[level-membership-for-pathology-category]
CHAPTER 25 Myeloproliferative neoplasms with eosinophilia
In the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues1 the term ‘myeloproliferative neoplasm’ (MPN) is preferrred to myeloproliferative disorder. Within the group of MPN is chronic eosinophilic leukemia, not otherwise specified.2 Other neoplasms in which eosinophils constitute a dominant or major component are categorized according to the underlying molecular defect, a fusion gene incorporating part of PDGFRA, PDGFRB or FGFR1.3 In the first and last of these groups of disorders there can also be a lymphoid component to the neoplasm, indicating that the responsible mutation occurred in a pluripotent lymphoid-myeloid stem cell. There are other MPN in which eosinophils are part of the neoplastic population but they are not usually a dominant feature. These include chronic myelogenous leukemia (BCR-ABL1 positive) and systemic mastocytosis (SM). In addition, other well-defined MPN can undergo an eosinophilic transformation. The WHO categories of MPN with eosinophilia and a well defined genetic abnormality will be discussed first since a diagnosis of eosinophilic leukemia, not otherwise specified, can be made only when they have been excluded. Non-neoplastic disorders with eosinophilia are discussed in Chapter 16.
Myeloid and lymphoid neoplasms with PDGFRA rearrangement
Most neoplasms resulting from rearrangement of PDGFRA present as chronic eosophilic leukemia (CEL).3,4 The natural history of this condition includes blastic transformation including myeloid sarcoma. A minority of patients present with acute leukemia, either myeloid or T-lymphoblastic with preceding eosinophilia being documented or being present at the time of diagnosis.5
The prototype of this group of neoplasms is CEL associated with a FIP1L1-PDGFRA fusion gene that has resulted from a cryptic interstitial deletion at 4q12. Until the discovery of this recurrent molecular abnormality6 these patients were regarded as having the idiopathic hypereosinophilic syndrome (HES). The diagnosis of idiopathic HES can now not be made without exclusion of FIP1L1-PDGFRA.
Clinicopathological features result from tissue infiltration by eosinophils and organ damage by release of eosinophil granule contents (Table 25.1). Endocardial and myocardial damage is prominent, as is thrombosis.
Table 25.1 Possible clinicopathological features of chronic eosinophilic leukemias
| Organ or system | Features |
|---|---|
| General | Fatigue, fever, weight loss, night sweats, headache |
| Hematological | Eosinophilia (by definition), variable increase in other leukocytes and their precursors, anemia, thrombocytopenia, increased bone marrow mast cells (in some subtypes), increased bone marrow reticulin, clonal cytogenetic or molecular abnormalities, increased serum vitamin B12, increased serum tryptase (in some subtypes) |
| Spleen | Splenomegaly, splenic infarction |
| Liver | Hepatomegaly |
| Lymph nodes | Lymphadenopathy (some subtypes) |
| Cardiovascular | Endocardial, myocardial and pericardial infiltration and damage, increased serum troponin, restrictive cardiomegaly, dilated cardiomyopathy, valvular incompetence, intracardiac thrombi and systemic embolization, cardiac failure, vasculitis, arterial thrombosis, Raynaud’s phenomenon, digital necrosis, venous thromboembolism and pulmonary embolism |
| Respiratory | Cough, dyspnea as a result of infiltration, restrictive or obstructive pulmonary disease, pleural effusion |
| Skin | Pruritis, urticaria, angioedema, papules or nodules due to infiltration, vasculitis |
| Central and peripheral nervous system | Cranial nerve palsies, paraspinal masses, cerebral embolism, peripheral neuropathy |
| Gastrointestinal | Diarrhea as a result of infiltration, malabsorption, mucosal ulcers |
| Musculoskeletal | Myalgia, bone pain |
Precise diagnosis of this condition is very important because of its sensitivity to treatment with imatinib and other tyrosine kinase inhibitors (TKI). Even patients who present in acute transformation can respond to imatinib. Because of the possibility of cardiac damage, rapid diagnosis can also be important. An algorithm showing a diagnostic approach to suspected eosinophilic leukemia is shown in Fig. 25.1.
