Chapter 92 Muscular Dystrophies
Introduction
Muscular dystrophies are progressive, inherited skeletal muscle disorders resulting in muscle degeneration and loss of strength. This heterogeneous group of disorders has been further characterized at the clinical and molecular level since the 1980s, giving rise to a complex classification based on phenotype and molecular genetic correlates. The onset of clinical symptoms ranges from the neonatal period to late adulthood. The distribution of predominant muscle weakness helps identify six major phenotypes [Emery, 2002]:






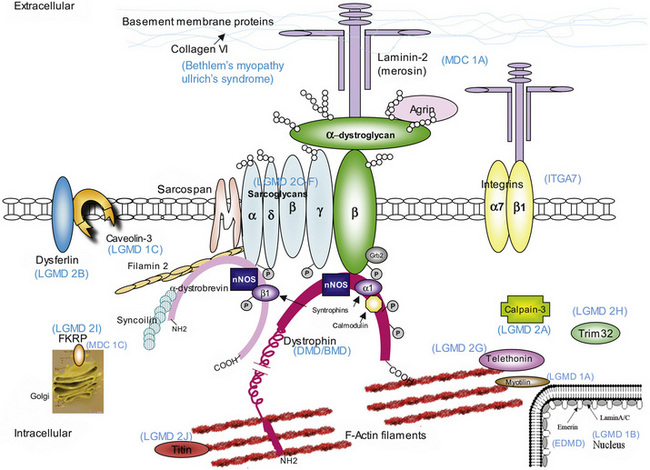
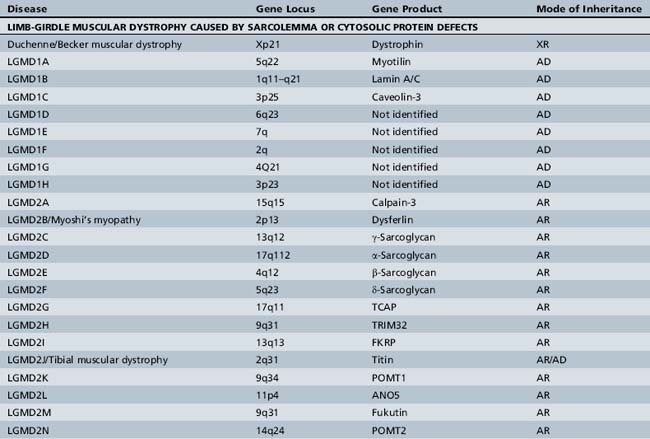
The mode of inheritance in muscular dystrophies can be autosomal-dominant, autosomal-recessive, or X-linked. Spontaneous mutation is responsible for many cases of muscular dystrophy usually transmitted by autosomal-dominant and X-linked inheritance. The underlying molecular defects responsible for these disorders are diverse, including extracellular matrix proteins (laminin-2, collagen VI), transmembrane- and sarcolemma-associated proteins (dystrophin, sarcoglycans, caveolin-3, α7 integrin, dysferlin), cytoplasmatic proteases (calpain-3), cytoplasmatic proteins associated with organelles and sarcomeres (titin, fukutin, telethonin), and nuclear membrane proteins (lamin, emerin). The molecular characterization of the different muscular dystrophies has advanced rapidly since the 1990s and has increased our understanding of sarcolemmal organization and underlying muscle biology responsible for the maintenance of muscle cells in health and disease. Figure 92-1 is a cartoon of the muscle membrane. Binding relationships among proteins are intricate, and the specific relation of these proteins, as well as the presence of phosphorylation and glycosylation sites, provides the basis for the pathophysiology of many of these disorders. Diseases caused by mutations of these proteins are listed in Table 92-1.
In view of the variable manifestation of the muscular dystrophies, the broad spectrum of other organs that might be involved in addition to muscle (i.e., myotonic dystrophies, syndromic congenital muscular dystrophies), and the diverse groups of proteins that cause these disorders, it may seem that the major uniting feature is the characteristic pathologic changes in muscle that allow characterization of these disorders as a “dystrophy.” Neuromuscular specialists have become increasingly dependent on molecular genetic testing to diagnose and counsel patients with muscular dystrophy and their families. DNA testing is expensive and access to this diagnostic tool is uneven. An invaluable resource is the GENETESTS website maintained by the University of Washington. This site offers excellent updated review articles on many genetic disorders (including the muscular dystrophies), as well as current information regarding molecular genetic testing available commercially or through research centers (http://www.genetests.org).
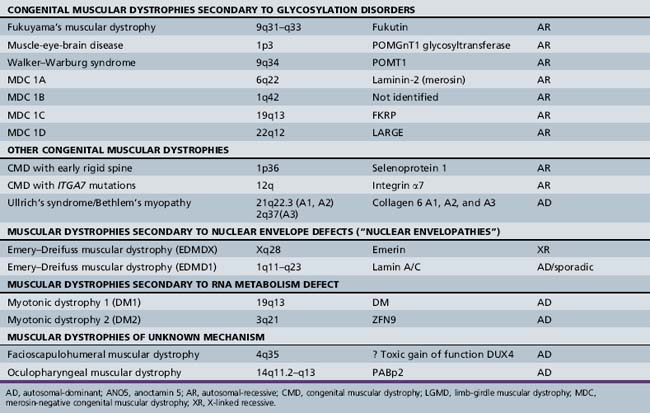
A molecular-based classification of the muscular dystrophies is presented in Table 92-2. Several of these disorders first manifest clinically in adulthood and are not covered in detail in this chapter. The most common muscular dystrophy in children is Duchenne muscular dystrophy, and it is the main focus of this review. Extensive investigations of this disorder have shed light on the pathophysiology of other muscular dystrophies, especially those related to the dystrophin-associated glycoprotein complex (DGC). The principles of clinical delineation of phenotype, categorization of functional deficits, and supportive therapies utilized for the Duchenne patient are applicable for nearly all the disorders discussed in this chapter.
Dystrophinopathies (Duchenne and Becker Muscular Dystrophies and Clinical Variants)
Dystrophinopathies are a group of muscular dystrophies resulting from mutations in the dystrophin gene, located in the Xp21 region [Baumbach et al., 1989]. Of these, Duchenne muscular dystrophy (DMD) is the most common dystrophinopathy, resulting from complete absence of the dystrophin gene product: the subsarcolemmal protein, dystrophin. It affects 1 in 3500 live male births and respects no ethnic or racial boundaries worldwide. Its allelic variant, Becker muscular dystrophy (BMD), is rarer. BMD usually results from in-frame mutations of the dystrophin gene that cause decreased quantity and decreased quality of the dystrophin protein, giving rise to a disease with varied severity and time of onset, ranging from childhood to adulthood. Whereas DMD typically manifests before age 4 years, BMD patients can be asymptomatic for decades.
Duchenne Muscular Dystrophy
History
In a communication to the Royal Medical and Chirurgical Society of London in December 1851, Edward Meryon, an English physician, described in detail a disease affecting eight males in three families. He noted the predilection for male family members, the familial nature of the disease, and the fact that the progressive muscle wasting and weakness resulted from a disease of muscle and not the central nervous system. He appears to have been the first physician to make a detailed clinical, genetic, and pathologic study of the disorder, which he accomplished several years before the brilliant work of Guillaume-Benjamin Duchenne [Emery, 1993]. Duchenne, a French neurologist and pioneer of electrical stimulation of muscle, described a syndrome characterized by muscular paralysis associated with muscle hypertrophy that resulted from accumulation of large amounts of fat and connective tissue in muscle. Duchenne described the first instrument for procuring muscle for biopsy and analysis. The disease he described as pseudohypertrophic, X-linked muscular dystrophy bears his name.
The discovery of the molecular defect in this disorder constituted the first example of reverse cloning. Restriction fragment length polymorphisms were used to track the co-segregation of the disease phenotype with DNA markers within a family. The initial cloning of the gene responsible for DMD occurred in 1985 and 1986 [Kunkel et al., 1985; Monaco et al., 1985; Ray et al., 1985], followed by isolation of the DNA clones complementary to the Duchenne dystrophy gene [Burghes et al., 1987; Cross et al., 1987; Monaco et al., 1986]. These complementary DNA clones, produced from RNA transcripts from the DMD locus, contain the coding exon sequences of the Duchenne gene. Subsequently, the protein product of the human DMD locus was identified through the use of polyclonal antibodies directed against fusion proteins containing two distinct regions of the DMD locus’s complementary DNA. This protein was named dystrophin [Hoffman et al., 1987a].
Molecular Pathogenesis
DMD is a relentlessly progressive skeletal muscle disorder, caused by a mutation in the X-linked dystrophin gene, resulting in absence of a critical protein, dystrophin [Hoffman et al., 1987a, 1988; Koenig et al., 1987]. The dystrophin gene includes 86 exons (including seven promoters linked to unique first exons), which make up only 0.6 percent of the gene; the rest consists of introns. The gene spans a distance of more than 2.5 million basepairs and is the largest human gene isolated to date [Burmeister and Lehrach, 1986]. In more than 90 percent of males with the DMD genotype, there is an absence of dystrophin resulting from an “out-of-frame” mutation that disrupts normal dystrophin transcription [Gillard et al., 1989]. These mutations include deletions, duplications, point mutations and splice site mutations, which cause early termination of messenger RNA transcription. As a result, an unstable RNA is produced, undergoes rapid decay, and then leads to production of absent or very low concentrations of abnormal dystrophin. If the mutation maintains translational reading as seen with an “in-frame” deletion, the BMD phenotype, with variably decreased amounts of abnormal molecular weight dystrophin, results [Hoffman et al., 1988]. The reading frame rule holds true for over 90 percent of afflicted individuals and is used both for diagnostic confirmation of dystrophinopathies and to assist in differentiating DMD from BMD; this distinction is occasionally difficult due to the spectrum of clinical involvement in these allelic disorders.
Exceptions to the reading frame rule occur in approximately 10 percent of patients. Out-of-frame deletions affecting exons 3–7, 5–7, 3–6, or downstream at exons 51, 49–50, 47–52, 44, or 45, can result in a milder BMD phenotype. A likely explanation for the production of at least some dystrophin in these patients is “exon skipping,” which occurs through alternative splicing [Nicholson et al., 1992; Patria et al., 1996]. In this form of BMD, the carboxy-terminus is always preserved [Arahata et al., 1991]. Exon skipping is also the probable underlying mechanism for revertant fibers (muscle fibers exhibiting dystrophin immunostaining in muscle biopsies), evident in about 50 percent of males with DMD [Winnard et al., 1995]. Limited expression of dystrophin may result in a slower progression of muscle weakness than in the usual Duchenne phenotype [Arahata et al., 1989; Baumbach et al., 1989; Koenig et al., 1989; Malhotra et al., 1988]. A new therapeutic strategy in DMD is being developed, utilizing antisense oligonucleotides to induce exon skipping in patients to restore the dystrophin reading frame and allow the production of enough dystrophin to ameliorate the phenotype (see Therapeutic Approaches) [Hoffman, 2007; Kinali et al., 2009; van Deutekom et al., 2007].
Exceptions to the reading frame rule may occur with large in-frame deletions in the 5′ end of the dystrophin gene, extending to the middle of the rod domain (i.e., deletions of exons 3–31, 3–25, 4–41, and 4–18) [Nevo et al., 2003], as well as with small in-frame deletions of exons 3–13 that cause the severe DMD phenotype [Muntoni et al., 1994]. These small and large mutations affect another actin-binding domain in the N-terminus of dystrophin and therefore may have significant functional consequences, resulting in the more severe phenotype. Other mechanisms, such as an unexpected effect of the deletion on splicing behavior, might also be implicated in determining the phenotypic outcome [Muntoni et al., 1994]. After the complete dystrophin complementary DNA (cDNA) was isolated [Koenig et al., 1987], it became evident that about 60 percent of patients with Duchenne and Becker muscular dystrophies manifest structural rearrangements due to deletion mutations [Darras et al., 1988; den Dunnen et al., 1987; Forrest et al., 1987; Koenig et al., 1987; Kunkel, 1986]. Deletions and, more rarely, duplications can arise almost anywhere in the dystrophin gene; however, two deletion hot spots are known. The most commonly mutated region includes deletions in exons 45 to 55 with genomic breakpoints (i.e., the end points of where the deletions actually occur) lying within intron 44. The second hot spot is located toward the 5′ end and includes exons 2 to 19 with genomic breakpoints commonly found in introns 2 and 7 [Beggs et al., 1990; den Dunnen et al., 1989; Nobile et al., 1995; Oudet et al., 1992]. Forty percent of patients have dystrophinopathy that results from small mutations (point mutations resulting in frameshift or nonsense mutations) or duplications.
