[level-membership-for-neurology-category]
Chapter 56 Myoclonic Seizures and Infantile Spasms
Introduction
Myoclonus, myoclonic seizures, and infantile spasms share many common features yet are seen in a wide variety of neurologic conditions. Myoclonus is not a diagnosis, but rather a sign that can have many underlying etiologies. The precise definitions of each of these terms has also been somewhat controversial, but for the purpose of this chapter we will use the definition proposed by Victor and Adams [Ropper and Samuels, 2009] and define myoclonus simply as a “shocklike irregular jerk,” with myoclonic seizures having a similar clinical appearance but also accompanied by neurophysiologic evidence of being cortically generated. While these definitions each have their own potential flaws, the distinction between a cortically generated movement (myoclonic seizure) and a subcortically generated movement (myoclonus) is extremely important, especially when thinking about some of the progressive myoclonic epilepsies, as many of these patients can manifest both myoclonus and myoclonic seizures. Infantile spasms, however, should not be confused with either, as the spasm itself is often a more complex constellation of movements with a myoclonic component.
Epilepsy Syndromes with Prominent Myoclonic Seizures
Myoclonic seizures can occur in a wide variety of pediatric epilepsy syndromes, including Lennox–Gastaut syndrome and childhood absence epilepsy, which are discussed elsewhere in Part VIII. Our focus will be on those syndromes in which myoclonus is a critical feature for the diagnosis. This coverage includes benign myoclonic epilepsy in infants (BME), severe myoclonic epilepsy in infancy (SMEI/Dravet’s syndrome), idiopathic epilepsy with myoclonic-astatic seizures (IEMAS), and juvenile myoclonic epilepsy (JME) (Table 56-1).
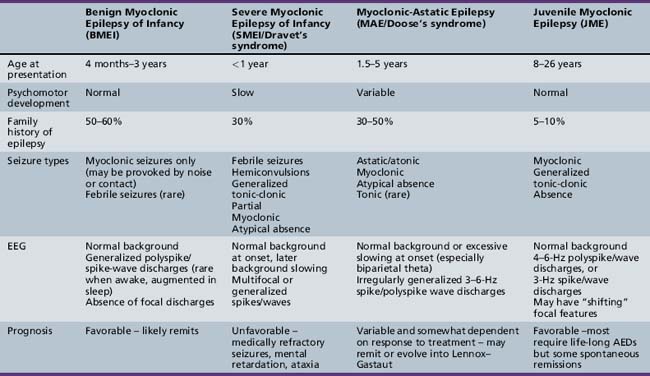
Benign Myoclonic Epilepsy of Infancy
BMEI was first described by Dravet and Bureau in 1981 [Dravet and Bureau, 1981]. The syndrome is characterized by brief, generalized myoclonic seizures, with the predominant area of muscle involvement being the proximal upper extremities [Hirano et al., 2009], and usually occurs in children between the ages of 4 months and 3 years, although cases have been described with onset up to 4 years 8 months [Rossi et al., 1997]. These attacks often occur multiple times per day. Detailed electroencephalography (EEG) and polygraphic electromyography (EMG) recordings have shown that the myoclonic seizures are often associated with a flexor postural change and approximately 80 percent of attacks involve the upper limbs [Hirano et al., 2009]. A history of febrile seizures (30 percent) and a family history of epilepsy (39 percent) is relatively common [Roger et al., 2002].
EEG Findings
The ictal EEG discharge is often a generalized spike wave (GSW) that may be slower than 3 Hz [Hirano et al., 2009]. Occasionally, the myoclonic seizures can be massive and result in a fall, or they may occur in a cluster. Some infants may exhibit photosensitivity at the onset of the syndrome, and the photic-induced myoclonic seizures are often more prominent than those that occur spontaneously [Capovilla et al., 2007]. Other infants may be sensitive to acoustic or tactile stimuli, leading some to argue for a distinctly separate epilepsy syndrome [Ricci et al., 1995]. We and others [Roger et al., 2002] do not believe this is a clinically distinct syndrome, but rather that the syndrome of BME may include both photosensitive and stimulus-provoked seizures in addition to spontaneous myoclonic seizures. In both groups, the waking EEG in between seizures is often normal but may contain rare generalized spike/wave discharges [Ricci et al., 1995; Capovilla et al., 2007], and the ictal EEG findings are indistinguishable.
Treatment and Outcome
The overall prognosis of myoclonic seizures is excellent, with complete resolution in almost all children usually within 1–2 years of diagnosis. In those who demonstrate photosensitivity, these seizures may be more difficult to control and may persist for a longer period of time [Roger et al., 2002]. The majority of children also have normal neurodevelopmental outcomes [Caraballo et al., 2009], although some studies have reported impaired psychomotor development and behavioral disturbances if the child is not treated or if onset of the syndrome is at less than 2 years of age [Mangano et al., 2005]. Those children with a prominent reflex component may have a more favorable neurodevelopmental outcome [Zuberi and O’Regan, 2006].
The medication of choice appears to be valproic acid (VPA), with 80–90 percent responding to VPA monotherapy, although high serum levels (>100 mg/L) may be necessary. When VPA monotherapy does not provide complete control, adjunctive use of a benzodiazepine, such as clonazepam or clobazam, can be helpful [Rossi et al., 1997].
Severe Myoclonic Epilepsy in Infancy
First described in 1978 by Charlotte Dravet, SMEI occurs in normally developing children who experience prolonged febrile (>20 minutes) or afebrile seizures, including hemiconvulsions, during the first year of life. Afebrile, mixed seizures follow, and the emergence of myoclonic seizures is common but not necessary for the diagnosis (borderline variant). For this reason, the syndrome is better referred to as Dravet’s syndrome, as the name severe myoclonic epilepsy of infancy may mislead clinicians and cause them not to suspect the diagnosis if myoclonic seizures are absent. The syndrome was once thought to be exceedingly rare, with an incidence of 1 in 40,000 children [Hurst, 1990], but with heightened awareness of the clinical spectrum and the availability of genetic testing, this may be an underestimation of the true incidence.
Mutations in the SCN1A Gene
The importance of SCN1A gene mutations as an underlying etiology in epilepsy first became apparent when Scheffer and Berkovic reported missense mutations in two families with the syndrome of generalized epilepsy with febrile seizures plus (GEFS+) [Scheffer and Berkovic, 1997]. Due to the common occurrence of febrile seizures in both GEFS+ and Dravet’s syndrome, Claes et al. screened seven patients with Dravet’s syndrome and found de novo missense mutations in all seven patients [Claes et al., 2001]. Testing for mutations in the SCN1A gene is clinically available, and hundreds of patients with Dravet’s syndrome with SCN1A mutations have been reported [Depienne et al., 2009]. The main difference between individuals with a GEFS+ phenotype and a Dravet phenotype appears to be that the former often have a missense mutation with reduced penetrance, whereas patients with Dravet have mutations that occur in isolated patients (arise de novo) [Claes et al., 2001].
Mutations in the SCN1A gene are seen in approximately 70–85 percent of patients with Dravet’s syndrome [Harkin et al., 2007], and therefore it remains a clinical diagnosis based on age of onset and clinical phenotype. One should not exclude the diagnosis on the basis of a negative result on SCN1A mutation testing. Approximately 200 different point mutations in the SCN1A gene were identified in 271 probands with a strict clinical diagnosis of Dravet’s syndrome [Depienne et al., 2009]. Of those probands without a point mutation, the multiplex ligation-dependent probe amplification (MPLA) technique found an additional 14 micro-rearrangements of SCN1A [Depienne et al., 2009]. An SCN1A variant database was published in 2009, listing each type of mutation and the associated epilepsy syndrome [Claes et al., 2009]. As of 2009, 648 point mutations have been reported, of which 582 (90 percent) are in subjects with Dravet’s syndrome. An additional 67 genomic rearrangements were reported, with 46 (7 percent) of these occurring in patients with Dravet’s syndrome. The remaining clinical phenotypes with SCN1A mutations are wide and varied [Harkin et al., 2007]. In Dravet’s syndrome, the end result of each of these mutations appears to be a total loss of function of the mutated allele, most commonly caused by a missense mutation that leads to an abnormality in the pore-forming unit of the sodium channel [Claes et al., 2009]. While specific mutation genotype–phenotype correlations within Dravet’s syndrome are not yet available, further research in this area may lead to particular treatment strategies for specific genotypes.
Seizure Semiologies
Patients with Dravet’s syndrome have many different seizure semiologies, often occurring in the same patient. The most common presenting seizure type is a prolonged febrile convulsion lasting longer than 25 minutes, but afebrile seizures can be the presenting seizure type in as many as 35 percent [Roger et al., 2002]. Japanese authors have observed that hot water immersion can trigger seizures in these patients [Fujiwara et al., 1990], and this feature is one part of a clinical screening test aimed at predicting the diagnosis of Dravet’s syndrome [Hattori et al., 2008]. Other convulsive seizure types include generalized tonic-clonic seizures, hemiclonic seizures, and “falsely generalized,” seizures as described by Dravet [Roger et al., 2002]. The latter has a complex semiology, with a high degree of discrepancy between the clinical and EEG findings. Seizures often start in one part of the body, spread to another (on the opposite side), and then potentially return to the original side of origin, only to involve a new body part.
Nonconvulsive seizures are also common and include simple partial, complex partial, atypical absence, and myoclonic types. The specific semiologies of each of these seizure types are similar to that seen in other forms of childhood epilepsy. Tonic seizures are rare. A unique seizure type, termed “obtundation status,” is unique to the syndrome. The semiology of this seizure type consists of variable impairment in consciousness, and fragmentary and erratic segmental myoclonias involving the limbs and face. Patients may still be able to engage in simple activities, such as eating or playing with a toy. These episodes may last several hours to days and can either be initiated by, or conclude with, a convulsion [Roger et al., 2002]. The EEG during these episodes is not a pattern of classical atypical absence status, but rather is characterized by a diffuse dysrhythmia with focal or diffuse spikes [Roger et al., 2002]. We have observed one patient, who would begin with this type of clinical phenomenon, including prominent eyelid myoclonia that was dramatically accentuated with eye closure, and would remain in this state for 2–3 days, culminating in a generalized convulsion, after which her mental status cleared. She would remain clear for 2–3 weeks, and then repeat the same cycle over again.
EEG Findings
The EEG is invariably normal early in the course of the disease, and therefore, in contrast with other epilepsy syndromes, the diagnosis must be suspected on clinical grounds. As the disease progresses, the EEG background becomes slow but often continues to manifest normal sleep architecture [Roger et al., 2002]. Some authors have observed rhythmic 5–6-Hz centroparietal theta but this is not specific to the syndrome [Doose et al., 1998]. Over time, the interictal EEG can show both generalized and multifocal spikes, or may not show any of these features despite frequent seizures of multiple types, further highlighting the lack of specificity of EEG findings. Given this lack of specificity, a screening test for predicting the diagnosis of Dravet’s syndrome under 1 year of age has been developed, with a positive predictive value as high as 94 percent [Hattori et al., 2008]. This screening test uses simple clinical features, as shown in Table 56-2. A sum score of 6 or greater is associated with a high risk of having Dravet’s syndrome, and therefore SCN1A testing should be done in those cases. Even if the hot water-induced risk factor is excluded, a score of 5 or higher is still indicative of a high-risk patient.
| Clinical Score | Risk Score |
|---|---|
| Onset <7 months | 2 |
| More than 5 seizures prior to 12 months of age | 3 |
| Hemiconvulsion | 3 |
| Focal seizure | 1 |
| Myoclonic seizure | 1 |
| Prolonged seizure (>10 minutes) | 3 |
| Hot water-induced seizure | 2 |
(Adapted from Hattori J et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia 2008;49(4):626–633.)
Imaging
Magnetic resonance imaging (MRI) studies are usually normal but may show nonspecific cerebral or cerebellar atrophy or isolated ventricular enlargement. Interestingly, these findings, specifically diffuse brain atrophy, appear to be more common in those children without associated SCN1A mutations. Furthermore, despite having frequent prolonged febrile and afebrile convulsions, mesial temporal sclerosis is rare, reported in only 1 of 58 patients [Striano et al., 2007].
Treatment
Until recently, treatment strategies were generally unsuccessful, with poor seizure control despite polytherapy. Many have observed that certain antiepileptic drugs (AEDs) exacerbate seizures, with the most notorious agents being carbamazepine (CBZ) and lamotrigine (LTG) [Wakai et al., 1996; Guerrini et al., 1998]. Some authors have recommended using CBZ as a means of “confirming” the diagnosis when there is a high index of clinical suspicion [Wakai et al., 1996]. Given the availability of genetic testing, we do not endorse this practice.
A study by Chiron et al. and a subsequent meta-analysis pooling these data with unpublished data showed that combination therapy with valproate (VPA) + stiripentol (STP) + clobazam (CLB) resulted in a 70 percent decrease in seizure frequency, with 43 percent of patients becoming seizure-free [Chiron et al., 2000; Kassai et al., 2008]. In this study, the STP dose began at 50 mg/kg/day divided in 2–3 doses, but could be titrated up to 100 mg/kg/day. The maximum recommended dose is 3500 mg/day [Chiron et al., 2000].
One proposed mechanism for the effect of STP is the potent inhibition of the p450 cytochrome system, resulting in higher levels of VPA and CLB. Plasma levels of CLB were significantly higher but levels of VPA were not [Chiron et al., 2000]. Interestingly, one other result of p450 inhibition is lower levels of metabolites, some of which are thought to explain some of the adverse toxic effects of AEDs. This finding perhaps explains why patients are able to tolerate the higher doses of certain AEDs [Chiron, 2005]. These observations, however, raise the possibility that STP has other mechanisms of action independent of its effects on the p450 system, including enhancement of gamma-aminobutyric acid (GABA)ergic transmission [Quilichini et al., 2006].
Of the newer agents, topiramate (TPM) appears to be very effective, especially if added to STP, resulting in 50 percent reduction of seizures in 78 percent of patients at a modest optimal dose of 3.2 mg/kg/day. Seventeen percent of patients remained seizure-free for at least 4 months [Kroll-Seger et al., 2006].
Bromide therapy has also shown promise in several studies [Oguni et al., 1994], specifically in reducing the number of convulsions. The ketogenic diet may be helpful in some patients [Caraballo et al., 1998].
Aggressive treatment of acute seizures and prevention of status epilepticus is critical, as some small studies have shown a trend towards improved neuropsychologic outcomes in those children with less frequent convulsive seizures [Chipaux et al., 2008]. Each patient should have a clearly defined “emergency plan,” and the clinician should ask each family if a particular treatment regimen has been particularly effective for their child [Nolan et al., 2006]. Parents should be educated about the inevitability of seizures, especially prolonged seizures in the setting of fever. Early use of high doses of benzodiazepines, either in the form of rectal diazepam (as high as 1 mg/kg per dose) or buccal midazolam, should be the first-line therapy. Some physicians have recommended insertion of a central venous port for those children who have proven to have difficult intravenous access and multiple episodes of status epilepticus [Dooley et al., 1995]. In these specific cases, families are able to cope much better, knowing that multiple intravenous attempts will not be necessary and appropriate treatment can be initiated to terminate the seizure faster.
Outcome
Long-term outcome with regard to seizure control and neuropsychological development is variable but generally poor. By definition, all children with Dravet’s syndrome have normal development at onset. Around 50 percent of children walk unsupported by a mean of 16 months of age; however, it is rare for children to utilize two-word sentences at the normal age of around 2 years [Wolff et al., 2006]. At around 2 years of age, there appears to be a gradual decline, and then there is a relative stabilization between the ages of 4 and 6 years [Wolff et al., 2006]. A trend was noted that fewer convulsive seizures often resulted in higher developmental quotients. Subsequent small cohorts have reported patients with normal or near-normal IQs and this favorable outcome is attributed to early diagnosis and appropriate management [Chipaux et al., 2008]. It remains to be seen in larger prospective studies whether early diagnosis and appropriate management with the medications outlined above will lead to improved seizure control and a subsequent improvement in neurodevelopmental outcome.
Myoclonic-Astatic Epilepsy of Doose
Etiology
Genetic factors clearly play a role in the pathogenesis of MAE, based on the high incidence of seizures (32 percent) in probands’ siblings and parents; the most common seizure type is absence seizures. Despite the initial discovery of the SCN1A gene in a family with GEFS+ that included a family member with MAE [Scheffer and Berkovic, 1997], only a small number of patients with mutations in the SCN1A gene have been identified in patients with MAE [Ebach et al., 2005; Harkin et al., 2007]. Further genotype–phenotype correlation studies are necessary to determine if there are underlying genetic factors that would be able to predict response to treatment and outcome.
