Chapter 20 Myoclonus
Phenomenology, etiology, physiology, and treatment
Literally, myoclonus means “a quick movement of muscle.” Sudden, brief jerks may be caused not only by active muscle contractions, positive myoclonus, but also by sudden, brief lapses of muscle contraction in active postural muscles, negative myoclonus or asterixis (Shibasaki, 1995).
The history of myoclonus has been described by Marsden and colleagues (1982), Hallett (1986), and Fahn (2002). Friedreich first defined myoclonus as a discrete entity in a case report published in 1881 of a patient with essential myoclonus. He wanted to separate the involuntary movement that he saw from epileptic clonus, a single jerk in patients with epilepsy, and chorea, which was the only previously described type of involuntary movement. For the next 10–20 years, many other types of involuntary movements, such as tic and myokymia, were also called myoclonus, but in 1903 Lundborg proposed a classification of myoclonus that cleared up much of the confusion. Lundborg classified myoclonus into three groups: symptomatic myoclonus, essential myoclonus, and familial myoclonic epilepsy.
Myoclonus is a common movement disorder. Caviness and Maraganore (Caviness et al., 1999) reviewed the record linkage system for Olmsted County at the Mayo Clinic, Rochester, Minnesota for the years 1976–1990 and found an average annual incidence of myoclonus of 1.3 cases per 100 000, and a prevalence in 1990 of 8.6 cases per 100 000.
Classification of myoclonus
Myoclonus can be classified on the basis of its clinical characteristics, its pathophysiology, or its cause (Table 20.1) (Marsden et al., 1982; Hallett et al., 1987; Fahn, 2002).
| Clinical | Pathophysiology | Etiology |
|---|---|---|
| Spontaneous Action Reflex Focal Axial Multifocal Generalized Irregular Repetitive Rhythmic |
Cortical Focal Multifocal Generalized Epilepsia partialis continua Thalamic Brainstem Reticular Startle Palatal Spinal Segmental Propriospinal Peripheral Ballistic |
Physiological Essential Epileptic Symptomatic Storage diseases Cerebellar degenerations Basal ganglia degenerations Dementias Viral encephalopathies Metabolic encephalopathies Toxic encephalopathies Hypoxia Focal damage |

Pathophysiology
The clinical features of myoclonus and the results of electrophysiologic investigation allow a relatively precise prediction as to its site of origin in the nervous system (Shibasaki and Hallett, 2005; Hallett and Shibasaki, 2008). On this basis, myoclonus may be shown to arise in the cerebral cortex (cortical myoclonus); in the brainstem (brainstem myoclonus); or in the spinal cord (spinal myoclonus). Rarely, lesions of spinal roots, nerve plexi, or peripheral nerves can cause myoclonus (peripheral myoclonus). Hemifacial spasm might be considered a form of peripheral myoclonus, due most often to neurovascular compression.
Cortical myoclonus, in which the abnormal activity originates in the sensorimotor cortex and is transmitted down the spinal cord in pyramidal pathways, may manifest as focal jerks, sometimes repetitive (epilepsia partialis continua), which can propagate into focal motor seizures; with or without secondary generalization (Hallett et al., 1979; Shibasaki and Hallett, 2005; Hallett and Shibasaki, 2008).
Myoclonus arising in the brainstem may take different forms (Hallett, 2002). One employs the pathways responsible for the startle reflex, causing exaggerated startle syndromes and the hyperekplexias. Another is independent of startle mechanisms, but causes generalized muscle jerks (brainstem reticular myoclonus). A third is the palatal myoclonus (tremor) syndrome.
Finally, one pathophysiologic type of essential myoclonus takes the form of spontaneous or action-induced ballistic electromyographic (EMG) bursts in muscles, with inappropriate overflow into other muscles (ballistic movement overflow myoclonus) (Hallett et al., 1977b).
Cause
With regard to etiology, so many neurologic conditions may produce myoclonus that such a classification runs to a textbook of neurology (Table 20.2) (Marsden et al., 1982; Hallett et al., 1987; Fahn, 2002; Hallett and Shibasaki, 2008). It is, however, useful to consider several broad categories.
| I. Physiological myoclonus (normal subjects) |
Physiologic myoclonus refers to muscle jerks occurring in certain circumstances in normal subjects. These include sleep jerks (hypnic jerks) and hiccup. Essential myoclonus consists of multifocal myoclonus in which there is no other neurologic deficit or abnormality on investigation. Epileptic myoclonus refers to conditions in which the major clinical problem is one of epilepsy, but one of the manifestations of the epileptic attacks is myoclonic jerks. Symptomatic generalized myoclonus refers to those many conditions in which generalized or multifocal muscle jerking is a manifestation of an underlying identifiable neurologic disease. Psychogenic myoclonus refers to myoclonus produced as a conversion symptom or as “voluntary” or “simulated” myoclonus (Thompson et al., 1992; Monday and Jankovic, 1993).
In the survey by Caviness et al. (1999), symptomatic myoclonus was most common, followed by epileptic myoclonus and essential myoclonus. Dementing illnesses were the commonest cause of symptomatic myoclonus.
Neurophysiologic assessment
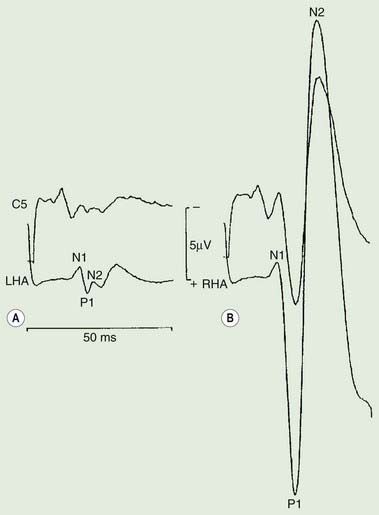
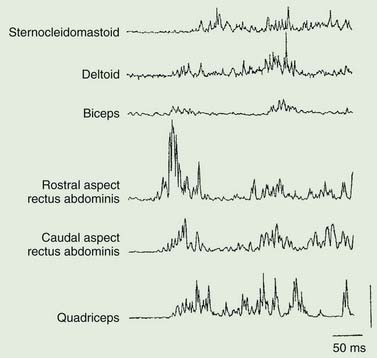
Polymyography (recording the duration, distribution, and stimulus sensitivity of EMG activity in affected muscles) is the first step in assessing a patient with myoclonus (Toro and Hallett, 2004; Shibasaki and Hallett, 2005; Hallett and Shibasaki, 2008). Most myoclonic jerks are due to brief EMG bursts of 10–50 ms. EMG bursts in the 100 ms range are seen in some situations such as essential myoclonus. Longer jerks of more than 100 ms are likely to be dystonic. Agonists and antagonists usually fire synchronously (Fig. 20.1).
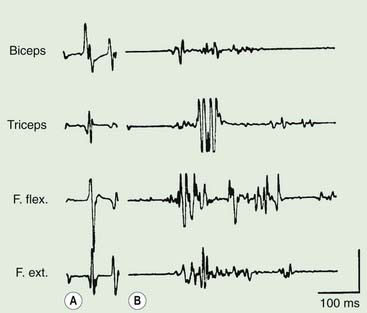
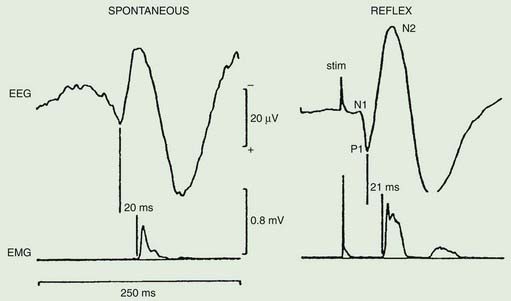
The distribution of muscles involved may suggest that it arises as a result of a lesion of a peripheral nerve, part of a plexus, a spinal root or a restricted number of segments of the spinal cord (segmental myoclonus). Myoclonic muscle jerks affecting axial muscles (neck, shoulders, trunk, and hips) may arise in the brainstem as an exaggerated startle response or brainstem reticular myoclonus, or in the spinal cord as propriospinal myoclonus. In brainstem myoclonus, there is no preceding cortical discharge. Cranial nerve muscles are usually activated from the XI nucleus up the brainstem; limb and axial muscles are activated in descending order. In propriospinal myoclonus, the first muscles activated are usually in the thoracic cord, with slow upward and downward spread. Cortical myoclonus is indicated when somatosensory evoked potentials produced by peripheral nerve stimulation are pathologically enlarged, and a cortical correlate can be back-averaged in the ongoing EEG by triggering from the EMG of the muscle jerk (Hallett et al., 1979; Shibasaki and Hallett, 2005). Stimuli generating giant somatosensory evoked potentials often provoke a subsequent EMG burst of myoclonic activity (the C reflex), at a latency compatible with conduction through fast corticomotoneuron pathways from the motor cortex to muscle. The giant somatosensory evoked potentials usually consist of an enlarged P25/N33 component; the first major cortical negative peak (N20), reflecting arrival of the sensory volley in the cortex, usually is of normal size. The motor volleys in cortical myoclonus activate the cranial and limb musculature in descending order via fast conducting corticospinal pathways. Abnormal corticomuscular and intermuscular coupling can also be a sensitive physiologic feature in cortical myoclonus (Grosse et al., 2003). The increased cortical excitability in cortical myoclonus may well be due to loss of inhibitory interneurons (Hanajima et al., 2008). Cortical reflex myoclonus usually consists of positive EMG discharges, but negative cortical reflex myoclonus also occurs (Shibasaki, 1995; Tassinari et al., 1998), where a giant somatosensory cortical potential is time-locked to EMG silence. Subcortical myoclonus is suggested when reflex myoclonus triggered by peripheral stimuli occurs after a latency that is too short to involve cortical pathways (Thompson et al., 1994; Cantello et al., 1997).
Psychogenic myoclonus is suggested if stimulus-evoked jerks are of very variable latency and longer than a voluntary reaction time (Thompson et al., 1992; Brown, 2006), and when the Bereitschaftspotential is evident prior to EMG bursts on jerk-locked back-averaging of the EEG, as in voluntary movement (Terada et al., 1995). Monday and Jankovic (1993) reported the clinical features of 18 such patients. There were 13 women and 5 men with an age range of 22–75 years. The myoclonus was present for 1–110 months; and it was segmental in 10, generalized in 7, and focal in 1. Stress precipitated or exacerbated the myoclonic movements in 15 patients; 14 had a definite increase in myoclonic activity during periods of anxiety. The following findings helped to establish the psychogenic nature of the myoclonus: clinical features incongruous with “organic” myoclonus, evidence of underlying psychopathology, an improvement with distraction or placebo, and the presence of incongruous sensory loss or false weakness. Over half of all patients with adequate follow-up improved after gaining insight into the psychogenic mechanisms of their movement disorder.
Focal myoclonus
Jerking of one body part may arise anywhere from the peripheral nerve to the motor cortex (Table 20.3). With peripheral nerve lesions, the myoclonus may well arise because of secondary central nervous system changes (Shin et al., 2007). Another possibility is that there is a peripheral ectopic generator that triggers the myoclonus (Tyvaert et al., 2009). Spontaneous rhythmic focal myoclonus is likely to be epilepsia partialis continua or spinal segmental myoclonus. Stimulus-sensitive myoclonus, particularly affecting the distal limbs, is most likely to arise in the cerebral cortex. Polymyography, somatosensory evoked potentials, and back-averaging from spontaneous jerks will usually suffice to define the site of origin.
| Category | Source | Etiology |
|---|---|---|
| Peripheral lesions | Peripheral nerve | Trauma |
| Plexus | Tumor | |
| Nerve roots | Electrical injury | |
| Surgery | ||
| Hemifacial spasm | ||
| Spinal lesions | (a)Spinal segmental myoclonus | Trauma |
| Inflammation | ||
| Infection | ||
| Demyelination | ||
| Tumor | ||
| Arteriovenous malformation | ||
| Ischemic myelopathy | ||
| Spondylitic myelopathy | ||
| Spinal anesthesia | ||
| Idiopathic | ||
| (b)Propriospinal myoclonus | Trauma | |
| Tumor | ||
| Idiopathic | ||
| Brainstem lesions | Palatal myoclonus | See Table 20.5 |
| Cortical lesions | Sensorimotor cortex | See Table 20.6 |
| Idiopathic |
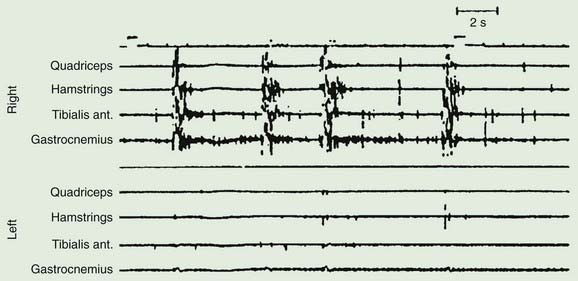
A variety of lesions of the peripheral nerve and spinal cord have been described as causing focal myoclonus (Frenken et al., 1976; Jankovic and Pardo, 1986; Massimi et al., 2009). These include peripheral nerve tumors, trauma or radiation, and spinal cord trauma, tumor, vascular lesions, multiple sclerosis and other inflammatory myelitis. Such spinal segmental myoclonus characteristically is rhythmic (0.5–3 Hz), is confined to muscles innervated by a few spinal segments, and persists during sleep (Fig. 20.2 and Video 20.3). Spinal segmental myoclonus appears to be due to loss of inhibitory interneurons in the posterior horns, which may be demonstrated physiologically (Di Lazzaro et al., 1996). As a result, there is spontaneous bursting of groups of anterior horn cells. Usually it is not stimulus-sensitive, but it can be (Davis et al., 1981). Clonazepam is most likely to help. One case was responsive to topiramate (Siniscalchi et al., 2004). 