As noted, the incidence of DMD is approximately 1 per 3500 male births [Brooks and Emery, 1977; Jeppesen et al., 2003]. The most common mode of inheritance is X-linked recessive, with inheritance from a mother who is a known or unidentified carrier. However, DMD is associated with a high spontaneous mutation rate, which accounts for approximately 30 percent of cases [Brooks and Emery, 1977; Moser, 1984; Scheuerbrandt et al., 1986; van Essen et al., 1992]. Therefore, almost one-third of males with DMD have no family history of muscular dystrophy. This mutation rate is estimated to be 10 times higher than for any other genetic disorder [Hoffman et al., 1992], due at least in part to the extremely large size of the dystrophin gene [Hoffman and Kunkel, 1989]. The 2.5 million basepairs constituting the gene (a full 1 percent of the X chromosome) provide a large target for random mutational events. Nonfamilial DMD patients might be the product of germinal mosaicism on the X chromosome (a mutation occurring before the birth of the mother) [Bakker et al., 1989; Bunyan et al., 1994; Lanman et al., 1987; Smith et al., 1999], in which case the mother is a DMD carrier but no other family member is affected with DMD. Another possibility is maternal or paternal gonadal mosaicism: a new mutation in maternal or paternal germ cells in an unaffected father or mother, whose other cell lines (i.e., muscle, lymphocytes) are normal [Darras et al., 1988; Hurko et al., 1989; Lanman et al., 1987]. This mother neither is an obligate carrier, nor does she have the neuromuscular characteristics of a DMD carrier. The remaining new mutations are partial gene duplications [Hu et al., 1989, 1990].
As the genetic defect is an X-linked recessive trait, dystrophinopathies are expressed primarily in boys and young men. However, females may manifest signs and symptoms of DMD if they also exhibit skewed X-inactivation, wherein the abnormal X chromosome is expressed in an excessive proportion of cells [Kinoshita et al., 1990; Lesca et al., 2003; Pena et al., 1987; Yoshioka et al., 1986]. Females with both DMD and Turner’s syndrome have been described. This rare combination arises when the single X chromosome of the Turner patient contains a mutant gene [Bjerglund Nielsen and Nielsen, 1984; Ferrier et al., 1965; Lescaut et al., 2004].
Genotype/phenotype correlation
There is no simple relationship between the size of a dystrophin gene deletion and the resultant clinical phenotype. Large deletions, which may involve nearly 50 percent of the gene, have been described in patients with BMD [England et al., 1990], whereas deletions of small exons, such as exon 44, typically result in a severe DMD phenotype. The central and distal rod domains of the dystrophin protein seem to be nonessential; some deletions in this region are associated with a syndrome characterized only by myalgia and muscle cramps or by an isolated increase in creatine kinase [Beggs et al., 1991; Gospe et al., 1989]. This finding has been reported in patients with in-frame deletions in exons 32–44, 48–51, or 48–53, all of whom had normal or near-normal dystrophin expression [Melis et al., 1998]. The effect of the genetic mutations on the phenotype depends on whether or not it disrupts the reading frame and on specific essential signaling or binding sites in the dystrophin protein that might be affected by the mutation. Good correlation is generally found between the severity of the phenotype and the effect of the deletion on the reading frame.
Deletions that disrupt the reading frame result in a severe phenotype, whereas in-frame deletions are associated with a milder disease course [Gillard et al., 1989]. These assumptions hold true for about 90 percent of the cases in which mutations are in the rod domain of the dystrophin protein. Rare exceptions to this rule exist, mainly resulting from frameshift mutations in the 5′ region of the gene (in particular, deletions involving exons 3–7), which are associated with a milder phenotype than expected [Malhotra et al., 1988]. When mutations arise in the 5′ region of the gene, especially in the first 13 exons of the dystrophin gene, one-third of patients have a phenotype different from that predicted [Muntoni et al., 1994]. Affected patients can have a severe clinical phenotype despite the presence of a small in-frame deletion, or they can have a mild phenotype with an out-of-frame deletion. In the first case, a Duchenne phenotype is associated with in-frame deletions of exon 5, of exon 3, and of exons 3–13. In the second case, an intermediate Duchenne or Becker phenotype is seen, with out-of-frame deletions involving not only the usual exons 3–7, but also 5–7 and 3–6 [Muntoni et al., 1994]. Therefore a proportion of patients with a deletion in the 5′ end of the gene have a phenotype that is not predictable on the basis of the effect of the deletion on the reading frame.
Very large chromosomal deletions, such as the Xp21 chromosomal microdeletion syndrome, cause multisystem disorders along with the DMD phenotype. Such contiguous deletions may result in a boy manifesting congenital adrenal hypoplasia, glycerate kinase deficiency, chronic granulomatous disease and/or McCleod’s syndrome [Francke et al., 1985]. The Leiden database (http://www.dmd.nl/) is a useful resource for phenotype/genotype correlation in situations in which genetic testing and phenotype do not appear to be clearly correlated.
Pathophysiology
Dystrophin protein
The normal dystrophin gene creates a 14-kilobase dystrophin messenger RNA that encodes 3685 amino acids, producing a 427-kD protein called dystrophin. Dystrophin localizes to the subsarcolemmal region in skeletal and cardiac muscle, and constitutes 0.002 percent of total muscle protein [Hoffman et al., 1987a, 1987b; Knudson et al., 1988]. Dystrophin binds to cytoskeletal actin and to the cytoplasmic tail of the transmembrane DGC protein, β-dystroglycan, and thus forms a link from the cytoskeleton to the extracellular matrix (see Figure 92-1).
Dystrophin contains four domains: the N-terminus, the rod domain, a cysteine-rich area, and the C-terminus [Koenig et al., 1987]. The N-terminus consists of the first 240 amino acids and provides an F-actin binding site at three distinct regions with α-actinin homology. The rod region is a succession of 25 triple-helical spectrinlike repeats and contains about 3000 residues, including four proline-rich regions that act as hinges. Another F-actin binding site is located near the middle of the dystrophin rod domain but with significantly lower affinity than the N-terminus site [Rybakova et al., 1996]. A cysteine-rich area with amino acids 3080–3360 has a WW domain, a protein domain with two highly conserved tryptophans that binds proline-rich peptide motifs, which binds to the protein β-dystroglycan, an essential protein through which dystrophin links to other integral membrane components of the DGC [Ozawa, 1995; Ozawa et al., 1995]. The site at which dystrophin binds to β-dystroglycan extends to the first half of the carboxy-terminal domain (C-terminus) [Watkins et al., 2000]. The C-terminus comprises the last 420 amino acids and has homology to utrophin and dystrobrevin. This area contains many potential phosphorylation sites and also binds to syntrophins at exon 74, and possibly to dystrophin-associated glycoproteins [Rybakova et al., 1996].
Besides the full dystrophin protein just described, seven different dystrophin transcripts can be identified. These transcripts have different promoter regions and initial exons. The N-terminus region is excluded in smaller dystrophins, but the C-terminus region is found in all seven dystrophin transcripts. The transcripts are specific to cell types. Three full-length isoforms have the same number of exons but are derived from three independent promoters in brain, muscle, and Purkinje cerebellar neurons. The best characterized of these transcripts is the previously described muscle protein, expressed not only in skeletal muscle but also in cardiac muscle, smooth muscle, and retina. A cortical 427-kDa transcript is found in cortical postsynaptic densities, retina, and skeletal muscle [Boyce et al., 1991]. A Purkinje cell 427-kDa transcript is found in the cerebellum [Gorecki et al., 1992]. There is also a retinal 260-kDa transcript, a brain and kidney 140-kDa transcript, and a Schwann cell 116-kDa protein resulting from transcription beginning on exon 56 [Byers et al., 1993]. A glial 71-kDa transcript resulting from transcription beginning at exon 63 can be found in glia, viscera, and cardiac muscle [Rapaport et al., 1992]. The presence or absence of these seven dystrophin transcripts in DMD depends on the location and size of deletion in the dystrophin gene, which determines phenotypic expression.
Primary and secondary (downstream) events
Muscle cell death in the muscular dystrophies (by apoptosis and necrosis) is conditional and reflects a propensity that varies between muscles and changes with age [Rando, 2001b]. The fact that adjacent muscle groups in DMD can be completely normal and others are undergoing active necrosis not only supports this concept but also argues against the concept of inevitability. If endogenous biochemical mechanisms alter the ability of a muscle cell to live or die while the genetic and biochemical defects remain constant, then pharmacologic modulation of these pathways might result in successful therapies for DMD and other muscular dystrophies [Rando, 2001b]. Although dystrophin deficiency is the primary cause of DMD, multiple secondary pathways are responsible for the progression of muscle necrosis, abnormal fibrosis, and failure of regeneration that results in progressive clinical deterioration [Tidball and Wehling-Henrichs, 2004]. Since the mid-1990s, hypothesis-driven research has dissected several abnormal pathways involved in muscular dystrophy progression. There is ample literature establishing evidence of oxidative radical damage to myofibers [Baker and Austin, 1989; Haycock et al., 1996a, 1996b; Murphy and Kehrer, 1986; Rando et al., 1998], inflammation [Cai et al., 2000; Kissel et al., 1993; Lagrota-Candido et al., 2002; McDouall et al., 1990; Nahirney et al., 1997; Porter et al., 2002; Shaw et al., 1996; Spencer et al., 2000; Spencer and Tidball, 2001], abnormal calcium homeostasis [Baker and Austin, 1989; De Luca et al., 2002; Leijendekker et al., 1996; Murphy and Kehrer, 1986; Pulido et al., 1998; Ruegg et al., 2002; Wrogemann and Pena, 1976], myonuclear apoptosis [Adams et al., 2001; Rando, 2001b; Sandri et al., 1995, 1997, 1998a, 1998b, 2001; Smith et al., 2000; Spencer et al., 1997; Tews, 2002; Tews and Goebel, 1997; Tindall et al., 1987], and abnormal fibrosis and failure of regeneration [Bernasconi et al., 1995, 1999; D’Amore et al., 1994; Iannaccone et al., 1995; Luz et al., 2002; Melone et al., 2000; Morrison et al., 2000; Murakami et al., 1999; Passerini et al., 2002; Rando, 2001b; Yamazaki et al., 1994]. This evidence has been validated by cross-sectional genome-wide approaches that allow an overall analysis of multiple defective mechanisms in DMD [Chen et al., 2000; Porter et al., 2002]. The increasing understanding of these events has led to the discovery of potential pharmacologic targets designed to reverse, stop, or slow down muscle damage and progression of disease, even if the primary genetic defect cannot be repaired at present. Thus, it is important to understand these mechanisms as the basis of present and future therapies for this otherwise fatal disease.
Mechanical Membrane Fragility
Dystrophin is a link between the intracellular cytoskeleton and the extracellular matrix. All evidence supports a structural function for dystrophin and the DGC, establishing the physical link for force transduction between the actin skeleton and the extracellular matrix. The carboxy-terminal of dystrophin is attached to the sarcolemma, the surface membrane of striated muscle cells [Arahata et al., 1988; Bonilla et al., 1988; Watkins et al., 1988; Zubrzycka-Gaarn et al., 1988], binding to β-dystroglycan [Jung et al., 1995] and other dystrophin-associated glycoproteins [Ervasti and Campbell, 1991]. When dystrophin is lost, disconnection of the link between contractile proteins to β-dystroglycan results in loss of β-dystroglycan and DGC from the sarcolemma, expressed as reduced sarcoglycan complex immunoreactivity. The location of dystrophin supports the concept of fragile membranes in DMD and BMD, with membrane gaps that allow extracellular leakage of cytoplasmic components, such as creatine kinase, and the influx of excessive Ca2+. Initially, it was thought that the main function of dystrophin was to provide mechanical reinforcement to the sarcolemma and thereby protect it from the mechanical stress of muscle contraction [Petrof et al., 1993]. Indeed, dystrophin is a load-bearing element, and deficiency in dystrophin leads to muscle membrane fragility and aberrant mechanotransduction [Kumar et al., 2004]. In the absence of dystrophin, there is disruption to normal force transmission and greater stress is placed on myofibrillar and membrane proteins, leading to muscle damage [Lynch, 2004], especially during lengthening (eccentric) contractions, during which a muscle contracts against a mechanical force pulling in the opposite direction [Lynch et al., 2000]. The consequent muscle membrane fragility and abnormal permeability characteristics allow increased intracellular Ca2+ influx [Mallouk and Allard, 2000; Mallouk et al., 2000] and initiate the pathologic cascade of events, resulting in muscle necrosis and fibrosis [Ruegg and Gillis, 1999; Ruegg et al., 2002]. However, it is likely that Ca2+ enters the cell through both “broken” membrane and physiologic Ca2+ channels.