Seizure Semiologies
Onset of MAE is often between the ages of 2 and 6 years, and children are usually developmentally normal [Kaminska et al., 1999]. These children may begin experiencing unexplained “jerks” and “falls,” which occur multiple times per day. These myoclonic seizures can take place in isolation, but often are followed by a brief period of atonia that results in a dramatic fall in which the child appears to be propelled to the ground. Polygraphic studies have documented that atonic falls can occur with or without the preceding myoclonus, and occur in about 64 percent of patients [Oguni et al., 2002]. Although atonic falls are not necessarily seen in all patients, more subtle myoclonic/atonic seizures are common. The only manifestation of these may be a brief head nod or “head drop,” with or without a myoclonic jerk of the upper extremities. When head drops occur in clusters with an associated alteration in level of consciousness, nonconvulsive status should be suspected. EEG findings during these episodes are always abnormal, with frequent runs of generalized spike/wave and slow spike-and-wave complexes. Absence seizures are quite common and may be similar to those seen in typical childhood absence epilepsy, but often are accompanied by more prominent myoclonus of the proximal upper extremities.
Treatment
Early recognition of the syndrome allows treatment to be focused on the broad-spectrum agents that have been shown to be particularly effective. The most commonly used and most effective AEDs have been VPA, LTG, levatiracetam, TPM and various benzodiazepines (clonazepam, clobazam, clorazepate). When absence seizures are frequent, ethosuximide (ETX) can be effective and has also been reported to decrease the frequency of myoclonic seizures; however, it is rarely used as monotherapy for this indication [Oguni et al., 2002]. The combination of VPA/ETX may be particularly effective, especially if high doses of VPA are used.
In those children who are refractory to medical management, a trial of the ketogenic diet should be considered early. The ketogenic diet has been shown to be as effective as other first-line agents in children with MAE [Kilaru and Bergqvist, 2007], and given this efficacy, we consider the ketogenic diet early in our treatment algorithm, especially if there is associated neurodevelopmental decline.
Outcome
The outcome with regard to cognitive development is highly variable but appears to be somewhat dependent on degree of seizure control. Some children may have fairly frequent seizures of multiple types and still have spontaneous remission, usually after approximately 3 years [Kaminska et al., 1999; Oguni et al., 2002]. In some children, the multiple seizure types remain intractable and these children have a higher risk of both cognitive and behavioral disturbances [Oguni et al., 2002]. The occurrence of tonic seizures has been described as a negative prognostic sign, although it remains unclear if these children may actually have an atypical form of Lennox–Gastaut syndrome rather than an atypical form of MAE [Kaminska et al., 1999].
Juvenile Myoclonic Epilepsy
JME is an idiopathic generalized epilepsy (IGE) syndrome characterized by myoclonic seizures, generalized tonic-clonic seizures, and absence seizures. It is extremely common, accounting for 26 percent of IGE and 10 percent of all epilepsies [Janz and Durner, 1998], although this may still be an underestimation; it is very likely underdiagnosed, as many clinicians may not inquire about the presence of myoclonic seizures [Renganathan and Delanty, 2003]. Mean age at onset is highly variable and difficult to determine truly, as some children with childhood absence epilepsy may evolve into the syndrome of JME. Excluding this population, the average age of onset is 15.1 years (7–28 years), with a slight female predominance [Martinez-Juarez et al., 2006].
Seizure Semiologies
In the classic syndrome of JME, myoclonic seizures may precede the first generalized tonic-clonic (GTC) seizure by 6–12 months, although GTCs occur as the first seizure type in approximately one-third of patients [Martinez-Juarez et al., 2006]. Some have proposed specific subgroups of JME, separating those patients who present with typical childhood absence or juvenile absence seizures, although this is much less common than the classical presentation of JME, accounting for only 10 percent of cases. The impact this distinction has on outcome will be discussed below. Photosensitivity is relatively common, occurring in approximately 30 percent of patients [Zifkin et al., 2005].
Myoclonic seizures often occur in the early morning hours shortly after awaking. Transcranial magnetic stimulation studies in patients with JME have demonstrated increased cortical excitability that is not present in other patients with focal epilepsy [Badawy et al., 2006]. Provocation of myoclonic seizures has been linked to higher cognitive tasks requiring higher-order thinking, such as writing or written calculation [Matsuoka et al., 2000].
Specific lifestyle features have been commonly associated with breakthrough seizures, including sleep deprivation, stress, alcohol, and menses [Martinez-Juarez et al., 2006].
EEG Findings
Patients with JME often have abnormal interictal EEGs, with the most common finding being generalized 4–6-Hz polyspike-and-wave (61 percent) or 3-Hz spike-and-wave discharges (14 percent) [Martinez-Juarez et al., 2006]. More prolonged (1–6 days; average 1–2 days) video EEG studies have shown EEG abnormalities of JME in 88 percent of patients [Park et al., 2009]. Other series have reported on the increased yield of an early morning EEG compared to an afternoon EEG (generalized epileptiform activity in 69 percent and 25 percent, respectively) [Labate et al., 2007]. A photoparoxysmal response in treatment-naive patients is also common and has been seen in as many as 35 percent of patients [Specchio et al., 2008].
Pathophysiology
It has long been accepted that the IGEs have a strong genetic component [Berkovic et al., 1998], and given the incidence of JME as a subtype of IGE, much research has been done to understand the underlying genetic basis of JME better. While JME appears to be a relatively homogeneous epilepsy syndrome, there are a number of studies implicating various genetic abnormalities. These include mutations in many different ion channels, including calcium, potassium, and chloride channels linked to at least seven different loci. There are also nonionic mechanisms that relate to neural migration during cortical development, and may explain some of the imaging and postmortem pathologic findings seen in these patients [de Nijs et al., 2009]. For further details about the specific genetic mutations and channels involved, please refer to Chapter 52.
Imaging
Imaging studies in patients with JME have indicated subtle structural changes in mesiofrontal cortex, with an increase in cortical gray matter [Woermann et al., 1999], although this has not been replicated in subsequent studies [Roebling et al., 2009]. These findings are interesting, given that the generalized discharges seen on routine EEGs often have a frontal predominance. This has been further investigated with dense array EEG, which further localized typical 4–6-Hz epileptiform activity to the orbitofrontal/medial frontopolar cortex, whereas some patients also demonstrated basal-medial-temporal sources [Holmes et al., 2010]. These electrophysiologic and anatomic abnormalities appear to be concordant with some of the neuropsychological deficits that are seen in patients with JME. Specific attention has been paid to executive dysfunction that has been demonstrated to correlate with smaller thalamic volumes and more frontal cerebrospinal fluid volumes in children with recent-onset JME when compared to children with benign rolandic epilepsy [Pulsipher et al., 2009].
Treatment
Valproate has long been considered the first-line agent, with reports of up to 86 percent of patients becoming seizure-free for at least 1 year [Penry et al., 1989]. Valproate has also been shown to be particularly effective in those patients with photosensitivity [Covanis et al., 2004]. Despite this efficacy, valproate has many side effects that may lead to significant comorbidities, including weight gain, hyperactivity, transaminitis, thrombocytopenia, pancreatitis. Women of child-bearing age must also consider the possible long-term cognitive effects on a fetus [Meador et al., 2009]. Due to these concerns, many newer AEDs are also commonly used, including lamotrigine, topiramate, zonisamide, and levetiracetam.
Lamotrigine is now accepted to be a broad-spectrum AED that is effective against both partial and generalized seizures [Biton et al., 2005]. Small studies have also shown it to be effective specifically in the management of patients with JME, both as monotherapy and polytherapy, resulting in seizure-free rates between 40 and 83 percent [Buchanan, 1996; Prasad et al., 2003]. LTG is also very well tolerated; however, it does have the potential for exacerbating myoclonic seizures [Prasad et al., 2003; Crespel et al., 2005]. The combination of LTG and VPA has been reported to be particularly effective in both partial and generalized epilepsy, including JME, and suggests the possibility of a synergistic effect [Brodie and Yuen, 1997].
Topiramate is also a broad-spectrum AED that has been shown to be effective in patients with JME, although there are no published studies on its use as monotherapy [Biton and Bourgeois, 2005]. It appears to be equally effective as LTG and VPA when used as polytherapy, although there is a suggestion that tolerability may be inferior [Prasad et al., 2003].
Zonisamide (ZNS) is another broad-spectrum AED with multiple proposed mechanisms of action. Small studies have shown ZNS to be broadly effective against all of the seizure types in JME, with 69, 62, and 38 percent of patients being free of GTC, myoclonic, and absence seizures, respectively [Kothare et al., 2004].
Levetiracetam is gaining a reputation as a broad-spectrum agent, as it has shown to be effective in patients with various forms of idiopathic generalized epilepsies including epilepsy with eyelid myoclonias and absence [Striano et al., 2009], as well as JME [Specchio et al., 2006; Noachtar et al., 2008]. In a study of 120 patients (93 percent of whom had JME) randomized to add-on levetiracetam or placebo, 25 percent were free of myoclonic seizures and 21.7 percent were free of all seizures, including GTCs, during the 12-week treatment phase [Noachtar et al., 2008]. In another open-label study of 48 patients with JME (10 newly diagnosed and 38 resistant to prior treatment), 73 percent were free of GTCs and 37.5 percent were free of myoclonic seizures over the study period, with a mean follow-up of 19 months [Specchio et al., 2006].
Benzodiazepines are widely accepted as having excellent antimyoclonic effects. This is also true of the myoclonic seizures seen in JME, with up to 88 percent of patients on clonazepam having complete control of myoclonic seizures but only 43 percent becoming free of GTCs [Obeid and Panayiotopoulos, 1989]. For this reason, benzodiazepines are not recommended as monotherapy for patients with JME, but can be extremely successful as add-on therapy when myoclonic seizures persist.
Outcomes
It has been widely accepted that JME is a lifelong condition with a high rate of relapse if weaned off AED therapy; however, some recent long-term follow-up studies have shown conflicting results [Martinez-Juarez et al., 2006; Baykan et al., 2008; Camfield and Camfield, 2009]. Furthermore, one must look at specific seizure types, as it appears the natural history of myoclonic seizures decreases after the fourth decade of life, independent of the on-going occurrence of the other seizure types [Baykan et al., 2008].
In contrast, patients in the CAE evolving into JME group fared much worse, with only 3 of 35 individuals achieving complete seizure freedom [Martinez-Juarez et al., 2006]. Although GTCs were controlled with medications in about 66 percent of patients, absence seizures persisted in 63 percent. These data support the conclusion that JME does appear to be a lifelong disease, specifically in the subgroup of patients with CAE evolving into JME. However, there is still a small subgroup (9 percent) of patients in whom the disease may not be lifelong, and based on these data, it is uncertain whether some of the patients who have been seizure-free on monotherapy for many years would be able to come off AED treatment successfully.
The study by Camfied and Camfield seems to suggest that this indeed may be a possibility. In 24 JME patients with a follow-up period of 25.8 years, 4 individuals (17 percent) were free of all seizures and off AEDs. The mean duration of therapy was 6.5 years before discontinuation. Two additional patients attempted to come off AEDs; they relapsed but later were able to be successfully weaned off medication and remain seizure-free. Another three patients (12.5 percent) had myoclonus only [Camfield and Camfield, 2009]. These results suggest that some patients with JME can successfully discontinue medications. Unfortunately, there do not appear to be clinical or EEG variables that can predict which patients will achieve this result. The decision to discontinue AEDs should be individualized for every patient, and a detailed discussion, with specific attention to the frequency of lifestyle provoking factors, is necessary in order to arrive at a safe and well-informed decision.
Progressive Myoclonic Epilepsies
The PME’s are a group of genetically inherited disorders characterized by both epileptic and nonepileptic myoclonus, generalized tonic-clonic seizures, and progressive neurological deterioration, resulting in dementia, ataxia, and various forms of tremor. Recent advances in the genetic basis of these disorders are leading to a better understanding of how each of the disease processes differs. The four most common PMEs will be discussed (Table 56-3).
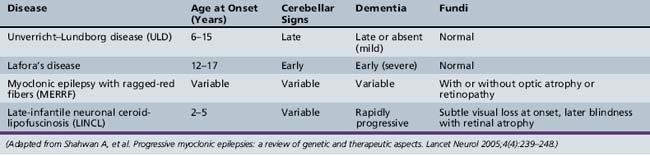
Unverricht–Lundborg Disease (ULD)
Unverricht–Lundborg disease (ULD) is the most common PME. It usually presents between the ages of 6 and 15 years, with the hallmark presenting symptom being stimulus-sensitive or action myoclonic jerks, which occur in up to half of patients [Lehesjoki, 2002]. Generalized tonic-clonic seizures are also a common feature early in the disease. At initial presentation, neurological examination and EEG studies may be normal, or the EEG may show generalized spike/wave discharges similar to those seen in IGE. It is not until the patient shows signs of progressive ataxia, intention tremor. and dysarthria that one begins to suspect PME clinically. As the disease progresses, the EEG becomes slow, with more frontally predominant polyspike/wave or spike-wave discharges, ranging in frequency between 2 and 6 Hz [Kyllerman et al., 1991]. MRI findings may be normal or may show nonspecific findings, such as cerebellar atrophy and reduced bulk of the basis pontis and medulla [Mascalchi et al., 2002].
The underlying cause of ULD is linked to an abnormality in the cystatin B gene on chromosome 21q22.3 [Lehesjoki et al., 1991]. Although the exact pathophysiology is unknown, mutations in cystatin B appear to lead to accelerated apoptosis, which may explain the progressive neurologic decline [Delgado-Escueta et al., 2001].
The diagnosis of ULD can be made by mutation testing of the cystatin B gene. There is no specific treatment for ULD, but improvements in seizure management with newer-generation AEDs has improved overall prognosis. VPA has been the mainstay of initial treatment; however, recent studies have shown a beneficial effect of levetiracetam, especially if started early [Magaudda et al., 2004].
Lafora’s Disease
Lafora’s disease (LD) has slightly later onset than ULD but has a more rapid decline, with many patients dying within 10 years of onset. Patients share similar clinical characteristics with other PMEs featuring seizures and myoclonus, but have a more prominent dementing component. Seizure types can be varied, but one peculiar type is that of occipital seizures with hallucinations and transient blindness [Minassian, 2001]. All patients are initially normal neurologically, but may have seizures indistinguishable from IGE until progressive neurological decline is noted. EEG findings early in the course may also resemble IGE, including photosensitivity, but the photosensitivity appears to be maximal at low frequencies between 1 and 6 Hz [Kobayashi et al., 1990]. As the disease progresses, the EEG background becomes slow and disorganized, and there is an evolution of the spike/wave discharges, the 3 Hz discharges becoming faster 6–12 Hz ones [Yen et al., 1991].
The underlying cause of LD is linked to two different sites on chromosome 6 – 6q24 and 6p22 – coding for two different genes: EPM2A (larforin) and NHLRC1 respectively [Minassian et al., 1998; Chan et al., 2003]. The former appears to be involved in regulation of protein folding and the latter may play a role in dendritic transport in neurons. How dysfunction in each of these functions leads to the disease remains unclear.
The diagnosis of LD can be made by testing for mutations in the EPM2A gene that accounts for 80 percent of cases [Minassian et al., 1998]. In those cases where mutation testing is negative but the clinical phenotype is giving cause for concern, Lafora bodies can be detected in skin biopsy specimens. Treatment remains palliative.
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF)
Myoclonic epilepsy with ragged-red fibers (MERRF) is a mitochondrial disease characterized by myoclonic epilepsy, ataxia, and ragged-red fibers on muscle biopsy. Multiple other clinical signs commonly seen in mitochondrial disorders may also be seen, such as myopathy, hearing loss, short stature, neuropathy, and optic atrophy [Chinnery et al., 1997]. EEG findings early in the disease are similar to the other PMEs, with 2–5-Hz generalized spike-wave discharges, and gradual background slowing as the disease progresses [So et al., 1989]. Imaging findings include basal ganglia calcifications and brain atrophy [DiMauro et al., 2002]. The key to differentiating MERFF from other PMEs is the constellation of other peripheral nervous system findings.
Diagnosis is made by genetic testing for the most common mutation in the tRNA (MTTK) gene of the mitochondrial DNA, which is seen in 90 percent of typical patients [Shoffner et al., 1990]. There is no specific treatment for MERRF although various antioxidants are commonly prescribed, as is customary in other mitochondrial disorders. VPA can be used, although l-carnitine supplementation is recommended [Tein et al., 1993].
Neuronal Ceroid-Lipofuscinoses
There are multiple types of neuronal ceroid-lipofuscinoses (NCLs) that vary in their age and mode of presentation. The most common type that presents with myoclonic seizures is the classical late-infantile type (LINCL; Jansky–Bielschowsky disease). Typical age at presentation is between 2 and 5 years. Multiple types of generalized seizures may occur, including myoclonic, atypical absence, atonic, and generalized tonic-clonic. Most children are previously normal, although nonspecific speech delay commonly precedes the onset of seizures. Shortly after or coincident with onset of seizures, patients become ataxic and psychomotor regression begins. Blindness eventually develops, due to retinal atrophy, but visual impairment is often subtle or incomplete early in the course of the disease [Williams et al., 2006]. Disease progression is variable but children are often bedridden and die within 5 years of the diagnosis.
