormality.
Glutaric acidemia: striatal degeneration; specific biochemical abnormality.
 Propionic acidemia: striatal gliosis and/or marbling; specific biochemical abnormality.
Propionic acidemia: striatal gliosis and/or marbling; specific biochemical abnormality.
 Methylmalonic acidemia: infarcts; hemorrhages; specific biochemical abnormality.
Methylmalonic acidemia: infarcts; hemorrhages; specific biochemical abnormality.

MACROSCOPIC AND MICROSCOPIC APPEARANCES
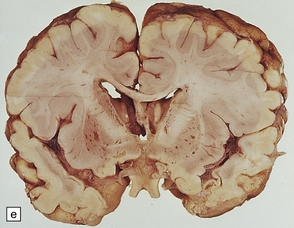
Two patterns are observed (Fig. 6.6):

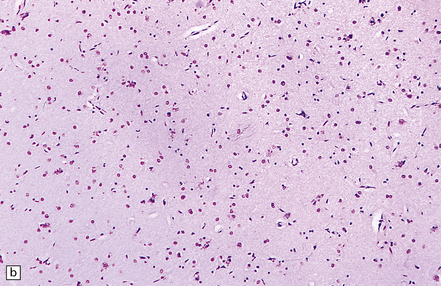
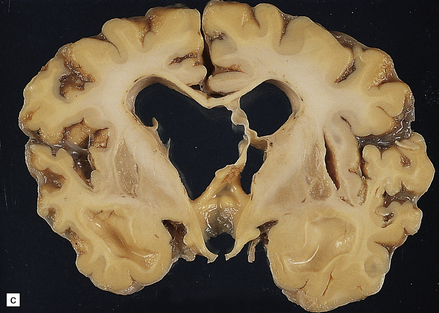
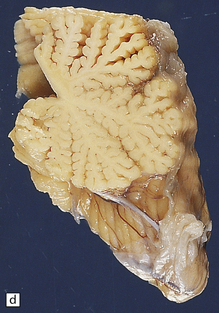
6.6 Holotopistic striatal necrosis.
(a) Histologic section demonstrating shrinkage and pallor of the dorsal halves of both caudate and putamen. (b) Marked neuronal loss and gliosis in the striatum. (c) In another patient the caudate nuclei are atrophic brown crescents and the putamina are replaced by cavities outlined by grayish membranes. Ventricular dilatation and cortical atrophy are pronounced. (d) In this child, there is also cerebellar cortical atrophy, seen here in the vermis.


NEURODEGENERATION WITH BRAIN IRON ACCUMULATION-1 (HALLERVORDEN–SPATZ DISEASE)
 HOLOTOPISTIC STRIATAL NECROSIS
HOLOTOPISTIC STRIATAL NECROSIS
 Some cases are familial (usually autosomal recessive, but a similar disorder can occur in association with Leber’s hereditary optic neuropathy due to mitochondrial DNA mutations).
Some cases are familial (usually autosomal recessive, but a similar disorder can occur in association with Leber’s hereditary optic neuropathy due to mitochondrial DNA mutations).
 Onset may be acute (following a febrile illness) or insidious.
Onset may be acute (following a febrile illness) or insidious.
 Symptoms include choreoathetosis, abnormal eye movements, seizures, and mental retardation.
Symptoms include choreoathetosis, abnormal eye movements, seizures, and mental retardation.
 NEURODEGENERATION WITH BRAIN IRON ACCUMULATION-1
NEURODEGENERATION WITH BRAIN IRON ACCUMULATION-1




MACROSCOPIC AND MICROSCOPIC APPEARANCES
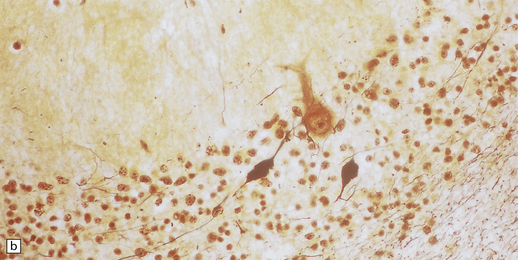
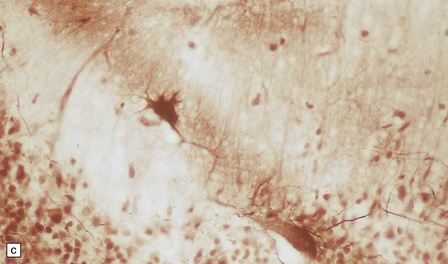
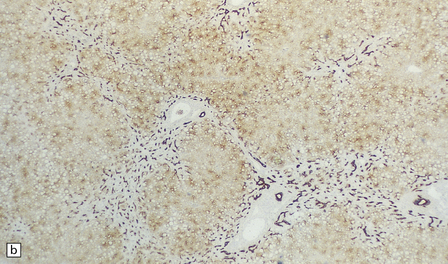
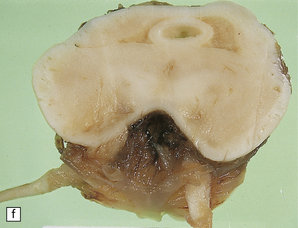
Yellow–brown discoloration of the globus pallidus and substantia nigra are evident (Fig. 6.7). Neuronal loss, gliosis, and deposition of iron pigment occur bilaterally in the internal segment of the globus pallidus and the pars reticularis of the substantia nigra. There is also a more widespread distribution of swollen axons (spheroids). Neurochemical studies indicate abnormal cysteine metabolism in the pallidum and it is suggested that cysteine chelates iron, which in turn induces tissue damage mediated by free radicals (see also neuroaxonal dystrophy, in Chapter 33).
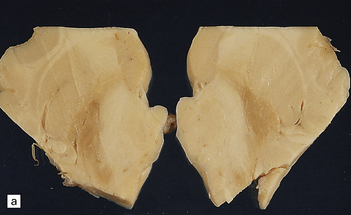
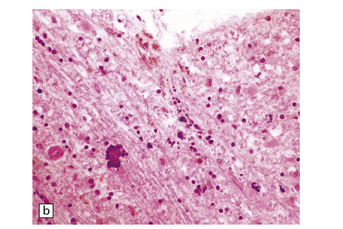
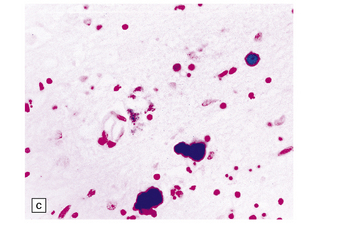
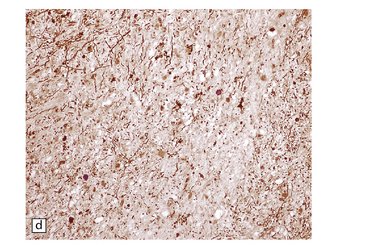
6.7 Neurodegeneration with brain iron accumulation-1.
(a) Coronal sections at the level of the anterior thalamus demonstrate a remarkable yellow–brown pigmentation in both pallida. (b) Microscopically, there are hematoxyphilic mineralizations, eosinophil axonal spheroids, and deposits of brown pigment in the pallidum. (c) These deposits stain positively for iron. (d) Neuroaxonal spheroids demonstrated with antibody to phosphorylated neurofilaments, and mineral concretions (stained blue) within the pallidum. (e) Neuroaxonal spheroids, labeled here with an antibody to neurofilament protein, are also plentiful in the gray matter bridges between caudate and putamen.
CEREBELLUM
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Microscopically there are neuronal loss and myelin deficiency in the temporal cortex and hippocampus. In the cerebellum marked depletion of the granular layer is combined with displacement of Purkinje cells, which show ‘torpedoes’, dendritic distortions, and somal sprouting (Fig. 6.8).
ATAXIA–TELANGIECTASIA
CARBOHYDRATE-DEFICIENT GLYCOPROTEIN SYNDROME TYPE 1 (CDG 1)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 MENKES’ DISEASE
MENKES’ DISEASE