Abnormal Permeability to Calcium and Chronic Increase of Intracellular Calcium
Although the theory of increased membrane microdisruptions with subsequent increased Ca2+ permeability cannot be refuted, there is accumulating evidence that abnormal Ca2+ handling may be related to direct dystrophin regulation of mechanosensitive transient receptor potential channels [Vandebrouck et al., 2002b; Yeung and Allen, 2004], as well as abnormal Ca2+ intracellular cycling [Doran et al., 2004; Dowling et al., 2004; Woods et al., 2004]. Several nifedipine-insensitive, voltage-independent Ca2+ channels might be involved in the initial abnormal Ca2+ entry. This evidence is extremely important because modulation of these channels could be a new therapeutic target. Although there is concordant evidence of increased Ca2+ influx to the cell, there is no consensus on an overall increase in resting intracellular Ca2+ concentration [Gillis, 1996]. One line of evidence involves abnormal function of “stretch-activated” and “stretch-inactivated” Ca2+ channels, a subfamily of the transient receptor potential channels [Clapham, 2003]. These stretch-activated channels are abnormally active under mechanical stimulation in myotubes of the mdx mouse, a murine model of DMD, and result in an increase in intracellular Ca2+ [Mallouk and Allard, 2000; Vandebrouck et al., 2002a, 2002b]. In addition, there is evidence that a large number of these channels, when subjected to mild mechanical stress, irreversibly shift to a stretch-inactivated channel form [Franco and Lansman, 1990] that remains open at rest and allows a chronic increase of calcium currents [Franco-Obregon and Lansman, 2002]. Accumulation of abnormally active Ca2+ leak channels over time results in a gradual loss of Ca2+ homeostasis and eventual cell death [Alderton and Steinhardt, 2000]. This increase of Ca2+ current might also be initiated by other types of Ca2+-permeable, growth factor-activated channels. The latter are normally localized in the cytoplasm of skeletal muscle, and translocate to the plasma membrane only under insulin-like growth factor 1 and possibly other growth factor stimuli [Iwata et al., 2003]. In dystrophic muscle, these channels are abnormally increased in the sarcolemma and seem to be involved directly with the increase in intracellular Ca2+ and abnormal creatine kinase efflux seen during mechanical stress of dystrophic muscle membrane [Iwata et al., 2003].
Of practical importance is the fact that blockade of these stretch-activated channels has been achieved by the nonselective transient receptor potential blockers gadolinium and mycin, as well as by the selective cationic transient receptor potential blocker GsMTx4 (spider venom toxin), and has resulted in normalization of intracellular calcium and muscle force generation ex vivo in mdx mouse muscle [Yeung et al., 2003, 2005]. Furthermore, treatment of mdx mice with oral streptomycin resulted in decreased muscle necrosis [Yeung et al., 2005]. Forced expression of the full-length and mini-dystrophin protein in dystrophin-deficient So18 skeletal myotubes also rectified steady-state levels of subcellular concentrations of Ca2+ and of Ca2+ transients [Marchand et al., 2001, 2004].
The L-type, voltage-gated Ca2+ channels appear to be abnormal in the absence of dystrophin; it was demonstrated that the Ca2+ currents in response to an action potential were much smaller in mdx mice than in normal control subjects. A disrupted direct or indirect linkage of dystrophin with these channels may be crucial for proper excitation–contraction coupling to initiate Ca2+ release from the sarcoplasmic reticulum. This linkage seems to be restored fully in the presence of mini-dystrophin [Friedrich et al., 2004].
Calcium cycling within the dystrophic muscle cell is also altered in mdx mice. Although the subsarcolemmal Ca2+ concentration might be elevated [Mallouk et al., 2000], release of Ca2+ by the sarcoplasmic reticulum in response to an action potential is much decreased in this model [Plant and Lynch, 2003; Woods et al., 2004] and might contribute to abnormal activation–contraction coupling and subsequent muscle weakness. The Ca2+ buffering system in dystrophin myotubes is equally affected. A key luminal Ca2+-binding protein, sarcalumenin, is affected in mdx skeletal muscle, which results in an abnormal shuttling of Ca2+ between the Ca2+-uptake sarco-endoplasmic reticulum Ca2+-adenosine triphosphatase and calsequestrin, and might amplify indirectly the Ca2+ leak channel-induced increase in cytosolic Ca2+ levels [Dowling et al., 2004]. Abnormal intracellular Ca2+ levels result in abnormal activation of Ca2+-activated proteases (e.g., calpain), with subsequent abnormal degradation of intracellular proteins that likely contribute to the abnormal functioning of the leak channels [Turner et al., 1993]. There is evidence that exercise worsens the abnormalities in calcium homeostasis in mdx mice [Fraysse et al., 2004]. This supports the clinical observation that excessive exercise by DMD patients may be deleterious and exacerbate muscle weakness [Allen, 2004; Ansved, 2003].
Abnormal Immunologic Response
In normal skeletal muscle, contraction-induced damage is followed by an inflammatory response involving multiple cell types that subsides over several days. This transient inflammatory response is a normal homeostatic reaction to muscle damage. In contrast, a persistent inflammatory response is observed in dystrophic skeletal muscle, which leads to an altered extracellular environment that includes an increased presence of inflammatory cells (i.e., macrophages) and elevated levels of various inflammatory cytokines (i.e., tumor necrosis factor-α, transforming growth factor-β). Therefore, the signals that lead to successful muscle repair in healthy muscle may promote muscle wasting and fibrosis in dystrophic muscle. Evidence supporting the role of the immune system in promoting muscle pathology in DMD, as well as the active role of cytotoxic T cells and myeloid cells in the pathophysiology of progressive muscle necrosis and fibrosis in DMD and mdx mice, has accumulated since the mid-1990s [Spencer and Tidball, 2001]. More recently, genome-wide approaches investigating the gene expression profile of DMD and mdx mouse muscles have revealed that an abnormal immunologic response is induced very early and is maintained at a low level in DMD patients and mdx mice from early in the neonatal period [Chen et al., 2000; Porter et al., 2003a]. Microarray experiments conducted longitudinally in mdx mice have allowed identification of abnormally increased cytokines and their receptors within the muscle cells, which implies that the dystrophic muscle might contribute to abnormal chemotaxis [Porter et al., 2002, 2003a]. These cytokine pathways could be new therapeutic targets.
Several pathways are involved in abnormal immune response in DMD. First, invasion of antigen-presenting cells activates cytotoxic and helper T cells [Banchereau and Steinman, 1998; Hart, 1997]. These release interleukin-2, which initiates polyclonal expansion of cytotoxic T cells [Gussoni et al., 1994; Spencer et al., 1997, 2001]; this, in turn, mediates muscle necrosis by perforin-mediated [Cai et al., 2000; Spencer et al., 1997, 2001] and cytokine-mediated (tumor necrosis factor-α and transforming growth factor-β) killing mechanisms [Isenberg et al., 1986; Lundberg et al., 1995; Morrison et al., 2000; Spencer et al., 2000; Tews and Goebel, 1996]. Another pathway is through circulating dermal dendritic cells, which are abundant in DMD muscle [Chen et al., 2000], and through CD4+ cells, which in turn activate (through interleukin-2) CD8+ cells. Another immune pathway starts with the invasion of muscle by myeloid cells, including macrophages, neutrophils, eosinophils, and mast cells [Arahata and Engel, 1988; Cai et al., 2000; Gorospe et al., 1994b; McDouall et al., 1990]. Most of these can kill target cells by liberating cytokines (tumor necrosis factor-α; macrophage inflammatory protein-1αβ, RANTES), and by generating high concentrations of oxidative radicals. Mast cells can also promote muscle fibrosis [Gorospe et al., 1994a; Granchelli et al., 1996]. Muscle cells might become autoreactive as they liberate tumor necrosis factor-α and could be an additional source of antigen-presenting cells [Behrens et al., 1998]. Many of these pathways are known to be blocked by prednisone, including induction of the transcription of I B (inhibitor), which keeps nuclear factor κβ in the inactive state; decreased production of proinflammatory cytokines; and induction of genes that inhibit cyclo-oxygenase-2, adhesion molecules, and other inflammatory mediators.
Abnormal Signaling Functions
Although the mechanical theory of muscle membrane disruption remains important and well documented [Kumar et al., 2004], there is accumulating evidence that the DGC also has important muscle cell-signaling functions, and its integrity is essential for muscle cell viability [Rando, 2001a]. These functions include transmembrane signaling (through β-dystroglycan), docking of signal transduction molecules (e.g., caveolin-3), and interaction with or regulation of other transmembrane complexes (e.g., integrins) [Rando, 2001a]. The absence of dystrophin causes extensive abnormalities and disruption of the DGC, in which sarcoglycans, neuronal nitric oxide synthetase [Rando, 2001b], and other members of the DGC lose their association with the sarcolemma. Many of the putative signaling cascades and molecules associated with this complex are known to regulate the balance between pro- and anti-apoptotic pathways [Rando, 2001a]. In addition to direct regulation of apoptosis, the defective DGC signaling might alter the metabolic pathways necessary to modulate cell susceptibility to injury. Of the downstream pathways involved in DMD pathogenesis leading to cell death, those involving oxidative radical metabolism [Baker and Austin, 1989; Haycock et al., 1996b; Kumar and Boriek, 2003; Ragusa et al., 1997; Rando, 2002; Spencer et al., 2001] and intracellular Ca2+ regulation are of particular interest because they could be targets of pharmacologic modulation. In addition, mechanical stretch has been demonstrated to activate the classic nuclear factor κβ pathway abnormally (in a Ca2+– independent manner) in the mdx mouse, resulting in increased expression of inflammatory cytokines interleukin-1β and tumor necrosis factor-α which precedes the onset of symptomatic myopathy [Kumar and Boriek, 2003].
The “vascular” theory of DMD pathogenesis is supported by morphologic findings in DMD and mdx mice of muscle fiber group necrosis, which occurs very early in the disease, presumably secondary to ischemia. Recent findings indicate that the mislocalization and reduction of neuronal nitric oxide synthetase in dystrophic muscle affects smooth vessel vasodilatation in response to alpha-adrenergic stimuli during exercise [Thomas et al., 2003], and results in muscle ischemia [Sander et al., 2000]. Dystrophin-associated α-syntrophin appears to be essential for the membrane localization of neuronal nitric oxide synthetase [Thomas et al., 2003]. The phosphodiesterase-5 inhibitor, sildenafil, has been shown to be cardioprotective in mdx mice [Adamo et al., 2010], prompting a clinical trial in DMD boys [Wagner, 2010].
Abnormal Fibrosis and Muscle Regeneration
Fibrosis (excessive deposition of endomysial and perimysial extracellular matrix) is a phenomenon known to be secondary to chronic muscle inflammation and fiber degeneration in DMD [Porter et al., 2002]. However, the amount of fibrosis in DMD seems disproportionate in relation to the clinical severity in the earlier stages of the disease, raising the question that an abnormal fibrotic process might be related directly to the absence of dystrophin and might occur in parallel to (as well as after) muscle necrosis and degeneration. There is evidence that both enhanced fibrinogenesis and decreased fibrinolysis [von Moers et al., 2005] are implicated in the development of muscle fibrosis in DMD. There is an increase in expression of the fibrogenic cytokine transforming growth factor-β1 in muscle of DMD patients [Bernasconi et al., 1995] and in serum samples from DMD boys [Bernasconi et al., 1999]. Preclinical studies in mdx mice have revealed that transforming growth factor-β1 levels are also significantly elevated in the diaphragm, the muscle in this model that best correlates with human skeletal muscle pathology [Hartel et al., 2001; Iannaccone et al., 1995; Morrison et al., 2000; Passerini et al., 2002; Porter et al., 2002]. Similar findings of abnormal cytokine levels and fibrosis early in the disease process are found in the golden retriever model of DMD [Passerini et al., 2002]. The level of messenger RNA transcript for transforming growth factor-β was found to be increased in mononucleated cells around areas of fiber necrosis in 6- and 9-week-old mdx mice, but not in 12-week-old mdx mice [Gosselin et al., 2004]. These findings suggest a role of transforming growth factor-β1 during the early stages of fibrogenesis in dystrophic diaphragmatic muscle. Transforming growth factor-β1 induces organ fibrosis by increasing extracellular matrix synthesis [Gosselin et al., 2004] and by simultaneously inhibiting matrix-degradation proteases, such as matrix metalloprotease 1 [Herbst et al., 1997; von Moers et al., 2005]. Another abnormal fibrotic pathway in DMD relates to an increase in levels of platelet-derived growth factor and its receptors, which have an important modulating role in the active stage of tissue destruction, as well as initiation and promotion of muscle fibrosis [Zhao et al., 2003]. Of concern is the fact that fibroblasts of DMD patients appear to have a paracrine function, inhibiting satellite cell growth [Melone et al., 2000], in which case abnormal fibrosis would decrease muscle regeneration directly.
