[level-membership-for-pathology-category]291
27.2 CNS trauma and raised intracranial pressure293
27.3 Cerebrovascular disease294
27.4 Degenerative and demyelinating diseases295
27.5 Congenital malformations and genetic disease of the CNS297
Diseases of the central nervous system (CNS) – particularly strokes, head injuries and dementia – are major causes of morbidity and mortality. Neurones do not divide in post-natal life (they are ‘permanent’ cells), and neuronal cell death is repaired by proliferation of supporting cells and ‘gliosis’ (the CNS equivalent of scarring). Children and young adults are not immune to serious CNS pathology – meningitis, congenital malformations, birth injuries, multiple sclerosis and certain neoplasms can particularly affect this population group.
The fixed available volume within the rigid skull means that an expanding mass lesion often results in raised intracranial pressure, which is frequently fatal without prompt medical intervention. Primary and secondary tumours are not uncommon in the CNS, and again the anatomical confines of the skull and vertebral column are important factors in prognosis, due to the effects of raised intracranial pressure and the physical limitation imposed on surgical resection.
27.1. Infection and inflammation
You should:
• know the spectrum of organisms that can cause meningitis and encephalitis
• understand the pathology of acute bacterial meningitis and the information that can be obtained from investigation of cerebrospinal fluid in suspected meningitis
• be aware of how human immunodeficiency virus (HIV) infection can manifest in the CNS
• understand the basic pathology of spongiform encephalopathies.
Intracranial infection
Intracranial infection can affect the arachnoid and pial membranes (meningitis) or the underlying brain itself (encephalitis). Viral, bacterial, protozoal, fungal and protein (prion) agents all contribute. Routes of entry into the CNS include:
• blood-borne spread from distant site of infection
• direct inoculation of organisms (traumatic or iatrogenic)
• local extension of sepsis (e.g. dental or sinus infection)
• via the peripheral nervous system (viral agents including herpes simplex and rabies).
Meningitis can be:
• pyogenic (bacterial)
• aseptic (viral)
• chronic (bacterial or fungal).
The microorganisms likely to be responsible vary with age and immunocompetence. Clinical symptoms include headache, neck stiffness, photophobia, irritability and altered consciousness. A skin rash may only be present with certain strains of meningococcal bacteria causing systemic sepsis. Biochemical analysis and microscopic examination of cerebrospinal fluid (CSF) obtained at lumbar puncture is helpful in discriminating the causative agent (Table 64). Bacteria and protozoa may be directly identified by microscopy.
| Infectious agent | Predominant cell content | Protein | Glucose |
|---|---|---|---|
| Pyogenic bacteria | Neutrophil polymorphs | Increased | Marked decrease |
| Viral | Lymphocytes | Mild increase | Normal, occasionally decreased |
| Tuberculosis | Lymphocytes | Marked increase | Decreased or normal |
Acute bacterial meningitis
This can be fatal if not treated at an early stage. Complications include:
• extension of infection into brain tissue with abscess formation
• venous thrombosis with cerebral infarction
• meningeal fibrosis with hydrocephalus.
The infective organisms vary with the age of patients and immunisation status but include: Escherichia coli, group B streptococci and Haemophilus influenzae in infants; Neisseria meningitidis and Streptococcus pneumoniae in older children and young adults; and Listeria monocytogenes in the elderly.
Viral meningitis
Viral meningitis is clinically less important and usually self-limiting.
Chronic meningitis
Tuberculous meningitis is rare, but is increasing in frequency among acquired immune deficiency syndrome (AIDS) patients. The onset is clinically more insidious with non-specific symptoms such as headache, confusion and vomiting. Chronic meningeal inflammation with granuloma formation and fibrosis can cause hydrocephalus and cranial nerve damage. Syphilis is a rare cause of neurological disease in the UK today, but a small percentage of those with tertiary syphilis develop chronic meningitis, sensory spinal cord damage (tabes dorsalis) or brain infection (dementia, Argyll Robertson pupils). The risk of developing neurosyphilis is increased in AIDS.
Viral encephalitis
Viral infections can cause encephalitis, with lymphocytic inflammation of the brain parenchyma and proliferation of glial cells. Intraneuronal inclusions may be seen in herpes simplex type 1 (HSV-1), cytomegalovirus (CMV) and rabies encephalopathy. Viral infections can be particularly damaging if acquired in fetal life (e.g. rubella, CMV-related congenital malformations) or during delivery (e.g. HSV-2-related neonatal sepsis).
Specific areas of the brain may be damaged, for example HSV-1 encephalitis particularly affects the temporal lobe.
Human immunodeficiency virus infection
HIV infects CNS macrophages and microglial cells. HIV infection and AIDS can cause numerous neurological lesions including:
• mild meningitis at seroconversion
• dementia-like illness
• spinal cord damage
• neuropathies
• congenital AIDS (microcephaly, motor delay and learning disabilities)
• opportunistic infections
• toxoplasmosis
• CMV
• cryptococcal meningitis
• progressive multifocal leucoencephalopathy (PML) – a papovavirus infection, which affects oligodendrocytes, causing demyelination and multifocal white-matter damage.
Rabies virus
The rabies virus gains entry to the brain by ascending along peripheral nerves. Symptoms arise weeks after initial infection and include abnormal CNS excitability (excessive pain on light touch, convulsions), paralysis, mania, stupor and coma. Local paraesthesia around the entry wound is a diagnostic pointer.
Poliomyelitis
Poliomyelitis is now very rare in industrialised countries. It initially occurs in the gut but in a small number of cases there is spread to lower motor neurones in the spinal cord, leading to muscle wasting and paralysis. Death can occur acutely due to a myocarditis or chronically due to respiratory muscle involvement.
Fungal infections of the CNS
Fungal infections of the CNS mainly occur in immunocompromised patients. The responsible organisms include Candida, Mucor, Aspergillus and Cryptococcus.
Spongiform encephalopathies
Spongiform encephalopathies are characterised by spongiform change (vacuolation) in the cerebral white matter. They are transmitted by prion protein, an abnormal form of a cellular protein that has undergone a conformational change and is able to induce a further conformational change in native protein when inoculated into previously normal cells. The prion protein gene is located on chromosome 20. It is highly conserved across species, and infectious particles in one species can corrupt the normal protein in other species. Spongiform encephalopathies include:
• scrapie in sheep
• bovine spongiform encephalopathy (BSE) in cattle
• transmissible encephalopathy in mink
• Creutzfeldt–Jakob disease (CJD), new variant CJD and kuru in humans.
