Chapter 5 Muscular Tone and Gait Disturbances
Pathology
The central and peripheral nervous systems modify tone, but intrinsic physical characteristics of the tendons, joints, and muscles and the anatomic interrelationships of these structures also contribute significantly to tone. In childhood central nervous system (CNS) dysfunction, upper motor neuron (unit) disease may cause increased or decreased muscle tone [Teddy et al., 1984]. Disease involving the lower motor neuron (unit) results in hypotonia and weakness.
The final common pathway of upper or lower motor unit modification of tone is through the gamma loop (fusimotor) system [Gordon and Ghez, 1991; Granit, 1975]. Intimately involved with monitoring and effecting tone are the two stretch-sensitive muscle receptors – the muscle spindles and the Golgi tendon organs (Figure 5-1). It also has become evident that nonreflex, mechanical mechanisms are involved in the maintenance of resting muscle tone. Spinal cord reflex responses depend on ongoing activity in interneurons [Davidoff, 1992].
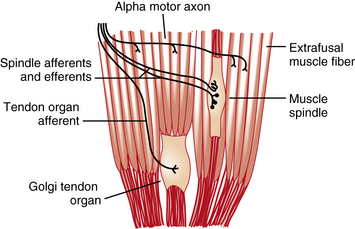
Fig. 5-1 Muscle spindles and Golgi tendon organs are encapsulated structures found in skeletal muscle.
(Adapted from Houck JC, Crago PE, Rymer WZ. Functional properties of the Golgi tendon organs. In: Desmedt JE, ed. Spinal and supraspinal mechanisms of voluntary motor control and locomotion. Progress in Clinical Neurophysiology, vol. 8. Basel: Karger, 1980:33.)
Stationed in all areas of the skeletal muscle is the muscle spindle, a fusiform-shaped receptor structure (Figure 5-2). The spindle is composed of contractile fibers at each end and a capsule covering a central fluid-filled dilatation. Sensory endings wrap around the central sections of the intrafusal fibers and monitor the stretch of these fibers; they communicate through the afferent axons that are described later in this chapter. Through efferent axons, gamma neurons within the anterior horn of the spinal cord innervate the contractile muscle portions on each end of the intrafusal fiber and enhance the sensitivity of the sensory endings to stretch [Gordon and Ghez, 1991]. Gamma motor neurons that innervate muscle spindles comprise the fusimotor system.
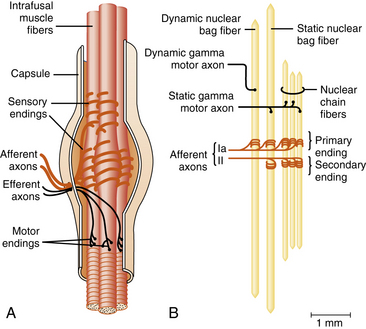
Fig. 5-2 The main components of the muscle spindle are intrafusal fibers, sensory endings, and motor axons.
(A, Adapted from Hullinger M. The mammalian muscle spindle and its central control. Rev Physiol Biochem Pharmacol 1984;101:1; B, Adapted from Boyd IA. The isolated mammalian muscle spindle. Trends Neurosci 1980;3:258. Reproduced with permission from Elsevier.)
The intrafusal muscle fibers are divided into three types: nuclear chain fibers, dynamic nuclear bag fibers, and static nuclear bag fibers. These fibers derive their names from the configuration of their nuclei in the fiber center. Chain fibers have nuclei arranged in a single column, whereas bag fibers have nuclei aligned in rows of two or three. A solitary Ia afferent fiber provides primary sensory innervation for all three types of intrafusal fibers. A group II afferent fiber innervates chain and static bag fibers providing secondary sensory endings. The various sensory endings on the different types of intrafusal fibers have different sensitivities to rate of change of length. Dynamic gamma motor axons innervate the contractile portions of dynamic nuclear bag fibers, and static gamma motor axons innervate the contractile portions of the static bag fibers [Gordon and Ghez, 1991]. This intricate system of muscle spindle innervation allows the muscle stretch receptors to monitor muscle tension, length, and velocity of stretch, and provide input for maintenance of tone [Carew, 1985].
It is through their effect on the gamma motor neuron that portions of the CNS (i.e., motor cortex, thalamus, basal ganglia, vestibular nuclei, reticular formation, and cerebellum) modify tone, with ensuing hypotonia or hypertonia (i.e., spasticity) [Alexander and Delong, 1985; Brooks and Stoney, 1971; Carew, 1985; Ghez, 1985].