Temporal gene expression profiling studies in DMD have demonstrated that inflammatory and profibrinogenic pathways predominate in the presymptomatic stages of the disease, whereas the acute activation of transforming growth factor-β and failure of metabolic pathways occur later in the disease [Chen and Nagaraju, 2005]. In hind limb mdx muscle, inflammation, proteolysis, and extracellular matrix upregulation and fibrosis are initiated very early in the course of the disease, represent a substantial component of the transcriptional response at that early age, and are tightly coordinated [Porter et al., 2003b]. Pharmacologic blockade of these pathways may have promising therapeutic implications, especially in symptomatic subjects [De Luca et al., 2005; Porter et al., 2002].
Clinical Features
The disease is on-going in infancy, with muscle fiber necrosis demonstrable on muscle biopsy and high serum creatine kinase enzyme levels. However, clinical manifestations frequently are not recognized until the child is 3 years of age or older. This potential “presymptomatic therapeutic window” has been underemphasized previously; however, it lends itself to the development of early therapeutic interventions to prevent or delay the onset of symptoms secondary to more advanced muscle degeneration. Walking frequently begins later than in normal children; independent ambulation often is achieved after 15 months of age. Affected children often experience more falls than their peers. Gait abnormality, difficulty rising from the floor, and problems negotiating stairs usually become apparent at 3–4 years of age, prompting clinical evaluation. Muscle weakness is present initially in neck flexor muscles, with less than antigravity power. As a result, children need to turn on their side when getting up from a supine position on the floor, which is the initial component of the Gowers’ maneuver (Figure 92-2). Hypertrophy of calf muscles typically occurs, usually being very prominent by age 3 or 4 years (Figure 92-3). Hypertrophy of other muscles also may develop, especially the vastus lateralis, infraspinous, deltoid, tongue, and, less frequently, the gluteus maximus, triceps, and masseter muscles. Muscle mass is usually decreased in later stages of the disease in the pectoral, peroneal, and anterior tibial muscles. Hip-girdle muscles are affected earlier than shoulder-girdle muscles. Because of weakness of the hip extensor muscles that act as “shock absorbers” when weight is placed on one leg, children tend to rock from side to side when walking, producing a waddling gait. Increased lumbar lordosis ensues, which is necessary to keep the center of balance stable, with shoulders lined up over hips, knees, and ankles. The preschooler has difficulty rising from the floor, turning 45, then 90, and, finally, 180 degrees, and placing the hands on the floor to get up. As pelvic weakness increases, the complete Gowers’ sign (see Figure 92-2) is exhibited. The patient assumes a locked-knee, buttocks-first position, followed by pushing off the floor with the hands, literally pushing the trunk erect by bracing the arms against the anterior thighs. The maneuver is necessary because of pronounced weakness of the hip muscles – predominantly, the gluteus maximus – and is present by age 5 or 6 years. As muscle deterioration proceeds, climbing stairs becomes difficult, necessitating use of both hands on a railing or crawling on all four extremities. Distal muscles of the arms and legs exhibit weaken as the disease progresses.
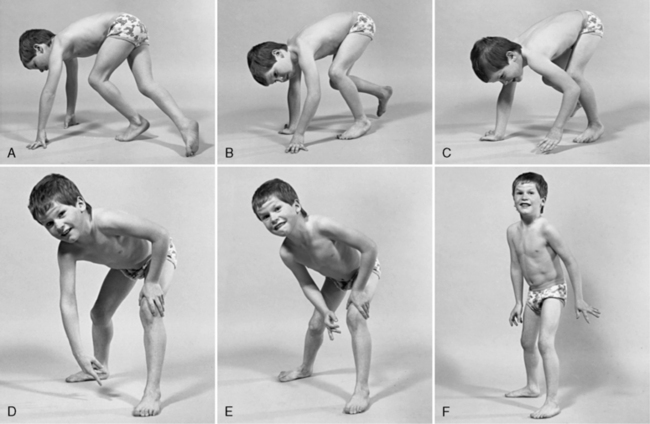
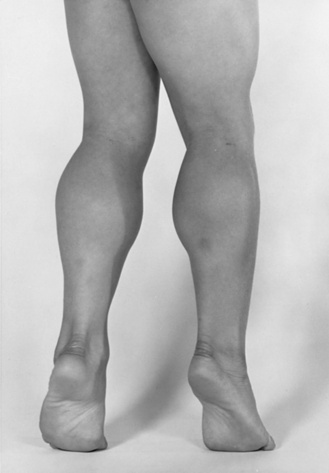
Fig. 92-3 Pseudohypertrophy of the gastrocnemius muscles is usually profound.
The large muscle mass is secondary to replacement of normal muscle with adipose and collagen tissue.
Pulmonary involvement
Pulmonary function becomes compromised because of weakness of intercostal and diaphragmatic muscles. Scoliosis occurs later in the disease in nonambulatory patients. Respiratory failure is the primary cause of mortality in DMD. Prolonging ambulation and standing with corticosteroid treatment [Moxley et al., 2005] and the use of stationary standers can have a significant impact on the development of scoliosis and respiratory function [Galasko et al., 1995]. Use of noninvasive respiratory support and aggressive pulmonary and cardiac care have increased survival [Gomez-Merino and Bach, 2002], even in patients who have not been treated with corticosteroids [Eagle et al., 2002].
Muscle weakness affects all aspects of lung function, including mucociliary clearance, gas exchange at rest and during exercise, and respiratory control during wakefulness and sleep [Gozal, 2000]. DMD is often associated with sleep-disordered breathing, which may be asymptomatic or only mildly symptomatic. Patients can have normal waking oxygen saturation and capillary blood gas levels and normal or near-normal forced vital capacity. Overnight polysomnography is useful for detecting abnormalities [Kirk et al., 2000].
Cardiac involvement
Children with DMD are at risk for cardiomyopathy, especially if they have deletion of exons 48–53 [Nigro et al., 1994]. Early screening for cardiomyopathy at ages 5–6 years, followed by cardiac surveillance with electrocardiography and echocardiography, allows detection of impaired cardiac output before signs of heart failure are apparent. If heart failure occurs, rigorous intervention to improve cardiac function and output is necessary. Mild degrees of cardiac compromise in DMD may occur in up to 95 percent of affected males [Melacini et al., 1996]. Chronic heart failure may affect up to 50 percent [Melacini et al., 1996; Wahi et al., 1971]. Sudden cardiac failure can occur, especially during adolescence. In one series of 19 patients, postmortem studies revealed that 84 percent had demonstrable cardiac involvement [Leth and Wulff, 1976]. Characteristic electrocardiographic changes include sinus tachycardia; tall R1 waves in lead V1; and prominent Q waves in leads II, III, aVF, and V6. Autonomic dysfunction may contribute to tachycardia [Danzig et al., 2003; Finsterer and Stollberger, 2003]. Routine echocardiography may yield normal findings. However, early abnormalities in cardiac function can be detected with newer echocardiography techniques in children between the ages of 4 and 10 years [Giglio et al., 2003]. Some young children with DMD might exhibit regional wall motion abnormalities in areas of fibrosis [Melacini et al., 1996]. With development of cardiac fibrosis, left ventricular dysfunction and ventricular dysrhythmias can occur. In the late stages of the disease, systolic dysfunction may lead to heart failure and sudden death. Subclinical or clinical cardiac insufficiency is present in about 90 percent of patients with DMD and BMD, and is the cause of death in 20 percent of those with DMD and 50 percent of those with BMD [Finsterer and Stollberger, 2003; Melacini et al., 1996].
Neuropsychologic involvement
Males with DMD have an intelligence quotient (IQ) curve shifted to the left. The mean IQ score in one study was 83 (range, 46–134) [Ogasawara, 1989]. However, other investigators have not been able to demonstrate a decrement in full-scale IQ relative to normal males [Bushby et al., 1995; Felisari et al., 2000; Hinton et al., 2000; Roccella et al., 2003]. Some cognitive areas are more affected than others, especially verbal memory [Hinton et al., 2000]. Genotype/phenotype studies have revealed that deletions localized in central and 3′ parts of the gene are preferentially associated with mental impairment, some of these directly affecting the regulatory and coding sequences for the three central nervous system-specific carboxy-terminal isoforms [Giliberto et al., 2004]. Another study of 137 males with DMD/BMD confirmed these findings, demonstrating that all patients with deletions upstream of the 5′end of the gene were mentally normal, whereas all patients with mental retardation or autism had deletions containing the 3′ end of the gene. Some researchers have found average intelligence [Sollee et al., 1985] but difficulties with immediate memory [Wicksell et al., 2004]. DMD boys are more likely to exhibit poor performance on measures of story recall, digit span, and auditory comprehension, a profile suggesting selective involvement of verbal working memory [Hinton et al., 2001]. Morphologic changes in the central nervous system and brain imaging studies have been inconsistent. Although many investigators have found no pathologic or radiologic brain abnormalities in DMD, others, using positron emission tomographic scanning, have reported hypometabolism in the cerebral and cerebellar hemispheres [Bresolin et al., 1994; Rae et al., 1998]. Overall, there is no firm evidence that an abnormality of the brain, either gross or histologic, is common in patients with DMD, and a correlation between abnormality and intellectual impairment has yet to be clearly established [Anderson et al., 2002].
Diagnosis
Clinical laboratory tests
The serum creatine kinase level is the most valuable and universally used screening test for Duchenne dystrophinopathy. Levels of creatine kinase muscle isozyme are greatly elevated, typically from 10,000 to 30,000 International Units/L, early in the disease. A normal or minimally elevated creatine kinase level effectively excludes the possibility of DMD. Gaps in the sarcolemma allow efflux of creatine kinase into the circulation. Serum creatine kinase levels can vary greatly with activity and decrease as muscle mass is lost with disease progression. There is no correlation between the serum creatine kinase level and clinical severity, and the use of creatine kinase levels as a surrogate marker of treatment response is not well supported. Due to the leakage of intracellular muscle proteins, other muscle isoenzyme levels also increase in the circulation. These include lactate dehydrogenase (LDH), alanine aminotransferase (ALT), and, to a lesser degree, aspartate aminotransferase (AST), all tested routinely as markers of liver function. Physicians may perform extensive investigations, including liver biopsy, before it is realized that these “liver enzymes” are of muscle derivation. Similar diagnostic confusion may occur in presymptomatic BMD patients [Korones et al., 2001; Lin et al., 1999; Tay et al., 2000; Zamora et al., 1996]. Obtaining a truly “liver-specific” gamma glutamyl transferase (GGT) level eliminates diagnostic error.