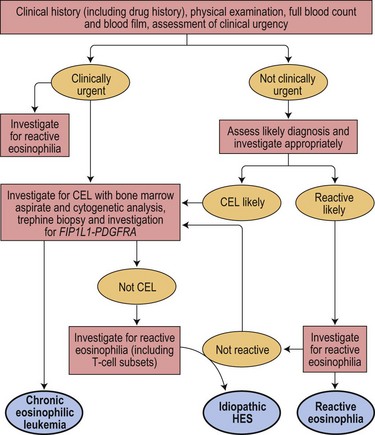
Fig. 25.1 An algorithm showing a diagnostic approach to eosinophilia and suspected eosinophilic leukemia
Pathologic features
Peripheral blood
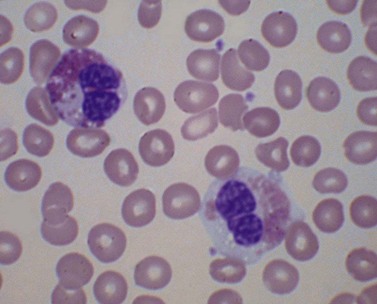
There is eosinophilia and often neutrophilia. Some patients are anemic or thrombocytopenic. The eosinophils may be cytologically normal but often there is degranulation, cytoplasmic vacuolation or nuclear abnormalities – either lack of segmentation or hypersegmentation (Fig. 25.2). Eosinophils may be sufficiently abnormal that they are not recognized by automated blood analyzers.
Bone marrow
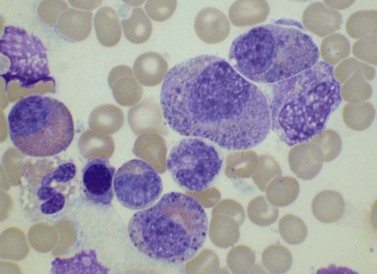
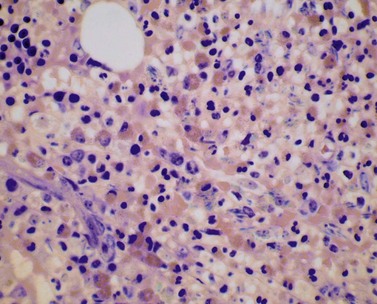
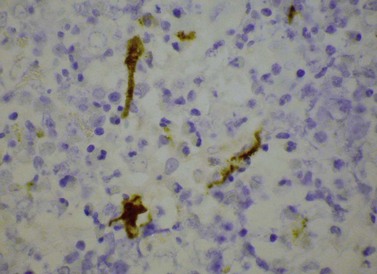
The bone marrow (BM) aspirate is hypercellular as the result of an increase in eosinophils and their precursors. Eosinophil precursors may have purple-staining proeosinophilic granules (Fig. 25.3). There may also be an increase in neutrophils and precursors. Bone marrow trephine biopsy (BMTB) sections show dominance of granulopoiesis with prominent eosinophilia (Fig. 25.4). Reticulin is increased. The majority of patients also have increased mast cells (Fig. 25.5). These are often spindle-shaped and usually dispersed or in loose clusters but in occasional patients they form cohesive infiltrates similar to those of SM. The mast cells can express CD25, an abnormality that is also a feature of SM; they may or may not express CD2, which is usually expressed in SM.3,7 CEL with increased BM mast cells has sometimes been misinterpreted as SM but it must be appreciated that SM with a KIT mutation and CEL with rearrangement of PDGFRA are distinct and different diseases.
Supplementary investigations
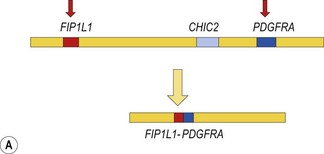
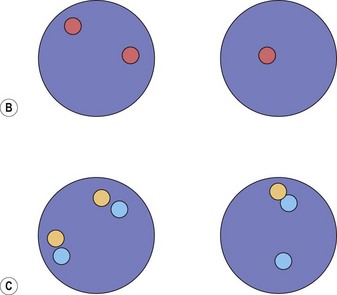
Cytogenetic analysis is usually normal but a minority of patients have a translocation with a 4q12 breakpoint or an unrelated abnormality such as trisomy 8. A secondary cytogenetic abnormality such as trisomy 8 may appear for the first time at evolution to AML.8 Diagnosis is dependent on molecular analysis or fluorescence in situ hydridization (FISH) analysis. The reverse transcriptase polymerase chain reaction (RT-PCR) may demonstrate the FIP1L1-PDGFRA fusion gene but sometimes RT-PCR is negative and nested RT-PCR is required for demonstration of the fusion gene. Various FISH techniques are used. Mainly they rely on demonstration of deletion of the CHIC2 gene, which is located between FIP1L1 and PDGFRA and is thus included in the cryptic deletion (Fig. 25.6).9 Probes flanking FIP1L1 and PDGFRA can also be used with a dual-color break-apart technique.
