Chapter 16 Neonatal Seizures
Epidemiology
Estimates of the incidence of neonatal seizures range from 1 to 3.5/1000 term births in the United States and similar rates have been reported from other developed countries. The incidence of neonatal seizures is substantially higher in premature infants, approaching rates as high as 5.8 percent in preterm neonates with birth weights less than 1500 g [Lanska et al., 1995].
Diverse case detection methods have been utilized in epidemiologic studies to determine the incidence of and associated risk factors for neonatal seizures. Lanska et al. [1995] attempted to identify all potential cases in one Kentucky county over 4 years [Lanska et al., 1995; Lanska and Lanska, 1996]. Their strategy was to search hospital-based medical record systems, regional birth certificate data files, and National Center for Health Statistics multiple-cause-of-death mortality data files, and couple these data with independent review of abstracted medical records for potential cases by three neurologists using prospectively determined case-selection criteria; they found 3.5 cases/1000 live births. Saliba et al. [1999] estimated the incidence of clinical neonatal seizures among newborns born between 1992 and 1994 in Harris County, Texas, by incorporating cases from four sources: hospital discharge diagnoses, birth certificates, death certificates, and a clinical study conducted concurrently at a tertiary care center in Houston, Texas; they reported 1.8 cases/1000 live births [Saliba et al., 1999]. In contrast, Ronen et al. performed a prospective study that included all obstetrical and neonatal units in Newfoundland, Canada [Ronen et al., 1999]. All units were given educational sessions on neonatal seizure symptoms, and detailed questionnaires were prospectively collected for all infants with probable neonatal seizures over a 5-year period; their reported rate was 2.6/1000 births. Glass et al. [2009] used a California Office of Statewide Planning and Development-linked Vital Statistics/Patient Discharge Data file created specifically to study perinatal outcomes, based on a cohort of 2.3 million California children born at >36 weeks’ gestation between 1998 and 2002 [Glass et al., 2009]. The matched file linked 97 percent of California birth certificates to the corresponding newborn and maternal hospital discharge record. The resulting data set included demographic data and up to 25 International Classification of Diseases, Ninth Revision, Clinical Modifications (ICD-9-CM) maternal and infant discharge diagnoses [Martin et al., 2008]. These approaches have all yielded rates in a relatively similar range. An important caveat that must be recognized in interpretation of all these reports is that they are invariably based on the clinical diagnoses of seizures. Later in this chapter, we will discuss the limitations inherent in reliance on clinical (rather than electroencephalographic) criteria for the diagnosis of neonatal seizures. None the less, there is substantial concordance among many studies regarding the relatively high incidence of neonatal seizures and the increased risk in premature infants. Among the studies cited above, the most recent one, that in which California births from 1998 to 2002 were analyzed, reported that the incidence of seizures during the birth admission was 0.95/1000 live births, i.e., at the low end of prior estimates [Glass et al., 2009]. Whether this rate reflects a significant decline in neonatal seizure frequency remains to be determined.
A consistently identified trend is the greater risk for seizures in premature than in term infants. Some studies have reported an inverse relationship between birth weight category (or degree of prematurity) and seizure risk. Neonatal seizure rates of 57.5/1000 live births have been reported among very low birth weight infants (less than 1500 g), compared with 4.4/1000 for infants with moderately low birth weight (1500–2499 g), 2.8/1000 for those with normal birth weight (2500–3999 g), and 2.0/1000 for those with high birth weight (4000 or more grams) [Lanska et al., 1995; Lanska and Lanska, 1996]. This was corroborated by another study that found that seizure incidence was highest among infants weighing less than 1500 g (19/1000) and decreased as birth weight increased [Saliba et al., 1999]. In the prospective study from Newfoundland, the rates reported were 11.1/1000 for preterm neonates, and 13.5/1000 for infants weighing less than 2500 g at birth (presumably including both preterm and small-for-gestational-age neonates) [Ronen et al., 1999]. Analysis of a recent population-based cohort of very low birth weight infants in Israel reported that the incidence of neonatal seizures was 5.6 percent (i.e. the same as in the study from Kentucky cited above, in the preceding decade). In the study from Israel, male sex, and major systemic and neurological comorbidities (e.g., intraventricular hemorrhage or periventricular leukomalacia) were independent predictors of neonatal seizures. Male gender was also identified as a risk factor in preterm infants with seizures in the Harris County, Texas, cohort (relative risk = 1.8, 95 percent confidence interval: 1.0, 3.4) [Saliba et al., 2001]. Gender differences in seizure incidence have also been identified in term infants; in the northern California cohort cited earlier, the proportion of males was higher in the seizure group than in the overall cohort (57.3 percent vs. 50.9 percent) [Glass et al., 2009]. Ethnicity-related risk factors for neonatal seizures have not been reported.
In the Texas cohort [Saliba et al., 2001], multivariate analysis identified several additional risk factors for term infants, including birth by cesarean section, low birth weight for gestational age, birth in a private/university hospital, and maternal age of 18–24, compared with 25–29 years. In the California cohort study, using multivariable logistic regression analysis, neonates of women aged 40 years and older who were nulliparous, or had diabetes mellitus, intrapartum fever, or infection, or delivered at > 42 weeks were at increased risk for seizures [Glass et al., 2009].
Pathophysiology
Since neonatal seizures are refractory to conventional antiepileptic drugs (AEDs) and can have severe consequences for long-term neurologic status, there is a growing body of research directed at defining age-specific mechanisms of this disorder to identify new therapeutic targets and biomarkers. There have been substantial advances with regard to understanding pathophysiology, particularly with respect to identification of the developmental stage-specific factors that influence mechanisms of seizure generation, responsiveness to anticonvulsants, and the impact of seizures on central nervous system (CNS) development (for detailed review, see Rakhade and Jensen [2009]). In addition, experimental data have raised concerns about the potential adverse effects of current treatments with barbiturates and benzodiazepines on brain development. Improved understanding of the unique age-specific mechanisms should yield new therapeutic targets with clinical potential. However, to date, no novel compounds have been specifically developed or achieved Food and Drug Administration (FDA) approval for treatment of neonatal seizures [Sankar and Painter, 2005].
Developmental age-specific mechanisms influence the generation and phenotype of seizures, the impact of seizures on brain structure and function, and the efficacy of anticonvulsant therapy. Factors governing neuronal excitability combine to create a relatively hyperexcitable state in the neonatal period, as evidenced by the extremely low threshold for seizures and reflected by the fact that, across the life span, this is the period when seizures occur most commonly [Hauser et al., 1993; Aicardi and Chevrie, 1970]. Similarly, in the rodent, seizure susceptibility peaks in the second postnatal week in many models, a time period compatible with the neonatal period in humans [Sanchez and Jensen, 2001; Sanchez et al., 2005b; Rakhade and Jensen, 2009]. In addition, the incomplete development of neurotransmitter systems results in a lack of “target” receptors for conventional AEDs. Finally, the relatively limited degree of cortical and subcortical myelination results in the multifocal nature or unusual behavioral correlates of seizures at this age [Haynes et al., 2005; Talos et al., 2006a].
The neonatal period is a period of intense physiological synaptic excitability, as synaptogenesis is wholly dependent upon activity. In the human, synapse and dendritic spine density are peaking around birth and into the first months of life [Takashima et al., 1980; Huttenlocher et al., 1982]. In addition, the balance between excitatory versus inhibitory synaptic activity is tipped in favor of excitation to permit robust activity-dependent synaptic formation, plasticity, and remodeling [Rakhade and Jensen, 2009]. Glutamate is the major excitatory neurotransmitter in the CNS, while gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter. There is considerable and growing evidence from animal models and human tissue studies that neurotransmitter receptors are highly developmentally regulated [Sanchez and Jensen, 2001; Rakhade and Jensen, 2009; Johnston, 1995] (Figure 16-1). Studies of cell morphology, myelination, metabolism and, more recently, neurotransmitter receptor expression suggest that the first 1–2 weeks of life in the rodent constitute an analogous stage to the human neonatal brain, and Figure 16-1 shows that both species reveal similar patterns of changes in neurotransmitters during this stage.
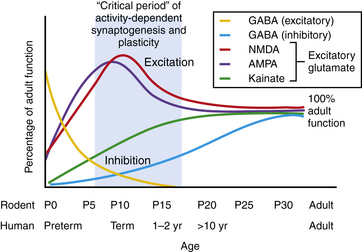
Enhanced Excitability of the Neonatal Brain
Glutamate receptors are critical for plasticity and are transiently overexpressed during development in animal models and human tissue studies. A relative overexpression of certain glutamate receptor subtypes in rodent and human developing cortex coincides with the ages of increased seizure susceptibility (see Figure 16-1) [Sanchez and Jensen, 2001; Talos et al., 2006b; Sanchez et al., 2001]. Glutamate receptors include ligand-gated ion channels, permeable to sodium, potassium, and in some cases calcium, and metabotropic subtypes [Hollmann and Heinemann, 1994]. They are localized to synapses and extrasynaptic sites on neurons, and are also expressed on glia. The ionotropic receptor subtypes are classified based on selective activation by specific ligands:



NMDA receptors are heteromeric, including an obligate NR1 subunit, and their structure is developmentally regulated. In the immature brain, the NR2 subunits are predominantly those of the NR2B subunit type, with the functional correlate of longer current decay time compared to the NR2A subunit, which is the form expressed in later life on mature neurons [Jiang et al., 2007]. Other developmentally regulated subunits with functional relevance include the NR2C, NR2D, and NR3A subunits. Rodent studies show that these are all increased in the first 2 postnatal weeks, and this is associated with lower sensitivity to magnesium, the endogenous receptor channel blocker that in turn results in increased excitability (Figure 16-1 and Figure 16-2) [Hollmann and Heinemann, 1994; Wong et al., 2002]. NMDA receptor antagonists administered to immature rat pups are highly effective against a variety of hypoxic/ischemic insults and seizures in the immature brain [Stafstrom et al., 1997; Chen et al., 1998; Mares and Mikulecka, 2009]. However, the clinical potential of NMDA antagonists may be limited due to their severe sedative effects and a propensity possibly to induce apoptotic death in the immature brain [Ikonomidou et al., 1999; Bittigau et al., 2003]. Importantly, memantine, an agent currently in clinical use as a neuroprotectant in Alzheimer’s disease, may be an exception with fewer side effects, owing to its use-dependent mechanism of action [Chen et al., 1998; Manning et al., 2008; Mares and Mikulecka, 2009].