Prions are neither destroyed by most normal disinfectants nor by formalin fixation.
Until the past decade, the incidence of CJD was approximately 1 per million, occurring sporadically in older adults. Iatrogenic transmission via corneal grafts, cadaveric growth hormone extracts or implanted electrodes has been documented. Recently there has been extensive debate about new variant (nv)CJD, which is thought to represent human infection by BSE transmitted by ingestion of contaminated meat products. Both CJD and new variant disease are characterised by rapidly progressive dementia with movement disorder (myoclonic jerks) and death usually occurs within 2years of symptom onset. The eventual number of individuals likely to be affected by nvCJD remains unknown.
Brain abscesses
The bacteria responsible are usually streptococcal or staphylococcal. Clinical symptoms include progressive focal neurological deficit and raised intracranial pressure.
27.2. CNS trauma and raised intracranial pressure
Mechanisms of traumatic brain injury
Contusion
Brain tissue is bruised on impact with the bony skull surface. A ‘coup’ injury occurs to the brain tissue underlying the point of external injury. A ‘contre-coup’ injury affects an area of brain directly opposite the impact. For example, a fall backwards onto the occiput causes contre-coup injury to inferior frontal lobes and inferior poles of temporal lobes (which is often more severe and clinically significant than the coup injury to the occipital lobe directly underlying the point of impact).
Laceration
Brain substance is torn, usually as a result of penetrating injury.
Diffuse axonal injury
Deceleration and rotational forces to the brain cause shearing injury to neurones and axonal processes. If extensive, this can cause coma and death. Diffuse axonal injury (DAI) is graded according to the extent of damage and the presence of grossly visible brain haemorrhage. Severe DAI can occur in the absence of any externally evident head trauma.
Tissue displacement following head injury damages blood vessels, causing haemorrhage and oedema with consequent mass effect (see below). Vascular injury and subsequent haemorrhage can be extradural, subdural, subarachnoid or intraparenchymal.
Intracranial haemorrhage
Extradural haemorrhage
Extradural haemorrhage is classically seen in association with a skull fracture involving the temporal bone with laceration of the middle meningeal artery. The typical clinical history includes a ‘lucid interval’ of several hours between the injury and neurological deterioration.
Subdural haemorrhage
Subdural haemorrhage usually originates from tearing of bridging veins that pass through the subdural space between the brain and the dural sinuses. The elderly are at increased risk, as brain atrophy increases stretching of the bridging veins and allows greater movement of the smaller brain within the skull following trauma. The precipitating head injury may be so trivial so as to have gone unnoticed. Subdural haemorrhage usually becomes clinically evident within hours, with non-specific signs (diminishing consciousness, headache) but may present chronically. Re-bleeding is common, and may occur from vascular granulation tissue within the organising haematoma.
Subarachnoid haemorrhage (SAH)
Subarachnoid haemorrhage occurring after trauma is usually secondary to brain tissue disruption. Non-traumatic SAH is more common and is discussed later in Section 27.3.
Raised intracranial pressure
The fixed skull volume allows very little room for the intracranial contents to expand in the presence of haemorrhage, tumour, abscess or oedema. Therefore a mild increase in intracranial mass can lead to increased intracranial pressure with serious consequences. Brain tissue becomes displaced (herniation). The site of herniation partly depends on whether mass increase is focal or diffuse (Figure 72):
• uncal gyrus (inferior temporal lobe) can herniate under the tentorium cerebelli and compress the midbrain (leading to altered consciousness), oculomotor nerve, contralateral cerebral peduncle, aqueduct and posterior cerebral artery
• cerebellar tonsils can herniate through the foramen magnum, compressing the brainstem (‘coning’).
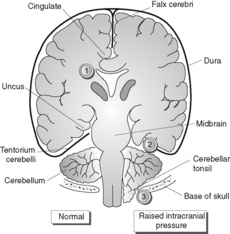
Compression of blood vessels supplying the midbrain causes venous stagnation with haemorrhage, necrosis and irreversible neuronal injury in this vital area.
Cerebral oedema
Cerebral oedema can be focal or diffuse. Oedema often makes a significant contribution to the mass effect of tumours and abscesses. The development of cerebral oedema indicates impaired function of blood–brain barrier, which normally tightly controls fluid movement within the brain. Mechanisms of cerebral oedema include:
• increased vascular permeability (vasogenic oedema)
• altered cell regulation of fluid (cytotoxic oedema)
• movement of fluid from the ventricular system into the brain.
Hydrocephalus
Hydrocephalus describes an increased volume of cerebrospinal fluid, usually due to a blockage in the CSF pathway. If occurring prior to the fusion of skull bone sutures in young children, hydrocephalus will result in head enlargement. The development of hydrocephalus may result from blood, post-inflammatory fibrosis or tumour blocking cerebrospinal fluid (CSF) flow. Apparent hydrocephalus in older adults due to atrophy of brain tissue and expansion of the ventricular system is sometimes called ‘hydrocephalus ex vacuo’.
27.3. Cerebrovascular disease
Cerebral infarction
Normal brain function ceases within a few seconds of loss of oxygen supply; irreversible neuronal damage occurs after 6–8 minutes of anoxia. Most cerebrovascular disease results from focal impairment of blood supply causing cerebral infarction. This manifests clinically as a ‘stroke’ – a neurological deficit of sudden onset but lasting more than 24 hours, caused by vascular insufficiency (neurological symptoms caused by lack of blood flow but resolving within 24 hours are known as transient ischaemic attacks or ‘TIA’). Stroke can be due to thrombosis or embolus.
Thrombotic stroke usually complicates atherosclerosis in the basilar artery, proximal middle cerebral artery or at the carotid bifurcation. As thrombotic stroke arises on a background of atheroma, risk factors include hypercholesterolaemia, hypertension, diabetes mellitus and ischaemic heart disease.
When stroke occurs secondary to embolism, the source of the embolus is usually the heart. Cardiac mural thrombus can complicate myocardial infarction and atrial fibrillation. Thrombus may also embolise from abnormal heart valves, arterial walls (especially atherosclerotic carotid arteries) or from sites of cardiac surgery. Less commonly the embolus may consist of fat, air or tumour. Cerebral fat embolus should be suspected when neurological signs develop following non-head trauma with multiple bone fractures. Most embolic strokes affect the middle cerebral artery territory. Identification of the embolus at post mortem is often not possible, as a high percentage will have lysed.