A minority of patients have a different rearrangement of PDGFRA, usually as a result of a translocation. These include single patients with the hematological features of CEL associated with t(2;4)(p24;q12) and STRN-PDGFRA, t(4;12)(q12;p13) and ETV6-PDGFRA fusion, ins(9;4)(q33;q12q25) with CDK5RAP2-PDGFRA fusion and a complex rearrangement involving chromosomes 3, 4, 10 and probably 13 with KIF5B-PDGFRA. In addition a small number of patients have been described with hematological features intermediate between CEL and chronic myelogenous leukemia (CML) associated with t(4;22)(q12;q11) and BCR-PDGFRA fusion; T and B lymphoid transformations have occurred in this subgroup suggesting that the variant chromosomal rearrangements involving PDGFRA also occur in a pluripotent stem cell. Acquisition of a further mutation in the FIP1L1-PDGFRA fusion gene conveying imatinib resistance may be associated with refractoriness to imatinib and in some cases with evolution to AML.8 The specific second mutation is of relevance since with T674I there is responsiveness to second line tyrosine kinase inhibitors whereas S801P is associated with resistance at least to imatinib and sorafenib10 and D842V is associated with multiresistant disease.11
It should be noted that neither demonstration of a clonal rearrangement of a T-cell receptor gene12 nor the occasional observation of an elevated serum immunoglobulin E7 excludes FIP1L1-PDGFRA-related CEL.
Myeloid neoplasms with PDGFRB rearrangement
The partner genes of myeloid neoplasms with a PDGFRB rearrangement are more numerous (Table 25.2) than is the case for myeloid neoplasms associated with rearrangement of PDGFRA and their clinicopathological features are more heterogeneous.3 The most frequently observed causative cytogenetic/molecular abnormality is t(5;12)(q31-q33;p12) with ETV6-PDGFRB fusion. These neoplasms are responsive to TKI.
Table 25.2 Chronic eosinophilic leukemia and related conditions associated with rearrangement of PDGFRB
| Chromosomal rearrangement | Fusion gene | Hematological presentation |
|---|---|---|
| t(5;12)(q31-q33;p12) or variant | ETV6-PDGFRB | CEL or other MPN (CML, primary myelofibrosis transforming to AML) or MDS/MPN (CMML, aCML), usually with eosinophilia; bone marrow mast cells may be increased; AML |
| t(1;3;5)(p36;p21;q33) | WDR48-PDGFRB (WDR48 is at 3p22) | CEL |
| der(1)t(1;5)(p34;q33), der(5)t(1;5)(p34;q15), der(11)ins(11;5)(p12;q15q33) |
GPIAP1-PDGRFB (GPIAP1 is at 11p13) | CEL |
| t(1;5)(q21;q33) | TPM3-PDGFRB | CEL |
| t(1;5)(q23;q33) | PDE4DIP-PDGFRB | MDS/MPN with eosinophilia |
| t(2;5)(p21;q33) | SPTBN1-PDGFRB | MPN with eosinophilia |
| t(3;5)(p21-25;q31-35) | GOLGA4-PDGFRB | CEL |
| t(4;5;5)(q23;q31;q33), t(4;5)(q21.2;q31.3) or t(4;5)(q21;q33) | PRKG2-PDGFRB | Two cases with chronic basophilic leukemia and abnormal bone marrow mast cells, one case with MPN with abnormal mast cells and an eosinophil count of 1.1 × 109/l |
| t(5;7)(q33;q11.2) | HIP1-PDGFRB | CMML with eosinophilia |
| t(5;10)(q33;q21) | CCDC6-PDGFRB | MPN with eosinophilia or aCML |
| t(5;12)(q31-33;q24) | GIT2-PDGFRB | CEL |
| t(5;12)(q33;p13.3) | ERC1-PDGFRB | AML (without eosinophilia) |
| t(5;14)(q33;q24) | NIN-PDGFRB | Ph-negative CML (13% eosinophils) |
| t(5;14){q33;q32) | TRIP11-PDGFRB | Occurred at relapse of AML, associated with appearance of eosinophilia |
| t(5;14)(q33;q32) | CCDC88C-PDGFRB | CMML with eosinophilia |
| t(5;15)(q33;q22) | TP53BP1-PDGFRB | Ph-negative CML with prominent eosinophilia |
| t(5;16)(q33;p13) | NDE1-PDGFRB | CMML with eosinophilia |
| t(5;17)(q33;p11.2) | SPECC1-PDGFRB | JMML with eosinophilia |
| t(5;17)(q33;p13) | RABEP1-PDGFRB | CMML (without eosinophilia) |
| t(5;17)(q33-34;q11.2) | MYO18A-PDGFRB | MPN with eosinophilia |
aCML, atypical chronic myeloid leukemia; CEL, chronic eosinophilic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; JMML, juvenile myelomonocytic leukemia; MDS/MPN, myelodysplastic/myeloproliferative neoplasm; MPN, myeloproliferative neoplasm.
