[level-membership-for-critical-care-medicine-category]64
Muscular Paralysis
Myasthenia Gravis and Guillaine-Barré Syndrome
Clinical Presentation
Myasthenia Gravis
MG is an autoimmune disease of defective neurotransmission leading to fatigable muscle weakness. The incidence is 0.5 to 5 cases per 100,000. Its pathogenesis can be summarized briefly as an antibody reaction at the antigen epitopes of the acetylcholine receptor (AChR), eventually leading to destruction and simplification of the junctional fold and widening synaptic cleft.1 Onset may be at any age but tends to be earlier in women (mean age 28 years) than in men (mean age 42 years). MG results in either transient or persistent focal or generalized fatigability or weakness. In more than half of the cases, the initial symptoms involve the eyes, presenting as ptosis, diplopia, or ophthalmoparesis.
Myasthenic crisis is arbitrarily defined as MG complicated by respiratory failure requiring mechanical ventilation or delayed extubation for more than 24 hours in an already intubated myasthenic patient. It is seen in 10% to 60% of MG patients typically within the first 12 months of disease onset.2,3 The need for ventilatory assistance usually follows onset of weakness of diaphragmatic or accessory respiratory muscles, but mechanical ventilation also may become necessary because of airway collapse from oropharyngeal muscle weakness, stridor from vocal cord weakness, or the inability to clear secretions.4 One third of the patients may have recurrent myasthenic crises.5
Common precipitating causes include viral or bacterial infection in 48% (pneumonia, respiratory infection, or sepsis), aspiration in 10%, reduction of pyridostigmine in 19%, and initiation of steroids in 7% patients.6,7 Changes in medication may relate to the recent addition of corticosteroid, dose reduction, or high-dose acetylcholinesterase inhibitors. Other precipitating factors may be physical trauma, surgical procedures (particularly thyroidectomy), pregnancy, emotional stress, or exposure to drugs with neuromuscular blocking action such as aminoglycosides. No precipitating factor can be identified for myasthenic crisis in 30% patients.6,7
Myasthenic crisis requires urgent evaluation and treatment. The mortality rate for myasthenic crisis has declined from a nearly always fatal outcome in the 1920s to 4.5% in the new millennium with the discovery and application of acetylcholinesterase compounds, mechanical ventilation, immunosuppressive agents, and more recently, plasma exchange and intravenous immunoglobulin (IVIG).8 Timely diagnosis and appropriate treatment with IVIG or plasma exchange for patients with bulbar and respiratory muscle weakness have helped to reduce the mortality rate and total hospital stay from myasthenic crisis, but care of a patient with MG may be prolonged and complicated. Median hospital stay after a myasthenic crisis is 4 to 6 weeks.
Guillain-Barré Syndrome
GBS is an acute, monophasic, inflammatory, demyelinating polyneuropathy, also known as acute inflammatory demyelinating polyneuropathy (AIDP). Its incidence is 1 to 2 cases per 100,000 people and remains fairly constant across the continents. GBS commonly is precipitated by an infection, but the immunopathogenesis has remained elusive since its original description in 1916. Campylobacter jejuni, cytomegalovirus, and Epstein-Barr virus predominate as preceding infection pathogens.9 Symptoms and signs usually progress within 1 to 2 weeks.
The diagnosis of GBS often is straightforward. Severe back pain and limb paresthesias, starting in the ankles and wrists with a “tight band” feeling, are typical presenting signs. The paresthesias gradually scatter over the limbs and move proximally. Although paresthesias are often the presenting symptoms, sensory modalities remain normal to mildly impaired. Weakness begins in the more proximal muscles, causing difficulty with climbing stairs and getting out of a chair, and is notable 1 or 2 days after the onset of paresthesias. Symmetrical weak muscles are accompanied by depressed or absent deep tendon reflexes. Mostly legs are more involved than arms, creating the impression of an ascending paralysis. Facial and oropharyngeal muscles are affected in 50% of the cases, and weakness of these muscle groups may be the initial manifestation. Patients may also have staccato speech (ability to speak only short sentences) and small tidal volume with increased respiratory rate. Respiratory failure, if it occurs, commonly appears within 1 week after the onset of paresthesias.10 Dysautonomia occurs in up to 70% of the cases, manifesting as arrhythmias, tachycardia, diaphoresis, labile blood pressures, urinary retention, or ileus.
Cerebrospinal fluid (CSF) analysis typically shows high protein content with a normal white blood cell count (the classic albuminocytologic dissociation). More than 10 white blood cells may be seen although this number is unusual but is more common in associated disorders such as Lyme disease, sarcoidosis, and AIDS.11 Electrophysiologic studies are more useful for diagnosis and less useful for prognostication. The typical finding involves demyelination seen as conduction block or increased conduction velocities. Specific tests for proximal nerve involvement seen early in the disease course include recording of F waves and nerve signal abnormalities like dispersion or dropout in signal. Prolonged F wave may be the only finding early on. Needle electromyography may be normal in the first 2 weeks. Recovery often starts after the second week. Progression over 8 weeks more likely suggests the diagnosis to chronic inflammatory demyelinating polyneuropathy (CIDP).
Acute Neuromuscular Respiratory Failure
Pathophysiology
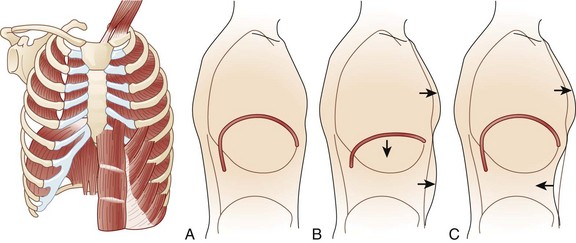
During respiration, lungs can expand and recoil in two ways: by downward and upward movement of the diaphragm to lengthen and shorten the chest cavity, and by elevation and depression of the ribs to increase and decrease the anteroposterior diameter of the chest (Fig. 64.1). Normal quiet breathing is largely accomplished by contraction of the diaphragm.