Palatal myoclonus (alternately referred to as palatal tremor) describes the syndrome of rhythmic palatal movements at about 1.5–3 Hz, sometimes synchronously affecting the eyes, face, tongue and larynx, and even the head, trunk, intercostal muscles and diaphragm (Deuschl et al., 1990, 1994a, 1994b). The movements usually are bilateral and symmetric, occurring between 100 and 150 times per minute, and, in some circumstances, persist during sleep. There are two forms, essential palatal myoclonus and symptomatic palatal myoclonus (Table 20.4 and Video 20.4). The main symptom caused by these movements, seen only in the essential form, is clicking in the ear due to rhythmic contractions of tensor veli palatini which opens the eustachian tube. The tensor veli palatini is innervated by the trigeminal nerve. 
Table 20.4 Differences between essential and symptomatic palatal myoclonus
| Essential palatal myoclonus |
In many cases, a focal brainstem lesion can be identified (symptomatic palatal myoclonus/tremor), usually a stroke, encephalitis, multiple sclerosis, tumor, trauma, or degenerative disease (Table 20.5). Often the palatal myoclonus appears some months after the acute lesion. Such patients will have symptoms appropriate to the brainstem damage and to the underlying cause, in addition to the palatal myoclonus. They also may have pendular vertical nystagmus (ocular myoclonus), as well as facial, intercostal, and diaphragmatic jerks in synchrony with the palatal myoclonus. The pathology in symptomatic palatal myoclonus damages the dentato-olivary pathway, often in the brainstem central tegmental tract (Fig. 20.3); the resulting denervation of the inferior olive leads to hypertrophy (Fig. 20.4), which can be seen on brain MRI (Deuschl et al., 1994b). In this situation, the palatal movement is due to contractions of the levator veli palatini (innervated by the nucleus ambiguus). Only rarely can the ear clicking be caused by spontaneous contractions of the levator veli palatini (Jamieson et al., 1996).
Table 20.5 Causes of palatal myoclonus in 287 patients
| Condition | Number | |
|---|---|---|
| (A) | Primary (essential) palatal myoclonus | 77 |
| (B) | Secondary (symptomatic) palatal myoclonus | 210 |
| 1.Vascular disease | 115 | |
| 2.Trauma | 23 | |
| 3.Tumor (brainstem) | 19 | |
| 4.Multiple sclerosis | 9 | |
| 5.Degenerations | 7 | |
| 6.Encephalitis | 5 | |
| 7.Other; e.g., arteriovenous malformation, herpes zoster | 32 | |
From Deuschl G, Mischke G, Schenck E, Schulte-Monting J, Lucking CH. Symptomatic and essential rhythmic palatal myoclonus. Brain 1990;113(Pt 6):1645–1672.
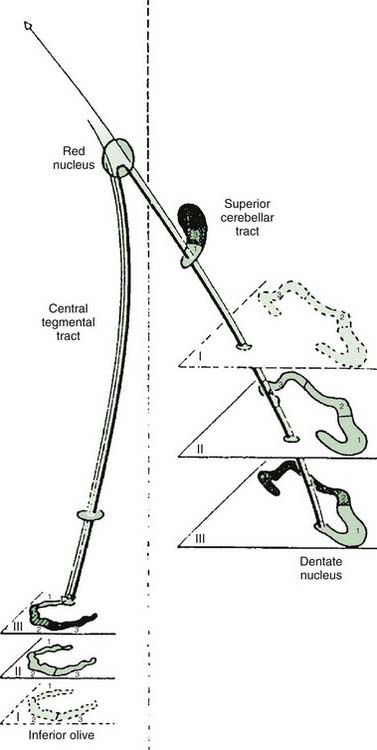
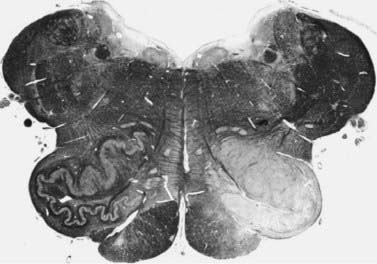
Figure 20.4 Unilateral hypertrophy of the inferior olive in a patient with unilateral symptomatic palatal myoclonus.
From Deuschl G, Toro C, Valls-Solé J, et al. Symptomatic and essential palatal tremor. 1. Clinical, physiological, and MRI analysis. Brain 1994;117:775–88, with permission.
In other cases, no cause is evident (essential palatal myoclonus/tremor). The complaint of these patients is the clicking; the eye and other structures are not involved; and there are no other symptoms or signs. These patients tend to be younger and do not appear to develop other diseases. Clonazepam, anticholinergics, or carbamazepine may help some patients with palatal myoclonus (Sakai and Murakami, 1981; Jabbari et al., 1987). Sumatriptan can be effective (Scott et al., 1996), but not in patients with symptomatic palatal myoclonus. Ear clicking can be relieved by injection of botulinum toxin into the appropriate muscles (Deuschl et al., 1991; Jamieson et al., 1996). Ear clicking occasionally may be due to simple partial seizures (Ebner and Noachtar, 1995). Some of these patients may be psychogenic (Pirio Richardson et al., 2006).
Cortical myoclonus produces spontaneous muscle jerks (spontaneous cortical myoclonus), jerks triggered by external stimuli (cortical reflex myoclonus), or jerks on movement (cortical action myoclonus) (Hallett et al., 1979; Obeso et al., 1985). Such patients have neurophysiologic evidence of an abnormal discharge in the sensory motor cortex generating the myoclonic jerks via fast conducting corticomotoneuron pathways (Figs 20.5 and 20.6). Myoclonus arising in the cerebral cortex can be focal affecting one body part, such as a hand or foot, but multiple cortical discharges can cause multifocal jerks, each jerk being due to a discrete discharge in one part of the motor cortex. In addition, cortical discharges can cause generalized muscle jerks, either by intracortical and transcallosal spread to activate both motor cortices (Brown et al., 1991a, 1996; Brown and Marsden, 1996), or by cortico-reticular pathways activating brainstem myoclonic generators. Such multifocal and generalized cortical myoclonus is discussed below. Patients with focal cortical myoclonus also exhibit epilepsia partialis continua (Juul-Jensen and Denny-Brown, 1966), partial motor seizures, and secondary generalization with tonic-clonic grand mal seizures (Cowan et al., 1986) (Fig. 20.7). Focal slow-frequency repetitive transcranial magnetic stimulation has suppressed focal cortical myoclonus in a patient with cortical dysplasia (Rossi et al., 2004).
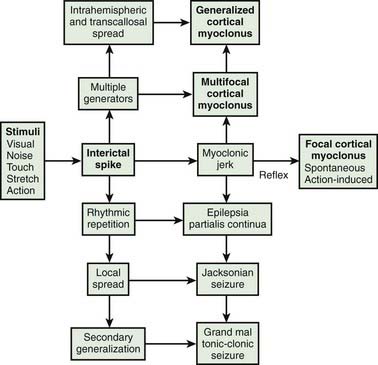
Epilepsia partialis continua is defined clinically as a syndrome of continuous focal jerking of a body part, usually localized to a distal limb, occurring over hours, days or even years, due to a cerebral cortical abnormality (Cockerell et al., 1996) (Fig. 20.8). The most common etiologies now are Rasmussen encephalitis and cerebrovascular disease. Most, but not all patients, have epileptic or other EEG abnormalities, and over half have identifiable cortical lesions on brain MRI. A similar clinical picture can occur with subcortical lesions, in which case it is suggested that the term “myoclonia continua” be employed (Cockerell et al., 1996). Cortical myoclonus sometimes is so rhythmic as to produce a tremor (Ikeda et al., 1990; Toro and Hallett, 2004) (Video 20.5). A focal cortical lesion produces focal myoclonus in the opposite appropriate body part. Such lesions include those due to vascular disease, tumor, granulomas, and focal encephalitis (Table 20.6) (Thomas et al., 1977; Cockerell et al., 1996). Chronic, prolonged focal myoclonic jerking in children suggests the possibility of Rasmussen encephalitis. 
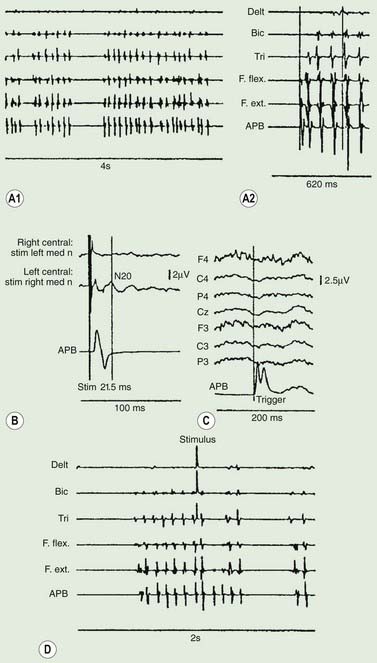
Table 20.6 Causes of focal cortical myoclonus and epilepsia partialis continua
| Etiology | Number | |
|---|---|---|
| Thomas et al. (1977) | Cockerell et al. (1996) | |
| Infarct or cerebral hemorrhage | 8 | 9 |
| Tumor | 5 | 4 |
| Astrocytoma | ||
| Hemangioma | ||
| Lymphoma | ||
| Uncertain | ||
| Encephalitis (including Rasmussen encephalitis) | 5 | 7 |
| Trauma | 2 | 1 |
| Hepatic encephalopathy | 2 | – |
| Subarachnoid hemorrhage | 1 | – |
| Unknown | 9 | 9 |
| Total | 32 | 30 |
| Others | ||
| Subdural hematoma | ||
| Abscess | ||
| Granuloma (TB) | ||
| Multiple sclerosis | ||
| Meningitis | ||
| Nonketotic hyperglycemia | ||
| Focal gliosis | ||
| Spinocerebellar degeneration | ||
| Mitochondrial disease | ||
Rasmussen encephalitis is a disorder of childhood and adolescence in which a unilateral focal seizure disorder is accompanied by a progressive hemiplegia due to focal cortical inflammation and destruction (Hart, 2004; Freeman, 2005). The seizures are severe, often with epilepsia partialis continua, partial motor seizures, and secondary generalization. The EEG may show focal epileptiform activity, or periodic lateralized discharges (PLEDs), or both. In addition to the progressive hemiplegia, there often is cognitive decline. The cause appears due to an autoimmune process, specifically to the glutamate receptor (GluR) 3 subunit, with such antibodies acting as a GluR agonist to cause an excitotoxic cascade (Gahring et al., 2001). Immunotherapy (steroids, plasmapheresis, or intravenous human immunoglobulin (IVIg)) may help some, but hemispherectomy may be necessary.
Axial myoclonus
Axial myoclonic jerks may arise in the spinal cord or brainstem. Propriospinal myoclonus involves long propriospinal fibers in the spinal cord distributed to axial muscles (Brown et al., 1991c) (Fig. 20.9). The most prominent movement of propriospinal myoclonus is truncal flexion, and it can be either spontaneous or stimulus induced (Video 20.6). A review of 60 patients noted a middle-aged male predominance (Roze et al., 2009). The myoclonus tended to be worse when lying down and at wake–sleep transitions. A premonitory sensation might be present before the jerks. The etiology of many of these cases is not clear, although injury to the spinal cord from trauma, infection, tumor, and disk herniation has been described (Capelle et al., 2005; Shprecher et al., 2010). Diffusion tensor imaging with fiber tracking of the spinal cord may reveal abnormalities (Roze et al., 2009). The EMG pattern of propriospinal myoclonus can be mimicked voluntarily, indicating that psychogenic myoclonus should be in the differential diagnosis in these cases (Kang and Sohn, 2006), and several such cases have now been reported (Williams et al., 2008; Slawek et al., 2010). In a series of 35 patients referred with axial jerks, 34 of them were considered to be psychogenic (van der Salm et al., 2010). More work is needed in this area to understand this disorder better. The most effective drug for treatment has been clonazepam (Roze et al., 2009). 