(reproduced with minor modifications from9)
Pathologic features
Peripheral blood
The hematological presentation may be as CEL but often there is more prominent involvement of other lineages so that the condition is better described as chronic myelomonocytic leukemia (CMML) with eosinophilia or, sometimes, atypical CML with eosinophilia. In a minority of patients eosinophilia has been absent. In the case of t(4;5) and related chromosomal rearrangements associated with PRKG2-PDGFRB, differentiation may be predominantly basophilic.13,14 Otherwise there is a variable increase of neutrophils, monocytes, eosinophils and granulocyte precursors. There may be anemia or thrombocytopenia and trilineage dysplasia.
Supplementary investigations
Serum tryptase and serum vitamin B12 may be elevated. Cytogenetic analysis is essential for confirmation of the diagnosis. Unless there is a chromosomal rearrangement with a breakpoint it does not seem worthwhile to carry out molecular analysis to look for PDGFRB rearrangement, since cryptic rearrangements have rarely been described. It should be noted that FISH has occasionally been negative when Southern blot analysis was positive. The causative cytogenetic molecular lesions that have been described are summarized in Table 25.2.9
Myeloid and lymphoid neoplasms with FGFR1 rearrangement
These rare neoplasms result from a number of translocations involving FGFR1 occurring in a pluripotent lymphoid myeloid stem cell.3,15,16 The most frequently observed translocations are t(8;13)(p11;q12), t(8;9)(p11;q33), t(6;8)(q27;p11-12) and t(8;22)(p11;q11). Common presentations are with CEL or other CML with eosinophilia, T lymphoblastic leukemia/lymphoma and myeloid sarcoma or acute myeloid leukemia (AML). However, B-lineage lymphoblastic leukemia/lymphoma also occurs. Mixed phenotype (B-myeloid) acute leukemia has been described. Often patients who present with AML or lymphoblastic leukemia/lymphoma also have eosinophilia with the eosinophils being part of the leukemic clone. Patients who present in chronic phase as CEL may subsequently undergo lymphoid or myeloid transformation or even both.
Clinical features
The age range is wide with peak incidence being in young adults. There is a moderate male preponderance. The most frequently observed clinical features are lymphadenopathy and splenomegaly. Tonsillar involvement has been prominent among patients with t(8;9).17 Systemic symptoms may be present. Prognosis is poor with those patients who present in chronic phase usually showing disease transformation within a year.