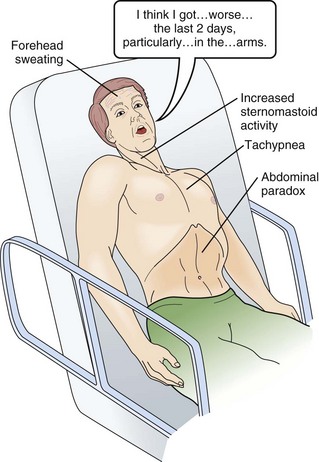
In neuromuscular respiratory failure, ventilatory function is compromised through two mechanisms: (1) respiratory muscle weakness or fatigue (involving the diaphragm and the intercostal muscles), and (2) oropharyngeal weakness, which leads to obstruction of the upper airway and inability to clear secretions. The neuromuscular respiratory failure follows the following pattern: failure of diaphragm and intercostal muscles followed by use of accessory muscles resulting in hypoventilation and atelectasis, further leading to shunting and hypoxia. Respiratory muscle weakness leads to shallow low tidal volume breathing and poor gas exchange leading to tachypnea and later hypercapnia. Oropharyngeal muscle weakness leads to aspiration pneumonitis and could further worsens the hypoxia. Clinically, the patient will pause frequently during speech and breathlessness improves in an upright position (Fig. 64.2).
The first indication of diaphragmatic weakness is alveolar hypoventilation and impaired CO2 exchange. These changes are followed by an increase in respiratory rate as a compensatory mechanism to attempt to maintain minute ventilation. Later, accessory muscles of ventilation are recruited in response to increased ventilatory demand. Paradoxical breathing, also known as thoracoabdominal asynchrony, occurs with severe respiratory weakness. Normally, the abdomen and chest expand and contract in a synchronized fashion. During inspiration, a downward movement of the diaphragm pushes the abdominal contents down and out as the rib margins are lifted and moved out, causing both chest and abdomen to rise. With diaphragmatic weakness or paralysis, the diaphragm moves up rather than down during inspiration, and the abdomen moves in, contracting during chest rise. This is known as “paradoxical breathing” and can be visualized with fluoroscopy (see Fig. 64.1). The ventilatory drive response to an increase in CO2 in patients with GBS and MG has been studied and was found to be unlikely to contribute to hypoventilation during ventilatory failure.12 Ventilatory drive increases during acute hypoventilation, and the ventilatory drive response to CO2 remains intact, even when the minute ventilation response to CO2 is poor.
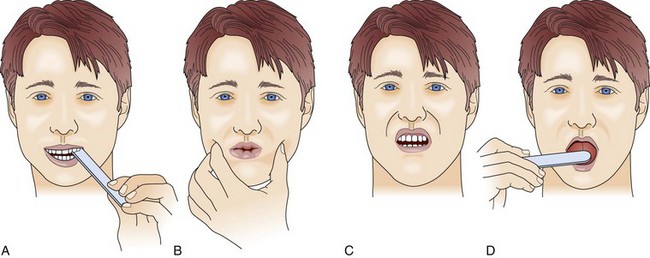
Although the upper airway muscles do not contribute directly to chest expansion or collapse, they are essential for keeping the airways open during respiration. They play an important role in preventing collapse of the pharynx during inspiration and preventing aspiration during swallowing. With the exception of laryngeal muscles, oropharyngeal muscles have a higher proportion of fast fibers. Their weakness can be seen early on in MG and later in GBS (Fig. 64.3). Slow fibers have a higher fatigue resistance than fast fibers due to their highly oxidative metabolism. The diaphragm has an equal proportion of slow and fast muscle fibers, which in association with small fiber size, high aerobic oxidative enzyme activity, and large number of capillaries make it resistant to fatigue.13
Clinical Evaluation
Frequent (at least four times daily) assessment of pulmonary function should be instituted using bedside spirometry. Although bedside spirometry is often done for respiratory evaluation in both MG and GBS, it is less reliable for MG as patients’ results often vary depending on their activity level prior to the testing. Patients should be coached for spirometry as they may have difficulty making an adequate seal around the mouthpiece of the spirometer. Poor effort and leak around the mouthpiece may lead to lower vital capacity (VC), maximal inspiratory pressure (PImax), and maximal expiratory pressure (PEmax) than expected based on arterial blood gas (ABG) and chest x-ray studies. VC, the volume of exhaled air after maximal inspiration, is 60 to 70 mL/kg in normal persons, and is determined primarily by the size of the thorax and lungs. In neuromuscular respiratory failure, reduction of VC to 30 mL/kg is associated with weak cough, accumulation of oropharyngeal secretions, atelectasis, and hypoxemia. Another measure of respiratory muscle strength is the ability to generate negative pressure with inspiratory effort. In normal persons, respiratory muscles cause pleural and alveolar pressures to change by approximately 3 cm H2O during the breathing cycle. Maximal pressure generation can be determined by blocking the upper airway and recording mouth pressure changes during inspiratory effort. PImax measures the strength of the diaphragm and other muscles of inspiration and reflects the ability to maintain normal lung expansion and avoid atelectasis. A maximal PImax is −114 cm H2O in young men and −67 cm H2O in young women (normal, exceeding −70 cm H2O). PEmax measures strength of the muscles of expiration and correlates with strength of cough and ability to clear secretions from the airway12 (Box 64.1). PEmax averages 160 cm H2O in young men and 95 cm H2O in young women (normal, greater than 100 cm H2O). This means that the respiratory muscles are capable of generating more than 30 times the amount of force necessary for tidal breathing.14 Worsening bedside spirometry can identify MG and GBS patients requiring invasive or noninvasive mechanical ventilation (NMV). An easy way to remember bedside pulmonary indicators of respiratory failure in GBS is the so-called 20/30/40 rule, with VC less than 20 mL/kg, a PImax less negative than −30 cm H2O, and a PEmax less than 40 cm H2O.
Noninvasive Ventilation: Bilevel Positive Airway Pressure
Traditionally, patients with acute neuromuscular respiratory failure are ventilated by invasive mechanical ventilation. Prolonged endotracheal intubation, however, is associated with discomfort and ventilator-associated complications.7 In both MG and GBS, mortality and morbidity rates are strongly tied to the duration of invasive mechanical ventilation. NMV is being used increasingly to manage acute deterioration in patients with neuromuscular failure.15 Downsides of NMV are, in patients with associated bulbar weakness, upper airway collapse with increased airway resistance, and lack of airway protection from secretions.