Brainstem reticular myoclonus (Table 20.7 and Fig. 20.10) may follow cerebral anoxia, and probably is responsible for the generalized muscle jerks that occur in a number of toxic myoclonic syndromes, as for example in uremia and other metabolic disturbances, as well as those precipitated by drugs. It is characterized by a generalized axial myoclonic jerk which starts in muscles innervated by the lower brainstem, then spreading up the brainstem and down the spinal cord (Hallett et al., 1977a). There is no time-locked cortical correlate and sensory evoked potentials are normal. Such brainstem reticular myoclonus may occur after cerebral anoxia, and can be seen in some patients with the stiff-man syndrome (Leigh et al., 1980). Brainstem lesions, such as occur in multiple sclerosis, also may be responsible (Smith and Scheinberg, 1990).
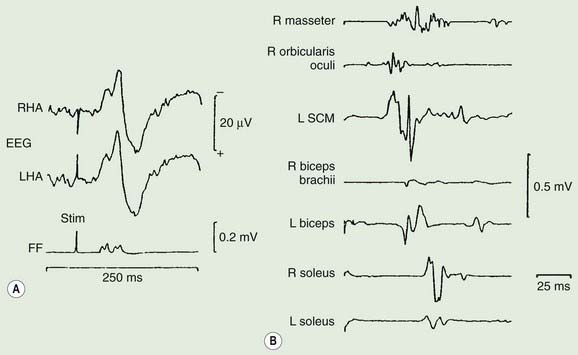
Exaggerated startle syndromes (hyperekplexia) consist of an excessive motor response or jump, to unexpected auditory and, sometimes, somesthetic and visual stimuli (Tables 20.7 and 20.8) (Suhren et al., 1966; Andermann and Andermann, 1988; Brown et al., 1991b; Matsumoto et al., 1992, Matsumoto and Hallett, 1994; Bakker et al., 2006). The jump consists of a blink, contortion of the face, flexion of the neck and trunk, and abduction and flexion of the arm (Fig. 20.11). Such patients jump in response to sound and crash to the ground, injuring themselves. There is no loss of consciousness. An exaggerated startle syndrome may be due to local brainstem pathology (anoxia, inflammatory lesions including sarcoidosis and multiple sclerosis, and hemorrhage), and also occurs as an inherited condition (hereditary hyperekplexia), transmitted as an autosomal dominant trait (Video 20.7). The first abnormal gene found for this disorder was a point mutation in the alpha-1 subunit of the glycine-receptor (Shiang et al., 1993, 1995; Tijssen et al., 1995). This may lead to altered ligand binding or disturbance of the chloride ion-channel part of the receptor. Subsequently other mutations were found in the glycine receptor and glycine receptor complex. Additionally, responsible mutations are also found presynaptically in the glycine transporter 2 (Rees et al., 2006). Glycine is an inhibitory neurotransmitter in several spinal interneurons, including Renshaw cells, Ia inhibitory interneurons, and some Ib inhibitory interneurons. Physiologic studies suggest normal recurrent inhibition, but abnormal Ia reciprocal inhibition in hyperekplexia (Floeter et al., 1996). Affected babies are hypertonic and stiff when handled, and have difficulty subsequently with walking. They develop excessive startles, which take two forms; the minor form is a simple brief startle jerk; the major form is a more prolonged tonic startle spasm. In these situations, the normal startle reflex is exaggerated, both in amplitude, and in its failure to rapidly habituate (Brown et al., 1991b; Matsumoto et al., 1992; Matsumoto and Hallett, 1994). The afferent and efferent systems of the startle reflex in hyperekplexia are identical to those of the normal startle response, involving a similar or the same generator in the lower brainstem, probably in the medial bulbopontine reticular formation. However, there may be differences between the minor form and major form of hyperekplexia. First, only the major form appears to be linked to the glycine receptor gene (Tijssen et al., 1995, 2002). Second, the physiologic abnormalities in the minor and major forms appear to differ (Tijssen et al., 1996, 1997). Treatment with clonazepam may be very effective in some patients (Matsumoto and Hallett, 1994). Others have responded to sodium valproate or piracetam. 
Table 20.8 Causes of the startle syndrome
| A. Pathologic exaggeration of the normal startle reflex |
| Static encephalopathies |
| Brainstem encephalitis |
| Structural |
| B. Brainstem reticular reflex myoclonus |
| C. Unknown physiology |
From Brown P, Rothwell JC, Thompson PD, et al. The hyperekplexias and their relationship to the normal startle reflex. Brain 1991;114:1903–1928.
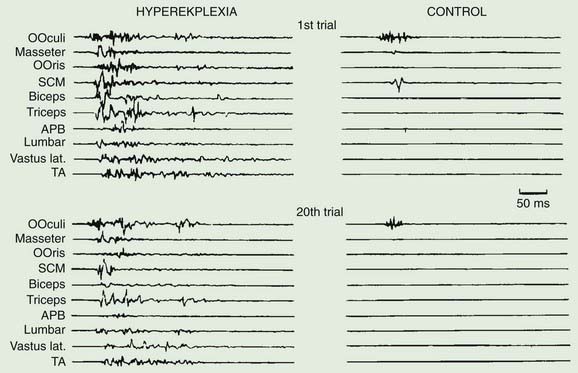
An exaggerated startle response may be responsible for the motor manifestations of “jumping Frenchmen” and other culturally determined startle syndromes (Andermann and Andermann, 1988; Brown et al., 1991b; Matsumoto and Hallett, 1994). In these syndromes the startle is followed by stereotyped behaviors such as a vocalization or striking out (Video 20.8). Hyperekplexia must be distinguished from startle-evoked epileptic seizures (Matsumoto and Hallett, 1994; Manford et al., 1996). 
Multifocal and generalized cortical myoclonus
Multifocal myoclonus of cortical origin typically affects many parts of the body bilaterally, but not synchronously. Each myoclonic jerk involves only a few adjacent muscles. The movements of the fingers or toes may be small twitches (minipolymyoclonus) (Wilkins et al., 1985); those of more proximal and axial muscles cause bigger movements. Multifocal cortical myoclonus is due to a generalized excitability of the cerebral cortex, particularly of the sensorimotor cortex. The EEG may show multifocal spike discharges, and back-averaging reveals the typical cortical correlate to each focal myoclonic jerk. Somatosensory evoked potentials often are enlarged. The jerks are frequently stimulus-sensitive.
Many patients with multifocal cortical myoclonus also exhibit generalized myoclonic jerks, synchronous in many muscles at the same time. Some of these are of brainstem origin (brainstem reticular myoclonus), perhaps driven by a cortical origin. However, discharge in the sensorimotor cortex may produce a generalized jerk as a result of transcallosal and intracortical spread (Brown et al., 1991a).
Many conditions cause multifocal and generalized cortical myoclonus. These include epileptic myoclonus, in which epilepsy dominates and there is no progressive disease of the brain, progressive myoclonus epilepsy and progressive myoclonic ataxia, post-anoxic myoclonus, viral encephalopathies, metabolic disease, and toxic encephalopathies (Table 20.2).
Epileptic myoclonus refers to those epilepsies characterized exclusively, or predominantly by brief myoclonic, atonic, or tonic seizures. Epileptic myoclonus can be positive (with active muscle contractions) or negative (lapses of postural tone) (Guerrini et al., 1993; Tassinari et al., 1995, 1998). Conditions subsumed under this category include infantile spasms, the Lennox–Gastaut syndrome, cryptogenic myoclonic epilepsy, the myoclonus associated with petit mal, and juvenile myoclonic epilepsy of adolescence (Janz). The key features of these various conditions are summarized in Table 20.9 (Aicardi, 1986; Shields, 2004; Nabbout and Dulac, 2008).
Table 20.9 Characteristics of major primary epileptic myoclonus syndromes
| Cryptogenic myoclonic epilepsy | Juvenile myoclonic epilepsy (Janz) |
| 1.Massive myoclonic jerks as the only or major seizure type | 1.Myoclonus, mainly in arms, especially in the morning |
| 2.Bursts of bilateral irregular slow spike/wave at 2.5 Hz or more | 2.Spikes, polyspikes and slow waves in EEG |
| 3.Onset 6 months to 5 years | 3.Onset around puberty |
| 4.No signs of brain damage prior to seizures | 4.No signs of brain damage |
| 5.Tonic-clonic seizures, but no tonic seizures | 5.Tonic-clonic seizures, especially at night. Absences in 10% |
| 6.Relatively good prognosis | 6.Very good response to sodium valproate |
| Eyelid myoclonus with absences | Absences in petit mal |
Of these conditions, juvenile myoclonic epilepsy (JME) is the most frequent epileptic syndrome presenting with myoclonus, usually in adolescence. The main symptom is myoclonic jerks, usually without loss of consciousness, predominantly in the morning after awakening from sleep. Generalized tonic-clonic seizures also tend to occur in the morning. Linkage studies have identified genetic loci for some patients with JME and other myoclonic epilepsies (Lu and Wang, 2009).
Progressive myoclonus epilepsy refers to a combination of severe myoclonus (spontaneous, action, and stimulus-sensitive), severe generalized tonic-clonic and other seizures, and progressive neurological decline, particularly dementia and ataxia (Berkovic and Andermann, 1986; Berkovic et al., 1986; Marseille Consensus Group, 1990; Ramachandran et al., 2009; Shahwan et al., 2005; Zupanc and Legros, 2004). Progressive myoclonic ataxia (sometimes known as the Ramsay Hunt syndrome) (Marsden et al., 1990) is distinguished from progressive myoclonus epilepsy by seizures being mild or absent, and dementia being mild or nonexistent, but myoclonus and ataxia are the major problems (Fig. 20.12).
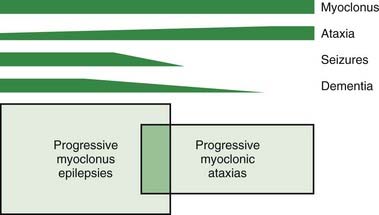
The differential diagnosis of progressive myoclonus epilepsy (Table 20.10) and progressive myoclonic ataxia (Table 20.11) is dominated by five main conditions.
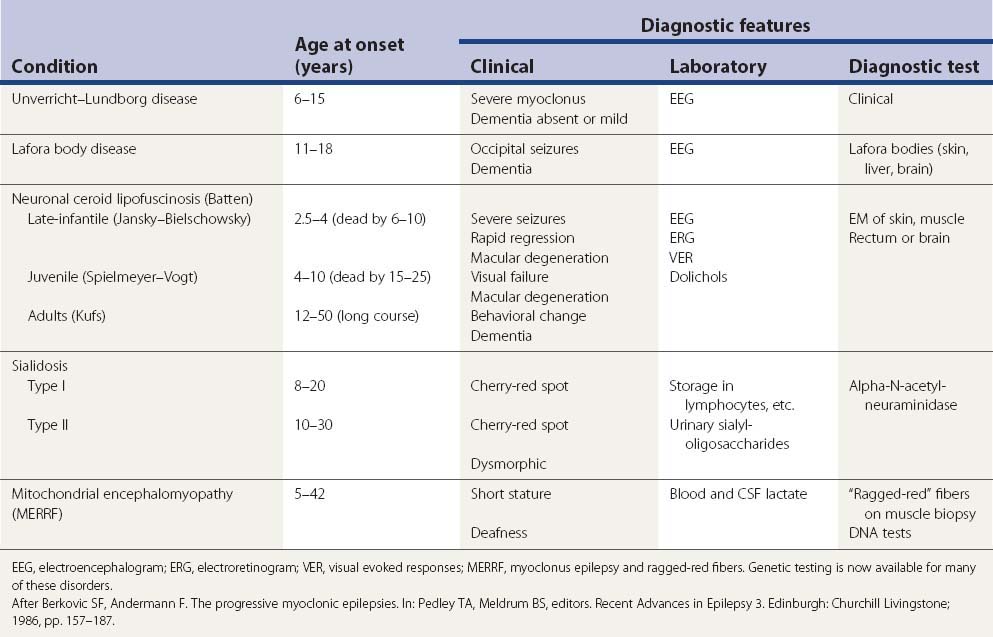
Table 20.11 Causes of progressive myoclonic ataxia
| Major causes |
| Rarer causes |
* Dementia often prominent, so not typical of progressive myoclonic ataxia.