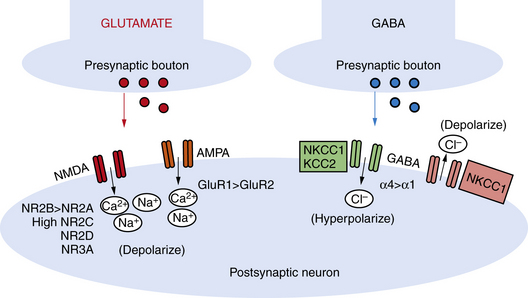
While the NMDA receptor is selectively activated in processes related to plasticity and learning, the AMPA subtype of glutamate receptor subserves most fast excitatory synaptic transmission. In addition, unlike the NMDA receptor, most AMPA receptors are not calcium-permeable in the adult. AMPA receptors are heteromeric and consist of four subunits, including combinations of the GluR1, GluR2, GluR3, or GluR4 subunits [Hollmann and Heinemann, 1994]. However, in the immature rodent and human brain, AMPA receptors are calcium-permeable because they lack the GluR2 subunit (see Figure 16-2) [Sanchez et al., 2001; Kumar et al., 2002]. AMPA receptor subunits are developmentally regulated, with GluR2 expressed only at low levels until the third postnatal week in rodents and later in the first year of life in human cortex [Talos et al., 2003, 2006b]. Hence AMPA receptors in the immature brain, owing to their enhanced calcium permeability, may play an important role in contributing not only to excitability but also to activity-dependent signaling downstream of the receptor. Both NMDA and AMPA receptors are expressed at levels and with subunit composition that enhance excitability of neuronal networks around term in the human and in the first 2 postnatal weeks in the rodent (see Figure 16-2).
Rodent studies show that AMPA receptor antagonists are potently effective against neonatal seizures, even superior to NMDA receptor antagonists or conventional AEDs and GABA agonists. Topiramate, which is FDA-approved for the treatment of seizures in children and adults, is an AMPA receptor antagonist, in addition to several other potential anticonvulsant mechanisms [Shank et al., 2000]. Topiramate is effective in suppressing seizures and long-term neurobehavioral deficits in a rodent neonatal seizure model, even when administered following seizures [Koh and Jensen, 2001; Koh et al., 2004]. In addition, topiramate in combination with hypothermia was neuroprotective in a rodent neonatal hypoxia-ischemia model [Liu et al., 2004]. Finally, the specific AMPA receptor antagonist talampanel, currently in phase II trials for epilepsy in children and adults, as well as for amyotrophic lateral sclerosis, was recently shown to protect against neonatal seizures in a rodent model [Aujla et al., 2009].
Decreased Efficacy of Inhibitory Neurotransmission in the Immature Brain
Expression and function of the inhibitory GABAA receptors are also developmentally regulated. Rodent studies show that GABA receptor binding, synthetic enzymes, and overall receptor expression are reduced early in life compared to later [Sanchez and Jensen, 2001; Swann et al., 1989]. GABA receptor function is regulated by subunit composition, and the α4 and α2 subunits are relatively overexpressed in the immature brain compared to the α1 subunit (see Figure 16-2) [Brooks-Kayal et al., 1998]. Notably, when the α4 subunit is expressed, the receptor is less sensitive to benzodiazepines compared to receptors containing α1 [Kapur and Macdonald, 1999], and as is often the case clinically, seizures in the immature rat respond poorly to benzodiazepines [Jensen et al., 1995; Swann et al., 1997].
Receptor expression and subunit composition can partially explain the resistance of seizures in the immature brain to conventional AEDs that act as GABA agonists. However, inhibition of neuronal excitability via GABA agonists relies on the ability of GABAA receptors to cause a net influx of chloride (Cl−) from the neuron, resulting in hyperpolarization [Dzhala and Staley, 2003]. In the immature rodent forebrain, GABA receptor activation can cause depolarization rather than hyperpolarization [Khazipov et al., 2004; Loturco et al., 1995; Owens et al., 1996]. GABAA-mediated depolarization occurs because the chloride (Cl−) gradient is reversed in the immature brain: chloride (Cl−) levels are high in the immature brain due to a relative underexpression of the Cl− exporter KCC2 compared to the mature brain (see Figure 16-1 and Figure 16-2) [Dzhala et al., 2005]. Recent studies in human brain have shown that KCC2 is virtually absent in cortical neurons until late in the first year of life, and gradually increases, while the Cl− importer NKCC1 is overexpressed in the neonatal human brain and during early life in the rat when seizures are resistant to GABA agonists [Dzhala et al., 2005]. The NKCC1 inhibitor, bumetanide, shows efficacy against kainate-induced seizures in the immature brain [Dzhala et al., 2008]. This agent, already FDA-approved as a diuretic in neonates, is currently under investigation in a phase I/II clinical trial as an add-on agent for the treatment of neonatal seizures (www.clinicaltrials.gov; trial ID: NCT00830531).
Ion Channel Configuration Favors Depolarization in Early Life
Ion channels also regulate neuronal excitability and, like neurotransmitter receptors, are developmentally regulated. Mutations in the K+ channels KCNQ2 and KCNQ3 are associated with benign familial neonatal convulsions [Cooper and Jan, 2003]. These mutations interfere with the normal hyperpolarizing K+ current that prevents repetitive action potential firing [Yue and Yaari, 2004]. Hence, at the time when there is an overexpression of GluRs and incomplete network inhibition, a compensatory mechanism is not available in these mutations. Another K+-channel superfamily member, the HCN (or h) channels, is also developmentally regulated. The h currents are important for maintenance of resting membrane potential and dendritic excitability [Pape, 1996], and their function is regulated by isoform expression. The immature brain has a relatively low expression of the HCN1 isoform, which serves to reduce dendritic excitability in the adult brain [Bender et al., 2001]. Ion channel maturation can thus also contribute to the hyperexcitability of the immature brain and can also have a cumulative effect when occurring in combination with the aforementioned differences in ligand-gated channels. Recently, selective blockers of HCN channels have been shown to disrupt synchronous epileptiform activity in the neonatal rat hippocampus [Bender et al., 2005], suggesting that these developmentally regulated channels may also represent a target for therapy in neonatal seizures. Both N- and P/Q-type voltage-sensitive calcium channels regulate neurotransmitter release [Iwasaki et al., 2000]. With maturation, this function is exclusively subsumed by the P/Q-type channels, formed by Cav2.1 subunits, a member of the Ca2+ channel superfamily [Noebels, 2003]. Mutations in Cav2.1 may be involved in absence epilepsy, suggesting a failure in the normal maturational profile [Chen et al., 2003].
A Role for Neuropeptides in the Hyperexcitability of the Immature Brain
Neuropeptide systems are also dynamically fluctuating in the perinatal period. An important example is corticotropin releasing hormone (CRH), which elicits potent neuronal excitation [Baram and Hatalski, 1998; Ju et al., 2003]. CRH and its receptors are expressed at higher levels in the perinatal period, specifically in the first 2 postnatal weeks in the rat, than later in life [Brunson et al., 2001b]. CRH levels increase during stress, and seizure activity in the immature brain may exacerbate subsequent seizure activity. Notably, adrenocorticotropic hormone, which has demonstrated efficacy in infantile spasms, also downregulates CRH gene expression [Brunson et al., 2001a]. Neuropeptide modulation may be an area of future clinical import in developing novel treatments for neonatal seizure.
Enhanced Potential for Inflammatory Response to Seizures in the Immature Brain
Neonatal seizures can occur in the setting of inflammation, either due to an intercurrent infection or secondary to hypoxic-ischemic injury. Experimental and clinical evidence exists for early microglial activation and inflammatory cytokine production in the developing brain when hypoxia-ischemia [Ivacko et al., 1996; Dommergues et al., 2003] or inflammation [Saliba and Henrot, 2001; Debillon et al., 2003] occurs. Importantly, microglia are highly expressed in immature white matter in rodents and humans during cortical development [Billiards et al., 2006]. Anti-inflammatory compounds or agents that inhibit microglial activation, such as minocycline, attenuate neuronal injury in some models of excitotoxicity and hypoxia-ischemia [Tikka et al., 2001]. During the term period, microglia density in deep gray matter is higher than at later ages; that is likely due to migration of the population of cells en route to more distal cortical locations. Experimental models demonstrate microglia activation, as seen by morphologic changes and rapid production of proinflammatory cytokines, occurring after acute seizures in different epilepsy animal models [Shapiro et al., 2008; Vezzani et al., 2008]. During development, microglia show maximal density simultaneous with the period of peak synaptogenesis [Dalmau et al., 2003]. During normal development, as well as in response to injury, microglia participate in “synaptic stripping” by detaching presynaptic terminals from neurons [Pfrieger and Barres, 1997; Stevens et al., 2007]. Importantly, the microglial inactivators, minocycline and doxycycline, are protective against seizure-induced neuronal death [Heo et al., 2006] and also protective in some neonatal stroke models [Jantzie et al., 2005; Lechpammer et al., 2008].