Genetic testing
A precise molecular genetic diagnosis is essential in all patients with DMD and BMD, even in those whose diagnosis has been confirmed by immunostaining for dystrophin on muscle biopsy. Genetic testing for DMD and BMD is widely available. Initial screening for deletions and duplications within the dystrophin gene confirms the diagnosis of dystrophinopathy in most patients. Direct sequencing of the entire dystrophin gene will define the vast majority of DMD/BMD patients not identified by deletion/duplication testing. The screening of only 19 exons by multiplex polymerase chain reaction identifies about 98 percent of all deletions [Beggs et al., 1990]. Southern blot analysis of these samples frequently can predict whether the deletion, when in the rod domain, will shift the reading frame, and thus is confirmatory for dystrophinopathy. This technique is very effective for the molecular diagnosis of common deletions (60 percent of patients); however, it cannot be used to identify duplications or to establish genotype in female carriers. Other diagnostic approaches, such as quantitative polymerase chain reaction [Abbs and Bobrow, 1992; Yau et al., 1996], multiplex amplifiable probe hybridization, and multiplex ligation-dependent probe amplification [White et al., 2002], can be used for detecting patients with either gene deletions or duplications, as well as carrier diagnosis. With more recent technology, it is possible to sequence the entire dystrophin gene for the specific molecular defects responsible in the other 30–40 percent of patients with DMD and BMD in whom a genetic abnormality has not been detected. These techniques include a protein truncation test [Tuffery-Giraud et al., 1999a, 1999b], single-condition amplification/internal primer sequencing [Mendell et al., 2001], and denaturing high-performance liquid chromatography followed by sequencing, which enables the detection of small mutations in nearly all the 79 exons [Bennett et al., 2001]. In a large series with single-condition amplification/internal primer sequencing, deletions of one or more exons were found in 66 percent of probands, which was consistent with the frequency of deletions in the previously reported literature. Point mutations were found in 18 percent, including premature stop codons in 13 percent and missense mutations in 4 percent of the total mutations. Frameshift mutations were found in 3 percent of patients. Fourteen subexonic mutations were also found, and these included nonsense mutations in 9 (64 percent), missense mutations in 3 (21 percent), and frameshift mutations in 2 (14 percent). Of the 45 deletions detected, 3 (7 percent) either were undetected or would have been undetected by that technique [Dent et al., 2005]. Duplications were detected in 6 percent of patients, and no disease-causing mutation was identified in 7 percent of the evaluated patients [Dent et al., 2005].
Several commercial laboratories offer gene sequencing for the diagnosis of suspected dystrophinopathy patients, as well as for carrier detection and prenatal diagnosis. (See GENETESTS website: www.genetests.org.)
Muscle biopsy
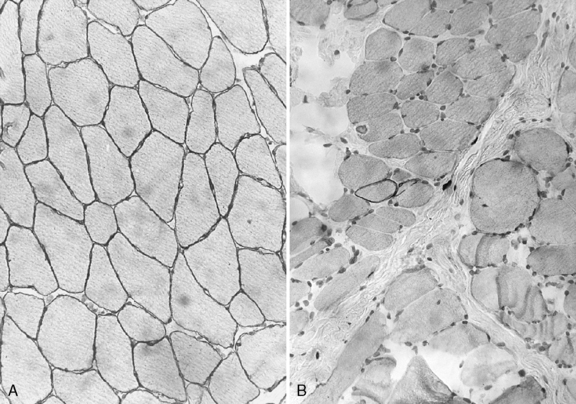
Histologic study reveals fiber size variation, degenerating and regenerating fibers, clusters of smaller fibers, endomysial fibrosis, and a few scattered lymphocytes. Large, opaque fibers, distinguished by intense staining with the modified Gomori trichrome, are prominent (Figure 92-4). As the disease progresses and degeneration exceeds regeneration, a decrease in the number of muscle fibers is apparent, with replacement of muscle with fat and connective tissue. Fiber typing of type 1 and type 2 muscle fibers with ATP histochemistry is less distinct than normal. Oxidative histochemistry shows that the intramyofibrillary network is maintained. On electron microscopic study, gaps in the sarcolemma with preservation of the basement lamina are seen in non-necrotic fibers (Figure 92-5 and Figure 92-6). Absence of immunoreactivity for dystrophin, with monoclonal antibodies against the C-terminal, rod domain, and N-terminal, is important for an accurate diagnosis of dystrophinopathy (Figure 92-7). However, quantitative dystrophin analysis by immunoblot correlates more closely with the diagnoses of DMD than immunostaining, with levels of dystrophin being less than 5 percent of normal in DMD patients.
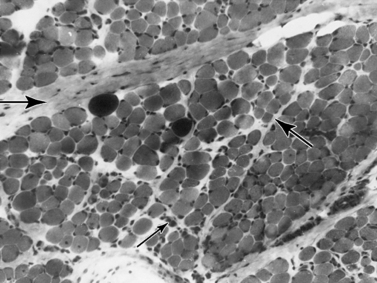
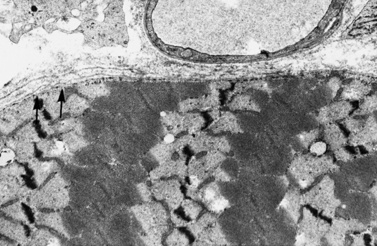
Fig. 92-5 Electron micrograph of a specimen from a 6-year-old child with Duchenne muscular dystrophy.
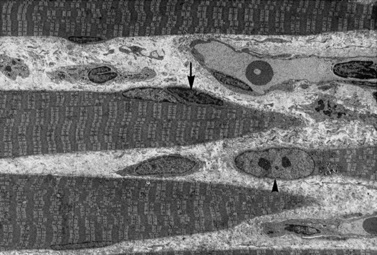
Management
Comprehensive standard of care guidelines for the diagnosis and treatment of patients with DMD have been recently published [Bushby et al., 2010a, 2010b]. The needs of children and adults with DMD and BMD change substantially with disease progression. The information presented in this section, although largely similar to that presented by Bushby, reflects the bias of the authors of this chapter.
Pharmacologic treatment
In the era before widespread treatment with corticosteroids and assisted ventilation of DMD patients, death usually occurred late in the second decade of life (median age of 18 years), and only 25 percent of males with DMD lived beyond 21 years of age Gardner-Medwin, 1970]. This natural history has changed [Eagle et al., 2002], and in current studies, researchers are trying to characterize the impact of corticosteroid treatment and age at start of treatment, as well as noninvasive ventilation, rehabilitation, scoliosis surgery, and pulmonary care, on the quality of life and survival of DMD patients. Daily corticosteroid therapy stabilizes or improves the strength of children with DMD, and it is the only treatment proven to be effective for this disease [Moxley et al., 2005]. Drachman and associates [1974] first reported the beneficial effect of prednisone in an open trial of 2 mg/kg/day. This finding was duplicated in other open-design trials [Brooke et al., 1987; DeSilva et al., 1987] and subsequently, through the collaboration of the Clinical Investigation of Duchenne Dystrophy investigators, in double-blinded placebo-controlled trials [Fenichel et al., 1991; Griggs et al., 1993; Mendell et al., 1989]. To date, the most effective, well-studied prednisone dose is 0.75 mg/kg/day. There is a dose–response effect, the lowest proven effective dose being 0.3 mg/kg/day [Mendell et al., 1989]. Effects on strength are seen as soon as 10 days after treatment starts, with a peak at 3 months, followed by slowing of disease progression [Griggs et al., 1991]. In a 3-year follow-up study, improvement was maintained in children who were kept on doses of at least 0.5 and 0.6 mg/kg/day [Fenichel et al., 1991]. It is hoped that long-term monitoring of these patients will confirm continuous benefit in muscle strength and respiratory function. Children with DMD treated with prednisone from an early age often remain ambulatory into their teens, have a lower incidence of scoliosis [Alman et al., 2004; Biggar et al., 2004] and of contractures [Yilmaz et al., 2004], and maintain better respiratory muscle function [Angelini et al., 1994; Biggar et al., 2004; Campbell and Jacob, 2003; Merlini et al., 2003]. The mechanism(s) of action of prednisone in DMD is not entirely known and may involve several mechanisms, including immunosuppressive actions with reduction in cytotoxic T cells, anti-inflammatory effects, gene transcription regulation, increase of laminin expression and membrane repair, and modulation of intracellular calcium [Kissel et al., 1993; Liu and Sun, 1996; Passaquin et al., 1998; Ruegg et al., 1998, 2002; St-Pierre et al., 2004; Wershil et al., 1995]. Steroids may help prevent exercise-induced apoptosis [Lim et al., 2004] and thus lessen exercise-related muscle damage.
Deflazacort, an oxazoline derivative of prednisone, is also used in the countries in which it is available. Deflazacort yields a similar benefit in improving and maintaining strength, with a decreased incidence of excessive weight gain relative to prednisone-treated patients. It carries an increased risk of asymptomatic cataract formation [Angelini et al., 1994; Biggar et al., 2001; Bonifati et al., 2000; Campbell and Jacob, 2003; Mesa et al., 1991; Moxley et al., 2005]. The suggested dosage is 0.9–1.2 mg/kg/day. Deflazacort is not Food and Drug Administration-approved and is not available in the United States. Families wishing to institute deflazacort therapy must import the drug from other countries. Alternative dosage regimens, including alternate-day corticosteroids, have not demonstrated sustained efficacy [Fenichel et al., 1991; Sansome et al., 1993]. A small study suggests benefit with corticosteroids administered for 10 consecutive days, followed by 10 days off [Kinali et al., 2002]. Clinical trials to determine the efficacy of other corticosteroid dosing regimens and head-to-head trials comparing the benefits of prednisone to those of deflazacort are in progress. Other immunosuppressive medications have produced mixed results. Whereas azathioprine yields no benefit [Griggs et al., 1993], there is uncontrolled evidence that cyclosporine is efficacious in DMD [Mendell et al., 1995; Sharma et al., 1993]. Prednisone remains the only drug that has been proven effective in 80 percent of children with DMD. An American Academy of Neurology Practice Parameter paper on the use of steroids in treating DMD [Moxley et al., 2005] and a Cochrane Database review of all trials [Manzur et al., 2004; Manzur et al., 2008] support the use of corticosteroids for DMD. Despite the many potential side effects of corticosteroid treatment, the benefits of steroid therapy outweigh the risks. There is accumulating evidence that treatment should start as soon as the diagnosis is made [Dubowitz et al., 2002; Kinali et al., 2002; Merlini et al., 2003]. However, early steroid treatment is associated with significant decrease in linear growth. A pilot study of 10 mg/kg of prednisone, given over 2 consecutive days of the week (Friday and Saturday), demonstrated that males with DMD had improved muscle strength and function while maintaining linear growth, and did not have an increase in their body mass index in comparison with males with untreated DMD [Connolly et al., 2002]. A larger trial has been completed and awaits publication. With the standard treatment of daily prednisone, the most common side effects are increased appetite with weight gain, irritability and neurobehavioral changes, hirsutism, cushingoid facies, and decreased linear growth [Moxley et al., 2005]. The most serious side effects of prednisone seen in older children and adults (e.g., diabetes, hypertension, ulcers, and infections) are extremely rare in the DMD population. The effect of steroids on bone metabolism and subsequent osteoporosis is difficult to discern, because baseline decreased bone density is nearly universally present in DMD boys secondary to decreased activity [Aparicio et al., 2002; Bianchi et al., 2003; Larson and Henderson, 2000]. A study of deflazacort in children with DMD revealed that treated patients had better bone density than did untreated patients [Biggar et al., 2004]. However, other studies have yielded conflicting results [Bianchi et al., 2003], leaving this an area for further research [Biggar et al., 2005]. Children with DMD who are treated with prednisone or deflazacort should be supplemented orally with calcium and vitamin D. There is no consensus regarding the need for monitoring bone density with dual-energy x-ray absorptiometric (DEXA) scans, inasmuch as the relationship of decreased Z scores and the incidence of fractures in this population (and the general population) has not been established. However, annual DEXA monitoring of DMD patients on chronic corticosteroid therapy and those patients defined as being at high risk of fractures is recommended. There is no proven benefit of bisphosphonates for treatment of asymptomatic children with DMD who have decreased bone density. However, in the setting of spontaneous pathologic fractures and osteoporosis, treatment with alendronate or similar drugs is often given empirically; the safety of these drugs has not been proven in children [Biggar et al., 2005]. Obesity is often a major problem in patients with DMD, even if steroids are not used. Children with DMD may gain excessive weight, which often becomes apparent before ambulation is lost. Diminished caloric expenditure and excessive caloric input are likely explanations. Obesity becomes more prominent after independent ambulation is lost. Boredom, diminished physical activity, and depression may lead to inappropriate food intake [Zanardi et al., 2003], and rigorous measures are required to forestall weight gain. Obesity reduces the period of independent walking and may contribute to respiratory and cardiac insufficiency. When steroid treatment is prescribed, patients need to follow a strict dietary caloric intake to prevent excessive weight gain. A nutritionist should be part of the medical team for patients with DMD, especially those who receive continuous steroid treatment. Experience suggests that the patient and the entire family must embrace these healthy eating habits. Obesity in the parents is a bad prognostic factor for the child’s compliance with diet.