Early in the course of the disease, the EEG often shows background slowing with generalized epileptiform discharges. The EEG response to photic stimulation is very characteristic, revealing posterior predominant spikes during low-frequency photic stimulation; a photoconvulsive response may be elicited. Imaging findings include cerebellar and cerebral atrophy with nonspecific T2 hyperintensity of the white matter [D’Incerti, 2000].
The gene responsible for LINCL has been mapped to chromosome 11p15, which codes for a protein called tripeptidyl peptidase 1 (TTP1) [Gardiner, 2002]. The protein is involved in lysosomal protein degradation, but it remains unclear how dysfunction in this protein specifically leads to the phenotype of LINCL. Diagnosis is made possible by testing for TTP1 enzyme activity; if this is absent or severely reduced, it is diagnostic of the disease.
Treatment at this time remains supportive. Trials of valproate (particularly at low doses), together with a benzodiazepine (usually clonazepam), can be helpful. Avoidance of carbamazepine and phenytoin is recommended. Trials are under way utilizing both stem cells and gene transfer vectors, with the aim of restoring some TTP1 activity (http://www.clinicaltrials.gov). These studies involve direct intracranial administration of stem cells or a viral vector. The preliminary studies have shown each of these techniques to be safe; however, disease progression appears to continue at the same rate.
Infantile Spasms
Infantile spasms constitute an age-dependent epilepsy syndrome that presents during infancy and is typically identified by clusters of spasms and an interictal EEG pattern known as hypsarrhythmia. As William West first described in his own son, in a letter to the Lancet in 1841 [Lux, 2001], the onset of spasms is often accompanied by psychomotor deterioration, thus meeting criteria of an epileptic encephalopathy. West’s syndrome refers to the classic triad of spasms, hypsarrhythmia, and psychomotor arrest or regression. With a worldwide incidence of approximately 1 per 3000 live births, infantile spasms is the most prevalent epilepsy syndrome of infancy, and its emotional and financial costs to society are enormous [Pellock et al., 2008].
Electroclinical Features
Regardless of etiology, the syndrome manifests during a specific period of brain maturation, most often between 4 and 8 months of life and nearly always before 2 years [Pellock et al., 2008; Riikonen, 1982]. The unique clinical spasms in the setting of hypsarrhythmia or its variants, lack of response to conventional antiepileptic agents, and generally poor outcome distinguish infantile spasms from similar epilepsies of infancy [Aicardi, 1986; Lombroso, 1990].
Spasms
The classic seizure type, referred to as an epileptic spasm, is characterized by symmetric, bilateral, brief contraction of the axial muscle groups. The EMG tracing during a spasm reveals an abrupt phasic contraction lasting less than 2 seconds, which may be followed by a less intense tonic contraction lasting from 2 to 10 seconds [Kellaway et al., 1979]. Therefore, this unique seizure type is longer than a myoclonic jerk and yet shorter than a tonic seizure.
Depending on which muscle groups are predominantly involved, a spasm is classified as mixed flexor-extensor, flexor, or extensor, in order of their relative frequency [Kellaway et al., 1979]. Most affected infants have more than one type. The flexor spasm consists of sudden flexion of the neck, trunk, and/or extremities. When involved, the legs are commonly held in adduction, whereas the arms may be adducted or abducted. The specific flexor muscles involved and force of contraction vary between spasms, leading to a range of clinical manifestations from massive “jackknife” spasms to more subtle spasms consisting of head-bobbing or shoulder-shrugging. Extensor spasms are marked by abrupt extensor muscle contractions of the same muscle groups. Mixed flexor-extensor spasms reveal a combination of flexor and extensor contractions, most commonly flexion of the neck, trunk, and arms, with extension of the legs. Whether a spasm is flexor, extensor, or mixed does not suggest etiology or influence prognosis. In contrast, asymmetric spasms, which occur in approximately 5–25 percent of all patients, are more likely to be associated with a structural brain abnormality [Kellaway et al., 1979; Gaily et al., 1995, 2001; Fusco and Vigevano, 1993].
Spasms typically occur in clusters, with a 5- to 30-second interval between successive spells [Kellaway et al., 1979]. Often, the intensity of the spasms within a cluster builds to a peak and then declines. The frequency of spasms may vary, from only a few to several hundred per day [Guzzetta et al., 2007]. One common feature to note when taking a history is the common occurrence of spasms upon awakening and their rare occurrence during sleep [Kellaway et al., 1979; King et al., 1985], although the frequency of spasms during day and night is similar, owing to frequent awakenings in affected infants. No obvious precipitating stimuli or circumstances are observed in most patients.
Various clinical phenomena are seen in association with spasms. A behavioral arrest may follow a spasm for up to 90 seconds, but rarely occurs as an independent ictal event without an associated spasm [Kellaway et al., 1979; King et al., 1985]. Crying frequently follows a spasm, which, together with the preceding movements, often leads parents and general practitioners mistakenly to suspect abdominal colic or an exaggerated startle response. Eye movements, consisting of deviation alone or followed by rhythmic nystagmoid movements, are also commonly seen during spasms. Changes in respiratory rhythm occur in the majority of patients, while alterations in heart rate are rare [Kellaway et al., 1979].
Hypsarrhythmia and the ictal EEG
The EEG hallmark of infantile spasms is hypsarrhythmia, a disorganized interictal pattern consisting of “random high-voltage slow waves and spikes” [Gibbs et al., 1954] (Figure 56-1). The spike discharges are usually multifocal, but when generalized, they are never rhythmically repetitive. A state-dependent EEG pattern, hypsarrhythmia is often present during wakefulness and quiet (non-rapid eye movement [REM]) sleep, and may be reduced or even absent during active (REM) sleep [Hrachovy et al., 1981]. The classic pattern is usually seen in the early stages of infantile spasms, and in patients younger than 1 year of age. In addition to the classic pattern, there are several hypsarrhythmia variants, which have been grouped together and given the term “modified hypsarrhythmia,” all of which have some elements of the classic pattern seen with typical epileptic spasms [Hrachovy et al., 1984]. These modified patterns include increased interhemispheric synchronization, asymmetrical or unilateral hypsarrhythmia, focal features, slow waves without spikes, and generalized background burst suppression. Modified hypsarrhythmia may occur more frequently than classic hypsarrhythmia [Kramer et al., 1997]. Hypsarrhythmia or modified hypsarrhythmia is seen in about two-thirds of cases, while other patterns, such as multifocal independent spike discharges, are present in the remainder [Shields, 2006]. Children with severe brain abnormalities, such as tuberous sclerosis, Aicardi’s syndrome, and lissencephaly, do not usually generate the typical hypsarrhythmic pattern. Asymmetric hypsarrhythmia may indicate the presence of a focal central nervous system lesion [Drury et al., 1995]. Regardless of the specific pattern, however, any significant EEG background abnormality in a patient with clinical spasms may contribute to the epileptic encephalopathy, and all patterns should be viewed equally when considering approach to treatment. The chaotic pattern becomes more organized with time and, by the first several years of life, may evolve into the pattern of Lennox–Gastaut syndrome.
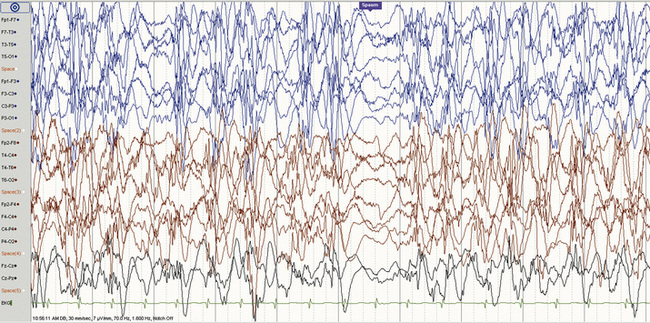
The ictal EEG pattern varies, although typically depicts a generalized, high-voltage, slow-wave transient followed by an abrupt voltage attenuation (see Figure 56-1) [Fusco and Vigevano, 1993]. There is no correlation between the ictal pattern and type of spasm, although longer ictal episodes are usually associated with behavioral arrest. Some children have partial seizures that are temporally related to clusters of spasms, a phenomenon that should raise suspicion for a structural brain abnormality [Pachatz et al., 2003], and possibly a focal abnormality with EEG concordance [Ohtsuka et al., 1996]. Among 92 patients with infantile spasms, 36 had partial seizures at some point (before spasm onset, concurrent with spasms, or after spasm cessation). The presence of partial seizures was associated with asymmetrical spasms, hemiparesis, asymmetric hypsarrhythmia, and persistence of seizures, and there was a trend toward increased focal structural imaging findings among these patients.
Classification
Symptomatic group
The symptomatic group consists of children who have evidence of neurologic injury at the time of onset or a known associated disorder. This group accounts for approximately 80 percent of patients with infantile spasms [Matsumoto et al., 1981; Riikonen, 2009; Partikian and Mitchell, 2009], although a study of 140 affected patients who were evaluated by computed tomography (CT), MRI, and/or positron emission tomography (PET) reported that 96 percent were symptomatic [Chugani and Conti, 1996], highlighting the importance of multimodal neuroimaging in the evaluation of these infants, especially if they do not respond to therapy.
Cryptogenic and idiopathic groups
Cryptogenic and idiopathic are often used interchangeably to represent children without an apparent cause on history, examination, or diagnostic studies. Without other neurologic disorders that alone predict poor prognosis, children with cryptogenic or idiopathic infantile spasms have the best outlook for normal future development. At this point, most investigators favor the term cryptogenic, with the idea that all cases must have an underlying cause, even when the cause is unknown [Lux and Osborne, 2006]. However, Vigevano et al. found that 55 percent of 31 patients with cryptogenic infantile spasms had normal neurodevelopmental outcomes and either a family history of idiopathic epilepsy or febrile convulsions or an EEG genetic trait, thus arguing for a truly idiopathic group [Vigevano et al., 1993]. Other investigators have proposed that an idiopathic group can be defined by specific EEG findings, including reappearance of hypsarrhythmia between consecutive spasms of a cluster, and normal neurodevelopmental outcomes [Dulac et al., 1993]. Even if an idiopathic group does exist, however, an idiopathic classification can only be assigned retrospectively, which limits its use at the time of diagnosis.
Etiology
Although the unifying epileptogenic mechanism is unknown, various underlying disorders give rise to infantile spasms. These disorders are often categorized into prenatal, perinatal, and postnatal groups. Accounting for over 40 percent of total cases [Ohtahara et al., 1993], prenatal etiologies include central nervous system malformations (focal cortical dysplasia, lissencephaly, holoprosencephaly, hemimegalencephaly, callosal agenesis/Aicardi’s syndrome), chromosomal abnormalities (trisomy 21, Miller–Dieker syndrome), single-gene errors (ARX, CDKL5/STK9), neurocutaneous syndromes (tuberous sclerosis, neurofibromatosis type 1, incontinentia pigmenti), congenital central nervous system infections (TORCH, toxoplasmosis, rubella, cytomegalovirus, herpes simplex), and rarely, inborn errors of metabolism (phenylketonuria*, mitochondrial disorders, nonketotic hyperglycinemia, pyridoxine dependency*, sulfite oxidase deficiency, Menkes’ disease*, biotinidase deficiency*) (the asterisked conditions represent treatable metabolic disorders). Perinatal precipitants include hypoxic ischemic encephalopathy (including periventricular leukomalacia in infants who were born prematurely) and hypoglycemia. Finally, postnatal factors include intracranial hemorrhage from trauma, acquired central nervous system infections, hypoxic-ischemic insults (near-drowning, cardiac arrest, stroke), and brain tumors [Frost et al., 2003; Watanabe, 1998]. Overall, cortical malformations, tuberous sclerosis, and hypoxia-ischemia are the most common known associated disorders.
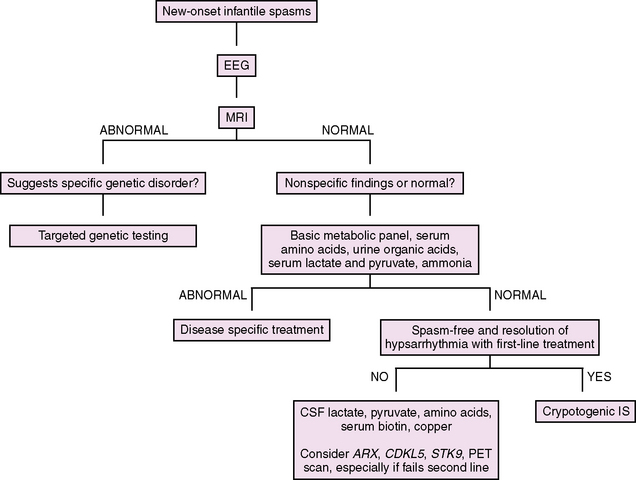
Diagnostic Evaluation
Our approach to the evaluation of infantile spasms is outlined in Figure 56-2. First, in a child with suspected infantile spasms, an EEG is needed to confirm the presence of hypsarrhythmia. Given the state-dependence of hypsarrhythmia, an extended EEG recording that captures at least one full sleep–wake cycle should be performed in all cases. If the EEG remains normal with no features of hypsarrhythmia or its variants, the EEG should be repeated in 1–2 weeks, again capturing at least one full sleep–wake cycle. Once the diagnosis of infantile spasms is established, the evaluation shifts to classification and determination of underlying etiology. When the history is being taken, special attention should be paid to perinatal issues and prior development. Examination may reveal dysmorphic features, neurologic signs, or neurocutaneous stigmata. Neuroimaging is the most important diagnostic test, leading to confirmation of etiology in approximately 70 percent of cases [Wyllie et al., 2005]. MRI is the initial neuroimaging modality of choice, with higher sensitivity in detecting subtle structural abnormalities than CT [van Bogaert et al., 1993]. In addition, an MRI itself may inform prognosis, as patients with a normal MRI have been shown to have better motor outcomes compared to those with abnormalities [Saltik et al., 2002]. While not indicated in the initial evaluation of infantile spasms, PET imaging is helpful in identifying abnormalities that are not appreciated by MRI or CT [Chugani et al., 1993; Chugani and Conti, 1996]. This technique should be considered in medically intractable cases when focal EEG or clinical exam findings raise suspicion for a focal central nervous system process that may be amenable to surgical resection. If neuroimaging or clinical examination raises suspicion for a genetic disorder, then targeted genetic testing may be indicated (for instance, in the case of Down syndrome or lissencephaly). Results of metabolic (serum and cerebrospinal fluid) and genetic testing combined may determine the etiology in an additional 10 percent of cases [Wyllie et al., 2005]. Of these, genetic testing has been found to be most helpful, while metabolic testing is often low-yield [Trasmonte and Barron, 1998; Jacobson and Conry, 2009]. However, if the examination and neuroimaging are unrevealing, we recommend a basic metabolic screen, including electrolytes, glucose, lactate, pyruvate, ammonia, plasma amino acids, and urine organic acids. If a patient without a known cause fails initial medical therapy, a lumbar puncture (cell count, glucose, protein, lactate, pyruvate, and amino acids – specifically glycine) should be performed, and serum testing for rare but treatable metabolic disorders that are not diagnosed on first-pass screening (copper for Menkes’ disease, biotin for biotinidase deficiency) should be considered. Finally, genetic testing may be revealing in refractory cases without a known cause. Specifically, ARX (in male infants) and CDKL5/STK9 (in female infants, particularly those with Rett’s syndrome-like features) should be considered. While the frequency of these gene mutations in infantile spasms is unknown, their contribution to this syndrome is likely appreciable. A recent study screened 177 patients with early-onset seizures of unknown etiology for CDKL5 mutations, including 30 men and 10 girls with Aicardi’s syndrome, for which screening was negative. Of 32 female patients with a history of infantile spasms, 9 (28 percent) had a CDKL5 mutation [Nemos et al., 2009].
Course and Prognosis
Spontaneous remission of spasms and disappearance of hypsarrhythmia in untreated patients have been reported in 25 percent of children by 1 year of age [Hrachovy et al., 1991]. Natural history is incompletely understood, as most studies involve patients who have been treated with medications that alter the course of infantile spasms. Regardless of treatment, however, it appears that clinical spasms and hypsarrhythmia disappear in one-half of children by 2 years of age and in nearly all children by 5 years of age [Jeavons et al., 1973]. Approximately 50 percent of children will develop other seizure types, although rates are lower for children with cryptogenic infantile spasms [Koo et al., 1993]. In many patients who exhibit diffuse or multifocal cerebral dysfunction, the disorder evolves into Lennox–Gastaut syndrome. Among infants with focal lesions, such as those seen in tuberous sclerosis, the spasms often evolve into a symptomatic partial epilepsy [Curatolo et al., 2008].
