[level-membership-for-pathology-category]
Neural (Sensory) Retina
Normal Anatomy
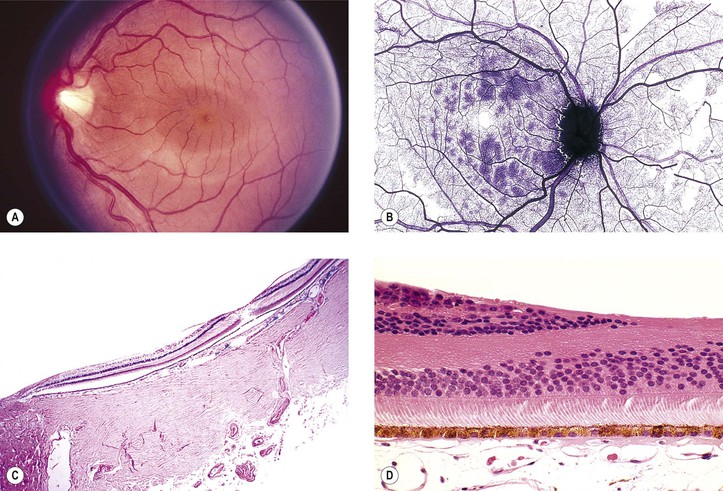
I. The neural retina (Figs. 11.1–11.3) is a highly specialized nervous tissue, in reality a part of the brain that has become exteriorized.
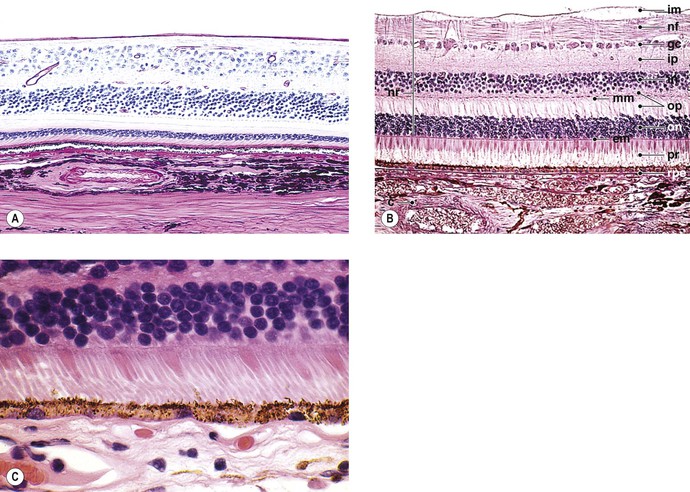
A. Traditionally, the retina, from the RPE externally to the internal limiting membrane internally, is divided into 10 layers (see Fig. 11.2B).
II. Foveomacular region of the neural retina
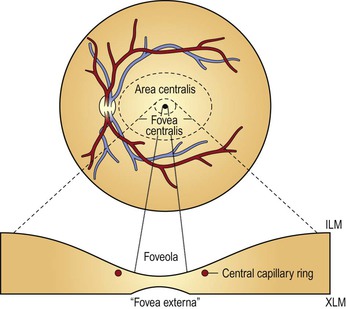
A. Clinicians often confuse the proper use of the terms fovea, macula, and posterior pole (see Fig. 11.3).
4. The anatomic macula (which corresponds to the clinical posterior pole) comprises an area larger than the anatomic fovea.
a. The term macula is derived from the term macula lutea.
c. The anatomic macula actually encompasses an area contained just within the optic nerve and the superior and inferior retinal temporal arcades, and extends temporally approximately two disc diameters beyond the central fovea.
Congenital Anomalies
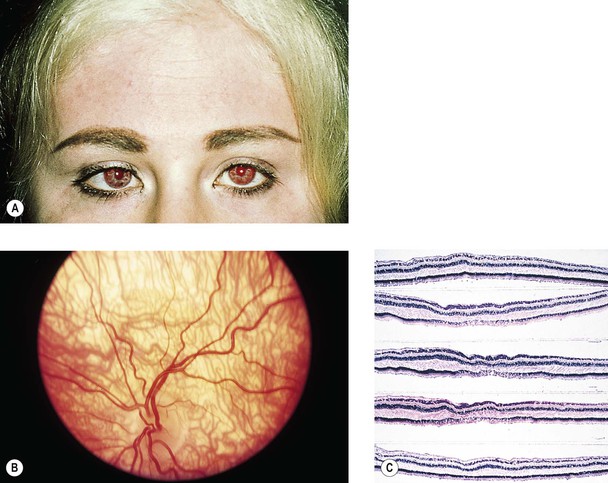
Albinism (Fig. 11.4)
III. The conditions are noted at birth.
B. Color vision is normal. Nyctalopia does not occur.
IV. Histology
A. The RPE is depleted of pigment.
B. Macromelanosomes may be present within the RPE.
A. Inheritance is autosomal recessive.
B. The condition is one of the immunodeficiency diseases (see Chapter 1) and is associated with a lack of resistance to infection and generalized lymphadenopathy. The condition is fatal from generalized infection, usually during the second decade of life.
Grouped Pigmentation (Bear Tracks)
I. Grouped pigmentation (see Fig. 17.22) is unilateral (85% of cases), nonprogressive, and presents as multiple, discrete, well-circumscribed, flat, uniformly pigmented areas resembling a bear’s footprints.
Coloboma
I. The typical coloboma “of the choroid” (see Fig. 9.8) involves the region of the embryonic cleft (fetal fissure; i.e., inferonasally) and is bilateral in 60% of patients. The coloboma may involve the total region, a large part of it, or one or more small isolated parts.
II. Histologically, the RPE seems to be primarily involved and is absent in the area of the coloboma.
A. The neural retina is atrophic and gliotic and may contain rosettes.
Retinal Dysplasia
See Chapter 18.
Lange’s Fold
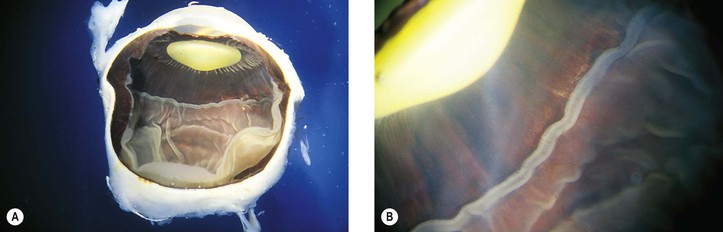
I. Lange’s fold (Fig. 11.5; see also Fig. 10.6) is an inward fold of neural retina at the ora serrata only seen in infants’ and children’s eyes.
Congenital Nonattachment of the Retina
I. Nonattachment of the neural retina (see Fig. 18.24) is normal in the embryonic eye; persistence after birth is abnormal.
Neural Retinal Cysts
I. A cyst of the neural retina (see Fig. 11.56) is defined arbitrarily as an intraneural retinal space whose internal–external diameter is greater than the thickness of the surrounding neural retina and of approximately equal dimension in any direction.
Myelinated (Medullated) Nerve Fibers
I. Myelinated nerve fibers (MNF; Fig. 11.6) usually occur as a unilateral condition, somewhat more common in men than in women.
B. Rarely, MNF occur after infancy and can progress.
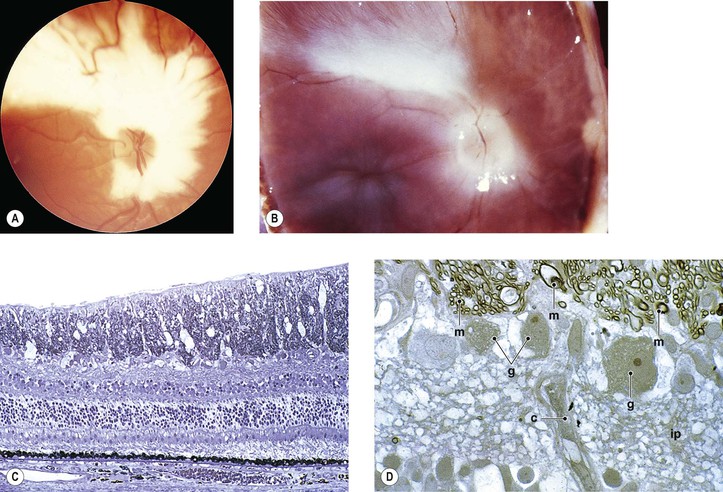
II. Clinically, they appear as an opaque white patch or arcuate band with feathery edges.
Leber’s Congenital Amaurosis
Inherited Retinal Arteriolar Tortuosity
Vascular Diseases
Definitions
Retinal Ischemia
Causes
I. Choroidal vascular insufficiency
II. Retinal vascular insufficiency
A. Large-artery disease anywhere from aortic arch to central retinal artery
1. Atherosclerosis shows patchy subendothelial lipid deposits and erosion of media.
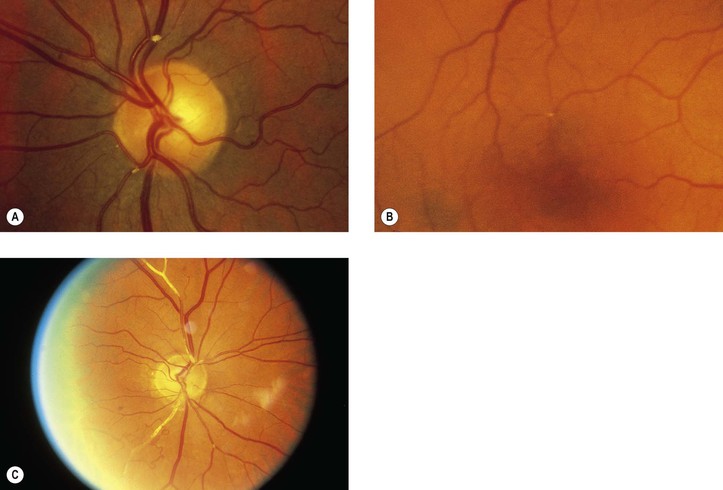
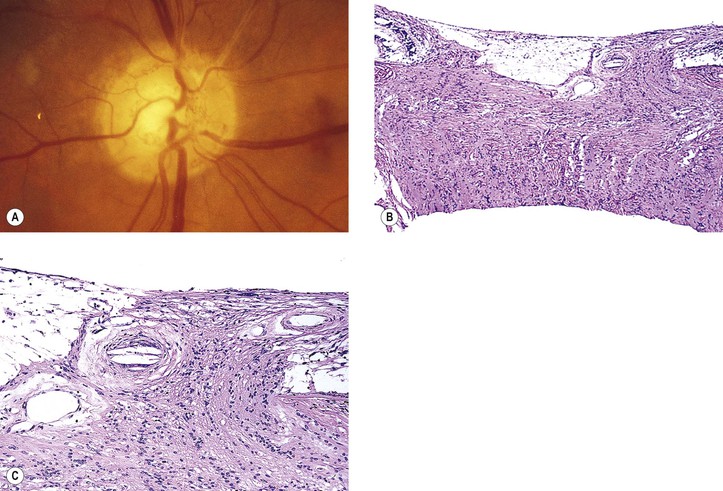
b. Embolic manifestations (Figs. 11.7 and 11.8; see also Fig. 5.54)
3) Rarely, ocular emboli cause a condition masquerading as temporal (cranial; giant cell) arteritis.
