CHAPTER 36 Natural anticoagulants and thrombophilia
Coagulation
Classic cascade mechanism of coagulation vs. cell-based models of thrombin generation
In the mid 1960s two different investigators conceived an original concept of the coagulation mechanisms triggered as a ‘cascade’ of proteolytic reactions acting as biological amplifiers.1,2 According to those classic concepts, coagulation mechanisms were activated through intrinsic and extrinsic pathways converging on a common pathway that will finally activate prothrombin into thrombin that would cause soluble fibrinogen to become polymerized into a solid fibrin clot (Fig. 36.1).
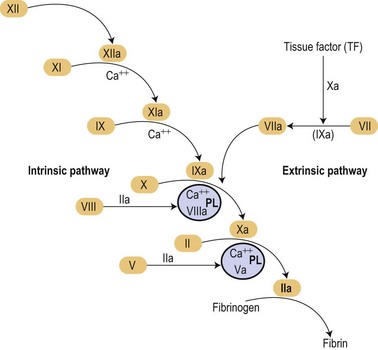
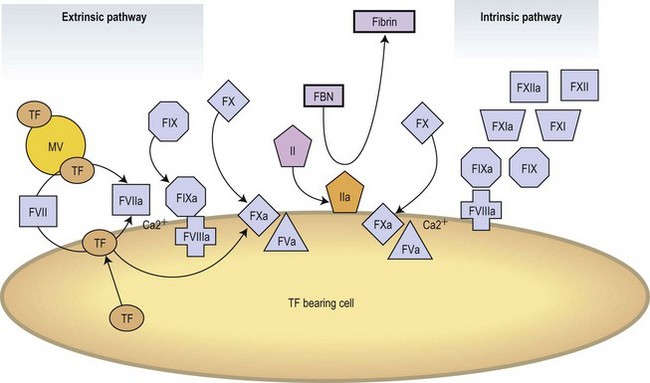
Modern concepts have integrated the classic stepwise cascade into a more comprehensive scheme in which blood coagulation is initiated by cellular components in vivo.3 Actual models contemplate the implication of cellular and enzymatic mechanisms in three differentiated steps: Initial activation, propagation and thrombin generation (Fig. 36.2). Fibroblasts, smooth muscle cells of the vascular wall, activated monocytes or endothelial cells, or even platelets are potential cellular sources of TF.4,5 According to current knowledge, the activation of the coagulation is initiated at the very moment in which TF is exposed on cellular surfaces. TF is a transmembrane protein that binds plasma factor VII/VIIa. In a first step, the TF exposed on damaged vascular areas forms a complex with FVII/FVIIa. FVIIa is present in reduced amounts in circulating blood. The TF : FVIIa complexes will activate FIX and FX. FXa generated will activate small quantities of prothrombin (FII) into thrombin (FIIa). Small amounts of thrombin generated in this short loop will be able to promote further activation of the coagulation by further activating FV and FVIII in the presence of platelet phospholipids and FIX.3 During the propagation step FIXa bound to FVIIa on the surface of activated platelets form the tenase complex that will further activate FX into FXa. FXa binds to FVa on the phospholipid surface provided by activated platelets and form the prothrombinase complex. During the final step of the cell-based model, the prothrombinase complex will further magnify the generation of thrombin facilitating the conversion of prothrombin into thrombin. Large amounts of thrombin generated during the final step will convert fibrinogen into fibrin. Thrombin itself activates FXIII thus stabilizing the fibrin formed.
Molecular mechanism
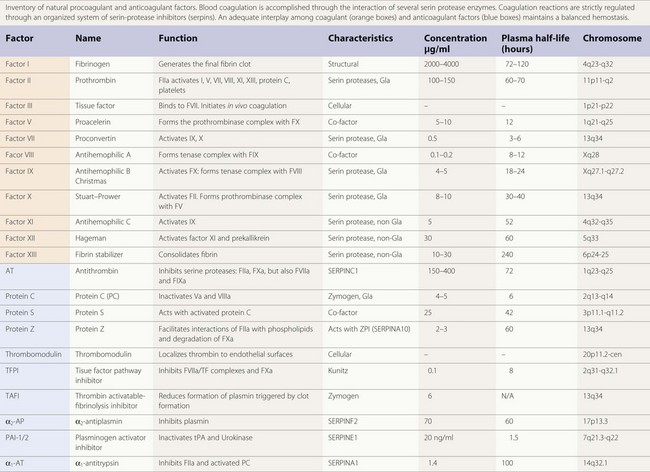
The majority of enzymes in the coagulation cascade are members of the same group of proteins called serine proteases (Table 36.1). They exhibit their function by limited proteolysis of peptide bounds of their substrates. The cascade design of the coagulation system allows a great amplification of signal in each step, since the enzymes activate more than one molecule of substrate. However, the enzymes by themselves do not have high catalytic efficiency and therefore the assembly of enzyme complexes is of crucial importance. This is for instance demonstrated by the bleeding diathesis experienced by hemophilia A patients, where the co-factor FVIII is deficient. In the enzyme complexes the activated membrane, exposing negatively charged phosphatidyl-serine plays a key role. Both the substrate and enzyme bind to the membrane concentrating the proteins at the surface. The correct orientation of the proteins on the cell surface facilitates the interaction. The importance of the membrane is shown in studies of the prothrombinase complex where the addition of phospholipid membrane gives an about 1000-fold increase in catalytic efficiency. Addition of co-factor FVa gives an additional increase and thus the prothrombinase complex is 106 times more efficient than the enzyme alone.6
 |
Vitamin K-dependent proteins and their binding to the membrane
The vitamin K dependent plasma proteins are a group of Gla domain containing proteins that require vitamin K for their synthesis7 (Table 36.1). The Gla domains contain 9–12 Glu residues that are γ-carboxylated in a post-translational process that require vitamin K. The γ-carboxylated Glu residues (Gla residues) mediate the binding of Ca2+ ions and are crucial for the membrane interaction. Thus, in patients on treatment with vitamin K antagonist (coumarin) the γ-carboxylation is impaired leading to loss of calcium and membrane binding. Due to high sequence similarity, the fold and phospholipid binding mechanism are thought to be similar for all the Gla domains. Although the sequence of the Gla domains are very similar for all the vitamin K zymogens, their affinities for the membrane surface vary 100 to 1000-fold. It has been suggested that this is caused by amino acid sequence differences in just a few positions.8
Regulatory mechanisms
Activation of coagulation switches on a series of mechanisms aimed at controlling and correcting fibrin formation. Plasminogen is converted into plasmin to initiate fibrinolysis. Mechanisms of coagulation and fibrinolysis are regulated at different steps by inhibitory mechanisms that maintain an adequate balance. This chapter is mainly focused on the implication of natural anticoagulants in thrombophilia. Certainly genetic or acquired alterations of fibrinolysis may result in delayed cleavage of the fibrin clot formed resulting in an indirect hypercoagulable state or in persistency of the associated symptoms.9,10 Despite the critical role of the fibrinolytic system in the resolution of thrombotic events, the overall impression is that its contribution to thrombophilia is relatively low when compared with deficiencies of natural anticoagulants.