The Golgi tendon organs, unlike the muscle spindles, are found in series with the skeletal muscle fibers (Figure 5-3), and are attached at one end to the muscle and at the other to the tendon. A number of individual skeletal muscle fibers enter a Golgi tendon organ through a constricted collar. The muscle fibers are attached to collagen fibers within the Golgi tendon organ. A single Ib axon enters each capsule and forms branches that are interlaced among the collagen fibers. The afferent axon branches are compressed when muscle contraction occurs and impulses are transmitted. Tendon organs are much more sensitive to muscle contraction than muscle spindles. Conversely, tendon organs are much less sensitive to stretch than muscle spindles. Each of these relative sensitivities plays a specific role during the performance of various motor tasks [Gordon and Ghez, 1991].
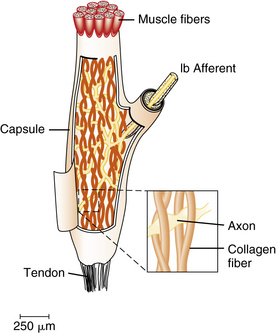
Fig. 5-3 Golgi tendon organs are specialized structures found at the junctions between muscle and tendon.
(Adapted from Schmidt RF. Motor systems. In: Schmidt RF, Thews G, eds. Human physiology [Biederman-Thorson MA, translator]. Berlin: Springer, 1983:81; inset, adapted from Swett JE, Schoultz TW. Mechanical transduction in the Golgi tendon organ: a hypothesis. Arch Ital Biol 1975;113:374.)
Evaluation of the Patient
History
The age at which hypotonia is first evident may be diagnostically crucial. The presence of hypotonia at birth or shortly thereafter can help differentiate among a number of conditions [Brooke et al., 1979; Dubowitz, 1985]. The presence or absence of weak fetal movements or the change from apparently normal fetal movements to those of decreased amplitude and vigor should be determined. Hypotonia in the setting of polyhydramnios signals prenatal interference with swallowing.
A careful genetic history must be sought because several conditions characterized by hypotonia are hereditary. In a retrospective review of hypotonic infants, Birdi et al. reported a family history of neuromuscular or neurologic disorders in almost half [Birdi et al., 2005].
Examination
Tone should be assessed both in the active state and when the neonate is at rest; active tone of the extremities is normally higher than passive tone. Passive pronation, supination, flexion, and extension of the limbs and gently shaking the hands and feet are the best ways to assess tone. The hands move over a large amplitude when the arms are shaken gently at the wrists. Often, in the hypotonic infant, the elbows can be extended beyond their normal range. The scarf sign involves wrapping the infant’s arm across the chest toward the neck on the contralateral side and is positive when the elbow can be readily moved beyond the midline. While abnormal in the term infant, it can normally be seen in preterm infants. The traction maneuver is one of the best means to evaluate tone, since it allows simultaneous evaluation of head control, flexion of elbows during infant participation, and general body and back posture (see Figure 3-7). The hypotonic infant’s foot can be brought to the opposite ear, and extreme passive foot dorsiflexion may be possible when hypotonia is profound.
The pectus excavatum deformity and a bell-shaped chest indicate relative weakness of intercostal muscles compared to better-preserved strength of the diaphragm during respiratory efforts. Skeletal deformities and fixed contractures are often present in congenital myotonic dystrophy and some congenital myopathies. Fixed contractures of the limbs may signal the presence of arthrogryposis multiplex congenita, which may result from dysfunction at a number of lower motor neuron unit sites [Lebenthal et al., 1970; Yuill and Lynch, 1974] (see Chapter 88).
Acute onset of progressive, profound weakness and hypotonia in previously normal infants suggests the possibility of infantile botulism [Infant botulism, 2003; Kao et al., 1976; Pickett et al., 1976; Ravid et al., 2000; Thompson et al., 1980]. There is almost always accompanying constipation, poor feeding, and bulbar involvement.
Weakness is often readily diagnosed in the infant and younger child by observation; in the older child, more formal and discrete muscle testing is possible, as described in Chapter 2. Examination also includes deep tendon reflexes, plantar reflexes, myotonic response to percussion and scrutiny of muscles for evidence of fasciculations.