5. Temporal (cranial; giant cell) arteritis (see Chapter 13)
B. Arteriolar and capillary disease of neural retinal vasculature
1. Arteriolosclerosis is associated with hypertension.
2. Branch retinal artery occlusion has many causes, including emboli (see Figs. 11.7 and 11.8), arteriolosclerosis, diabetes mellitus, arteritis, dysproteinemias, collagen diseases, and malignant hypertension.
3. Diabetes mellitus (see Chapter 15)
4. Malignant hypertension, toxemia of pregnancy, hemoglobinopathies, collagen diseases (Fig. 11.9), dysproteinemias, carbon monoxide poisoning, and blood dyscrasias of many kinds may involve the small retinal vessels and cause neural retinal ischemia.
Histology of Retinal Ischemia
I. Early (Fig. 11.10; see also Figs. 11.9 and 11.15D)
A. The neural retina shows coagulative necrosis of its inner layers, which are supplied by the retinal arterioles.
1. The neuronal cells become edematous during the first few hours after occlusion of the artery.
2. The intracellular swelling accounts for the clinical gray neural retinal opacity.
B. If the area of coagulative necrosis (see Chapter 1) is small and localized, it appears clinically as a cotton-wool spot.
1. The cotton-wool spot observed clinically (Fig. 11.11; see also Fig. 11.9) is a result of a microinfarct of the nerve fiber layer of the neural retina.
2. The cytoid body, observed microscopically (see Figs. 11.9 and 11.11), is a swollen, interrupted axon in the neural retinal nerve fiber layer.
II. Late (see Fig. 11.10)
Retinal Hemorrhagic Infarction (Fig. 11.12)
Causes and Risk Factors of Hemorrhagic Infarction
Types of Hemorrhagic Infarction
I. Occlusion of central retinal vein, branch retinal vein, or venule
A. CRVO may be considered to consist of two distinct types.
1. Nonischemic retinopathy (~65% of cases, but approximately one-fourth of these eyes will convert to the ischemic type)
2. Ischemic (hemorrhagic) retinopathy
a. It is caused by occlusion of the central retinal vein at, or anterior to, the lamina cribrosa, associated with retinal ischemia that leads to significant retinal hypoxia.
4. Bilateral CRVO may occur as part of the acquired immunodeficiency syndrome (AIDS).
B. Branch retinal vein occlusion (BRVO)
1. BRVO occurs approximately three times more frequently than CRVO. In approximately two-thirds of cases, the superior temporal neural retinal vein is involved. Most of the remaining cases show involvement of the inferior temporal retinal vein.
b. A hemispheric vein occlusion behaves like an ischemic CRVO.
II. Papillophlebitis (retinal vasculitis, mild and moderate papillary vasculitis, benign retinal vasculitis, optic disc vasculitis)
Complications of Hemorrhagic Infarction
I. Macular hemorrhagic infarction may result in permanent loss of vision.
II. Leakage of fluid into the macula may result in cystoid macular edema.
III. Iris neovascularization (clinically seen rubeosis iridis)
A. Iris neovascularization (see Figs. 9.13 and 9.14) occurs mainly with ischemic CRVO; it rarely occurs with nonischemic CRVO or BRVO.
IV. Neovascularization of the neural retina (Fig. 11.13) occurs mainly with BRVO; it rarely occurs with ischemic or nonischemic CRVO.
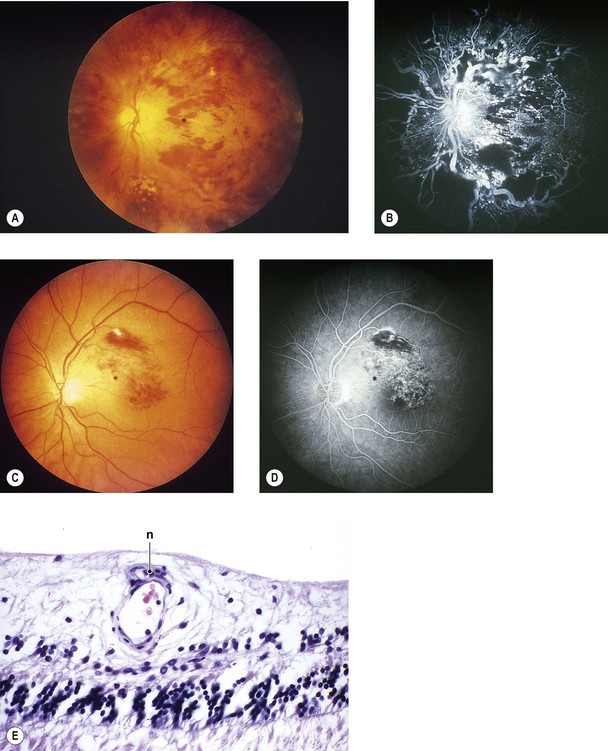
Histology of Retinal Hemorrhagic Infarction (See Fig. 11.12)
I. Early—hemorrhagic necrosis of neural retina
A. Massive intraneural retinal hemorrhage involves all the neural retinal layers.
1. The hemorrhage frequently spreads within the nerve fiber layer of the neural retina (see Fig. 15.18) and appears clinically in sheets or flame-shaped.
B. Cytoid bodies (see Fig. 11.11) are common histologically but may be masked by the hemorrhages clinically. As the hemorrhages resorb, hard, waxy exudates may appear (see Chapter 15 and later in this chapter).
II. Late—organization of hemorrhage and gliosis
B. Gliosis (“scarring”) is usually seen in the inner neural retinal layers.
C. Hemosiderosis of the retina (see Fig. 11.12C) is present, with the hemosiderin often located in macrophages.
Hypertensive and Arteriolosclerotic Retinopathy1
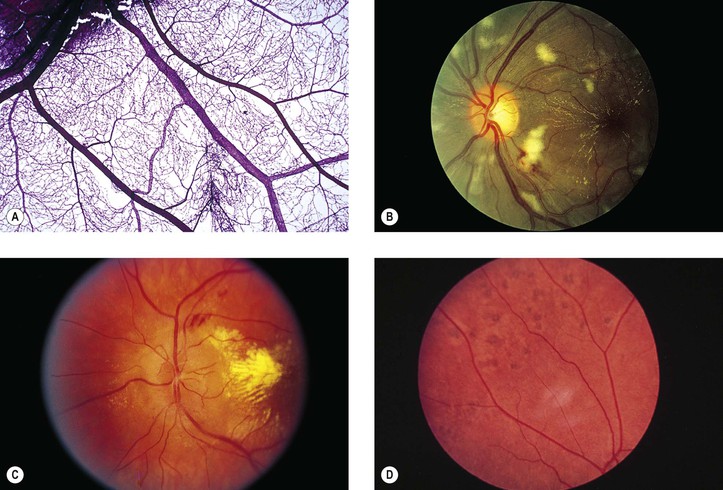
I. Hypertensive retinopathy (Fig. 11.14)
A. Grade I: a generalized narrowing of the arterioles
B. Grade II: grade I changes plus focal arteriolar spasms
C. Grade III: grade II changes plus hemorrhages and exudates
1. Flame-shaped (splinter) hemorrhages (see Figs. 11.12–11.14; see also Fig. 15.18) are characteristic and present in the nerve fiber layer.
2. Dot-and-blot hemorrhages (see Figs. 11.13 and 15.18) may be seen in the inner nuclear layer with spreading to the outer plexiform layer.
3. Cotton-wool spots (see Figs. 11.11 and 11.14; see earlier in this chapter) are characteristic.
4. Hard (waxy) exudates may be seen; these are lipophilic exudates located in the outer plexiform layer (see Figs. 11.14, 15.13, and 15.16).
D. Grade IV: all the changes of grade III plus optic disc edema
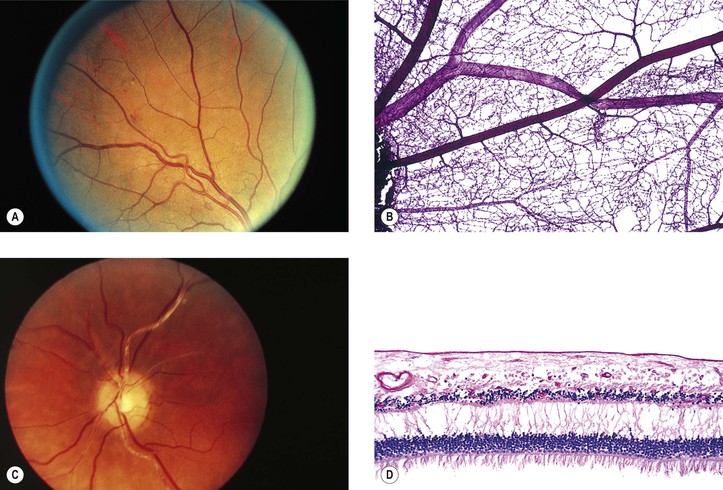
II. Arteriolosclerotic retinopathy (Fig. 11.15)
A. Grade I: an increase in the arteriolar light reflex
B. Grade II: grade I changes plus arteriolovenular crossing defects
C. Grade III: grade II changes plus “copper-wire” arterioles
Hemorrhagic Retinopathy
I. Neural retinal hemorrhages (see Figs. 11.12 and 15.18) may be caused by many diseases, such as diabetes mellitus (see Chapter 15), sickle-cell disease, retinal venous diseases, hypertension, blood dyscrasias, leukemias, polycythemia vera, subacute bacterial endocarditis, cytomegalovirus retinitis, acute retinal necrosis (ARN), lymphomas, idiopathic thrombocytopenia, trauma, multiple myeloma, pernicious anemia, collagen diseases, carcinomatosis, anemia, and many others.