 ATAXIA–TELANGIECTASIA
ATAXIA–TELANGIECTASIA Normal early development precedes gait and truncal ataxia, which becomes apparent when the child starts to walk.
Normal early development precedes gait and truncal ataxia, which becomes apparent when the child starts to walk.

 Independent walking is lost by 12 years of age.
Independent walking is lost by 12 years of age.
 Telangiectasias of conjunctivae are seen after 3 years of age.
Telangiectasias of conjunctivae are seen after 3 years of age.
 Skin lesions develop later in sun-exposed areas.
Skin lesions develop later in sun-exposed areas.
 Increased incidence of respiratory infection is related to abnormal humoral and cellular immunity.
Increased incidence of respiratory infection is related to abnormal humoral and cellular immunity.


 CARBOHYDRATE-DEFICIENT GLYCOPROTEIN SYNDROME TYPE 1
CARBOHYDRATE-DEFICIENT GLYCOPROTEIN SYNDROME TYPE 1





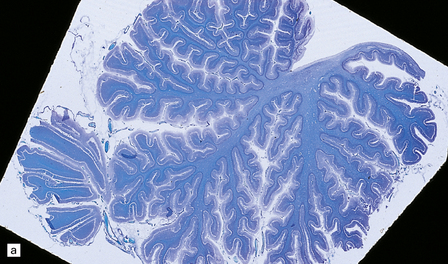
globally small with narrow, prominent, and hard folia (Fig. 6.10). The pontine base is flattened.
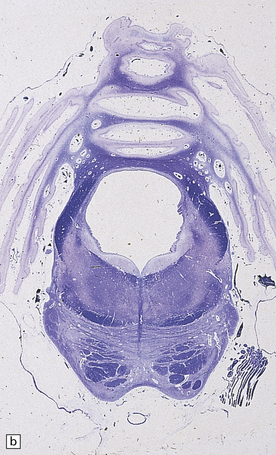
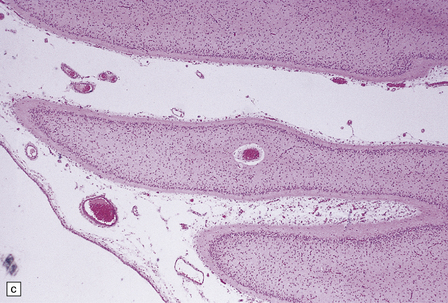
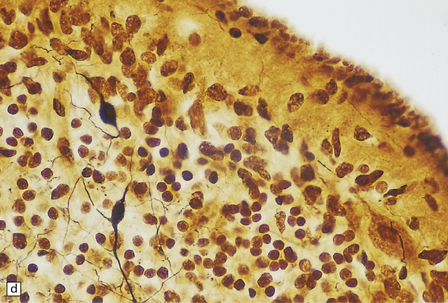
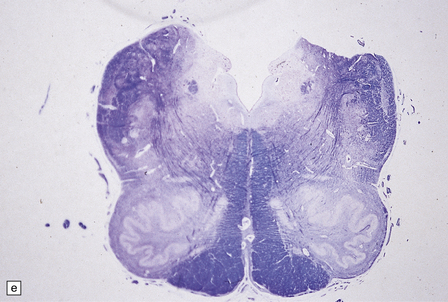
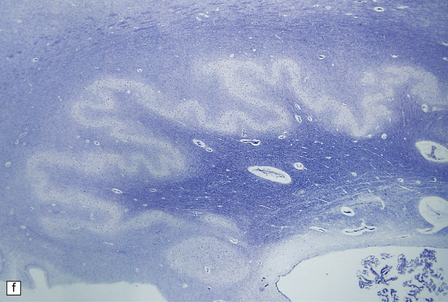
6.10 CDG1.
(a) Seen from below, the cerebral hemispheres of this infant (one of two affected brothers) are quite normal, but the cerebellum is extremely shrunken, the narrow folia standing out. The base of the pons appears excavated. (b) Horizontal section through the hindbrain. The cerebellar folia are reduced to thin unmyelinated plates. The pontine base is shallow and concave in outline; the descending tracts are prominent, but nuclei pontis and transverse fibers are lacking. Compared with the minuscule middle cerebellar peduncles, the superior cerebellar peduncles are well preserved. (c) Histologic detail of the cerebellar folia. There is extreme atrophy leaving gliotic slivers of tissue devoid of all cortical neurons. (d) Occasional axonal ‘torpedoes’ are encountered in the atrophic cortex. (e) Horizontal section through the medulla showing moderate fall-out of olivary neurons and loss of hilar and olivocerebellar fibers. (f) By comparison the dentate neurons are well preserved along with their hilar output to the superior cerebellar peduncles, although the amiculum is quite pale.
At necropsy, pleural and pericardial effusions and ascites are common. The liver shows macrovesicular fatty infiltration (Fig. 6.11), abnormal bile duct plates, and portal fibrosis. The kidneys show pronounced cystic dilatation of tubules and collecting ducts.
CEREBELLOCORTICAL DEGENERATION (JERVIS)
 DIFFERENTIAL DIAGNOSIS OF DISORDERS CHARACTERIZED BY CHILDHOOD INVOLVEMENT OF THE OLIVOPONTOCEREBELLAR AXIS
DIFFERENTIAL DIAGNOSIS OF DISORDERS CHARACTERIZED BY CHILDHOOD INVOLVEMENT OF THE OLIVOPONTOCEREBELLAR AXISThe following discriminating clinicopathological features should be sought:
 Menkes’ disease: internal granular cell depletion; abnormal dendrites on Purkinje cells; hair abnormality; X-linked defect of copper metabolism.
Menkes’ disease: internal granular cell depletion; abnormal dendrites on Purkinje cells; hair abnormality; X-linked defect of copper metabolism.