When the lower motor unit is involved, the deep tendon reflexes range from hypoactive to absent. The reflexes are uniformly absent in infantile spinal muscular atrophy [Bundey and Lovelace, 1975; Smith and Swaiman, 1983]. Reflexes tend to be increased or normal when the upper motor unit is involved, but may be decreased in the case of acute injury or concomitant involvement of the basal ganglia or their output tracts.
Further neurologic examination is necessary and should include the search for fasciculations, ptosis, squint, myotonia, and extensor plantar reflexes. The presence of squint or ptosis suggests the possibility of congenital myopathies [Clancy et al., 1980; McComb et al., 1979; Riggs et al., 2003], myotonic dystrophy, myasthenia gravis [Holmes et al., 1980; Namba et al., 1970], or mitochondrial myopathies (see Chapters 37 and 93). Knowledge about the congenital myopathies has grown considerably during the past decade, and the clinical and genetic complexities have become increasingly evident [Bruno and Minetti, 2004; Kirschner and Bonnemann, 2004].
Diagnosis
It is important to distinguish whether hypotonia is derived from a central or peripheral etiology. While there can be difficulty in distinguishing the localization, one study examined sensitivity and specificity of findings predictive of primary neuromuscular disorders. These included a history of reduced fetal movements with polyhydramnios, significant impairment or absence of antigravity movements, and presence of contractures [Vasta et al., 2005]. Congenital hypotonia may be extremely difficult to categorize; however, certain characteristics may prove helpful in diagnosis (Table 5-1) [Harris, 2008].
Table 5-1 Differentiation of Central versus Peripheral Causes of Congenital Hypotonia
| Characteristic | Central | Peripheral |
|---|---|---|
| Weakness | Mild to moderate | Significant (“paralytic”) |
| Deep tendon reflexes | Decreased or increased | Absent |
| Placing reaction | Sluggish | Absent |
| Motor delays | Yes | Yes |
| Antigravity movements in prone and supine | Some (less than normal) | Often absent |
| Pull to sit | Head lag (more than normal) | Marked head lag |
| Cognition/affect | Delayed | Typical |
| Ability to “build up” tone, e.g., tapping under knees with infant in supine to assist him/her in holding hips in adduction | Yes | No |
(From Harris S. Congenital hypotonia. Dev Med Child Neurol 2008; 50:889.)
The presence of hypothyroidism is suggested by decreased length and weight, large tongue, and developmental delay. Some conditions associated with hypotonia are listed in Box 5-1 and are discussed in detail elsewhere in this book.



































































Clinical Laboratory Studies
The diagnosis of peripheral neuropathy, particularly in conditions that involve the central and peripheral nervous systems, may be readily overlooked without the determination of nerve conduction velocities. Normative data are available for all age groups [Gamstorp, 1963].
Gait Impairment
Gait disturbances in children are often caused by neurologic disease, but it is overly simplistic to attribute all such abnormalities to neurologic dysfunction. Foot deformities are present in 4 percent of neonates, but the natural course of such congenital deformities is favorable, except for clubfoot which often requires surgical repair [Widhe, 1997]. Congenital abnormalities such as hamstring muscle or plantar foot flexor tightness may result in difficulties with posture and back pain [Jozwiak et al., 1997]. Clinical indices are available for the evaluation of gait pathology in children [Romei et al., 2004].
Gait is a demanding, complicated skill that requires integration of many functional components of the nervous system and is the result of a repetitive sequence of limb movements. The walking pendulum mechanism seen in older children and adults is not yet developed when the toddler first learns to walk independently, but by 2 years of age mechanical energy data are already similar to adult patterns [Ivanenko et al., 2004]. Significant changes in plantar pressure of the foot occur during the first year of standing and walking and the age of development of a mature pattern is highly variable [Bertsch et al., 2004]. Sophisticated evaluation of foot kinetics during walking is useful in providing information about gait impairment [MacWilliams et al., 2003]. Development of mature displacement of center of mass of the body during independent walking is a gradual neural process that evolves until the age of 7 years [Dierick et al., 2004]. Optimal gait requires the least expenditure of energy possible. Mechanical energy must be generated and then dissipated in a controlled fashion during each cycle [Gage et al., 1984; Õunpuu et al., 1991]. The encumbrance of additional weight can interfere with optimal walking posture; backpack load and walking distance do not affect stride, but a load of above 15 percent of body weight induces a significant increase in trunk inclination [Hong and Cheung, 2003]. During the gait cycle, posture and balance must be maintained, and the feet must clear the ground without scraping. Quantitative gait evaluation is increasingly precise and useful [Schwartz et al., 2004]. When children with pathologic gait characteristics are being compared with normal children, testing should involve patients and normal controls should be tested at the same walking speed [van der Linden et al., 2002].