Pathologic features
Bone marrow
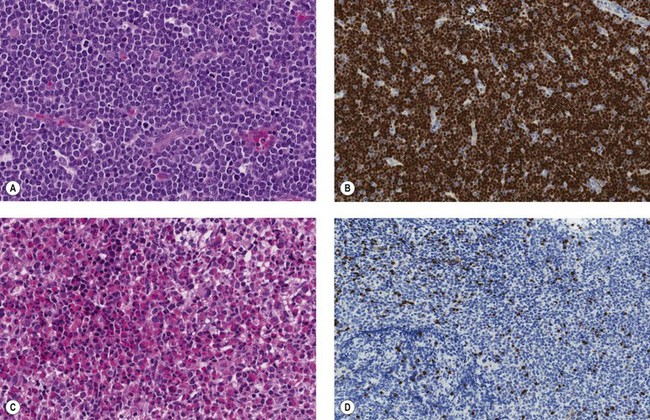
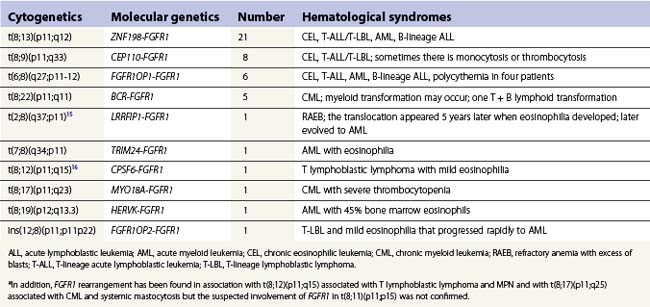
The BM may show only increased cellularity with an increase in eosinophils and precursors or there may be infiltration by lymphoblasts or myeloblasts (Fig. 25.7).
Chronic eosinophilic leukemia, not otherwise specified
This diagnosis is made when there is eosinophilia (eosinophil count at least 1.5 × 109/l) with evidence that the disorder is leukemic in nature and there is no BCR-ABL1 fusion or rearrangement of PDGFRA. PDGFRB or FGFR1. Cases with RUNX1-RUNX1T1 and CBFB-MYH11 fusion are also excluded. In the WHO classification2 the evidence that the condition is leukemic may be either that blast cells are increased (more than 2% in the blood or more than 5% in the BM) or that there is a clonal cytogenetic or molecular genetic abnormality.
Pathologic features
Peripheral blood
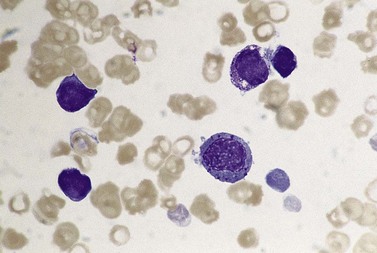
There is eosinophilia as the dominant feature but there may be some increase of neutrophils or monocytes or the presence of granulocyte precursors including blast cells (Fig. 25.8). There may be anemia or thrombocytopenia.
Bone marrow
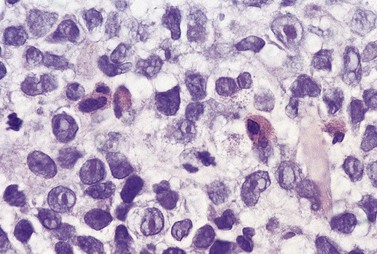
The BM shows an increase of eosinophils and precursors. Blast cells may be increased but are less than 20%. BMTB may show increased blast cells in addition to increased eosinophils and precursors (Fig. 25.9) as well as increased reticulin.
Supplementary investigations
Cytogenetic analysis may show a clonal cytogenetic abnormality such as trisomy 8, del(20q), i(17)(q10), trisomy 10, −Y (not necessarily a clonal abnormality), monosomy 11 plus monosomy 1918 or del(16)(q22).19 A few patients have t(8;9)(p23;p24) with PCM1-JAK2 fusion.20 Occasional patients have other translocations such as t(1;4)(q24;q35)21 and a variety of translocations involving chromosome 9 producing an ETV6-ABL1 fusion gene (at least four patients reported).22 Other patients have a clonal molecular abnormality. JAK2V617F has been described in at least seven patients18,23 but this is an uncommon finding in eosinophilic leukemia (2/36 of cases in one systematic study and 2/134 in another18).
Eosinophilia in other myeloproliferative neoplasms
Dominant eosinophilia is uncommon in MPN but well documented cases have been reported.
Chronic myelogenous leukemia
Philadelphia-positive, BCR-ABL1-positive CML usually has an absolute eosinophilia but it is very unusual for eosinophilia to be the predominant manifestation. Occasional such cases have been reported.24,25 The poor prognosis that has been reported suggests that these cases may represent disease already in evolution. Interpretation of dominant eosinophilia as a manifestation of accelerated phase disease is suggested by a patient in whom only 60% of BM metaphases showed t(9;22).25
Primary myelofibrosis
Eosinophilic leukemia has been reported in myelofibrosis but without detailed documentation of the nature of the condition;26,27 these two cases were not investigated for a JAK2 or MPL mutation or for FIP1L1-PDGRA fusion although the first case did have typical clinicopathological features of primary myelofibrosis.