Noninvasive mechanical ventilation using BiPAP ventilation (inspiration and expiration pressure application) in acute respiratory failure caused by MG was shown to be effective in a group of patients with MG from the Mayo Clinic.15 The mean duration of BiPAP was 4.3 days. NMV averted endotracheal intubation in 14 cases of severe MG exacerbation, even in the presence of bulbar weakness. In this study, the presence of hypercapnia (PaCO2 greater than 45 mm Hg) at onset predicted NMV failure and subsequent intubation. Lower PEmax on arrival was associated with longer ventilator duration.15 In contrast with the findings with MG, data on use of NMV in GBS is scarce. One study warned against its use because improvement following initiation of NMV did not prevent emergency intubation in patients with GBS.16
Mechanical Ventilation: Endotracheal Intubation
Myasthenia Gravis
Although several parameters traditionally have been suggested for intubation in MG patients (VC less than 15 mL/kg, PImax less than −30), the fluctuating nature of the disease and frequently associated facial muscle weakness give these parameters limited positive predictive power.17,18 PImax worsening of 30% or more may predict patients at higher risk of requiring mechanical or noninvasive ventilation.18 Ventilatory support is unlikely to be needed when VC is more than 20 mL/kg.
Assessment of respiratory function in MG patients should use a range of criteria including symptoms of breathlessness, paradoxical abdominal wall motion, bulbar nerve palsy, and ABG analysis to look for the presence of hypercapnia or hypoxia. As discussed earlier, patients with PaCO2 greater than 45 mm Hg usually are unresponsive to noninvasive ventilation and should be managed with endotracheal intubation. However, hypercapnia alone is not always a mandatory indication for mechanical ventilation, and this decision should take into consideration the patient’s ease of respiration, level of consciousness, and stability of PaCO2.18 MG patients who are positive for anti-MuSK (muscle-specific kinase) antibodies are at a greater risk for the development of respiratory compromise and bulbar symptoms and should be watched more closely.19–21
Invasive mechanical ventilation in patients with MG can be avoided in a majority of cases with early implementation of rapid immunomodulatory treatments and noninvasive ventilation.15 Mechanical ventilation has been associated with atelectasis and cardiac, infectious, and veno-occlusive complications.8 MG patients requiring mechanical ventilation have high mortality rates of 4% to 17%, with common causes of death identified as acute respiratory distress syndrome, disseminated intravascular coagulation, cardiac failure, and multiorgan failure.6 Duration of mechanical ventilation usually ranges from 1 to 4 weeks. Age more than 50 years, preintubation serum bicarbonate level of 30 mmol/L or more, and peak VC less than 25 mL/kg in the first week after intubation were noted to be the three independent predictors of prolonged intubation.7 Major medical complications secondary to myasthenic crisis are related to days on mechanical ventilation. Fever is the most common complication (occurring in 70% of patients), followed by pneumonia (in 50%) and atelectasis (in 40%). Half of those who survive an MG crisis are functionally dependent at discharge, more commonly with intubation period more than 2 weeks.7
Guillain-Barré Syndrome
Mechanical ventilation is required in 20% to 30% of patients with GBS.22,23 A majority of complications in GBS occur during this stage. The decision of when to intubate GBS patients with respiratory failure has been discretionary. It requires a clinical choice between premature intubation with possible secondary risk of tracheal and pulmonary injury or watchful observation, which could lead to the need for emergency intubation. GBS patients requiring emergency intubation had prolonged mechanical ventilation and anoxic brain injury if associated with respiratory arrest.24
Several clinical factors have been proposed to predict when to intervene. Predictive factors for intubation include rapid progression, dysautonomia, bilateral facial palsy, and oropharyngeal weakness.25 Serial measurements on pulmonary function testing are essential in anticipating the need for mechanical ventilation. As previously mentioned, the 20/30/40 rule may help to predict the need for mechanical ventilation.5 Profound abnormalities of phrenic nerve conduction time and findings on diaphragmatic electromyography may predict the need for mechanical ventilation.26
Duration of mechanical ventilation in patients with GBS ranges from 18 to 49 days.27–29 Inspiratory pressures and ABG tests immediately preceding intubation do not predict the need for prolonged ventilation.23,29 IVIG and plasma exchange have been shown to reduce the duration of ventilation in randomized trials.30 Prolonged intubation (14 days) was found to be associated with increased morbidity and mortality rates, mainly due to increased risk of ventilator-associated pneumonia and longer duration of mechanical ventilation.31 Mechanical ventilation–related respiratory complications such as pneumonia and tracheobronchitis occur in more than half of GBS patients. An increase in ICU stay also increased the number of systemic infections.10 Complications are less common if the ICU stay is less than 3 weeks. Independent ambulation can be seen in up to 80% of patients who survive from the acute phase of GBS.10 Patients may take 2 months to 10 years for independent ambulation.32 ICU complications, prolonged mechanical ventilation, and early axonal abnormalities are associated with slower recovery.7 Advanced age and delay in transfer to ICU are independent factors predictive of poor outcome.33
Aggressive respiratory therapy, including frequent suctioning and use of ventilatory strategies aiming at minimizing atelectasis, should be emphasized. If no improvement is noted, aggressive respiratory management should include tracheostomy to allow more efficient bronchial clearing and reduction in the work of breathing imposed by the endotracheal tube. Tracheostomy and its timing are debated topics. The need for tracheostomy is more likely in the elderly and in the presence of preexisting pulmonary disease.23 A majority of patients with GBS-associated neuromuscular failure and mechanical ventilation undergo tracheostomy (up to 89%).31 Tracheostomy should be postponed until the third week in patients who show some clinical improvement. This provides a period of assessment during which neuromuscular respiratory function may improve in response to treatment, and up to 50% of these patients (those showing improvement) could be spared from tracheostomy by waiting until the third week.