Gait must be assessed when the patient’s chief complaint focuses on walking or running; however, assessment of gait affords the clinician rapid appraisal of a number of significant nervous system units when patient complaints are other than those relating to gait. Acquired idiopathic gait difficulties may occur with some frequency in children admitted to some children’s hospitals. Some acquired gait disorders have a definite physical cause, and some are idiopathic [Wassmer et al., 2002]. Further data documenting the incidence of these disorders is needed to evaluate their economic and social impact.
Physiologic Considerations
Skills required for standing must be synthesized into the walking procedure. The walking sequence requires that the non–weight-bearing leg moves forward while weight is shifted smoothly from leg to leg. The definitive components of support and forward movement require separate consideration; the rhythm and duration of each phase require monitoring [Gage and Õunpuu, 1989; Paine and Oppe, 1966]. Conventionally, the period from one heel–ground contact to the next heel–ground contact of one foot is one gait cycle; walking can be divided into stance and swing phases. The instant from which heel–ground contact occurs until the instant when contact terminates is the stance phase. The stance phase can be divided into four parts: initial contact, loading response, midstance, and terminal stance (Figure 5-4 and Figure 5-5). The period beginning immediately after the toe leaves the ground until the heel contacts the ground is the swing phase (see Figure 5-4 and Figure 5-5) [Burnett and Johnson, 1971a, 1971b; Norlin et al., 1981]. The swing phase can also be divided into four parts: preswing, initial swing, midswing, and terminal swing. Decreased knee flexion during the swing phase (i.e., stiff-knee gait) may be caused by overactivity of the rectus femoris [Piazza and Delp, 1996]. Normally, the stance phase occupies 60 percent of the duration of the cycle, and the swing phase occupies 40 percent.
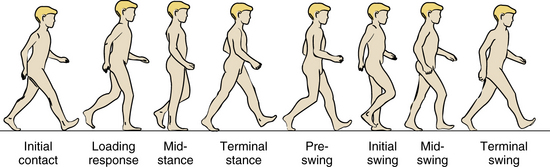
Fig. 5-4 Schematic representation of various phases of a child walking.
(Adapted from Õunpuu S, Gage JR, Davis RB. Three-dimensional lower extremity joint kinetics in normal pediatric gait. J Pediatr Orthop 1991;11:341.)
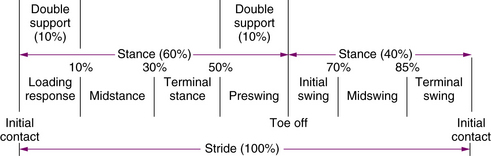
Fig. 5-5 Graphic representation of the phases of gait and their duration.
(From Õunpuu S, Gage JR, Davis RB. Three-dimensional lower extremity joint kinetics in normal pediatric gait. J Pediatr Orthop 1991;11:341.)
Elaborate methods for the assessment of phases of gait have been devised [Burnett and Johnson, 1971a; Õunpuu et al., 1991]. The center of gravity is affected by several factors during ambulation: pelvic rotation, pelvic tilt, knee flexion at midstance, foot and knee mechanics, and lateral displacement of the pelvis [Saunders et al., 1953]. The center of gravity in older children shifts approximately 4.5 cm during the gait cycle. Pelvic rotation and pelvic tilt are usually necessary for development of independent gait [Burnett and Johnson, 1971a, 1971b]. Children usually acquire adult patterns of walking within 55 weeks of independent gait being achieved [Burnett and Johnson, 1971a, 1971b].
Because walking is such a complex skill, the normal participation of many motor system parts is vital; these include the basal ganglia; sensory cortex; neck proprioceptors; visual receptors; cerebellum; spinal cord motor and sensory tracts, and gray matter masses; peripheral nerves; neuromuscular junctions; and muscles [Norlin et al., 1981; Winter, 1990]. The participation of various muscles varies greatly during each portion of the gait cycle (Figure 5-6).