 Werdnig–Hoffman variant: cerebellar cortical degeneration; associated anterior horn cell disease.
Werdnig–Hoffman variant: cerebellar cortical degeneration; associated anterior horn cell disease.
 ADCA II: olivopontocerebellar atrophy with retinal degeneration; autosomal dominant inheritance.
ADCA II: olivopontocerebellar atrophy with retinal degeneration; autosomal dominant inheritance.
microcephaly from late infancy, epilepsy, and ataxia are the main clinical features, with symptomatic or electrophysiologic evidence of visual failure. Death occurred between 9 months and 16 years of age. These cases may have diverse etiology (see Chapter 29 for a discussion of the modern nosology of cerebellar degenerations).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Microcephaly and marked cerebellar atrophy are seen macroscopically (Fig. 6.12). Histologic features are cerebellar cortical degeneration, severe Purkinje cell loss accompanied by ‘torpedo’ axonal swellings, asteroid dendritic expansions, and variable granule cell involvement. The inferior olivary nuclei are always involved, but the pontine nuclei are spared. Atrophy of the optic nerves, superior colliculi, lateral geniculate nuclei, and visual cortex can also occur.
6.12 Cerebellocortical degeneration.
(a) Mesial view of the hemisected brain. There is severe cerebellar atrophy as well as shrinkage of the optic nerve. (b) In another patient there is global cerebellar cortical atrophy. (c) Glees silver impregnation shows many empty baskets and some ‘torpedo’ swellings associated with surviving Purkinje cells. (d) Another feature of this type of cerebellar degeneration that is demonstrated here with antineurofilament antibodies is the presence of abnormal dendritic swellings on Purkinje cells.
AUTOSOMAL DOMINANT CEREBELLAR ATAXIA TYPE II (ADCA II)
 CHILDHOOD NEURODEGENERATIVE DISORDERS AFFECTING THE CEREBELLUM
CHILDHOOD NEURODEGENERATIVE DISORDERS AFFECTING THE CEREBELLUM













MACROSCOPIC AND MICROSCOPIC APPEARANCES
Microcephaly, cerebellar atrophy, a shallow pons, and flat inferior olives are accompanied by a subtotal loss of Purkinje cells, lesser depletion of granule cells, Bergmann cell gliosis, severe olivary atrophy, and variable depletion of the nuclei pontis (Fig. 6.13).
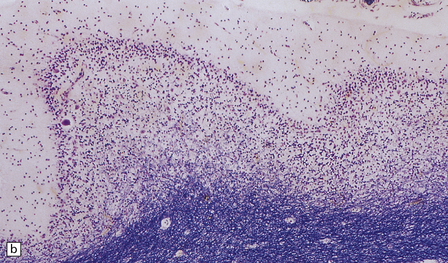
6.13 ADCA II.
(a) Horizontal section showing prominent folial atrophy throughout the cerebellum. (b) Histology shows global devastation of the cerebellar cortex. An occasional Purkinje cell remains among Bergmann glia.
 AUTOSOMAL DOMINANT CEREBELLAR ATAXIA TYPE II
AUTOSOMAL DOMINANT CEREBELLAR ATAXIA TYPE II