II. Histologically, the size and anatomic location of the hemorrhage determine its clinical appearance (see Fig. 15.18).
III. Roth’s spots
Exudative Retinopathy
I. Retinal exudates (see Figs. 11.14, 15.13, and 15.16)
II. Circinate retinopathy consists of a circular deposit of masses of hard, waxy exudates around a clear area, often within the anatomic macula.
A. It is degeneration secondary to ischemic vascular disease (e.g., diabetes mellitus).
B. Histologically, the exudates are identical to isolated, small, hard, waxy exudates.
III. Histology
A. Hard, waxy exudates (see Chapter 15)
B. Cotton-wool spots (cytoid bodies; see earlier in this chapter and see Chapter 15)
Diabetes Mellitus
See Chapter 15.
Coats’ Disease, Leber’s Miliary Aneurysms, and Retinal Telangiectasia
See Chapter 18.
Idiopathic Macular Telangiectasia (Idiopathic Juxtafoveolar Retinal Telangiectasis)
See Chapter 18.
Retinal Arterial and Arteriolar Macroaneurysms
I. Macroaneurysms of the retinal arteries and arterioles (Fig. 11.16) may be congenital or acquired.
A. Congenital entities include angiomatosis retinae and the diseases of Eales, Leber, and Coats.
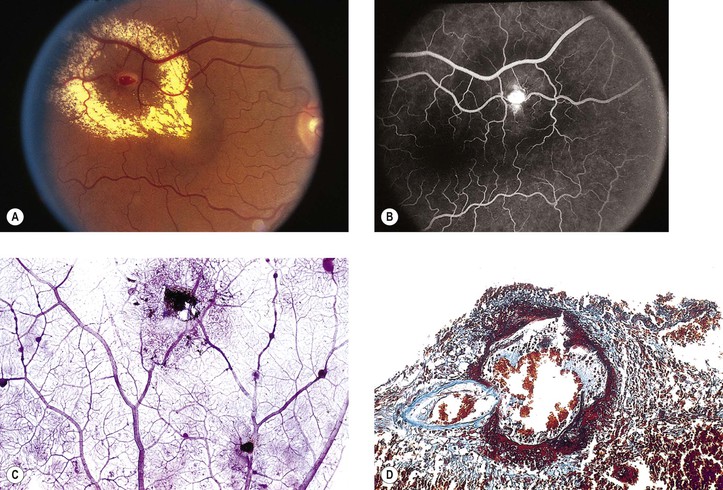
II. Most macroaneurysms involute spontaneously without sequelae.
IV. Histologically, spherical or fusiform aneurysms arise from arteriolar walls.
Sickle-Cell Disease
I. Sickle-cell disease (Figs. 11.17 and 11.18) is caused by a point mutation in the hemoglobin gene.
B. The sickled erythrocytes are much more rigid than normal ones and cause occlusions of small vessels.

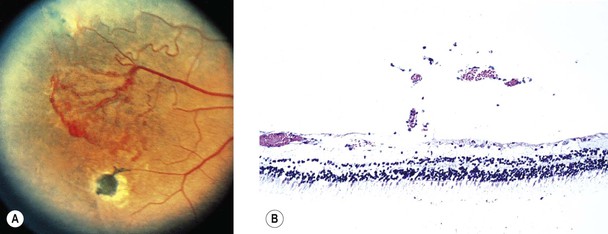
III. Classification of retinopathy
A. Stage I: peripheral arteriolar occlusion (between the equator and the ora serrata)
1. The primary site of occlusion appears to be at the precapillary arteriole level.
B. Stage II: peripheral arteriolovenular anastomoses (AVA; most commonly in the temporal quadrant)
C. Stage III: neovascular and fibrous proliferations
1. New vessels arise from pre-existing AVA, on the venular side.
4. Salmon-patch hemorrhage, single or multiple, may occur in sickle-cell disease and SC disease.
a. It is usually found in the mid-periphery adjacent to a retinal arteriole.
D. Stage IV: vitreous hemorrhage (usually arising from a neovascular patch)
Eales’ Disease (Primary Perivasculitis of the Retina)
Retinopathy of Prematurity
See Chapter 18.
Hereditary Hemorrhagic Telangiectasia (Rendu–Osler–Weber Disease)
See Chapter 7.
Disseminated Intravascular Coagulation
I. Disseminated intravascular coagulation (DIC; Fig. 11.19) is a syndrome in which a physiologic imbalance occurs between clotting and lysis of clot.
C. Coincident with microthrombi formation, a gradual depletion of coagulation factors, platelets, and fibrin ensues, resulting in a change from a hypercoagulable to a hypocoagulable state.
2. Detection of the cross-linked fibrin degradation fragment, D-dimer, in patients at risk for DIC is strong evidence for the diagnosis.
a. D-dimer confirms that both thrombin and plasmin generation have occurred.
Inflammations
Nonspecific Retinal Inflammations
Secondary retinitis is usually caused by a vasculitis.
Specific Retinal Inflammations (See Chapters 2–4)
I. A toxic, exudative retinopathy may occur with carbon monoxide intoxication.
III. Endogenous mycotic retinitis (e.g., candidiasis) results from fungus infection.
IV. Viral retinitis
A. Herpes simplex retinitis (see Chapter 8)
1. Type 1 herpes simplex virus produces lesions in nongenital sites, including the mouth, cornea (see Fig. 8.13), skin above the waist, and in the central nervous system.
a. Type 1 virus is a rare cause of retinitis in children and adults.
b. The virus may cause encephalitis.
2. Type 2 herpes simplex virus (see Fig. 3.6) is transmitted as a venereal infection, usually producing lesions below the waist, except in newborns, in whom it may infect any organ. Approximately 20% of neonates infected with type 2 virus have ocular manifestations, including retinitis.
B. Cytomegalic inclusion disease (see Chapter 4)
A. The lesions resolve rapidly but may leave permanent retinal pigment epithelial alterations.
VI. Acute retinal pigment epitheliitis is characterized by an acute onset, mainly in the posterior pole, that resolves fairly rapidly, usually in 6–12 weeks.
B. The cause and histology are unknown.
VII. Acute macular neuroretinopathy
A. Acute macular neuroretinopathy, a rare condition, tends to occur bilaterally in young women and shows subtle, reddish-brown, wedge- or tear-shaped (pointing to the fovea) lesions in the fovea.
B. The cause and histology are unknown.
VIII. Birdshot retinopathy (vitiliginous retinochoroidopathy; diffuse inflammatory salmon-patch choroidopathy)
B. The condition, which has an ultimately poor visual prognosis, is characterized by:
5. Enhanced depth OCT may show extramacular retinal and choroidal changes not seen in conventional OCT.
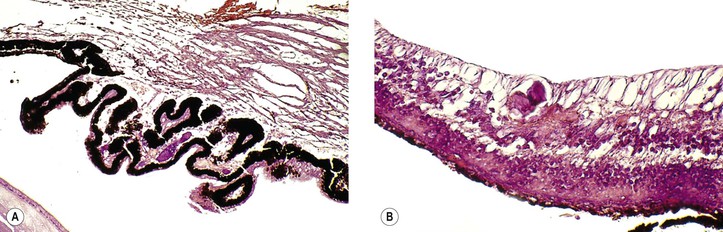
IX. Acute retinal necrosis (ARN) (Fig. 11.20)
B. After 1–3 months, neural retinal detachments develop, followed by proliferative vitreoretinopathy.
C. The condition is caused most commonly by the varicella–zoster virus (46%) and also by herpes simplex virus types 1 (25%) and 2 (21%).
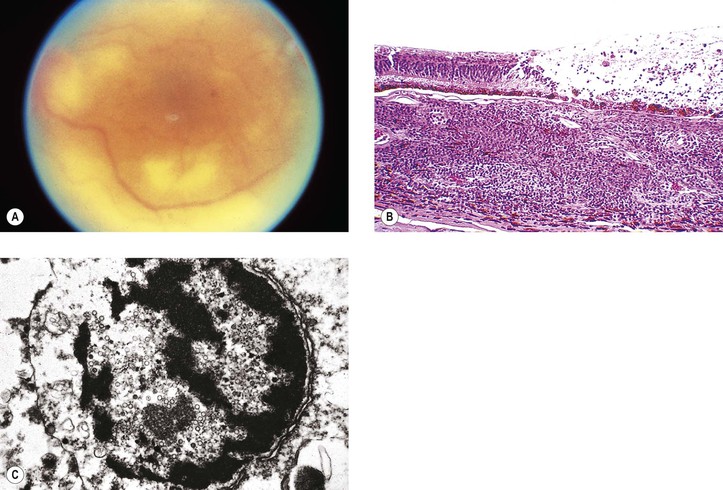
X. Multiple evanescent white dot syndrome (MEWDS)
C. Vitreal cells, reduced visual acuity, and abnormalities in the ERG and early receptor potential may be found.
D. The cause and histology are unknown.
XI. Unilateral acute idiopathic maculopathy (UAIM)
A. UAIM occurs in young adults who experience sudden, severe visual loss (to 20/200 or worse), often after a flu-like illness, caused by an exudative maculopathy.
3. A rapid and complete resolution usually takes place (vision 20/25 or better).
XII. Diffuse unilateral subacute neuroretinitis (see Chapter 4)
XIII. Retinal pigment epitheliopathy associated with the amyotrophic lateral sclerosis/parkinsonism–dementia complex (ALS/PDC) of Guam, especially in the native Chamorro population
B. Histologically, focal areas of attenuation of the RPE and a reduced amount of intracellular pigment correlate with the fundus lesions.
2. The pathogenesis is unknown.
XIV. Acute multifocal retinitis
B. The areas of retinitis tend to be posterior and localized to the inner retina, varying in size from 100 to 500 µm in diameter. The retinal lesions are often multiple and bilateral.
1. Vision usually returns to normal without treatment in 1–4 weeks.
2. Optic disc edema may occur.
3. Fluorescein angiography shows early hypofluorescence and late staining of retinal lesions.
Injuries
See Chapter 5.