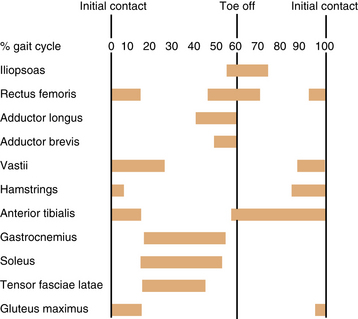
Fig. 5-6 Graphic representation of the involvement of various muscles during the phases of gait.
(From Õunpuu S, Gage JR, Davis RB. Three-dimensional lower extremity joint kinetics in normal pediatric gait. J Pediatr Orthop 1991;11:341.)
Indices have been devised to quantify deviations from normal gait and prove helpful in overall characterization of the patient’s degree of abnormal gait [Schutte et al., 2000; Schwartz and Rozumalski, 2008].
Evaluation of the Patient
Associated movements of the arms should be carefully observed. The fingers and hands may flex in association with infolding of the thumbs, indicating possible corticospinal tract dysfunction. The arms should move so that the contralateral arm swings forward synchronously with the swing phase of each leg (see Figure 5-4). When the child runs, abnormal arm and hand postures and movements are frequently accentuated. It is important in gait evaluation to measure leg length accurately. Discrepancies of less than 3 percent are not associated with compensatory movements [Song et al., 1997].
More precise methods of gait analysis are available. Three-dimensional bilateral kinematic data can be obtained for analysis of the various facets of gait in children. The child is studied from several aspects: sagittal (i.e., the subject is viewed and monitored from the side), coronal (i.e., the subject is viewed and monitored from the front), and transverse (i.e., the subject is viewed and monitored from above). Data are generated from the changes in relationships as measured in angles (degrees) of various skeletal parts during the gait cycle [Gage, 1991; Myers et al., 2004; Õunpuu et al., 1991; Schwartz et al., 2004].
Electromyographic patterns of extrinsic ankle muscles in healthy children between 4 and 11 years old demonstrate the significant effect of walking speed changes but are independent of growth over this age range. This information can also be reduced to and retrieved from a nomograph [Detrembleur et al., 1997].
Differential Diagnosis
Spastic Paraplegic Gait
Toe walking may be the only indication of spastic paraplegia. However, isolated toe walking has a broad differential diagnosis, including spinal cord lesions (e.g., tethered cord syndrome), and can even be an early sign of autism. Sometimes, it is idiopathic, benign, or on a familial basis without relevance to future development. The clinician should establish the presence of other manifestations of upper motor neuron unit dysfunction before attributing pathologic significance to toe walking [Kelly et al., 1997; Volpe, 1997].
Cerebellar Gait
Conditions affecting the cerebellum range from congenital malformations to infections, acute and chronic metabolic diseases, and progressive degenerative disorders. Among these conditions are congenital malformations, inherited cerebellar atrophies, aminoacidurias, mitochondrial diseases, lipid storage diseases, anoxic episodes, demyelinating diseases, posterior fossa tumors, hydrocephalus, and intoxication. Many hereditary cerebellar diseases have been described; these conditions often affect only a small number of pedigrees [Brown, 1980].
Acute Cerebellar Ataxia
The sudden, isolated appearance of ataxia without obvious cause requires systematic evaluation. Although the process is most often benign and self-limiting, the differential diagnosis is broad and includes some serious conditions [Hayakawa and Katoh, 1995; Sunaga et al., 1995]. In many cases, the cause is postinfectious, often following an influenza-like syndrome within the preceding few weeks. The patient is usually between 1 and 4 years old with a peak in the second year of life [Weiss and Carter, 1959]. The attacks may be so severe that the patient is bedridden, but more often present with unsteadiness and truncal ataxia. Nystagmus is present in one-half of these children [Cotton, 1957]. Other inconstant features are hypotonia, tremor, and scanning speech. Noncerebellar symptoms may include headaches, photophobia, and lightheadedness.
Lumbar puncture usually includes normal opening pressure, mild pleocytosis and normal glucose and protein, although a slight increase in protein content may be evident after several weeks. Acute cerebellar ataxia has been linked with numerous bacterial and viral infections, including diphtheria, pertussis, typhoid fever, rubella, mumps, varicella, coxsackievirus A9, echovirus 9, and poliomyelitis [King et al., 1958; Mendez-Cashion et al., 1962].
Full recovery, even with corticosteroid treatment, may require several months, but some children return to normal within 10 days, even without treatment. If no improvement in the ataxia occurs after several weeks, the clinician should be alert to the possible existence of a serious underlying cause. Approximately 30 percent of children retain a neurologic deficit, including ataxia and speech impairment [Weiss and Carter, 1959]. The differential diagnosis of ataxia is found in Chapter 67.