Percutaneous tracheostomy may allow transfer of unstable patients to the operating room, thereby decreasing the incidence of wound infection, reducing cost, and providing better cosmetic results. Tracheostomy-related complications include short-term complications such as bleeding from the tracheostomy site, early cuff leak, wound infection, wound breakdown, and self-decannulation; long-term complications include minor voice change.34 Percutaneous tracheostomy decreases the incidence of complications such as tracheal obstruction and pneumothorax, which are seen more often in open tracheostomy.35 With the current neurocritical care availability for GBS patients, the mortality rate has decreased to less than 5%.32,33
Extubation Trials
The weaning process in patients with MG often is challenging because of the fluctuating nature of the disease. Reintubation is not uncommon. In a review of 26 episodes of myasthenic crisis, the reintubation rate was 27%. In selected patients, noninvasive ventilation can be used for bridging during the weaning process to prevent reintubation.36 Older age, pneumonia, and atelectasis are major risk factors for poor outcome.37 Weaning trials may begin when VC exceeds 15 mL/kg, PImax exceeds −30 cm H2O, and oxygenation is adequate on inspired oxygen concentrations (FIO2) of 40% or less. It is important to reintroduce cholinesterase inhibitors before extubation trials are initiated. Weaning methods may vary. Patients can be switched to continuous positive airway pressure (CPAP) with pressure support ventilation (PSV) and the level decreased 1 to 3 cm H2O each day. A decrease in tidal volume and an increase in respiratory and heart rates are indicators of fatigue. Once the patient demonstrates good endurance at low pressure support (5 cm H2O), usually for more than 2 hours, extubation can be accomplished. After extubation, incentive spirometry is helpful to reduce the risk of atelectasis and reintubation.38
In GBS, weaning from mechanical ventilation should be undertaken as early as possible because of the number of significant complications related to prolonged intubation.39 After intubation, however, respiratory function parameters often continue to fall. Reducing intermittent mandatory ventilation rate or reducing pressure support level can be used as weaning approaches, at the discretion of the treating physician. However, one should anticipate weeks on the ventilator. The weaning process can be initiated once VC reaches 25 mL/kg and spontaneous tidal volumes of 10 to 12 mL/kg are attained. PImax exceeding −50 cm H2O and VC improvement by 4 mL/kg from preintubation to pre-extubation are associated with successful extubation.40 Extubation often is delayed if dysautonomia is still present. Electrophysiologic testing can be helpful while deciding for extubation. Established risk factors for poor outcome in GBS are electrophysiologic evidence of axonal degeneration, preceding diarrheal illness, and rapid disease progression.29
Management
Myasthenia Gravis
Treatment of MG is geared toward identifying the typical causes of crisis (Fig. 64.4). Common causes of myasthenic crisis as mentioned earlier include infection, aspiration, surgery (especially thymectomy), and changes in medication. A careful review of medications, including over-the-counter preparations, is needed. Postoperative complications may be reduced in MG patients with baseline minimal bulbar or respiratory muscle weakness by preoperative plasma exchange or IVIG.2
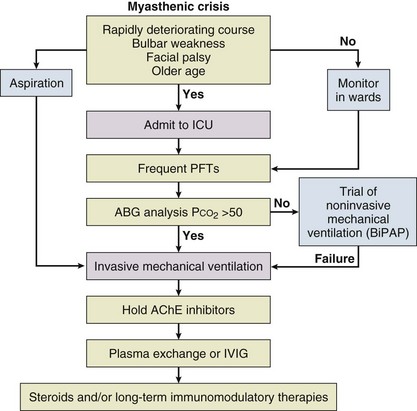
The issue of cholinergic crisis is controversial. Cholinergic crisis is believed to occur as a result of overtreatment with acetylcholinesterase inhibitors. It may manifest with copious respiratory secretions and fasciculations from nicotinic toxicity. Historically, when the treatment of MG was limited to use of acetylcholinesterase inhibitors, overdose could lead to MG crisis, but this entity is now infrequently encountered, and its importance may have been overstated.41 In a retrospective review of 73 episodes of myasthenic crisis, no instances of cholinergic crisis were identified.7 In another review of 27 patients, only one patient had cholinergic crisis.6 To avoid any confusion during the acute management of myasthenic crisis, it is recommended to discontinue acetylcholinesterase inhibitors during invasive mechanical ventilation. During NMV, this decision should be made on a case-by-case basis, depending on the patient’s response to acetylcholinesterase inhibitors.
The mainstay of immunotherapy for the short-term treatment of myasthenic crisis is plasma exchange. Plasma exchange directly removes AChR antibodies from the circulation, and clinical improvement roughly correlates with the degree of elimination as reflected in AChR antibody levels. Although widely used and considered effective, plasmapheresis for MG crisis has not been compared with sham exchange in a controlled trial. A total of 5 to 7 exchanges typically are done over 2 weeks, and 2 to 4 L of plasma is removed with each exchange. One randomized controlled trial did not show any difference in efficacy when plasma exchange was done daily versus every other day.42 Improvement usually is noted within days and can be quite dramatic, although some patients may not show evidence of response until weeks after therapy. The effects of plasma exchange may last up to 3 months. Major complications of plasma exchange are problems with vascular access (including infection, local thrombosis, and vascular perforation), hypotension, transient electrolyte disturbances, and heparin-induced thrombocytopenia.
IVIG is also used in the management of MG but much less in myasthenic crisis. Its mechanism of action in MG is less certain, but it is believed to work by introduction of anti-idiotype antibodies and reduced AChR antibody production achieved through negative feedback. IVIG when compared to placebo clearly showed benefit for MG patients in a randomized controlled trial.43 It often is employed in a dose of 0.4 g/kg of body weight given over 5 days (total dose of 2 g/kg), although a recent randomized clinical trial found no significant superiority of 2 g/kg over 1 g/kg of IVIG in MG exacerbation.44 IVIG is better tolerated as compared to a plasma exchange. Side effects related to IVIG treatment are often mild and include headache, chills, fever, and nausea. These manifestations are related to the rate of infusion. Major side effects, independent of infusion rate, are acute renal failure, thrombotic events, and anaphylaxis.
A recent randomized controlled trial comparing IVIG and plasma exchange found both therapies to be equally efficacious in MG patients, but it did not include severely affected MG patients or patients in myasthenic crisis.45 Trials comparing plasma exchange and IVIG found more rapid onset of improvement in clinical measures of MG, as well as earlier time to extubation, with early (in the first 2 weeks) institution of plasmapheresis, but did not find any significant difference on long-term follow-up evaluation.46,47 One study found plasma exchange also to be effective in patients in whom IVIG has failed.48 Plasma exchange is generally preferred as first-line therapy in MG crisis. High-dose corticosteroids can transiently worsen the neuromuscular weakness associated with MG during early stages of treatment in up to 50% of patients. For this reason, during myasthenic crisis, it is recommended to start steroids only after initiation of plasma exchange or IVIG treatments and then to continue until a remission occurs.17
Complete thymectomy is indicated in cases of thymoma-associated MG. Thymectomy is controversial in patients without thymoma. In a 17-year follow-up study of 110 MG patients, post-thymectomy myasthenic crisis episodes were fewer, were less severe, required less ventilatory support, and had reduced ICU stay when compared to a nonsurgical group.49 Desflurane plus remifentanil was found to be a better anesthetic combination than propofol plus remifentanil in MG patients undergoing video-assisted thoracoscopic-extended thymectomy due to its reversible muscle relaxation effect and faster recovery with no increase in side effects.50 Empiric thymectomy in the elderly is likely to be less effective because of atrophy of the thymus.