Degenerations
Definitions
Degenerations are a result of previous disease (i.e., ocular “fingerprints” left by prior disease).
Microcystoid Degeneration
I. Typical peripheral microcystoid degeneration (PMD; Blessig–Iwanoff cysts) (Figs. 11.21 and 11.22)
C. The tendency is toward relatively equal bilateral involvement.
E. Histologically, spaces within the neural retina (cysts) are located in the outer plexiform and adjacent nuclear layers.
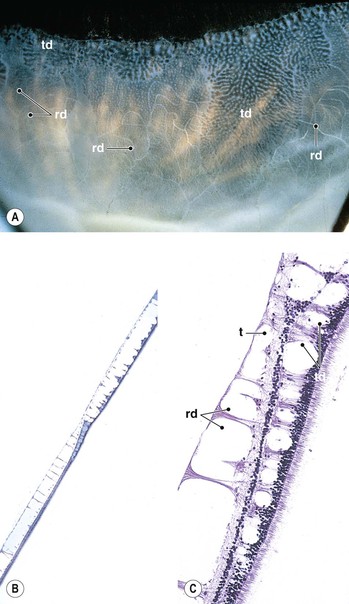
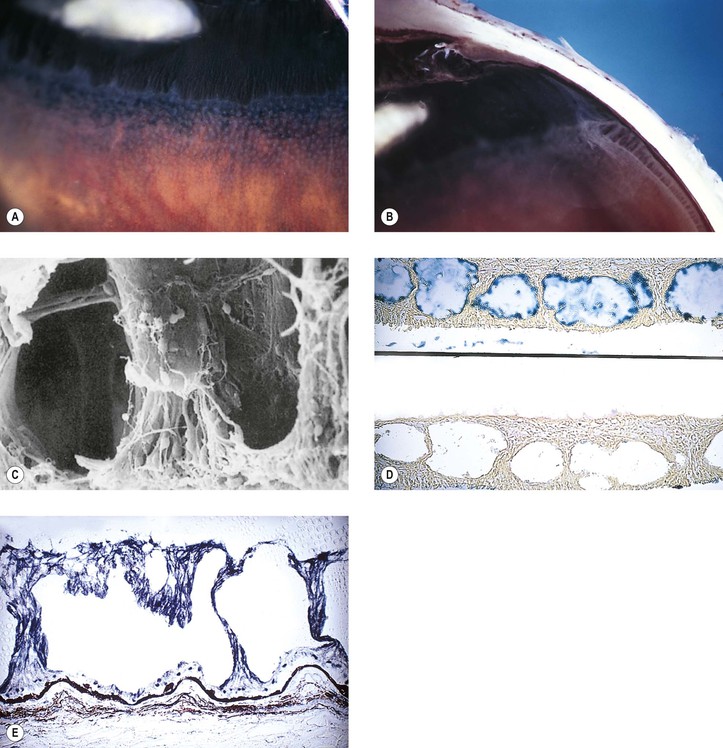
II. Reticular peripheral cystoid degeneration (RPCD; see Fig. 11.21)
D. Histologically, the neural retinal cysts of RPCD are located in the nerve fiber layer of the neural retina.
Degenerative Retinoschisis
I. Retinoschisis—typical degenerative senile (adult) type (Fig. 11.23)
E. Histologically, a splitting is seen in the outer plexiform layer and adjacent nuclear layers. The cavity is filled with a hyaluronidase-sensitive acid mucopolysaccharide, presumably hyaluronic acid.
1. As the area of the retinoschisis enlarges, the involved neural retina is destroyed.
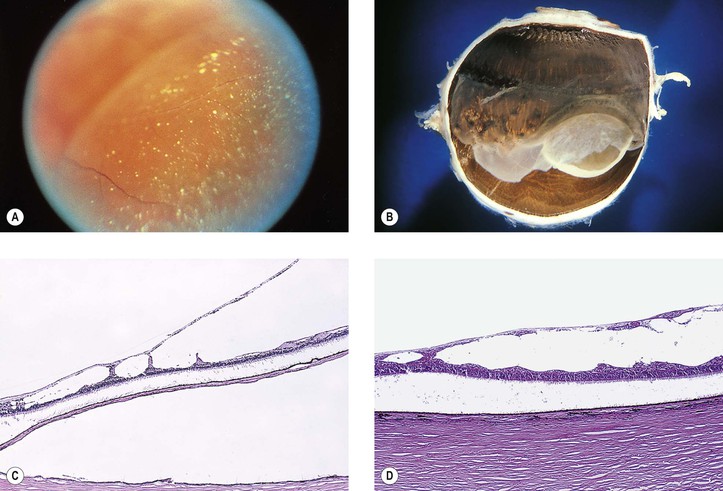
II. Retinoschisis—reticular degenerative (adult) type (see Fig. 11.23)
C. Round or oval holes may be present in the outer wall but rarely in the inner wall.
D. Histologically, the inner wall of the schisis is composed of the neural retinal internal limiting membrane and minimal remnants of the nerve fiber layer.
1. The outer wall is made up of receptors and outer nuclear and plexiform layers.
Secondary Microcystoid Degeneration and Retinoschisis
Paving Stone (Cobblestone) Degeneration (Peripheral Chorioretinal Atrophy; Equatorial Choroiditis)
I. The lesions of paving stone degeneration (Fig. 11.24) tend to increase in incidence and in size with age and with axial length of the eye. They are present in approximately 25% of autopsy cases and bilateral in approximately 38%.
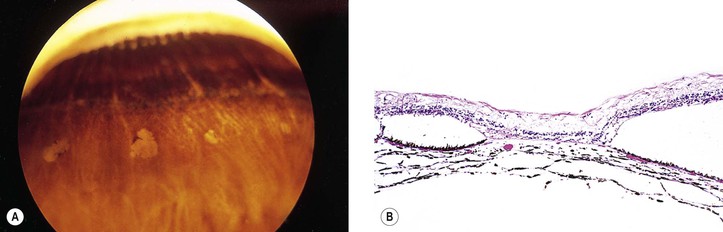
II. The lesions are located primarily between the ora serrata and equator and are separated from the ora serrata by normal neural retina.
III. Histologically, the lesions are characterized by:
C. Hypertrophy and hyperplasia of the pigment epithelium is present at the lesion’s margin.
Macular Degeneration
Idiopathic Serous Detachment of the RPE (Fig. 11.25)
II. Serous RPE detachments have a good prognosis and probably are a variant of idiopathic central serous choroidopathy.
B. Most resorb or flatten, leaving behind a disturbance of pigmentation.
Idiopathic Central Serous Choroidopathy (Central Serous Retinopathy; Central Angiospastic Retinopathy) (See Fig. 11.25)
Drusen
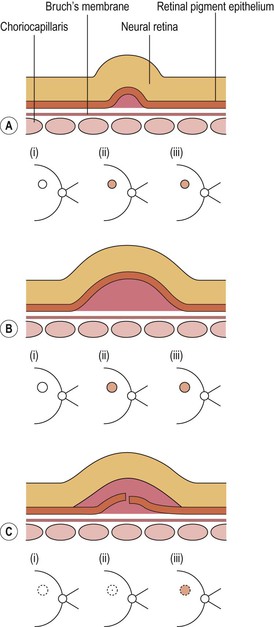
I. Drusen (Figs. 11.26 and 11.27) are focal or diffuse basement membrane products produced by the RPE and admixed with other materials that may become trapped in the drusen as they pass through them in transit between the RPE and choriocapillaris.
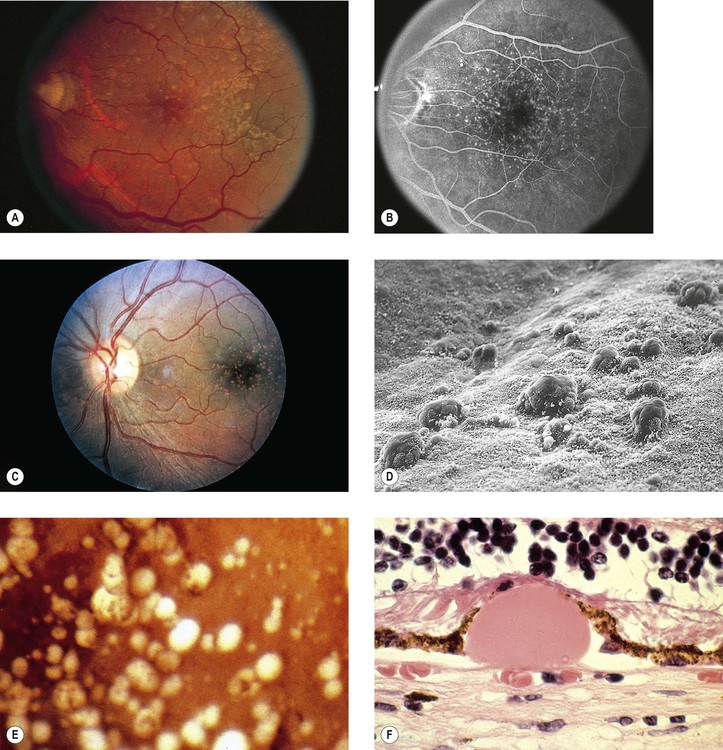
II. Drusen consist of at least two fundamentally different focal types, which by fluorescein angiography, some show early fluorescence and late staining.2
A. The first focal type, nodular (“hard,” discrete) drusen, consists of small, yellow or yellow-white spots or discrete RPE lesions measuring approximately 50 µm (see Fig. 11.26).
4. Histologically (see Fig. 11.26), nodular drusen have an eosinophilic, PAS-positive, amorphous appearance and are located external and contiguous to, or replace, the original, thin basement membrane of the RPE (i.e., between RPE basement membrane and Bruch’s membrane). The overlying RPE is usually atrophic, whereas the adjacent RPE is frequently hyperplastic.
5. The so-called basal laminar (cuticular) drusen (see Fig. 11.26) are one form of nodular drusen and appear yellowish, punctiform, and uniform in size. Nodular drusen may become calcified, lipidized, cholesterolized, or, infrequently, vascularized.
B. The second, focal type is a limited separation of the relatively normal basement membrane of the RPE from its attachment to Bruch’s membrane at the inner collagenous zone by a wide variety of materials that differ in consistency from bone to fluid.