Lafora body disease and neuronal ceroid lipofuscinosis generally produce severe neurologic decline with dementia or regression, along with myoclonus and fits (Rapin, 1986). Lafora body disease (EPM2), inherited as an autosomal recessive trait, is characterized by polyglucosan acid–Schiff (PAS)-positive inclusions in cells of brain, liver, muscle, and skin (eccrine sweat glands). Onset usually is in childhood with behavioral and cognitive change, dementia and seizures, as well as myoclonus; late-onset forms with a more benign onset also occur (Footitt et al., 1997). The gene responsible in about 75% of cases has been localized to chromosome 6q and has now been identified (Minassian et al., 1999) as encoding a protein tyrosine phosphatase (PTP) (Minassian et al., 1998; Serratosa et al., 1999). The protein, called laforin (or EPM2A), localizes at the plasma membrane and the endoplasmic reticulum (Minassian et al., 2001). Laforin presumably metabolizes polyglucosans preventing their aggregation (Chan et al., 2004a) with a mode of action of regulating autophagy (Aguado et al., 2010). A second gene is associated with Lafora body disease, NHLRC1 (also called EPM2B), which encodes malin, a putative E3 ubiquitin ligase with a RING finger domain and six NHL motifs (Chan et al., 2003; Ganesh et al., 2006; Singh and Ganesh, 2009). Laforin is a substrate for malin (Gentry et al., 2005). Both laforin and malin colocalize to the ER, suggesting they operate in a related pathway protecting against polyglucosan accumulation (Delgado-Escueta, 2007; Singh et al., 2008; Moreno et al., 2010). A third locus is predicted on the basis of a family without linkage to the two known sites (Chan et al., 2004b). There is a mouse model of the disorder (Ganesh et al., 2002). Zonisamide has been described as very useful for the myoclonus and epilepsy in these patients (Yoshimura et al., 2001).
Neuronal ceroid lipfuscinosis (Batten disease), inherited as an autosomal recessive condition, also presents with seizures and myoclonus with cognitive impairment and dementia, along with blindness in the late infantile and juvenile forms (Jalanko and Braulke, 2009). The adult form is often dominated by psychiatric and cognitive impairment. Lipopigment accumulates in lysosomes in brain, eccrine glands, skin, muscle, and gut with characteristic inclusions (curvilinear bodies and finger print profiles). Both Lafora body disease and neuronal ceroid lipofuscinosis usually can be diagnosed in axillary skin biopsies with electron microscopy.
Unverricht–Lundborg disease (EPM1) is characterized by stimulus-sensitive myoclonus, tonic-clonic seizures, a characteristic EEG (paroxysmal generalized spike-wave activity, and photosensitivity), and a progressive course with ataxia but only mild intellectual decline (Eldridge et al., 1983; Koskiniemi, 1986; Lehesjoki and Koskiniemi, 1998; Lehesjoki and Koskiniemi, 1999; Kalviainen et al., 2008). In some patients there might be oculomotor apraxia, dystonia, or rapidly progressive dementia (Chew et al., 2008). It is inherited as an autosomal recessive trait, with onset around the age of 6–15 years, and occurs worldwide. Autopsy shows widespread degeneration, with prominent loss of cerebellar Purkinje cells without storage material. The gene for Unvericht–Lundborg disease has now been linked to the long arm of chromosome 21q22.3 in a number of Finnish and Mediterranean families, and the gene involved codes for cystatin B, a small protein that is a member of a superfamily of cysteine protease inhibitors (Lehesjoki and Koskiniemi, 1998, 1999). The most common mutation is an unstable expansion of a dodecamer minisatellite repeat unit in the promoter region of the cystatin B gene. The exact role of cystatin B is not known, but it has been found primarily localized in the nucleus (Riccio et al., 2001). Cystatin B interacts with a variety of proteins and these multiprotein complexes are found in the cerebellum (Di Giaimo et al., 2002). One possible pathophysiologic mechanism is sensitization to oxidative stress (Lehtinen et al., 2009). An animal model has been developed (Shannon et al., 2002). In an Arab family with similar clinical appearance, a cystatin B mutation was ruled out, linkage was identified on chromosome 12, and the disorder named EPM1B (Berkovic et al., 2005).
It is of critical importance in these cases to recognize that phenytoin treatment may be associated with worsening of the condition (Eldridge et al., 1983). Patients can be treated successfully with other anticonvulsants, as described later.
Mitochondrial encephalomyopathy presents in many guises. One phenotype is the myoclonus epilepsy and ragged-red fiber syndrome (MERRF) (Canafoglia et al., 2001; Traff et al., 1995). Symptoms typically commence in the second decade, but onset as late as the age of 40 has been described. Myoclonus and ataxia are typical features, while tonic-clonic seizures and dementia often also occur. Deafness and short stature may be a clue to the diagnosis. Muscle weakness is variable. A raised blood or CSF lactate concentration may suggest the diagnosis. Muscle biopsy reveals the characteristic “ragged-red” fibers. Maternal inheritance can sometimes be evident, and a number of mutations of mitochondrial genes have been identified.
Sialidosis (cherry-red spot myoclonus syndrome) also is inherited as an autosomal recessive condition with onset in childhood or adolescence (Rapin, 1986). The sialidoses are lysosomal storage disorders associated with a deficiency of alpha-N-acetylneuraminidase and, in some, with additional deficiency of alpha-galactosidase.
In adults, another group of conditions, namely spinocerebellar degenerations, may cause myoclonus. For example, one presentation of dentatorubro-pallidoluysian atrophy (DRPLA) is with myoclonus and epilepsy (Becher et al., 1997). Some patients with the autosomal dominant cerebellar ataxias, including type 1, also may have prominent myoclonus. So, too, may patients with multiple system atrophy, where the myoclonus tends to affect the fingers and toes, and is stimulus-sensitive (Rodriguez et al., 1994).
There is an interesting but unexplained association between progressive myoclonic ataxia and celiac disease (Bhatia et al., 1995; Lu et al., 1986). The onset of the neurologic syndrome followed the gastrointestinal and other manifestations of celiac disease while on a gluten-free diet, in the absence of overt features of malabsorption or nutritional deficiency. The neurologic syndrome was dominated by action and stimulus-sensitive myoclonus of cortical origin with mild ataxia and infrequent seizures. The condition progressed despite strict adherence to diet. No treatment is known.
Action myoclonus–renal failure syndrome (AMRF) is a distinctive form of progressive myoclonus epilepsy associated with renal dysfunction. Badhwar et al. (2004) have described 15 individuals who presented with either renal or neurologic features. Segregation analyses suggested autosomal recessive inheritance. Tremor (onset 17–26 years), action myoclonus (onset 14–29 years), infrequent generalized seizures (onset 20.28 years), and cerebellar features were characteristic. Brain autopsy in two patients revealed extraneuronal pigment accumulation. Proteinuria, detected between ages 9 and 30 years in all cases, progressed to renal failure in most patients within 8 years. Renal biopsies showed collapsing glomerulopathy, a severe variant of focal glomerulosclerosis. Dialysis and renal transplantation were effective for the renal but not the neurologic features, which continued to progress even when kidney function was improved.
Other causes of progressive myoclonus epilepsy and progressive myoclonic ataxia are much rarer (Table 20.11). Investigation of patients with these clinical syndromes should include neurophysiology, white cell enzyme assays, and muscle and axillary skin biopsy.
Myoclonus after cerebral anoxia is usually the result of an anesthetic accident, cardiac arrest, or respiratory failure often due to asthma (Lance and Adams, 1963; Werhahn et al., 1997; Frucht, 2002) (Table 20.12). In acute post-hypoxic myoclonus, there is an initial period of coma, sometimes with myoclonic storms. The first three days are crucial to long-term prognosis; generalized myoclonic status epilepticus carries a very poor prognosis (Wijdicks et al., 1994). In those who survive, spontaneous, action-induced and stimulus-sensitive myoclonus may emerge on recovery (Video 20.9). Intellect may be normal, but there may be additional cerebellar ataxia as well as the disabling myoclonus. Seizures may persist. Post-anoxic myoclonus may consist of multifocal jerks of cortical origin, or generalized jerks due to intrahemispheric and transcallosal spread, or brainstem reticular myoclonus, or a mixture of all these types (Werhahn et al., 1997; Hallett, 2000). Many patients have prominent negative myoclonus particularly of the legs, causing sudden falls (Video 20.10). An animal model has been developed that should be useful in understanding the pathophysiology of this disorder (Kanthasamy et al., 2000; Nguyen et al., 2000; Truong et al., 2000). 
Table 20.12 Causes of post-hypoxic myoclonus
| Cause | Number of patients |
|---|---|
| Associated with anesthesia and surgery | 32 |
| Myocardial infarct | 12 |
| Obstructed airway | 11 |
| Drug intoxication | 9 |
| Miscellaneous | 21 |
| Not stated | 3 |
| Total | 88 |
From Fahn S. Posthypoxic action myoclonus: literature review update. Adv Neurol 1986;43:157–169.
The clinical syndrome as reported by Lance and Adams noted the precipitating feature of action, and the association with cerebellar ataxia, postural lapses, gait disturbance, and grand mal seizures. Werhahn and colleagues (1997) reported the clinical and neurophysiologic features of 14 patients with chronic post-hypoxic myoclonus. All patients had had a cardiorespiratory arrest, most caused by an acute asthmatic attack. All patients had multifocal action myoclonus, and 11 had additional stimulus-sensitive myoclonus. There was late improvement in the myoclonic syndrome and the level of disability in all but one patient. Cognitive deficits were found in seven patients and were usually mild. Electrophysiologic investigation confirmed cortical action myoclonus in every case, although this could be combined with cortical reflex myoclonus, an exaggerated startle response, or brainstem reticular reflex myoclonus. The site of the responsible lesion in the brain is not certain, but there does appear to be a disorder of serotonin metabolism supported not only by the therapeutic response to 5-hydroxytryptophan, but also by the reduction in CSF levels of 5-HIAA which improves with successful therapy. A study using glucose PET showed a bilateral hypermetabolism in the ventrolateral thalamus and pontine tegmentum relative to controls, but the significance of this is not clear (Frucht et al., 2004).
Multifocal and generalized myoclonus also may occur in a variety of acute encephalitic illnesses or postinfectious disseminated encephalomyelitis (Bhatia et al., 1992). It is characteristic of some of the slow viral encephalopathies such as subacute sclerosing panencephalitis (SSPE) and Creutzfeldt–Jakob disease (Shibasaki et al., 1981; Brown et al., 1986). In SSPE, the myoclonus is often periodic at long intervals (Video 20.11). Carbamazepine might be considered for the myoclonus of SSPE (Yigit and Sarikaya, 2006). In these latter conditions, the myoclonus often has a characteristic hung-up jerk, and periodic EEG discharges are evident in the majority of patients. Myoclonus also is seen in patients with the AIDS–dementia complex (Maher et al., 1997). 
The myoclonic encephalopathy of infants (opsoclonus–myoclonus, or dancing eyes–dancing feet syndrome) appears between the ages of 6 and 18 months and often responds dramatically to steroid or ACTH treatment (Pranzatelli, 1992, 1996; Tate et al., 2005) (Table 20.13). Plasmapheresis may also be effective (Yiu et al., 2001). A recent approach is B cell depletion therapy with rituximab, ACTH, and IVIg (Pranzatelli et al., 2010a, 2010b). About a half of such cases have an underlying neuroblastoma (De Grandis et al., 2009). An autoimmune origin seems likely since it is associated with a distinctive pattern of serum IgM and IgG binding to neural tissues and antigens (Connolly et al., 1997; Blaes et al., 2008; Raffaghello et al., 2008). Reduced levels of the metabolites of serotonin (5-HIAA) and dopamine (HVA) have been found in the CSF (Pranzatelli et al., 1995) and some patients may benefit from treatment with the serotonin precursor 5-hydroxy-L-tryptophan. The incidence of subsequent developmental or neurologic disturbance is high (Rudnick et al., 2001; Krug et al., 2010). A similar syndrome of opsoclonus and myoclonus may occur in adults, whether inflammatory or paraneoplastic (Bataller et al., 2001; Markakis et al., 2008; Sabater et al., 2008) (Table 20.14 and Video 20.12). The “idiopathic” cases have a more benign course than those with cancer. If the cancer is treated, the opsoclonus–myoclonus may resolve. One defined syndrome is breast cancer and anti-Ri antibody (Weizman and Leong, 2004). A patient with large-cell carcinoma of the lung and ANNA-2 antibodies responded well to treatment of the tumor and antibody levels declined (Erlich et al., 2004). Whipple disease also may produce such a clinical picture (Schwartz et al., 1986). A case was seen in a young woman with anti-N-methyl-D-aspartate receptor encephalitis (Kurian et al., 2010). Symptomatic therapy that should be considered includes steroids or ACTH. Plasmapheresis may be effective (Yiu et al., 2001). Trazadone may also be helpful. 
Table 20.13 The opsoclonus–myoclonus syndrome (dancing eyes and dancing feet)
Table 20.14 “Brainstem encephalitis”
|
Subacute onset of ataxia, dysarthria, diplopia and ophthalmoplegia, nystagmus, opsoclonus, vertigo, deafness and myoclonus
|
Antibodies to voltage-gated potassium channels (VGKC) may also be found in patients with myoclonus. In a study from the Mayo Clinic where they screened sera from 130 000 patients for markers of paraneoplastic neurologic autoimmunity, 80 patients were identified with VGKC immunoreactivity, and 29% manifested myoclonus (Tan et al., 2008).