Other Dyskinetic Gaits
Antalgic Gait (Painful Gait)
Pain can arise from any leg and foot structure, including nails, skin, joints, bone, and muscles. The associated limp is caused by a decreased weight support on the painful leg and increased duration of weight support on the unaffected leg [Chung, 1974]. The examiner may require prolonged observation to determine the precise nature of the limp. The exact limp pattern is determined by the location of the pain [Hensinger, 1977]. Bilateral antalgic gait can be the result of rickets.
Conversion Reaction Gait
The gait of a child with conversion reaction may be outrageously intricate and may vary during the course of the examination. Slowness of gait is common in psychogenic gait disorders [Baik and Lang, 2007]. Short periods of normal walking activity may occur at times. Tremulousness of the fingers and hands during standing or walking may be associated findings. Patients with conversion reactions resembling hemiplegia or monoplegia usually drag the foot along the floor or push it ahead, in contradistinction to patients with corticospinal tract difficulty who elevate and circumduct the leg during each step. When both legs are involved, the patient may be bedridden or use crutches. On occasion, the child lurches out of control but does not fall, demonstrating remarkable coordination and strength (astasia-abasia). In patients with a positive Romberg sign, the swaying is often at the hips with a tendency to separate the legs despite instructions to stand with feet together. Patients with conversion reactions usually do not separate their feet. They may have associated rapid random movements of the head, hands, and hips. If the patient falls, there may be a dramatic aspect to the mishap. Despite dramatic unsteadiness, the patient may be able to run or walk backward without difficulty. One caveat: patients with dystonia musculorum deformans may walk backward smoothly, although they have problems with forward ambulation.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Alexander G., Delong M. Organization of supraspinal motor systems. In: McKhann A., McDonald W., editors. Diseases of the nervous system: Clinical neurology. Philadelphia: WB Saunders, 1985.
Baik J.S., Lang A.E. Gait abnormalities in psychogenic movement disorders. Mov Disord. 2007;22:395-399.
Bertsch C., Unger H., Winkelmann W., et al. Evaluation of early walking patterns from plantar pressure distribution measurements. First year results of 40 children. Gait Posture. 2004;19:235.
Birdi K., Prasad A.N., Prasad C., et al. The floppy infant: retrospective analysis of clinical experience (1990–2000) in a tertiary care facility. J Child Neurol. 2005;20:803.
Boyd I.A. The isolated mammalian muscle spindle. Trends Neurosci. 1980;3:258.
Brooke M.H., Carroll J.E., Ringel S.P. Congenital hypotonia revisited [Review]. Muscle Nerve. 1979;2:84.
Brooks V.B., Stoney S.D. Motor mechanisms: The role of the pyramidal system in motor control. Annu Rev Physiol. 1971;33:337.
Brown J.R. Diseases of the cerebellum. In Baker A.B., Baker L.H., editors: Clinical neurology, ed 4, Baltimore: Harper & Row, 1980.
Bruno C., Minetti C. Congenital myopathies. Curr Neurol Neurosci Rep. 2004;4:68.
Bundey S., Lovelace R.E. A clinical and genetic study of chronic proximal spinal muscular atrophy. Brain. 1975;98:455.
Burnett C.N., Johnson E.W. Development of gait in childhood. I. Method. Dev Med Child Neurol. 1971;13:196.
Burnett C.N., Johnson E.W. Development of gait in childhood. II. Dev Med Child Neurol. 1971;13:207.
Carew T. Posture and locomotion. In Kandel E., Schwartz J., editors: Principles of neural science, ed 2, New York: Elsevier Science Publishing, 1985.
Chung S.M. Identifying the cause of acute limp in childhood: Some informal comments and observations. Clin Pediatr (Phila). 1974;13:769.
Clancy R.R., Kelts K.A., Oehlert J.W. Clinical variability in congenital fiber type disproportion. J Neurosci. 1980;46:257.
Cotton D.G. Acute cerebellar ataxia. Arch Dis Child. 1957;32:181.
Davidoff R.A. Skeletal muscle tone and the misunderstood stretch reflex. Neurology. 1992;42:951.
Detrembleur C., Willems P., Plaghki L. Does walking speed influence the time pattern of muscle activation in normal children? Dev Med Child Neurol. 1997;39:803.