2. One form of this second type is large (“soft,” exudative, fluffy) drusen (see Fig. 11.27).
3. Histologically, large drusen consist of an amorphous, PAS-positive material, which is indistinguishable from an RPE detachment.
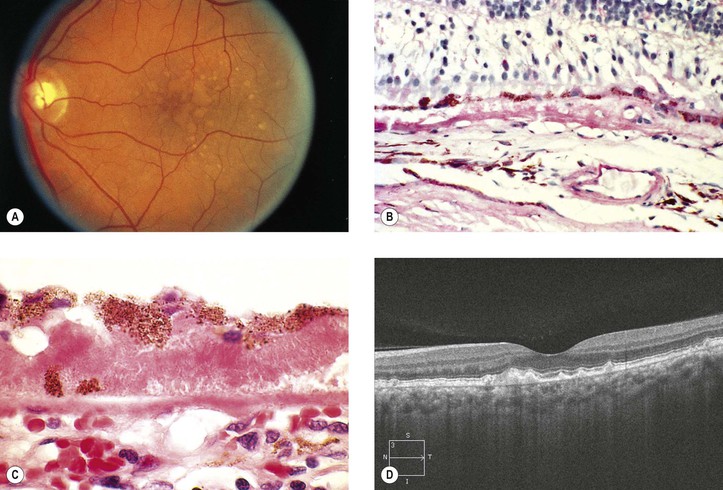
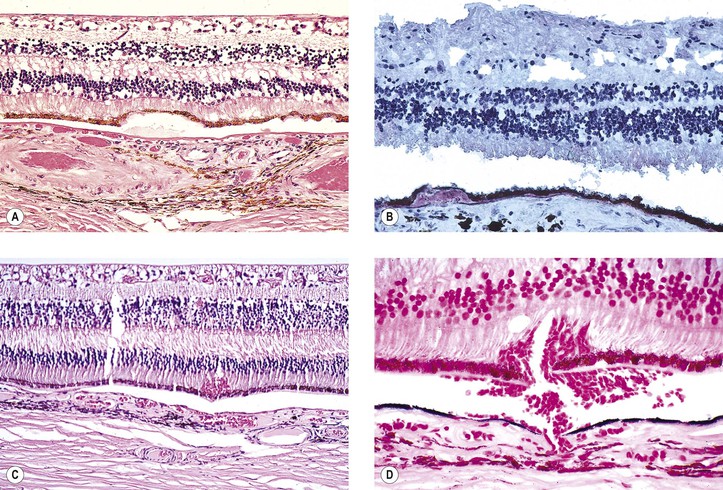
Dry Age-Related Macular Degeneration (Dry, Atrophic, or Senile Atrophic Macular Degeneration)
I. Dry ARMD (Figs. 11.28 and 11.29) is characterized by a gradual reduction of central vision.
B. The risk increases with age, especially 75 years and older, and in women.
A. Pigment disturbances (e.g., increased and decreased pigmentation) are seen in the macula.
B. The RPE atrophy tends to spread and form well-demarcated borders, called geographic atrophy. The underlying choriocapillaris is atrophic.
IV. Histologically, the following changes may be seen:
B. The RPE may show atrophy with depigmentation, hypertrophy, or even hyperplasia.
C. In both dry and wet ARMD, RPE, photoreceptors, and inner nuclear cells die by apoptosis.
D. The neural retina often shows microcystoid or even macrocystoid (retinoschisis) degeneration.
1. Hole formation may occur in the inner wall of the macrocyst.
2. Rarely, total hole formation may occur, leaving the macular retinal ends with rounded, smooth edges.
E. In addition to the well-known aging changes of the RPE, including drusen and an increase in cell lipofuscin, the cell, in situ, can also undergo a change known as lipidic degeneration, the noxious stimulus for which is unknown (see Fig. 17.44).
F. With aging, Bruch’s membrane shows, in addition to drusen, increased amounts of calcium and lipid.
Age-Related Exudative Macular Degeneration (Exudative, Wet, or Senile Disciform Macular Degeneration; Kuhnt–Junius Macular Degeneration)
I. Typically, exudative ARMD (Fig. 11.30) occurs mainly in people 60 years of age or older. The cause is unknown.
B. No sex predilection exists, and the degeneration is often bilateral.
G. Hyperopia may also be a risk factor.
K. The presence of CFH CC genotype increases the risk 144-fold.
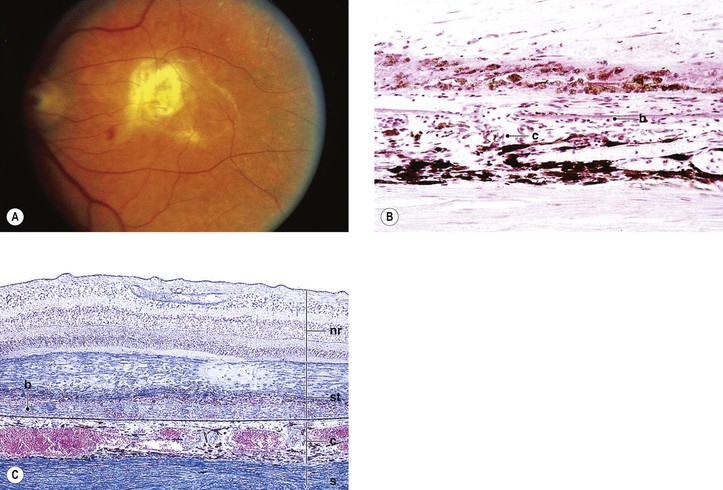
II. Evolution of exudative ARMD
A. Early degenerative changes are seen in the choriocapillaris and in Bruch’s membrane in the macular area, manifested clinically as drusen, especially soft drusen; collectively, these changes are called age-related macular choroidal degeneration.
1. Large, soft drusen seem to predispose the eye to exudative ARMD.
2. Each year, 16,000 people in the United States become blind from ARMD.
B. The age-related macular choroidal degeneration becomes complicated by neovascular invasion.
2. Two fundamentally different types of CNV arise from the choroid: types 1 and 2.
D. Finally, a hemorrhage between Bruch’s membrane and RPE occurs (hematoma of the choroid).
E. Although the hemorrhage may remain localized, it usually breaks through the RPE under the neural retina; rarely, it may extend into the choroid, the neural retina, or even the vitreous. Organization of the hemorrhage is accompanied by RPE proliferation and fibrous metaplasia.
1. Ingrowth of mesenchymal tissue forms granulation tissue.
F. Retinal angiomatous proliferation (RAP)
3. Three stages have been described:
b. Stage II—IRN extends posteriorly into the subneural space (subneural retinal neovascularization)
c. Stage III—IRN anastamosing with CNV
III. Histologically, the following features are noted:
B. The subretinal (sub-RPE or subneural retinal) membranes consist of a cellular and an extracellular matrix component.
Exudative Macular Degeneration Secondary to Focal Choroiditis (Juvenile Disciform Degeneration of the Macula)
II. Most cases probably occur secondary to focal inflammatory cell infiltration of the choroid.
III. Five subdivisions have been identified:
A. Exudative (wet) macular detachment secondary to multifocal choroiditis (POHS; Fig. 11.31)—the most common type
Overlying RPE disturbances soon appear, resulting in a small, dark greenish macular ring.
4. Serous and hemorrhagic disciform detachment of the neural retina may ensue.
6. An irregular area of peripapillary degeneration of the choroid and RPE is frequently seen.
7. HLA-B7 is found in association with POHS.
8. Histologically, the peripheral lesions show either a chronic nongranulomatous or a granulomatous inflammatory infiltrate in the choroid. The typical acute macular lesions have not been examined histologically.
a. Overlying Bruch’s membrane and RPE may or may not be involved.
b. Excised subretinal membranes are composed of fibrovascular tissue between Bruch’s membrane and RPE and probably represent nonspecific granulation tissue.
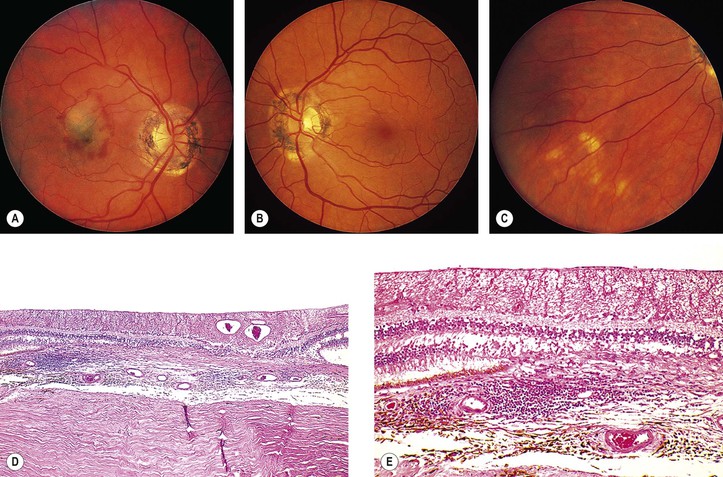
B. Idiopathic subretinal (choroidal) neovascularization
E. Exudative macular detachment secondary to Toxocara canis (see Chapter 4).
Idiopathic Polypoidal Choroidal Vasculopathy
Cystoid Macular Edema (Irvine–Gass Syndrome)
See Chapters 5 and 15.
Toxic Retinal Degenerations
I. Chloroquine (Fig. 11.32) and hydroxychloroquine
A. The characteristic but nonspecific “bull’s-eye” macular degeneration appears to be directly related to the total dosage of chloroquine.