Selective Neuronal Injury in the Developing Brain
While many studies suggest that seizures, or status epilepticus, induce less death in the immature than in the adult brain, there is evidence that some neuronal populations are vulnerable. Similar to the sensitivity of subplate neurons, hippocampal neurons in the perinatal rodent undergo selective cell death, as well as oxidative stress, following chemoconvulsant-induced cell death [Wasterlain et al., 2002]. Stroke studies in neonatal rodents also suggest that there can be selective vulnerability of specific cell populations in early development [Stone et al., 2008]. Subplate neurons are present in significant numbers in the deep cortical regions during the preterm and neonatal period [Kinney et al., 2004]. These neurons are critical for the normal maturation of cortical networks [Lein et al., 1999; Kanold et al., 2003]. Importantly, in both humans and rodents, these cells possess high levels of both AMPA receptors and NMDA receptors [Talos et al., 2006a, b]. These cells may also lack oxidative stress defenses present in mature neurons. Animal models have revealed that these neurons are selectively vulnerable compared to overlying cortex following an hypoxic-ischemic insult [McQuillen et al., 2003]. Indeed, chemoconvulsant-induced seizures in rats, provoked by the convulsant kainate in early postnatal life, produced a similar loss of subplate neurons with consequent abnormal development of inhibitory networks [Lein et al., 1999].
A number of studies have shown that the application of clinically available antioxidants, such as erythropoietin, is protective against neuronal injury in neonatal stroke [Chang et al., 2005; Gonzalez et al., 2007]. Recently, erythropoietin was shown to reduce later increases in seizure susceptibility of hippocampal neurons following hypoxia-induced neonatal seizures in rats [Mikati et al., 2007].
Seizure-Induced Neuronal Network Dysfunction: Potential Interaction Between Epileptogenesis and Development of Neurocognitive Disability
Given that there is minimal neuronal death in most models of neonatal seizures, the long-term outcome of neonatal seizures is thought to be due to seizure-induced alterations in surviving networks of neurons. Evidence for this theory comes from several studies that reveal disordered synaptic plasticity and impaired long-term potentiation, as well as learning later in life, in rodents following brief neonatal seizures [Sayin et al., 2004; Ben Ari and Holmes, 2006]. The neonatal period represents a stage of naturally enhanced synaptic plasticity when learning occurs at a rapid pace [Silverstein and Jensen, 2007; Maffei and Turrigiano, 2008]. A major factor in this enhanced synaptic plasticity is the predominance of excitation over inhibition, which also increases susceptibility to seizures, as mentioned above. However, seizures that occur during this highly responsive developmental window appear to access signaling events that are central to normal synaptic plasticity. There are rapid increases in synaptic potency that appear to mimic long-term potentiation, and this pathologic activation may contribute to enhanced epileptogenesis [Rakhade et al., 2008]. In addition, GluR-mediated molecular cascades associated with physiological synaptic plasticity may be overactivated by seizures, especially in the developing brain [Cornejo et al., 2007; Rakhade et al., 2008]. Rodent studies demonstrate a reduction in synaptic plasticity in neuronal networks such as hippocampus following seizures early in life, suggesting that the pathologic plasticity may have occluded normal plasticity, and this mechanism could contribute to the impaired learning [Rakhade et al., 2008]. Many models reveal that neonatal seizures alter synaptic plasticity [Stafstrom et al., 2006], and recent studies are delineating the molecular signaling cascades that are altered following early-life seizures [Sanchez et al., 2005a; Raol et al., 2006]. In addition to glutamate receptors, inhibitory GABAA receptors can also be affected by seizures in early life, resulting in long-term functional impairments. Early and immediate functional decreases in inhibitory GABAergic synapses mediated by post-translational changes in GABAA subunits are seen following hypoxia-induced seizures in rat pups [Sanchez et al., 2005a]. Flurothyl-induced seizures result in a selective impairment of GABAergic inhibition within a week [Isaeva et al., 2006]. Importantly, there is evidence that some of these changes may be downstream of Ca2+ permeable glutamate receptors and Ca2+ signaling cascades, and that early postseizure treatment with GluR antagonists or phosphatase inhibitors may interrupt these pathologic changes that underlie the long-term disabilities and epilepsy [Sanchez et al., 2005a; Rakhade et al., 2008].
Diagnosis
Neonatal seizures can be difficult to diagnose, as there are often no clinical correlates of the electrographic seizures, a phenomenon called electroclinical dissociation. Regional interconnectivity, including interhemispheric as well as corticospinal, is not fully mature due to incomplete myelination of white matter tracts, leading to only modest behavioral manifestations of these seizures. Infants can show no signs or very subtle tonic or clonic movements, often limited to only one limb, making the diagnosis difficult to discern from myoclonus or other automatisms [Mizrahi and Kellaway, 1998]. A recent study revealed that approximately 80 percent of EEG-documented seizures were not accompanied by observable clinical seizures [Clancy, 2006a]. Hence, EEG is essential for diagnosis and for assessing treatment efficacy in this group (see Chapter 12). Full 20-lead EEGs are most sensitive in detecting these, often multifocal, seizures (Figure 16-3). As full-lead EEGs can be difficult to obtain on an emergent basis in many neonatal intensive care units, amplitude-integrated EEG (aEEG) devices are becoming increasingly utilized. aEEG is usually obtained from a pair or limited number of leads and is displayed as a fast Fourier spectral transform. With aEEG, seizures are detected by acute alterations in spectral width, and a raw EEG from the single channel can be accessed by the viewer for confirmation [Lawrence et al., 2009]. Several reports now indicate that aEEG has relatively high specificity but compromised sensitivity, detecting approximately 75 percent of that of conventional full-lead montage EEG [Tekgul et al., 2005a, b; Clancy, 2006b; Navakatikyan et al., 2006; Shellhaas and Clancy, 2007; Shellhaas et al., 2007; de Vries and Toet, 2006]. Chapter 12 discusses in detail the ontogeny of EEG development, the types of EEG patterns that can occur, and characteristic EEG abnormalities reported in various neonatal disorders.
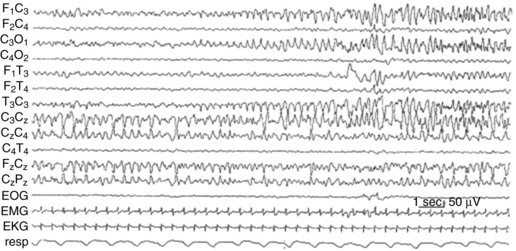
Fig. 16-3 Electroencephalographic appearance of neonatal seizures.
(Reprinted with permission from Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology 1987;37:1837–1844.)
Once neonatal seizures are confirmed, treatable metabolic, genetic, or symptomatic causes need to be identified. Serologic studies include blood and serologic studies to assess for systemic infection, and metabolic derangements such as electrolyte disturbances, acidosis and hypoglycemia. The timing of seizures can be a helpful indicator, such as in the case of “fifth day fits” or seizures due to hypocalcemia. Pyridoxine-dependent seizures present as refractory early neonatal seizures that uniquely respond to pyridoxine administration [Baxter, 2001; Grillo et al., 2001]. Seizures that continue to be refractory in the setting of a history consistent with hypoxic-ischemic encephalopathy (HIE) manifest within the first 24–48 hours of life, and persist over several days, then may gradually remit. Chapter 20 reviews metabolic disorders that can cause neonatal seizures.
Brain imaging is often the next step in the diagnostic evaluation. The type of brain imaging acquired depends on the clinical setting, institutional resources, and the infant’s medical status. In premature infants, who are at greatest risk for intraventricular hemorrhage, cranial ultrasound is often the most appropriate initial neuroimaging modality. Computed tomography (CT) can provide useful complementary radiological information, but magnetic resonance imaging (MRI) may yield the most information concerning the etiology of neonatal seizures (see Chapter 11). Figure 16-4 illustrates how MRI, in some cases complemented by MR spectroscopy, can be informative with regard to the etiology of seizures.
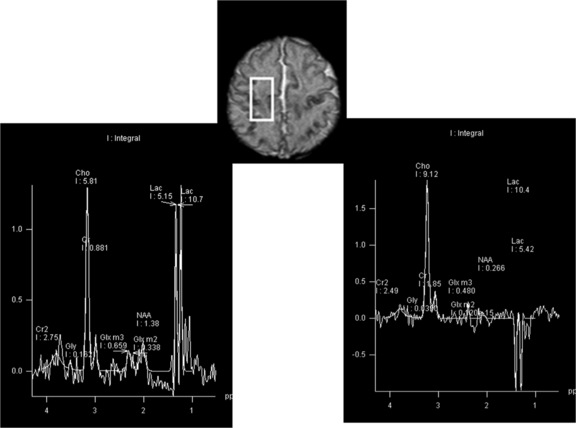
Fig. 16-4 An MRI performed on day 10.
(Images were generously provided by Drs. K. Poskitt and S. Miller, University of British Columbia.)
MRI provides an important assessment of risk in infants with neonatal seizures. Imaging can identify cerebral dysgenesis and gross structural malformations that can be associated with neonatal seizures, such as that seen in association with tuberous sclerosis, hemimegalencephaly, or cortical dysplasias. For symptomatic seizures due to hypoxia-ischemia, abnormal T2, fluid-attenuated inversion-recovery (FLAIR) and diffusion-weighted imaging can localize regional injury and determine its severity [Grant and Yu, 2006]. MR spectroscopy can provide data estimating the severity of neuronal injury and may be an important prognostic indicator. A previous study has shown that in neonates with hypoxic-ischemic injury and seizures, an increased lactate to choline ratio, as well as reduced N-acetyl-aspartate ratios, was more abnormal in patients with a greater seizure burden [Miller et al., 2005]. Another study of term infants with asphyxia and/or seizures [Glass et al., 2009] demonstrated that, after adjusting for the degree of MRI abnormality, seizure severity was associated with a higher risk of neuromotor abnormalities at 4 years of age than in those without seizures. These results suggest that neonatal seizures may independently be associated with a poorer outcome, even when MRI lesions are present.
Etiology
Box 16-1 summarizes the general categories of disorders that can cause neonatal seizures. A step-wise diagnostic approach to determine the etiology of neonatal seizures will prioritize identification of treatable causes; however, clinical judgment and resource availability, among many factors, will influence the sequence, extent, and rapidity of testing decisions. Assessment of the cause(s) and initiation of treatment(s) should occur concurrently. The cause of neonatal seizures is sometimes easily determined and in these cases little additional laboratory testing may be needed. Neurodiagnostic testing should proceed in a step-wise fashion, geared initially to confirm the diagnosis of seizures (discussed in the preceding section) and to identify disorders with specific effective treatments. The highest priorities are the identification of reversible, acute metabolic derangements and treatable infections. In neonates with unexplained seizures and depressed mental status, a lumbar puncture is an essential component of the initial diagnostic evaluation. Interpretation of cerebrospinal fluid abnormalities and the range of bacterial, viral, and other pathogens that cause neonatal CNS infections and their treatments are discussed in Chapters 10 and 80–82.