Myoclonus and dementia in adults occurs not only in Creutzfeldt–Jakob disease but also in Alzheimer disease (Wilkins et al., 1984). Parkinson disease rarely manifests myoclonus. While the myoclonus appears to be cortical in origin, there is no reflex myoclonus or a giant somatosensory evoked potential (Caviness et al., 1998, 2002). Myoclonus is a well-described feature of dopa-dyskinesias (Luquin et al., 1992), and this would be its most frequent cause in patients with Parkinson disease. Patients with Parkinson disease and unsteadiness when standing may have orthostatic myoclonus (Leu-Semenescu et al., 2007). Myoclonus is more common in multiple system atrophy. Myoclonus can be seen in patients with dystonia, the myoclonus in this situation being a short “burst” of dystonia (Kinugawa et al., 2008) (Video 20.13). It may also be a feature of Huntington disease, and in one case it was shown to be cortical myoclonus (Caviness and Kurth, 1997). Myoclonus is a prominent, characteristic feature of corticobasal degeneration (Kompoliti et al., 1998). The myoclonus is stimulus-sensitive with a short latency that may be helpful in differential diagnosis. Its pathophysiology is still debated as to whether it is cortical or subcortical in origin (Carella et al., 1997; Strafella et al., 1997). 
A wide variety of drugs and toxins may provoke multifocal and generalized myoclonus (Gordon, 2002), as may renal failure (Chadwick and French, 1979). Among drugs, antidepressants (particularly the selective serotonin reuptake inhibitors), anesthetics, anticonvulsants (particularly at toxic levels), withdrawal of benzodiazepines and propranolol, lithium (Caviness and Evidente, 2003), monoamine oxidase inhibitors, and levodopa can all cause myoclonus. Among toxins, bismuth, heavy metals, glue and gasoline sniffing, and toxic cooking oil in Spain can cause prominent myoclonus. Myoclonus is prominent in the “serotonin syndrome,” along with confusion, agitation, diarrhea, fever, and sweating. This syndrome has been reported after treatment with tryptophan, monoamine oxidase inhibitors, selective serotonin reuptake inhibitors, and tricyclic antidepressants, alone or in combination (Mason et al., 2000). Myoclonus is also associated with the use of gabapentin, but is generally mild and may not cause any symptomatic problem (Asconape et al., 2000).
Essential myoclonus refers to a syndrome of nonprogressive multifocal myoclonus, without other cognitive or neurologic symptoms or signs, or fits. It may be inherited as an autosomal dominant trait (Fahn and Sjaastad, 1991). Essential myoclonus usually presents in the first or second decade and the course is benign. There are no seizures, dementia or ataxia, and the EEG is normal. In some with essential myoclonus the physiologic abnormality appears to be that of overflow of ballistic movement patterns (Hallett et al., 1977b).
Sporadic cases of essential myoclonus also occur (Bressman and Fahn, 1986). In some families with essential myoclonus, there also are manifestations of dystonia; some individuals have both, others have myoclonus or dystonia. The myoclonus in such families may be very responsive to alcohol – hence the description as alcohol-sensitive myoclonus dystonia (Quinn, 1996). Most families with myoclonus dystonia have an abnormal gene for ε-sarcoglycan (Zimprich et al., 2001). The clinical presentation is relatively homogeneous with this gene defect with myoclonus predominantly of neck and upper limbs and dystonia presenting as cervical dystonia and/or writer’s cramp (Asmus et al., 2002; Nardocci et al., 2008; Roze et al., 2008). In one family, however, epilepsy was also seen in affected members so the phenotype may be broad (Foncke et al., 2003). There is clearly genetic heterogeneity (Grimes et al., 2002). See also Chapter 12 for more details on myoclonus–dystonia.
Myoclonus is a frequent presentation of a psychogenic movement disorder, and it is important to keep this in mind in the differential diagnosis (Video 20.14). Psychogenic movement disorders are discussed in Chapter 25, but in the context of discussing myoclonus, it is noteworthy that the clinical neurophysiology is distinctive and can help to resolve the differential diagnosis (Brown, 2006; Esposito et al., 2009; Hallett, 2010). 
Drug treatment of generalized or multifocal myoclonus
Myoclonus, particularly action myoclonus, can be very disabling. It distorts speech, interferes with manual function, and prevents walking. The drugs used to treat myoclonus generally possess anticonvulsant properties (Table 20.15) (Obeso et al., 1989; Brown, 1995; Obeso, 1995; Pranzatelli and Nadi, 1995; Frucht, 2000), usually by enhancing gamma-aminobutyric (GABA) inhibitory activity. In epilepsy, it is currently fashionable to try and manage patients using a single drug rather than polytherapy. However, there are good reasons for the opposite approach in the treatment of myoclonus. Electrophysiologic evidence suggests that antimyoclonic drugs may exert different actions on the sequence of events responsible for myoclonus, at least for those concerned with cortical myoclonus. Some drugs which decrease cortical myoclonus increase the size of the giant sensory evoked potential, while others have the opposite effect. Myoclonus thus often responds best to a combination of drugs (Obeso et al., 1989).
Table 20.15 Appropriate dosages of agents for the treatment of myoclonus
| Drug | Dosage (mg/day) |
|---|---|
| Baclofen | 15–100 |
| Benzatropine | 4–9 |
| Carbamazepine | 800–1600 |
| Clonazepam | Up to 15 |
| Diazepam | 5–30 |
| 5-hydroxytryptophan | Up to 1500* |
| Levetiracetam | 1000–3000 |
| Phenobarbital | 60–180 |
| Phenytoin | 100–300 |
| Piracetam | 2400–16 800 |
| Primidone | 500–750 |
| Tetrabenazine | 50–200 |
| Trihexyphenidyl (benzhexol) | Up to 35 |
| Valproic acid (sodium valproate) | 1000–1500 |
* In combination with a peripheral aromatic amino acid decarboxylase inhibitor (such as carbidopa 100–300 mg/day).
Modified from Brown P. Myoclonus: A practical guide to drug therapy. CNS Drugs 1995;3:22–29.
Table 20.16 summarizes the drugs used to treat specific types of myoclonus arising in different parts of the neuraxis.
Table 20.16 Drug treatment for specific types of myoclonus
| Type of myoclonus | Drug(s) of first choice | Other agents |
|---|---|---|
| Cortical myoclonus | Valproic acid (sodium valproate or clonazepam) | Primidone or phenobarbital, levetiracetam, piracetam, 5-HTP* |
| Brainstem reticular myoclonus | Valproic acid or clonazepam† | 5-HTP* |
| Hyperekplexia | Clonazepam | Carbamazepine, phenytoin |
| Ballistic overflow myoclonus | Benzatropine or trihexyphenidyl (Benzhexol) | Alcohol (ethanol), clonazepam, 5-HTP* |
| Palatal myoclonus | Phenytoin, carbamazepine, clonazepam, diazepam, trihexyphenidyl or baclofen† | 5-HTP*, sumatriptan |
| Propriospinal myoclonus | Clonazepam | |
| Segmental spinal myoclonus | Clonazepam | Diazepam, carbamazepine, tetrabenazine |
5-HTP, 5-hydroxytryptophan.
* Combined with a peripheral aromatic amino acid decarboxylase inhibitor (such as carbidopa). Usefulness limited by poor tolerability.
† Treatment with any agent is often unsuccessful.
Epileptic myoclonus and cortical myoclonus respond best to drugs such as sodium valproate and clonazepam, often used in combination in maximum tolerated anticonvulsant doses (Obeso et al., 1989). Sodium valproate is the drug of choice to treat juvenile myoclonic epilepsy (Mantoan and Walker, 2011).
It is conventional to start with sodium valproate in patients with severe myoclonus, then to add clonazepam. If disability is not adequately improved, piracetam or levetiracetam can then be added. The mechanism of action of piracetam is uncertain, but it undoubtedly possesses antimyoclonic activity (Obeso et al., 1988; Brown et al., 1993; Ikeda et al., 1996; Koskiniemi et al., 1998; Genton et al., 1999; Fedi et al., 2001). Levetiracetam, a molecule related to piracetam that has been approved for the treatment of epilepsy, also is very effective for myoclonus (Genton and Van Vleymen, 2000; Genton and Gelisse, 2000, 2001; Frucht et al., 2001; Krauss et al., 2001; Schauer et al., 2002; Magaudda et al., 2004). There is one report of tizanidine helping two of three patients (Mukand and Giunti, 2004).
Primidone also may be of value as an additional drug in severely affected patients, as may clobazam and acetazolamide (Vaamonde et al., 1992). The potential antimyoclonic activity of newer anticonvulsants, such as vigabatrin, gabapentin, and lamotrogine, remains to be established. However, both vigabatrin and gabapentin may, paradoxically, worsen some types of myoclonus (Asconape et al., 2000). Milacemide, a glycine precursor, has been shown to be ineffective (Brown et al., 1991d; Gordon et al., 1993). One patient with alcohol-responsive post-hypoxic myoclonus responded well to gamma-hydroxybutryic acid (Frucht et al., 2005).
Unfortunately, while spontaneous, action, and reflex positive myoclonus often is helped by drug therapy, negative myoclonus frequently is resistant. As a result, disabling postural lapses in antigravity leg muscles usually persist to cause the typical bouncy unsteady stance and gait, often with falls. Two patients have been described with an excellent response to levetiracetam (Gelisse et al., 2003; Yu et al., 2009). In the setting of childhood partial epilepsy, negative myoclonus can be effectively treated with ethosuximide (Oguni et al., 1998; Capovilla et al., 1999). It is not clear whether this treatment would be useful in other circumstances.
The antioxidant N-acetylcysteine has been claimed to have considerable beneficial effect on the myoclonus in Unverricht–Lundborg disease (Hurd et al., 1996; Ben-Menachem et al., 2000). Alcohol also has been found to be a potent antimyoclonic agent in post-hypoxic myoclonus as well as Unverricht–Lundborg disease (Genton and Guerrini, 1992).
Those with brainstem myoclonus seem to respond best to clonazepam. 5-hydroxytrytophan with a peripheral decarboxylase inhibitor also has been used with some success, although gastrointestinal side effects may be prominent (Chadwick et al., 1977).
Spinal and other segmental myoclonus also responds best to treatment with clonazepam (Obeso, 1995; Devetag Chalaupka and Bernardi, 1999), but tetrabenazine and baclofen occasionally may be helpful. Levetiracetam can be useful (Keswani et al., 2002). One case responded to valproate and serotonergic therapy (Jimenez-Jimenez et al., 1991). If all else fails, botulinum toxin can be used (Lagueny et al., 1999).
Essential myoclonus sometimes improves with alcohol, a beta-blocker such as propranolol, or an anticholinergic agent (Duvoisin, 1984; Chokroverty et al., 1987). In myoclonus–dystonia syndrome, alcohol can be of benefit to the myoclonus, but not to the dystonia with which it may be associated (Quinn, 1996). Myoclonus–dystonia can be improved with bilateral deep brain stimulation of the globus pallidus interna (Cif et al., 2004; Azoulay-Zyss et al., 2011).
Aguado C., Sarkar S., Korolchuk V.I., et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 2010;19(14):2867-2876.
Aicardi J. Myoclonic epilepsies of infancy and childhood. Adv Neurol. 1986;43:11-31.
Andermann F., Andermann E. Startle disorders of man: hyperekplexia, jumping and startle epilepsy. Brain Dev. 1988;10:214-222.
Asconape J., Diedrich A., DellaBadia J. Myoclonus associated with the use of gabapentin. Epilepsia. 2000;41(4):479-481.
Asmus F., Zimprich A., Tezenas Du Montcel S., et al. Myoclonus-dystonia syndrome: epsilon-sarcoglycan mutations and phenotype. Ann Neurol. 2002;52(4):489-492.
Azoulay-Zyss J., Roze E., Welter M.L., et al. Bilateral deep brain stimulation of the pallidum for myoclonus-dystonia due to ε-sarcoglycan mutations: a pilot study. Arch Neurol. 2011;68(1):94-98.
Badhwar A., Berkovic S.F., Dowling J.P., et al. Action myoclonus-renal failure syndrome: characterization of a unique cerebro-renal disorder. Brain. 2004;127(Pt 10):2173-2182.
Bakker M.J., van Dijk J.G., van den Maagdenberg A.M., Tijssen M.A. Startle syndromes. Lancet Neurol. 2006;5(6):513-524.
Bataller L., Graus F., Saiz A., Vilchez J.J. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain. 2001;124(Pt 2):437-443.
Becher M.W., Rubinsztein D.C., Leggo J., et al. Dentatorubral and pallidoluysian atrophy (DRPLA). Clinical and neuropathological findings in genetically confirmed North American and European pedigrees. Mov Disord. 1997;12(4):519-530.