Eosinophilic transformation of other myeloid neoplasms
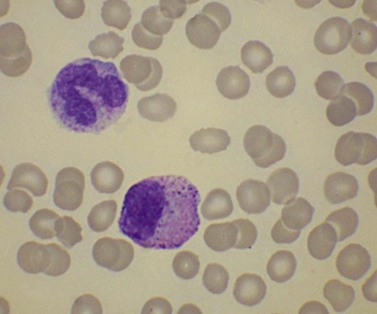
CEL can occur as part of the disease evolution of MPN, e.g. in BCR-ABL1-positive CML, polycythemia vera28 or primary myelofibrosis (Fig. 25.10). Eosinophilic transformation of myelodysplastic/myeloproliferative neoplasms (MDS/MPN) such as CMML can also occur.29 Eosinophilic transformation can occur in MDS but is uncommon. In one survey of 322 patients defined as MDS by French-American-British (FAB) criteria (therefore including CMML and refractory anemia with excess of blasts in transformation) one patient with CMML developed an eosinophil/basophil transformation (both cell types at least 20%) and a further seven patients (2.1%) developed an eosinophil transformation (eosinophils at least 20%).29
1 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphatic Tissues. Lyon: IARC; 2008.
2 Bain B, Gilliland DG, Vardiman J, Horny HP. Chronic eosinophilic leukaemia, not otherwise specified. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. Lyon: IARC Press; 2008. p. 51–3
3 Bain B, Gilliland DG, Horny HP, Vardiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB and FGFR1. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. Lyon: IARC Press; 2008.
4 Gleich GJ, Leiferman KM. The hypereosinophilic syndromes: current concepts and treatments. Br J Haematol. 2009;145:271-285.
5 Metzgeroth G, Walz C, Score J, et al. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophilia-associated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia. 2007;21:1183-1188.
6 Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201-1214.
7 Metzgeroth G, Walz C, Erben P, et al. Safety and efficacy of imatinib in chronic eosinophilic leukaemia and hypereosinophilic syndrome: a phase-II study. Br J Haematol. 2008;143:707-715.
8 von Bubnoff N, Sandherr M, Schlimok G, et al. Myeloid blast crisis evolving during imatinib treatment of an FIP1L1-PDGFR alpha-positive chronic myeloproliferative disease with prominent eosinophilia. Leukemia. 2005;19:286-287.
9 Bain B. Leukemia Diagnosis. Oxford: Blackwell Publishing; 2010.
10 Salemi S, Yousefi S, Simon D, et al. A novel FIP1L1-PDGFRA mutant destabilizing the inactive conformation of the kinase domain in chronic eosinophilic leukemia/hypereosinophilic syndrome. Allergy. 2009;64:913-918.
11 Lierman E, Michaux L, Beullens E, et al. FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009;23:845-851.
12 Helbig G, Moskwa A, Swiderska A, et al. Weekly imatinib dosage for chronic eosinophilic leukaemia expressing FIP1L1-PDGFRA fusion transcript: extended follow-up. Br J Haematol. 2009;145:132-134.
13 Walz C, Metzgeroth G, Haferlach C, et al. Characterization of three new imatinib-responsive fusion genes in chronic myeloproliferative disorders generated by disruption of the platelet-derived growth factor receptor beta gene. Haematologica. 2007;92:163-169.
14 Lahortiga I, Akin C, Cools J, et al. Activity of imatinib in systemic mastocytosis with chronic basophilic leukemia and a PRKG2-PDGFRB fusion. Haematologica. 2008;93:49-56.
15 Soler G, Nusbaum S, Varet B, et al. LRRFIP1, a new FGFR1 partner gene associated with 8p11 myeloproliferative syndrome. Leukemia. 2009;23:1359-1361.
16 Hidalgo-Curtis C, Chase A, Drachenberg M, et al. The t(1;9)(p34;q34) and t(8;12)(p11;q15) fuse pre-mRNA processing proteins SFPQ (PSF) and CPSF6 to ABL and FGFR1. Genes Chromosomes Cancer. 2008;47:379-385.