Post-thymectomy myasthenic crisis is not uncommon and can be seen in up to 30% of patients. Most of these patients (65%) present within the first 6 months.51 Alternate day oral corticosteroids reduce immediate postoperative myasthenic crisis to 5% based on a retrospective series.52 Plasma exchange and IVIG can also be useful to prevent deterioration preoperatively in patients undergoing thymectomy or other surgery. Preoperative plasma exchange was found to reduce the incidence of postoperative myasthenic crisis and improve long-term outcome.53 In a retrospective study, predictors of postoperative myasthenic crisis were preoperative oropharyngeal weakness, preoperative serum anti-AChR antibody level greater than 100 nmol/L, and intraoperative blood loss greater than 1 L.54 Previous history of myasthenic crisis and presence of thymoma are other significant predictors for post-thymectomy myasthenic crisis.55 Delaying thymectomy until immunosuppressive treatment has been initiated and bulbar symptoms are resolved may be beneficial in avoiding postoperative myasthenic crisis. Postoperative myasthenic crisis leads to prolonged mechanical ventilation and need for tracheostomy. Improvement after thymectomy may not be seen for months or even years.
Guillain-Barré Syndrome
The management of patients with GBS can be challenging because of its unpredictable course, potential for rapid deterioration, and high chances for respiratory failure (Fig. 64.5). Any patient with worsening weakness on initial evaluation or presentation will need admission and observation in an ICU. However, only 1 in 3 patients will deteriorate significantly enough to mandate further or prolonged ICU monitoring and possibly intubation.
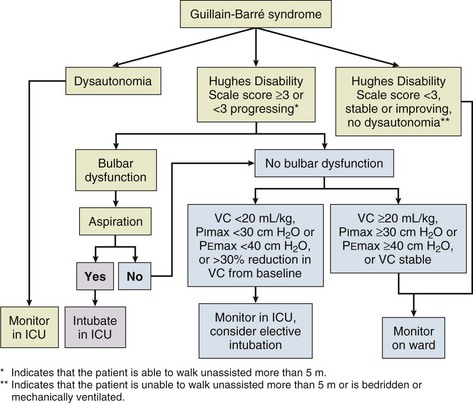
Main modalities of acute treatment are plasma exchange or IVIG, both of which are recommended for nonambulatory patients with GBS who present within 4 weeks of disease onset. Large multicenter trials have established the effectiveness of plasma exchange in GBS.56,57 Earlier clinical improvement, reduced need for mechanical ventilation, and faster recovery have been shown with plasma exchange. A systematic review of six randomized trials in 2002 found that plasma exchange was superior to supportive care.58 Plasma exchange was most effective when started within 7 days of symptom onset; however, an improvement in outcome was still observed if treatment was instituted up to 30 days after onset of symptoms in the North American study. Two plasma exchanges are superior to none in mild GBS, and four exchanges are superior to two in moderately severe GBS. However, six exchanges were not superior to four in severe GBS requiring mechanical ventilation.59
No randomized trials have been conducted to compare IVIG with placebo for the treatment of GBS, but IVIG was shown to be as effective as plasma exchange for the treatment of GBS by a Cochrane systematic review of five trials.60 Analysis found no significant difference between plasma exchange and IVIG in disability scores at 4 weeks; however, treatment effect was faster with plasma exchange as compared to IVIG.46 Combining IVIG with plasma exchange also has been tried and was found not to have additional benefit.61 Corticosteroids alone have no significant benefit in GBS. Intravenous methylprednisolone in combination with IVIG may hasten recovery but does not significantly affect the long-term outcome or neuropathic pain from GBS.62,63 One third of the patients felt completely cured at 12 months, changed their job due to GBS, and did not function at home as well as before in a self-administered questionnaire containing questions on their physical status at homecoming and at 12 months.64
References
1. Engel, AG. Myasthenia gravis and myasthenic syndromes. Ann Neurol. 1984; 16:519–534.
2. Juel, VC. Myasthenia gravis: Management of myasthenic crisis and perioperative care. Semin Neurol. 2004; 24:75–81.
3. Bedlack, RS, Sanders, DB. On the concept of myasthenic crisis. J Clin Neuromusc Dis. 2002; 4:40–42.
4. Keesey, JC. “Crisis” in myasthenia gravis: An historical perspective. Muscle Nerve. 2002; 26:1–3.
5. Lawn, ND, Fletcher, DD, Henderson, RD, et al. Anticipating mechanical ventilation in Guillain-Barré syndrome. Arch Neurol. 2001; 58:893–898.
6. O’Riordan, JI, Miller, DH, Mottershead, JP, et al. The management and outcome of patients with myasthenia gravis treated acutely in a neurological intensive care unit. Eur J Neurol. 1998; 5:137–142.
7. Thomas, CE, Mayer, SA, Gungor, Y, et al. Myasthenic crisis: Clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997; 48:1253–1260.
8. Alshekhlee, A, Miles, JD, Katirji, B, et al. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology. 2009; 72:1548–1554.
9. Hadden, RDM, Karch, H, Hartung, H-P, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001; 56:758–765.
10. Henderson, RD, Lawn, ND, Fletcher, DD, et al. The morbidity of Guillain-Barré syndrome admitted to the intensive care unit. Neurology. 2003; 60:17–21.
11. Fulgham, JR, Wijdicks, EF. Guillain-Barré syndrome. Crit Care Clin. 1997; 13:1–15.
12. Borel, CO, Teitelbaum, JS, Hanley, DF. Ventilatory drive and carbon dioxide response in ventilatory failure due to myasthenia gravis and Guillain-Barré syndrome. Crit Care Med. 1993; 21:1717–1726.
13. Polla, B, D’Antona, G, Bottinelli, R, Reggiani, C. Respiratory muscle fibres: Specialisation and plasticity. Thorax. 2004; 59:808–817.
14. Hicks, GH. Cardiopulmonary Anatomy and Physiology. Philadelphia: WB Saunders; 2000.
15. Seneviratne, J, Mandrekar, J, Wijdicks, EFM, Rabinstein, AA. Noninvasive ventilation in myasthenic crisis. Arch Neurol. 2008; 65:54–58.