SERPINs: a family of coagulation inhibitors
An important proportion of the natural anticoagulants exert their inhibitory action through inhibition of the enzymatic action of procoagulant serine proteases. This family of inhibitory proteins is called SERPINs (SERine Protease INhibitors). Antithrombin (SERPINC1), heparin co-factor II (SERPIND1), protein C inhibitor (SERPINA5) and PAI-1 (SERPINE1), are representative members of this family of inhibitors (Table 36.1). Although their primary sequence similarity is low, these proteins have a conserved three-dimensional structure that is crucial for their function in the regulation of hemostasis, thrombosis and fibrinolysis.11 Their mechanism of action can be compared to a mousetrap, where the serine protease to be inhibited is lured into an irreversible binding leading to a distortion of the enzyme and loss of activity.12 The inhibition is irreversible and one molecule of serpin can only inhibit one molecule of serine protease.
Antithrombin
Antithrombin (AT) is a α2-globulin, member of the serpin family of protease inhibitors (SERPINC1), synthesized in the liver and is the major inhibitor of blood coagulation. Human AT is a single chain glycoprotein of 432 aminoacids with a reactive site cleaved by target enzymes located at Arg393-Ser 394. Antithrombin is a very powerful inactivator of thrombin (FIIa) and FXa, but does also inhibit FVIIa, FIXa, FXIa and FXIIa to a lesser extent.13 Antithrombin has a molecular weight of 58 200 kDa. There are two isoforms, α and β, of circulating AT. The α represents 90–95% and the β, 5–10% of the total. Under physiological conditions AT circulates in a form that expresses low inhibitory affinity. AT possess a binding site for heparin, being the β isoform the fraction with a higher affinity for heparin.14 Under physiological conditions heparin sulfates present on the surface of endothelium will provide a background level of activation on AT. Interaction of specific glycosaminoglycan sequences with the specific heparin-binding domain induces a conformational change in AT and accelerates by an order of a thousand times its inhibitory action on activated serin proteases.15 Inactivation of thrombin (FIIa) by AT produces thrombin–antithrombin complexes (TAT) that can be determined and used as a surrogate marker of the activation of the coagulation. The anticoagulant action of conventional heparins, low molecular weight heparins, and pentasaccharide is mediated by AT. Congenital or acquired deficiencies of AT, may reduce the therapeutic action of the previous anticoagulants. In addition to its anticoagulant action AT has an important anti-inflammatory effect that seems closely dependent on its ability to bind to endothelial glycosaminoglycans.
Protein C system
Protein C structure
Protein C is a vitamin K-dependent protein of 62 kDa that circulates in blood as a zymogen in a concentration of about 4 µg/ml. The protein is synthesized in the liver. The mature protein C molecule is composed of a light and a heavy chain, the two chains being disulfide-linked. The light chain consists of a Gla domain and two epidermal growth factor (EGF)-like domains. The heavy chain contains a short activation peptide and a serine protease domain. Similar to the procoagulant co-factors, protein C circulates as an inactive proenzyme and needs to be activated to gain anticoagulant activity. Like for the other vitamin K-dependent proteins, the Gla domain of protein C mediates the membrane binding. Compared to the other vitamin K-dependent coagulation proteins, protein C has a quite low affinity for the membrane.8
Protein C activation complex
Protein C is activated by thrombin in complex with thrombomodulin.16 Thrombomodulin is a transmembrane glycoprotein consisting of a short cytoplasmic tail, a well-conserved membrane spanning region and a serine-threonine rich domain followed by 6 EGF-like repeats.17 Upon binding to thrombomodulin, thrombin changes its substrate specificity and is able to activate protein C. The efficiency of the thrombomodulin/thrombin complex is low and an additional co-factor, the endothelial receptor EPCR is needed for fully efficient protein C activation.
Protein C function
Activated protein C (APC) down-regulates the blood coagulation by limited proteolytic cleavage of co-factors FVIIIa and FVa. FVa and FVIIIa are homologous co-factors with the same domain structure: A1-A2 (the heavy chain), the B domain (only present in the non-activated FV and FVIII) and A3-C1-C2 (the light chain). APC cleaves at sites in the heavy chains of the proteins, at Arg306, Arg506 and Arg679 in FVa and Arg336 and Arg562 in FVIIIa.18 For the inactivation of FVa, the Arg506 cleavage is kinetically favored, but leads only to a partial loss of pro-coagulant function, due to decreased affinity for FXa. For complete loss of activity the cleavage at Arg306 is also needed, which leads to dissociation of the fragments. The FVIIIa is by itself an unstable molecule and is spontaneously degraded by dissociation of the A2 domain. Cleavage of FVIIIa at Arg336 is kinetically favored over cleavage at Arg562 in APC-mediated inactivation of FVIIIa. The cleavage at Arg336 does however not lead to complete loss of activity and the additional cleavage at Arg562 is needed.19
Protein S
Protein S is a 70 kDa large vitamin K-dependent protein that circulates in the blood in a concentration of 20–25 mg/ml. More than 60% of the protein S circulates in blood bound to a complement regulator protein called C4b-binding protein (C4BP).20 Protein S is mainly synthesized in the liver but synthesis has also been observed in endothelial cells, by testicular Leydig cells and by osteoclasts.21 Protein S has the following domain structure: an N-terminal vitamin K-dependent Gla domain, a thrombin-sensitive region (TSR), four EGF-like domains and two laminin G-type (LamG) domains (also called the SHBG-like domain). Like the other vitamin K-dependent blood proteins, the Gla domain of protein S mediates the membrane binding. Of the vitamin K-dependent proteins, protein S has the highest affinity for the membrane surface.8 The TSR is not found in the other vitamin K-dependent proteins. The region is sensitive to thrombin cleavage and cleavage leads to loss of APC co-factor function.22 Factor Xa has also been reported to cleave in the TSR, with the same result.