BRAIN STEM


MACROSCOPIC AND MICROSCOPIC APPEARANCES
 HEREDITARY BULBAR PALSIES
HEREDITARY BULBAR PALSIES



Bulbar hereditary motor neuropathy type II (Fazio–Londe disease)
 There are three clinical subtypes:
There are three clinical subtypes:
• Dominant inheritance; presents at 4–20 years of age with dysphagia and dysarthria; progressive and fatal.
• Recessive inheritance; presents <5 years of age with respiratory symptoms and stridor; fatal within 2 years.
• Recessive inheritance; onset >5 years of age with dysphagia, dysarthria, and facial weakness; survival for many years.
 EMG shows a neurogenic process in bulbar muscles that may also be seen in limb muscles.
EMG shows a neurogenic process in bulbar muscles that may also be seen in limb muscles.
 Motor cranial nerve nuclei, including oculomotor nerve nuclei, in the brain stem.
Motor cranial nerve nuclei, including oculomotor nerve nuclei, in the brain stem.
 Anterior horn cells in the spinal cord (Fig. 6.14) with consequent widespread neurogenic atrophy of muscles, including the tongue.
Anterior horn cells in the spinal cord (Fig. 6.14) with consequent widespread neurogenic atrophy of muscles, including the tongue.
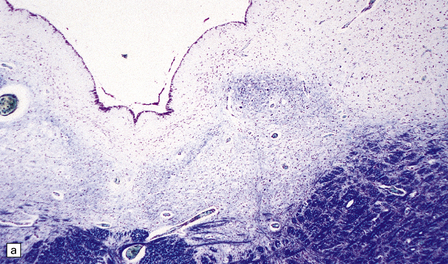
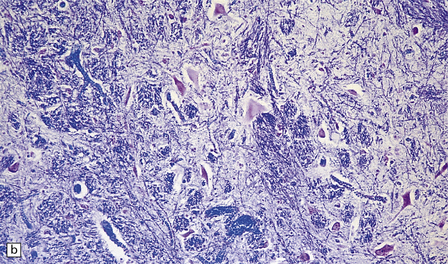
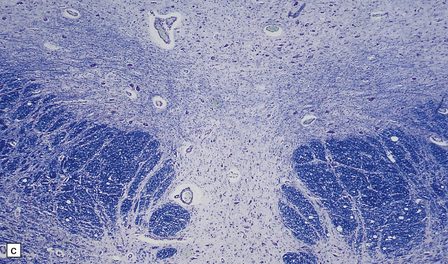
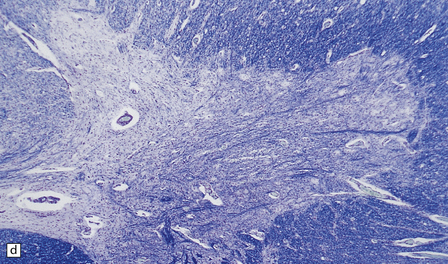
6.14 Fazio–Londe disease (bulbar hereditary motor neuropathy type II).
(a) Neuronal degeneration and depletion is widespread in cranial nerve nuclei such as the 10th and 12th in the medulla. (b) It also affects the motor 5th in the pons. (c) The oculomotor nuclei in the midbrain are involved. (d) There is anterior horn cell loss in the cord.
INFANTILE NEURO-AXONAL DYSTROPHY (INAD)
 INFANTILE NEURO-AXONAL DYSTROPHY (INAD)
INFANTILE NEURO-AXONAL DYSTROPHY (INAD)





disorders, experimental toxicology, and certain inherited disorders (see also Chapter 33). The main examples of the latter are:


MACROSCOPIC AND MICROSCOPIC APPEARANCES
Apart from cerebellar atrophy, macroscopic abnormality in INAD is minimal. Axonal spheroids are widely distributed throughout the CNS (Fig. 6.15), but are most easily found in the cerebellum, brain stem, and spinal cord, notably among the long sensory tracts. They are also present in cerebellar and cerebral white matter, basal ganglia, thalamus, and in the cerebral cortex where their small size makes detection difficult with conventional histology.
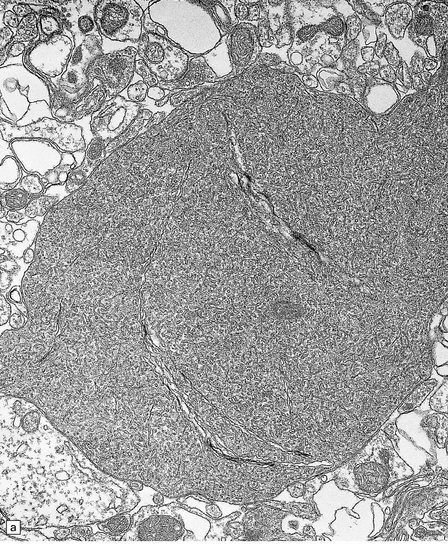
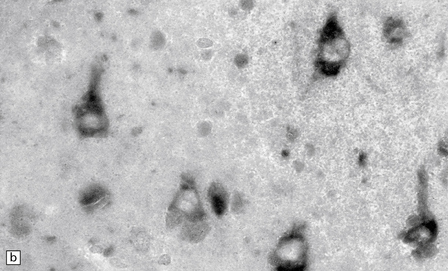
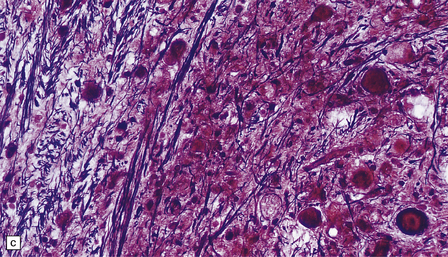
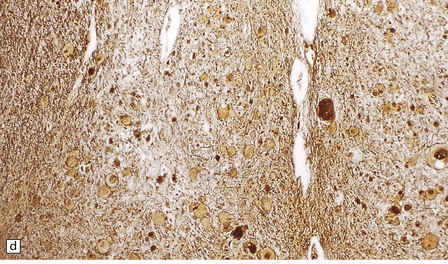
6.15 INAD.
(a) Ultrastructurally the axonal spheroids contain closely packed tubulovesicular membranes interrupted by narrow clefts. (b) The nonspecific esterase technique is a useful method for detecting spheroids, particularly in the cerebral cortex where they are relatively small. Note that the small spheroid to the left of this micrograph contains a curved cleft. Surrounding neurons are also positive with this method. (c) Axonal spheroids are particularly common in the dorsal medulla. Silver impregnation gives a variable and irregular result. (d) Spheroids are usually numerous in the dorsal funiculi, but note the variable immunoreactivity with neurofilament antibody. (e) Cerebellar cortical degeneration is often associated with INAD and is characterized by marked Purkinje cell fallout and torpedo swellings of proximal axons, which are visualized here with an antibody to neurofilament protein.
LEIGH’S DISEASE (SUBACUTE NECROTIZING ENCEPHALOMYELOPATHY)
MACROSCOPIC APPEARANCES
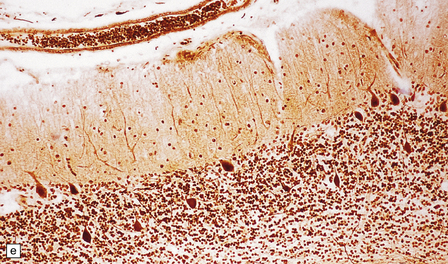
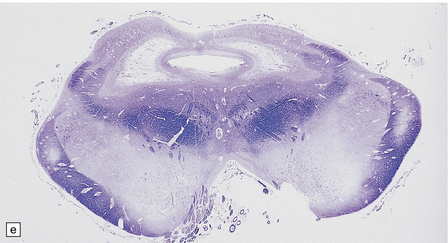
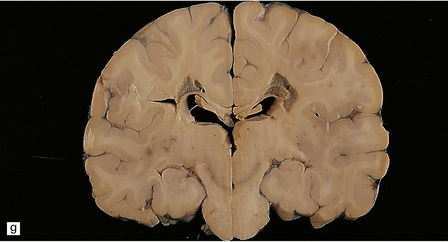
There are characteristic symmetric circumscribed brown patches or gray lesions, which are sometimes cavitated (Fig. 6.16). These lesions are present in the striatum, subthalamic nucleus, substantia nigra, inferior olives, and hindbrain tegmentum, and often run in a linear fashion below the ventricle.
6.16 Leigh’s disease.
(a) The lesions are usually bilateral and are most easy to recognize in the brain stem as brown patches or linear streaks in the tegmentum, inferior colliculi, and substantia nigra. In this 8-month-old child, the nigra should not be pigmented. (b) Other lesions can be found in the corpus striatum. (c) Lesions can also be found surrounding the cerebellar dentate nuclei. (d) An extensive lesion may completely transect the spinal cord (Luxol fast blue/cresyl violet). (e) Older lesions become cavitated, as in this example within the striatum. (f) Bilateral cavitated lesions in the midbrain within the periaqueductal gray matter and substantia nigra.
 LEIGH’S DISEASE
LEIGH’S DISEASE