Other immunosuppressive and other pharmacologic treatments have undergone limited investigation. Cyclosporine (5 mg/kg/day) produced improvement in strength, tested on an isolated muscle [Sharma et al., 1993]. The anabolic steroid oxandrolone was tried in a randomized trial, with a trend toward improvement of muscle strength [Fenichel et al., 2001]. A randomized, controlled, blinded trial of creatine monohydrate and glutamine supplementation in ambulant DMD patients (aged 5–11) also yielded negative findings based on manual muscle testing (primary outcome). However, there were trends toward improvement in isometric muscle strength in the older patients with creatine, and increase in function in the younger children with creatine and glutamine [Escolar et al., 2005]. Other studies have demonstrated increases in muscle strength and body mass index with creatine [Tarnopolsky et al., 2004; Walter et al., 2000]. No significant side effects were seen with doses of 5 g/day for at least 6 months [Escolar et al., 2005].
Experimental therapies
Approximately 15 percent of DMD patients have stop codon mutations as the molecular genetic basis of their dystrophinopathy. In an attempt to repair mutant dystrophin genes in vivo, gentamicin, an aminoglycoside antibiotic that binds the ribosome and causes “read-through” of premature stop codon (nonsense) mutations, was tried first in the mdx murine model of DMD [Barton-Davis et al., 1999], and then in clinical trials in patients with DMD or BMD [Wagner et al., 2001; Malik et al., 2010]. Although the mdx experiments revealed positive dystrophin measurements in about 15 percent of previously dystrophin-negative muscle fibers, the investigators did not find consistent dystrophin expression in humans. A recently completed study utilizing PTC124 for stop codon read-through is currently under analysis [Finkel, 2010]. Other approaches involving gene repair mechanisms are being evaluated in experimental models. These include delivery to dystrophin-deficient cells of RNA–DNA oligonucleotides that target the specific mutation and cause it to revert to the normal sequence (chimeraplasts) [Rando et al., 2000] and the delivery of antisense RNA molecules to dystrophin-deficient cells so that semifunctional dystrophin can be produced [Errington et al., 2003; Gebski et al., 2003; Lu et al., 2003; Mann et al., 2001; Wells et al., 2003]. This method forces the cell-splicing machinery to skip the dystrophin gene exon that contains the gene mutation, which results in the full translation of dystrophin messenger RNA (minus the mutant exon) into an “in-frame” semifunctional dystrophin protein. Antisense oligonucleotides are small molecules and can be systemically delivered. Proof of concept for exon skipping has been advanced in recent studies designed to restore the reading frame by skipping exon 51 of the dystrophin gene [Van Deutekom et al., 2007; Kinali et al., 2009].
Preclinical studies involving delivery of functional mini-dystrophin genes to replace the missing dystrophin, through adeno-associated viral vectors, are also in progress. In this model, dystrophin can be produced, the immune response can be prevented, and there is evidence of improved function and amelioration of pathology [Wang et al., 2000] in young and older animals [DelloRusso et al., 2002; Hartigan-O’Connor et al., 2001; Scott et al., 2002]. A major hurdle was crossed when systemic delivery of AAV type 6 vector with a mini-dystrophin gene was achieved in older mdx mice [Gregorevic et al., 2004]. Preliminary studies of AAV administration to patients afflicted with α-sarcoglycan deficiency support a possible role for gene transfer in several forms of muscular dystrophy [Mendell et al., 2010].
Despite encouraging preclinical studies [Huard et al., 1994], the results of myoblast transfer are not encouraging [Partridge, 2002]. Myoblast transplantation was attempted in children with DMD without success [Miller et al., 1997; Munsat, 1990; Neumeyer et al., 1998]. Preclinical studies of mesangioblasts as muscle progenitor cells are in progress.
Respiratory care
With the progression of muscle weakness, loss of respiratory muscle strength, with associated ineffective cough and hypoventilation, leads to pneumonia, atelectasis, and respiratory insufficiency, initially in sleep and later in the waking state [Gozal, 2000]. These complications are often treatable with careful monitoring and assessments of respiratory function. Patients with DMD should have routine immunizations. In addition, these patients should receive the pneumococcal vaccine and an annual influenza vaccine. There is debate as to whether DMD patients on chronic corticosteroid therapy should receive live virus immunizations.
Older ambulatory patients with DMD should undergo annual spirometry. Once a child is nonambulatory, if either the forced vital capacity falls below 80 percent of predicted or the child is 12 years of age, or both, the child should be seen twice a year by a pediatric pulmonary specialist [Finder et al., 2004]. Patients in more advanced stages of the disease who require mechanically assisted airway clearance therapy (Cough Assist) or noninvasive assisted ventilation (bilevel positive airway pressure or positive pressure ventilation) should see a pulmonologist every 3–6 months. Routine evaluations at these visits should include oxyhemoglobin saturation by pulse oximetry, spirometry, and measures of inspiratory and expiratory pressures and peak cough flow [Bach et al., 1997].
Maintaining good pulmonary toilet is essential; hence, these patients should be taught strategies to improve airway clearance and how to employ these techniques early and aggressively. The use of assisted cough technologies should be recommended when peak cough flow is below 270 L/min or when the maximal expiratory pressures are less than 60 cm H2O, or both [Finder et al., 2004].
Patients with DMD have an increased risk for sleep apnea, nocturnal hypopnea, and hypoxemia. Treatment of these problems with noninvasive nocturnal ventilation can significantly increase the quality of life [Baydur et al., 2000; Vianello et al., 1994].
A polysomnographic study with continuous CO2 monitoring is a sensitive way to assess the need for ventilatory support. Pulse oximetry, especially during the waking state, fails to identify many patients with significant hypoventilation. Decisions regarding long-term ventilation, either invasive or noninvasive, should involve the patient, caregivers, and medical teams. End-of-life decision-making should be discussed earlier, with all possible information available to the patient [Hilton et al., 1993].
Rehabilitation
Exercise
On the basis of the preclinical data in the mdx murine model of DMD, and clinical experience in boys with DMD, there is general consensus that high-resistance exercises, especially those involving eccentric contractions, such as weight lifting, are damaging to the muscle cell membrane and should be avoided [Ansved, 2003; Petrof, 1998]. However, a sedentary life should also be discouraged [McDonald, 2002]. Furthermore, another line of evidence in mdx mice indicates that sustained, nonresistive activity might produce a histochemical shift of the muscle cells toward fatigue resistance, with benefits in activity and fatigability measures [Carter et al., 1995; Hayes et al., 1993; Petrof, 1998]. Therefore, active nonresistive exercises are encouraged. Swimming is widely recommended for these children. Maintaining an active lifestyle will also help prevent excessive weight gain. Regular periods of daily walking enhance maintenance of strength and may retard contracture formation. Swivel walkers may be used to provide low-energy ambulation and improve the quality of life [Sibert et al., 1987]. Both the nature and degree of activity should be modified to avoid myalgia and prolonged fatigue. Wheelchair games can be played when ambulation is lost. Children confined to bed because of intercurrent disease, injury, or surgery require physical therapy, including range-of-motion exercises, with return to more active exercise, including walking, as soon as possible; if such exercise is not resumed, loss of independent ambulation may be hastened. In the event of leg fractures, walking casts should be used as soon as possible [Siegel, 1977].
Contractures
Contractures of the Achilles tendons and, later, development of more widespread contractures occur in all DMD patients. With early hip-girdle weakness, anterior rotation of the hips and posterior displacement of the shoulders evolve to keep the center of gravity perpendicular to the ground through the shoulders, hips, and ankles. Active range-of-motion exercises supplemented by passive stretching are important for preventing contractures. Night-time stretching orthoses are probably useful and are recommended at age 5–6. A standing board tilted up 20 degrees may be used for 20 minutes twice per day to provide constant stretching of the Achilles tendons. Keeping the heel cords stretched through vigorous passive stretching by parents and physical therapists helps the patient maintain better gait mechanics. This program requires stretching of the tensor fascia lata, hamstrings, knee flexors, and ankle plantar flexors. If strenuous stretching is not effective, surgical release of tight heel cords may be beneficial [Do, 2002], even if both the quadriceps and gastrocnemius muscle groups have less than antigravity strength. In the latter case, long leg bracing can be offered to maintain assisted standing and modest assisted ambulation after contractures are corrected. Mobilization in a walking cast immediately after surgery is essential for preventing loss of strength. Temporary bracing after surgery is necessary for optimal results after tenotomy procedures (Figure 92-8).

The iliotibial bands may also tighten because of the broad-based gait used to maintain stability. Hip flexors may become contracted when ambulation is still present as a result of the anterior rotation of the hips or, later, because of sitting for prolonged periods in a wheelchair. Hip flexion contractures may benefit from surgical release, followed by application of long leg braces. Resection of the tensor fascia lata (Rideau procedure) may be beneficial for some patients [Do, 2002].
Scoliosis
Most patients with DMD develop scoliosis after losing independent ambulation. The use of solid seat and back inserts in properly fitted wheelchairs is helpful in preventing scoliosis by keeping truncal posture erect. Baseline thoracolumbar spine radiographs to document the degree of curvature should be obtained for comparison with future films. The use of corticosteroids lowers the risk of severe scoliosis requiring surgical intervention, most likely due to prolongation of ambulation [Alman et al., 2004; Yilmaz et al., 2004].
Once a scoliotic curve reaches 30 degrees, further progression is nearly universal [Yamashita et al., 2001a]. Failure to address scoliosis in DMD can result in increased hospitalization rates, worsening of pulmonary function, and poor quality of life [Finder et al., 2004; Yamashita et al., 2001b]. Surgical intervention should be undertaken when lung and cardiac function are satisfactory (with best recovery when the forced vital capacity exceeds 40 percent). However, there are no absolute contraindications to scoliosis surgery that are based on pulmonary function [Finder et al., 2004]. Surgery is usually considered once the Cobb angle measured on scoliosis films is between 30 and 50 degrees [Brook et al., 1996].
Cardiac management
Cardiac involvement is nearly inevitable in DMD and BMD. The mechanisms of skeletal muscle damage discussed previously appear to hold true for cardiac muscle degeneration. In DMD and BMD, the left posterobasal and lateral cardiac walls are more extensively affected, with sparing of the left and right atria and the right ventricle [Finsterer and Stollberger, 2003]. The conduction system does not appear to be involved until there is advanced fibrosis; most cardiac deaths occur secondary to left ventricular cardiac failure [Corrado et al., 2002]. With the advent of aggressive noninvasive and invasive respiratory support, DMD and BMB patients are facing symptomatic cardiomyopathy as their cause of death. Dystrophinopathies may manifest cardiomyopathy with relative sparing of skeletal muscle. X-linked cardiomyopathy due to deletions in the muscle promoter has been reported [Muntoni et al., 1993]. Two families with promoter and exon 1 involvement presenting with X-linked dilated cardiomyopathy enabled the discovery of a novel regulatory element, DEM2, which appears to be involved with the expression of dystrophin in the heart [Bastianutto et al., 2002]. Although electrocardiographic abnormalities are common in DMD, a better correlation of cardiac involvement and prognosis is obtained with left echocardiography [Corrado et al., 2002]. Guidelines for the study of cardiac involvement in DMD [Bushby et al., 2003; Bushby and Finkel, 2010b; Finsterer and Stollberger, 2003] recommend that DMD patients undergo electrocardiography and echocardiography at the time of diagnosis, and then be screened every 2 years up to age 10 and subsequently at yearly intervals. Early preventive use of angiotensin-converting enzyme (ACE) inhibitors and, later, beta blockers may be needed [Bushby et al., 2003; Finsterer and Stollberger,2003]. The introduction of ACE inhibitors prior to the onset of clinical and echocardiographic abnormalities may be cardioprotective [Duboc et al., 2007], and many neuromuscular centers utilize prophylactic treatment.