Overall, the developmental outcome in infantile spasms is poor. In a cohort study of 214 patients treated with adrenocorticotropic hormone (ACTH) who were followed for 20–35 years or until death, cognitive outcome was normal or mildly abnormal in 24 percent of survivors [Riikonen, 1996]. Approximately 50 percent have long-term motor impairment [Jeavons et al., 1973; Matsumoto et al., 1981]. Several predictive factors of outcome have been proposed. Most certainly, the pathological process underlying the syndrome heavily influences the prognosis of infantile spasms. Of children classified as symptomatic, only 5–20 percent have normal or mildly impaired development, compared to 50–80 percent who are classified as cryptogenic [Riikonen, 2009; Cowan and Hudson, 1991]. Among the symptomatic group, children with severe brain malformations tend to have worse prognosis. Better initial control of spasms has been associated with improved neurodevelopmental outcome [Riikonen, 1996; Partikian and Mitchell, 2009], even when only evaluating cryptogenic infantile spasms without confounding underlying diagnoses [Lux et al., 2005]. Observational studies have suggested that neurodevelopmental outcomes are improved in infants who have had shorter duration of spasms at time of initial treatment, specifically in infants with cryptogenic infantile spasms treated within 1 month of spasm onset [Kivity et al., 2004; Riikonen, 2009; Cohen-Sadan et al., 2009], and children with infantile spasms and Down syndrome treated within 2 months [Eisermann et al., 2003]. Finally, it has been proposed that a classic hypsarrhythmic pattern in infants with cryptogenic infantile spasms may be associated with improved outcomes, although this remains uncertain [Pellock et al., 2008].
Pathophysiology
Several hypotheses have been proposed, including brainstem dysfunction due to either disruption of serotonergic neurons [Langlais et al., 1991], or abnormal interaction between the brainstem and a focal or diffuse cortical abnormality [Chugani et al., 1990, 1992], abnormalities in cortical-subcortical interactions [Lado and Moshe, 2002], and immunologic dysfunction.
Alteration in the hypothalamic-adrenal-pituitary (HPA) axis has also been explored. One hypothesis proposes that stress from variable causes during early development results in the release of corticotropin-releasing hormone (CRH), which then leads to increased neuronal excitability and seizures [Brunson et al., 2001]. Supporting evidence for this CRH excess hypothesis includes decreased levels of ACTH found in the cerebrospinal fluid of patients with infantile spasms [Baram et al., 1992b, 1995], and the known effectiveness of ACTH and glucocorticoids in the treatment of infantile spasms. In addition, it has been shown that the CRH receptors are most abundant during infancy, which argues for increased susceptibility to stress during this developmental period [Avishai-Eliner et al., 1996]. However, while it has been shown that injecting CRH into the brains of infant rats produces acute seizures, the ictal semiology and EEG features are not typical of the human infantile spasms phenotype [Baram et al., 1992a]. In addition, CRH levels are not elevated in the cerebrospinal fluid of patients with infantile spasms. While this model accounts for the phenotypic convergence of various etiologies and is supported by the efficacy of hormonal therapy, its usefulness as an animal model is limited. Another hypothesis involving the HPA axis proposes that the protective action of ACTH in infantile spasms could be related, at least in part, to neurosteroids [Rho et al., 2004]. ACTH stimulates adrenal synthesis of both cortisol and deoxycorticosterone (DOC), the latter resulting in increased levels of circulating DOC-derived neurosteroids. In a nonrandomized study, ganaxolone (a neuroactive steroid that modulates GABA receptors via a unique recognition site) as add-on therapy was associated with a 50 percent reduction of spasms in 33 percent of patients with intractable infantile spasms [Kerrigan et al., 2000]. However, corticosteroids are not converted into neurosteroids and yet are also effective in stopping spasms, arguing for a broader underlying mechanism.
In recent years, several new animal models have emerged, spanning the etiological spectrum from acquired to genetic factors. The N-methyl-d-aspartate (NMDA) model (injection of NMDA after prenatal exposure to betamethasone) [Velisek et al., 2007] and the Ts65Dn Down syndrome model (injection of GABA agonists) [Cortez et al., 2009] both produced acute spasms with associated electrodecremental EEG changes. However, spasms did not occur spontaneously in either of these models, and therefore they do not address the underlying question of how spasms are generated. Three other animal models do have spontaneous spasmlike seizures. First, the tetrodoxin (TTX) model [Lee et al., 2008] tests the developmental desynchronization hypothesis, which states that infantile spasms result from a particular temporal desynchronization of two or more developmental processes, resulting in brain dysfunction [Frost and Hrachovy, 2005]. TTX is a sodium channel blocker that blocks normal neuronal activity. While the seizures do not develop until the animals are in late adolescence, this model does support the concept of a final common pathway, as multiple genetic and environmental factors could disrupt the developmental process. Another interesting animal model utilizes three toxic compounds (doxorubicin, lipopolysaccharide, and p-chloro-phenylalanine), which, given in short succession, result in cortical and subcortical brain injury and serotonin depletion [Scantlebury et al., 2009]. These pups manifest early spontaneous spasms with associated EEG changes, as well as behavioral changes after spasm onset. Although this model mimics common etiologies, such as neonatal hypoxic-ischemic encephalopathy, the induced brain injury is a confounding factor.
Finally, two new ARX mouse models have recently been published [Marsh and Golden, 2009; Price et al., 2009]. The Aristaless-related homeobox (ARX) gene encodes a transcription factor involved in cortical development, including GABAergic interneuron migration in the brain [Kitamura et al., 2002; Friocourt et al., 2008]. ARX mutations have been found to be associated with a spectrum of neurologic disorders involving mental retardation and epilepsy, including X-linked lissencephaly with ambiguous genitalia, nonsyndromic X-linked mental retardation, and X-linked infantile spasms [Sherr, 2003]. An apparent phenotype–genotype correlation exists, with loss-of-function mutations typically resulting in cortical malformations, and with insertion/duplication mutations resulting in the other ARX-related diseases, including infantile spasms. The prevalence of ARX mutations in infantile spasms is unknown. Previously, it had been shown that ARX knockout mice exhibit a profound GABAergic interneuron migration defect [Kitamura et al., 2002], resulting in a significant deficit of GABAergic interneurons in the neocortex, hippocampus, and striatum. This finding gave rise to the “interneuronopathy” hypothesis, which proposes that the severe epilepsy phenotype in children with ARX mutations results largely from cortical interneuron dysfunction [Kato and Dobyns, 2005]. Since the previous ARX knockout mice die at birth, limiting postnatal evaluation, investigators have created two new viable mouse models. One is a conditional knockout mouse model (selective deletion of ARX from interneurons) [Marsh and Golden, 2009], which produces spasms with associated electrodecrement on EEG, although spasms occur at a later age than in human equivalents and no cognitive changes have been shown. The second is a polyalanine knockin mouse model (targeted expansion of the first polyalanine tract from 16 to 23 alanine codons, the human mutation most commonly associated with X-linked infantile spasms), which produces typical spasms and EEG changes, as well as cognitive sequelae [Price et al., 2009]. Further studies are required to elucidate the role of interneurons in brain development and developmental epilepsies better.
Treatment
The goal of treatment is to prevent or ameliorate the encephalopathy by stopping the spasms and improving the EEG background. Despite numerous published reports on the treatment of infantile spasms over the past half-century, interpretation has been limited due to methodological shortcomings [Lux and Osborne, 2006]. Therefore, at present, there is no consensus regarding optimal treatment, and there is no conclusive evidence that current medical therapies alter neurocognitive or epilepsy outcomes [Hancock et al., 2008]. The most commonly used medical therapies for treatment of infantile spasms are ACTH or oral corticosteroids in the United States, vigabatrin in the UK, and pyridoxine in Japan. With the recent Food and Drug Administration (FDA) approval of vigabatrin in 2009, its use may increase, particularly for infants with tuberous sclerosis or pre-existing cortical blindness. Conventional antiepileptic agents, as well as the ketogenic diet, have been used with incomplete success when first-line agents fail. Finally, fueled by advances in neuroimaging, surgical resection of focal lesions has emerged as a promising option for patients with medically intractable infantile spasms.
We propose a treatment algorithm for infantile spasms based on available evidence and potential known side effects (Figure 56-3). Considering some evidence that suggests improved cognitive outcomes in children with cryptogenic infantile spasms or Down syndrome who are treated promptly, we recommend prompt evaluation and treatment of all patients with infantile spasms. In addition, it has been reported that infantile spasms in tuberous sclerosis are independently associated with a reduced IQ, and that the risk of mental retardation increases with prolonged duration of spasms, highlighting the importance of prompt and effective therapy, even in symptomatic cases [O’Callaghan et al., 2004; Goh et al., 2005]. Following the UKISS trial looking at medication schedules for hormonal therapy and vigabatrin, our proposed algorithm supports rapid crossover to an alternate therapy if a complete response has not been achieved with initial therapy. This strategy is supported by the UKISS trial, which showed a high response rate for those who initially did not respond to the allocated treatment but who subsequently received the alternate therapy (74 percent and 75 percent for crossover to hormonal therapy and vigabatrin, respectively) [Lux et al., 2004].
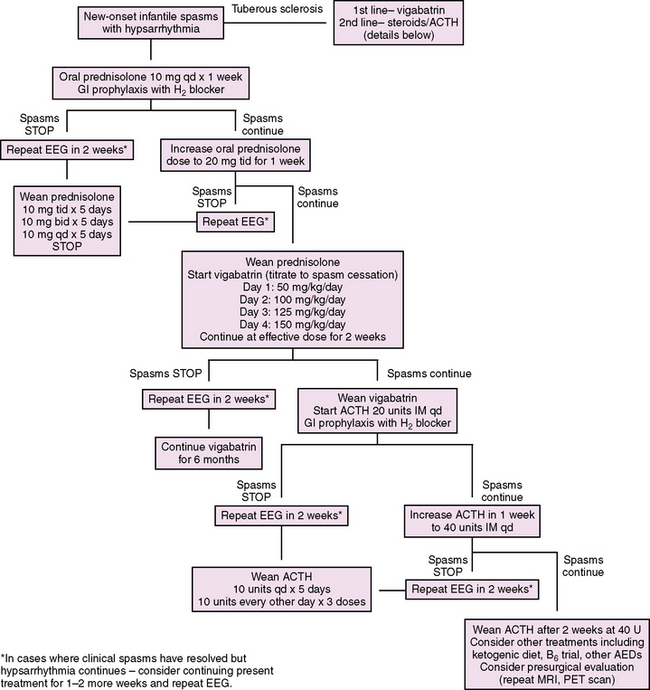
Hormonal therapy
Since initial reports in 1958 describing the effectiveness of ACTH and corticosteroids in stopping the spasms and improving the EEG, hormonal therapy has been the mainstay of treatment for infantile spasms in the United States [Sorel and Dusaucy-Bauloye, 1958]. The recent UKISS trial reported that hormonal therapy (either synthetic ACTH or corticosteroids) stopped the spasms in 73 percent of infants (without tuberous sclerosis) after 2 weeks, compared with vigabatrin in 54 percent [Lux et al., 2004]. While absence of spasms did not differ by treatment group in the follow-up study at 14 months (75 percent versus 76 percent, respectively), there was a clinically significant trend toward better developmental outcomes at 14 months in cryptogenic patients initially treated with hormonal therapy compared to vigabatrin [Lux et al., 2005], highlighting the importance of early spasm cessation and EEG improvement to mitigate the associated encephalopathy. In 2004, a Practice Parameter by the American Academy of Neurology and Child Neurology Society deemed ACTH “probably effective,” reaffirming the prevailing consensus that ACTH was the gold standard [Mackay et al., 2004]. In recent years, however, considering the subanalysis results from UKISS (see below), the escalating cost of ACTH, and the severe known potential side effects of ACTH therapy, a national discussion about oral corticosteroids as first-line therapy has emerged.
ACTH
Several prospective, randomized trials of ACTH or cosyntropin of varying doses have shown that 42–87 percent of patients experienced cessation of spasms within 2 weeks of initiating therapy [Baram et al., 1996; Hrachovy et al., 1983, 1994; Vigevano and Cilio 1997; Yanagaki et al., 1999]. Hypsarrhythmia resolved in 20–90 percent of patients, and relapse rates were 15–33 percent. While there is no agreement about dose or duration of treatment, two randomized controlled trials comparing high-dose ACTH with low-dose ACTH did not show a significant difference in terms of spasm cessation. In a long-term follow-up study, there was no clear benefit of 150 IU/day compared with 40 IU/day [Riikonen, 1982]. Given the lack of evidence to support high-dose ACTH, and considering the dose-dependent potential severe side effects, we recommend lower doses and a short treatment course. A prospective study comparing ACTH and vigabatrin for all patients with infantile spasms showed superior short-term spasm cessation in the ACTH group (74 percent versus 48 percent, respectively) [Vigevano and Cilio, 1997], and the UKISS study showed similar short-term results comparing hormonal therapy with vigabatrin in non-tuberous sclerosis patients [Lux et al., 2004]. Based on these and other studies, a recent Cochrane review concluded that hormonal treatment is more effective than vigabatrin in the short-term treatment of non-tuberous sclerosis-related infantile spasms [Hancock et al., 2008]. ACTH side effects are significant and should be carefully considered when determining the most appropriate form of hormonal therapy for the patient. Cushingoid obesity and irritability develop in most patients. More serious side effects appear to be dose-dependent and include arterial hypertension, cardiomyopathy, electrolyte imbalance, gastric ulcer, immunosuppression, and growth retardation [Shields 2006].
Corticosteroids
Corticosteroids result in short-term spasm cessation in 29–70 percent of patients with infantile spasms [Mackay et al., 2004] [Lux et al., 2004]. Except for UKISS, these studies evaluated low-dose corticosteroids. One randomized controlled trial reported that low-dose oral corticosteroids was less effective than ACTH in stopping spasms (29 percent vs 87 percent, respectively) [Baram et al., 1996], while another similar study did not show a significant difference between the two groups (33 percent vs 42 percent, respectively) [Hrachovy et al., 1983]. Based on varying study results, the 2004 Practice Parameter concluded that there was insufficient evidence to recommend oral corticosteroids. Since that time, the UKISS trial subanalysis showed that oral prednisolone was equivalent to synthetic ACTH (70 percent vs 76 percent spasm-free after 14 days, respectively). While this study was not powered to determine a significant difference between the two groups, it had more subjects than prior studies and represents the only randomized controlled trial evaluating the effectiveness of high-dose oral corticosteroid. Incorporating UKISS results, a 2008 Cochrane review concluded that, should oral prednisolone be used, “high dose” is recommended [Hancock et al., 2008]. The difference in dose may explain the significantly lower spasm-free rate of oral prednisone reported in the 1983 and 1996 studies mentioned above. In those studies, prednisone was used at a dose of 2 mg/kg/day, which is roughly half of the UKISS starting dose of 10 mg four times daily.
Of note is the fact that, in August 2007, the price of H.P. Acthar Gel increased 15-fold and a full course of treatment now costs roughly $100,000 [Gettig et al., 2009]; this does not include the specialized nursing training and sometimes brief inpatient admission necessary to train caregivers on how to administer an injectable medication. In comparison, a course of oral prednisolone costs a few hundred dollars and is much easier to administer. Given these circumstances, we and many centers have moved towards using oral prednisolone as first-line therapy [Kossoff et al., 2009], but an adequately powered study comparing prednisolone and ACTH is still warranted.
Vigabatrin
The first AED to be developed on the basis of a targeted mechanism of action, vigabatrin is an irreversible inhibitor of the enzyme gamma-aminobutyric acid transaminase. Inhibition of this enzyme results in increased levels of GABA in the brain. Vigabatrin has been shown to be effective in the short-term treatment of infantile spasms, particularly in patients with tuberous sclerosis as the underlying cause. In a multicenter retrospective study, suppression of spasms was achieved within a mean of 6 days in 69 percent of all infants and in 96 percent of those with tuberous sclerosis [Aicardi et al., 1996]. In a small study, Chiron et al. reported a 100 percent response rate in patients with tuberous sclerosis treated with vigabatrin (150 mg/kg/day), compared to 45 percent treated with hydrocortisone at 1-month follow-up [Chiron et al., 1997]. Later studies have confirmed the particular efficacy of vigabatrin in patients with infantile spasms due to tuberous sclerosis. In contrast, short-term efficacy rates for all or non-TS infantile spasms range from 35 to 54 percent [Lux et al., 2004; Vigevano and Cilio, 1997; Appleton et al., 1999]. We recommend vigabatrin as first-line therapy for patients with infantile spasms due to tuberous sclerosis. Considering that 75 percent of patients in the UKISS trial, who initially failed hormonal therapy but then were treated with vigabatrin, were spasm-free at 14 months [Lux et al., 2005], we support the use of vigabatrin as second-line therapy in patients without tuberous sclerosis.