Protein S function
Protein S is an important co-factor for APC both in the inactivation of FVa and FVIIIa. In the FVa-inactivation protein S stimulates the proteolytic cleavage at Arg306 about 20-fold and only minor stimulation of the Arg506-cleavage is observed.18 However, when FVa participates in the prothrombinase complex protein S also influences the cleavage at Arg506 by counteracting the FXa-mediated inhibition of this cleavage site. It was originally thought that only the free form of protein S stimulates the APC-mediated cleavage of FVa; however, a recent report indicates that protein S in complex with C4BP can also stimulate the cleavage at Arg306.23 In the inactivation of FVIIIa protein S functions as a co-factor synergistically with the intact form of factor V.18 Protein S stimulates both the cleavage sites in FVIIIa.
In addition to being a co-factor for APC, protein S has been shown to exhibit an APC-independent anticoagulant activity.24 It has been demonstrated that the APC-independent anticoagulant function of protein S is an effect on the TFPI inhibition of the extrinsic pathway.25 As there is covariation of plasma TFPI and protein S levels both in health and in disease, these findings suggest that the risk of DVT associated with protein S deficiency states might be in part explained by the accompanying low plasma TFPI levels.24
Tissue factor pathway inhibitor TFPI
TFPI, also known as lipoprotein-associated coagulation inhibitor or extrinsic pathway inhibitor, is a Kunitz-type inhibitor with a molecular weight of 34 kD.26 The molecule possesses an acidic amino terminus, three Kunitz-type domains and a basic carboxy terminus. Circulating TFPI is synthesized by endothelial cells and smooth muscle cells.27 Approximately 10% of the blood TFPI is present in platelets. TFPI neutralizes FVIIa-TF complexes, but is also capable of neutralizing FVIIa and FXa, though at a much slower rate. In fact, the inhibitory action of TFPI on FVIIa-TF complexes requires certain levels of FXa. In experimental animals, administration of TFPI reduces mortality in a model of septic shock induced by Escherichia coli.28
Abnormal levels of TFPI or mutation in the respective gene are not strongly associated with thrombophilia. Treatment with exogenous hormones has a profound lowering effect on TFPI levels in women, with lower levels in oral contraceptive users than in premenopausal non-users. Levels of TFPI are strongly reduced levels in patients with abetalipoproteinemia, though they do not seem to be exposed to an enhanced risk of thrombosis. Recent studies indicate that low levels of TFPI, especially low TFPI-free and total antigen in plasma, may constitute a risk factor for DVT.29
Protein Z-dependent protease inhibitor
Protein Z is a vitamin K-dependent protein with a molecular weight of 62 kD, plasma concentration ranging from 2 to 3 µg/ml and with an approximate half-life of 2.5 days. Although the structure of protein Z contains a Gla domain it does not possess protease activating action on other coagulation factors. Protein Z promotes FXa inhibition induced by ZPI.30 Protein Z-dependent protease inhibitor (ZPI) is another member of the serpin family (SERPINA10) with known inhibitory action on FXa and FXIa.31 Heparin enhances the interaction of ZPI with FXIa.
The prothrombotic phenotype associated to protein Z-ZPI deficiency is controversial. Knock out mice for the protein Z gene do not seem to develop a thrombotic tendency unless there is another associated condition such as factor V Leiden.32 It has been postulated that thrombotic potential of the protein Z and ZPI deficiencies studies in humans question the thrombotic relevance of both ZPI and PZ deficiencies except when they act in combination with other prothrombotic factors.30
Thrombophilia
Thrombophilic states have been gaining attention as health professionals have realized the epidemiological impact of thrombotic complications on our societies. Venous thromboembolism (VTE), comprising deep-vein thrombosis (DVT) and pulmonary embolism (PE), are becoming a major public health concern since they are bound to a high incidence of major disability and a leading cause of morbidity and mortality for the general population. Comprehensive epidemiological data have been generated from studies on US and EC populations reveal a VTE incidence of ranging from 70 to 180 per 100 000 person-years with slight differences among geographical areas.33 Similar data are already reported for the European population.34,35 Rates of recurrence after a primary thrombotic event reach 30%. The mortality related to VTE estimated from one epidemiological analysis in six European countries including 300 million inhabitants predicted more than 350 000 deaths per year.35 Still these analyses underestimate the total medical and social cost of the disease because they do not include undiagnosed or misdiagnosed clinical thrombotic events, associated complications, unrecognized VTE-related or post-thrombotic syndromes.
The pathogenesis of thrombosis involves alterations in the blood vessel and a hypercoagulable condition developing in the circulating blood.36 The hypercoagulable condition is multifactorial and may involve genetic or acquired deficiencies of natural anticoagulants, abnormal function of coagulation factors and often combinations of these.37–40 Genetic risk factors range from rare anticoagulant deficiencies with high risk of VTE to common genetic polymorphism with moderate risk (Table 36.2). DVT and PE are obvious manifestations of thrombophilia, but miscarriage, repeated abortions and occasional arterial thrombosis should also be considered related to a thrombophilic state. Thrombophilia should also be suspected in patients who present with idiopathic thrombosis of the portal vein. Incidence of thrombotic complications are common in cases of coinheritance of two hereditary factors, especially when they are associated with other acquired risks such as immobilization, pregnancy, hormonal replacement, antiphospholipid syndrome or hyperhomocysteinemia. The following text will attempt to review the contribution of genetic deficiencies of natural anticoagulants, polymorphisms producing coagulant factors with abnormal functions and other conditions known to be associated with elevated risk of thrombotic complications. Emphasis would be placed on those thrombophilic states known to be associated with elevated risk of thrombosis or those with a high prevalence in the population.
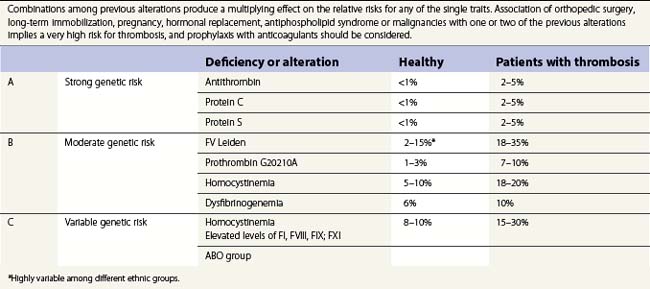
Hereditary deficiencies of natural anticoagulants
Antithrombin deficiency
Antithrombin deficiencies are uncommon with prevalence in the general population ranging from 1 in 500 to 1 in 5000 (Table 36.2). Hereditary AT deficiency was first reported by Olav Egeberg in several members of a family with a clear history of thrombotic events.41 Although plasma levels range from 112 to 140 µg/ml, laboratories usually refer levels as percentages with normality ranging from 80% to 120%. Most patients with congenital heterozygous AT deficiencies have levels below 60%. Deficiencies are associated with increased risk of VTE and pregnancy loss and rarely with arterial thrombosis. There is no definite relationship between levels and risk of thrombosis.42
Inherited quantitative (type I) and qualitative (type II) deficiencies have been described. In type I AT deficiency, functional and antigenic levels are proportionally decreased. Normal antigenic levels and abnormally reduced functional activities are found in type II. Several mutations are responsible for the type II deficiencies that can be further classified into subtypes.43 Acquired deficiencies of AT are observed in patients with cirrhosis, nephrotic syndrome and patients with sepsis and disseminated intravascular coagulation. Treatments with L-asparginase and heparins may also reduce levels of AT.