Genetic counseling
DMD and BMD are inherited as X-linked recessive traits, and the risk to siblings of a patient depends on the carrier status of the mother. Female carriers have a 50 percent chance of transmitting the dystrophin mutation in each pregnancy. Sons who inherit the abnormal gene are affected, whereas daughters who inherit it are carriers. Male patients with DMD rarely reproduce. However, male patients with BMD and those with dystrophinopathy-associated X-linked dilated cardiomyopathy do. All their daughters are carriers, but none of the sons inherit a father’s dystrophin mutation. Until the molecular genetics of DMD and BMD were understood, diagnosis of maternal and female sibling carriers was based on pedigree analysis and indirect assays. These included serum creatine kinase determinations [Griggs et al., 1985; Milhorat and Goldstone, 1965], the occasional finding of histologic abnormalities in muscle obtained from carriers [Maunder-Sewry and Dubowitz, 1981], and in vitro muscle ribosomal protein synthesis [Ionasescu et al., 1971a, 1971b, 1980]. Today, the specific molecular characterization of a patient makes genetic counseling and prenatal diagnosis far more accurate. If a specific mutation is identified in a DMD or BMD patient, genetic testing of the mother or sister for the same mutation determines whether she is or is not a carrier; appropriate counseling for future pregnancies can be performed. In cases where DNA is not available, full gene sequencing can be performed. The possibility of germline mutations precludes giving absolute clearance for risk of recurrence of DMD/BMD for simplex mothers who have undergone negative molecular genetic investigations. When DNA analysis in the patient is not informative and an at-risk mother is pregnant with a second male fetus, chorionic villus biopsy can be used for diagnosis [Kuller et al., 1992]. A genetic counselor is an essential part of the multidisciplinary team that diagnoses and monitors patients with DMD. The genetic counselor should give the family a full explanation of the X-linked recessive inheritance pattern of DMD, and molecular genetic testing should be made available to the patient and other family members as necessary.
Drug precautions
Use of anticholinergic drugs and ganglionic blocking agents should be avoided because of their tendency to decrease muscle tone. Patients with DMD have increased susceptibility to malignant hyperthermia, and proper evaluation and preparation before administration of general anesthesia are recommended [Heiman-Patterson et al., 1986]. Cardiotoxic drugs, such as halothane, should not be used, and caution is advised in inducing general anesthesia [Smith and Bush, 1985].
Emotional and behavioral abnormalities
Dysthymic disorder and major depressive disorder can occur in children with DMD, especially during adolescence [Fitzpatrick et al., 1986; Witte, 1985]. An affected child’s preoccupation with his disease and subsequent emotional withdrawal may lead many families to seek counseling. Depression may develop in a patient who has lost an older sibling or close friend with DMD. Depression is often associated with intellectual limitation, which may induce low tolerance for frustration. Psychologic evaluation and counseling may be necessary. Neuropsychologic screening for intellectual deficits and behavioral problems is ideally conducted at time of school entry and should be repeated in preadolescence. Appropriate follow-up with the family and school officials is important. Mothers identified as carriers of DMD may suffer from feelings of guilt and require psychologic support [Nereo et al., 2003]. Depression is also common among parents and should be recognized [Abi Daoud et al., 2004]. Early discussions regarding end-of-life care, including home mechanical ventilation, are nearly always appropriate [Hilton et al., 1993].
Becker Muscular Dystrophy
A “late-onset X-linked muscular dystrophy” was reported by Becker and Kiener in 1955. Like DMD, BMD is an X-linked recessive condition with mutation at Xp21. When the dystrophin reading frame is maintained, dystrophin is translated but with altered quality and quantity. Distal rod domain dystrophin gene deletions involving exons 45–48 result in a relatively high level of dystrophin, whereas N-terminus deletions are associated with lower levels of dystrophin and more severe clinical expression. Distal rod in-frame deletions result in a moderate Becker phenotype. The most severe phenotype, resembling DMD with minimal dystrophin expression, results from carboxy-terminus deletions. At times, routine immunohistochemistry studies demonstrate normal dystrophin in patients with increased creatine kinase and only mild or no muscle weakness. In these patients, immunostaining for neuronal nitric oxide synthetase has been found useful, revealing complete absence in three patients who were later found to have in-frame deletions in the common rod domain exons (in these cases, exons 48, 45–51, and 47–53) consistent with BMD [Torelli et al., 2004]. In one family, deletion of exon 48 was demonstrated in five members (a 4-year-old female and four male relatives, aged 8–58), all of whom had normal muscle strength. These individuals by definition have BMD but represent one end of the clinical spectrum [Morrone et al., 1997]. With in-frame deletions in the proximal third of the rod domain involving exons 13–17, the clinical manifestations are cramps and myalgia. In this particular Becker phenotype, immunoreactivity over the sarcolemma for dystrophin often appears normal. Creatine kinase levels in BMD are usually between 2000 and 20,000 U/L but may be in the normal range for mildly affected male patients. Because these patients can be asymptomatic for decades, it is not uncommon that they receive misdiagnoses of a liver disorder when routine laboratory testing indicates elevated transaminase levels (aspartate aminotransferase and alanine aminotransferase), which are, in reality, muscle isoenzymes, abnormally elevated in parallel with the creatine kinase serum levels [Tay et al., 2000; Zamora et al., 1996]. Muscle biopsy histology is similar to that seen in DMD, but findings are less pronounced. The sarcolemmal gaps displayed ultrastructurally in DMD are not as readily seen in BMD. When monoclonal antibodies are directed to separate regions of dystrophin, immunoreactivity over the sarcolemma has a variety of staining patterns, ranging from intact to absent with one or more antibodies [Jay et al., 1993].
BMD occurs in 3–6 per 100,000 male births [Gardner-Medwin, 1970]. The onset of clinical symptoms usually occurs after age 7 years and often in the second decade. Ambulation is, by definition, maintained beyond age 15 and often decades longer. Calf muscle hypertrophy, proximal hip weakness resulting in Gowers’ sign, and electrocardiographic abnormalities as seen in DMD are also common in BMD (Figure 92-9). A few patients with earlier onset of symptoms of BMD have virtually the same phenotype as patients with DMD but prove to have Western blot findings of muscle dystrophin and molecular genetic testing consistent with BMD [Hoffman, 1991]. The phenotype–genotype correlation of DMD and BMD may make it difficult to diagnose one over the other neatly. The clinical picture should be the determinant of therapeutic intervention, including corticosteroid treatment. A number of children and adolescents with BMD experience postexertional calf pain or muscle cramps, which may be their initial symptoms [Samaha and Quinlan, 1996]. In others, strength may be nearly normal, but vigorous exercise produces severe cramps and occasionally rhabdomyolysis/myoglobinuria [Figarella-Branger et al., 1997]. BMD may present with heart failure caused by a dilated cardiomyopathy before weakness is recognized. Sudden cardiac failure caused by heart block may occur in BMD. Early-onset cardiomyopathy with mild muscle weakness has been associated with muscle promoter and exon 1 deletion.
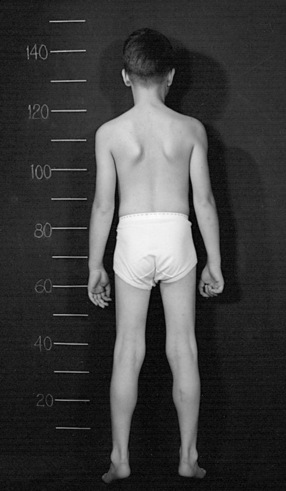
The likelihood of intellectual disabilities, although greater than that in the general population, is less than that in DMD. However, intellectual disabilities (IQ range, 60–68), psychiatric disturbance and autistic spectrum neurobehavioral symptoms in the absence of muscle weakness have been reported in BMD [North et al., 1996].
Therapeutic suggestions and measures appropriate for DMD are, in some measure, appropriate for BMD. This approach includes the use of corticosteroids in the more severely affected patients. Patients with BMD may be predisposed to malignant hyperthermia, and appropriate precautions should be taken when general anesthesia is necessary [Breucking et al., 2000; Kleopa et al., 2000].
Chromosome Xp21 Microdeletion Syndromes
The Xp21 microdeletion syndromes are a series of syndromes that include DMD, Aland’s eye disease, adrenal hypoplasia, glycerol kinase deficiency, retinitis pigmentosa (RP3), mental retardation (MRX1), and ornithine transcarbamylase deficiency. The combination of several of these conditions in a boy with DMD and the confirmation of contiguous gene deletions on the short arm of the X chromosome led to the localization of the DMD gene on Xp21 [Francke et al., 1985].
Female Duchenne Muscular Dystrophy, Carriers, and Manifesting Carriers
Female DMD carriers are heterozygous with a normal dystrophin gene on one X chromosome and a mutant gene on the other. More than 90 percent of female carriers are asymptomatic. However, female carriers may manifest various degrees of muscle weakness because random inactivation of one X chromosome, according to the Lyon hypothesis, may leave more than half of mutant X chromosomes operant in muscle cells (skewed X-inactivation). When substantially more than half the X chromosomes in a given muscle fiber express a mutant gene for DMD, the muscle fiber is prone to degeneration, presumably by the same mechanisms seen in classic DMD. In the presence of muscle fiber degeneration resulting from a burden of more than half mutant X chromosomes in muscle nuclei, muscle fiber degeneration is followed by regeneration. The new fiber, formed from fused myoblasts, may have less than half or more than half mutant X chromosomes and be susceptible to degeneration. Overall, individual muscle groups may express less than normal strength. Each muscle responds separately. The tendency in muscles with a high percentage of mutant X chromosomes is that, with remodeling of the muscle through cycles of degeneration and regeneration, there is an increase in the number of normal X chromosomes but not necessarily a parallel increase in dystrophin content [Pegoraro et al., 1995]. Immunohistochemical and immunoblot analysis of muscle biopsies in these female carriers reveals a mosaic of dystrophin-normal and dystrophin-abnormal muscle fibers [Minetti et al., 1991].
Signs and symptoms of female dystrophinopathy include muscle weakness, myopathic muscle biopsy specimens, elevated levels of creatine kinase, and partial absence of dystrophin in the muscle [Hoffman et al., 1992]. Abnormal dystrophin immunostaining is seen in about one-fifth of DMD carriers [Hoogerwaard et al., 1999a]. A search for female dystrophinopathy should be initiated in women with elevated creatine kinase enzyme levels and muscle weakness, even in the absence of a DMD-positive family history [Hoffman, 1996].
DMD carriers appear to have an increased incidence of cardiomyopathy. Different studies have revealed an incidence of asymptomatic cardiomyopathy in 8–48 percent among adult DMD carriers [Grain et al., 2001; Hoogerwaard et al., 1999a, 1999b], and in 0–15 percent among DMD carrier girls younger than 16 years [Nolan et al., 2003].
Limb-Girdle Muscular Dystrophies
Most gene loci have been identified and specific genes sequenced, but the protein products associated with the autosomal-dominant disorders LGMD1D to 1H have not yet been defined. The majority of cases of LGMD are autosomal-recessive. Autosomal-dominant forms are found only in a few families, except for LGMD1B, which results in the autosomal-dominant form of Emery–Dreifuss muscular dystrophy and allelic neuromuscular and non-neuromuscular disorders. An integrated classification taking into account the type and localization of the proteins has been proposed [Emery, 2002]. Table 92-1 summarizes the current classification of the LGMDs and other muscular dystrophies. Table 92-2 subcategorizes the disorders by the age at onset and the key clinical findings and proposed function of the affected proteins, to provide a clinical-biochemical classification more useful to physicians evaluating these disorders. Many of these diseases are extremely rare, with only a few affected families described in the literature. These rarer forms are not reviewed in detail in this chapter. For additional information, the reader is referred to the article by Laval and Bushby [2004], or the superb text, Disorders of Voluntary Muscle, by Karpati et al. (8th edition [2010]). The classification by proteins causing a congenital muscular dystrophy phenotype have been summarized in Tables 92-3 and 92-4 and are described in the section on congenital muscular dystrophies. Over the past decade, it has become painfully clear that a patient with suspected limb girdle muscular dystrophy may have a phenotype subserved by multiple genotypes. Conversely, a single genotype with a well-defined mutation may manifest markedly different phenotypes, especially with regard to age of onset of symptoms and clinical severity. For example, patients with mutations in the Fukutin gene, the Fukutin-related protein gene, and the lamin A/C gene may manifest early signs and symptoms consistent with congenital muscular dystrophy, or have a later onset of milder symptoms consistent with LGMD. The wide variations in genotype-phenotype observations likely reflect differences in genetic background and (perhaps) environmental factors influencing downstream pathophysiology.