In August 2009, vigabatrin was approved by the FDA to treat infantile spasms in children aged 1 month to 2 years. It had been available in many countries for nearly two decades, but was previously unavailable in the U.S. due to concern about an irreversible, cumulative dose-dependent peripheral visual field defect (VFD). In various studies, the prevalence of the vigabatrin-induced peripheral VFD ranged from 25 to 50 percent in adults, 15 percent in children, and 15 to 31 percent in infants [Willmore et al., 2009]. The earliest onset of VFD in infants was found to be 3.1 months. The VFD is a bilateral, concentric, predominantly nasal constriction of the visual field. Visual acuity is not affected, and patients are often asymptomatic until the defect has encroached into the central vision. Perimetry, visual-evoked potentials, and electroretinography testing has been used in adults and children to detect peripheral VFD. In infants, however, electroretinography is the only sensitive technique, and the required sedation limits its use. A recent report recommends using “age-appropriate” visual field testing in infants at baseline and then every 3 months until 18 months of treatment, and then every 6 months thereafter [Willmore et al., 2009]. Unfortunately, there is no viable age-appropriate option for visual field testing in this population, and emphasis is instead placed on informed consent and weighing the risks of anesthesia required for electroretinography against unidentified VFD with drug continuation. To avoid unnecessary exposure, we recommend discontinuation of vigabatrin if a response is not obtained within 2 weeks. If a response is achieved, the infant should have a formal ophthalmologic examination at 3 months and electroretinography should be discussed with the family and ophthalmologist. Some centers that follow electroretinography every 3–4 months also recommend an electroretinogram 6 months after the vigabatrin has been stopped [AES, 2009]. Given the natural history of the disorder, we generally recommend discontinuation of vigabatrin at 6 months, although the benefits of continuation may outweigh the risks in some cases such as pre-existing cortical blindness.
Surgical therapy
Facilitated by advances in structural and functional neuroimaging techniques, surgical resection of focal or unilateral central nervous system lesions has emerged as an effective epilepsy treatment option in selected patients with intractable infantile spasms. Epilepsy surgery for infantile spasms was fueled by the observation that PET of glucose metabolism could identify focal cortical lesions localized to epileptogenic regions in infants with cryptogenic infantile spasms whose functional imaging was unrevealing, and that surgical resection was associated with a favorable outcome [Chugani et al., 1990]. Following this report, Chugani et al. reported on 23 patients with infantile spasms who underwent cortical resection or hemispherectomy. In 14 of the 23 patients, PET was the only imaging modality that showed the regional abnormality [Chugani et al., 1993]. These and later series have shown that surgical resection of a focal cortical lesion for intractable infantile spasms results in complete seizure control in 50–60 percent of patients [Chugani et al., 1993; Wyllie et al., 1998]. When focal resections are performed on cortical areas showing abnormal metabolism or perfusion on PET or single photon emission computed tomography (SPECT), neuropathological examination of the resected tissue usually reveals cortical dysplasia [Chugani et al., 1990; Vinters, 2002]. The impact of surgery on long-term development is uncertain, although limited evidence points toward improved outcomes, particularly in cases of early surgical intervention [Asano et al., 2001]. While further studies are required to determine the impact of surgery on developmental outcomes, surgical resection of focal cortical regions is an important therapeutic option, and specialized presurgical evaluation should be considered in patients with localizing clinical or EEG signs, specifically asymmetrical spasms, hemihypsarrhythmia, and partial seizures, even in the absence of a well-defined MRI lesion.
Other treatments
As an effective broad-spectrum agent for generalized seizure types that may follow infantile spasms, valproate is a reasonable choice after first-line treatments fail, although the increased risk of hepatotoxicity in children under 2 years of age should be considered. Valproate may benefit 40 percent of patients who do not respond to ACTH [Bachman, 1982]. In a recent abstract, 5 out of 5 patients with Down syndrome treated with valproate (doses 30–45 mg/kg/day) after failing at least one other medication were spasm-free at 2 years, and 4 of 5 had resolution of hypsarrhythmia within 2–4 weeks [Patterson et al., 2009]. Further studies are required to determine if valproate is particularly effective in this group of patients. In terms of valproate monotherapy for infantile spasms, one uncontrolled prospective open-label study reported cessation of spasms in 73 percent and resolution of hypsarrhythmia in 91 percent of 22 children at 6 months follow-up, with cessation of spasms in 50 percent within 4 weeks [Siemes et al., 1988]. The 6-month cessation rate is difficult to interpret, however, because dexamethasone therapy was added at 4 weeks if valproate monotherapy was ineffective.
Several open-label studies evaluating topiramate as monotherapy or adjunctive therapy in the treatment of infantile spasms [Peltzer et al., 2009; Glauser et al., 1998] have reported seizure cessation rates of 20–50 percent within variable follow-up periods, with mean maintenance doses of approximately 15 mg/kg/day. A survey of 41 U.S. pediatric epileptologists reported that topiramate is the most commonly used agent after hormonal therapy in patients with symptomatic infantile spasms not associated with tuberous sclerosis [Wheless et al., 2005].
Zonisamide has also been used for infantile spasms. In the largest series, 33 percent of the 27 patients treated with zonisamide (mean dose 8 mg/kg/day) monotherapy or add-on therapy had spasm cessation and 75 percent had resolution of hypsarrhythmia at a mean of 5 days after initiation of treatment, although the relapse rate was 44 percent [Yanai et al., 1999]. Another similar study reported spasm cessation in 26 percent of 23 patients treated with zonisamide monotherapy or add-on therapy, without any relapses at 6 months [Lotze and Wilfong, 2004]. Suzuki et al. reported that 20 percent of 54 infants given zonisamide after failing vitamin B6 had cessation of spasms within 3 weeks, although 27 percent relapsed shortly after initial cessation [Suzuki, 2001]. High-dose vitamin B6 has been the treatment of choice for symptomatic and cryptogenic infantile spasms in Japan, although there are no randomized controlled trials. In two uncontrolled, prospective open-label studies, spasm cessation was achieved in 13–29 percent of patients treated with variable doses of pyridoxine within several weeks [Pietz et al., 1993]. With a mild side effect profile and the possibility of pyridoxine-dependent seizures, a brief trial of pyridoxine should be considered when first-line therapy fails. While an intravenous bolus of pyridoxine during the initial EEG recoding is optimal, this is often not technically feasible. Given the recent evidence that pyridoxine-dependent seizures and folinic-responsive seizures are allelic [Gallagher et al., 2009], folinic acid should be co-administered with pyridoxine. Response to this treatment should be re-evaluated at 1–2 weeks. Finally, there is limited evidence that the ketogenic diet may help control spasms in refractory cases, and possibly even as first-line therapy, although additional studies are required [Kossoff et al., 2002, 2008]. We recommend consideration of the ketogenic diet after hormonal therapy and vigabatrin have failed; whether it is attempted prior to initiation of another medication is based on family preferences and an informed discussion about known side effects of each therapy.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
AES CR. Infantile Spasms. 2009. Boston
Aicardi J. Myoclonic epilepsies of infancy and childhood. Adv Neurol. 1986;43:11-31.
Aicardi J., Mumford J.P., Dumas C., et al. Vigabatrin as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996;37(7):638-642.
Appleton R.E., Peters A.C., Mumford J.P., et al. Randomised, placebo-controlled study of vigabatrin as first-line treatment of infantile spasms. Epilepsia. 1999;40(11):1627-1633.
Asano E., Chugani D.C., Juhasz C., et al. Surgical treatment of West syndrome. Brain Dev. 2001;23(7):668-676.
Avishai-Eliner S., Yi S.J., Baram T.Z. Developmental profile of messenger RNA for the corticotropin-releasing hormone receptor in the rat limbic system. Brain Res Dev Brain Res. 1996;91(2):159-163.
Bachman D.S. Use of valproic acid in treatment of infantile spasms. Arch Neurol. 1982;39(1):49-52.
Badawy R.A., Curatolo J.M., Newton M., et al. Sleep deprivation increases cortical excitability in epilepsy: syndrome-specific effects. Neurology. 2006;67(6):1018-1022.
Baram T.Z., Hirsch E., Snead O.C.3rd, et al. Corticotropin-releasing hormone-induced seizures in infant rats originate in the amygdala. Ann Neurol. 1992;31(5):488-494.
Baram T.Z., Mitchell W.G., Hanson R.A., et al. Cerebrospinal fluid corticotropin and cortisol are reduced in infantile spasms. Pediatr Neurol. 1995;13(2):108-110.
Baram T.Z., Mitchell W.G., Snead O.C.3rd, et al. Brain-adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology. 1992;42(6):1171-1175.
Baram T.Z., Mitchell W.G., Tournay A., et al. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics. 1996;97(3):375-379.
Baykan B., Altindag E.A., Bebek N., et al. Myoclonic seizures subside in the fourth decade in juvenile myoclonic epilepsy. Neurology. 2008;70(22 Pt 2):2123-2129.
Berkovic S.F., Howell R.A., Hay D.A., et al. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43(4):435-445.
Biton V., Bourgeois B.F. Topiramate in patients with juvenile myoclonic epilepsy. Arch Neurol. 2005;62(11):1705-1708.
Biton V., Sackellares J.C., Vuong A., et al. Double-blind, placebo-controlled study of lamotrigine in primary generalized tonic-clonic seizures. Neurology. 2005;65(11):1737-1743.
Brodie M.J., Yuen A.W. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 Study Group. Epilepsy Res. 1997;26(3):423-432.
Brunson K.L., Eghbal-Ahmadi M., Baram T.Z. How do the many etiologies of West syndrome lead to excitability and seizures? The corticotropin releasing hormone excess hypothesis. Brain Dev. 2001;23(7):533-538.
Buchanan N. The use of lamotrigine in juvenile myoclonic epilepsy. Seizure. 1996;5(2):149-151.
Camfield C.S., Camfield P.R. Juvenile myoclonic epilepsy 25 years after seizure onset: a population-based study. Neurology. 2009;73(13):1041-1045.
Capovilla G., Beccaria F., Gambardella A., et al. Photosensitive benign myoclonic epilepsy in infancy. Epilepsia. 2007;48(1):96-100.
Caraballo R., Tripoli J., Escobal L., et al. Ketogenic diet: efficacy and tolerability in childhood intractable epilepsy. Rev Neurol. 1998;26(149):61-64.
Caraballo R.H., Capovilla G., Vigevano F., et al. The spectrum of benign myoclonus of early infancy: Clinical and neurophysiologic features in 102 patients. Epilepsia. 2009;50(5):1176-1183.
Chan E.M., Young E.J., Ianzano L., et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125-127.
Chinnery P.F., Howell N., Lightowlers R.N., et al. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain. 1997;120(Pt 10):1713-1721.
Chipaux M., Dubouch C., Bouis C., et al. Cognitive and Behavioral Outcome in 64 Patients witih Severe Myoclonic Epilepsy in Infancy (SMEI) or Dravet Syndrome. American Epilepsy Society. Philadelphia. Epilepsia. (Suppl 0):2008. (Abst. 1.336)
Chiron C. Stiripentol. Expert Opin Investig Drugs. 2005;14(7):905-911.
Chiron C., Dumas C., Jambaque I., et al. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res. 1997;26(2):389-395.
Chiron C., Marchand M.C., Tran A., et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356(9242):1638-1642.
Chugani H.T., Conti J.R. Etiologic classification of infantile spasms in 140 cases: role of positron emission tomography. J Child Neurol. 1996;11(1):44-48.
Chugani H.T., Shewmon D.A., Sankar R., et al. Infantile spasms: II. Lenticular nuclei and brain stem activation on positron emission tomography. Ann Neurol. 1992;31(2):212-219.
Chugani H.T., Shewmon D.A., Shields W.D., et al. Surgery for intractable infantile spasms: neuroimaging perspectives. Epilepsia. 1993;34(4):764-771.
Chugani H.T., Shields W.D., Shewmon D.A., et al. Infantile spasms: I. PET identifies focal cortical dysgenesis in cryptogenic cases for surgical treatment. Ann Neurol. 1990;27(4):406-413.
Claes L., Del-Favero J., Ceulemans B., et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327-1332.
Claes L.R., Deprez L., Suls A., et al. The SCN1A variant database: a novel research and diagnostic tool. Hum Mutat. 2009;30(10):E904-E920.
Cohen-Sadan S., Kramer U., Ben-Zeev B., et al. Multicenter long-term follow-up of children with idiopathic West syndrome: ACTH versus vigabatrin. Eur J Neurol. 2009;16(4):482-487.
Cortez M.A., Shen L., Wu Y., et al. Infantile spasms and Down syndrome: a new animal model. Pediatr Res. 2009;65(5):499-503.
Covanis A., Stodieck S.R., Wilkins A.J. Treatment of photosensitivity. Epilepsia. 2004;45(Suppl 1):40-45.
Cowan L.D., Hudson L.S. The epidemiology and natural history of infantile spasms. J Child Neurol. 1991;6(4):355-364.
Crespel A., Genton P., Berramdane M., et al. Lamotrigine associated with exacerbation or de novo myoclonus in idiopathic generalized epilepsies. Neurology. 2005;65(5):762-764.
Curatolo P., Bombardieri R., Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657-668.
D’Incerti L. MRI in neuronal ceroid lipofuscinosis. Neurol Sci. 2000;21(Suppl 3):S71-S73.
Delgado-Escueta A.V., Ganesh S., Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet. 2001;106(2):129-138.
de Nijs L., Leon C., Nguyen L., et al. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat Neurosci. 2009;12(10):1266-1274.
Depienne C., Trouillard O., Saint-Martin C., et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46(3):183-191.
DiMauro S., Hirano M., Kaufmann P., et al. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. Adv Neurol. 2002;89:217-229.
Dooley J., Camfield P., Gordan K., et al. Severe polymorphic epilepsy of infancy. J Child Neurol. 1995;10(4):339-340.
Doose H., Lunau H., Castiglione E., et al. Severe idiopathic generalized epilepsy of infancy with generalized tonic-clonic seizures. Neuropediatrics. 1998;29(5):229-238.
Dravet C., Bureau M. The benign myoclonic epilepsy of infancy (author’s transl). Rev Electroencephalogr Neurophysiol Clin. 1981;11(3–4):438-444.
Drury I., Beydoun A., Garofalo E.A., et al. Asymmetric hypsarrhythmia: clinical electroencephalographic and radiological findings. Epilepsia. 1995;36(1):41-47.
Dulac O., Plouin P., Jambaque I. Predicting favorable outcome in idiopathic West syndrome. Epilepsia. 1993;34(4):747-756.
Ebach K., Joos H., Doose H., et al. SCN1A mutation analysis in myoclonic astatic epilepsy and severe idiopathic generalized epilepsy of infancy with generalized tonic-clonic seizures. Neuropediatrics. 2005;36(3):210-213.
Eisermann M.M., DeLaRaillere A., Dellatolas G., et al. Infantile spasms in Down syndrome–effects of delayed anticonvulsive treatment. Epilepsy Res. 2003;55(1–2):21-27.
Friocourt G., Kanatani S., Tabata H., et al. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J Neurosci. 2008;28(22):5794-5805.
Frost J., Frost J., Hrachovy R., editors. Infantile Spasms: Diagnosis, Management and Prognosis. Norwell: Kluwer Academic Publishers, 2003.
Frost J.D.Jr, Hrachovy R.A. Pathogenesis of infantile spasms: a model based on developmental desynchronization. J Clin Neurophysiol. 2005;22(1):25-36.
Fujiwara T., Nakamura H., Watanabe M., et al. Clinicoelectrographic concordance between monozygotic twins with severe myoclonic epilepsy in infancy. Epilepsia. 1990;31(3):281-286.
Fusco L., Vigevano F. Ictal clinical electroencephalographic findings of spasms in West syndrome. Epilepsia. 1993;34(4):671-678.
Gaily E., Liukkonen E., Paetau R., et al. Infantile spasms: diagnosis and assessment of treatment response by video-EEG. Dev Med Child Neurol. 2001;43(10):658-667.
Gaily E.K., Shewmon D.A., Chugani H.T., et al. Asymmetric and asynchronous infantile spasms. Epilepsia. 1995;36(9):873-882.
Gallagher R.C., Van Hove J.L., Scharer G., et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65(5):550-556.
Gardiner R.M. Clinical features and molecular genetic basis of the neuronal ceroid lipofuscinoses. Adv Neurol. 2002;89:211-215.
Gettig J., Cummings J.P., Matuszewski K. H.P. Acthar Gel and Cosyntropin Review: Clinical and Financial Implications. P T. 2009;34(5):250-257.
Gibbs E.L., Fleming M.M., Gibbs F.A. Diagnosis and prognosis of hypsarhythmia and infantile spasms. Pediatrics. 1954;13(1):66-73.
Glauser T.A., Clark P.O., Strawsburg R. A pilot study of topiramate in the treatment of infantile spasms. Epilepsia. 1998;39(12):1324-1328.
Goh S., Kwiatkowski D.J., Dorer D.J., et al. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology. 2005;65(2):235-238.
Guerrini R., Dravet C., Genton P., et al. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39(5):508-512.