1. The bull’s-eye macular degeneration indicates advanced, irreversible damage.
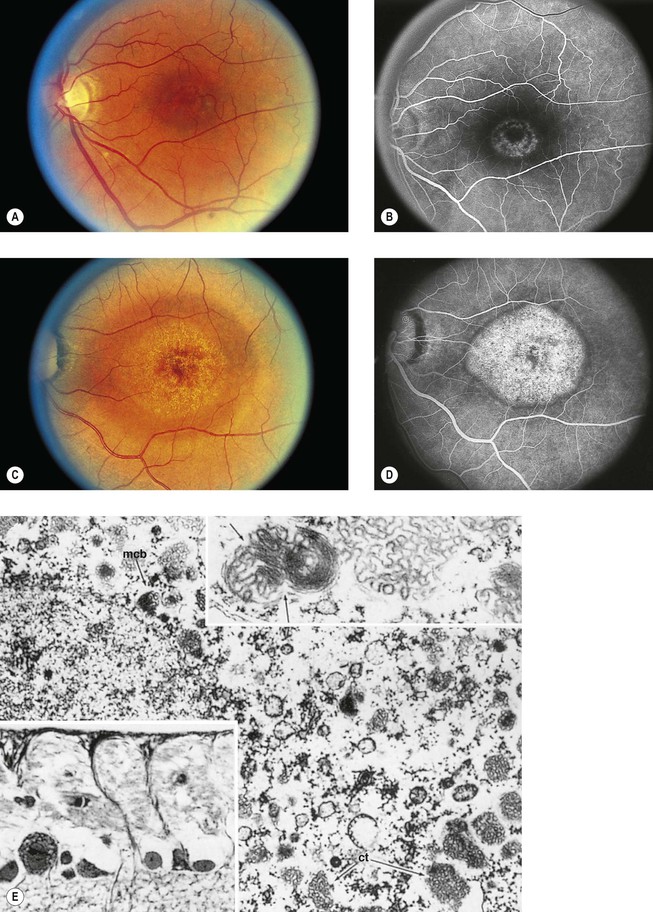
Postirradiation Retinopathy
Bone Marrow Transplant Retinopathy
Cancer-Associated Retinopathy (Paraneoplastic Syndrome; Paraneoplastic Retinopathy; Paraneoplastic Photoreceptor Retinopathy; Melanoma-Associated Retinopathy)
Idiopathic Macular Holes
I. The pathogenesis of idiopathic macular holes (IMH) is not known.
A. IMH may be bilateral and tend to occur in eyes that do not have a posterior vitreous detachment.
2. If the first eye has an incomplete hole, a 19–24% chance exists of eventual bilaterality.
B. It appears that vitreous contraction or separation may play an important role in the development of IMH. Reduced choroidal thickness, as seen with OCT, may also play a role.
2. Spontaneous complete vitreous separation from the fovea could reverse the process.
II. The development of IMH can be divided into three stages:
A. Stage 1: tractional detachment or impending macular hole
B. Stage 2: small hole formation
1. Increased traction causes a tangential tear, usually at the foveal edge.
C. Stage 3: large hole formation
III. Histologically, in membranes stripped out during vitrectomy for stage 1 IMH, an acellular collagenous tissue layer is found.
Light Energy Retinopathy
See Chapter 5.
Traumatic Retinopathy
See Chapter 5.
Hereditary Primary Retinal Dystrophies
Definitions
I. Dystrophies are primary phenomena that are inherited and tend to be bilateral and symmetric.
II. They may remain stationary or be slowly progressive.
III. Many retinal dystrophies may be genetically determined by the process of apoptosis (see Chapter 1).
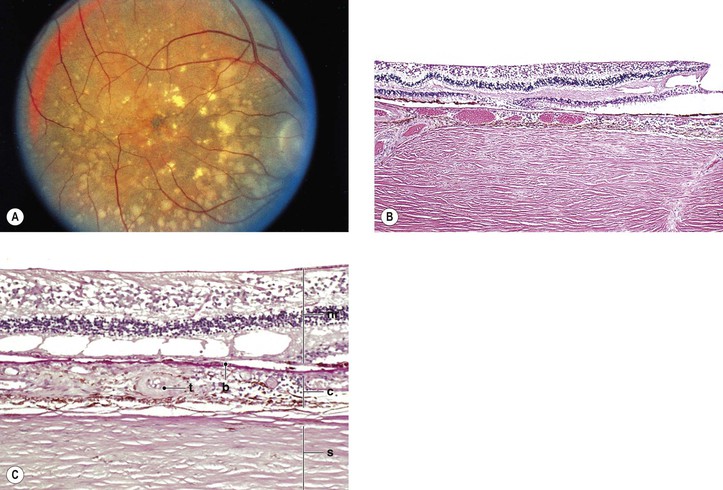
Juvenile Retinoschisis (Vitreous Veils; Congenital Vascular Veils; Cystic Disease of the Retina; Congenital Retinal Detachment)
I. Juvenile retinoschisis (Fig. 11.33) is a bilateral condition, tends to be slowly progressive, and often culminates in extensive chorioretinal atrophy with macular involvement.
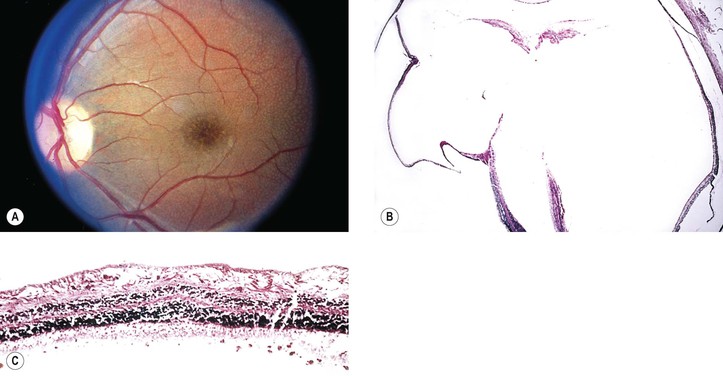
III. Ophthalmoscopic appearance
B. Foveal retinoschisis is present in almost all cases. It appears clinically much like the polycystic fovea seen in Irvine–Gass syndrome, but without fluorescein leakage.
1. OCT shows foveomacular retinoschisis is most frequent in the inner nuclear layer.
IV. Histologically, in the region of the retinoschisis, the neural retina shows a cleavage at the level of the nerve fiber and ganglion cell layers; in areas away from the schisis, the neural retina shows a “looseness” or microcystic degeneration that mainly involves the nerve fiber layer and, to a lesser extent, the ganglion cell layer.
V. Goldmann–Favre vitreoretinal dystrophy has an autosomal-recessive heredity, and it consists of juvenile retinoschisis and the following:
B. Cataracta complicata lens opacities and leakage of fluorescein from retinal vessels
C. Hemeralopia with abolition of the ERG response and a progressive decrease in vision function
VI. Wagner’s vitreoretinal dystrophy (Table 11.1) consists of:
TABLE 11.1
Clinical Findings in Syndromes with Wagner-Like Vitreoretinal Degenerations
| Disease | Systemic Findings | Refractive Error | Vitreous Veils and Perivascular Lattice | Retinal Detachments | Cataract |
| A. With Ocular Symptomatology Only | |||||
| Normal | Moderate myopia; occasionally severe myopia | Yes | None | Mild childhood; mature by 35–40 years | |
| Normal | Low hypermetropia or moderate myopia; occasionally severe myopia | Yes | Yes | Cortical opacities in teens; mature in fourth decade | |
| B. With Associated Systemic Anomalies | |||||
|
1. Hereditary arthro-ophthalmopathy with marfanoid habitus (Stickler’s syndrome) |
Micrognathia, cleft palate, joint laxity, epiphyseal dysplasia | Moderate to severe myopia; occasionally mild myopia | Yes | Yes | Common |
|
2. Hereditary arthro-ophthalmopathy with Weill–Marchesani-like habitus |
Low normal stature, cleft palate, joint stiffness, epiphyseal dysplasia, deafness | Moderate to severe myopia; occasionally mild myopia | Yes | Occasional | Common |
| Below third percentile in stature, cleft palate, epiphyseal dysplasia, deafness | Severe myopia | Yes | None | No | |
| Adult height 106–145 cm, abnormal facies, cleft palate, joint limitation, epiphyseal dysplasia, deafness | Severe myopia | Yes | Occasional | Occasional | |
| Adult height 102 cm, bifid uvula, torus planitus, loose joints, epiphyseal dysplasia, deafness | Severe myopia | Yes | Yes | Yes | |
| Adult height 84–128 cm, loose joints, epiphyseal dysplasia | Mild hypermetropia to severe myopia | Yes | None | No | |
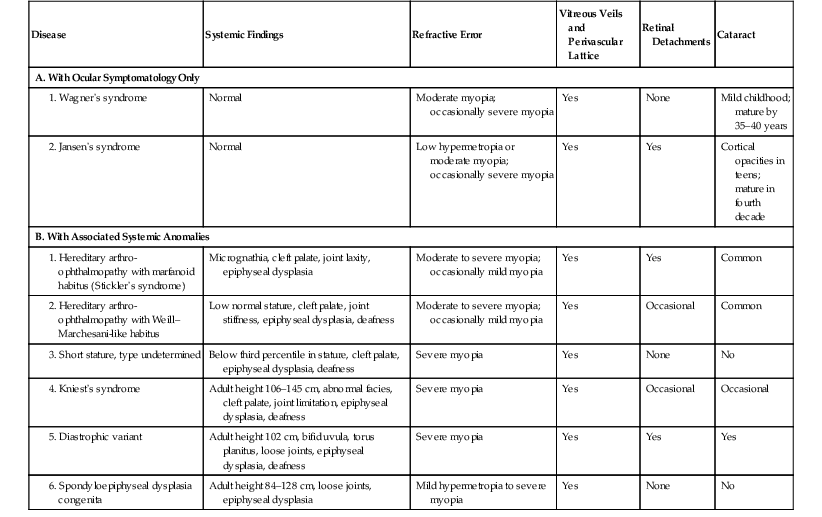
(Modified from Maumenee IH: Am J Ophthalmol 88:432. © Elsevier 1979.)
Choroidal Dystrophies
See Chapter 9.
Stargardt’s Disease (Fundus Flavimaculatus)
I. Stargardt’s disease (Fig. 11.34) is inherited as an autosomal-recessive trait [the photoreceptor gene ABCR or ABCA4 (also known as STGD1) on chromosome 1p21 is mutated in Stargardt’s disease].