It is important to be cognizant of rare, potentially treatable conditions that can present with neonatal seizures (e.g. pyridoxine-dependent seizures), particularly if the seizures do not respond to conventional AED treatment and the underlying cause is not readily identified. In some cases (if seizures persist and are associated with unexplained encephalopathy), continuing the neurodiagnostic evaluation is warranted, including consideration of very rare disorders – identification of which may help illuminate prognosis and/or genetic recurrence risk. The general principles discussed in Chapter 20 on perinatal metabolic encephalopathies are applicable to this diagnostic approach.
Acute Metabolic Abnormalities
Other acute metabolic derangements that can result in neonatal seizures are withdrawal syndromes associated with maternal therapeutic or illicit drug use. Rarely, neonatal seizures can result from abrupt withdrawal from a drug that was administered therapeutically to the mother; for example, otherwise unexplained seizures were described in a neonate whose mother was being treated for spasticity with baclofen [Ratnayaka et al., 2001]. Convulsions are an uncommon but well-recognized complication of the neonatal abstinence syndrome that is attributable to withdrawal from maternally administered opiates [Kaltenbach and Finnegan, 1986]; other neurological findings include frequent myoclonic jerks, tremor, irritability, poor sleep, a high-pitched cry, and autonomic instability. The combination of a relevant maternal history and associated clinical findings raises the index of suspicion for this etiology.
Exposure to anesthetics such as lidocaine and mepivacaine, accidentally injected into the fetal scalp with administration of local anesthesia during labor, has been described as a cause of neonatal seizures [Hillman et al., 1979; Kim et al., 1979].
Rare Inborn Errors of Metabolism and Genetic Disorders
There is an ever-expanding list of very rare metabolic and other genetic disorders that can present with neonatal seizures. Of particular clinical importance are those disorders for which specific biochemically targeted treatments are available (reviewed in Pearl [2009]). The prototypical disorder in this category is pyridoxine-dependent epilepsy (PDE) due to antiquitin deficiency (see Chapter 52). The clinical syndrome of refractory neonatal seizures that rapidly abated with intravenous administration of pyridoxine was well described for many years. The underlying genetic abnormality, alpha-aminoadipic semialdehyde dehydrogenase (antiquitin) deficiency, was recently delineated, and it was subsequently reported that folinic acid-dependent seizures were allelic with pyridoxine dependency. These disorders can be diagnosed by measurement of alpha-aminoadipic semialdehyde in urine; alternatively, seizure cessation in response to an empiric trial of intravenous pyridoxine with EEG (and EKG) monitoring can provide the initial diagnosis. A related condition, pyridox(am)ine-5′-phosphate oxidase (PNPO) deficiency, also presents in neonates with a severe epileptic encephalopathy and is responsive to pyridoxal phosphate but not to pyridoxine. Consideration must be given in those neonates who have medically refractory epilepsy and who do not respond to pyridoxine, to using pyridoxal phosphate, which is not readily available for clinical use. Serine-dependent seizures (see Chapters 20 and 32) and glucose transporter deficiency (see Chapters 20 and 34) are similarly very rare causes of neonatal seizures that have specific treatments.
One of the best-characterized inborn errors of metabolism that can present with refractory neonatal seizures is glycine encephalopathy (nonketotic hyperglycinemia) [Hoover-Fong et al., 2004]. This disorder is caused by a defect in the glycine cleavage system; typical features in affected neonates include apnea, hypotonia, and persistent seizures. The diagnosis is established by detection of marked elevation in cerebrospinal fluid glycine concentrations, and more specifically, elevated ratio of cerebrospinal fluid:plasma glycine concentration (>0.08). Seizures may be attributable to overactivation of N-methyl-D-aspartate-type excitatory amino acid receptors (at a glycine regulatory site), and in some cases treatment with dextromethorphan, a clinically available weak NMDA antagonist, along with sodium benzoate, can help control refractory seizures in affected infants.
Peroxisomal biogenesis disorders (Zellweger’s syndrome, neonatal adrenoleukodsytrophy, infantile Refsum’s syndrome) can also present with persistent neonatal seizures and severe encephalopathy; there are typically associated subtle dysmorphic features (see Chapter 38) that prompt consideration of these disorders. The initial diagnostic testing includes measurement of serum very long chain fatty acids.
There are also other neurometabolic disorders that elude even the most advanced newborn screening assays and that can result in persistent neonatal seizures. Molybdenum co-factor deficiency, a very rare inherited metabolic disorder in which there is combined deficiency of aldehyde oxidase, xanthine dehydrogenase, and sulfite oxidase, typically presents soon after birth with intractable seizures; isolated sulfite oxidase deficiency is similarly very rare, and can also present with intractable neonatal seizures. An interesting feature of these two disorders is that their clinical manifestations, as well as neuroimaging features, can mimic hypoxic-ischemic injury, and this may lead to misdiagnosis and delayed awareness of genetic recurrence risks [Eichler et al., 2006]. However, neuroimaging with proton MR spectroscopy can provide important diagnostic clues [Basheer et al., 2007]. Proton MR spectroscopy demonstrates profound metabolic abnormalities with very high lactate peaks (see Figure 16-4), and MRI may demonstrate evolution of cystic white matter damage.
Benign Familial Neonatal Convulsions
These genetic disorders, typically inherited in an autosomal-dominant fashion, present with early neonatal seizures that typically remit within a few months of life, and are commonly associated with good neurological outcomes (see Chapter 52). Several mutations of potassium channel genes KCNQ2 and KCNQ3 have been identified in affected individuals [Coppola et al., 2003, 2006; Singh et al., 2003]. Another benign syndrome possibly associated with a mutation in KCNQ2 is that of “fifth day fits,” which transiently occur for a day or so around the fifth or sixth postnatal day [Claes et al., 2004]. Berkovic et al. have also identified mutations in the sodium channel gene SCN2A in families with autosomal-dominant “benign familial neonatal-infantile seizures.” In these families, seizures began as early as day 2, but also as late as 7 months of age; in all reported cases, the seizures stopped by age 12 months [Berkovic et al., 2004].
Infection
Infections of the CNS, acquired prenatally, can also present in the neonatal period with seizures. These diverse infections, collectively classified as “TORCH” infections, are suspected in neonates with intracranial calcifications, unexplained hydrocephalus, and specific disease-associated systemic abnormalities. These are discussed in Chapters 80–82.
Hypoxic-Ischemic Encephalopathy
Term infants with neonatal encephalopathy as a result of hypoxia-ischemia are at particularly high risk for seizures (see Chapter 17). In two recent clinical trials of hypothermia for treatment of neonatal HIE, seizures were frequent at the time of randomization (40–60 percent) [Gluckman et al., 2005; Shankaran et al., 2005]. Clinically apparent seizures typically begin 6–12 hours after the asphyxial event; the pathophysiological mechanisms underlying this lag period are uncertain. Seizures are often difficult to control with medication for 2–3 days, and then often wane.
A subject of considerable controversy is whether seizures contribute to the progression of hypoxic-ischemic brain injury, and whether more rapid and effective control of seizures could improve neurodevelopmental outcome. Several studies provide evidence that seizures do, in fact, contribute to poor outcomes in affected neonates; however, to date, there is no evidence that seizure treatment is of long-term benefit [Booth and Evans, 2004].
Cerebrovascular Disorders
There is increasing recognition of the broad range of cerebrovascular pathology in neonates, and seizures are often the initial symptom of ischemic or hemorrhagic brain injury. Neonatal cerebrovascular disorders include arterial and venous infarction, and intracranial hemorrhage (intraparenchymal, intraventricular, subarachnoid, and/or subdural). These disorders are discussed in Chapter 18 and 19.
Intraparenchymal hemorrhages, as a result of trauma, coagulopathy, vascular malformation, or unknown causes, may also present with seizures. There is an unusual reported association between temporal lobe hemorrhages and apneic seizures in full-term neonates [Sirsi et al., 2007]. Progression of intraventricular hemorrhage may provoke seizures in premature infants, sometimes in association with periventricular hemorrhagic infarction.
Congenital Heart Disease
Neonates with complex congenital heart disease are at high risk for seizures. Risk factors include coexistent CNS developmental anomalies, pre- and postnatal cerebrovascular compromise, hypoparathyroidism with DiGeorge syndrome complex, complications of cardiac catheterization including therapeutic interventions such as atrial septostomy, cardiopulmonary bypass intraoperatively, and diverse postoperative complications. These are reviewed in Chapter 101.
In the acute postoperative period, accurate seizure detection requires electrophysiological monitoring. In a study of 183 consecutive infants undergoing heart surgery who were monitored by video-EEG for 48 hours postoperatively, electrographic neonatal seizures occurred in 21 (11.5 percent); none had clinically visible seizure, while some of the affected infants had numerous seizures [Clancy et al., 2005]. Whether more rapid and effective treatment of seizures would improve neurodevelopmental outcome in these infants is an important unanswered question.
Developmental Disorders
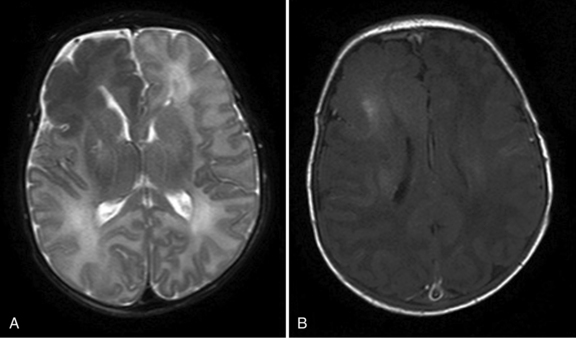
Many types of cerebral dysgenesis present with neonatal seizures. Infants with CNS developmental anomalies may tolerate labor and delivery poorly, and initially, their seizures and encephalopathy may be attributable to perinatal asphyxia. Often, it is persistence of seizures or atypical manifestations (e.g., sustained unilateral focal motor seizures) that raise suspicion of underlying neurodevelopmental anomalies, and these are best delineated with MRI (as illustrated in Figure 16-5). These disorders are discussed in Part IV.