Protein C deficiency
The importance of the protein C system is demonstrated by the thrombotic disorders associated with protein C or protein S deficiency.44 Homozygous protein C deficiency is a rare condition that causes severe and fatal thrombosis already in the neonatal period. The clinical picture is characterized by purpura fulminans, thrombosis in the brain and disseminated intravascular coagulation. Heterozygous protein C deficiency is more common and is estimated to be around 1/300.45 The condition gives a higher risk for venous thrombosis while the risk for arterial thrombosis seems unaffected. Protein C deficiencies can be classified as type I and type II deficiencies. Type I deficiencies are the most common of the two and results in a reduction in both activity and antigen level whereas in type II deficiency the antigen levels are normal while the activity of APC is decreased. In heterozygous patients a very variable penetrance is observed, probably dependent on the coexistence of other risk factors. For instance, combined heterozygous protein C deficiency and APC resistance gives a high rate of thrombosis.46
Protein S deficiency
Homozygous protein S deficiency is very rare and gives a clinical picture similar to that of homozygous protein C deficiency.47 Heterozygous protein S deficiency is associated with venous thrombosis48 and late fetal loss. The prevalence of protein S deficiency is difficult to assess because of diagnostic difficulties and the rareness of the disease. The protein S levels are influenced by age, sex, pregnancy, oral contraceptive use, vitamin K intake and polymorphism in the PS gene. Also, the protein S genome is large and a pseudogene, PROSP, exists with about 97% sequence similarity to the protein S gene (PROS1), complicating mutational diagnostics.
Protein S deficiency is divided into three subclasses.47 Type I is characterized by a decreased amount of total and free protein S in plasma as well as decreased APC-co-factor activity. Type II is a qualitative defect where the protein S levels are normal but the anticoagulant APC co-factor activity is impaired. In type III defects the APC-co-factor activity is impaired and the amount free protein S is decreased; however, the total protein S is normal. There is a debate if the distinction between type I and type III deficiencies has any biological basis. Often type I and type III deficiencies coexist in the same families. An often occurring glycosylation variant, protein S Heerlen, seems, however, to solely be associated with type III deficiency.
Genetic polymorphisms causing abnormal function of coagulant factors
Activated protein C resistance and factor V Leiden
Activated protein C resistance was detected in 1993 and shown to give a predisposition for venous thrombosis.49 It is characterized by less prolongation of the APTT-based clotting time in the presence of APC compared to normal. The condition is in at least 90% of the cases caused by a point mutation in FV at nucleotide 1691, rendering the amino acid Arg506 to a Gln (FV Leiden),50 thereby impairing the APC-mediated cleavage at the Arg506 site, the fast phase of the FVa inactivation. It also leads to a loss of anticoagulant co-factor function of FV, since the cleavage at Arg506 is needed for full expression of this activity.18 The mutation is only present in Caucasians and the prevalence varies between 2% and 15%.51 In Europe the prevalence is highest in the northern parts. The polymorphism occurred from a common source about 30 000 years ago (founder effect). The most common clinical manifestations of FV Leiden are superficial and deep venous thrombosis. Pulmonary embolism can also occur, though to a lesser extent than for deficiencies of antithrombin, protein S and protein C. No increased risk for arterial thrombosis has been observed in FV Leiden carriers.
Two other naturally occurring mutations that affect the Arg306 site have been reported, FV Hong Kong and FV Cambridge.18 While FV Cambridge is very rare, FV Hong Kong is found in a prevalence of 4.7%, in the Hong Kong population. The association to DVT for FV Cambridge is complicated by the low prevalence. None of the studies performed for FV Hong has convincingly shown any association with increased risk for venous thrombosis.
Another polymorphism in FV gene that has been suggested to be associated with thrombotic disease is the HR2 haplotype.52 This consists of numerous point mutations in one allele and is found to be frequent in many populations both in Europe, Asia and Africa. Several functional defects have been reported for this polymorphism. The factor V levels are moderately low, it has been suggested to give a mild APC-resistance and the co-factor function of FV in the APC-mediated inactivation of FVIIIa has been reported to be moderately impaired. There is an ongoing debate on the clinical significance of the HR2 haplotype. However, compound heterozygous individuals for FV Leiden and the HR2 haplotype have a higher risk for venous thrombosis compared to individuals heterozygous for FV Leiden.
Prothrombin polymorphism
In 1996 another polymorphism was identified that is associated with venous thrombosis, the prothrombin polymorphism.53 This is a substitution in the 3′-untranslated region of prothrombin at nucleotide 20210, G to A. Since the mutation is in the untranslated region, the mutation does not give any amino acid substitution. However, it is associated with elevated prothrombin levels, which is thought to be the reason behind the hypercoagulability. The molecular mechanism behind the increased prothrombin levels has been suggested to be an increased process of the pre-mRNA to the ready translatable RNA as well as a higher translation efficiency.54 Just as for FV Leiden, the 20210 A allele arose from a founder about 20 000–30 000 years ago. Several studies have estimated the prevalence of the 20210 A allele in the white population to around 2%, with an opposite distribution as compared to FV Leiden, with the highest prevalence in southern Europe and a lower prevalence in northern Europe. Studies have confirmed that the 20210 A allele is a risk factor for DVT, including cerebral vein thrombosis. The increased risk for DVT has been estimated to be approximately threefold.
Dysfibrinogenemia
Abnormal fibrinogen molecules (dysfibrinogenemia) are found in some cases of patients presenting with venous thrombosis.55 Dysfibrinogenemia is caused by many different mutations in all the three different fibrinogen polypeptides, Aα, Bβ and γ. Only some of the known cases (about 20%) are associated with an increased risk of venous thrombosis; the majority of cases (about 55%) are asymptomatic and the rest (25%) are associated with bleeding. An online database exists listing the known fibrinogen variants and their reported association with symptoms (http://www.geht.org). The molecular mechanism behind the association to thrombophilia is often obscure. Proposed mechanisms include resistance to plasmin degradation and decreased thrombin binding affinity with higher thrombin levels.