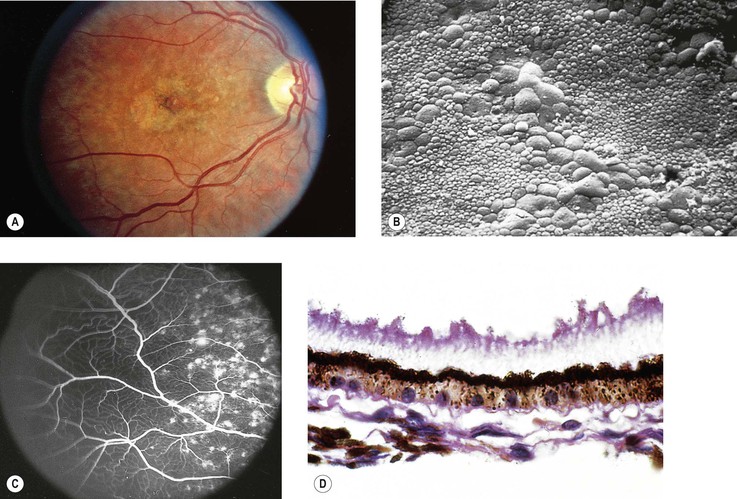
II. Stargardt’s disease consists of two parts that may occur simultaneously or independently: a macular dystrophy and a flecked neural retina (fundus flavimaculatus).
A. The macular dystrophy component
1. Initially, it is confined to the posterior pole, eventually leading to loss of central vision.
4. Ophthalmoscopically, the macula takes on a “beaten-bronze atrophy” caused by a sharply defined RPE atrophy.
b. The peripheral neural retina may show areas that resemble RP.
6. Fluorescein angiography shows fluorescence of the central fovea without leakage, often in a bull’s-eye configuration, suggesting defects in the RPE but an intact Bruch’s membrane.
b. An increased amount of lipofuscin-like material in the RPE causes the decreased fluorescence.
7. Histologically, there is a complete disappearance of the RPE and of the visual elements in the macular area.
The inner layers of the neural retina may show cystoid degeneration and calcium deposition.
B. The flecked neural retina (fundus flavimaculatus) component
2. Approximately 50% of eyes show a Stargardt-type macular dystrophy with a decrease in visual acuity.
3. The peripheral neural retina may show areas that resemble RP.
4. Dark adaptation and ERG are usually normal but may be subnormal.
5. Fluorescein angiography does not cause fluorescence of the spots in early lesions, and not all the spots fluoresce in late lesions.
b. The fluorescein pattern clearly differentiates the spots from drusen.
6. Histologically, the RPE is solely involved—hence the abnormal electro-oculogram (EOG)—and shows the following:
a. PAS positivity and increased autofluorescence
b. Displacement of the nucleus from near the base of the cell to the center or apical surface
d. Great variation in RPE cell size, from much larger than normal to normal
Dominant Drusen of Bruch’s Membrane
Doyne’s Honeycomb Dystrophy; Malattia Lèventinese; Hutchinson–Tay Choroiditis; Guttate Choroiditis; Holthouse–Batten Superficial Choroiditis; Family Choroiditis; Crystalline Retinal Degeneration; Iridescent Crystals of the Macula; Hyaline Dystrophies
I. Dominant drusen of Bruch’s membrane is a bilateral, symmetric, progressive disease whose onset is usually between 20 and 30 years of age.
A. It involves the posterior pole predominantly and results in loss of vision.
1. The posterior polar lesion consists of moderate- to large-sized drusen, resembling soft drusen.
B. It is inherited as an autosomal-dominant trait.
Best Vitelliform Disease
Vitelliform Foveal Dystrophy; Vitelliform Macular Degeneration; Vitelliruptive Macular Degeneration; Exudative Central Detachment of the Retina—Macular Pseudocysts; Cystic Macular Degeneration; Exudative Foveal Dystrophy
I. Best vitelliform disease (BVD; Fig. 11.35) is a bilateral, symmetric, progressive disease involving the RPE of the macular area with resultant loss of vision. Its onset is usually before 15 years of age. Juvenile and adult forms exist.
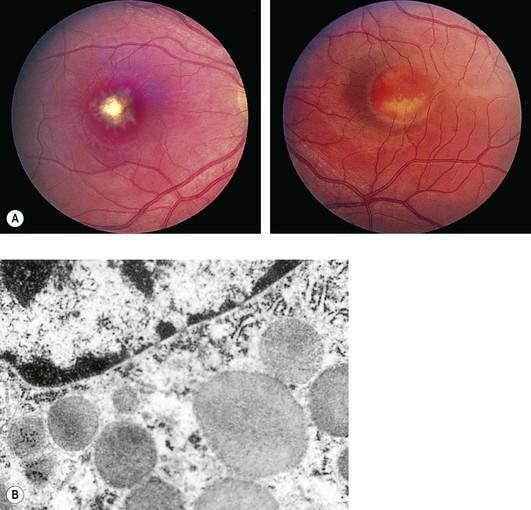
B. Approximately 1% of all cases of macular degeneration are attributable to BVD.
III. Dark adaptation and ERG are usually normal.
A. EOG shows an abnormal light-peak/dark-trough ratio in affected patients as well as in carriers.
V. Lesions similar to those seen in BVD may occur in patients as part of ARMD, a condition called pseudovitelliform or adult vitelliform macular degeneration.
Dominant Progressive Foveal Dystrophy
Dominant Cystoid Macular Dystrophy
I. Dominant cystoid macular dystrophy, inherited as an autosomal-dominant trait, consists of macular cystoid edema with central bull’s-eye (see previously in this chapter for differential diagnosis of bull’s-eye macula) and peripheral neural retinal pigmentary disturbance, cells in the vitreous body, wrinkling of the internal limiting membrane, and axial hypermetropia.
Fenestrated Sheen Macular Dystrophy
North Carolina Macular Dystrophy
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Neural (Sensory) Retina
Normal Anatomy
I. The neural retina (Figs. 11.1–11.3) is a highly specialized nervous tissue, in reality a part of the brain that has become exteriorized.
A. Traditionally, the retina, from the RPE externally to the internal limiting membrane internally, is divided into 10 layers (see Fig. 11.2B).
II. Foveomacular region of the neural retina
A. Clinicians often confuse the proper use of the terms fovea, macula, and posterior pole (see Fig. 11.3).
4. The anatomic macula (which corresponds to the clinical posterior pole) comprises an area larger than the anatomic fovea.
a. The term macula is derived from the term macula lutea.
c. The anatomic macula actually encompasses an area contained just within the optic nerve and the superior and inferior retinal temporal arcades, and extends temporally approximately two disc diameters beyond the central fovea.
Congenital Anomalies
Albinism (Fig. 11.4)
III. The conditions are noted at birth.
B. Color vision is normal. Nyctalopia does not occur.
IV. Histology
A. The RPE is depleted of pigment.
B. Macromelanosomes may be present within the RPE.
A. Inheritance is autosomal recessive.
B. The condition is one of the immunodeficiency diseases (see Chapter 1) and is associated with a lack of resistance to infection and generalized lymphadenopathy. The condition is fatal from generalized infection, usually during the second decade of life.
Grouped Pigmentation (Bear Tracks)
I. Grouped pigmentation (see Fig. 17.22) is unilateral (85% of cases), nonprogressive, and presents as multiple, discrete, well-circumscribed, flat, uniformly pigmented areas resembling a bear’s footprints.
Coloboma
I. The typical coloboma “of the choroid” (see Fig. 9.8) involves the region of the embryonic cleft (fetal fissure; i.e., inferonasally) and is bilateral in 60% of patients. The coloboma may involve the total region, a large part of it, or one or more small isolated parts.
II. Histologically, the RPE seems to be primarily involved and is absent in the area of the coloboma.
A. The neural retina is atrophic and gliotic and may contain rosettes.
Retinal Dysplasia
See Chapter 18.
Lange’s Fold
I. Lange’s fold (Fig. 11.5; see also Fig. 10.6) is an inward fold of neural retina at the ora serrata only seen in infants’ and children’s eyes.
Congenital Nonattachment of the Retina
I. Nonattachment of the neural retina (see Fig. 18.24) is normal in the embryonic eye; persistence after birth is abnormal.
Neural Retinal Cysts
I. A cyst of the neural retina (see Fig. 11.56) is defined arbitrarily as an intraneural retinal space whose internal–external diameter is greater than the thickness of the surrounding neural retina and of approximately equal dimension in any direction.
Myelinated (Medullated) Nerve Fibers
I. Myelinated nerve fibers (MNF; Fig. 11.6) usually occur as a unilateral condition, somewhat more common in men than in women.
B. Rarely, MNF occur after infancy and can progress.
II. Clinically, they appear as an opaque white patch or arcuate band with feathery edges.
Leber’s Congenital Amaurosis
Inherited Retinal Arteriolar Tortuosity
Vascular Diseases
Definitions
Retinal Ischemia
Causes
I. Choroidal vascular insufficiency
II. Retinal vascular insufficiency
A. Large-artery disease anywhere from aortic arch to central retinal artery
1. Atherosclerosis shows patchy subendothelial lipid deposits and erosion of media.
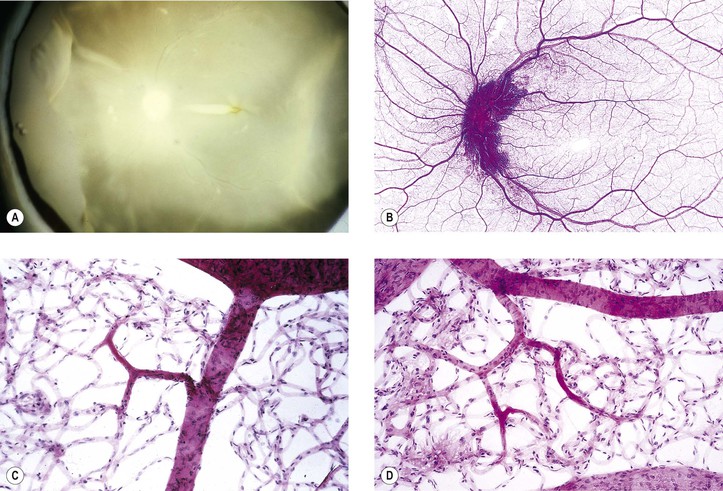
b. Embolic manifestations (Figs. 11.7 and 11.8; see also Fig. 5.54)
3) Rarely, ocular emboli cause a condition masquerading as temporal (cranial; giant cell) arteritis.
5. Temporal (cranial; giant cell) arteritis (see Chapter 13)
B. Arteriolar and capillary disease of neural retinal vasculature
1. Arteriolosclerosis is associated with hypertension.
2. Branch retinal artery occlusion has many causes, including emboli (see Figs. 11.7 and 11.8), arteriolosclerosis, diabetes mellitus, arteritis, dysproteinemias, collagen diseases, and malignant hypertension.