Guzzetta F., Bernandina B., Guerrini R., editors. Progress in Epileptic Spasms and West Syndrome. Montrouge: John Libbey Eurotext, 2007.
Hancock E.C., Osborne J.P., Edwards S.W. Treatment of infantile spasms. Cochrane Database Syst Rev. (4):2008. CD001770
Harkin L.A., McMahon J.M., Iona X., et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(Pt 3):843-852.
Hattori J., Ouchida M., Ono J., et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia. 2008;49(4):626-633.
Hirano Y., Oguni H., Funatsuka M., et al. Differentiation of myoclonic seizures in epileptic syndromes: a video-polygraphic study of 26 patients. Epilepsia. 2009;50(6):1525-1535.
Holmes M.D., Quiring J., Tucker D.M. Evidence that juvenile myoclonic epilepsy is a disorder of frontotemporal corticothalamic networks. Neuroimage. 2010;49(1):80-93.
Hrachovy R.A., Frost J.D.Jr, Glaze D.G. High-dose, long-duration versus low-dose, short-duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124(5 Pt 1):803-806.
Hrachovy R.A., Frost J.D.Jr, Kellaway P. Sleep characteristics in infantile spasms. Neurology. 1981;31(6):688-693.
Hrachovy R.A., Frost J.D.Jr, Kellaway P. Hypsarrhythmia: variations on the theme. Epilepsia. 1984;25(3):317-325.
Hrachovy R.A., Frost J.D.Jr, Kellaway P., et al. Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103(4):641-645.
Hrachovy R.A., Glaze D.G., Frost J.D.Jr. A retrospective study of spontaneous remission and long-term outcome in patients with infantile spasms. Epilepsia. 1991;32(2):212-214.
Hurst D.L. Epidemiology of severe myoclonic epilepsy of infancy. Epilepsia. 1990;31(4):397-400.
Jacobson D., Conry J.. Infantile Spasms: Etiology and Predictors of Outcome. 2009. (Abstract)
Janz D, Durner M: Juvenile Myoclonic Epilepsy. In Engel JJR, Pedley TA eds. Epilepsy: A Comprehensive Textbook. Lippencott-Raven, USA
Jeavons P.M., Bower B.D., Dimitrakoudi M. Long-term prognosis of 150 cases of “West syndrome”. Epilepsia. 1973;14(2):153-164.
Kaminska A., Ickowicz A., Plouin P., et al. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res. 1999;36(1):15-29.
Kassai B., Chiron C., Augier S., et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta-analysis of individual patient data. Epilepsia. 2008;49(2):343-348.
Kato M., Dobyns W.B. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol. 2005;20(4):392-397.
Kellaway P., Hrachovy R.A., Frost J.D.Jr, et al. Precise characterization and quantification of infantile spasms. Ann Neurol. 1979;6(3):214-218.
Kerrigan J.F., Shields W.D., Nelson T.Y., et al. Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000;42(2–3):133-139.
Kilaru S., Bergqvist A.G. Current treatment of myoclonic astatic epilepsy: clinical experience at the Children’s Hospital of Philadelphia. Epilepsia. 2007;48(9):1703-1707.
King D.W., Dyken P.R., Spinks I.L.Jr, et al. Infantile spasms: ictal phenomena. Pediatr Neurol. 1985;1(4):213-218.
Kitamura K., Yanazawa M., Sugiyama N., et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32(3):359-369.
Kivity S., Lerman P., Ariel R., et al. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. 2004;45(3):255-262.
Kobayashi K., Iyoda K., Ohtsuka Y., et al. Longitudinal clinicoelectrophysiologic study of a case of Lafora disease proven by skin biopsy. Epilepsia. 1990;31(2):194-201.
Koo B., Hwang P.A., Logan W.J. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology. 1993;43(11):2322-2327.
Kossoff E.H., Hartman A.L., Rubenstein J.E., et al. High-dose oral prednisolone for infantile spasms: an effective and less expensive alternative to ACTH. Epilepsy Behav. 2009;14(4):674-676.
Kossoff E.H., Hedderick E.F., Turner Z., et al. A case-control evaluation of the ketogenic diet versus ACTH for new-onset infantile spasms. Epilepsia. 2008;49(9):1504-1509.
Kossoff E.H., Pyzik P.L., McGrogan J.R., et al. Efficacy of the ketogenic diet for infantile spasms. Pediatrics. 2002;109(5):780-783.
Kothare S.V., Valencia I., Khurana D.S., et al. Efficacy and tolerability of zonisamide in juvenile myoclonic epilepsy. Epileptic Disord. 2004;6(4):267-270.
Kramer U., Sue W.C., Mikati M.A. Focal features in West syndrome indicating candidacy for surgery. Pediatr Neurol. 1997;16(3):213-217.
Kroll-Seger J., Portilla P., Dulac O., et al. Topiramate in the treatment of highly refractory patients with Dravet syndrome. Neuropediatrics. 2006;37(6):325-329.
Kyllerman M., Sommerfelt K., Hedstrom A., et al. Clinical and neurophysiological development of Unverricht-Lundborg disease in four Swedish siblings. Epilepsia. 1991;32(6):900-909.
Labate A., Ambrosio R., Gambardella A., et al. Usefulness of a morning routine EEG recording in patients with juvenile myoclonic epilepsy. Epilepsy Res. 2007;77(1):17-21.
Lado F.A., Moshe S.L. Role of subcortical structures in the pathogenesis of infantile spasms: what are possible subcortical mediators? Int Rev Neurobiol. 2002;49:115-140.
Langlais P.J., Wardlow M.L., Yamamoto H. Changes in CSF neurotransmitters in infantile spasms. Pediatr Neurol. 1991;7(6):440-445.
Lee C.L., Frost J.D.Jr, Swann J.W., et al. A new animal model of infantile spasms with unprovoked persistent seizures. Epilepsia. 2008;49(2):298-307.
Lehesjoki A.E. Clinical features and genetics of Unverricht-Lundborg disease. Adv Neurol. 2002;89:193-197.
Lehesjoki A.E., Koskiniemi M., Sistonen P., et al. Localization of a gene for progressive myoclonus epilepsy to chromosome 21q22. Proc Natl Acad Sci USA. 1991;88(9):3696-3699.
Lombroso C.T. Early myoclonic encephalopathy, early infantile epileptic encephalopathy, and benign and severe infantile myoclonic epilepsies: a critical review and personal contributions. J Clin Neurophysiol. 1990;7(3):380-408.
Lotze T.E., Wilfong A.A. Zonisamide treatment for symptomatic infantile spasms. Neurology. 2004;62(2):296-298.
Lux A.L. West & son: the origins of West syndrome. Brain Dev. 2001;23(7):443-446.
Lux A.L., Edwards S.W., Hancock E., et al. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet. 2004;364(9447):1773-1778.
Lux A.L., Edwards S.W., Hancock E., et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol. 2005;4(11):712-717.
Lux A.L., Osborne J.P. The influence of etiology upon ictal semiology, treatment decisions and long-term outcomes in infantile spasms and West syndrome. Epilepsy Res. 2006;70(Suppl 1):S77-S86.
Mackay M.T., Weiss S.K., Adams-Webber T., et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62(10):1668-1681.
Magaudda A., Gelisse P., Genton P. Antimyoclonic effect of levetiracetam in 13 patients with Unverricht-Lundborg disease: clinical observations. Epilepsia. 2004;45(6):678-681.
Mangano S., Fontana A., Cusumano L. Benign myoclonic epilepsy in infancy: neuropsychological and behavioural outcome. Brain Dev. 2005;27(3):218-223.
Marsh E.D., Golden J.A. Developing an animal model for infantile spasms: pathogenesis, problems and progress. Dis Model Mech. 2009;2(7–8):329-335.
Martinez-Juarez I.E., Alonso M.E., Medina M.T., et al. Juvenile myoclonic epilepsy subsyndromes: family studies and long-term follow-up. Brain. 2006;129(Pt 5):1269-1280.
Mascalchi M., Michelucci R., Cosottini M., et al. Brainstem involvement in Unverricht-Lundborg disease (EPM1): An MRI and (1)H MRS study. Neurology. 2002;58(11):1686-1689.
Matsumoto A., Watanabe K., Negoro T., et al. Infantile spasms: etiological factors, clinical aspects, and long term prognosis in 200 cases. Eur J Pediatr. 1981;135(3):239-244.
Matsuoka H., Takahashi T., Sasaki M., et al. Neuropsychological EEG activation in patients with epilepsy. Brain. 2000;123(Pt 2):318-330.
Meador K.J., Baker G.A., Browning N., et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med. 2009;360(16):1597-1605.
Minassian B.A. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25(1):21-29.
Minassian B.A., Lee J.R., Herbrick J.A., et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171-174.
Nemos C., Lambert L., Giuliano F., et al. Mutational spectrum of CDKL5 in early-onset encephalopathies: a study of a large collection of French patients and review of the literature. Clin Genet. 2009;76(4):357-371.
Noachtar S., Andermann E., Meyvisch P., et al. Levetiracetam for the treatment of idiopathic generalized epilepsy with myoclonic seizures. Neurology. 2008;70(8):607-616.
Nolan K.J., Camfield C.D., Camfield P.R. Coping with Dravet Syndrome: parental experiences with a catastrophic epilepsy. Dev Med Child Neurol. 2006;48(9):761-765.
O’Callaghan F.J., Harris T., Joinson C., et al. The relation of infantile spasms, tubers, and intelligence in tuberous sclerosis complex. Arch Dis Child. 2004;89(6):530-533.
Obeid T., Panayiotopoulos C.P. Clonazepam in juvenile myoclonic epilepsy. Epilepsia. 1989;30(5):603-606.
Oguni H., Tanaka T., Hayashi K., et al. Treatment and long-term prognosis of myoclonic-astatic epilepsy of early childhood. Neuropediatrics. 2002;33(3):122-132.
Oguni H., Hyashi K., Oguni M., et al. Treatment of severe myoclonic epilepsy in infants with bromide and its borderline variant. Epilepsy. 1994;35(6):1140-1145.
Ohtahara S., Ohtsuka Y., Yamatogi Y., et al. Prenatal etiologies of West syndrome. Epilepsia. 1993;34(4):716-722.
Ohtsuka Y., Murashima I., Asano T., et al. Partial seizures in West syndrome. Epilepsia. 1996;37(11):1060-1067.
Pachatz C., Fusco L., Vigevano F. Epileptic spasms and partial seizures as a single ictal event. Epilepsia. 2003;44(5):693-700.
Park K.I., Lee S.K., Chu K., et al. The value of video-EEG monitoring to diagnose juvenile myoclonic epilepsy. Seizure. 2009;18(2):94-99.
Partikian A., Mitchell W.G. Neurodevelopmental and Epilepsy Outcomes in a North American Cohort of Patients With Infantile Spasms. J Child Neurol. 2009.
Patterson C., Resnick T., Holder D., et al. Does valproic acid have a specific benefit in the treatment of infantile spasms in patients with Down Syndrome?. 2009. Abstract
Pellock J., Bourgeois B., Dodson W., editors. Pediatric Epilepsy: Diagnosis and Therapy. New York: Demos Medical Publishing, LLC, 2008.
Peltzer B., Alonso W.D., Porter B.E. Topiramate and adrenocorticotropic hormone (ACTH) as initial treatment for infantile spasms. J Child Neurol. 2009;24(4):400-405.
Penry J.K., Dean J.C., Riela A.R. Juvenile myoclonic epilepsy: long-term response to therapy. Epilepsia. 1989;30(Suppl 4):S19-S23. discussion S24–S17
Pietz J., Benninger C., Schafer H., et al. Treatment of infantile spasms with high-dosage vitamin B6. Epilepsia. 1993;34(4):757-763.
Prasad A., Kuzniecky R.I., Knowlton R.C., et al. Evolving antiepileptic drug treatment in juvenile myoclonic epilepsy. Arch Neurol. 2003;60(8):1100-1105.
Price M.G., Yoo J.W., Burgess D.L., et al. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci. 2009;29(27):8752-8763.
Pulsipher D.T., Seidenberg M., Guidotti L., et al. Thalamofrontal circuitry and executive dysfunction in recent-onset juvenile myoclonic epilepsy. Epilepsia. 2009;50(5):1210-1219.
Quilichini P.P., Chiron C., Ben-Ari Y., et al. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABA-A receptor channels. Epilepsia. 2006;47(4):704-716.
Renganathan R., Delanty N. Juvenile myoclonic epilepsy: under-appreciated and under-diagnosed. Postgrad Med J. 2003;79(928):78-80.
Rho J., Sankar R., Cavazos J., editors. Epilepsy: Scientific Foundations of Clinical Practice. New York: Marcel Dekker, Inc, 2004.
Ricci S., Cusmai R., Fusco L., et al. Reflex myoclonic epilepsy in infancy: a new age-dependent idiopathic epileptic syndrome related to startle reaction. Epilepsia. 1995;36(4):342-348.
Riikonen R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982;13(1):14-23.
Riikonen R. Long-term outcome of West syndrome: a study of adults with a history of infantile spasms. Epilepsia. 1996;37(4):367-372.
Riikonen R.S. Favourable prognostic factors with infantile spasms. Eur J Paediatr Neurol. 2009.
Roebling R., Scheerer N., Uttner I., et al. Evaluation of cognition, structural, and functional MRI in juvenile myoclonic epilepsy. Epilepsia. 2009;50(11):2456-2465.
Roger J., Bureau M., Dravet C., Genton P., Tassinari C.A., Wolf P., editors. Epileptic Syndromes in Infancy, Childhood, and Adolscence. Eastleigh: John Libbey & Co Ltd, 2002.
Ropper A.H., Samuels M.A. Adams and Victors Principles of Neurology, 9E, New York. 2009.
Rossi P.G., Parmeggiani A., Posar A., et al. Benign myoclonic epilepsy: long-term follow-up of 11 new cases. Brain Dev. 1997;19(7):473-479.
Saltik S., Kocer N., Dervent A. Informative value of magnetic resonance imaging and EEG in the prognosis of infantile spasms. Epilepsia. 2002;43(3):246-252.
Scantlebury M.H., Galanopoulou A.S., Chudomelova L., et al. A model of symptomatic infantile spasms syndrome. Neurobiol Dis. 2009.
Scheffer I.E., Berkovic S.F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120(Pt 3):479-490.
Sherr E.H. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr. 2003;15(6):567-571.
Shields W.D. Infantile Spasms: Little Seizures, BIG Consequences. Epilepsy Curr. 2006;6(3):63-69.
Shoffner J.M., Lott M.T., Lezza A.M., et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61(6):931-937.
Siemes H., Spohr H.L., Michael T., et al. Therapy of infantile spasms with valproate: results of a prospective study. Epilepsia. 1988;29(5):553-560.
So N., Berkovic S., Andermann F., et al. Myoclonus epilepsy and ragged-red fibres (MERRF). 2. Electrophysiological studies and comparison with other progressive myoclonus epilepsies. Brain. 1989;112(Pt 5):1261-1276.
Sorel L., Dusaucy-Bauloye A. Findings in 21 cases of Gibbs’ hypsarrhythmia; spectacular effectiveness of ACTH. Acta Neurol Psychiatr Belg. 1958;58(2):130-141.
Specchio L.M., Gambardella A., Giallonardo A.T., et al. Open label, long-term, pragmatic study on levetiracetam in the treatment of juvenile myoclonic epilepsy. Epilepsy Res. 2006;71(1):32-39.
Specchio N., Boero G., Michelucci R., et al. Effects of levetiracetam on EEG abnormalities in juvenile myoclonic epilepsy. Epilepsia. 2008;49(4):663-669.
Striano P., Mancardi M.M., Biancheri R., et al. Brain MRI findings in severe myoclonic epilepsy in infancy and genotype-phenotype correlations. Epilepsia. 2007;48(6):1092-1096.
Striano S., Capovilla G., Sofia V., et al. Eyelid myoclonia with absences (Jeavons syndrome): a well-defined idiopathic generalized epilepsy syndrome or a spectrum of photosensitive conditions? Epilepsia. 2009;50(Suppl 5):15-19.
Suzuki Y. Zonisamide in West syndrome. Brain Dev. 2001;23(7):658-661.
Tein I., DiMauro S., Xie Z.W., et al. Valproic acid impairs carnitine uptake in cultured human skin fibroblasts. An in vitro model for the pathogenesis of valproic acid-associated carnitine deficiency. Pediatr Res. 1993;34(3):281-287.
Trasmonte J.V., Barron T.F. Infantile spasms: a proposal for a staged evaluation. Pediatr Neurol. 1998;19(5):368-371.
van Bogaert P., Chiron C., Adamsbaum C., et al. Value of magnetic resonance imaging in West syndrome of unknown etiology. Epilepsia. 1993;34(4):701-706.
Velisek L., Jehle K., Asche S., et al. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61(2):109-119.