Alterations of fibrinolytic mechanisms
The investigation of fibrinolysis has an uncertain place in thrombosis. In the Multiple Environmental and Genetic Assessment (MEGA), an association of overall hypofibrinolysis and venous thrombosis was observed (see 56 for review). On the other hand, when the individual components of the fibrinolytic system were assessed only high TAFI showed evidence of being associated with venous thrombosis, while no association was observed for plasminogen, α2-antiplasmin, tPA or PAI-1. It is uncertain if the association between overall fibrinolytic potential and venous thrombosis reflects the combined effects of all the fibrinolytic components, the effect of TAFI alone or a mechanism other than fibrin clot lysis.
Excess of other procoagulant factors
High levels of fibrinogen have been found to confer a slightly increased risk of thrombosis.57 Several studies have demonstrated an impact of high FVIII levels on the risk for venous thrombosis. Factor XI levels have, in three larger studies, the Leiden thrombophilia study (LETS), the Multiple Environmental and Genetic Assessment (MEGA) and the Longitudinal Investigation of Thromboembolism (LITE), shown a weak association with venous thrombosis. In contrast, in the case of FIX and FX the results of different studies have been conflicting.58
Acquired thrombophilic states
Lupus anticoagulants and the antiphospholipid syndrome
The antiphospholipid syndrome (APLS) cannot be considered as a single entity but rather as a combination of clinical and laboratory manifestations that are associated with elevated risks of thrombosis. Antibodies against phospholipids are relatively frequent and often detected as a prolongation of routine coagulation tests. Since these prolongations in coagulation tests were initially observed in patients with lupus erythematosus, antiphospholipid antibodies have also been named lupus anticoagulants.59 Antiphospholipid antibodies (APA) are a heterogeneous family of immunoglobulins that includes lupus anticoagulants and anticardiolipin antibodies. Lupus anticoagulants behave as acquired inhibitors of coagulation, prolonging phospholipid-dependent in vitro coagulation tests. Antiphospholipid antibodies can be measured by immunoassay, using cardiolipin or other anionic phospholipids in solid phase.60 Elevated presence of antibodies against cardiolipin or phosphatidylserine is more frequently associated with thrombosis. Despite their name, APA do not recognize phospholipids, but plasma proteins bound to suitable anionic surfaces. This may explain their interference with routine coagulation tests.
The antiphospholipid syndrome is a clinical condition characterized by enhanced risk of arterial and venous thrombosis, recurrent miscarriage and occasional thrombocytopenia. Although antiphospholipid antibodies have not been conclusively shown to be causal in thrombosis and miscarriage, they are useful laboratory markers for the antiphospholipid syndrome. Alpha2-glycoprotein I, and prothrombin are the most widely investigated antigenic targets.61
Hyperhomocysteinemia
Causes of hyperhomocysteinemia are diverse; they include genetic defects in homocysteine metabolism, vitamin deficiency, kidney disease, malabsorption and age. Severe hyperhomocysteinemia (homocystinuria) is a rare autosomal disease with early onset caused by mutations in homocysteine metabolism. The symptoms are many but include DVT and arterial thrombosis. Mild to moderate homocystinemia can be caused by both acquired and genetic defects.62 A polymorphism (MTHFR C677T) in the gene for methylene tetrahydrofolate reductase (MTHFR), which participates in the homocysteine–methionine cycle, has a frequency of 10–15% in white North Americans and a frequency as high as 25% in the Hispanic American population (homozygous form). Several studies have demonstrated a weak association of mild to moderate hyperhomocysteinemia and venous thrombosis. However, it is not clear if the association is causative.63
Malignancies and inflammatory conditions
The overall risk of venous thrombosis increases by a factor of seven in patients with cancer compared with those without malignancy (odds ratio 6.7, 95% confidence interval 5.2 to 8.6). Patients with hematological malignancies had the highest risk of venous thrombosis, adjusted for age and sex, followed by patients with lung cancer and gastrointestinal cancers. Patients with distant metastases are at higher risk than those without and the risk of thrombosis seems to be highest in the first few months after the diagnosis.64 The association of thrombotic symptomatology with underlying malignancies is straightforward. Thrombotic complications often precede the diagnosis of malignancy. Approximately 10% of patients presenting with VTE will be found to have malignancy in the subsequent year. Despite this fact, extensive search for malignancies in patients who present with idiopathic thrombosis has been found unproductive and should be disregarded. A thrombophilic state has been recognized in hematological malignancies. Myeloproliferative disorders (MPD), especially polycythemia vera (PV) or essential thrombocythemia (ET), are known to be associated with elevated risk of thrombosis.65 Alteration of platelet function and increased blood viscosity have been classically related to this elevated risk of thrombosis. Recent investigations are exploring the role of the JAK2 V617F mutation modifying the thrombotic risk in these patients.
Inflammatory and metabolic disorders also contribute to an enhanced thrombotic tendency. Tissue factor (TF) expression is up-regulated by multiple inflammatory proteins, and its expression results in activation of both the extrinsic and intrinsic blood coagulation cascades.66 Elevated levels of C reactive protein (CRP) and cytokines are frequently observed in patients with DVT. Moreover, the acute phase reaction observed in chronic inflammatory disorders results in the elevation of several procoagulant proteins such as fibrinogen and factor VIII that have also been involved in the generation of a thrombophilic state. Underlying inflammatory mechanisms may also explain the increased rate of thrombosis among patients with infections, malignancies or antiphospholipid antibodies.
Associations among congenital and/or acquired conditions
As shown in Table 36.2, although single hereditary deficiencies have been identified as the underlying cause for thrombophilia, the prevalence of these alterations is relatively low in the healthy population.67 There is considerable evidence that in most instances the thrombophilic state arises from the interaction of a number of different factors, including hereditary or acquired deficiencies in natural anticoagulants and dysfunctional coagulation factors combined simultaneously with a precipitating clinical situation such as orthopedic surgery, trauma or immobilization. Age above 50 years and obesity are both strongly associated with an elevated incidence of thrombotic events.68
Coinheritance of the more prevalent heterozygous mutations FV Leiden and the prothrombin G20210A increases four times the initial odds ratio for VTE of the single mutation.69 Combinations of FV Leiden, antithrombin, protein C or protein S deficiencies and hyperhomocysteinemia, have been found to result in a five to tenfold increase in the relative risk of thrombosis, but also in enhanced rates of recurrence and its appearance of thrombotic complications at an early age. Hormonal changes resulting from pregnancy, treatment with oral contraceptives and hormone replacement therapy substantially enhance the risk of VTE in women with thrombophilias. Exposure to oral contraceptives of a woman carrying the prothrombin G20210A mutation increases sixfold the risk of thrombosis. Deficiencies of antithrombin, protein C or protein S also significantly enhance the risk of venous thrombosis in women under oral contraceptives.70
Carriers of the factor V Leiden or the prothrombin 20210 A mutation who also had cancer are at 12 times the risk of a venous thrombosis compared with those who only had the mutation. But the research team considered that there was little point in screening for these mutations in patients with cancer, because malignancy was itself associated with a greatly increased risk of venous thromboembolism.64
Evaluation, diagnosis and treatment
Evaluation and diagnosis: screening for thrombophilias?