3. Diabetes mellitus (see Chapter 15)
4. Malignant hypertension, toxemia of pregnancy, hemoglobinopathies, collagen diseases (Fig. 11.9), dysproteinemias, carbon monoxide poisoning, and blood dyscrasias of many kinds may involve the small retinal vessels and cause neural retinal ischemia.
Histology of Retinal Ischemia
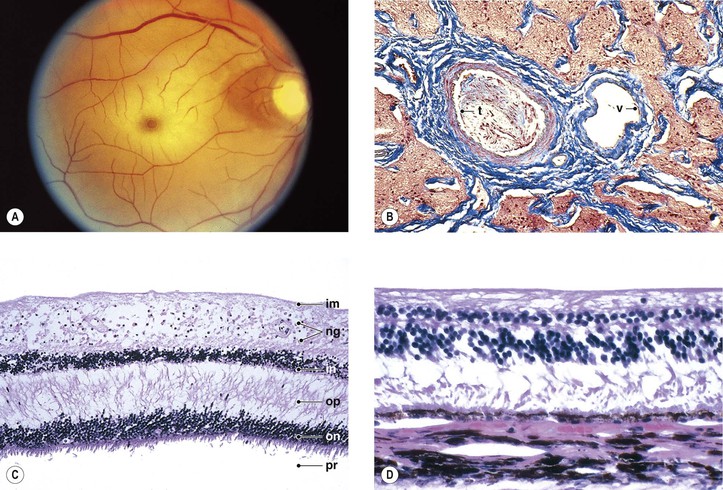
I. Early (Fig. 11.10; see also Figs. 11.9 and 11.15D)
A. The neural retina shows coagulative necrosis of its inner layers, which are supplied by the retinal arterioles.
1. The neuronal cells become edematous during the first few hours after occlusion of the artery.
2. The intracellular swelling accounts for the clinical gray neural retinal opacity.
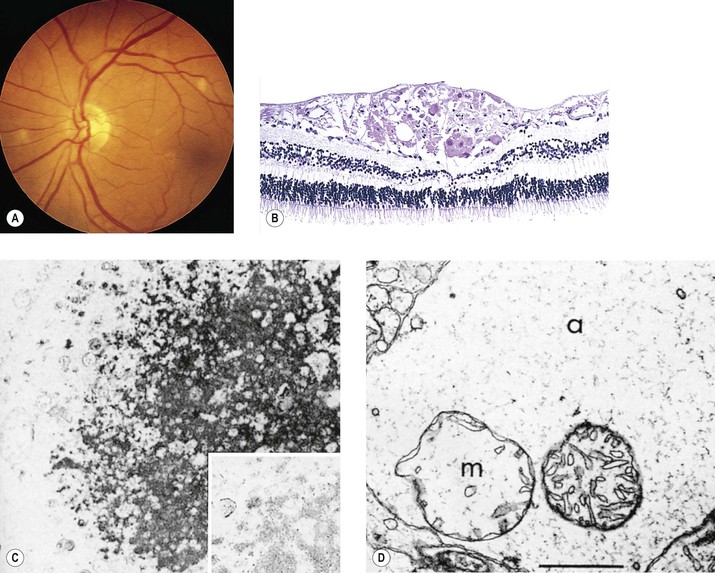
B. If the area of coagulative necrosis (see Chapter 1) is small and localized, it appears clinically as a cotton-wool spot.
1. The cotton-wool spot observed clinically (Fig. 11.11; see also Fig. 11.9) is a result of a microinfarct of the nerve fiber layer of the neural retina.
2. The cytoid body, observed microscopically (see Figs. 11.9 and 11.11), is a swollen, interrupted axon in the neural retinal nerve fiber layer.
II. Late (see Fig. 11.10)
Retinal Hemorrhagic Infarction (Fig. 11.12)
Causes and Risk Factors of Hemorrhagic Infarction
Types of Hemorrhagic Infarction
I. Occlusion of central retinal vein, branch retinal vein, or venule
A. CRVO may be considered to consist of two distinct types.
1. Nonischemic retinopathy (~65% of cases, but approximately one-fourth of these eyes will convert to the ischemic type)
2. Ischemic (hemorrhagic) retinopathy
a. It is caused by occlusion of the central retinal vein at, or anterior to, the lamina cribrosa, associated with retinal ischemia that leads to significant retinal hypoxia.
4. Bilateral CRVO may occur as part of the acquired immunodeficiency syndrome (AIDS).
B. Branch retinal vein occlusion (BRVO)
1. BRVO occurs approximately three times more frequently than CRVO. In approximately two-thirds of cases, the superior temporal neural retinal vein is involved. Most of the remaining cases show involvement of the inferior temporal retinal vein.
b. A hemispheric vein occlusion behaves like an ischemic CRVO.
II. Papillophlebitis (retinal vasculitis, mild and moderate papillary vasculitis, benign retinal vasculitis, optic disc vasculitis)
Complications of Hemorrhagic Infarction
I. Macular hemorrhagic infarction may result in permanent loss of vision.
II. Leakage of fluid into the macula may result in cystoid macular edema.
III. Iris neovascularization (clinically seen rubeosis iridis)
A. Iris neovascularization (see Figs. 9.13 and 9.14) occurs mainly with ischemic CRVO; it rarely occurs with nonischemic CRVO or BRVO.
IV. Neovascularization of the neural retina (Fig. 11.13) occurs mainly with BRVO; it rarely occurs with ischemic or nonischemic CRVO.
Histology of Retinal Hemorrhagic Infarction (See Fig. 11.12)
I. Early—hemorrhagic necrosis of neural retina
A. Massive intraneural retinal hemorrhage involves all the neural retinal layers.
1. The hemorrhage frequently spreads within the nerve fiber layer of the neural retina (see Fig. 15.18) and appears clinically in sheets or flame-shaped.
B. Cytoid bodies (see Fig. 11.11) are common histologically but may be masked by the hemorrhages clinically. As the hemorrhages resorb, hard, waxy exudates may appear (see Chapter 15 and later in this chapter).
II. Late—organization of hemorrhage and gliosis
B. Gliosis (“scarring”) is usually seen in the inner neural retinal layers.
C. Hemosiderosis of the retina (see Fig. 11.12C) is present, with the hemosiderin often located in macrophages.
Hypertensive and Arteriolosclerotic Retinopathy1
I. Hypertensive retinopathy (Fig. 11.14)
A. Grade I: a generalized narrowing of the arterioles
B. Grade II: grade I changes plus focal arteriolar spasms
C. Grade III: grade II changes plus hemorrhages and exudates
1. Flame-shaped (splinter) hemorrhages (see Figs. 11.12–11.14; see also Fig. 15.18) are characteristic and present in the nerve fiber layer.
2. Dot-and-blot hemorrhages (see Figs. 11.13 and 15.18) may be seen in the inner nuclear layer with spreading to the outer plexiform layer.
3. Cotton-wool spots (see Figs. 11.11 and 11.14; see earlier in this chapter) are characteristic.
4. Hard (waxy) exudates may be seen; these are lipophilic exudates located in the outer plexiform layer (see Figs. 11.14, 15.13, and 15.16).
D. Grade IV: all the changes of grade III plus optic disc edema
II. Arteriolosclerotic retinopathy (Fig. 11.15)
A. Grade I: an increase in the arteriolar light reflex
B. Grade II: grade I changes plus arteriolovenular crossing defects
C. Grade III: grade II changes plus “copper-wire” arterioles
Hemorrhagic Retinopathy
I. Neural retinal hemorrhages (see Figs. 11.12 and 15.18) may be caused by many diseases, such as diabetes mellitus (see Chapter 15), sickle-cell disease, retinal venous diseases, hypertension, blood dyscrasias, leukemias, polycythemia vera, subacute bacterial endocarditis, cytomegalovirus retinitis, acute retinal necrosis (ARN), lymphomas, idiopathic thrombocytopenia, trauma, multiple myeloma, pernicious anemia, collagen diseases, carcinomatosis, anemia, and many others.
II. Histologically, the size and anatomic location of the hemorrhage determine its clinical appearance (see Fig. 15.18).
III. Roth’s spots
Exudative Retinopathy
I. Retinal exudates (see Figs. 11.14, 15.13, and 15.16)
II. Circinate retinopathy consists of a circular deposit of masses of hard, waxy exudates around a clear area, often within the anatomic macula.
A. It is degeneration secondary to ischemic vascular disease (e.g., diabetes mellitus).
B. Histologically, the exudates are identical to isolated, small, hard, waxy exudates.
III. Histology
A. Hard, waxy exudates (see Chapter 15)
B. Cotton-wool spots (cytoid bodies; see earlier in this chapter and see Chapter 15)
Diabetes Mellitus
See Chapter 15.
Coats’ Disease, Leber’s Miliary Aneurysms, and Retinal Telangiectasia
See Chapter 18.
Idiopathic Macular Telangiectasia (Idiopathic Juxtafoveolar Retinal Telangiectasis)
See Chapter 18.
Retinal Arterial and Arteriolar Macroaneurysms
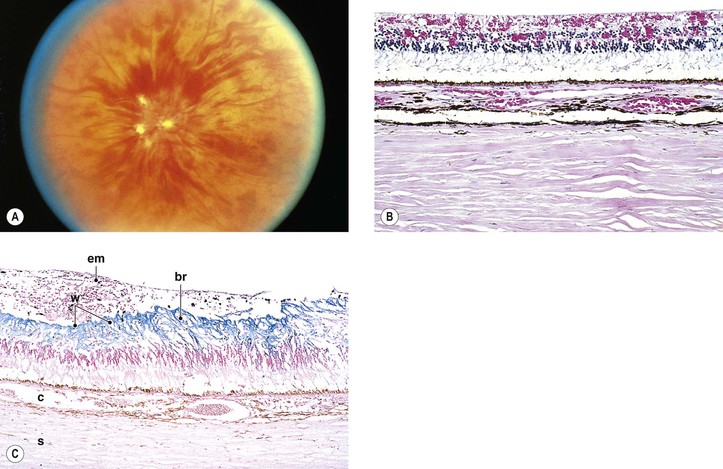
I. Macroaneurysms of the retinal arteries and arterioles (Fig. 11.16) may be congenital or acquired.
A.
[/not-level-membership-for-pathology-category]