Cerebral infarction can be haemorrhagic or non-haemorrhagic, as demonstrated by computed tomography (CT) or magnetic resonance imaging (MRI). The distinction is of therapeutic importance, as anticoagulation therapy is contraindicated in the presence of intracerebral bleeding. Thrombotic strokes are not usually complicated by bleeding. Haemorrhage can be secondary to reperfusion following an embolic stroke, but is more commonly seen in spontaneous intracerebral haemorrhage (ICH) associated with hypertension. ICH usually arises in:
• deep white matter of cerebral hemispheres/basal ganglia (>50%)
• pons (10%)
• cerebellum (10%).
Microscopically, the small arterial branches that rupture may show fibrinoid necrosis of the vessel wall and formation of microaneurysms known as Charcot–Bouchard aneurysms. Severe bleeding can cause rapid mass effect with an acute fatal rise in intracranial pressure. Haemorrhage may rupture internally into the ventricular system or externally into the subarachnoid space.
Hypertension can also result in small, often multiple lacunar infarctions (<15mm in diameter) in the basal ganglia, deep white matter and pons. In accelerated or malignant hypertension, an encephalopathy may develop with headaches, vomiting, convulsions and altered conscious level.
The sequelae of stroke depend on the area of the brain involved and the extent of the infarction. Anterior circulation infarcts (internal carotid supply) affect cerebral hemispheres and can cause hemiparesis, sensory loss, dysphasia, incontinence and hemianopia. Posterior circulation infarcts (vertebrobasilar circulation) affect the brainstem and cerebellum. Neurological deficits may include ataxia and gaze abnormalities. Even small posterior circulation infarcts can cause coma and death due to involvement of vital regulatory centres in the brainstem.
Rarer causes of intracerebral haemorrhage include:
• arteriovenous malformations
• tumours
• bleeding diatheses or anticoagulation therapy
• amyloid angiopathy.
Cerebral amyloid angiopathy occurs in the elderly and is associated with Alzheimer’s disease (see Section 27.4). Haemorrhage is usually peripheral (lobar) in distribution.
Diffuse cerebral ischaemia can result following profound systemic hypertension. Clinically this may manifest as transient confusion with no permanent damage, or may result in widespread cerebral infarction with coma and death. The watershed areas of the brain – at the margins of perfusion by the major circle of Willis arterial branches – are particularly susceptible to such global ischaemia. Other brain regions particularly sensitive to hypoxia include:
• cerebellar Purkinje cells
• pyramidal neurones of hippocampus
• deeper neuronal layers of the cerebral cortex.
Subarachnoid haemorrhage
Subarachnoid haemorrhage is a relatively frequent natural cause of sudden unexpected death in young and middle-aged adults. The pathogenesis involves a congenital defect in the muscle wall of the cerebral arteries, which becomes manifest in later life as saccular dilatations known as ‘berry aneurysms’. There is an association with adult polycystic renal disease (see Ch. 23) and hypertension. The aneurysms develop mainly in the anterior part of the circle of Willis (Figure 73), around the origin and proximal branches of the anterior and middle cerebral arteries. In 25% of cases there are multiple aneurysms. Rupture causes subarachnoid haemorrhage and clinically manifests with a sudden very severe headache. Bleeding may be precipitated by an acute rise in blood pressure (e.g. during sexual intercourse). Females are more frequently affected than males.
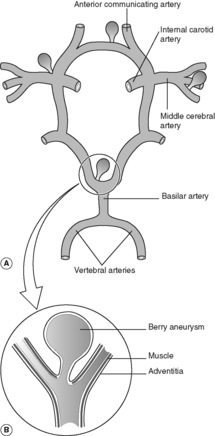
As with aneurysms elsewhere in the vascular system, the risk of rupture increases in proportion with the aneurysm size. Re-bleeding is common in those who survive the initial insult. As the haemorrhage resolves, fibrosis of the meninges at the base of the brain can lead to subsequent hydrocephalus.
27.4. Degenerative and demyelinating diseases
Dementia
Alzheimer’s disease
Dementia is the progressive loss of cognitive function, manifest by memory loss and global intellectual impairment without diminished consciousness. The commonest cause of dementia in the UK is Alzheimer’s disease (AD). AD is uncommon in people under 75 but thereafter its incidence increases dramatically. Up to 10% of cases are familial. Down’s syndrome patients can develop AD changes at an earlier age, but seem to progress at a slower rate. CT head scan and brain examination at autopsy show cortical atrophy with widened sulci and compensatory dilatation of the ventricular system (‘hydrocephalus ex vacuo’). Microscopically, three lesions are seen:
Neurofibrillary tangles are bundles of intracytoplasmic filaments present within neurones. The tangles are often rounded or flame shaped. They can be seen on routine histological staining (haematoxylin and eosin (H&E)) but are better demonstrated with silver impregnation stains or by immunocytochemistry. Senile (neuritic) plaques are collections of neuronal processes surrounding an amyloid core. The main protein component of the core is β-amyloid protein, derived from amyloid precursor protein (APP). β-Amyloid protein is also present in the vascular amyloid deposits of amyloid angiopathy.
Plaques, tangles and amyloid angiopathy can all be seen to a certain extent in ‘normal’ brains and in individuals without clinical signs of AD. Amyloid angiopathy is a cause of peripheral intracerebral haemorrhage (see Section 27.3). It is the increased number and wide distribution of plaques and tangles that correlates with clinical dementia. In AD, cortical neurones, hippocampus and the amygdala are commonly involved sites.
Pick’s disease
Pick’s disease is a much less common form of dementia. There is severe atrophy of the frontal and temporal lobes, with basal ganglia involvement. Cytoplasmic Pick’s bodies (filamentous structures similar to those seen in AD) are present within neurones.
Huntington’s disease
This is a progressive inherited dementia associated with uncontrolled movements (chorea). Onset is in middle age. Atrophy occurs in the caudate, putamen and frontal lobes. The involuntary movements result from loss of GABA-utilising inhibitory neurones, which regulate motor output in the basal ganglia. Huntington’s is an autosomal dominant condition with full penetrance, and new mutations are very rare. The gene responsible lies on chromosome 4 and contains repeats of the trinucleotide base sequence CAG. The normal allele contains 11–34 CAG copies, but in Huntington’s there is amplification of this base sequence, with up to several hundred copies. The age of onset of disease decreases with each successive generation as the number of CAG copies increases (this phenomenon is known as ‘anticipation’). A similar mechanism of disease is seen in X-linked spinal muscular atrophy, which is linked to trinucleotide repeats in the androgen receptor gene.
Parkinson’s disease
Parkinson’s disease is a movement disorder characterised by:
• abnormal gait and facial expression
• rigidity
• pill-rolling tremor
• difficulty in initiating movement
• dementia in a proportion of cases.