Despite its high prevalence, VTE is often clinically silent, frequently misdiagnosed, and may go unrecognized.35 Advances in the understanding of the biochemistry of the hemostatic mechanism have led to the development of sensitive methods for measuring peptides, enzyme-inhibitor complexes and enzymes that are liberated with the activation of the coagulation system in vivo.71 Prothrombin fragment F1+2, which is released when prothrombin is converted to thrombin, remains in the plasma and can be used to detect that thrombin has been generated. Fibrinopeptide A is the product of the enzymatic cleavage from the N-terminal ends of the α and β chains of the fibrinogen molecule and is an indirect indicator of fibrin generation. Thrombin–antithrombin (TAT) complexes are generated as a result of natural anticoagulant action of antithrombin neutralizing thrombin (FIIa). TAT complexes can be also quantified in the laboratory and used as indirect markers of the activation of coagulation mechanisms. Despite these tests being highly sensitive in the detection of early activation of the coagulation system and very helpful for research purposes, they possess a low diagnostic value in the confirmation of thrombosis.
The D-dimer is a marker of degradation product of polymerized fibrin. D-dimer levels are elevated in the plasma of the majority of patients with DVT or PTE.72 A combination of a clinical probability score with well defined cut-off values for the D-dimer can be used to rule out patients clinically unlikely to have DVT.73 Ultrasound testing and further imaging techniques can be safely used to confirm or discard a diagnosis of DVT or PE. It has been proposed that patients with a low clinical suspicion of a PE and a normal D-dimer do not need additional testing to exclude this disease.74
Laboratory tests for detection of hereditary or acquired thrombophilia are available. Controversy exists on how these tests should be applied.75,76 The overall impression is that an extensive screening after a first thrombotic event is not likely to confer a clinical benefit to the patient. Laboratory testing must be considered after the initial anticoagulant treatment has been completed. Testing for thrombophilia should be performed: 1) in patients younger than 50 years who develop spontaneous VTE; 2) in patients with a family history of venous thrombosis or with recurrent thrombotic events; or 3) in patients who develop thrombosis in unexpected locations or after being exposed to hormonal treatment. Repeated unexplained abortions must be considered as recurrent thrombotic events.
Phenotypic testing should be performed no sooner than 3 months after acute thrombotic events and at least 2–3 weeks after discontinuation of oral anticoagulant treatment. Testing should be based on the phenotype for antithrombin, protein C and protein S; on the phenotype and genotype (factor V Leiden mutation) for activated protein C resistance; and on the genotype (G20210A mutation) for hyperprothrombinemia. If testing for inherited thrombophilia is positive, the study should be extended to other family members even though they were still asymptomatic. The cost-effectiveness of indiscriminate screening of the general population of asymptomatic women before prescribing oral contraceptives is questionable. Due to their relative increased prevalence, some acquired prothrombotic states should be identified. These states include antiphospholipid syndrome, myeloproliferative disorders and cancer.77 Confirmation of some of these acquired risks may influence the duration and intensity of the anticoagulant therapy.40
It is accepted that genetic or biological causes for thrombophilia are identified in only 30% of the patients. The failure to identify a risk factor in many patients and the belief that genetic factors play an important role in the development of venous thrombosis stimulate the search for novel predictive genetic variants. Genome-wide linkage analysis may provide a key tool for future discovery of new thrombophilic conditions.78
Prevention and treatment
As mentioned earlier, VTE is a prevalent medical complication associated with elevated morbidity and mortality rates with inherent social and economical costs.33,35 Medical approaches to prevent and treat VTE derived from thrombophilic conditions imply three well established levels: prophylaxis of thrombotic events, acute treatment of the thrombotic complications and prevention of further recurrence. Although thrombophilic states are evidently the underlying cause for thromboembolic complications VTE is often clinically silent, misdiagnosed and unrecognized. Epidemiological studies reveal that three quarters of all VTE-related deaths were related to hospital-acquired conditions.35 Hip or knee arthroplasty, surgery after hip fracture, major trauma or surgery in hospitalized patients with associated thrombophilic conditions are at the highest risk for thrombosis. Surgery in in-patients older than 60 or 40–60 years old with additional hypercoagulable states are also at elevated risk for VTE.33 The incidence of VTE in these patients can be greatly reduced by appropriate prophylaxis with low molecular weight heparins before and after hospital discharge. Extensive evidence-based guidelines exist for prophylaxis of VTE in patients with variable risk profiles.79
Patients with a known thrombophilia who present with VTE should be treated with a standard regimen of heparin overlapped with classic oral anticoagulants (vitamin K antagonists) until an international normalized ratio (INR) of 2.0–3.0 is achieved. The main goal of continued treatment with oral anticoagulants is to prevent recurrent venous thromboembolism which occurs in 30% of untreated patients and 50% untreated patients with a preexisting thrombophilia. Treatment with anticoagulants reduces the risk of recurrence by 90–95%, with an annual risk of fatal hemorrhage of 0.25%.80 Duration of the anticoagulation to prevent recurrence is still an unresolved issue in thrombophilia. Although discrepancies exist among different guidelines, it is recommended that patients with the lowest risks or those who present with distal vein thrombosis after surgery or trauma should be treated with anticoagulants for 3 months. All other patients should receive treatment with oral anticoagulants for at least 6 months. Extension of oral anticoagulation for 6–18 months should be considered for patients with associated thrombophilias. There is increasing evidence that a normal D-dimer level and the absence of residual venous thrombosis after discontinuation of oral anticoagulation could be used as a predictor of a lower risk of recurrent VTE events.81 Based on the risk of thrombophilia or associated hereditary or acquired conditions (see Table 36.2), anticoagulant treatment should be maintained indefinitely. The coexistence of malignancies, antiphospholipid antibodies and coinheritance of two or more hereditary deficiencies are situations where indefinite anticoagulant therapy should be considered.