Vigevano F., Cilio M.R. Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospective study. Epilepsia. 1997;38(12):1270-1274.
Vigevano F., Fusco L., Cusmai R., et al. The idiopathic form of West syndrome. Epilepsia. 1993;34(4):743-746.
Vinters H.V. Histopathology of brain tissue from patients with infantile spasms. Int Rev Neurobiol. 2002;49:63-76.
Wakai S., Ito N., Sueoka H., et al. Severe myoclonic epilepsy in infancy and carbamazepine. Eur J Pediatr. 1996;155(8):724.
Watanabe K. West syndrome: etiological and prognostic aspects. Brain Dev. 1998;20(1):1-8.
Wheless J.W., Clarke D.F., Carpenter D. Treatment of pediatric epilepsy: expert opinion, 2005. J Child Neurol. 2005;20(Suppl 1):S1-S56. quiz S59–S60
Williams R.E., Aberg L., Autti T., et al. Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta. 2006;1762(10):865-872.
Willmore L.J., Abelson M.B., Ben-Menachem E., et al. Vigabatrin: 2008 update. Epilepsia. 2009;50(2):163-173.
Woermann F.G., Free S.L., Koepp M.J., et al. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain. 1999;122(Pt 11):2101-2108.
Wolff M., Casse-Perrot C., Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006;47(Suppl 2):45-48.
Wyllie E., Gupta A., Lachhwani D., editors. The Treatment of Epilepsy: Principles and Practice. Lippincott: Williams & Wilkins, 2005.
Wyllie E., Comair Y.G., Kotagal P., et al. Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol. 1998;44(5):740-748.
Yanagaki S., Oguni H., Hayashi K., et al. A comparative study of high-dose and low-dose ACTH therapy for West syndrome. Brain Dev. 1999;21(7):461-467.
Yanai S., Hanai T., Narazaki O. Treatment of infantile spasms with zonisamide. Brain Dev. 1999;21(3):157-161.
Yen C., Beydoun A., Drury I. Longitudinal EEG studies in a kindred with Lafora disease. Epilepsia. 1991;32(6):895-899.
Zifkin B., Andermann E., Andermann F. Mechanisms, genetics, and pathogenesis of juvenile myoclonic epilepsy. Curr Opin Neurol. 2005;18(2):147-153.
Zuberi S.M., O’Regan M.E. Developmental outcome in benign myoclonic epilepsy in infancy and reflex myoclonic epilepsy in infancy: a literature review and six new cases. Epilepsy Res. 2006;70(Suppl 1):S110-S115.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 56 Myoclonic Seizures and Infantile Spasms
Introduction
Myoclonus, myoclonic seizures, and infantile spasms share many common features yet are seen in a wide variety of neurologic conditions. Myoclonus is not a diagnosis, but rather a sign that can have many underlying etiologies. The precise definitions of each of these terms has also been somewhat controversial, but for the purpose of this chapter we will use the definition proposed by Victor and Adams [Ropper and Samuels, 2009] and define myoclonus simply as a “shocklike irregular jerk,” with myoclonic seizures having a similar clinical appearance but also accompanied by neurophysiologic evidence of being cortically generated. While these definitions each have their own potential flaws, the distinction between a cortically generated movement (myoclonic seizure) and a subcortically generated movement (myoclonus) is extremely important, especially when thinking about some of the progressive myoclonic epilepsies, as many of these patients can manifest both myoclonus and myoclonic seizures. Infantile spasms, however, should not be confused with either, as the spasm itself is often a more complex constellation of movements with a myoclonic component.
Epilepsy Syndromes with Prominent Myoclonic Seizures
Myoclonic seizures can occur in a wide variety of pediatric epilepsy syndromes, including Lennox–Gastaut syndrome and childhood absence epilepsy, which are discussed elsewhere in Part VIII. Our focus will be on those syndromes in which myoclonus is a critical feature for the diagnosis. This coverage includes benign myoclonic epilepsy in infants (BME), severe myoclonic epilepsy in infancy (SMEI/Dravet’s syndrome), idiopathic epilepsy with myoclonic-astatic seizures (IEMAS), and juvenile myoclonic epilepsy (JME) (Table 56-1).
Benign Myoclonic Epilepsy of Infancy
BMEI was first described by Dravet and Bureau in 1981 [Dravet and Bureau, 1981]. The syndrome is characterized by brief, generalized myoclonic seizures, with the predominant area of muscle involvement being the proximal upper extremities [Hirano et al., 2009], and usually occurs in children between the ages of 4 months and 3 years, although cases have been described with onset up to 4 years 8 months [Rossi et al., 1997]. These attacks often occur multiple times per day. Detailed electroencephalography (EEG) and polygraphic electromyography (EMG) recordings have shown that the myoclonic seizures are often associated with a flexor postural change and approximately 80 percent of attacks involve the upper limbs [Hirano et al., 2009]. A history of febrile seizures (30 percent) and a family history of epilepsy (39 percent) is relatively common [Roger et al., 2002].
EEG Findings
The ictal EEG discharge is often a generalized spike wave (GSW) that may be slower than 3 Hz [Hirano et al., 2009]. Occasionally, the myoclonic seizures can be massive and result in a fall, or they may occur in a cluster. Some infants may exhibit photosensitivity at the onset of the syndrome, and the photic-induced myoclonic seizures are often more prominent than those that occur spontaneously [Capovilla et al., 2007]. Other infants may be sensitive to acoustic or tactile stimuli, leading some to argue for a distinctly separate epilepsy syndrome [Ricci et al., 1995]. We and others [Roger et al., 2002] do not believe this is a clinically distinct syndrome, but rather that the syndrome of BME may include both photosensitive and stimulus-provoked seizures in addition to spontaneous myoclonic seizures. In both groups, the waking EEG in between seizures is often normal but may contain rare generalized spike/wave discharges [Ricci et al., 1995; Capovilla et al., 2007], and the ictal EEG findings are indistinguishable.
Treatment and Outcome
The overall prognosis of myoclonic seizures is excellent, with complete resolution in almost all children usually within 1–2 years of diagnosis. In those who demonstrate photosensitivity, these seizures may be more difficult to control and may persist for a longer period of time [Roger et al., 2002]. The majority of children also have normal neurodevelopmental outcomes [Caraballo et al., 2009], although some studies have reported impaired psychomotor development and behavioral disturbances if the child is not treated or if onset of the syndrome is at less than 2 years of age [Mangano et al., 2005]. Those children with a prominent reflex component may have a more favorable neurodevelopmental outcome [Zuberi and O’Regan, 2006].
The medication of choice appears to be valproic acid (VPA), with 80–90 percent responding to VPA monotherapy, although high serum levels (>100 mg/L) may be necessary. When VPA monotherapy does not provide complete control, adjunctive use of a benzodiazepine, such as clonazepam or clobazam, can be helpful [Rossi et al., 1997].
Severe Myoclonic Epilepsy in Infancy
First described in 1978 by Charlotte Dravet, SMEI occurs in normally developing children who experience prolonged febrile (>20 minutes) or afebrile seizures, including hemiconvulsions, during the first year of life. Afebrile, mixed seizures follow, and the emergence of myoclonic seizures is common but not necessary for the diagnosis (borderline variant). For this reason, the syndrome is better referred to as Dravet’s syndrome, as the name severe myoclonic epilepsy of infancy may mislead clinicians and cause them not to suspect the diagnosis if myoclonic seizures are absent. The syndrome was once thought to be exceedingly rare, with an incidence of 1 in 40,000 children [Hurst, 1990], but with heightened awareness of the clinical spectrum and the availability of genetic testing, this may be an underestimation of the true incidence.
Mutations in the SCN1A Gene
The importance of SCN1A gene mutations as an underlying etiology in epilepsy first became apparent when Scheffer and Berkovic reported missense mutations in two families with the syndrome of generalized epilepsy with febrile seizures plus (GEFS+) [Scheffer and Berkovic, 1997]. Due to the common occurrence of febrile seizures in both GEFS+ and Dravet’s syndrome, Claes et al. screened seven patients with Dravet’s syndrome and found de novo missense mutations in all seven patients [Claes et al., 2001]. Testing for mutations in the SCN1A gene is clinically available, and hundreds of patients with Dravet’s syndrome with SCN1A mutations have been reported [Depienne et al., 2009]. The main difference between individuals with a GEFS+ phenotype and a Dravet phenotype appears to be that the former often have a missense mutation with reduced penetrance, whereas patients with Dravet have mutations that occur in isolated patients (arise de novo) [Claes et al., 2001].
Mutations in the SCN1A gene are seen in approximately 70–85 percent of patients with Dravet’s syndrome [Harkin et al., 2007], and therefore it remains a clinical diagnosis based on age of onset and clinical phenotype. One should not exclude the diagnosis on the basis of a negative result on SCN1A mutation testing. Approximately 200 different point mutations in the SCN1A gene were identified in 271 probands with a strict clinical diagnosis of Dravet’s syndrome [Depienne et al., 2009]. Of those probands without a point mutation, the multiplex ligation-dependent probe amplification (MPLA) technique found an additional 14 micro-rearrangements of SCN1A [Depienne et al., 2009]. An SCN1A variant database was published in 2009, listing each type of mutation and the associated epilepsy syndrome [Claes et al., 2009]. As of 2009, 648 point mutations have been reported, of which 582 (90 percent) are in subjects with Dravet’s syndrome. An additional 67 genomic rearrangements were reported, with 46 (7 percent) of these occurring in patients with Dravet’s syndrome. The remaining clinical phenotypes with SCN1A mutations are wide and varied [Harkin et al., 2007]. In Dravet’s syndrome, the end result of each of these mutations appears to be a total loss of function of the mutated allele, most commonly caused by a missense mutation that leads to an abnormality in the pore-forming unit of the sodium channel [Claes et al., 2009]. While specific mutation genotype–phenotype correlations within Dravet’s syndrome are not yet available, further research in this area may lead to particular treatment strategies for specific genotypes.
Seizure Semiologies
Patients with Dravet’s syndrome have many different seizure semiologies, often occurring in the same patient. The most common presenting seizure type is a prolonged febrile convulsion lasting longer than 25 minutes, but afebrile seizures can be the presenting seizure type in as many as 35 percent [Roger et al., 2002]. Japanese authors have observed that hot water immersion can trigger seizures in these patients [Fujiwara et al., 1990], and this feature is one part of a clinical screening test aimed at predicting the diagnosis of Dravet’s syndrome [Hattori et al., 2008]. Other convulsive seizure types include generalized tonic-clonic seizures, hemiclonic seizures, and “falsely generalized,” seizures as described by Dravet [Roger et al., 2002]. The latter has a complex semiology, with a high degree of discrepancy between the clinical and EEG findings. Seizures often start in one part of the body, spread to another (on the opposite side), and then potentially return to the original side of origin, only to involve a new body part.
Nonconvulsive seizures are also common and include simple partial, complex partial, atypical absence, and myoclonic types. The specific semiologies of each of these seizure types are similar to that seen in other forms of childhood epilepsy. Tonic seizures are rare. A unique seizure type, termed “obtundation status,” is unique to the syndrome. The semiology of this seizure type consists of variable impairment in consciousness, and fragmentary and erratic segmental myoclonias involving the limbs and face. Patients may still be able to engage in simple activities, such as eating or playing with a toy. These episodes may last several hours to days and can either be initiated by, or conclude with, a convulsion [Roger et al., 2002]. The EEG during these episodes is not a pattern of classical atypical absence status, but rather is characterized by a diffuse dysrhythmia with focal or diffuse spikes [Roger et al., 2002]. We have observed one patient, who would begin with this type of clinical phenomenon, including prominent eyelid myoclonia that was dramatically accentuated with eye closure, and would remain in this state for 2–3 days, culminating in a generalized convulsion, after which her mental status cleared. She would remain clear for 2–3 weeks, and then repeat the same cycle over again.
EEG Findings
The EEG is invariably normal early in the course of the disease, and therefore, in contrast with other epilepsy syndromes, the diagnosis must be suspected on clinical grounds. As the disease progresses, the EEG background becomes slow but often continues to manifest normal sleep architecture [Roger et al., 2002]. Some authors have observed rhythmic 5–6-Hz centroparietal theta but this is not specific to the syndrome [Doose et al., 1998]. Over time, the interictal EEG can show both generalized and multifocal spikes, or may not show any of these features despite frequent seizures of multiple types, further highlighting the lack of specificity of EEG findings. Given this lack of specificity, a screening test for predicting the diagnosis of Dravet’s syndrome under 1 year of age has been developed, with a positive predictive value as high as 94 percent [Hattori et al., 2008]. This screening test uses simple clinical features, as shown in Table 56-2. A sum score of 6 or greater is associated with a high risk of having Dravet’s syndrome, and therefore SCN1A testing should be done in those cases. Even if the hot water-induced risk factor is excluded, a score of 5 or higher is still indicative of a high-risk patient.
| Clinical Score | Risk Score |
|---|---|
| Onset <7 months | 2 |
| More than 5 seizures prior to 12 months of age | 3 |
| Hemiconvulsion | 3 |
| Focal seizure | 1 |
| Myoclonic seizure | 1 |
| Prolonged seizure (>10 minutes) | 3 |
| Hot water-induced seizure | 2 |
(Adapted from Hattori J et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia 2008;49(4):626–633.)
Imaging
Magnetic resonance imaging (MRI) studies are usually normal but may show nonspecific cerebral or cerebellar atrophy or isolated ventricular enlargement. Interestingly, these findings, specifically diffuse brain atrophy, appear to be more common in those children without associated SCN1A mutations. Furthermore, despite having frequent prolonged febrile and afebrile convulsions, mesial temporal sclerosis is rare, reported in only 1 of 58 patients [Striano et al., 2007].
Treatment
Until recently, treatment strategies were generally unsuccessful, with poor seizure control despite polytherapy. Many have observed that certain antiepileptic drugs (AEDs) exacerbate seizures, with the most notorious agents being carbamazepine (CBZ) and lamotrigine (LTG) [Wakai et al., 1996; Guerrini et al., 1998]. Some authors have recommended using CBZ as a means of “confirming” the diagnosis when there is a high index of clinical suspicion [Wakai et al., 1996]. Given the availability of genetic testing, we do not endorse this practice.
A study by Chiron et al. and a subsequent meta-analysis pooling these data with unpublished data showed that combination therapy with valproate (VPA) + stiripentol (STP) + clobazam (CLB) resulted in a 70 percent decrease in seizure frequency, with 43 percent of patients becoming seizure-free [Chiron et al., 2000; Kassai et al., 2008]. In this study, the STP dose began at 50 mg/kg/day divided in 2–3 doses, but could be titrated up to 100 mg/kg/day. The maximum recommended dose is 3500 mg/day [Chiron et al., 2000].
One proposed mechanism for the effect of STP is the potent inhibition of the p450 cytochrome system, resulting in higher levels of VPA and CLB. Plasma levels of CLB were significantly higher but levels of VPA were not [Chiron et al., 2000]. Interestingly, one other result of p450 inhibition is lower levels of metabolites, some of which are thought to explain some of the adverse toxic effects of AEDs. This finding perhaps explains why patients are able to tolerate the higher doses of certain AEDs [Chiron, 2005]. These observations, however, raise the possibility that STP has other mechanisms of action independent of its effects on the p450 system, including enhancement of gamma-aminobutyric acid (GABA)ergic transmission [Quilichini et al., 2006].
Of the newer agents, topiramate (TPM) appears to be very effective, especially if added to STP, resulting in 50 percent reduction of seizures in 78 percent of patients at a modest optimal dose of 3.2 mg/kg/day. Seventeen percent of patients remained seizure-free for at least 4 months [Kroll-Seger et al., 2006].
Bromide therapy has also shown promise in several studies [Oguni et al., 1994], specifically in reducing the number of convulsions. The ketogenic diet may be helpful in some patients [Caraballo et al., 1998].
Aggressive treatment of acute seizures and prevention of status epilepticus is critical, as some small studies have shown a trend towards improved neuropsychologic outcomes in those children with less frequent convulsive seizures [Chipaux et al., 2008]. Each patient should have a clearly defined “emergency plan,” and the clinician should ask each family if a particular treatment regimen has been particularly effective for their child [Nolan et al., 2006]. Parents should be educated about the inevitability of seizures, especially prolonged seizures in the setting of fever. Early use of high doses of benzodiazepines, either in the form of rectal diazepam (as high as 1 mg/kg per dose) or buccal midazolam, should be the first-line therapy. Some physicians have recommended insertion of a central venous port for those children who have proven to have difficult intravenous access and multiple episodes of status epilepticus [Dooley et al., 1995]. In these specific cases, families are able to cope much better, knowing that multiple intravenous attempts will not be necessary and appropriate treatment can be initiated to terminate the seizure faster.