Ben-Menachem E., Kyllerman M., Marklund S. Superoxide dismutase and glutathione peroxidase function in progressive myoclonus epilepsies. Epilepsy Res. 2000;40(1):33-39.
Berkovic S.F., Andermann F. The progressive myoclonic epilepsies. In: Pedley T.A., Meldrum B.S., editors. Recent Advances in Epilepsy 3. Edinburgh: Churchill Livingstone; 1986:157-187.
Berkovic S.F., Andermann F., Carpenter S., Wolfe L.D. Progressive myoclonic epilepsies: specific causes and diagnosis. N Engl J Med. 1986;315:296-305.
Berkovic S.F., Mazarib A., Walid S., et al. A new clinical and molecular form of Unverricht-Lundborg disease localized by homozygosity mapping. Brain. 2005;128(Pt 3):652-658.
Bhatia K.P., Brown P., Gregory R., et al. Progressive myoclonic ataxia associated with coeliac disease. The myoclonus is of cortical origin, but the pathology is in the cerebellum. Brain. 1995;118(Pt 5):1087-1093.
Bhatia K., Thompson P.D., Marsden C.D. “Isolated” postinfectious myoclonus. J Neurol Neurosurg Psychiatry. 1992;55(11):1089-1091.
Blaes F., Pike M.G., Lang B. Autoantibodies in childhood opsoclonus-myoclonus syndrome. J Neuroimmunol. 2008;201–202:221-226.
Bressman S., Fahn S. Essential myoclonus. Adv Neurol. 1986;43:287-294.
Brown P. Myoclonus: A practical guide to drug therapy. CNS Drugs. 1995;3:22-29.
Brown P. Clinical neurophysiology of myoclonus. In: Hallett M., Fahn S., Jankovic J., Lang A.E., Cloninger C.R., Yudofsky S.C., editors. Psychogenic Movement Disorders: Neurology and Neuropsychiatry. Philadelphia: Lippincott Williams & Wilkins; 2006:262-264.
Brown P., Cathala F., Castaigne P., Gajdusek D.C. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol. 1986;20(5):597-602.
Brown P., Day B.L., Rothwell J.C., et al. Intrahemispheric and interhemispheric spread of cerebral cortical myoclonic activity and its relevance to epilepsy. Brain. 1991;114(Pt 5):2333-2351.
Brown P., Marsden C.D. Rhythmic cortical and muscle discharge in cortical myoclonus. Brain. 1996;119(Pt 4):1307-1316.
Brown P., Ridding M.C., Werhahn K.J., et al. Abnormalities of the balance between inhibition and excitation in the motor cortex of patients with cortical myoclonus. Brain. 1996;119(Pt 1):309-317.
Brown P., Rothwell J.C., Thompson P.D., et al. The hyperekplexias and their relationship to the normal startle reflex. Brain. 1991;114:1903-1928.
Brown P., Steiger M.J., Thompson P.D., et al. Effectiveness of piracetam in cortical myoclonus. Mov Disord. 1993;8(1):63-68.
Brown P., Thompson P.D., Rothwell J.C., et al. Axial myoclonus of propriospinal origin. Brain. 1991;114:197-214.
Brown P., Thompson P.D., Rothwell J.C., et al. A therapeutic trial of milacemide in myoclonus and the stiff-person syndrome. Mov Disord. 1991;6(1):73-75.
Canafoglia L., Franceschetti S., Antozzi C., et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56(10):1340-1346.
Cantello R., Gianelli M., Civardi C., Mutani R. Focal subcortical reflex myoclonus. A clinical and neurophysiological study. Arch Neurol. 1997;54(2):187-196.
Capelle H.H., Wohrle J.C., Weigel R., et al. Propriospinal myoclonus due to cervical disc herniation. Case report. J Neurosurg Spine. 2005;2(5):608-611.
Capovilla G., Beccaria F., Veggiotti P., et al. Ethosuximide is effective in the treatment of epileptic negative myoclonus in childhood partial epilepsy. J Child Neurol. 1999;14(6):395-400.
Carella F., Ciano C., Panzica F., Scaioli V. Myoclonus in corticobasal degeneration. Mov Disord. 1997;12(4):598-603.
Caviness J.N., Adler C.H., Beach T.G., et al. Small-amplitude cortical myoclonus in Parkinson’s disease: physiology and clinical observations. Mov Disord. 2002;17(4):657-662.
Caviness J.N., Adler C.H., Newman S., et al. Cortical myoclonus in levodopa-responsive parkinsonism. Mov Disord. 1998;13(3):540-544.
Caviness J.N., Alving L.I., Maranganore D.M., et al. The incidence and prevalence of myoclonus in Olmsted County. Minnesota. Mayo Clinic Proc. 1999;74(6):565-569.
Caviness J.N., Evidente V.G. Cortical myoclonus during lithium exposure. Arch Neurol. 2003;60(3):401-404.
Caviness J.N., Kurth M. Cortical Myoclonus in Huntington’s disease associated with an enlarged somatosensory evoked potential. Mov Disord. 1997;12(6):1046-1051.
Chadwick D., French A.T. Uraemic myoclonus: an example of reticular reflex myoclonus? J Neurol Neurosurg Psychiatry. 1979;42(1):52-55.
Chadwick D., Hallett M., Harris R., et al. Clinical, biochemical, and physiological features distinguishing myoclonus responsive to 5-hydroxytryptophan, tryptophan with a monoamine oxidase inhibitor, and clonazepam. Brain. 1977;100(3):455-487.
Chan E.M., Ackerley C.A., Lohi H., et al. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet. 2004;13(11):1117-1129.
Chan E.M., Omer S., Ahmed M., et al. Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology. 2004;63(3):565-567.
Chan E.M., Young E.J., Ianzano L., et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125-127.
Chew N.K., Mir P., Edwards M.J., Cordivari C., et al. The natural history of Unverricht-Lundborg disease: a report of eight genetically proven cases. Mov Disord. 2008;23(1):107-113.
Chokroverty S., Manocha M.K., Duvoisin R.C. A physiologic and pharmacologic study in anticholinergic-responsive essential myoclonus. Neurology. 1987;37(4):608-615.
Cif L., Valente E.M., Hemm S., et al. Deep brain stimulation in myoclonus-dystonia syndrome. Mov Disord. 2004;19(6):724-727.
Cockerell O.C., Rothwell J., Thompson P.D., et al. Clinical and physiological features of epilepsia partialis continua. Cases ascertained in the UK. Brain. 1996;119(Pt 2):393-407.
Connolly A.M., Pestronk A., Mehta S., et al. Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: an analysis of antigenic targets in neural tissues. J Pediatr. 1997;130(6):878-884.
Cowan J.M., Rothwell J.C., Wise R.J., Marsden C.D. Electrophysiological and positron emission studies in a patient with cortical myoclonus, epilepsia partialis continua and motor epilepsy. J Neurol Neurosurg Psychiatry. 1986;49(7):796-807.
Davis S.M., Murray N.M., Diengdoh J.V., et al. Stimulus-sensitive spinal myoclonus. J Neurol Neurosurg Psychiatry. 1981;44(10):884-888.
De Grandis E., Parodi S., Conte M., et al. Long-term follow-up of neuroblastoma-associated opsoclonus-myoclonus-ataxia syndrome. Neuropediatrics. 2009;40(3):103-111.
Delgado-Escueta A.V. Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 2007;7(5):428-433.
Deuschl G., Löhle E., Heinen F., Lücking C. Ear click in palatal tremor: its origin and treatment with botulinum toxin. Neurology. 1991;41:1677-1679.
Deuschl G., Mischke G., Schenck E., et al. Symptomatic and essential rhythmic palatal myoclonus. Brain. 1990;113(Pt 6):1645-1672.
Deuschl G., Toro C., Hallett M. Symptomatic and essential palatal tremor. 2. Differences of palatal movements. Mov Disord. 1994;9:676-678.
Deuschl G., Toro C., Valls-Solé J., et al. Symptomatic and essential palatal tremor. 1. Clinical, physiological, and MRI analysis. Brain. 1994;117:775-788.
Devetag Chalaupka F., Bernardi M. A case of segmental myoclonus in amputation stump: evidence for spinal generator and physiopathogenetic hypothesis. Ital J Neurol Sci. 1999;20(5):327-331.
Di Giaimo R., Riccio M., Santi S., et al. New insights into the molecular basis of progressive myoclonus epilepsy: a multiprotein complex with cystatin B. Hum Mol Genet. 2002;11(23):2941-2950.
Di Lazzaro V., Restuccia D., Nardone R., et al. Changes in spinal cord excitability in a patient with rhythmic segmental myoclonus. J Neurol Neurosurg Psychiatry. 1996;61(6):641-644.
Duvoisin R.C. Essential myoclonus: response to anticholinergic therapy. Clin Neuropharmacol. 1984;7:141-147.
Ebner A., Noachtar S. Ear clicking as the initial symptom of simple partial seizures. J Neurol. 1995;242(3):180-181.
Eldridge R., Iivanainen M., Stern R., et al. Baltic myoclonus epilepsy: hereditary disorder of childhood made worse by phenytoin. Lancet. 1983;2:838-842.
Erlich R., Morrison C., Kim B., et al. ANNA-2: an antibody associated with paraneoplastic opsoclonus in a patient with large-cell carcinoma of the lung with neuroendocrine features – correlation of clinical improvement with tumor response. Cancer Invest. 2004;22(2):257-261.
Esposito M., Edwards M.J., Bhatia K.P., et al. Idiopathic spinal myoclonus: A clinical and neurophysiological assessment of a movement disorder of uncertain origin. Mov Disord. 2009;24(16):2344-2349.
Fahn S. Posthypoxic action myoclonus: literature review update. Adv Neurol. 1986;43:157-169.
Fahn S. Overview, history and classification of myoclonus. In: Fahn S., Frucht S., Hallett M., Truong D.D., editors. Myoclonus and Paroxysmal Dyskinesias Advances in Neurology, vol. 89. Philadelphia: Lippincott Williams & Wilkins; 2002:13-17.
Fahn S., Sjaastad O. Hereditary essential myoclonus in a large Norwegian family. Mov Disord. 1991;6(3):237-247.
Fedi M., Reutens D., Dubeau F., et al. Long-term efficacy and safety of piracetam in the treatment of progressive myoclonus epilepsy. Arch Neurol. 2001;58(5):781-786.
Floeter M.K., Andermann F., Andermann E., et al. Physiological studies of spinal inhibitory pathways in patients with hereditary hyperekplexia. Neurology. 1996;46(3):766-772.
Foncke E.M., Klein C., Koelman J.H., et al. Hereditary myoclonus-dystonia associated with epilepsy. Neurology. 2003;60(12):1988-1990.
Footitt D.R., Quinn N., Kocen R.S., et al. Familial Lafora body disease of late onset: report of four cases in one family and a review of the literature. J Neurol. 1997;244(1):40-44.
Freeman J.M. Rasmussen’s syndrome: progressive autoimmune multi-focal encephalopathy. Pediatr Neurol. 2005;32(5):295-299.
Frenken C.W., Notermans S.L., Korten J.J., Horstink M.W. Myoclonic disorders of spinal origin. Clin Neurol Neurosurg. 1976;79(2):107-118.
Frucht S. Myoclonus. Curr Treat Options Neurol. 2000;2(3):231-242.
Frucht S.J. The clinical challenge of posthypoxic myoclonus. Adv Neurol. 2002;89:85-88.
Frucht S.J., Bordelon Y., Houghton W.H. Marked amelioration of alcohol-responsive posthypoxic myoclonus by gamma-hydroxybutyric acid (Xyrem). Mov Disord. 2005;20(6):745-751.
Frucht S.J., Louis E.D., Chuang C., Fahn S. A pilot tolerability and efficacy study of levetiracetam in patients with chronic myoclonus. Neurology. 2001;57(6):1112-1114.
Frucht S.J., Trost M., Ma Y., Eidelberg D. The metabolic topography of posthypoxic myoclonus. Neurology. 2004;62(10):1879-1881.
Gahring L., Carlson N.G., Meyer E.L., Rogers S.W. Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol. 2001;166(3):1433-1438.
Ganesh S., Delgado-Escueta A.V., Sakamoto T., et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11(11):1251-1262.
Ganesh S., Puri R., Singh S., et al. Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J Hum Genet. 2006;51(1):1-8.
Gelisse P., Crespel A., Genton P., Baldy-Moulinier M. Dramatic effect of levetiracetam on epileptic negative myoclonus. Acta Neurol Scand. 2003;107(4):302-303.
Genton P., Gelisse P. Antimyoclonic effect of levetiracetam. Epileptic Disord. 2000;2(4):209-212.
Genton P., Gelisse P. Suppression of post-hypoxic and post-encephalitic myoclonus with levetiracetam. Neurology. 2001;57(6):1144-1145.