16. Wijdicks, EF, Roy, TK. BiPAP in early Guillain-Barré syndrome may fail. Can J Neurol Sci. 2006; 33:105–106.
17. Rieder, P, Louis, M, Jolliet, P, Chevrolet, JC. The repeated measurement of vital capacity is a poor predictor of the need for mechanical ventilation in myasthenia gravis. Intensive Care Med. 1995; 21:663–668.
18. Thieben, MJ, Blacker, DJ, Liu, PY, et al. Pulmonary function tests and blood gases in worsening myasthenia gravis. Muscle Nerve. 2005; 32:664–667.
19. Deymeer, F, Gungor-Tuncer, O, Yilmaz, V, et al. Clinical comparison of anti-MuSK vs. anti-AChR-positive and seronegative myasthenia gravis. Neurology. 2007; 68:609–611.
20. Padua, L, Tonali, P, Aprile, I, et al. Seronegative myasthenia gravis: Comparison of neurophysiological picture in MuSK+ and MuSK− patients. Eur J Neurol. 2006; 13:273–276.
21. Stickler, DE, Massey, JM, Sanders, DB. MuSK-antibody positive myasthenia gravis: Clinical and electrodiagnostic patterns. Clin Neurophysiol. 2005; 116:2065–2068.
22. Rees, JH, Thompson, RD, Smeeton, NC, Hughes, RA. Epidemiological study of Guillain-Barré syndrome in southeast England. J Neurol Neurosurg Psychiatry. 1998; 64:74–77.
23. Lawn, ND, Wijdicks, EFM. Tracheostomy in Guillain-Barré syndrome. Muscle Nerve. 1999; 22:1058–1062.
24. Wijdicks, EFM, Henderson, RD, McClelland, RL. Emergency intubation for respiratory failure in Guillain-Barré syndrome. Arch Neurol. 2003; 60:947–948.
25. Lawn, ND, Fletcher, DD, Henderson, RD, et al. Anticipating mechanical ventilation in Guillain-Barré syndrome. Arch Neurol. 2001; 58:893–898.
26. Zifko, U, Chen, R, Remtulla, H, et al. Respiratory electrophysiological studies in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1996; 60:191–194.
27. Ropper, AH, Kehne, SM. Guillain-Barré syndrome: Management of respiratory failure. Neurology. 1985; 35:1662–1665.
28. Gracey, DR, McMichan, JC, Divertie, MB, Howard, FM, Jr. Respiratory failure in Guillain-Barré syndrome: A 6-year experience. Mayo Clin Proc. 1982; 57:742–746.
29. French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Efficiency of plasma exchange in Guillain-Barré syndrome: Role of replacement fluids. Ann Neurol. 1987; 22:753–761.
30. Lacomis, D, Petrella, JT, Giuliani, MJ. Causes of neuromuscular weakness in the intensive care unit: A study of ninety-two patients. Muscle Nerve. 1998; 21:610–617.
31. Ali, MI, Fernandez-Perez, ER, Pendem, S, et al. Mechanical ventilation in patients with Guillain-Barré syndrome. Respir Care. 2006; 51:1403–1407.
32. Dhar, R, Stitt, L, Hahn, AF. The morbidity and outcome of patients with Guillain-Barré syndrome admitted to the intensive care unit. J Neurol Sci. 2008; 264:121–128.
33. Fletcher, DD, Lawn, ND, Wolter, TD, Wijdicks, EF. Long-term outcome in patients with Guillain-Barré syndrome requiring mechanical ventilation. Neurology. 2000; 54:2311–2315.
34. Mittendorf, EA, McHenry, CR, Smith, CM, et al. Early and late outcome of bedside percutaneous tracheostomy in the intensive care unit. Am Surg. 2002; 68:342–346.
35. Griggs, WM, Myburgh, JA, Worthley, LI. A prospective comparison of a percutaneous tracheostomy technique with standard surgical tracheostomy. Intensive Care Med. 1991; 17:261–263.
36. Rabinstein, A, Wijdicks, EF. BiPAP in acute respiratory failure due to myasthenic crisis may prevent intubation. Neurology. 2002; 59:1647–1649.
37. Rabinstein, A, Mueller-Kronast, N. Risk of extubation failure in patients with myasthenic crisis. Neurocrit Care. 2005; 3:213–215.
38. Varelas, PN, Chua, HC, Natterman, J, et al. Ventilatory care in myasthenia gravis crisis: Assessing the baseline adverse event rate. Crit Care Med. 2002; 30:2663–2668.
39. Wijdicks, EF, Borel, CO. Respiratory management in acute neurologic illness. Neurology. 1998; 50:11–20.
40. Borel, CO, Guy, J. Ventilatory management in critical neurologic illness. Neurol Clin. 1995; 13:627–644.
41. Engel AG, ed. Myasthenia Gravis and Myasthenic Disorders. Oxford: Oxford University Press, 1999.
42. Trikha, I, Singh, S, Goyal, V, et al. Comparative efficacy of low dose, daily versus alternate day plasma exchange in severe myasthenia gravis. J Neurol. 2007; 254:989–995.
43. Zinman, L, Ng, E, Bril, V. IV immunoglobulin in patients with myasthenia gravis: A randomized controlled trial. Neurology. 2007; 68:837–841.
44. Gajdos, P, Tranchant, C, Clair, B, et al. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: A randomized double-blind clinical trial. Arch Neurol. 2005; 62:1689–1693.
45. Barth, D, Ng, E, Nwe, P, et al. Comparison of IVIG and PLEX in patients with myasthenia gravis. Neurology. 2011; 76:2017–2023.
46. Rønager, J, Ravnborg, M, Hermansen, I, Vorstrup, S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs. 2001; 25:967–973.
47. Qureshi, AI, Choudhry, MA, Akbar, MS, et al. Plasma exchange versus intravenous immunoglobulin treatment in myasthenic crisis. Neurology. 1999; 52:629.
48. Stricker, RB, Kwiatkowska, BJ, Habis, JA, Kiprov, DD. Myasthenic crisis: Response to plasmapheresis following failure of intravenous γ-globulin. Arch Neurol. 1993; 50:837–840.
49. Soleimani, A, Moayyeri, A, Akhondzadeh, S, et al. Frequency of myasthenic crisis in relation to thymectomy in generalized myasthenia gravis: A 17-year experience. BMC Neurol. 2004; 4:12.