Treatment
In neonates, single seizures are unusual, and recurrence is expected. Single seizures are treated in a similar fashion to status epilepticus in older age groups, i.e., with rapid intravenous administration of a full loading dose of an AED, most commonly phenobarbital (20–30 mg/kg) [Bartha et al., 2007; Blume et al., 2009]. There is no consensus regarding the optimal second-line therapy, if seizures persist. Options include additional phenobarbital, phenytoin or fosphenytoin, lorazepam or midazolam, and lidocaine (Table 16-1).
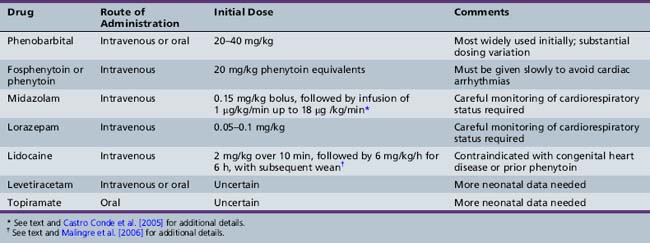
Systematic assessment of AED efficacy for treatment of neonatal seizures is extremely challenging and very limited data are available. There is consensus that such studies need to incorporate continuous EEG monitoring, both for confirmation of the diagnosis and also for assessment of the efficacy of treatment [Silverstein et al., 2008]. Studies that include evaluation of long-term outcomes also require selection of uniform or well-matched populations with respect to gestation, gender, and neurological and systemic disease. The classic study of Painter and colleagues compared the efficacy of loading doses of phenobarbital and phenytoin for treatment of EEG-confirmed seizures in term infants [Painter et al., 1999]. Their major findings were that these drugs were equally but incompletely effective, that either drug alone controlled seizures in fewer than half of the patients, and that adding the second drug conferred some additional benefit (overall rate of seizure control was about 60 percent). Although the two agents are equipotent, phenobarbital is used much more widely in neonatal intensive care units. In many centers, fosphenytoin has replaced phenytoin as the second-line intravenous drug, since it is considered much less likely to cause local injury if there is tissue extravasation.
Benzodiazepines are often used for treatment of refractory seizures. Hypotension and respiratory depression may complicate treatment, and midazolam has gained more widespread usage in intensive care unit settings because of its short half-life. Variable efficacy (ranging from minimal to 100 percent) has been reported for midazolam as a second-line or third-line medication. A recent study reported that in 10 of 13 neonates with EEG-confirmed status epilepticus that had been refractory to conventional therapy (40 mg/kg phenobarbital ± phenytoin 20 mg/kg), treatment with midazolam infusion was effective within an hour [Castro Conde et al., 2005]. A favorable response to treatment was defined as no more than two electrical seizures per hour lasting less than 30 seconds. Their midazolam dose was an initial intravenous bolus of 0.15 mg/kg, followed by continuous infusion (1 µg/kg/min), increasing by 0.5–1 µg/kg/min every 2 minutes until a favorable response or a maximum of 18 µg/kg/min was attained; if status epilepticus persisted after the initial bolus, another bolus of 0.10–0.15 mg/kg was administered 15–30 minutes later.
Lidocaine infusion can also be effective for treatment of refractory neonatal seizures, but concerns about cardiac toxicity have limited widespread adoption of this agent. A recent European study provided a detailed protocol [Malingre et al., 2006] that recommended a loading dose of 2 mg/kg in 10 minutes, followed by the continuous infusion of 6 mg/kg per hour for 6 hours, 4 mg/kg per hour for 12 hours, and finally 2 mg/kg per hour for 12 hours. The authors provided specific guidelines for its usage, including that it only be used in a neonatal intensive care unit with continuous cardiac monitoring, that the infusion be stopped if any cardiac arrhythmia develops, that it be used only for seizures that have not responded to phenobarbital and midazolam, and that it not be used in infants with congenital heart disease or prior treatment with phenytoin.
Metabolic Therapies
Administration of pyridoxine is often considered in neonates with unexplained refractory seizures. There are no widely accepted clinical criteria that mandate this treatment trial and practice varies. Intravenous administration of 100 mg pyridoxine with continuous EEG and cardiac monitoring should result in a marked attenuation of epileptiform activity within 15 minutes in responders; doses as high as 500 mg have been suggested. Rarely, treatment is associated with adverse effects, including onset of respiratory depression up to a day later [Pearl, 2009]. Of interest, a recent report described the novel strategy of antenatal pyridoxine supplementation in a pregnant woman with a family history of pyridoxine-dependent neonatal seizures [Bok et al., 2010].
Although recent work indicates genetic overlap between pyridoxine-responsive and folinic acid-responsive neonatal seizures, infants who respond preferentially to folinic acid (2.5–5 mg/day) have been described. There are recently described cases of mutations in the pyridox(am)ine-5′-phosphate oxidase gene, in which affected individuals presented with neonatal seizures unresponsive to pyridoxine and anticonvulsant treatment but responsive to pyridoxal phosphate. Pyridox(am)ine-5′-phosphate oxidase converts pyridoxine phosphate and pyridoxamine phosphate to pyridoxal phosphate, which is the biologically active form [Bagci et al., 2008].
Chronic Therapy
An important unresolved issue is whether chronic AED therapy is warranted in neonates. In many term neonates whose seizures are attributable to acute brain injury, seizures stop by the end of the first week of life. There is less information available about the natural history of seizures in premature infants. Although there is a significant risk for development of epilepsy after neonatal seizures, there is no evidence that AED therapy modifies this risk. None the less, continuation of AED therapy after hospital discharge is common [Bartha et al., 2007].
Hellstrom-Westas et al. evaluated the risk of seizure recurrence within the first year of life in 31 neonates for whom antiepileptic treatment was discontinued after 1–65 days (median 4.5 days) [Hellstrom-Westas et al., 1995]. Seizures recurred in only three – one infant receiving prophylaxis, one treated for 65 days, and one infant treated for six days – and in this small group, no clinical features were predictive of recurrence. A retrospective study that attempted to evaluate whether chronic phenobarbital treatment influenced seizure recurrence or neurological development found no benefit [Guillet and Kwon, 2007]. These investigators are now undertaking a prospective study to address this issue (www.clinicaltrials.gov; trial ID: NCT01089504).
The Interaction Between Anticonvulsants and the Developing Brain
Emerging identification of age-specific mechanisms for neonatal seizures is pointing to the use of novel therapeutic targets. Caution must be exercised when devising new therapies, as the target may indeed be essential for normal brain development, albeit a contributor to neuronal hyperexcitability. Over two decades ago, experimental data emerged demonstrating that phenobarbital exposure had adverse effects on survival and morphology of cultured neurons, derived from fetal mouse tissue, and these observations raised concerns about risks of this drug for treatment of neonatal seizures [Bergey et al., 1981; Serrano et al., 1988]. Subsequent studies in neonatal rats demonstrated that daily treatment with phenobarbital or diazepam in the first postnatal month resulted in measurable changes in regional cerebral metabolism and behavior [Pereira et al., 1990; Schroeder et al., 1995].
More recently, evidence emerged that brief systemic treatment with conventional AEDs, such as phenobarbital, diazepam, phenytoin, and valproate, increases apoptotic neuronal death in normal immature rodents [Bittigau et al., 2002]. Similarly, NMDA receptor antagonists also induce an increase in constitutive apoptosis in the developing rodent brain [Ikonomidou et al., 1999]. Yet, the AMPA receptor antagonists NBQX and topiramate do not cause such adverse effects [Ikonomidou et al., 1999; Glier et al., 2004], although the mechanism for this relative safety over the other agents is not understood. The novel AED levetiracetam also has no effect on apoptosis in the developing brain [Manthey et al., 2005].
Prognosis
Holden et al. (1982) reviewed the prognosis of 277 newborns with seizures who were enrolled in the National Collaborative Perinatal Project (data collected about 50 years ago) [Holden et al., 1982]; gestational age was less than 36 weeks in about 30 percent. There was 35 percent mortality, and 30 percent of survivors had adverse neurological outcomes at age 7 years; these included cerebral palsy (13 percent), intelligence quotient less than 70 (19 percent), and epilepsy (20 percent), alone or in combination. Bergman et al. [1983] evaluated outcomes in 131 children, aged 1–5 years old, who had neonatal seizures, and were treated in a single intensive care unit from 1976 to 1979. Half were born at less than 37 weeks’ gestation (28 percent at or below 31 weeks). Fifty-one of 131 were normal, 42 had minor to moderate disabilities, 30 had severe disabilities, and 6 had died (2 were lost to follow-up); 26 had epilepsy. Of 77 with neonatal asphyxia and seizures, 41 developed moderate or severe disabilities. Significant neonatal predictors of poor outcome included later onset of seizures, tonic seizures, and seizures lasting for many days. Although seizure frequency and neonatal mortality were greatest in very premature infants, the outcome in survivors did not differ from that in term infants.
In a study of 90 infants with prospectively diagnosed clinical neonatal seizures, born in Newfoundland between 1990 and 1995, trends were similar [Ronen et al., 2007]. Among term infants, 28 of 62 (45 percent) were normal, whereas only 3 of 15 (20 percent) surviving preterm infants were normal. Of survivors, 17 (27 percent) developed epilepsy, 16 (25 percent) had cerebral palsy, 13 (20 percent) had mental retardation, and 17 (27 percent) had learning disorders. Variables associated with poor prognosis were severe encephalopathy, cerebral dysgenesis, complicated intraventricular hemorrhage, infections in preterm infants, abnormal neonatal EEGs, and the need for multiple drugs to treat the neonatal seizures. Pure clonic seizures without facial involvement in term infants suggested favorable outcome, whereas generalized myoclonic seizures in preterm infants were associated with mortality.