Genton P., Guerrini R. Effect of alcohol on action myoclonus in Lance-Adams syndrome and progressive myoclonus epilepsy. Mov Disord. 1992;7(1):92.
Genton P., Guerrini R., Remy C. Piracetam in the treatment of cortical myoclonus. Pharmacopsychiatry. 1999;32(Suppl. 1):S49-S53.
Genton P., Van Vleymen B. Piracetam and levetiracetam: close structural similarities but different pharmacological and clinical profiles. Epileptic Disord. 2000;2(2):99-105.
Gentry M.S., Worby C.A., Dixon J.E. Insights into Lafora disease: Malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA. 2005;102:8501-8506.
Gordon M.F. Toxin and drug-induced myoclonus. Adv Neurol. 2002;89:49-76.
Gordon M.F., Diaz-Olivo R., Hunt A.L., Fahn S. Therapeutic trial of milacemide in patients with myoclonus and other intractable movement disorders. Mov Disord. 1993;8(4):484-488.
Grimes D.A., Han F., Lang A.E., et al. A novel locus for inherited myoclonus-dystonia on 18p11. Neurology. 2002;59(8):1183-1186.
Grosse P., Guerrini R., Parmeggiani L., et al. Abnormal corticomuscular and intermuscular coupling in high-frequency rhythmic myoclonus. Brain. 2003;126(Pt 2):326-342.
Guerrini R., Dravet C., Genton P., et al. Epileptic negative myoclonus. Neurology. 1993;43:1078-1083.
Hallett M. Early history of myoclonus. In: Fahn S., Marsden C.D., Van Woert M.H., editors. Myoclonus. New York: Raven Press; 1986:7-10.
Hallett M. Physiology of posthypoxic myoclonus. Mov Disord. 2000;15(Suppl 1):8-13.
Hallett M. Neurophysiology of brainstem myoclonus. Adv Neurol. 2002;89:99-102.
Hallett M. Physiology of psychogenic movement disorders. J Clin Neurosci. 2010;17(8):959-965.
Hallett M., Chadwick D., Adam J., Marsden C.D. Reticular reflex myoclonus: a physiological type of human post-hypoxic myoclonus. J Neurol Neurosurg Psychiatry. 1977;40(3):253-264.
Hallett M., Chadwick D., Marsden C.D. Ballistic movement overflow myoclonus a form of essential myoclonus. Brain. 1977;100(2):299-312.
Hallett M., Chadwick D., Marsden C.D. Cortical reflex myoclonus. Neurology. 1979;29(8):1107-1125.
Hallett M., Marsden C.D., Fahn S. Myoclonus. In: Vinken P.J., Bruyn G.W., Klawans H.L., editors. Handbook of Clinical Neurology. Amsterdam: Elsevier Science Publishers; 1987:609-625.
Hallett M., Shibasaki H. Myoclonus and myoclonic syndromes. In: Engel J.Jr, Pedley T.A., editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott, Williams & Wilkins; 2008:2765-2770.
Hanajima R., Okabe S., Terao Y., et al. Difference in intracortical inhibition of the motor cortex between cortical myoclonus and focal hand dystonia. Clin Neurophysiol. 2008;119(6):1400-1407.
Hart Y. Rasmussen’s encephalitis. Epileptic Disord. 2004;6(3):133-144.
Hurd R.W., Wilder B.J., Helveston W.R., Uthman B.M. Treatment of four siblings with progressive myoclonus epilepsy of the Unverricht-Lundborg type with N-acetylcysteine. Neurology. 1996;47(5):1264-1268.
Ikeda A., Kakigi R., Funai N., et al. Cortical tremor: a variant of cortical reflex myoclonus. Neurology. 1990;40:1561-1565.
Ikeda A., Shibasaki H., Tashiro K., et al. Clinical trial of piracetam in patients with myoclonus: nationwide multiinstitution study in Japan. The Myoclonus/Piracetam Study Group. Mov Disord. 1996;11(6):691-700.
Jabbari B., Rosenberg M., Scherokman B., et al. Effectiveness of trihexyphenidyl against pendular nystagmus and palatal myoclonus: evidence of cholinergic dysfunction. Mov Disord. 1987;2(2):93-98.
Jalanko A., Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793(4):697-709.
Jamieson D.R., Mann C., O’Reilly B., Thomas A.M. Ear clicks in palatal tremor caused by activity of the levator veli palatini. Neurology. 1996;46(4):1168-1169.
Jankovic J., Pardo R. Segmental myoclonus: clinical and pharmacological study. Arch Neurol. 1986;43:1025-1031.
Jimenez-Jimenez F.J., Roldan A., Zancada F., et al. Spinal myoclonus: successful treatment with the combination of sodium valproate and L-5-hydroxytryptophan. Clin Neuropharmacol. 1991;14(2):186-190.
Juul-Jensen P., Denny-Brown D. Epilepsia partialis continua. Arch Neurol. 1966;15(6):563-578.
Kalviainen R., Khyuppenen J., Koskenkorva P., et al. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia. 2008;49(4):549-556.
Kang S.Y., Sohn Y.H. Electromyography patterns of propriospinal myoclonus can be mimicked voluntarily. Mov Disord. 2006;21(8):1241-1244.
Kanthasamy A.G., Nguyen B.Q., Truong D.D. Animal model of posthypoxic myoclonus: II. Neurochemical, pathologic, and pharmacologic characterization. Mov Disord. 2000;15(Suppl 1):31-38.
Keswani S.C., Kossoff E.H., Krauss G.L., Hagerty C. Amelioration of spinal myoclonus with levetiracetam. J Neurol Neurosurg Psychiatry. 2002;73(4):457-458.
Kinugawa K., Vidailhet M., Clot F., et al. Myoclonus-dystonia: An update. Mov Disord. 2008;24(4):479-489.
Kompoliti K., Goetz C.G., Boeve B.F., et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol. 1998;55(7):957-961.
Koskiniemi M.L. Baltic myoclonus. Adv Neurol. 1986;43:57-64.
Koskiniemi M., Van Vleymen B., Hakamies L., et al. Piracetam relieves symptoms in progressive myoclonus epilepsy: a multicentre, randomised, double blind, crossover study comparing the efficacy and safety of three dosages of oral piracetam with placebo. J Neurol Neurosurg Psychiatry. 1998;64(3):344-348.
Krauss G.L., Bergin A., Kramer R.E., et al. Suppression of post-hypoxic and post-encephalitic myoclonus with levetiracetam. Neurology. 2001;56(3):411-412.
Krug P., Schleiermacher G., Michon J., et al. Opsoclonus-myoclonus in children associated or not with neuroblastoma. Eur J Paediatr Neurol. 2010;14(5):400-409.
Kurian M., Lalive P.H., Dalmau J.O., Horvath J. Opsoclonus-myoclonus syndrome in anti-N-methyl-D-aspartate receptor encephalitis. Arch Neurol. 2010;67(1):118-121.
Lagueny A., Tison F., Burbaud P., et al. Stimulus-sensitive spinal segmental myoclonus improved with injections of botulinum toxin type A. Mov Disord. 1999;14(1):182-185.
Lance J.W., Adams R.D. The syndrome of intention or action myoclonus as a sequel to hypoxic encephalopathy. Brain. 1963;86:111-136.
Lapresle J. Palatal myoclonus. Adv Neurol. 1986;43:265-273.
Lehesjoki A.E., Koskiniemi M. Clinical features and genetics of progressive myoclonus epilepsy of the Univerricht-Lundborg type. Ann Med. 1998;30(5):474-480.
Lehesjoki A.E., Koskiniemi M. Progressive myoclonus epilepsy of Unverricht-Lundborg type. Epilepsia. 1999;40(Suppl 3):23-28.
Lehtinen M.K., Tegelberg S., Schipper H., et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J Neurosci. 2009;29(18):5910-5915.
Leigh P.N., Rothwell J.C., Traub M., Marsden C.D. A patient with reflex myoclonus and muscle rigidity: “jerking stiff-man syndrome. J Neurol Neurosurg Psychiatry. 1980;43(12):1125-1131.
Leu-Semenescu S., Roze E., Vidailhet M., et al. Myoclonus or tremor in orthostatism: an under-recognized cause of unsteadiness in Parkinson’s disease. Mov Disord. 2007;22(14):2063-2069.
Lu C.S., Thompson P.D., Quinn N.P., et al. Ramsay Hunt syndrome and coeliac disease: a new association? Mov Disord. 1986;1(3):209-219.
Lu Y., Wang X. Genes associated with idiopathic epilepsies: a current overview. Neurol Res. 2009;31(2):135-143.
Luquin M.R., Scipioni O., Vaamonde J., et al. Levodopa-induced dyskinesias in Parkinson’s disease: clinical and pharmacological classification. Mov Disord. 1992;7(2):117-124.
Magaudda A., Gelisse P., Genton P. Antimyoclonic effect of levetiracetam in 13 patients with unverricht-lundborg disease: clinical observations. Epilepsia. 2004;45(6):678-681.
Maher J., Choudhri S., Halliday W., et al. AIDS dementia complex with generalized myoclonus. Mov Disord. 1997;12(4):593-597.
Manford M.R., Fish D.R., Shorvon S.D. Startle provoked epileptic seizures: features in 19 patients. J Neurol Neurosurg Psychiatry. 1996;61(2):151-156.
Mantoan L., Walker M. Treatment Options in Juvenile Myoclonic Epilepsy. Curr Treat Options Neurol. 2011. [epub ahead of print]
Markakis I., Alexiou E., Xifaras M., et al. Opsoclonus-myoclonus-ataxia syndrome with autoantibodies to glutamic acid decarboxylase. Clin Neurol Neurosurg. 2008;110(6):619-621.
Marsden C.D., Hallett M., Fahn S. The nosology and pathophysiology of myoclonus. In: Marsden C.D., Fahn S., editors. Movement Disorders. London: Butterworth Scientific; 1982:196-248.
Marsden C.D., Harding A.E., Obeso J.A., Lu C.S. Progressive myoclonic ataxia (the Ramsay Hunt syndrome). Arch Neurol. 1990;47(10):1121-1125.
Marseille Consensus Group. Classification of progressive myoclonus epilepsies and related disorders. Ann Neurol. 1990;28(1):113-116.
Mason P.J., Morris V.A., Balcezak T.J. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore). 2000;79(4):201-209.
Massimi L., Battaglia D., Paternoster G., et al. Segmental spinal myoclonus and metastatic cervical ganglioglioma: an unusual association. J Child Neurol. 2009;24(3):365-369.
Matsumoto J., Fuhr P., Nigro M., Hallett M. Physiological abnormalities in hereditary hyperekplexia. Ann Neurol. 1992;32:41-50.
Matsumoto J., Hallett M. Startle syndromes. In: Marsden C.D., Fahn S., editors. Movement Disorders 3. Oxford: Butterworth-Heinemann; 1994:418-433.
Minassian B.A., Andrade D.M., Ianzano L., et al. Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann Neurol. 2001;49(2):271-275.
Minassian B.A., Lee J.R., Herbrick J.A., et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171-174.
Minassian B.A., Sainz J., Serratosa J.M., et al. Genetic locus heterogeneity in Lafora’s progressive myoclonus epilepsy. Ann Neurol. 1999;45(2):262-265.
Monday K., Jankovic J. Psychogenic myoclonus. Neurology. 1993;43:349-352.
Moreno D., Towler M.C., Hardie D.G., et al. The Laforin-Malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol Biol Cell. 2010. Jun 9
Mukand J.A., Giunti E.J. Tizanidine for the treatment of intention myoclonus: a case series. Arch Phys Med Rehabil. 2004;85(7):1125-1127.
Nabbout R., Dulac O. Epileptic syndromes in infancy and childhood. Curr Opin Neurol. 2008;21(2):161-166.
Nardocci N., Zorzi G., Barzaghi C., et al. Myoclonus-dystonia syndrome: clinical presentation, disease course, and genetic features in 11 families. Mov Disord. 2008;23(1):28-34.
Nguyen B.Q., Kanthasamy A.G., Truong D.D. Animal models of myoclonus: an overview. Mov Disord. 2000;15(Suppl 1):22-25.
Obeso J.A. Therapy of myoclonus. Clin Neurosci. 1995;3(4):253-257.
Obeso J.A., Artieda J., Quinn N., et al. Piracetam in the treatment of different types of myoclonus. Clin Neuropharmacol. 1988;11:529-536.
Obeso J.A., Artieda J., Rothwell J.C., et al. The treatment of severe action myoclonus. Brain. 1989;112:765-777.
Obeso J.A., Rothwell J.C., Marsden C.D. The spectrum of cortical myoclonus: from focal reflex jerks to spontaneous motor epilepsy. Brain. 1985;108:193-224.
Oguni H., Uehara T., Tanaka T., et al. Dramatic effect of ethosuximide on epileptic negative myoclonus: implications for the neurophysiological mechanism. Neuropediatrics. 1998;29(1):29-34.