There is damage to dopamine-utilising neurones in the nigrostriatal tract. Loss of substantia nigra pigmentation can be identified at autopsy. Microscopically, Lewy bodies (cytoplasmic neuronal inclusions) can be seen in the brainstem nuclei, the cingulate gyrus and the hippocampus.
Motor neurone disease
This is clinically manifest by progressive muscular atrophy, fasciculation and symmetrical weakness. There is atrophy of anterior motor roots of the spinal cord (lower motor neurones) and of corticospinal tracts (upper motor neurones). Patients are usually middle-aged men. Death is often due to respiratory complications.
Toxic and metabolic disorders
Toxic and metabolic disorders of the nervous system include thiamine (vitamin B1) deficiency and B12 deficiency. The latter affects long tracts with lower-limb weakness, ataxia and sensory loss (known as subacute combined degeneration of the cord). Other uncommon but important causes of neurological damage include hypoglycaemia, hepatic encephalopathy and ethanol.
Fetal alcohol syndrome
Fetal alcohol syndrome, which is caused by excess maternal ethanol intake during pregnancy, comprises:
• growth restriction
• cardiac septal defects
• facial abnormalities
• learning disabilities.
Demyelinating disorders
Demyelinating disorders affect oligodendrocytes in the CNS or the myelin sheath of peripheral nerves.
Multiple sclerosis
Multiple sclerosis (MS) is characterised by repeated episodes of demyelination within the central nervous system. Neurological deficit is variable. It may be relatively mild, with relapses and remissions, or severe and chronically progressive. Unilateral optic neuropathy causing sudden visual deterioration is the commonest presentation, but the diagnosis is based on finding CNS lesions disseminated in time and space. The aetiology of MS is unknown. No specific infectious agent has been implicated, although there are increased serum and CSF antibody titres to common viruses. The pathogenesis may involve an immune reaction against myelin, possibly triggered by viral infection.
Epidemiology
• Peak onset age 20–35
• More common in women
• Increased risk for first degree relatives but no pattern of inheritance.
CSF analysis shows normal or increased protein and there is often an oligoclonal increase in IgG. Lymphocytes are usually increased in number. The pathological lesions are plaques, a few millimetres in diameter, which can be seen on MRI scans and on examination of the brain and spinal cord at autopsy. Typical sites include:
• periventricular cerebral white matter
• optic nerves
• brainstem
• cervical cord.
Myelin is destroyed and oligodendrocytes lost but axons are preserved. There is a chronic inflammatory infiltrate. Older plaques become scarred (‘gliotic’).
27.5. Congenital malformations and genetic disease of the CNS
Neural tube defects
Neural tube defects (NTDs) affect 2 per 1000 pregnancies in the UK, although their frequency in live births is much lower (many such pregnancies are terminated). Many NTDs can be detected antenatally by serum α-fetoprotein measurement and ultrasound. Defects include anencephaly, encephalocele and spinal fusion defects (Table 65). The rate of recurrence in subsequent pregnancies is approximately 5%. The incidence of NTDs varies in different ethnic groups. Folate deficiency is a recognised aetiological factor. Spinal defects are commonest and usually involve the lumbosacral region, predisposing to infection and cord damage. Table 66 details other more common CNS malformations.
| Anencephaly | Absence of brain and skull |
| Encephalocele | Occipital bone defect with herniation of brain tissue |
| Meningomyelocele | Spinal column bony defect with exposed CNS tissue |
| Meningocele | Spinal column defect with herniation of meninges but no involvement of neural tissue |
| Spina bifida | Vertebral body bony defects but no involvement of spinal cord tissue |
| Microcephaly (small brain) | Fetal alcohol syndrome |
| HIV | |
| Cytomegalovirus | |
| Holoprosencephaly (incomplete separation of cerebral hemispheres) | Trisomy 13 |
| Arnold–Chiari malformation | Multiple abnormalities involving cerebellum, medulla, aqueduct (stenosis) and lumbosacral spinal column (myelomeningocele) |
| Dandy–Walker malformation | Absent central cerebellum replaced by midline cyst |
Cerebral palsy
Is a non-progressive neurological defect arising in the perinatal period. The cause may not be apparent but intrauterine hypoxia, infection, birth trauma, kernicterus and hypoglycaemia may contribute. In premature babies, development of cerebral palsy is associated with cerebral haemorrhage and ischaemic damage to white matter (periventricular leucomalacia).
Inborn errors of metabolism in the CNS
Inborn errors of metabolism in the CNS include neuronal storage diseases and white matter diseases (leucodystrophies) involving damage to myelin or oligodendrocytes. Storage diseases usually have an autosomal recessive pattern of inheritance. Enzyme deficiencies cause accumulation of substrate (e.g. mucopolysaccharides, sphingolipids) within neuronal lysosomes and ultimately cell death.
Friedreich’s ataxia
Friedreich’s ataxia is an inherited spinocerebellar degeneration with axonal loss in long tracts. Onset is usually in late childhood, with boys more commonly affected than girls. Symptoms include ataxia, dysarthria, sensory loss and progressive paralysis. There may be associated cardiomyopathy.
27.6. Neoplasms
Central nervous system tumours
Intracranial tumours are 10 times more frequent than spinal neoplasms. About 20% of childhood cancers arise in the CNS. Primary tumours of the CNS cannot be labelled as ‘benign’ or ‘malignant’ using the same criteria as for tumours arising elsewhere in the body. Cytologically benign tumours can show locally extensive growth patterns preventing surgical excision. Slow-growing lesions can prove fatal due to their location or mass effect (e.g. benign tumour of the brainstem). Even microscopically high-grade tumours rarely metastasise outside the CNS, although dissemination can occur via the subarachnoid space and CSF pathways. Up to half of all CNS neoplasms are primary. The rest are metastases arising largely from carcinoma of the lung, breast, kidney, gastrointestinal tract and from malignant melanoma.