17 Park TS, Song J, Kim JS, et al. 8p11 myeloproliferative syndrome preceded by t(8;9)(p11;q33), CEP110/FGFR1 fusion transcript: morphologic, molecular, and cytogenetic characterization of myeloid neoplasms associated with eosinophilia and FGFR1 abnormality. Cancer Genet Cytogenet. 2008;181:93-99.
18 Helbig G, Majewski M, Wieczorkiewicz A, et al. Screening for JAK2 V617F point mutation in patients with hypereosinophilic syndrome – in response to ‘Hypereosinophilic syndrome: another face of Janus?’ by Dahabreh et al. published in Leukemia Research, in press. Leuk.Res. 2009;33:e1-e2.
19 Kamineni P, Baptiste A, Hassan M, et al. Case of chronic eosinophilic leukemia with deletion of chromosome 16 and hepatitis C. J Natl Med Assoc. 2006;98:1356-1360.
20 Cornfield DB, Gheith SM, Friedman EL. A third case of chronic eosinophilic leukemia with the (8;9)(p23;p24) translocation. Cancer Genet Cytogenet. 2008;185:60-61.
21 Arora RS. Chronic eosinophilic leukemia with a unique translocation. Indian Pediatr. 2009;46:525-527.
22 Keung YK, Beaty M, Steward W, et al. Chronic myelocytic leukemia with eosinophilia, t(9;12)(q34;p13), and ETV6-ABL gene rearrangement: case report and review of the literature. Cancer Genet Cytogenet. 2002;138:139-142.
23 Dahabreh IJ, Giannouli S, Zoi C, et al. Hypereosinophilic syndrome: another face of Janus? Leuk Res. 2008;32:1483-1485.
24 Gotlib V, Darji J, Bloomfield K, et al. Eosinophilic variant of chronic myeloid leukemia with vascular complications. Leuk Lymphoma. 2003;44:1609-1613.
25 Aggrawal DK, Bhargava R, Dolai TK, et al. An unusual presentation of eosinophilic variant of chronic myeloid leukemia (eoCML). Ann Hematol. 2009;88:89-90.
26 Cox MC, Panetta P, Venditti A, et al. New reciprocal translocation t(6;10) (q27;q11) associated with idiopathic myelofibrosis and eosinophilia. Leuk Res. 2001;25:349-351.
27 Ishii Y, Ito Y, Kuriyama Y, et al. Successful treatment with imatinib mesylate of hypereosinophilic syndrome (chronic eosinophilic leukemia) with myelofibrosis. Leuk Res. 2004;28(Suppl 1)):S79-S80.
28 Chim CS, Ma SK. Eosinophilic leukemic transformation in polycythemia rubra vera (PRV). Leuk Lymphoma. 2005;46:447-450.
29 Wimazal F, Baumgartner C, Sonneck K, et al. Mixed-lineage eosinophil/basophil crisis in MDS: a rare form of progression. Eur J Clin Invest. 2008;38:447-455.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 25 Myeloproliferative neoplasms with eosinophilia
In the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues1 the term ‘myeloproliferative neoplasm’ (MPN) is preferrred to myeloproliferative disorder. Within the group of MPN is chronic eosinophilic leukemia, not otherwise specified.2 Other neoplasms in which eosinophils constitute a dominant or major component are categorized according to the underlying molecular defect, a fusion gene incorporating part of PDGFRA, PDGFRB or FGFR1.3 In the first and last of these groups of disorders there can also be a lymphoid component to the neoplasm, indicating that the responsible mutation occurred in a pluripotent lymphoid-myeloid stem cell. There are other MPN in which eosinophils are part of the neoplastic population but they are not usually a dominant feature. These include chronic myelogenous leukemia (BCR-ABL1 positive) and systemic mastocytosis (SM). In addition, other well-defined MPN can undergo an eosinophilic transformation. The WHO categories of MPN with eosinophilia and a well defined genetic abnormality will be discussed first since a diagnosis of eosinophilic leukemia, not otherwise specified, can be made only when they have been excluded. Non-neoplastic disorders with eosinophilia are discussed in Chapter 16.