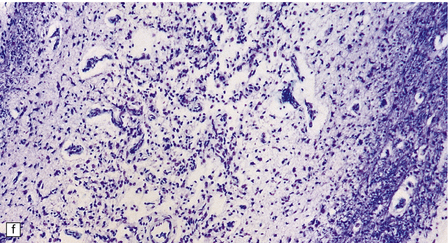
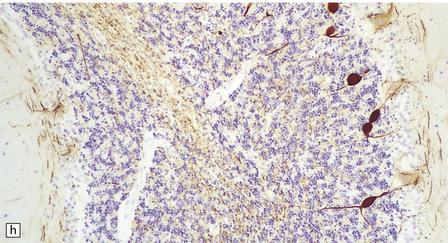
MICROSCOPIC APPEARANCES
In typical cases there are many vasculonecrotic lesions distributed symmetrically through the CNS. These lesions take three histologic forms (Fig. 6.17), which probably reflect varying chronicity, but the different appearances are often contiguous, suggesting repetitive damage to the area. The oldest lesions are fibrous or cavitated gliotic scars, equivalent to old infarcts. The youngest and least frequent lesions are composed of poorly cellular edematous neuropil with activated macrophages and eosinophilic neurons. The most typical lesion is characterized by numerous foamy macrophages and variable astrocytosis, prominent congested and hypertrophic capillaries, and collapse of the neuropil, within which there are often some normal neurons. This recent lesion (probably several weeks old) resembles the lesions of Wernicke’s encephalopathy, but there is never any hemosiderin deposition.
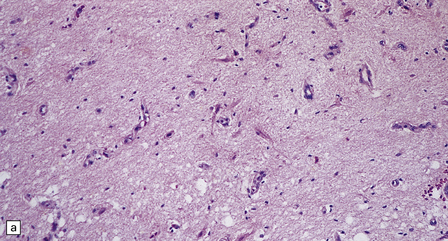
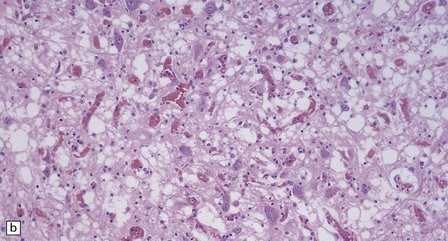
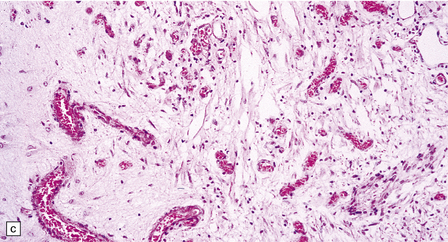
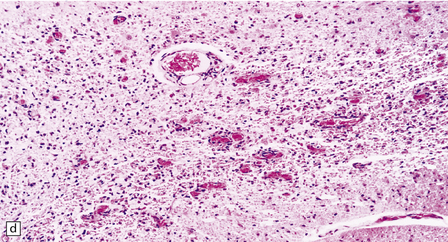
6.17 Leigh’s disease.
(a) In the youngest lesions subtle histologic changes include tissue edema and neuronal necrosis. (b–d) The more characteristic changes are a variable mixture of tissue necrosis, macrophage infiltration, hypertrophy of astrocytes, and a prominent vascularity. Characteristically, some neurons are intact within the areas of partial necrosis. (e) The oldest lesions are merely glial-lined cavities. Histologic section of the midbrain shown in Fig. 6.16 f. (f) Degeneration of the central white matter of the optic nerves. (g) Lesions may also involve the periventricular white matter, here showing very ancient cavities. (h) Cerebellar degeneration is quite common. In this example, loss of Purkinje cells is accompanied by profuse ‘torpedo’ formation, shown by neurofilament protein immunohistochemistry.
Symmetric lesions, often confined to the same part of a nucleus, are found in many gray areas (Table 6.1). Lesions are nearly always present in the substantia nigra, but in only about 50% of patients in the striatum and inferior olives, notably in those patients dying after the first year.
Table 6.1
Frequency of gray matter lesions in Leigh’s disease
| Affected region | Approximate frequency (%) |
| Caudate nucleus | 50 |
| Putamen | 50 |
| Globus pallidus | 25 |
| Substantia nigra | 95 |
| Thalamus | 33 |
| Periaqueductal gray | 60 |
| Mammillary bodies | Rare |
| Subthalamic nucleus | 40 |
| Red nucleus | 33 |
| Superior colliculus | 25 |
| Inferior colliculus | 80 |
| Pontine tegmentum | 60 |
| Cerebellar nuclei | 60 |
| Inferior olivary nucleus | 50 |
| Medullary tegmentum | 80 |
| Spinal gray matter | 75 |
 LEIGH’S DISEASE
LEIGH’S DISEASE