Astrocytomas
Astrocytomas are the most common adult primary brain tumours (Table 67). High-grade tumours (glioblastoma multiforme) have a very poor prognosis. Low-grade astrocytomas often dedifferentiate to high-grade tumours over the course of several years.
| Incidence | Commonest in middle-aged and elderly adults (cerebral hemispheres); cerebellar astrocytomas in children |
| Risk factors | Not established |
| Protective factors | Not established |
| Associated lesions | Occasional gliomas are part of neurofibromatosis or inherited cancer syndromes (e.g. Turcot syndrome) |
| Common clinical presentation | Seizures, focal neurological signs Headache and vomiting if raised intracranial pressure |
| Location | Cerebral hemispheres most common, but can arise in brainstem, cerebellum and spinal cord |
| Macroscopic appearance | Poorly defined mass infiltrating adjacent brain |
| Histological features | Vary from low to high grade according to mitotic activity, nuclear pleomorphism and necrosis |
| Pattern of spread | Local brain infiltration |
| Prognosis (per cent 5-year survival) | Approximately 5–10years’ survival for low-grade astrocytoma; 8–10months’ average survival in glioblastoma multiforme (high-grade tumour) |
Oligodendrocytomas
Oligodendrocytomas account for up to 15% of gliomas, and occur in middle-aged adults. Tumours arise in the cerebral hemispheres. Survival averages 5–10years post-diagnosis.
Ependymomas
Ependymomas most commonly occur in the fourth ventricle in children, causing hydrocephalus. Their prognosis is poor as surgery is difficult and there is a tendency to dissemination via the cerebrospinal fluid.
Choroid plexus papillomas
Choroid plexus papillomas mainly occur in the lateral ventricle during childhood. They cause hydrocephalus due to obstruction of CSF flow and increased CSF production.
Medulloblastoma
Medulloblastoma is a primitive neural tumour which accounts for 20% of childhood brain tumours. It is a rapidly growing lesion in the central cerebellum, which can cause hydrocephalus. Medulloblastoma spreads along CSF pathways, but is highly sensitive to radiotherapy.
Primary brain lymphoma
Primary brain lymphoma is increasing in incidence. The pathogenesis is linked to Epstein–Barr virus infection and immunosuppression, but the frequency of lymphoma is also rising in immunocompetent individuals. Tumour deposits are often multiple. Histologically, these are high-grade, B lymphocyte, non-Hodgkin’s lymphomas.
Meningiomas
Meningiomas are usually benign tumours of adulthood, arising from meningothelial cells of the arachnoid. Meningiomas are commoner in women and occur in young and middle-aged adults. Meningiomas can compress and invaginate into the brain substance but remain separate from it, which may permit surgical excision. Overlying bone can be eroded or infiltrated. There are many histological subtypes.
Metastatic tumours
Metastatic tumours frequently found in the CNS include lung and breast carcinomas and malignant melanomas.
Paraneoplastic symptoms
Paraneoplastic symptoms are defined as clinical phenomena associated with malignancy but not directly attributable to tumour infiltration. Paraneoplastic symptoms involving the CNS most commonly occur with small cell lung carcinoma. The pathogenesis is uncertain but may be immunologically mediated by antibodies against tumour expressed antigens cross-reacting against nervous system cells. CNS paraneoplastic syndromes include:
• spinal cord damage
• limbic encephalitis (a form of subacute dementia).
Peripheral nerve sheath tumours
Schwannoma
Schwannoma is a benign tumour of cells that produce myelin in the peripheral nervous system. Schwannomas can involve cranial nerves, especially the VIIIth nerve, which gives rise to the name ‘acoustic neuroma’. Schwannomas are encapsulated tumours attached to the nerve. Malignant change is very rare.
Neurofibroma
Neurofibroma is a common tumour composed of Schwann cells, fibroblasts and perineural cells. Neurofibromas are unencapsulated, and cannot be separated from nerve in which they arise. Neurofibromas occur sporadically or in patients with neurofibromatosis (Box 33).
Type I
• Mutation in tumour suppressor gene on chromosome 17.
• Autosomal dominantly inherited or spontaneous mutation.
Characterised by:
• multiple neurofibromas (cutaneous and visceral)
• pigmented skin macules (café-au-lait patches)
• pigmented nodules on the iris (Lisch nodules).
There is an increased incidence of other tumours including acoustic neuromas, gliomas, meningiomas, phaeochromocytomas. Learning disabilities and skeletal abnormalities are common.
Type II
• Bilateral acoustic schwannomas, with or without cutaneous neurofibromas.
• No iris nodules.
• Tumour suppressor gene involved is on chromosome 22.
Malignant peripheral nerve sheath tumours
Malignant peripheral nerve sheath tumours arise de novo or from transformation of neurofibromas in neurofibromatosis.
Self-assessment: questions
True-false questions
1. Prion protein:
a. Is degraded by formalin
b. is present in plaques of Alzheimer’s disease
c. is highly conserved across species
d. causes cell damage by inducing a conformational change in normal protein
e. can be transmitted by corneal grafts
2. Berry aneurysms:
a. are usually multiple
b. classically cause subdural haemorrhage
c. are associated with polycystic kidney disease
d. are present at birth
e. have a high risk of re-bleeding following rupture
3. In multiple sclerosis:
a. there is loss of both myelin and axons
b. unilateral optic nerve involvement is a common presentation
c. the incidence is highest in equatorial countries
d. the cerebrospinal fluid often contains an oligoclonal increase in IgM antibodies
e. plaques are characteristically found in the periventricular white matter of the cerebral hemispheres
4. Subdural haemorrhage:
a. is commonest in the elderly
b. may not become clinically evident until several weeks after the event
c. is always preceded by a significant head injury
d. usually originates from the subdural sinuses
e. is rarely complicated by re-bleeding
5. The following are correctly paired:
a. Huntington’s disease – trinucleotide repeats
b. Parkinson’s disease – neurofibrillary tangles
c. Pick’s disease – frontal lobe atrophy
d. Alzheimer’s disease – amyloid angiopathy
e. fetal alcohol syndrome – cardiac septal defects
6. The following are causes of non-traumatic intracranial haemorrhage:
a. hypertension
b. thrombocytopenia
c. cerebral embolism
d. arteriovenous malformations
e. glioblastoma multiforme
7. Concerning meningitis:
a. cerebrospinal fluid neutrophils are always increased
b. cerebral infarction is a complication
c. Neisseria meningitidis is the commonest causative organism in infants
d. cryptococcal meningitis is increasing in incidence
e. viral meningitis is associated with markedly low CSF glucose
8. Neural tube defects:
a. affect 2 per 1000 live births in the UK
b. can be detected by measurement of serum carcinoembryonic antigen
c. of the spine are commonest in the cervical region
d. recur in 10% of subsequent pregnancies with the same parents
e. are associated with folate deficiency
9. Concerning CNS tumours:
a. the commonest primary glial tumours are astrocytomas
b. 20% of childhood malignancy occurs in the CNS
c. glioblastoma multiforme metastasises widely outside the CNS
d. medulloblastoma is usually radiosensitive
e. primary brain lymphoma is associated with herpes simplex virus
10. The following are risk factors for cerebral infarction:
a. Myocardial infarction
b. infective endocarditis
c. fat embolism
d. diabetes insipidus
e. septic shock
Case history questions
Case history 1
1. What clinical diagnosis do these symptoms suggest?
2. What further information would you seek from the history and clinical examination to help determine the underlying cause?