Pregnant patients with thrombophilia who develop VTE should be treated similarly to pregnant women without thrombophilia.82 Classic oral anticoagulants have been related to embryopathy and should be avoided between 6 and 12 weeks’ gestation. Low molecular weight heparins may be considered for pregnant patients who require anticoagulant treatment.83
New orally active small molecules that directly target FIIa or FXa are being developed.84 This new generation of oral anticoagulants is expected to circumvent the slow onset of action, narrow therapeutic window, many food and drug interactions and need for monitoring of classic anti-vitamin K agents.85 Some of these new anticoagulants are already approved for the indication of thromboprophylaxis in surgical patients at elevated risk. Results of several ongoing clinical trials are likely to confirm new indications in the acute and continued treatment of DVT and VTE. The advent of these new anticoagulants is expected to have a dramatic impact on the management of patients who have developed thrombotic complications. The net balance between prevention of thrombotic events vs. bleeding risk of these new anticoagulants is expected to have a profound impact on the therapeutic approach to patients with known thrombophilic conditions.
1 MacFarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a bichemical amplifier. Nature. 1964 May 2;202:498-499.
2 Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science. 1964 Sep 18;145:1310-1312.
3 Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost. 2006 Apr;32(Suppl. 1):32-38.
4 Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007 Aug;27(8):1687-1693.
5 Lopez-Vilchez I, Escolar G, Diaz-Ricart M, et al. Tissue factor-enriched vesicles are taken up by platelets and induce platelet aggregation in the presence of factor VIIa. Thromb Haemost. 2007 Feb;97(2):202-211.
6 Rosing J, Tans G, Govers-Riemslag JW, et al. The role of phospholipids and factor Va in the prothrombinase complex. J Biol Chem. 1980 Jan 10;255(1):274-283.
7 Kalafatis M, Swords NA, Rand MD, Mann KG. Membrane-dependent reactions in blood coagulation: role of the vitamin K-dependent enzyme complexes. Biochim Biophys Acta. 1994 Nov 29;1227(3):113-129.
8 McDonald JF, Shah AM, Schwalbe RA, et al. Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry. 1997 Apr 29;36(17):5120-5127.
9 Kluft C. The fibrinolytic system and thrombotic tendency. Pathophysiol Haemost Thromb. 2003 Sep 20;33(5–6):425-429.
10 Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005 May;129(3):307-321.
11 Rau JC, Beaulieu LM, Huntington JA, Church FC. Serpins in thrombosis, hemostasis and fibrinolysis. J Thromb Haemost. 2007 Jul;5(Suppl. 1):102-115.
12 Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000 Oct 19;407(6806):923-926.
13 Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008 Nov;14(6):1229-1239.
14 Rosenberg RD, Damus PS. The purification and mechanism of action of human antithrombin-heparin cofactor. J Biol Chem. 1973 Sep 25;248(18):6490-6505.
15 Carrell RW, Huntington JA. How serpins change their fold for better and for worse. Biochem Soc Symp. 2003;70:163-178.
16 Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1982 Jan 25;257(2):859-864.
17 Esmon CT. The protein C pathway. Chest. 2003 Sep;124(Suppl. 3):26S-32S.
18 Nicolaes GA, Dahlback B. Factor V and thrombotic disease: description of a Janus-faced protein. Arterioscler Thromb Vasc Biol. 2002 Apr 1;22(4):530-538.
19 Gale AJ, Cramer TJ, Rozenshteyn D, Cruz JR. Detailed mechanisms of the inactivation of factor VIIIa by activated protein C in the presence of its cofactors, protein S and factor V. J Biol Chem. 2008 Jun 13;283(24):16355-16362.
20 Dahlback B, Stenflo J. High molecular weight complex in human plasma between vitamin K-dependent protein S and complement component C4b-binding protein. Proc Natl Acad Sci USA. 1981 Apr;78(4):2512-2516.
21 Griffin JH, Fernandez JA, Gale AJ, Mosnier LO. Activated protein C. J Thromb Haemost. 2007 Jul;5(Suppl. 1):73-80.
22 Brinkman HJ, Mertens K, van Mourik JA. Proteolytic cleavage of protein S during the hemostatic response. J Thromb Haemost. 2005 Dec;3(12):2712-2720.
23 Maurissen LF, Thomassen MC, Nicolaes GA, et al. Re-evaluation of the role of the protein S-C4b binding protein complex in activated protein C-catalyzed factor Va-inactivation. Blood. 2008 Mar 15;111(6):3034-3041.
24 Sere KM, Rosing J, Hackeng TM. Inhibition of thrombin generation by protein S at low procoagulant stimuli: implications for maintenance of the hemostatic balance. Blood. 2004 Dec 1;104(12):3624-3630.
25 Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost. 2009 Jul;7(Suppl. 1):165-168.
26 Broze GJJr. Tissue factor pathway inhibitor. Thromb Haemost. 1995 Jul;74(1):90-93.
27 Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost. 2001 Oct;86(4):959-972.
28 Lwaleed BA, Bass PS. Tissue factor pathway inhibitor: structure, biology and involvement in disease. J Pathol. 2006 Feb;208(3):327-339.
29 Dahm A, van Hylckamavlieg A, Bendz B, et al. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood. 2003;101:4387-4392.
30 Corral J, Gonzalez-Conejero R, Hernandez-Espinosa D, Vicente V. Protein Z/Z-dependent protease inhibitor (PZ/ZPI) anticoagulant system and thrombosis. Br J Haematol. 2007 Apr;137(2):99-108.
31 Han X, Fiehler R, Broze GJJr. Characterization of the protein Z-dependent protease inhibitor. Blood. 2000 Nov 1;96(9):3049-3055.
32 Yin ZF, Huang ZF, Cui J, et al. Prothrombotic phenotype of protein Z deficiency. Proc Natl Acad Sci USA. 2000 Jun 6;97(12):6734-6738.
33 Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008 Mar;28(3):370-372.
34 Fowkes FJ, Price JF, Fowkes FG. Incidence of diagnosed deep vein thrombosis in the general population: systematic review. Eur J Vasc Endovasc Surg. 2003 Jan;25(1):1-5.
35 Cohen AT, Agnelli G, Anderson FA, et al. Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. 2007 Oct;98(4):756-764.
36 Schafer AI. Hypercoagulable states: molecular genetics to clinical practice. Lancet. 1994 Dec 24;344(8939–8940):1739-1742.
37 Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001 Apr 19;344(16):1222-1231.