Brunquell et al. [2002] performed a retrospective review of 77 cases of neonatal seizures (2.2 percent of their neonatal intensive care unit admissions over 7 years), and also found that clinical seizure semiology (determined from review of medical records) was predictive of outcomes. Neonatal mortality was high (30 percent). Of 53 survivors with mean 3.5-year follow-up, 59 percent had abnormal neurological examinations, 40 percent were mentally retarded, 43 percent had cerebral palsy, and 21 percent had epilepsy. Compared with patients with other seizure types, those with subtle and generalized tonic seizures or two or more seizure types had a higher prevalence of adverse outcomes.
In a recent study of 89 term infants with clinical seizures evaluated at the Children’s Hospital in Boston, neonatal mortality was 7 percent, and long-term neurological outcome was poor in 28 percent. Cerebral dysgenesis and global hypoxia-ischemia were associated with poor outcome. Normal neonatal/early infancy neurological examination and normal/mildly abnormal neonatal EEG were associated with a favorable outcome, particularly if neonatal neuroimaging was normal [Tekgul et al., 2005b, 2006].
A recent study from Italy is noteworthy because enrollment was restricted to infants with video-EEG-confirmed seizures; 106 neonates, admitted to a single neonatal intensive care unit from 1999 to 2004, were enrolled (51 preterm, 55 full-term) [Pisani et al., 2007]. They also evaluated the prognostic significance of neonatal status epilepticus, defined as continuous seizure activity for 30 minutes or recurrent seizures lasting more than 30 minutes without definite return to baseline neurological condition between seizures. At age 24 months, 34 percent were normal, 19 percent had died, and 47 percent had an adverse outcome. Of those with neonatal status epilepticus, 25 of 26 had adverse outcomes. Birth weight, severely abnormal cerebral ultrasound scans, and status epilepticus were independent predictors of abnormal outcome.
A critical unanswered question is whether effective treatment of seizures could improve outcomes. A recent Cochrane analysis concluded that there was no evidence that treatment of neonatal seizures improved outcome; however, this analysis was based on evaluation of conventional AEDs with limited efficacy in neonates [Booth and Evans, 2004]. Moreover, it is evident that the underlying cause is a major determinant both of medical “refractoriness” and of neurodevelopmental outcome. The converse, however, is also true, and the deleterious effects of seizures may be most pronounced in the setting of underlying hypoxic-ischemic brain injury.
Miller et al. used MR spectroscopy to evaluate brain metabolism and integrity in neonates with asphyxia and (clinically detected) seizures [Miller et al., 2002]. This analysis demonstrated that seizures contributed to abnormal metabolism (as measured by the lactate/choline ratio) and tissue injury (as measured by the N-acetyl-aspartate/choline ratio). In neonates with neonatal hypoxic-ischemic brain injury (based on neonatal MRI measures), neonatal seizures (again, clinically detected) were an independent risk factor for adverse neurodevelopmental outcome at age 4 years [Glass et al., 2009]. Similarly, in the CoolCap neonatal asphyxia and hypothermia treatment trial, the absence of seizures was a predictor of better outcome [Wyatt et al., 2007]. Mechanisms that could account for seizure-induced amplification of ischemic injury include increases in brain temperature and metabolic demands, generation of mediators (such as reactive oxygen species) that contribute to tissue damage, and disruption of endogenous protection and repair mechanisms [Silverstein, 2009].
Underlying CNS pathology influences the prognosis of neonatal seizures, but it is also possible that neonatal seizures have complex effects on the impact of and recovery from neonatal brain injury (distinct from amplification of hypoxic-ischemic injury). In a recent study of cognitive outcome after unilateral perinatal ischemic stroke that included longitudinal testing of intellectual and cognitive abilities at two time points in 29 children, one of the most remarkable and unexpected findings was that the presence of seizures had an adverse effect on cognitive outcome. The mechanisms underlying this limitation of plasticity and reorganization after neonatal brain injury remain uncertain, and the effects of seizures and their treatment with AEDs could not be distinguished [Ballantyne et al., 2008].
Future Directions
Although the experimental data regarding the potential efficacy of agents such as bumetanide, topiramate, and levetiracetam are encouraging, the duration of use of these agents may be limited by safety concerns related to their effects on long-term brain development. Animal model trials and human studies must be aligned in order to understand how data obtained from rodent and nonhuman primates predict human responses. Whether early-life seizure models in which there are long-term deleterious effects on learning can be used to determine the effects of AED treatment on cognitive development in humans is uncertain. Indeed, there is now an FDA-approved investigational new drug (IND) for a clinical trial assessing the pharmacokinetics and safety of bumetanide in combination with standard phenobarbital treatment of early neonatal seizures (http://www.clinicaltrials.gov). In this study, neonates who have had a breakthrough seizure after a first loading dose of phenobarbital are enrolled for full-lead EEG monitoring, and receive randomized treatment with a second dose of phenobarbital in combination with bumetanide or vehicle.
New Therapeutic Targets
Refractory neonatal seizures remain a significant clinical problem, and no new treatments for this condition have been introduced for decades. Many new mechanisms and components of neonatal seizures have been uncovered and present important possibilities for novel therapeutic strategies in the population of neonates at risk for acute and long-term neurologic damage from neonatal seizures. Several major classes of agents with possible age-specific effects have emerged and are summarized in Table 16-2. These include modulators of neurotransmitter receptors and ion channels and transporters, anti-inflammatory compounds, neuroprotectants, and antioxidants. Interdisciplinary collaboration between neonatologists and neonatal neurologists is essential for the success of such studies. As basic research reveals new age-specific therapeutic targets, these targets can be validated with analysis of cell-specific gene and protein expression in human autopsy samples. Experimental data regarding the potential efficacy of agents such as bumetanide, topiramate, and levetiracetam are encouraging, but the duration of use of these agents may be limited by safety concerns related to their effects on long-term brain development. Incorporation of continuous EEG monitoring into clinical studies of neonatal seizure therapy will be essential. Seizure cessation is an important therapeutic goal; yet, improved neurodevelopmental outcome is clearly of critical importance.
Table 16-2 Candidate Potential Targets and Therapies from Emerging Experimental and Clinical Literature
| Temporal Profile | Mechanism Targeted | Potential Therapeutic Options |
|---|---|---|
| Acute changes | Immediate early genes | Chromatin acetylation modifiers/histone deacetylation inhibitors (valproate) |
| NMDA receptors | NMDA receptor inhibitors (memantine, felbamate) NR2B-specific inhibitors (ifenprodil) |
|
| AMPA receptors | AMPA receptor antagonists (topiramate, talampanel, GYKI compounds) | |
| NKCC1 chloride transporters | NKCC1 inhibitor (bumetanide – in combination with GABA agonists phenobarbital, benzodiazepines) | |
| GABA receptors | GABA receptor agonists (phenobarbital, benzodiazepines) | |
| Phosphatases (e.g., calcineurin) | Phosphatase inhibitors (FK-506) | |
| Kinases (activation of PKA, PKC, CaMKII, Src kinases, etc.) | Kinase inhibitors (CaMKII inhibitor KN-62, PKA inhibitor KT5720, PKC inhibitor chelerythrine) | |
| Subacute changes | Inflammation | Anti-inflammatory compounds (ACTH), microglial inactivators (minocycline, doxycycline) |
| Neuronal injury | Erythropoietin, antioxidants, NO inhibitors, NMDA receptor antagonists (memantine) | |
| HCN channels | I(h)-blocker ZD7288 | |
| CB1 receptor | CB1 receptor antagonists (SR 14176A, rimonabant) | |
| Chronic changes | Sprouting | Protein synthesis inhibitors (rapamycin, cycloheximide) |
| Gliosis | Anti-inflammatory agents, (Cox-2 inhibitors, minocycline, doxycycline) |
(Reprinted with permission from Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol 2009;5:380–391.)
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aicardi J., Chevrie J.J. Convulsive status epilepticus in infants and children. A study of 239 cases. Epilepsia. 1970;11:187-197.
Aujla P.K., Fetell M., Jensen F.E. Talampanel suppresses the acute and chronic effects of seizures in a rodent neonatal seizure model. Epilepsia. 2009. In Press
Bagci S., Zschocke J., Hoffmann G.F., et al. Pyridoxal phosphate-dependent neonatal epileptic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2008;93:F151-F152.
Ballantyne A.O., Spilkin A.M., Hesselink J., et al. Plasticity in the developing brain: intellectual, language and academic functions in children with ischaemic perinatal stroke. Brain. 2008;131:2975-2985.
Baram T.Z., Hatalski C.G. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21(11):471-476.
Bartha A., Shen J., Katz K.H., et al. Neonatal Seizures: Multi-Center Variability in Current Treatment Practices. Pediatr Res. 2007;37(2):85-90.
Basheer S.N., Waters P.J., Lam C.W., et al. Isolated sulfite oxidase deficiency in the newborn: lactic acidaemia and leukoencephalopathy. Neuropediatrics. 2007;38:38-41.
Baxter P. Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol. 2001;43:416-420.
Ben Ari Y., Holmes G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055-1063.
Bender R.A., Brewster A., Santoro B., et al. Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1-4 suggests evolving roles in the developing rat hippocampus. Neuroscience. 2001;106:689-698.
Bender R.A., Galindo R., Mameli M., et al. Synchronized network activity in developing rat hippocampus involves regional hyperpolarization-activated cyclic nucleotide-gated (HCN) channel function. Eur J Neurosci. 2005;22:2669-2674.
Bergey G.K., Swaiman K.F., Schrier B.K., et al. Adverse effects of phenobarbital on morphological and biochemical development of fetal mouse spinal cord neurons in culture. Ann Neurol. 1981;9:584-589.
Bergman I., Painter M., Hirsch R.P., et al. Outcome in neonates with convulsions treated in an intensive care unit. Ann Neurol. 1983;14:642-647.