Case history 2
You are a consultant histopathologist, and have been asked by the coroner to carry out a post-mortem examination on a 19-year-old man who died unexpectedly. The deceased had been out with friends the night before his death and had consumed several pints of lager. He had been involved in a fight and had briefly been knocked unconscious but appeared to recover within a few minutes and did not seek medical help. He had later felt tired and had been taken home to bed by his friends, where he was found dead the morning after.
1. What causes of death would you consider likely in this scenario?
2. On exposing the skull, you identify a fracture of the right temporal bone. What other abnormalities do you now expect to find inside the cranial cavity?
Case history 3
A 32-year-old woman presents with a 2-week history of headaches and recent onset of left-arm weakness. A CT head scan (with contrast) shows an intracerebral mass with rim enhancement and a low-density centre.
1. What is the differential diagnosis?
2. What further information would you seek from the history and clinical examination to help determine the underlying cause?
Viva question
1. How can tumour involvement of the CNS present clinically?
Self-assessment: answers
True-false answers
1.
a. False.
b. False.
c. True.
d. True.
e. True.
2.
a. False. Twenty-five per cent are multiple.
b. False. Subarachnoid.
c. True. In a proportion of cases.
d. False. The weakness of the arterial wall is present at birth but the aneurysms develop in later life.
e. True.
3.
a. False. Axons are not damaged.
b. True.
c. False.
d. False. There may be oligoclonal IgG antibody increase.
e. True.
4.
a. True. Alcoholics are another high-risk group.
b. True.
c. False. The injury may be trivial and may have gone unnoticed or not be remembered.
d. False. From bridging veins.
e. False. Re-bleeding from vascular granulation tissue in the organising thrombus is common.
5.
a. True.
b. False.
c. True.
d. True.
e. True.
6.
a. True.
b. True.
c. True.
d. True.
e. True.
7.
a. False. Neutrophil polymorphs are inconsistently present in tuberculous meningitis and are absent in viral meningitis.
b. True.
c. False. Haemophilus influenzae in young children and Escherichia coli in babies.
d. True. Through its association with HIV infection.
e. False. CSF glucose is usually normal in viral meningitis.
8.
a. False. Many are detected antenatally and may be aborted spontaneously or by medical intervention.
b. False. Maternal serum α-fetoprotein may be raised.
c. False. They are most common in the lumbosacral region.
d. False. Recurrence rate is less than 5%.
e. True.
9.
a. True.
b. True.
c. False.
d. True.
e. False. It is associated with Epstein–Barr virus infection in HIV-positive patients.
10.
a. True. Cardiac mural thrombi are a source of cerebral emboli.
b. True. Embolisation of valve vegetations.
c. True.
d. False.
e. True. Profound hypotension can cause global cerebral ischaemia.
Case history answers
Case history 1
1. The symptoms are those of a chronic decline in cognitive function, suggesting dementia. The commonest causes in the UK are Alzheimer’s disease and multiple cerebral infarctions. The age of onset in this patient is unusually young for Alzheimer’s but this certainly does not exclude the diagnosis. A family history of dementia, especially with decreasing age of onset in successive generations, should raise the suspicion of Huntington’s disease; a history of movement disorders should also be sought. CJD and nvCJD are very rare conditions (at least currently) and the typical history is of a more rapidly progressive dementia often with psychiatric symptoms and muscle disease (myoclonia or wasting).
Case history 2
1. The history raises the suspicion of traumatic injuries, either to the head or the viscera. Catastrophic haemorrhage could have occurred from a ruptured spleen. Acute alcohol intoxication certainly needs to be excluded (post-mortem blood samples for alcohol and drugs would be taken).
2. The history of brief loss of consciousness followed by a ‘lucid interval’ and the finding of a temporal bone fracture are highly suggestive of extradural haemorrhage. The bleeding is arterial in nature, with origin from a torn middle meningeal artery. At post mortem you would expect to see the extradural haematoma, and if this is the cause of death, there will almost certainly be signs of raised intracranial pressure.
Case history 3
1. The radiological appearances here suggest either an abscess or a tumour (primary or secondary).
2. You should specifically ask about and look for sepsis – either a local infection (middle ear or sinuses) or a distant source of septic embolism (infective endocarditis from congenital heart disease, intravenous drug use, etc.). You must establish whether the patient is immunosuppressed (diabetes, HIV, steroid therapy, organ transplant), which would increase the risk of brain abscess following infection with virulent and opportunistic organisms. Although metastatic tumour is rare in this age group, remember that breast cancer and melanoma in particular can affect young adults and are common tumours to metastasise to the CNS. Primary glial malignancies are most frequently seen in older adults but can present at any age.
Viva answer
Comment: Think logically:
• Localising neurological signs of gradual onset – focal weakness or sensory deficit, hearing loss (acoustic neuroma), spinal cord level of dysfunction for intravertebral tumours. Sudden severe neurological deficit may be caused by haemorrhage from the tumour.
• Focal or generalised epilepsy.
• Local mass of tumour with or without oedema causing raised intracranial pressure with headaches and vomiting.
• Remember that primary brain malignancies very rarely metastasise outside the CNS, but some will disseminate along the CSF pathway.
• Remember that some paraneoplastic syndromes occur in the CNS.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]291
27.2 CNS trauma and raised intracranial pressure293
27.3 Cerebrovascular disease294
27.4 Degenerative and demyelinating diseases295
27.5 Congenital malformations and genetic disease of the CNS297
Diseases of the central nervous system (CNS) – particularly strokes, head injuries and dementia – are major causes of morbidity and mortality. Neurones do not divide in post-natal life (they are ‘permanent’ cells), and neuronal cell death is repaired by proliferation of supporting cells and ‘gliosis’ (the CNS equivalent of scarring). Children and young adults are not immune to serious CNS pathology – meningitis, congenital malformations, birth injuries, multiple sclerosis and certain neoplasms can particularly affect this population group.
The fixed available volume within the rigid skull means that an expanding mass lesion often results in raised intracranial pressure, which is frequently fatal without prompt medical intervention. Primary and secondary tumours are not uncommon in the CNS, and again the anatomical confines of the skull and vertebral column are important factors in prognosis, due to the effects of raised intracranial pressure and the physical limitation imposed on surgical resection.