38 Christiansen SC, Cannegieter SC, Koster T, et al. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005 May 18;293(19):2352-2361.
39 Hron G, Kollars M, Binder BR, et al. Identification of patients at low risk for recurrent venous thromboembolism by measuring thrombin generation. JAMA. 2006 Jul 26;296(4):397-402.
40 Bezemer ID, Bare LA, Doggen CJ, et al. Gene variants associated with deep vein thrombosis. JAMA. 2008 Mar;19;299(11):1306-1314.
41 Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965 Jun 15;13:516-530.
42 Abildgaard U. Antithrombin – early prophecies and present challenges. Thromb Haemost. 2007 Jul;98(1):97-104.
43 Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs. 2007;67(10):1429-1440.
44 Marlar RA, Neumann A. Neonatal purpura fulminans due to homozygous protein C or protein S deficiencies. Semin Thromb Hemost. 1990 Oct;16(4):299-309.
45 Miletich J, Sherman L, Broze GJr. Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med. 1987 Oct 15;317(16):991-996.
46 Koeleman BP, Reitsma PH, Allaart CF, Bertina RM. Activated protein C resistance as an additional risk factor for thrombosis in protein C-deficient families. Blood. 1994 Aug 15;84(4):1031-1035.
47 Gandrille S, Borgel D, Sala N, et al. Protein S deficiency: a database of mutations – summary of the first update. Thromb Haemost. 2000 Nov;84(5):918.
48 Garcia de FP, Fuentes-Prior P, Hurtado B, Sala N. Molecular basis of protein S deficiency. Thromb Haemost. 2007 Sep;98(3):543-556.
49 Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993 Feb 1;90(3):1004-1008.
50 Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994 May 5;369(6475):64-67.
51 Dahlback B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008 Jul 1;112(1):19-27.
52 Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood. 1997 Aug 15;90(4):1552-1557.
53 Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996 Nov 15;88(10):3698-3703.
54 Danckwardt S, Hartmann K, Gehring NH, et al. 3′ end processing of the prothrombin mRNA in thrombophilia. Acta Haematol. 2006;115(3–4):192-197.
55 Hill M, Dolan G. Diagnosis, clinical features and molecular assessment of the dysfibrinogenaemias. Haemophilia. 2008 Sep;14(5):889-897.
56 Meltzer ME, Doggen CJ, de Groot PG, et al. The impact of the fibrinolytic system on the risk of venous and arterial thrombosis. Semin Thromb Hemost. 2009 Jul;35(5):468-477.
57 Soria JM, Almasy L, Souto JC, et al. A genome search for genetic determinants that influence plasma fibrinogen levels. Arterioscler Thromb Vasc Biol. 2005 Jun;25(6):1287-1292.
58 Cushman M, O’Meara ES, Folsom AR, Heckbert SR. Coagulation factors IX through XIII and the risk of future venous thrombosis: the Longitudinal Investigation of Thromboembolism Etiology. Blood. 2009 Oct 1;114(14):2878-2883.
59 Greaves M. Antiphospholipid antibodies and thrombosis. Lancet. 1999 Apr 17;353(9161):1348-1353.
60 Galli M, Luciani D, Bertolini G, Barbui T. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome: a systematic review of the literature. Blood. 2003 Mar 1;101(5):1827-1832.
61 Espinosa G, Cervera R. Antiphospholipid syndrome. Arthritis Res Ther. 2008;10(6):230.
62 Eldibany MM, Caprini JA. Hyperhomocysteinemia and thrombosis: an overview. Arch Pathol Lab Med. 2007 Jun;131(6):872-884.
63 den HM, Willems HP, Blom HJ, et al. Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: a randomized, placebo-controlled, double-blind trial. Blood. 2007 Jan 1;109(1):139-144.
64 Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005 Feb 9;293(6):715-722.
65 Tefferi A, Elliott M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost. 2007 Jun;33(4):313-320.
66 Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008 Feb 21;451(7181):914-918.
67 Bezemer ID, Rosendaal FR. Predictive genetic variants for venous thrombosis: what’s new? Semin Hematol. 2007 Apr;44(2):85-92.
68 Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999 Apr 3;353(9159):1167-1173.
69 Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism – pooled analysis of 8 case-control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001 Sep;86(3):809-816.
70 Bloemenkamp KW, Helmerhorst FM, Rosendaal FR, Vandenbroucke JP. Thrombophilias and gynaecology. Best Pract Res Clin Obstet Gynaecol. 2003 Jun;17(3):509-528.
71 Bauer KA. Activation markers of coagulation. Baillière’s. Best Pract Res Clin Haematol. 1999 Sep;12(3):387-406.
72 Adam SS, Key NS, Greenberg CS. D-dimer antigen: current concepts and future prospects. Blood. 2009 Mar 26;113(13):2878-2887.
73 Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med. 2003 Sep 25;349(13):1227-1235.
74 Kearon C, Ginsberg JS, Douketis J, et al. An evaluation of D-dimer in the diagnosis of pulmonary embolism: a randomized trial. Ann Intern Med. 2006 Jun 6;144(11):812-821.
75 Tripodi A. A review of the clinical and diagnostic utility of laboratory tests for the detection of congenital thrombophilia. Semin Thromb Hemost. 2005 Feb;31(1):25-32.
76 Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008 Jun;121(6):458-463.
77 Greaves M, Watson HG. Laboratory testing for prothrombotic states: clinical utility. Curr Hematol Rep. 2003 Sep;2(5):429-434.
78 Soria JM, Almasy L, Souto JC, et al. A genome search for genetic determinants that influence plasma fibrinogen levels. Arterioscler Thromb Vasc Biol. 2005 Jun;25(6):1287-1292.
79 Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008 Jun;133(Suppl. 6):381S-453S.
80 Hirsh J, Kearon C, Ginsberg J. Duration of anticoagulant therapy after first episode of venous thrombosis in patients with inherited thrombophilia. Arch Intern Med. 1997 Oct 27;157(19):2174-2177.
81 Zhu T, Martinez I, Emmerich J. Venous thromboembolism: risk factors for recurrence. Arterioscler Thromb Vasc Biol. 2009 Mar 1;29(3):298-310.
82 Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004 Sep;126(Suppl. 3):627S-644S.
83 Brenner B. Clinical management of thrombophilia-related placental vascular complications. Blood. 2004 Jun 1;103(11):4003-4009.
84 Eriksson BI, Quinlan DJ, Weitz JI. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clin Pharmacokinet. 2009;48(1):1-22.
85 Weitz JI, Hirsh J, Samama MM. New anticoagulant drugs: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004 Sep;126(Suppl. 3):265S-286S.