Outcome
Long-term outcome with regard to seizure control and neuropsychological development is variable but generally poor. By definition, all children with Dravet’s syndrome have normal development at onset. Around 50 percent of children walk unsupported by a mean of 16 months of age; however, it is rare for children to utilize two-word sentences at the normal age of around 2 years [Wolff et al., 2006]. At around 2 years of age, there appears to be a gradual decline, and then there is a relative stabilization between the ages of 4 and 6 years [Wolff et al., 2006]. A trend was noted that fewer convulsive seizures often resulted in higher developmental quotients. Subsequent small cohorts have reported patients with normal or near-normal IQs and this favorable outcome is attributed to early diagnosis and appropriate management [Chipaux et al., 2008]. It remains to be seen in larger prospective studies whether early diagnosis and appropriate management with the medications outlined above will lead to improved seizure control and a subsequent improvement in neurodevelopmental outcome.
Myoclonic-Astatic Epilepsy of Doose
Etiology
Genetic factors clearly play a role in the pathogenesis of MAE, based on the high incidence of seizures (32 percent) in probands’ siblings and parents; the most common seizure type is absence seizures. Despite the initial discovery of the SCN1A gene in a family with GEFS+ that included a family member with MAE [Scheffer and Berkovic, 1997], only a small number of patients with mutations in the SCN1A gene have been identified in patients with MAE [Ebach et al., 2005; Harkin et al., 2007]. Further genotype–phenotype correlation studies are necessary to determine if there are underlying genetic factors that would be able to predict response to treatment and outcome.
Seizure Semiologies
Onset of MAE is often between the ages of 2 and 6 years, and children are usually developmentally normal [Kaminska et al., 1999]. These children may begin experiencing unexplained “jerks” and “falls,” which occur multiple times per day. These myoclonic seizures can take place in isolation, but often are followed by a brief period of atonia that results in a dramatic fall in which the child appears to be propelled to the ground. Polygraphic studies have documented that atonic falls can occur with or without the preceding myoclonus, and occur in about 64 percent of patients [Oguni et al., 2002]. Although atonic falls are not necessarily seen in all patients, more subtle myoclonic/atonic seizures are common. The only manifestation of these may be a brief head nod or “head drop,” with or without a myoclonic jerk of the upper extremities. When head drops occur in clusters with an associated alteration in level of consciousness, nonconvulsive status should be suspected. EEG findings during these episodes are always abnormal, with frequent runs of generalized spike/wave and slow spike-and-wave complexes. Absence seizures are quite common and may be similar to those seen in typical childhood absence epilepsy, but often are accompanied by more prominent myoclonus of the proximal upper extremities.
Treatment
Early recognition of the syndrome allows treatment to be focused on the broad-spectrum agents that have been shown to be particularly effective. The most commonly used and most effective AEDs have been VPA, LTG, levatiracetam, TPM and various benzodiazepines (clonazepam, clobazam, clorazepate). When absence seizures are frequent, ethosuximide (ETX) can be effective and has also been reported to decrease the frequency of myoclonic seizures; however, it is rarely used as monotherapy for this indication [Oguni et al., 2002]. The combination of VPA/ETX may be particularly effective, especially if high doses of VPA are used.
In those children who are refractory to medical management, a trial of the ketogenic diet should be considered early. The ketogenic diet has been shown to be as effective as other first-line agents in children with MAE [Kilaru and Bergqvist, 2007], and given this efficacy, we consider the ketogenic diet early in our treatment algorithm, especially if there is associated neurodevelopmental decline.
Outcome
The outcome with regard to cognitive development is highly variable but appears to be somewhat dependent on degree of seizure control. Some children may have fairly frequent seizures of multiple types and still have spontaneous remission, usually after approximately 3 years [Kaminska et al., 1999; Oguni et al., 2002]. In some children, the multiple seizure types remain intractable and these children have a higher risk of both cognitive and behavioral disturbances [Oguni et al., 2002]. The occurrence of tonic seizures has been described as a negative prognostic sign, although it remains unclear if these children may actually have an atypical form of Lennox–Gastaut syndrome rather than an atypical form of MAE [Kaminska et al., 1999].
Juvenile Myoclonic Epilepsy
JME is an idiopathic generalized epilepsy (IGE) syndrome characterized by myoclonic seizures, generalized tonic-clonic seizures, and absence seizures. It is extremely common, accounting for 26 percent of IGE and 10 percent of all epilepsies [Janz and Durner, 1998], although this may still be an underestimation; it is very likely underdiagnosed, as many clinicians may not inquire about the presence of myoclonic seizures [Renganathan and Delanty, 2003]. Mean age at onset is highly variable and difficult to determine truly, as some children with childhood absence epilepsy may evolve into the syndrome of JME. Excluding this population, the average age of onset is 15.1 years (7–28 years), with a slight female predominance [Martinez-Juarez et al., 2006].
Seizure Semiologies
In the classic syndrome of JME, myoclonic seizures may precede the first generalized tonic-clonic (GTC) seizure by 6–12 months, although GTCs occur as the first seizure type in approximately one-third of patients [Martinez-Juarez et al., 2006]. Some have proposed specific subgroups of JME, separating those patients who present with typical childhood absence or juvenile absence seizures, although this is much less common than the classical presentation of JME, accounting for only 10 percent of cases. The impact this distinction has on outcome will be discussed below. Photosensitivity is relatively common, occurring in approximately 30 percent of patients [Zifkin et al., 2005].
Myoclonic seizures often occur in the early morning hours shortly after awaking. Transcranial magnetic stimulation studies in patients with JME have demonstrated increased cortical excitability that is not present in other patients with focal epilepsy [Badawy et al., 2006]. Provocation of myoclonic seizures has been linked to higher cognitive tasks requiring higher-order thinking, such as writing or written calculation [Matsuoka et al., 2000].
Specific lifestyle features have been commonly associated with breakthrough seizures, including sleep deprivation, stress, alcohol, and menses [Martinez-Juarez et al., 2006].
EEG Findings
Patients with JME often have abnormal interictal EEGs, with the most common finding being generalized 4–6-Hz polyspike-and-wave (61 percent) or 3-Hz spike-and-wave discharges (14 percent) [Martinez-Juarez et al., 2006]. More prolonged (1–6 days; average 1–2 days) video EEG studies have shown EEG abnormalities of JME in 88 percent of patients [Park et al., 2009]. Other series have reported on the increased yield of an early morning EEG compared to an afternoon EEG (generalized epileptiform activity in 69 percent and 25 percent, respectively) [Labate et al., 2007]. A photoparoxysmal response in treatment-naive patients is also common and has been seen in as many as 35 percent of patients [Specchio et al., 2008].
Pathophysiology
It has long been accepted that the IGEs have a strong genetic component [Berkovic et al., 1998], and given the incidence of JME as a subtype of IGE, much research has been done to understand the underlying genetic basis of JME better. While JME appears to be a relatively homogeneous epilepsy syndrome, there are a number of studies implicating various genetic abnormalities. These include mutations in many different ion channels, including calcium, potassium, and chloride channels linked to at least seven different loci. There are also nonionic mechanisms that relate to neural migration during cortical development, and may explain some of the imaging and postmortem pathologic findings seen in these patients [de Nijs et al., 2009]. For further details about the specific genetic mutations and channels involved, please refer to Chapter 52.
Imaging
Imaging studies in patients with JME have indicated subtle structural changes in mesiofrontal cortex, with an increase in cortical gray matter [Woermann et al., 1999], although this has not been replicated in subsequent studies [Roebling et al., 2009]. These findings are interesting, given that the generalized discharges seen on routine EEGs often have a frontal predominance. This has been further investigated with dense array EEG, which further localized typical 4–6-Hz epileptiform activity to the orbitofrontal/medial frontopolar cortex, whereas some patients also demonstrated basal-medial-temporal sources [Holmes et al., 2010]. These electrophysiologic and anatomic abnormalities appear to be concordant with some of the neuropsychological deficits that are seen in patients with JME. Specific attention has been paid to executive dysfunction that has been demonstrated to correlate with smaller thalamic volumes and more frontal cerebrospinal fluid volumes in children with recent-onset JME when compared to children with benign rolandic epilepsy [Pulsipher et al., 2009].
Treatment
Valproate has long been considered the first-line agent, with reports of up to 86 percent of patients becoming seizure-free for at least 1 year [Penry et al., 1989]. Valproate has also been shown to be particularly effective in those patients with photosensitivity [Covanis et al., 2004]. Despite this efficacy, valproate has many side effects that may lead to significant comorbidities, including weight gain, hyperactivity, transaminitis, thrombocytopenia, pancreatitis. Women of child-bearing age must also consider the possible long-term cognitive effects on a fetus [Meador et al., 2009]. Due to these concerns, many newer AEDs are also commonly used, including lamotrigine, topiramate, zonisamide, and levetiracetam.
Lamotrigine is now accepted to be a broad-spectrum AED that is effective against both partial and generalized seizures [Biton et al., 2005]. Small studies have also shown it to be effective specifically in the management of patients with JME, both as monotherapy and polytherapy, resulting in seizure-free rates between 40 and 83 percent [Buchanan, 1996; Prasad et al., 2003]. LTG is also very well tolerated; however, it does have the potential for exacerbating myoclonic seizures [Prasad et al., 2003; Crespel et al., 2005]. The combination of LTG and VPA has been reported to be particularly effective in both partial and generalized epilepsy, including JME, and suggests the possibility of a synergistic effect [Brodie and Yuen, 1997].
Topiramate is also a broad-spectrum AED that has been shown to be effective in patients with JME, although there are no published studies on its use as monotherapy [Biton and Bourgeois, 2005]. It appears to be equally effective as LTG and VPA when used as polytherapy, although there is a suggestion that tolerability may be inferior [Prasad et al., 2003].
Zonisamide (ZNS) is another broad-spectrum AED with multiple proposed mechanisms of action. Small studies have shown ZNS to be broadly effective against all of the seizure types in JME, with 69, 62, and 38 percent of patients being free of GTC, myoclonic, and absence seizures, respectively [Kothare et al., 2004].
Levetiracetam is gaining a reputation as a broad-spectrum agent, as it has shown to be effective in patients with various forms of idiopathic generalized epilepsies including epilepsy with eyelid myoclonias and absence [Striano et al., 2009], as well as JME [Specchio et al., 2006; Noachtar et al., 2008]. In a study of 120 patients (93 percent of whom had JME) randomized to add-on levetiracetam or placebo, 25 percent were free of myoclonic seizures and 21.7 percent were free of all seizures, including GTCs, during the 12-week treatment phase [Noachtar et al., 2008]. In another open-label study of 48 patients with JME (10 newly diagnosed and 38 resistant to prior treatment), 73 percent were free of GTCs and 37.5 percent were free of myoclonic seizures over the study period, with a mean follow-up of 19 months [Specchio et al., 2006].
Benzodiazepines are widely accepted as having excellent antimyoclonic effects. This is also true of the myoclonic seizures seen in JME, with up to 88 percent of patients on clonazepam having complete control of myoclonic seizures but only 43 percent becoming free of GTCs [Obeid and Panayiotopoulos, 1989]. For this reason, benzodiazepines are not recommended as monotherapy for patients with JME, but can be extremely successful as add-on therapy when myoclonic seizures persist.
Outcomes
It has been widely accepted that JME is a lifelong condition with a high rate of relapse if weaned off AED therapy; however, some recent long-term follow-up studies have shown conflicting results [Martinez-Juarez et al., 2006; Baykan et al., 2008; Camfield and Camfield, 2009]. Furthermore, one must look at specific seizure types, as it appears the natural history of myoclonic seizures decreases after the fourth decade of life, independent of the on-going occurrence of the other seizure types [Baykan et al., 2008].
In contrast, patients in the CAE evolving into JME group fared much worse, with only 3 of 35 individuals achieving complete seizure freedom [Martinez-Juarez et al., 2006]. Although GTCs were controlled with medications in about 66 percent of patients, absence seizures persisted in 63 percent. These data support the conclusion that JME does appear to be a lifelong disease, specifically in the subgroup of patients with CAE evolving into JME. However, there is still a small subgroup (9 percent) of patients in whom the disease may not be lifelong, and based on these data, it is uncertain whether some of the patients who have been seizure-free on monotherapy for many years would be able to come off AED treatment successfully.
The study by Camfied and Camfield seems to suggest that this indeed may be a possibility. In 24 JME patients with a follow-up period of 25.8 years, 4 individuals (17 percent) were free of all seizures and off AEDs. The mean duration of therapy was 6.5 years before discontinuation. Two additional patients attempted to come off AEDs; they relapsed but later were able to be successfully weaned off medication and remain seizure-free. Another three patients (12.5 percent) had myoclonus only [Camfield and Camfield, 2009]. These results suggest that some patients with JME can successfully discontinue medications. Unfortunately, there do not appear to be clinical or EEG variables that can predict which patients will achieve this result. The decision to discontinue AEDs should be individualized for every patient, and a detailed discussion, with specific attention to the frequency of lifestyle provoking factors, is necessary in order to arrive at a safe and well-informed decision.
Progressive Myoclonic Epilepsies
The PME’s are a group of genetically inherited disorders characterized by both epileptic and nonepileptic myoclonus, generalized tonic-clonic seizures, and progressive neurological deterioration, resulting in dementia, ataxia, and various forms of tremor. Recent advances in the genetic basis of these disorders are leading to a better understanding of how each of the disease processes differs. The four most common PMEs will be discussed (Table 56-3).
Unverricht–Lundborg Disease (ULD)
Unverricht–Lundborg disease (ULD) is the most common PME. It usually presents between the ages of 6 and 15 years, with the hallmark presenting symptom being stimulus-sensitive or action myoclonic jerks, which occur in up to half of patients [Lehesjoki, 2002]. Generalized tonic-clonic seizures are also a common feature early in the disease. At initial presentation, neurological examination and EEG studies may be normal, or the EEG may show generalized spike/wave discharges similar to those seen in IGE. It is not until the patient shows signs of progressive ataxia, intention tremor. and dysarthria that one begins to suspect PME clinically. As the disease progresses, the EEG becomes slow, with more frontally predominant polyspike/wave or spike-wave discharges, ranging in frequency between 2 and 6 Hz [Kyllerman et al., 1991]. MRI findings may be normal or may show nonspecific findings, such as cerebellar atrophy and reduced bulk of the basis pontis and medulla [Mascalchi et al., 2002].
The underlying cause of ULD is linked to an abnormality in the cystatin B gene on chromosome 21q22.3 [Lehesjoki et al., 1991]. Although the exact pathophysiology is unknown, mutations in cystatin B appear to lead to accelerated apoptosis, which may explain the progressive neurologic decline [Delgado-Escueta et al., 2001].
The diagnosis of ULD can be made by mutation testing of the cystatin B gene. There is no specific treatment for ULD, but improvements in seizure management with newer-generation AEDs has improved overall prognosis. VPA has been the mainstay of initial treatment; however, recent studies have shown a beneficial effect of levetiracetam, especially if started early [Magaudda et al., 2004].
Lafora’s Disease
Lafora’s disease (LD) has slightly later onset than ULD but has a more rapid decline, with many patients dying within 10 years of onset. Patients share similar clinical characteristics with other PMEs featuring seizures and myoclonus, but have a more prominent dementing component. Seizure types can be varied, but one peculiar type is that of occipital seizures with hallucinations and transient blindness [Minassian, 2001]. All patients are initially normal neurologically, but may have seizures indistinguishable from IGE until progressive neurological decline is noted. EEG findings early in the course may also resemble IGE, including photosensitivity, but the photosensitivity appears to be maximal at low frequencies between 1 and 6 Hz [Kobayashi et al., 1990]. As the disease progresses, the EEG background becomes slow and disorganized, and there is an evolution of the spike/wave discharges, the 3 Hz discharges becoming faster 6–12 Hz ones [Yen et al., 1991].
The underlying cause of LD is linked to two different sites on chromosome 6 – 6q24 and 6p22 – coding for two different genes: EPM2A (larforin) and NHLRC1 respectively [Minassian et al., 1998; Chan et al., 2003]. The former appears to be involved in regulation of protein folding and the latter may play a role in dendritic transport in neurons. How dysfunction in each of these functions leads to the disease remains unclear.
The diagnosis of LD can be made by testing for mutations in the EPM2A gene that accounts for 80 percent of cases [Minassian et al., 1998]. In those cases where mutation testing is negative but the clinical phenotype is giving cause for concern, Lafora bodies can be detected in skin biopsy specimens. Treatment remains palliative.
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF)
Myoclonic epilepsy with ragged-red fibers (MERRF) is a mitochondrial disease characterized by myoclonic epilepsy, ataxia, and ragged-red fibers on muscle biopsy. Multiple other clinical signs commonly seen in mitochondrial disorders may also be seen, such as myopathy, hearing loss, short stature, neuropathy, and optic atrophy [Chinnery et al., 1997]. EEG findings early in the disease are similar to the other PMEs, with 2–5-Hz generalized spike-wave discharges, and gradual background slowing as the disease progresses [So et al., 1989
[/not-level-membership-for-neurology-category]