SPINAL CORD



MACROSCOPIC AND MICROSCOPIC APPEARANCES
At necropsy, there is a striking atrophy of anterior spinal nerve roots and motor nerves (Fig. 6.18). There is usually a profound loss of anterior horn cells and gliosis at many levels of the spinal cord. In the early stages there are acute degenerative changes such as chromatolysis and microglial nodules. The degenerative process may also extend to the bulbar cranial nerve nuclei and sometimes to the dorsal root sensory ganglion cells.

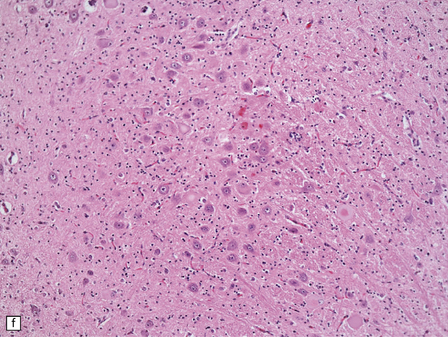
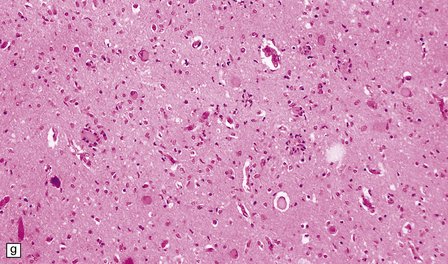
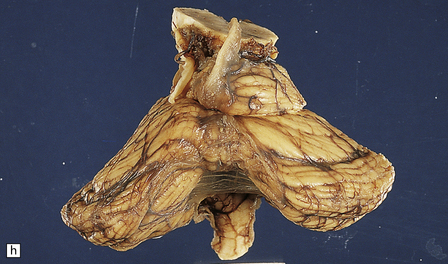
6.18 Spinal muscular atrophy.
(a) Dissection of the lower cord. Compare the gray atrophic anterior roots with the white, well myelinated posterior roots. The cardinal histologic changes are loss of motor neurons from the anterior horn, the process of degeneration being one of ballooning and chromatolysis, with microglial activation and neuronophagia (b). Usually a similar neuronal degeneration occurs in the thalamus (c) and occasionally in brain stem motor nuclei such as the 12th nucleus (d), the 7th nucleus (e) or the 3rd nucleus (f).Often overlooked by prosectors, examination of dorsal root ganglia regularly reveals degenerated ganglion cells or residual nodules of Nageotte (g). In one variant of SMA there is an associated severe cerebellar degeneration. Here the atrophic cerebellum is viewed from behind (h).
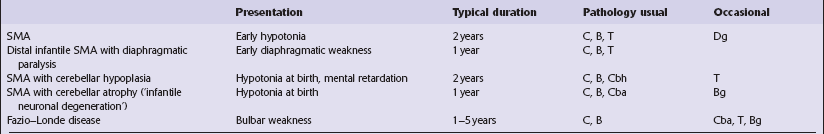
SMA VARIANTS
There are several similar clinical entities of diverse genetic background that cause anterior horn cell degeneration and these should be differentiated from the typical acute form of SMA. Table 6.2 places them in context alongside SMA and the progressive bulbar palsies of childhood.
 GENETIC ASPECTS OF SMA
GENETIC ASPECTS OF SMA