Myeloid and lymphoid neoplasms with PDGFRA rearrangement
Most neoplasms resulting from rearrangement of PDGFRA present as chronic eosophilic leukemia (CEL).3,4 The natural history of this condition includes blastic transformation including myeloid sarcoma. A minority of patients present with acute leukemia, either myeloid or T-lymphoblastic with preceding eosinophilia being documented or being present at the time of diagnosis.5
The prototype of this group of neoplasms is CEL associated with a FIP1L1-PDGFRA fusion gene that has resulted from a cryptic interstitial deletion at 4q12. Until the discovery of this recurrent molecular abnormality6 these patients were regarded as having the idiopathic hypereosinophilic syndrome (HES). The diagnosis of idiopathic HES can now not be made without exclusion of FIP1L1-PDGFRA.
Clinicopathological features result from tissue infiltration by eosinophils and organ damage by release of eosinophil granule contents (Table 25.1). Endocardial and myocardial damage is prominent, as is thrombosis.
Table 25.1 Possible clinicopathological features of chronic eosinophilic leukemias
| Organ or system | Features |
|---|---|
| General | Fatigue, fever, weight loss, night sweats, headache |
| Hematological | Eosinophilia (by definition), variable increase in other leukocytes and their precursors, anemia, thrombocytopenia, increased bone marrow mast cells (in some subtypes), increased bone marrow reticulin, clonal cytogenetic or molecular abnormalities, increased serum vitamin B12, increased serum tryptase (in some subtypes) |
| Spleen | Splenomegaly, splenic infarction |
| Liver | Hepatomegaly |
| Lymph nodes | Lymphadenopathy (some subtypes) |
| Cardiovascular | Endocardial, myocardial and pericardial infiltration and damage, increased serum troponin, restrictive cardiomegaly, dilated cardiomyopathy, valvular incompetence, intracardiac thrombi and systemic embolization, cardiac failure, vasculitis, arterial thrombosis, Raynaud’s phenomenon, digital necrosis, venous thromboembolism and pulmonary embolism |
| Respiratory | Cough, dyspnea as a result of infiltration, restrictive or obstructive pulmonary disease, pleural effusion |
| Skin | Pruritis, urticaria, angioedema, papules or nodules due to infiltration, vasculitis |
| Central and peripheral nervous system | Cranial nerve palsies, paraspinal masses, cerebral embolism, peripheral neuropathy |
| Gastrointestinal | Diarrhea as a result of infiltration, malabsorption, mucosal ulcers |
| Musculoskeletal | Myalgia, bone pain |
Precise diagnosis of this condition is very important because of its sensitivity to treatment with imatinib and other tyrosine kinase inhibitors (TKI). Even patients who present in acute transformation can respond to imatinib. Because of the possibility of cardiac damage, rapid diagnosis can also be important. An algorithm showing a diagnostic approach to suspected eosinophilic leukemia is shown in Fig. 25.1.
Fig. 25.1 An algorithm showing a diagnostic approach to eosinophilia and suspected eosinophilic leukemia
Pathologic features
Peripheral blood
There is eosinophilia and often neutrophilia. Some patients are anemic or thrombocytopenic. The eosinophils may be cytologically normal but often there is degranulation, cytoplasmic vacuolation or nuclear abnormalities – either lack of segmentation or hypersegmentation (Fig. 25.2). Eosinophils may be sufficiently abnormal that they are not recognized by automated blood analyzers.
Bone marrow
The bone marrow (BM) aspirate is hypercellular as the result of an increase in eosinophils and their precursors. Eosinophil precursors may have purple-staining proeosinophilic granules (Fig. 25.3). There may also be an increase in neutrophils and precursors. Bone marrow trephine biopsy (BMTB) sections show dominance of granulopoiesis with prominent eosinophilia (Fig. 25.4). Reticulin is increased. The majority of patients also have increased mast cells (Fig. 25.5). These are often spindle-shaped and usually dispersed or in loose clusters but in occasional patients they form cohesive infiltrates similar to those of SM. The mast cells can express CD25, an abnormality that is also a feature of SM; they may or may not express CD2, which is usually expressed in SM.3,7 CEL with increased BM mast cells has sometimes been misinterpreted as SM but it must be appreciated that SM with a KIT mutation and CEL with rearrangement of PDGFRA are distinct and different diseases.