27.1. Infection and inflammation
You should:
• know the spectrum of organisms that can cause meningitis and encephalitis
• understand the pathology of acute bacterial meningitis and the information that can be obtained from investigation of cerebrospinal fluid in suspected meningitis
• be aware of how human immunodeficiency virus (HIV) infection can manifest in the CNS
• understand the basic pathology of spongiform encephalopathies.
Intracranial infection
Intracranial infection can affect the arachnoid and pial membranes (meningitis) or the underlying brain itself (encephalitis). Viral, bacterial, protozoal, fungal and protein (prion) agents all contribute. Routes of entry into the CNS include:
• blood-borne spread from distant site of infection
• direct inoculation of organisms (traumatic or iatrogenic)
• local extension of sepsis (e.g. dental or sinus infection)
• via the peripheral nervous system (viral agents including herpes simplex and rabies).
Meningitis can be:
• pyogenic (bacterial)
• aseptic (viral)
• chronic (bacterial or fungal).
The microorganisms likely to be responsible vary with age and immunocompetence. Clinical symptoms include headache, neck stiffness, photophobia, irritability and altered consciousness. A skin rash may only be present with certain strains of meningococcal bacteria causing systemic sepsis. Biochemical analysis and microscopic examination of cerebrospinal fluid (CSF) obtained at lumbar puncture is helpful in discriminating the causative agent (Table 64). Bacteria and protozoa may be directly identified by microscopy.
| Infectious agent | Predominant cell content | Protein | Glucose |
|---|---|---|---|
| Pyogenic bacteria | Neutrophil polymorphs | Increased | Marked decrease |
| Viral | Lymphocytes | Mild increase | Normal, occasionally decreased |
| Tuberculosis | Lymphocytes | Marked increase | Decreased or normal |
Acute bacterial meningitis
This can be fatal if not treated at an early stage. Complications include:
• extension of infection into brain tissue with abscess formation
• venous thrombosis with cerebral infarction
• meningeal fibrosis with hydrocephalus.
The infective organisms vary with the age of patients and immunisation status but include: Escherichia coli, group B streptococci and Haemophilus influenzae in infants; Neisseria meningitidis and Streptococcus pneumoniae in older children and young adults; and Listeria monocytogenes in the elderly.
Viral meningitis
Viral meningitis is clinically less important and usually self-limiting.
Chronic meningitis
Tuberculous meningitis is rare, but is increasing in frequency among acquired immune deficiency syndrome (AIDS) patients. The onset is clinically more insidious with non-specific symptoms such as headache, confusion and vomiting. Chronic meningeal inflammation with granuloma formation and fibrosis can cause hydrocephalus and cranial nerve damage. Syphilis is a rare cause of neurological disease in the UK today, but a small percentage of those with tertiary syphilis develop chronic meningitis, sensory spinal cord damage (tabes dorsalis) or brain infection (dementia, Argyll Robertson pupils). The risk of developing neurosyphilis is increased in AIDS.
Viral encephalitis
Viral infections can cause encephalitis, with lymphocytic inflammation of the brain parenchyma and proliferation of glial cells. Intraneuronal inclusions may be seen in herpes simplex type 1 (HSV-1), cytomegalovirus (CMV) and rabies encephalopathy. Viral infections can be particularly damaging if acquired in fetal life (e.g. rubella, CMV-related congenital malformations) or during delivery (e.g. HSV-2-related neonatal sepsis).
Specific areas of the brain may be damaged, for example HSV-1 encephalitis particularly affects the temporal lobe.
Human immunodeficiency virus infection
HIV infects CNS macrophages and microglial cells. HIV infection and AIDS can cause numerous neurological lesions including:
• mild meningitis at seroconversion
• dementia-like illness
• spinal cord damage
• neuropathies
• congenital AIDS (microcephaly, motor delay and learning disabilities)
• opportunistic infections
• toxoplasmosis
• CMV
• cryptococcal meningitis
• progressive multifocal leucoencephalopathy (PML) – a papovavirus infection, which affects oligodendrocytes, causing demyelination and multifocal white-matter damage.
Rabies virus
The rabies virus gains entry to the brain by ascending along peripheral nerves. Symptoms arise weeks after initial infection and include abnormal CNS excitability (excessive pain on light touch, convulsions), paralysis, mania, stupor and coma. Local paraesthesia around the entry wound is a diagnostic pointer.
Poliomyelitis
Poliomyelitis is now very rare in industrialised countries. It initially occurs in the gut but in a small number of cases there is spread to lower motor neurones in the spinal cord, leading to muscle wasting and paralysis. Death can occur acutely due to a myocarditis or chronically due to respiratory muscle involvement.
Fungal infections of the CNS
Fungal infections of the CNS mainly occur in immunocompromised patients. The responsible organisms include Candida, Mucor, Aspergillus and Cryptococcus.
Spongiform encephalopathies
Spongiform encephalopathies are characterised by spongiform change (vacuolation) in the cerebral white matter. They are transmitted by prion protein, an abnormal form of a cellular protein that has undergone a conformational change and is able to induce a further conformational change in native protein when inoculated into previously normal cells. The prion protein gene is located on chromosome 20. It is highly conserved across species, and infectious particles in one species can corrupt the normal protein in other species. Spongiform encephalopathies include:
• scrapie in sheep
• bovine spongiform encephalopathy (BSE) in cattle
• transmissible encephalopathy in mink
• Creutzfeldt–Jakob disease (CJD), new variant CJD and kuru in humans.
Prions are neither destroyed by most normal disinfectants nor by formalin fixation.
Until the past decade, the incidence of CJD was approximately 1 per million, occurring sporadically in older adults. Iatrogenic transmission via corneal grafts, cadaveric growth hormone extracts or implanted electrodes has been documented. Recently there has been extensive debate about new variant (nv)CJD, which is thought to represent human infection by BSE transmitted by ingestion of contaminated meat products. Both CJD and new variant disease are characterised by rapidly progressive dementia with movement disorder (myoclonic jerks) and death usually occurs within 2years of symptom onset. The eventual number of individuals likely to be affected by nvCJD remains unknown.
Brain abscesses
The bacteria responsible are usually streptococcal or staphylococcal. Clinical symptoms include progressive focal neurological deficit and raised intracranial pressure.