50. Gritti, P, Carrara, B, Khotcholava, M, et al. The use of desflurane or propofol in combination with remifentanil in myasthenic patients undergoing a video-assisted thoracoscopic-extended thymectomy. Acta Anaesthesiol Scand. 2009; 53:380–389.
51. Nam T-S, Lee, S-H, Kim, B-C, et al. Clinical characteristics and predictive factors of myasthenic crisis after thymectomy. J Clin Neurosci. 2011; 18:1185–1188.
52. Sekine, Y, Kawaguchi, N, Hamada, C, et al. Does perioperative high-dose prednisolone have clinical benefits for generalized myasthenia gravis? Eur J Cardiothorac Surg. 2006; 29:908–913.
53. Nagayasu, T, Yamayoshi, T, Matsumoto, K, et al. Beneficial effects of plasmapheresis before thymectomy on the outcome in myasthenia gravis. Jpn J Thorac Cardiovasc Surg. 2005; 53:2–7.
54. Watanabe, A, Watanabe, T, Obama, T, et al. Prognostic factors for myasthenic crisis after transsternal thymectomy in patients with myasthenia gravis. J Thorac Cardiovasc Surg. 2004; 127:868–876.
55. Yu, L, Zhang, X-J, Ma, S, et al. Thoracoscopic thymectomy for myasthenia gravis with and without thymoma: A single-center experience. Ann Thorac Surg. 2012; 93:240–244.
56. Osterman, PO, Lundemo, G, Pirskanen, R, et al. Beneficial effects of plasma exchange in acute inflammatory polyradiculopathy. Lancet. 1984; 324:1296–1299.
57. The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985; 35:1096–1104.
58. Raphael, JC, Chevret, S, Hughes, RA, Annane, D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002.
59. Esperou, H, Jars-Guincestre, MC, Bolgert, F, et al. Cost analysis of plasma-exchange therapy for the treatment of Guillain-Barré syndrome. French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Intensive Care Med. 2000; 26:1094–1100.
60. Hughes, RA, Swan, AV, van Doorn, PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010.
61. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997; 349:225–230.
62. van Koningsveld, R, Schmitz, PIM, van der Meché, FGA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: Randomised trial. Lancet. 2004; 363:192–196.
63. Ruts, L, van Koningsveld, R, Jacobs, B, van Doorn, P. Determination of pain and response to methylprednisolone in Guillain-Barré syndrome. J Neurol. 2007; 254:1318–1322.
64. Bernsen, RA, de Jager, AE, van der Meché, FGA, Suurmeijer, TP. How Guillain-Barré patients experience their functioning after 1 year. Acta Neurol Scand. 2005; 112:51–56.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]64
Muscular Paralysis
Myasthenia Gravis and Guillaine-Barré Syndrome
Clinical Presentation
Myasthenia Gravis
MG is an autoimmune disease of defective neurotransmission leading to fatigable muscle weakness. The incidence is 0.5 to 5 cases per 100,000. Its pathogenesis can be summarized briefly as an antibody reaction at the antigen epitopes of the acetylcholine receptor (AChR), eventually leading to destruction and simplification of the junctional fold and widening synaptic cleft.1 Onset may be at any age but tends to be earlier in women (mean age 28 years) than in men (mean age 42 years). MG results in either transient or persistent focal or generalized fatigability or weakness. In more than half of the cases, the initial symptoms involve the eyes, presenting as ptosis, diplopia, or ophthalmoparesis.
Myasthenic crisis is arbitrarily defined as MG complicated by respiratory failure requiring mechanical ventilation or delayed extubation for more than 24 hours in an already intubated myasthenic patient. It is seen in 10% to 60% of MG patients typically within the first 12 months of disease onset.2,3 The need for ventilatory assistance usually follows onset of weakness of diaphragmatic or accessory respiratory muscles, but mechanical ventilation also may become necessary because of airway collapse from oropharyngeal muscle weakness, stridor from vocal cord weakness, or the inability to clear secretions.4 One third of the patients may have recurrent myasthenic crises.5
Common precipitating causes include viral or bacterial infection in 48% (pneumonia, respiratory infection, or sepsis), aspiration in 10%, reduction of pyridostigmine in 19%, and initiation of steroids in 7% patients.6,7 Changes in medication may relate to the recent addition of corticosteroid, dose reduction, or high-dose acetylcholinesterase inhibitors. Other precipitating factors may be physical trauma, surgical procedures (particularly thyroidectomy), pregnancy, emotional stress, or exposure to drugs with neuromuscular blocking action such as aminoglycosides. No precipitating factor can be identified for myasthenic crisis in 30% patients.6,7
Myasthenic crisis requires urgent evaluation and treatment. The mortality rate for myasthenic crisis has declined from a nearly always fatal outcome in the 1920s to 4.5% in the new millennium with the discovery and application of acetylcholinesterase compounds, mechanical ventilation, immunosuppressive agents, and more recently, plasma exchange and intravenous immunoglobulin (IVIG).8 Timely diagnosis and appropriate treatment with IVIG or plasma exchange for patients with bulbar and respiratory muscle weakness have helped to reduce the mortality rate and total hospital stay from myasthenic crisis, but care of a patient with MG may be prolonged and complicated. Median hospital stay after a myasthenic crisis is 4 to 6 weeks.
Guillain-Barré Syndrome
GBS is an acute, monophasic, inflammatory, demyelinating polyneuropathy, also known as acute inflammatory demyelinating polyneuropathy (AIDP). Its incidence is 1 to 2 cases per 100,000 people and remains fairly constant across the continents. GBS commonly is precipitated by an infection, but the immunopathogenesis has remained elusive since its original description in 1916. Campylobacter jejuni, cytomegalovirus, and Epstein-Barr virus predominate as preceding infection pathogens.9 Symptoms and signs usually progress within 1 to 2 weeks.
The diagnosis of GBS often is straightforward. Severe back pain and limb paresthesias, starting in the ankles and wrists with a “tight band” feeling, are typical presenting signs. The paresthesias gradually scatter over the limbs and move proximally. Although paresthesias are often the presenting symptoms, sensory modalities remain normal to mildly impaired. Weakness begins in the more proximal muscles, causing difficulty with climbing stairs and getting out of a chair, and is notable 1 or 2 days after the onset of paresthesias. Symmetrical weak muscles are accompanied by depressed or absent deep tendon reflexes. Mostly legs are more involved than arms, creating the impression of an ascending paralysis. Facial and oropharyngeal muscles are affected in 50% of the cases, and weakness of these muscle groups may be the initial manifestation. Patients may also have staccato speech (ability to speak only short sentences) and small tidal volume with increased respiratory rate. Respiratory failure, if it occurs, commonly appears within 1 week after the onset of paresthesias.10 Dysautonomia occurs in up to 70% of the cases, manifesting as arrhythmias, tachycardia, diaphoresis, labile blood pressures, urinary retention, or ileus.