Dierick F., Lefebvre C., van den Hecke A., et al. Development of displacement of center of mass during independent walking in children. Dev Med Child Neurol. 2004;46:533.
Dubowitz V. Evaluation and differential diagnosis of the hypotonic infant. Pediatr Rev. 1985;6:237.
Early clinical signs and imaging findings in Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu)
Gage J.R. Gait analysis in cerebral palsy. London: MacKeith Press; 1991.
Gage J.R., Fabian D., Hicks R., et al. Pre- and postoperative gait analysis in patients with spastic diplegia: A preliminary report. J Pediatr Orthop. 1984;4:715.
Gage J.R., Õunpuu S. Gait analysis in clinical practice. Semin Orthop. 1989;4:72.
Gamstorp I. Normal conduction velocity of ulnar, median and peroneal nerves in infancy, childhood and adolescence. Acta Paediatr Scand. 1963;146(Suppl):68.
Ghez C. Introduction to the motor systems. In Kandel E., Schwartz J., editors: Principles of neural science, ed 2, New York: Elsevier Science Publishing, 1985.
Gordon J., Ghez C. Muscle receptors and spinal reflexes: The stretch reflex. In Kandel E., Schwartz J., Jessell T., editors: Principles of neural science, ed 3, New York: Elsevier Science Publishing, 1991.
Granit R. The functional role of the muscle spindles–facts and hypotheses. Brain. 1975;98:531.
Harris S. Congenital hypotonia. Dev Med Child Neurol. 2008;50:889.
Hayakawa H., Katoh T. Severe cerebellar atrophy following acute cerebellitis. Pediatr Neurol. 1995;12:159.
Hensinger R.N. Limp. Pediatr Clin North Am. 1977;24:723.
Holmes L.B., Driscoll S.G., Bradley W.G. Contractures in a newborn infant of a mother with myasthenia gravis. J Pediatr. 1980;6:1067.
Hong Y., Cheung C.K. Gait and posture responses to backpack load during level walking in children. Gait Posture. 2003;17:28.
Houck J.C., Crago P.E., Rymer W.Z.. Functional properties of the Golgi tendon organs. Desmedt J.E., editor. Spinal and supraspinal mechanisms of voluntary motor control and locomotion. Progress in clinical neurophysiology, vol 8. Karger: Basel, 1980.
Hullinger M. The mammalian muscle spindle and its central control. Rev Physiol Biochem Pharmacol. 1984;101:1.
Infant botulism–New York City, 2001–2002. MMWR Morb Mortal Wkly Rep. 2003;52:21.
Ivanenko Y.P., Dominici N., Cappellini G., et al. Development of pendulum mechanism and kinematic coordination from the first unsupported steps in toddlers. J Exp Biol. 2004;207:3797.
Jozwiak M., Pietrzak S., Tobjasz F. The epidemiology and clinical manifestations of hamstring muscle and plantar foot flexor shortening. Dev Med Child Neurol. 1997;39:481.
Kao I., Drachman D.B., Price D.L. Botulinum toxin. Mechanism presynaptic blockade. Science. 1976;193:1256.
Kelly I.P., Jenkinson A., Stephens M., et al. The kinematic patterns of toe-walkers. J Pediatr Orthop. 1997;17:478.
King G., Schwarz G.A., Slade H.W. Acute cerebellar ataxia of childhood. Pediatrics. 1958;21:731.
Kirschner J., Bonnemann C.G. The congenital and limb-girdle muscular dystrophies: Sharpening the focus, blurring the boundaries. Arch Neurol. 2004;61:189.
Lebenthal E., Shochet S.B., Adam A., et al. Arthrogryposis multiplex congenita: Twenty-three cases in an Arab kindred. Pediatrics. 1970;46:891.
MacWilliams B.A., Cowley M., Nicholson D.E. Foot kinematics and kinetics during adolescent gait. Gait Posture. 2003;17:214.
McComb R.D., Markesbery W.R., O’Connor W.N. Fatal neonatal nemaline myopathy with multiple congenital anomalies. J Pediatr. 1979;94:47.
Mendez-Cashion D., Sanchez-Longo L.P., Valcarcel M., et al. Acute cerebellar ataxia in children associated with infection by polio virus I. Pediatrics. 1962;29:808.
Myers K.A., Wang M., Marks R.M., et al. Validation of a multisegment foot and ankle kinematic model for pediatric gait. IEEE Trans Neural Syst Rehabil Eng. 2004;12:122.