 SPINAL MUSCULAR ATROPHY
SPINAL MUSCULAR ATROPHY










REFERENCES
Bertini, E., Gadisseux, J.L., Palmieri, G., et al. Distal infantile spinal muscular atrophy associated with paralysis of the diaphragm: a variant of infantile spinal muscular atrophy. Am J Med Genet.. 1989;33:328–335.
Cavanagh, J.B., Harding, B.N. Pathogenic factors underlying the lesions in Leigh’s disease: tissue responses to cellular energy deprivation and their clinico-pathological consequences. Brain.. 1994;117:1357–1376.
Chou, S.M., Wang, H.S. Aberrant glycosylation/phosphorylation in chromatolytic motoneurons of Werdnig–Hoffmann disease. J Neurol Sci.. 1997;152:198–209.
Friede, R.L. Developmental neuropathology. Berlin: Springer-Verlag; 1989.
Gallai, V., Hockaday, J.M., Hughes, J.T., et al. Ponto-bulbar palsy with deafness (Brown–Vialetto–Van Laere syndrome). J Neurol Sci.. 1981;50:259–275.
Goutières, F., Aicardi, J., Farkas, E. Anterior horn cell disease associated with pontocerebellar hypoplasia in infants. J Neurol Neurosurg Psychiatry.. 1977;40:370–378.
Hagberg, B.A., Blennow, G., Kristriansson, B., et al. Carbohydrate-deficient glycoprotein syndromes: peculiar group of new disorders. Pediatr Neurol.. 1993;9:255–262.
Harding, B.N. Progressive neuronal degeneration of childhood with liver disease (Alpers–Huttenlocher syndrome) – a personal review. J Child Neurol.. 1990;5:273–287.
Harding, B.N., Alsanjari, N., Smith, S.J., et al. Progressive neuronal degeneration of childhood with liver disease (Alpers’ disease) presenting in young adults. J Neurol Neurosurg Psychiatry.. 1995;58:320–325.
Horslen, S.P., Clayton, P.T., Harding, B.N., et al. Neonatal onset olivopontocerebellar atrophy and disialotransferrin deficiency syndrome. Arch Dis Child.. 1991;66:1027–1032.
Ikemoto, A., Hirano, A., Matsumoto, S., et al. Synaptophysin expression in the anterior horn of Werdnig–Hoffmann disease. J Neurol Sci.. 1996;136:94–100.
Iwahashi, H., Eguchi, Y., Yasuhara, N., et al. Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature.. 1997;390:413–417.
Kamoshita, S., Takei, Y., Miyao, M., et al. Pontocerebellar hypoplasia associated with infantile motor neuron disease (Norman’s disease). Pediatr Pathol.. 1990;10:133–142.
McShane, M.A., Boyd, S., Harding, B., et al. Progressive bulbar paralysis of childhood: a reappraisal of Fazio–Londe disease. Brain.. 1992;115:1889–1900.
Mellins, R.B., Hays, A.P., Gold, A.P., et al. Respiratory distress as the initial manifestation of Werdnig–Hoffmann disease. Pediatrics.. 1974;53:33–40.
Moretto, G., Sparaco, M., Monarco, S., et al. Cytoskeletal changes and ubiquitin expression in dystrophic axons of Seitelberger’s disease. Clin Neuropathol.. 1993;12:34–37.
Morris, A.A., Singh, K.R., Perry, R.H., et al. Respiratory chain dysfunction in progressive neuronal degeneration of childhood with liver disease. J Child Neurol.. 1996;11:417–419.
Murayama, S., Bouldin, T.W., Suzuki, K. Immunocytochemical and ultrastructural studies of Werdnig–Hoffmann disease. Acta Neuropathol.. 1991;81:408–417.
Naviaux, R.K., Nyhan, W.L., Barshop, B.A., et al. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann Neurol.. 1999;45:54–58.
Norman, R.M., Kay, J.M. Cerebello-thalamo-spinal degeneration in infancy: an unusual variant of Werdnig–Hoffmann Disease. Arch Dis Child.. 1965;40:302–308.
Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD). 2000. http://www.ncbi.nlm.nih.gov/omim/.
Ozmen, M., Caliskan, M., Goebel, H.H., et al. Infantile neuroaxonal dystrophy:diagnosis by skin biopsy. Brain Dev.. 1991;13:256–259.
Ramaekers, V.T., Lake, B.D., Harding, B., et al. Diagnostic difficulties in infantile neuroaxonal dystrophy: a clinicopathological study of eight cases. Neuropediatrics.. 1987;18:170–175.
Rudnik Schoneborn, S., Forkert, R., Hahnen, E., et al. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics. 1996;27:8–15.
Schindler, D., Bishop, D.F., Wolfe, D.E., et al. Neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. N Engl J Med.. 1989;320:1735–1740.
Schmalbruch, H., Haase, G. Spinal muscular atrophy: present state. Brain Pathol.. 2001;11:231–247.
Schrank, B., Gotz, R., Gunnersen, J.M., et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A.. 1997;94:9920–9925.
Simic, G., Seso-Simic, D., Lucassen, P.J., et al. Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig–Hoffmann disease. J Neuropathol Exp Neurol.. 2000;59:398–407.
Steimann, G.S., Rorke, L.B., Brown, M.J. Infantile neuronal degeneration masquerading as Werdnig–Hoffmann disease. Ann Neurol.. 1980;8:317–324.
Stromme, P., Maehlen, J., Strom, E.H., et al. Post mortem findings in two patients with the carbohydrate-deficient glycoprotein syndrome. Acta Paediatr Scand.. 1991;375(suppl):55–62.
Summers, B.A., Swash, M., Schwartz, M.S., et al. Juvenile-onset bulbospinal muscular atrophy with deafness: Vialetta–van Laere syndrome or Madras-type motor neuron disease? J Neurol.. 1987;234:440–442.
Yamanouchi, Y., Yamanouchi, H., Becker, L.E. Synaptic alterations of anterior horn cells in Werdnig–Hoffmann disease. Pediatr Neurol.. 1996;15:32–35.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]6
Neurodegenerative disorders of gray matter in childhood
CEREBRAL CORTEX
ALPERS–HUTTENLOCHER SYNDROME OR PROGRESSIVE NEURONAL DEGENERATION OF CHILDHOOD (PNDC)
MACROSCOPIC APPEARANCES
Lesions may be minimal, patchy, or extensive (Fig. 6.1). In affected regions the cortical ribbon is thin, granular, and brown, and even dehiscent in some poorly fixed brains. The calcarine cortex is often picked out in a remarkably selective and characteristic way. Rarely, there is softening of the occipital white matter.
6.1 Alpers–Huttenlocher syndrome.
Selective involvement of the calcarine cortex is a helpful diagnostic pointer. (a) In this child, coronal sections of occipital lobe show the primary visual cortex in both hemispheres delineated by granularity and brown discoloration (arrow). (b) Similar discrete areas of cortical thinning and discoloration are present in the posterior frontal cortex at midthalamic level (arrow). (c) Although cortical lesions are usually patchy, in extreme cases the whole cortical ribbon may be diffusely and uniformly shrunken. In addition, there is ventricular dilatation, some thalamic atrophy, and marked shrinkage of Ammon’s horns.
MICROSCOPIC APPEARANCES
Histologic abnormalities are more widespread than expected from the macroscopic appearances. The patchy lesions do not conform to vascular territories or watershed zones and show a graded intensification and extension of the degenerative process through the depth of the cortical gray matter. Mild superficial spongiosis gives way to increasing sponginess, neuronal loss, and gliosis extending down through the cortex. In severe lesions, the whole ribbon is replaced by a narrow remnant of hypertrophic astrocytes devoid of nerve cells (Fig. 6.2). Neutral fat may be deposited in considerable amounts. Lesions may be symmetric or asymmetric, but there is a striking predilection for the striate cortex. Secondary changes are found in the white matter. Other variable findings include hippocampal sclerosis, cerebellar cortical infarcts, spinal cord tract degeneration, and spongiosis and gliosis in the thalamus (Fig. 6.3), amygdala, substantia nigra, and dentate nuclei.