Berkovic S.F., Heron S.E., Giordano L., et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550-557.
Billiards S.S., Haynes R.L., Folkerth R.D., et al. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199-208.
Bittigau P., Sifringer M., Genz K., et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99:15089-15094.
Bittigau P., Sifringer M., Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci. 2003;993:103-114. discussion 123–104
Blume H.K., Garrison M.M., Christakis D.A. Neonatal seizures: treatment and treatment variability in 31 United States pediatric hospitals. J Child Neurol. 2009;24:148-154.
Bok L.A., Been J.V., Struys E.A., et al. Antenatal treatment in two Dutch families with pyridoxine-dependent seizures. Eur J Pediatr. 2010;169:297-303.
Booth D., Evans D.J. Anticonvulsants for neonates with seizures. Cochrane Database Syst Rev. 2004. CD004218
Brooks-Kayal A., Jin H., Price M., et al. Developmental expression of GABA(A) receptor subunit mRNAs in individual hippocampal neurons in vitro and in vivo. J Neurochem. 1998;70(3):1017-1028.
Brunquell P.J., Glennon C.M., DiMario FJr, et al. Prediction of outcome based on clinical seizure type in newborn infants. J Paediatr. 2002;140:707-712.
Brunson K.L., Khan N., Eghbal-Ahmadi M., et al. Corticotropin (ACTH) acts directly on amygdala neurons to down-regulate corticotropin-releasing hormone gene expression. Ann Neurol. 2001;49:304-312.
Brunson K.L., Eghbal-Ahmadi M., Bender R., et al. Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci USA. 2001;98:8856-8861.
Castro Conde J.R., Hernandez Borges A.A., Domenech Martinez E., et al. Midazolam in neonatal seizures with no response to phenobarbital. Neurology. 2005;64:876-879.
Chang Y.S., Mu D., Wendland M., et al. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatr Res. 2005;58:106-111.
Chen H.S., Wang Y.F., Rayudu P.V., et al. Neuroprotective concentrations of the N-methyl-D-aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121-1132.
Chen Y., Lu J., Pan H., et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239-243.
Claes L.R., Ceulemans B., Audenaert D., et al. De novo KCNQ2 mutations in patients with benign neonatal seizures. Neurology. 2004;63:2155-2158.
Clancy R.R. Prolonged electroencephalogram monitoring for seizures and their treatment. Clin Perinatol. 2006;33:649-665. vi
Clancy R.R. Summary proceedings from the neurology group on neonatal seizures. Pediatrics. 2006;117:S23-S27.
Clancy R.R., Sharif U., Ichord R., et al. Electrographic neonatal seizures after infant heart surgery. Epilepsia. 2005;46:84-90.
Cooper E.C., Jan L.Y. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60:496-500.
Coppola G., Castaldo P., Miraglia del Giudice E., et al. A novel KCNQ2 K+ channel mutation in benign neonatal convulsions and centrotemporal spikes. Neurology. 2003;61:131-134.
Coppola G., Veggiotti P., Del Giudice E.M., et al. Mutational scanning of potassium, sodium and chloride ion channels in malignant migrating partial seizures in infancy. Brain Dev. 2006;28:76-79.
Cornejo B.J., Mesches M.H., Coultrap S., et al. A single episode of neonatal seizures permanently alters glutamatergic synapses. Ann Neurol. 2007:411-426.
Dalmau I., Vela J.M., Gonzalez B., et al. Dynamics of microglia in the developing rat brain. J Comp Neurol. 2003;458:144-157.
Debillon T., Gras-Leguen C., Leroy S., et al. Patterns of cerebral inflammatory response in a rabbit model of intrauterine infection-mediated brain lesion. Brain Res Dev Brain Res. 2003;145:39-48.
de Vries L.S., Toet M.C. Amplitude integrated electroencephalography in the full-term newborn. Clin Perinatol. 2006;33:619-632. vi
Dommergues M.A., Plaisant F., Verney C., et al. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121:619-628.
Dzhala V.I., Staley K.J. Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J Neurosci. 2003;23:1840-1846.
Dzhala V.I., Talos D.M., Sdrulla D.A., et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205-1213.
Dzhala V.I., Brumback A.C., Staley K.J. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222-235.
Eichler F., Tan W.H., Shih V.E., et al. Proton magnetic resonance spectroscopy and diffusion-weighted imaging in isolated sulfite oxidase deficiency. J Child Neurol. 2006;21:801-805.
Glass H.C., Glidden D., Jeremy R.J., et al. Clinical Neonatal Seizures are Independently Associated with Outcome in Infants at Risk for Hypoxic-Ischemic Brain Injury. J Pediatr. 2009;155(3):318-323.
Glier C., Dzietko M., Bittigau P., et al. Therapeutic doses of topiramate are not toxic to the developing rat brain. Exp Neurol. 2004;187:403-409.
Gluckman P.D., Wyatt J.S., Azzopardi D., et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663-670.
Gonzalez F.F., McQuillen P., Mu D., et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29:321-330.
Grant P.E., Yu D. Acute injury to the immature brain with hypoxia with or without hypoperfusion. Radiol Clin North Am. 2006;44:63-77. viii
Grillo E., da Silva R.J., Barbato J.H.Jr. Pyridoxine-dependent seizures responding to extremely low-dose pyridoxine. Dev Med Child Neurol. 2001;43:413-415.
Guillet R., Kwon J. Seizure recurrence and developmental disabilities after neonatal seizures: outcomes are unrelated to use of phenobarbital prophylaxis. J Child Neurol. 2007;22:389-395.
Hauser W.A., Annegers J.F., Kurland L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota:1935-1984. Epilepsia. 1993;34:453-468.
Haynes R.L., Borenstein N.S., DeSilva T.M., et al. Axonal development in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2005;484:156-167.
Hellstrom-Westas L., Blennow G., Lindroth M., et al. Low risk of seizure recurrence after early withdrawal of antiepileptic treatment in the neonatal period. Arch Dis Child Fetal Neonatal Ed. 1995;72:F97-101.
Heo K., Cho Y.J., Cho K.J., et al. Minocycline inhibits caspase-dependent and-independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci Lett. 2006;398:195-200.
Hillman L.S., Hillman R.E., Dodson W.E. Diagnosis, treatment, and follow-up of neonatal mepivacaine intoxication secondary to paracervical and pudendal blocks during labor. J Pediatr. 1979;95:472-477.
Holden K.R., Mellits E.D., Freeman J.M. Neonatal seizures. I. Correlation of prenatal and perinatal events with outcomes. Pediatrics. 1982;70:165-176.
Hollmann M., Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31-108.
Hoover-Fong J.E., Shah S., Van Hove J.L., et al. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63:1847-1853.
Huttenlocher P.R., deCourten C., Garey L.J., et al. Synaptogenesis in human visual cortex – evidence for synapse elimination during normal development. Neurosci Lett. 1982;33:247.
Ikonomidou C., Bosch F., Miksa M., et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70-74.
Isaeva E., Isaev D., Khazipov R., et al. Selective impairment of GABAergic synaptic transmission in the flurothyl model of neonatal seizures. Eur J Neurosci. 2006;23:1559-1566.
Ivacko J.A., Sun R., Silverstein F.S. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res. 1996;39:39-47.
Iwasaki S., Momiyama A., Uchitel O.D., et al. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59-65.
Jantzie L.L., Cheung P.Y., Todd K.G. Doxycycline reduces cleaved caspase-3 and microglial activation in an animal model of neonatal hypoxia-ischemia. J Cereb Blood Flow Metab. 2005;25:314-324.
Jensen F.E., Alvarado S., Firkusny I.R., et al. NBQX blocks the acute and late epileptogenic effects of perinatal hypoxia. Epilepsia. 1995;36(10):966-972.
Jiang Q., Wang J., Wu X., et al. Alterations of NR2B and PSD-95 expression after early-life epileptiform discharges in developing neurons. Int J Dev Neurosci. 2007;25:165-170.
Johnston M.V. Neurotransmitters and vulnerability of the developing brain. Brain Dev. 1995;17(5):301-306.
Ju W.K., Kim K.Y., Neufeld A.H. Increased activity of cyclooxygenase-2 signals early neurodegenerative events in the rat retina following transient ischemia. Exp Eye Res. 2003;77:137-145.
Kaltenbach K., Finnegan L.P. Neonatal abstinence syndrome, pharmacotherapy and developmental outcome. Neurobehav Toxicol Teratol. 1986;8:353-355.
Kanold P.O., Kara P., Reid R.C., et al. Role of subplate neurons in functional maturation of visual cortical columns. Science. 2003;301:521-525.
Kapur J., Macdonald R.L. Postnatal development of hippocampal dentate granule cell g-aminobutyric acid A receptor pharmacological properties. Mol Pharmacol. 1999;55:444-452.
Khazipov R., Khalilov I., Tyzio R., et al. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590-600.
Kim W.Y., Pomerance J.J., Miller A.A. Lidocaine intoxication in a newborn following local anesthesia for episiotomy. Pediatrics. 1979;64:643-645.
Kinney H.C., Haynes R.L., Folkerth R.D., et al. White matter lesions in the perinatal period (Invited). In: Pathology and Genetics: Acquired and Inherited Diseases of the Developing Nervous System. Basel: ISN Neuropathology Press; 2004.
Koh S., Jensen F. Topiramate Blocks Perinatal Hypoxia- Induced Seizures in Rat Pups. Ann Neurol. 2001;50:366-372.
Koh S., Tibayan F.D., Simpson J., et al. NBQX or topiramate treatment following perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569-575.
Kumar S.S., Bacci A., Kharazia V., et al. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005-3015.
Lanska M.J., Lanska D.J. Neonatal seizures in the United States: results of the National Hospital Discharge Survey, 1980–1991. Neuroepidemiology. 1996;15:117-125.
Lanska M.J., Lanska D.J., Baumann R.J., et al. A population-based study of neonatal seizures in Fayette County, Kentucky. Neurology. 1995;45:724-732.