27.2. CNS trauma and raised intracranial pressure
Mechanisms of traumatic brain injury
Contusion
Brain tissue is bruised on impact with the bony skull surface. A ‘coup’ injury occurs to the brain tissue underlying the point of external injury. A ‘contre-coup’ injury affects an area of brain directly opposite the impact. For example, a fall backwards onto the occiput causes contre-coup injury to inferior frontal lobes and inferior poles of temporal lobes (which is often more severe and clinically significant than the coup injury to the occipital lobe directly underlying the point of impact).
Laceration
Brain substance is torn, usually as a result of penetrating injury.
Diffuse axonal injury
Deceleration and rotational forces to the brain cause shearing injury to neurones and axonal processes. If extensive, this can cause coma and death. Diffuse axonal injury (DAI) is graded according to the extent of damage and the presence of grossly visible brain haemorrhage. Severe DAI can occur in the absence of any externally evident head trauma.
Tissue displacement following head injury damages blood vessels, causing haemorrhage and oedema with consequent mass effect (see below). Vascular injury and subsequent haemorrhage can be extradural, subdural, subarachnoid or intraparenchymal.
Intracranial haemorrhage
Extradural haemorrhage
Extradural haemorrhage is classically seen in association with a skull fracture involving the temporal bone with laceration of the middle meningeal artery. The typical clinical history includes a ‘lucid interval’ of several hours between the injury and neurological deterioration.
Subdural haemorrhage
Subdural haemorrhage usually originates from tearing of bridging veins that pass through the subdural space between the brain and the dural sinuses. The elderly are at increased risk, as brain atrophy increases stretching of the bridging veins and allows greater movement of the smaller brain within the skull following trauma. The precipitating head injury may be so trivial so as to have gone unnoticed. Subdural haemorrhage usually becomes clinically evident within hours, with non-specific signs (diminishing consciousness, headache) but may present chronically. Re-bleeding is common, and may occur from vascular granulation tissue within the organising haematoma.
Subarachnoid haemorrhage (SAH)
Subarachnoid haemorrhage occurring after trauma is usually secondary to brain tissue disruption. Non-traumatic SAH is more common and is discussed later in Section 27.3.
Raised intracranial pressure
The fixed skull volume allows very little room for the intracranial contents to expand in the presence of haemorrhage, tumour, abscess or oedema. Therefore a mild increase in intracranial mass can lead to increased intracranial pressure with serious consequences. Brain tissue becomes displaced (herniation). The site of herniation partly depends on whether mass increase is focal or diffuse (Figure 72):
• uncal gyrus (inferior temporal lobe) can herniate under the tentorium cerebelli and compress the midbrain (leading to altered consciousness), oculomotor nerve, contralateral cerebral peduncle, aqueduct and posterior cerebral artery
• cerebellar tonsils can herniate through the foramen magnum, compressing the brainstem (‘coning’).
Compression of blood vessels supplying the midbrain causes venous stagnation with haemorrhage, necrosis and irreversible neuronal injury in this vital area.
Cerebral oedema
Cerebral oedema can be focal or diffuse. Oedema often makes a significant contribution to the mass effect of tumours and abscesses. The development of cerebral oedema indicates impaired function of blood–brain barrier, which normally tightly controls fluid movement within the brain. Mechanisms of cerebral oedema include:
• increased vascular permeability (vasogenic oedema)
• altered cell regulation of fluid (cytotoxic oedema)
• movement of fluid from the ventricular system into the brain.
Hydrocephalus
Hydrocephalus describes an increased volume of cerebrospinal fluid, usually due to a blockage in the CSF pathway. If occurring prior to the fusion of skull bone sutures in young children, hydrocephalus will result in head enlargement. The development of hydrocephalus may result from blood, post-inflammatory fibrosis or tumour blocking cerebrospinal fluid (CSF) flow. Apparent hydrocephalus in older adults due to atrophy of brain tissue and expansion of the ventricular system is sometimes called ‘hydrocephalus ex vacuo’.
27.3. Cerebrovascular disease
Cerebral infarction
Normal brain function ceases within a few seconds of loss of oxygen supply; irreversible neuronal damage occurs after 6–8 minutes of anoxia. Most cerebrovascular disease results from focal impairment of blood supply causing cerebral infarction. This manifests clinically as a ‘stroke’ – a neurological deficit of sudden onset but lasting more than 24 hours, caused by vascular insufficiency (neurological symptoms caused by lack of blood flow but resolving within 24 hours are known as transient ischaemic attacks or ‘TIA’). Stroke can be due to thrombosis or embolus.
Thrombotic stroke usually complicates atherosclerosis in the basilar artery, proximal middle cerebral artery or at the carotid bifurcation. As thrombotic stroke arises on a background of atheroma, risk factors include hypercholesterolaemia, hypertension, diabetes mellitus and ischaemic heart disease.
When stroke occurs secondary to embolism, the source of the embolus is usually the heart. Cardiac mural thrombus can complicate myocardial infarction and atrial fibrillation. Thrombus may also embolise from abnormal heart valves, arterial walls (especially atherosclerotic carotid arteries) or from sites of cardiac surgery. Less commonly the embolus may consist of fat, air or tumour. Cerebral fat embolus should be suspected when neurological signs develop following non-head trauma with multiple bone fractures. Most embolic strokes affect the middle cerebral artery territory. Identification of the embolus at post mortem is often not possible, as a high percentage will have lysed.
Cerebral infarction can be haemorrhagic or non-haemorrhagic, as demonstrated by computed tomography (CT) or magnetic resonance imaging (MRI). The distinction is of therapeutic importance, as anticoagulation therapy is contraindicated in the presence of intracerebral bleeding. Thrombotic strokes are not usually complicated by bleeding. Haemorrhage can be secondary to reperfusion following an embolic stroke, but is more commonly seen in spontaneous intracerebral haemorrhage (ICH) associated with hypertension. ICH usually arises in:
• deep white matter of cerebral hemispheres/basal ganglia (>50%)
• pons (10%)
• cerebellum (10%).
Microscopically, the small arterial branches that rupture may show fibrinoid necrosis of the vessel wall and formation of microaneurysms known as Charcot–Bouchard aneurysms. Severe bleeding can cause rapid mass effect with an acute fatal rise in intracranial pressure. Haemorrhage may rupture internally into the ventricular system or externally into the subarachnoid space.
Hypertension can also result in small, often multiple lacunar infarctions (<15mm in diameter) in the basal ganglia, deep white matter and pons. In accelerated or malignant hypertension, an encephalopathy may develop with headaches, vomiting, convulsions and altered conscious level.
Buy Membership for Pathology Category to continue reading. Learn more here
[/not-level-membership-for-pathology-category]