Namba T., Brown S.B., Grob D. Neonatal myasthenia gravis: Report on two cases and review of the literature. Pediatrics. 1970;45:488.
Norlin R., Odenrick P., Sandlund B. Development of gait in normal children. J Pediatr Orthop. 1981;1:261.
Õunpuu S., Gage J.R., Davis R.B. Three-dimensional lower extremity joint kinetics in normal pediatric gait. J Pediatr Orthop. 1991;11:341.
Paine R., Oppe T. Posture and gait. In: Paine R., Oppe T., editors. Neurological examination of children. London: William Heinemann Medical Books, 1966.
Piazza S.J., Delp S.L. The influence of muscles on knee flexion during the swing phase of gait. J Biomech. 1996;29:723.
Pickett J., Berg B., Chaplin E. Syndrome of botulism in infancy: Clinical and electrophysiologic study. N Engl J Med. 1976;295:770.
Ravid S., Maytal J., Eviatar L. Biphasic course of infant botulism. Pediatr Neurol. 2000;23:338.
Riggs J.E., Bodensteiner J.B., Schochet S.S.Jr. Congenital myopathies/dystrophies. Neurol Clin. 2003;21:779.
Romei M., Galli M., Motta F., et al. Use of the normalcy index for the evaluation of gait pathology. Gait Posture. 2004;19:85.
Saunders J.B., Inmann V.T., Eberhart H.D. The major determinants in normal and pathological gait. J Bone Joint Surg Am. 1953;35:543.
Schmidt R.F. Motor systems. In: Schmidt R.F., Thews G., editors. Human physiology. Berlin: Springer; 1983:81. [Biederman-Thorson MA, Trans.]
Schutte L.M., Narayanan U., Stout J.L., et al. An index for quantifying deviations from normal gait. Gait Posture. 2000;11(1):25-31.
Schwartz M.H., Rozumalski A. The Gait Deviation Index: a new comprehensive index of gait pathology. Gait Posture. 2008;28(3):351-357. Epub 2008 Jun 18
Schwartz M.H., Trost J.P., Wervey R.A. Measurement and management of errors in quantitative gait data. Gait Posture. 2004;20:196.
Smith S., Swaiman K. Hypotonic infant. In: Moss A., editor. Pediatrics update. New York: Elsevier Biomedical, 1983.
Song K.M., Halliday S.E., Little D.G. The effect of limb-length discrepancy on gait. J Bone Joint Surg Am. 1997;79:1690.
Sunaga Y., Kikima A., Ostuku T., et al. Acute cerebellar ataxia with abnormal MRI lesions after varicella vaccinations. Pediatr Neurol. 1995;13:340.
Swett J.E., Schoultz T.W. Mechanical transduction in the Golgi tendon organ: A hypothesis. Arch Ital Biol. 1975;113:374.
Teddy P.J., Silver J.R., Baker J.H., et al. Traumatic cerebral flaccid paraplegia. Paraplegia. 1984;22:320.
Thompson J.A., Glasgow L.A., Warpinski J.R., et al. Infant botulism: Clinical spectrum and epidemiology. Pediatrics. 1980;66:936.
van der Linden M.L., Kerr A.M., Hazlewood M.E., et al. Kinematic and kinetic gait characteristics of normal children walking at a range of clinically relevant speeds. J Pediatr Orthop. 2002;22:800.
Vasta I., Kinali M., Messina S., et al. Can clinical signs identify infants with neuromuscular disorders? J Pediatr. 2005;146:73.
Volpe R.G. Evaluation and management of in-toe gait in the neurologically intact child. Clin Pediatr Med Surg. 1997;14:57.
Wassmer E., Wright E., Rideout S., et al. Idiopathic gait disorder among in-patients with acquired gait disorders admitted to a children’s hospital. Pediatr Rehabil. 2002;5:21.
Weiss S., Carter S. Course and prognosis of acute cerebellar ataxia in children. Neurology. 1959;9:711.
Widhe T. Foot deformities at birth: A longitudinal prospective study over a 16-year period. J Pediatr Orthop. 1997;17:20.
Winter D.A. The biomechanics and motor control of human gait, ed 2. Toronto: John Wiley & Sons; 1990.
Yuill G.M., Lynch P.G. Congenital non-progressive peripheral neuropathy with arthrogryposis multiplex. J Neurol Neurosurg Psychiatry. 1974;37:316.