Lawrence R., Mathur A., Nguyen The Tich S., et al. A pilot study of continuous limited-channel aEEG in term infants with encephalopathy. J Pediatr. 2009;154:835-841. e831
Lechpammer M., Manning S.M., Samonte F., et al. Minocycline treatment following hypoxic/ischaemic injury attenuates white matter injury in a rodent model of periventricular leucomalacia. Neuropathol Appl Neurobiol. 2008;34(4):379-393.
Lein E.S., Finney E.M., McQuillen P.S., et al. Subplate neuron ablation alters neurotrophin expression and ocular dominance column formation. Proc Natl Acad Sci USA. 1999;96:13491-13495.
Liu Y., Barks J.D., Xu G., et al. Topiramate extends the therapeutic window for hypothermia-mediated neuroprotection after stroke in neonatal rats. Stroke. 2004;35:1460-1465.
Loturco J.J., Owens D.F., Heath M.J., et al. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287-1298.
Maffei A., Turrigiano G. The age of plasticity: developmental regulation of synaptic plasticity in neocortical microcircuits. Prog Brain Res. 2008;169:211-223.
Malingre M.M., Van Rooij L.G., Rademaker C.M., et al. Development of an optimal lidocaine infusion strategy for neonatal seizures. Eur J Pediatr. 2006;165:598-604.
Manning S.M., Talos D.M., Zhou C., et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670-6678.
Manthey D., Asimiadou S., Stefovska V., et al. Sulthiame but not levetiracetam exerts neurotoxic effect in the developing rat brain. Exp Neurol. 2005;193:497-503.
Mares P., Mikulecka A. Different effects of two N-methyl-D-aspartate receptor antagonists on seizures, spontaneous behavior, and motor performance in immature rats. Epilepsy Behav. 2009;14:32-39.
Martin J.A., Kung H.C., Mathews T.J., et al. Annual summary of vital statistics: 2006. Pediatrics. 2008;121:788-801.
McQuillen P.S., Sheldon R.A., Shatz C.J., et al. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23:3308-3315.
Mikati M.A., El Hokayem J.A., El Sabban M.E. Effects of a single dose of erythropoietin on subsequent seizure susceptibility in rats exposed to acute hypoxia at P10. Epilepsia. 2007;48:175-181.
Miller S.P., Vigneron D.B., Henry R.G., et al. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. J Magn Reson Imaging. 2002;16:621-632.
Miller S.P., Ramaswamy V., Michelson D., et al. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146:453-460.
Mizrahi E.M., Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37:1837-1844.
Mizrahi E.M., Kellaway P. Diagnosis and Management of Neonatal Seizures. Philadelphia: Lippincott-Raven; 1998.
Navakatikyan M.A., Colditz P.B., Burke C.J., et al. Seizure detection algorithm for neonates based on wave-sequence analysis. Clin Neurophysiol. 2006;117:1190-1203.
Noebels J.L. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599-625.
Owens D.F., Boyce L.H., Davis M.B., et al. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414-6423.
Painter M.J., Scher M.S., Stein A.D., et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485-489.
Pape H.C. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299-327.
Pearl P.L. New treatment paradigms in neonatal metabolic epilepsies. J Inherit Metab Dis. 2009;32:204-213.
Pereira dV., Colin C., Desor D., et al. Influence of early neonatal phenobarbital exposure on cerebral energy metabolism and behavior. Exp Neurol. 1990;108:176-187.
Pfrieger F.W., Barres B.A. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684-1687.
Pisani F., Cerminara C., Fusco C., et al. Neonatal status epilepticus vs recurrent neonatal seizures: clinical findings and outcome. Neurology. 2007;69:2177-2185.
Rakhade S.N., Jensen F.E. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380-391.
Rakhade S.N., Zhou C., Aujla P.K., et al. Early Alterations of AMPA Receptors Mediate Synaptic Potentiation Induced by Neonatal Seizures. J Neurosci. 2008;28:7979-7990.
Raol Y.H., Lund I.V., Bandyopadhyay S., et al. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342-11346.
Ratnayaka B.D., Dhaliwal H., Watkin S. Drug points: Neonatal convulsions after withdrawal of baclofen. BMJ. 2001;323:85.
Ronen G.M., Penney S., Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr. 1999;134:71-75.
Ronen G.M., Buckley D., Penney S., et al. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816-1822.
Saliba E., Henrot A. Inflammatory mediators and neonatal brain damage. Biol Neonate. 2001;79:224-227.
Saliba R.M., Annegers J.F., Waller D.K., et al. Incidence of neonatal seizures in Harris County, Texas, 1992–1994. Am J Epidemiol. 1999;150:763-769.
Saliba R.M., Annegers F.J., Waller D.K., et al. Risk factors for neonatal seizures: a population-based study, Harris County, Texas, 1992–1994. Am J Epidemiol. 2001;154:14-20.
Sanchez R.M., Jensen F.E. Maturational aspects of epilepsy mechanisms and consequences for the immature brain. Epilepsia. 2001;42:577-585.
Sanchez R.M., Koh S., Rio C., et al. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154-8163.
Sanchez R.M., Dai W., Levada R.E., et al. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25:3442-3451.
Sanchez R.M., Jensen F.E., Pitanken A., et al. Modeling hypoxia-induced seizures and hypoxic encephalopathy in the neonatal period. In: Models of Seizures and Epilepsy. San Diego: Elsevier Press; 2005.
Sankar R., Painter M.J. Neonatal seizures: After all these years we still love what doesn’t work. Neurology. 2005;64:776-777.
Sayin U., Sutula T.P., Stafstrom C.E. Seizures in the developing brain cause adverse long-term effects on spatial learning and anxiety. Epilepsia. 2004;45:1539-1548.
Schroeder H., Humbert A.C., Koziel V., et al. Behavioral and metabolic consequences of neonatal exposure to diazepam in rat pups. Exp Neurol. 1995;131:53-63.
Serrano E.E., Kunis D.M., Ransom B.R. Effects of chronic phenobarbital exposure on cultured mouse spinal cord neurons. Ann Neurol. 1988;24:429-438.
Shankaran S., Laptook A.R., Ehrenkranz R.A., et alNational Institute of Child Health and Human Development Neonatal Research Network. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574-1584.
Shank R.P., Gardocki J.F., Streeter A.J., et al. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41(Suppl 1):S3-S9.
Shapiro L.A., Wang L., Ribak C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia. 2008;49(Suppl 2):33-41.
Shellhaas R.A., Clancy R.R. Characterization of neonatal seizures by conventional EEG and single-channel EEG. Clin Neurophysiol. 2007;118:2156-2161.
Shellhaas R.A., Soaita A.I., Clancy R.R. Sensitivity of amplitude-integrated electroencephalography for neonatal seizure detection. Pediatrics. 2007;120:770-777.
Silverstein F.S. Do seizures contribute to neonatal hypoxic-ischemic brain injury. J Pediatr. 2009;155:305-306.
Silverstein F.S., Jensen F.E. Neonatal seizures. Ann Neurol. 2007;62:112-120.
Silverstein F.S., Jensen F.E., Inder T., et al. Improving the treatment of neonatal seizures: National Institute of Neurological Disorders and Stroke workshop report. J Pediatr. 2008;153:12-15.
Singh N.A., Westenskow P., Charlier C., et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726-2737.
Sirsi D., Nadiminti L., Packard M.A., et al. Apneic seizures: a sign of temporal lobe hemorrhage in full-term neonates. Pediatr Neurol. 2007;37:366-370.
Stafstrom C.E., Tandon P., Hori A., et al. Acute effects of MK801 on kainic acid-induced seizures in neonatal rats. Epilepsy Res. 1997;26:335-344.
Stafstrom C.E., Moshe S.L., Swann J.W., et al. Models of pediatric epilepsies: strategies and opportunities. Epilepsia. 2006;47:1407-1414.
Stevens B., Allen N.J., Vazquez L.E., et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164-1178.
Stone B.S., Zhang J., Mack D.W., et al. Delayed neural network degeneration after neonatal hypoxia-ischemia. Ann Neurol. 2008;64:535-546.
Swann J., Moshe S.L., Engel J.Jr, et al. Developmental issues in animal models. In: Epilepsy: A comprehensive textbook. Philadelphia: Lippincott-Raven Publishers; 1997:467-480.
Swann J.W., Brady R.J., Martin D.L. Postnatal development of GABA-mediated synaptic inhibition in rat hippocampus. Neuroscience. 1989;28(3):551-561.
Takashima S., Chan F., Becker L.E., et al. Morphology of the developing visual cortex of the human infant: a quantitative and qualitative Golgi study. J Neuropathol Exp Neurol. 1980;39:487-501.
Talos D.M., Follett P.L., Jensen F.E. Developmental regulation of AMPA receptor subunits GluR1 and GluR2 in human neocortex. Soc Neurosci Abs. 2003.
Talos D.M., Follett P.L., Folkerth R.D., et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006;497:61-77.
Talos D.M., Fishman R.E., Park H., et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol. 2006;497:42-60.
Tekgul H., Bourgeois B.F., Gauvreau K., et al. Electroencephalography in neonatal seizures: comparison of a reduced and a full 10/20 montage. Pediatr Neurol. 2005;32:155-161.
Tekgul H., Bourgeois B.F., Gauvreau K., et al. Electroencephalography in neonatal seizures: comparison of a reduced and a full 10/20 montage. Pediatr Neurol. 2005;32:155-161.
Tekgul H., Gauvreau K., Soul J., et al. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics. 2006;117:1270-1280.
Tikka T., Fiebich B.L., Goldsteins G., et al. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580-2588.
Vezzani A., Balosso S., Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797-803.
Wasterlain C.G., Niquet J., Thompson K.W., et al. Seizure-induced neuronal death in the immature brain. Prog Brain Res. 2002;135:335-353.
Wong H.K., Liu X.B., Matos M.F., et al. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J Comp Neurol. 2002;450:303-317.
Wyatt J., Gluckman P., Liu P., et alCool Cap Study Group. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119:912-921.
Yue C., Yaari Y. KCNQ/M channels control spike after depolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614-4624.