6.2 Alpers–Huttenlocher syndrome.
(a) The mildest histologic changes consist of a fine spongiosis in the superficial cortical layers. (b) Neuronal loss and gliosis gradually extend more deeply through the cortical ribbon. (c) Eventually, the whole cortex is replaced by hypertrophic astrocytes. This shows the striate cortex, which is especially prone to such severe destruction. (d) The neurons and neuropil of the striate cortex are entirely replaced by a cystic meshwork of glial processes emanating from plump hypertrophic astrocytes. (e) Massive amounts of neutral fat can be deposited in the degenerating cortex. (f) The chronic end stage of this process is a thin, poorly cellular gliotic remnant. This shows striate cortex from the same case as in Fig. 6.1c.
HEPATIC PATHOLOGY
Nearly all patients show characteristic changes in the liver: the hepatocytes undergo severe microvesicular fatty or oncocytic change (Fig. 6.4). There are hepatocyte necrosis, diffuse haphazard bile duct proliferation, and bridging fibrosis, with disorganization and regeneration that amount to cirrhosis at one end of the histologic spectrum (Fig. 6.5), or end-stage collapse and fibrosis at the other.
6.4 Alpers–Huttenlocher syndrome: liver pathology.
Histologic changes in the liver are characteristic and essential for a confident diagnosis. (a) The principal features are hepatic steatosis and oncocytic change along with hepatocyte necrosis and a profuse and haphazard proliferation of bile ductules. (b) Fibrosis and nodular regeneration often give rise to a cirrhotic pattern. (c) Fat may be heavily deposited within the regenerative nodules. (d) In some patients, regeneration does not occur and the liver is replaced by dense fibrous tissue and irregular bile ductules. Note the bile retention.
6.5 Alpers–Huttenlocher syndrome.
Liver pathology in two brothers who died from PNDC. One of these brothers received valproate therapy, but both showed typical liver pathology. (a) Macroscopic appearance of the finely nodular cirrhotic liver of one brother. (b) Liver histology shows the typical changes of PNDC in the other brother. The role of drug therapy and in particular valproate toxicity in the pathogenesis of PNDC is controversial.
The following discriminating features should be sought:
Hypoxic–ischemic encephalopathy: predilection for vascular boundary zones, relative preservation of gyral crests; striate cortex spared.
Hepatic encephalopathy: involvement of basal ganglia; cavitation of gray–white matter interface.
Neuronal storage disorders: neuronal abnormalities.
ETIOLOGY OF ALPERS–HUTTENLOCHER SYNDROME
Prominent spongiosis prompted speculation of a kinship with Creutzfeldt–Jakob disease but one apparently successful animal transmission experiment has not been duplicated.
Evidence of mitochondrial dysfunction in some patients includes cytochrome oxidase deficiency.
Most patients are compound heterozygotes with two mutations of the mtDNA polymerase gene POLG1.
BASAL GANGLIA
HOLOTOPISTIC STRIATAL NECROSIS (FAMILIAL STRIATAL DEGENERATION)
DIFFERENTIAL DIAGNOSIS OF BASAL GANGLIA DISORDERS IN CHILDHOOD
The following discriminating clinicopathological features should be sought.
Holotopistic striatal necrosis: acute necrosis or slow degeneration; recessive inheritance.
Wilson’s disease: striatal, retinal, and hepatic degeneration.
Juvenile Parkinson disease: degeneration of substantia nigra and locus ceruleus.
Glutaric acidemia: striatal degeneration; specific biochemical abnormality.
Propionic acidemia: striatal gliosis and/or marbling; specific biochemical abnormality.
Methylmalonic acidemia: infarcts; hemorrhages; specific biochemical abnormality.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Two patterns are observed (Fig. 6.6):
6.6 Holotopistic striatal necrosis.
(a) Histologic section demonstrating shrinkage and pallor of the dorsal halves of both caudate and putamen. (b) Marked neuronal loss and gliosis in the striatum. (c) In another patient the caudate nuclei are atrophic brown crescents and the putamina are replaced by cavities outlined by grayish membranes. Ventricular dilatation and cortical atrophy are pronounced. (d) In this child, there is also cerebellar cortical atrophy, seen here in the vermis.
NEURODEGENERATION WITH BRAIN IRON ACCUMULATION-1 (HALLERVORDEN–SPATZ DISEASE)
HOLOTOPISTIC STRIATAL NECROSIS
Some cases are familial (usually autosomal recessive, but a similar disorder can occur in association with Leber’s hereditary optic neuropathy due to mitochondrial DNA mutations).
Onset may be acute (following a febrile illness) or insidious.
Symptoms include choreoathetosis, abnormal eye movements, seizures, and mental retardation.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Yellow–brown discoloration of the globus pallidus and substantia nigra are evident (Fig. 6.7). Neuronal loss, gliosis, and deposition of iron pigment occur bilaterally in the internal segment of the globus pallidus and the pars reticularis of the substantia nigra. There is also a more widespread distribution of swollen axons (spheroids). Neurochemical studies indicate abnormal cysteine metabolism in the pallidum and it is suggested that cysteine chelates iron, which in turn induces tissue damage mediated by free radicals (see also neuroaxonal dystrophy, in Chapter 33).
6.7 Neurodegeneration with brain iron accumulation-1.
(a) Coronal sections at the level of the anterior thalamus demonstrate a remarkable yellow–brown pigmentation in both pallida. (b) Microscopically, there are hematoxyphilic mineralizations, eosinophil axonal spheroids, and deposits of brown pigment in the pallidum. (c) These deposits stain positively for iron. (d) Neuroaxonal spheroids demonstrated with antibody to phosphorylated neurofilaments, and mineral concretions (stained blue) within the pallidum. (e) Neuroaxonal spheroids, labeled here with an antibody to neurofilament protein, are also plentiful in the gray matter bridges between caudate and putamen.
CEREBELLUM
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Microscopically there are neuronal loss and myelin deficiency in the temporal cortex and hippocampus. In the cerebellum marked depletion of the granular layer is combined with displacement of Purkinje cells, which show ‘torpedoes’, dendritic distortions, and somal sprouting (Fig. 6.8).