Pirio Richardson S., Mari Z., Matsuhashi M., Hallett M. Psychogenic palatal tremor. Mov Disord. 2006;21(2):274-276.
Pranzatelli M.R. The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol. 1992;15:186-228.
Pranzatelli M.R. The immunopharmacology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol. 1996;19(1):1-47.
Pranzatelli M.R., Huang Y., Tate E., et al. Cerebrospinal fluid 5-hydroxyindoleacetic acid and homovanillic acid in the pediatric opsoclonus-myoclonus syndrome. Ann Neurol. 1995;37(2):189-197.
Pranzatelli M.R., Nadi N.S. Mechanism of action of antiepileptic and antimyoclonic drugs. Adv Neurol. 1995;67:329-360.
Pranzatelli M.R., Tate E.D., Swan J.A., et al. B cell depletion therapy for new-onset opsoclonus-myoclonus. Mov Disord. 2010;25(2):238-242.
Pranzatelli M.R., Tate E.D., Travelstead A.L., Colliver J.A. Long-term cerebrospinal fluid and blood lymphocyte dynamics after rituximab for pediatric opsoclonus-myoclonus. J Clin Immunol. 2010;30(1):106-113.
Quinn N.P. Essential myoclonus and myoclonic dystonia. Mov Disord. 1996;11(2):119-124.
Raffaghello L., Conte M., De Grandis E., Pistoia V. Immunological mechanisms in opsoclonus-myoclonus associated neuroblastoma. Eur J Paediatr Neurol. 2008;13(3):219-223.
Ramachandran N., Girard J.M., Turnbull J., Minassian B.A. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia. 2009;50(Suppl 5):29-36.
Rapin I. Myoclonus in neuronal storage and Lafora diseases. Adv Neurol. 1986;43:65-85.
Rees M.I., Harvey K., Pearce B.R., et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38(7):801-806.
Riccio M., Di Giaimo R., Pianetti S., et al. Nuclear localization of cystatin B, the cathepsin inhibitor implicated in myoclonus epilepsy (EPM1). Exp Cell Res. 2001;262(2):84-94.
Rodriguez M.E., Artieda J., Zubieta J.L., Obeso J.A. Reflex myoclonus in olivopontocerebellar atrophy. J Neurol Neurosurg Psychiatry. 1994;57(3):316-319.
Rossi S., Ulivelli M., Bartalini S., et al. Reduction of cortical myoclonus-related epileptic activity following slow-frequency rTMS. Neuroreport. 2004;15(2):293-296.
Rothwell J.C., Obeso J.A., Marsden C.D. On the significance of giant somatosensory evoked potentials in cortical myoclonus. J Neurol Neurosurg Psychiatry. 1984;47(1):33-42.
Rothwell J.C., Obeso J.A., Marsden C.D. Electrophysiology of somatosensory reflex myoclonus. Adv Neurol. 1986;43:385-398.
Roze E., Apartis E., Clot F., et al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology. 2008;70(13):1010-1016.
Roze E., Bounolleau P., Ducreux D., et al. Propriospinal myoclonus revisited: Clinical, neurophysiologic, and neuroradiologic findings. Neurology. 2009;72(15):1301-1309.
Rudnick E., Khakoo Y., Antunes N.L., et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children’s Cancer Group Study. Med Pediatr Oncol. 2001;36(6):612-622.
Sabater L., Xifro X., Saiz A., et al. Analysis of antibodies to neuronal surface antigens in adult opsoclonus-myoclonus. J Neuroimmunol. 2008;196(1–2):188-191.
Sakai T., Murakami S. Palatal myoclonus responding to carbamazepine. Ann Neurol. 1981;9(2):199-200.
Schauer R., Singer M., Saltuari L., Kofler M. Suppression of cortical myoclonus by levetiracetam. Mov Disord. 2002;17(2):411-415.
Schwartz M.A., Selhorst J.B., Ochs A.L., et al. Oculomasticatory myorhythmia: a unique movement disorder occurring in Whipple’s disease. Ann Neurol. 1986;20(6):677-683.
Scott B.L., Evans R.W., Jankovic J. Treatment of palatal myoclonus with sumatriptan. Mov Disord. 1996;11(6):748-751.
Serratosa J.M., Gomez-Garre P., Gallardo M.E., et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet. 1999;8(2):345-352.
Shahwan A., Farrell M., Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4(4):239-248.
Shannon P., Pennacchio L.A., Houseweart M.K., et al. Neuropathological changes in a mouse model of progressive myoclonus epilepsy: cystatin B deficiency and Unverricht-Lundborg disease. J Neuropathol Exp Neurol. 2002;61(12):1085-1091.
Shiang R., Ryan S.G., Zhu Y.Z., et al. Mutational analysis of familial and sporadic hyperekplexia. Ann Neurol. 1995;38(1):85-91.
Shiang R., Ryan S.G., Zhu Y.Z., et al. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351-358.
Shibasaki H. Pathophysiology of negative myoclonus and asterixis. In: Fahn S., Hallett M., Lüders H.O., Marsden C.D., editors. Negative Motor Phenomena. Philadelphia: Lippincott-Raven Publishers; 1995:199-209.
Shibasaki H., Hallett M. Electrophysiological studies of myoclonus. Muscle Nerve. 2005;31(2):157-174.
Shibasaki H., Motomura S., Yamashita Y., et al. Periodic synchronous discharge and myoclonus in Creutzfeldt-Jakob disease: diagnostic application of jerk-locked averaging method. Ann Neurol. 1981;9(2):150-156.
Shields W.D. Diagnosis of infantile spasms, Lennox-Gastaut syndrome, and progressive myoclonic epilepsy. Epilepsia. 2004;45(Suppl 5):2-4.
Shin H.W., Ye B.S., Kim J., et al. The contribution of a spinal mechanism in developing peripheral myoclonus: a case report. Mov Disord. 2007;22(9):1350-1352.
Shprecher D., Silberstein H., Kurlan R. Propriospinal myoclonus due to cord compression in the absence of myelopathy. Mov Disord. 2010;25(8):1100-1101.
Singh S., Ganesh S. Lafora progressive myoclonus epilepsy: A meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat. 2009;30(5):715-723.
Singh S., Satishchandra P., Shankar S.K., Ganesh S. Lafora disease in the Indian population: EPM2A and NHLRC1 gene mutations and their impact on subcellular localization of laforin and malin. Hum Mutat. 2008;29(6):E1-E12.
Siniscalchi A., Mancuso F., Russo E., et al. Spinal myoclonus responsive to topiramate. Mov Disord. 2004;19(11):1380-1381.
Slawek J., Wichowicz H.M., Cubala W.J., et al. Psychogenic axial myoclonus: report on two cases. Neurol Sci. 2010;31(2):219-222.
Smith C.R., Scheinberg L. Coincidence of myoclonus and multiple sclerosis: dramatic response to clonazepam. Neurology. 1990;40(10):1633-1634.
Strafella A., Ashby P., Lang A.E. Reflex myoclonus in cortical-basal ganglionic degeneration involves a transcortical pathway. Mov Disord. 1997;12(3):360-369.
Suhren O., Bruyn G.W., Tuynman J.A. Hyperexplexia: a hereditary startle syndrome. J Neurol Sci. 1966;3:577-605.
Tan K.M., Lennon V.A., Klein C.J., et al. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70(20):1883-1890.
Tassinari C.A., Rubboli G., Parmeggiani L., et al. Epileptic negative myoclonus. In: Fahn S., Hallett M., Lüders H.O., Marsden C.D., editors. Negative Motor Phenomena. Philadelphia: Lippincott-Raven Publishers; 1995:181-197.
Tassinari C.A., Rubboli G., Shibasaki H. Neurophysiology of positive and negative myoclonus. Electroencephalogr Clin Neurophysiol. 1998;107(3):181-195.
Tate E.D., Allison T.J., Pranzatelli M.R., Verhulst S.J. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J Pediatr Oncol Nurs. 2005;22(1):8-19.
Terada K., Ikeda A., Van Ness P.C., et al. Presence of Bereitschaftspotential preceding psychogenic myoclonus: clinical application of jerk-locked back averaging. J Neurol Neurosurg Psychiatry. 1995;58:745-747.
Thomas J.E., Reagan T.J., Klass D.W. Epilepsia partialis continua. A review of 32 cases. Arch Neurol. 1977;34(5):266-275.
Thompson P.D., Colebatch J.G., Brown P., et al. Voluntary stimulus-sensitive jerks and jumps mimicking myoclonus or pathological startle syndromes. Mov Disord. 1992;7(3):257-262.
Thompson P.D., Day B.L., Rothwell J.C., et al. The myoclonus in corticobasal degeneration. Evidence for two forms of cortical reflex myoclonus. Brain. 1994;117(Pt 5):1197-1207.
Tijssen M.A., Padberg G.W., van Dijk J.G. The startle pattern in the minor form of hyperekplexia. Arch Neurol. 1996;53(7):608-613.
Tijssen M.A., Shiang R., van Deutekom J., et al. Molecular genetic reevaluation of the Dutch hyperekplexia family. Arch Neurol. 1995;52(6):578-582.
Tijssen M.A., Vergouwe M.N., van Dijk J.G., et al. Major and minor form of hereditary hyperekplexia. Mov Disord. 2002;17(4):826-830.
Tijssen M.A., Voorkamp L.M., Padberg G.W., van Dijk J.G. Startle responses in hereditary hyperekplexia. Arch Neurol. 1997;54(4):388-393.
Toro C., Hallett M. Pathophysiology of myoclonic disorders. In: Watts R.L., Koller W.C., editors. Movement Disorders. second ed. New York: McGraw-Hill; 2004:671-681.
Traff J., Holme E., Ekbom K., Nilsson B.Y. Ekbom’s syndrome of photomyoclonus, cerebellar ataxia and cervical lipoma is associated with the tRNA(Lys) A8344G mutation in mitochondrial DNA. Acta Neurol Scand. 1995;92(5):394-397.
Truong D.D., Kanthasamy A., Nguyen B., et al. Animal models of posthypoxic myoclonus: I. Development and validation. Mov Disord. 2000;15(Suppl 1):26-30.
Tyvaert L., Krystkowiak P., Cassim F., et al. Myoclonus of peripheral origin: two case reports. Mov Disord. 2009;24(2):274-277.
Vaamonde J., Legarda I., Jimenez-Jimenez J., Obeso J.A. Acetazolamide improves action myoclonus in Ramsay Hunt syndrome. Clin Neuropharmacol. 1992;15:392-396.
van der Salm S.M., Koelman J.H., Henneke S., et al. Axial jerks: a clinical spectrum ranging from propriospinal to psychogenic myoclonus. J Neurol. 2010;257(8):1349-1355.
Weizman D.A., Leong W.L. Anti-Ri antibody opsoclonus-myoclonus syndrome and breast cancer: a case report and a review of the literature. J Surg Oncol. 2004;87(3):143-145.
Werhahn K.J., Brown P., Thompson P.D., Marsden C.D. The clinical features and prognosis of chronic posthypoxic myoclonus. Mov Disord. 1997;12(2):216-220.
Wijdicks E.F., Parisi J.E., Sharbrough F.W. Prognostic value of myoclonus status in comatose survivors of cardiac arrest. Ann Neurol. 1994;35(2):239-243.
Wilkins D.E., Hallett M., Berardelli A., et al. Physiologic analysis of the myoclonus of Alzheimer’s disease. Neurology. 1984;34:898-903.
Wilkins D.E., Hallett M., Erba G. Primary generalised epileptic myoclonus: a frequent manifestation of minipolymyoclonus of central origin. J Neurol Neurosurg Psychiatry. 1985;48(6):506-516.
Williams D.R., Cowey M., Tuck K., Day B. Psychogenic propriospinal myoclonus. Mov Disord. 2008;23(9):1312-1313.
Yigit A., Sarikaya S. Myoclonus relieved by carbamazepine in subacute sclerosing panencephalitis. Epileptic Disord. 2006;8(1):77-80.
Yiu V.W., Kovithavongs T., McGonigle L.F., Ferreira P. Plasmapheresis as an effective treatment for opsoclonus-myoclonus syndrome. Pediatr Neurol. 2001;24(1):72-74.
Yoshimura I., Kaneko S., Yoshimura N., Murakami T. Long-term observations of two siblings with Lafora disease treated with zonisamide. Epilepsy Res. 2001;46(3):283-287.
Yu H.Y., Kwan S.Y., Lirng J.F., et al. Epileptic negative myoclonus: SPECT, PET, and video/EEG studies and the dramatic effects of levetiracetam. Epilepsy Behav. 2009;14(4):687-690.
Zimprich A., Grabowski M., Asmus F., et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29(1):66-69.
Zupanc M.L., Legros B. Progressive myoclonic epilepsy. Cerebellum. 2004;3(3):156-171.