Cerebrospinal fluid (CSF) analysis typically shows high protein content with a normal white blood cell count (the classic albuminocytologic dissociation). More than 10 white blood cells may be seen although this number is unusual but is more common in associated disorders such as Lyme disease, sarcoidosis, and AIDS.11 Electrophysiologic studies are more useful for diagnosis and less useful for prognostication. The typical finding involves demyelination seen as conduction block or increased conduction velocities. Specific tests for proximal nerve involvement seen early in the disease course include recording of F waves and nerve signal abnormalities like dispersion or dropout in signal. Prolonged F wave may be the only finding early on. Needle electromyography may be normal in the first 2 weeks. Recovery often starts after the second week. Progression over 8 weeks more likely suggests the diagnosis to chronic inflammatory demyelinating polyneuropathy (CIDP).
Acute Neuromuscular Respiratory Failure
Pathophysiology
During respiration, lungs can expand and recoil in two ways: by downward and upward movement of the diaphragm to lengthen and shorten the chest cavity, and by elevation and depression of the ribs to increase and decrease the anteroposterior diameter of the chest (Fig. 64.1). Normal quiet breathing is largely accomplished by contraction of the diaphragm.
In neuromuscular respiratory failure, ventilatory function is compromised through two mechanisms: (1) respiratory muscle weakness or fatigue (involving the diaphragm and the intercostal muscles), and (2) oropharyngeal weakness, which leads to obstruction of the upper airway and inability to clear secretions. The neuromuscular respiratory failure follows the following pattern: failure of diaphragm and intercostal muscles followed by use of accessory muscles resulting in hypoventilation and atelectasis, further leading to shunting and hypoxia. Respiratory muscle weakness leads to shallow low tidal volume breathing and poor gas exchange leading to tachypnea and later hypercapnia. Oropharyngeal muscle weakness leads to aspiration pneumonitis and could further worsens the hypoxia. Clinically, the patient will pause frequently during speech and breathlessness improves in an upright position (Fig. 64.2).
The first indication of diaphragmatic weakness is alveolar hypoventilation and impaired CO2 exchange. These changes are followed by an increase in respiratory rate as a compensatory mechanism to attempt to maintain minute ventilation. Later, accessory muscles of ventilation are recruited in response to increased ventilatory demand. Paradoxical breathing, also known as thoracoabdominal asynchrony, occurs with severe respiratory weakness. Normally, the abdomen and chest expand and contract in a synchronized fashion. During inspiration, a downward movement of the diaphragm pushes the abdominal contents down and out as the rib margins are lifted and moved out, causing both chest and abdomen to rise. With diaphragmatic weakness or paralysis, the diaphragm moves up rather than down during inspiration, and the abdomen moves in, contracting during chest rise. This is known as “paradoxical breathing” and can be visualized with fluoroscopy (see Fig. 64.1). The ventilatory drive response to an increase in CO2 in patients with GBS and MG has been studied and was found to be unlikely to contribute to hypoventilation during ventilatory failure.12 Ventilatory drive increases during acute hypoventilation, and the ventilatory drive response to CO2 remains intact, even when the minute ventilation response to CO2 is poor.
Although the upper airway muscles do not contribute directly to chest expansion or collapse, they are essential for keeping the airways open during respiration. They play an important role in preventing collapse of the pharynx during inspiration and preventing aspiration during swallowing. With the exception of laryngeal muscles, oropharyngeal muscles have a higher proportion of fast fibers. Their weakness can be seen early on in MG and later in GBS (Fig. 64.3). Slow fibers have a higher fatigue resistance than fast fibers due to their highly oxidative metabolism. The diaphragm has an equal proportion of slow and fast muscle fibers, which in association with small fiber size, high aerobic oxidative enzyme activity, and large number of capillaries make it resistant to fatigue.13
Clinical Evaluation
Frequent (at least four times daily) assessment of pulmonary function should be instituted using bedside spirometry. Although bedside spirometry is often done for respiratory evaluation in both MG and GBS, it is less reliable for MG as patients’ results often vary depending on their activity level prior to the testing. Patients should be coached for spirometry as they may have difficulty making an adequate seal around the mouthpiece of the spirometer. Poor effort and leak around the mouthpiece may lead to lower vital capacity (VC), maximal inspiratory pressure (PImax), and maximal expiratory pressure (PEmax) than expected based on arterial blood gas (ABG) and chest x-ray studies. VC, the volume of exhaled air after maximal inspiration, is 60 to 70 mL/kg in normal persons, and is determined primarily by the size of the thorax and lungs. In neuromuscular respiratory failure, reduction of VC to 30 mL/kg is associated with weak cough, accumulation of oropharyngeal secretions, atelectasis, and hypoxemia. Another measure of respiratory muscle strength is the ability to generate negative pressure with inspiratory effort. In normal persons, respiratory muscles cause pleural and alveolar pressures to change by approximately 3 cm H2O during the breathing cycle. Maximal pressure generation can be determined by blocking the upper airway and recording mouth pressure changes during inspiratory effort. PImax measures the strength of the diaphragm and other muscles of inspiration and reflects the ability to maintain normal lung expansion and avoid atelectasis. A maximal PImax is −114 cm H2O in young men and −67 cm H2O in young women (normal, exceeding −70 cm H2O). PEmax measures strength of the muscles of expiration and correlates with strength of cough and ability to clear secretions from the airway12 (Box 64.1). PEmax averages 160 cm H2O in young men and 95 cm H2O in young women (normal, greater than 100 cm H2O). This means that the respiratory muscles are capable of generating more than 30 times the amount of force necessary for tidal breathing.14 Worsening bedside spirometry can identify MG and GBS patients requiring invasive or noninvasive mechanical ventilation (NMV). An easy way to remember bedside pulmonary indicators of respiratory failure in GBS is the so-called 20/30/40 rule, with VC less than 20 mL/kg, a PImax less negative than −30 cm H2O, and a PEmax less than 40 cm H2O.
[/not-level-membership-for-critical-care-medicine-category]