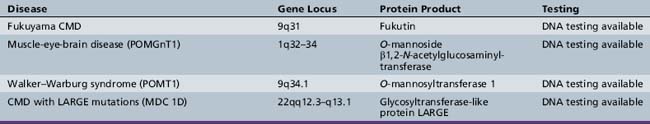
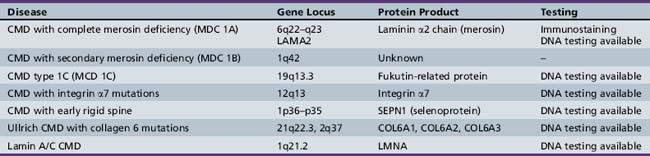
The pathophysiology and clinical picture of the syndromes caused by defects of sarcolemmal proteins constituting the DGC is similar to that described for dystrophinopathies. In contrast, other LGMDs have distinct pathophysiologic mechanisms. For example, dysferlin deficiency causes abnormal muscle membrane repair, whereas calpain-3 deficiency involves abnormal regulation of anti-apoptotic pathways and likely abnormal cytoskeletal remodeling. Taken in toto, the ever-growing number of disorders grouped under the umbrella of “limb girdle muscular dystrophy” often poses a formidable task for the clinician evaluating patients with proximal weakness. The differentiation of patients with LGMD from other forms of dystrophy, such as scapuloperoneal or facioscapulohumeral muscular dystrophy, from acquired autoimmune myopathy, as seen in polymyositis, from metabolic myopathies, such as late-onset Pompe’s disease, or from primary neurogenic disorders, such as type 3 spinal muscular atrophy, taxes the clinician’s diagnostic skills. Complementary studies, including serum creatine kinase levels, electrodiagnostic examination of nerve conduction and needle electromyography studies, and muscle biopsy interpretation, often fail to produce a definitive diagnosis. Even the best-equipped centers specializing in the diagnosis and treatment of the LGMD patient fail to arrive at a specific diagnosis in 25 percent or more of referred patients. In a study of 266 muscle biopsies supplemented with 121 Western blotting studies performed in major neuromuscular centers in the United States on patients with undefined LGMD, 12 percent of patients proved to have calpainopathy, 18 percent dysferlinopathy, 15 percent sarcoglycanopathy, 15 percent dystroglycanopathy, and 1.5 percent caveolinopathy [Moore et al., 2006]. The detail with which muscle biopsy tissue is processed varies tremendously among hospital and academic laboratories and all too often produces nondiagnostic or even misleading interpretations. Given the huge increase in knowledge of the molecular genetic underpinnings of many genetic neuromuscular diseases, the logical next step will be the utilization of the newest and most efficient gene sequencing technologies to screen for dozens of neuromuscular diseases concurrently.
Limb Girdle Dystrophy 1B: Lamin A/C
The reader is referred to the section of this chapter on Emery–Dreifuss muscular dystrophy.
Limb-Girdle Muscular Dystrophy 1C: Caveolinopathy
Caveolins are small transmembrane proteins with intracellular domains that undergo extensive oligomerization to form membrane complexes known as caveolae. Skeletal muscle disorders result from mutations on chromosome 3p25 in the gene that encodes caveolin-3, which interacts with neuronal nitric oxide synthase and the dystrophin-associated protein complex via the intracellular portion of β-dystroglycan [Song et al., 1996; Tang et al., 1996]. During muscle differentiation, caveolin-3 is associated with developing T tubules [Parton et al., 1997]. In the original reports of caveolinopathy, the disorders resulted from heterozygous changes in the membrane-spanning domain, which produced an autosomal-dominant LGMD phenotype [Minetti et al., 1998]. The clinical features of subsequently reported cases have been heterogeneous, and too few cases have been reported to define a predominant phenotype. Symptoms can begin in childhood with slowly progressive weakness. In a recent series of patients seen in the United Kingdom, myalgia and rippling muscle disease were the most common symptoms, often occurring in the first decade [Aboumousa et al., 2008]. Calf hypertrophy is present, and creatine kinase levels are elevated 4- to 20-fold. Other families may manifest only elevated serum creatine kinase levels (hyper-creatine kinase-emia) and postexertional myalgia and cramping, which may also begin in the first decade [Herrmann et al., 2000; Merlini et al., 2002]. Caveolin-3 mutations have been defined in autosomal-dominant rippling muscle disease [Betz et al., 2001], and severe rippling muscle symptoms have been reported in two unrelated patients with homozygous caveolin-3 mutations [Kubisch et al., 2003]. A case of distal myopathy has been described [Tateyama et al., 2002]. The broad spectrum of clinical symptoms and the possibility that spontaneous or germline mutations produce simplex cases frequently raise the possibility of caveolinopathy. The postulated dominant negative effect of a number of caveolin-3 mutations leads to a substantial or complete loss of caveolin-3 immunoreactivity, allowing for screening with immunohistochemical techniques [Carbone et al., 2000; Herrmann et al., 2000].
Limb-Girdle Muscular Dystrophy 2A: Calpainopathy
Calpainopathy is a relatively frequent, childhood-onset LGMD that was fully characterized, and the molecular and biochemical abnormalities were described, in 1995 [Chiannilkulchai et al., 1995; Richard et al., 1995]. Calpainopathy has been reported as the most frequent autosomal-recessive LGMD in several series [Chae et al., 2001; Dincer et al., 1997; Richard et al., 1997; Topaloglu et al., 1997]. The abnormal protein, calpain-3, is a nonlysosomal intracellular, muscle-specific, Ca2+-activated neutral protease [Johnson, 1990]. Before recognition of this new LGMD, the focus on calpain-3 was in relation to its abnormal activation in DMD secondary to increased levels of intracellular Ca2+. Type 2A was the first form of LGMD identified that is caused by deficiency of a nonstructural protein; this deficiency emphasizes the role of abnormal molecular signaling as the basis for muscle cell degeneration, as discussed before for DMD.
Molecular Pathogenesis
The gene responsible for LGMD2A (CAPN3) is located at chromosome region 15q15.1–q21.1 [Chiannilkulchai et al., 1995], and consists of 24 exons extending over a genomic region of 50 kilobases [Richard et al., 1995]. It is expressed as a 3.5-kilobase transcript, a 94-kDa translated protein. More than 140 different mutations have been reported, including different nonsense, splice site, frameshift, or missense calpain-3 mutations, most of which are “unique” mutations [Saenz et al., 2005]. In addition, several ethnic groups have unique mutations that co-segregate with the disease [Canki-Klain et al., 2004; Chae et al., 2001; Chrobakova et al., 2004; Cobo et al., 2004; Fanin et al., 2004; Jia et al., 2001; Kramerova et al., 2004; Passos-Bueno et al., 1999]. In a large series, it was found that 87 percent of mutant alleles are concentrated in seven exons (exons 1, 4, 5, 8, 10, 11, and 21), and 61 percent correspond to only eight mutations, which indicates the regions to which future molecular analysis could be restricted [Fanin et al., 2004]. In patients in Brazil, most mutations are found in exons 1, 2, 4, 5, 11, and 22 [Zatz et al., 2003]. However, other investigators have not found the same deletion pattern [Saenz et al., 2005], which suggests that these hot spots could be related to confounder mutations in the area. Mutations can be homozygous or compound heterozygous. Homozygous null-null mutations appear to confer the most severe phenotype, in terms of earlier loss of ambulation, whereas a heterozygous missense mutation confers a milder phenotype [Saenz et al., 2005].
Calpain-3 is a muscle-specific protein that belongs to the calpain family of proteins. This protein has many unique features and is considered a “modulator protease.” Several lines of evidence suggest that calpain-3 is mostly in an inactive state in muscle and needs a signal to be activated. Activated calpain-3 has several relationships with important cytoskeleton proteins. Some of its substrates include titin and filamin C [Taveau et al., 2003]. The interaction of calpain-3 with these cytoskeletal proteins may modulate remodeling during myofibrillogenesis by affecting scaffolding of titin for sarcomere assembling [Kramerova et al., 2004], and by cleaving filamin C and inhibiting its regulation of sarcoglycan assembly [Guyon et al., 2003]. There is also evidence that, when calpain-3 is activated, its proteolytic activity leads to disruption of the actin cytoskeleton. Thus, calpain-3 appears to be a cytoskeleton modulator with an important role in muscle maturation [Spencer et al., 2002]. Other pathogenic hypotheses are proposed to explain why mutations in this gene might cause muscular dystrophy. Calpain-3 could have a protective effect and be involved in muscle detoxification, preventing degradation of the muscle fiber [Zatz et al., 2003]. It may also play a role in intracellular signaling pathways and its deficiency might be associated with myonuclear apoptosis by deregulation of the nuclear factor κβ pathway [Baghdiguian et al., 1999].
The majority of the LGMD2A mutations appear to affect domain–domain interaction, which may be critical in the assembly and activation of the multidomain calpain-3 [Jia et al., 2001]. Under normal circumstances, calpain-3 undergoes rapid autocatalytic activity after translation, [Taveau et al., 2003]. During the short time (less than 5 minutes in muscle cultures) that the protein is present, it is in a resting state, unless it is activated by signaling mechanisms [Rey and Davies, 2002]. The autocatalytic activity resides in a Ca2+-sensitive region between domains II and III of the protein. In the 20–40 percent [Fanin et al., 2003; Saenz et al., 2005] of patients with genetically proven LGMD2A, in which Western blots fail to reveal any protein abnormality, the mutations are in these autocatalytic regions (R490Q, R489Q, R490W missense mutations), resulting in a reduced autocatalytic activity by lowering the Ca2+ sensitivity [Fanin et al., 2003].
Clinical Manifestations
LGMD2A has a characteristic phenotype in about 65 percent of the genetically confirmed patients. Interfamilial and intrafamilial variability are not uncommon [Zatz et al., 2003]. The age at onset of symptoms is between 2 and 49 years; the mean age at presentation is 14 (±8) years [Saenz et al., 2005]. The disease causes symmetric weakness of the pelvic and shoulder girdle muscles. There is preferential involvement of the posterior compartment of the thighs and posterior calves that can be seen clinically and by magnetic resonance imaging (MRI) studies. Facial, oculomotor, and cardiac muscles are not affected. Scapular winging can be observed early [Beckmann and Bushby, 1996]. Contractures, if present, occur late in the disease and do not extend into the spine [Beckmann and Bushby, 1996]. Most patients become unable to walk about 25 years after symptom onset. The creatine kinase level is 5- to 20-fold elevated. However, occasional patients (reported in Brazil) have normal creatine kinase levels, and a normal creatine kinase value should not be an exclusion criterion. Cognition is not impaired. The clinical phenotype can be confused with that of LGMD2I (fukutin-related protein gene [FKRP]) or facioscapulohumeral muscular dystrophy because of the presence of scapular winging. However, the distribution of weakness and the muscle MRI findings are very characteristic and distinct from those of these two disorders [Mercuri et al., 2005].
Diagnosis
Diagnosis of a calpainopathy is based on a typical phenotype, a muscle biopsy sample revealing Western blot abnormalities, and, more precisely, use of commercially available genetic testing indicating two mutations on two alleles. The combination of a typical phenotype with an abnormal Western blot result in muscle biopsy samples (at least two abnormal bands) increases the probability that an affected patient has a calpainopathy to 90 percent [Saenz et al., 2005]. However, if one of these variables is missing, the probability is reduced to about 75 percent [Saenz et al., 2005]. False-negative reports with Western blots in 20–40 percent of these patients dictate that DNA sequencing has to be pursued when a calpainopathy is clinically suspected on the basis of the typical phenotype [Fanin et al., 2003; Saenz et al., 2005].
Muscle pathology findings are positive for necrosis, regeneration, altered myofibrillar architecture, increased numbers of centrally placed nuclei, fibrosis, fiber type I predominance, and normal immunoreactivity for dystrophin and α-sarcoglycan. Muscle biopsy samples in preclinical cases have exhibited an unusual pattern of isolated fascicles of degenerating fibers in an almost normal muscle. This pattern has not been seen in patients early in the disease course who are affected by other forms of LGMD, which suggests that a peculiar pattern of focal degeneration occurs early in calpainopathy, independently of the type of mutation or the amount of calpain-3 in the muscle [Vainzof et al., 2003].
Western blot analysis of muscle calpain-3 can reveal total, partial, or no deficiency, with no relation between protein expression and severity of phenotype [Saenz et al., 2005; Zatz et al., 2003]. Secondary reduction of calpain-3 can be seen in other conditions – specifically, dysferlinopathies and LGMD2I due to FKRP deficiency [Vainzof et al., 2000a].
Treatment is symptomatic. Corticosteroids have not been tried in a controlled study. Gene transfer utilizing AAV vectors showed evidence of therapeutic effect in a murine model of calpain-3-deficient myopathy [Bartoli et al., 2006].
Limb-Girdle Muscular Dystrophy 2B: Dysferlinopathy
Mutations in the dysferlin gene on chromosome 2p13 cause distinct phenotypes of muscular dystrophy: LGMD type 2B (LGMD2B), Miyoshi’s myopathy, and distal anterior compartment myopathy, which are known by the term dysferlinopathies. Dysferlinopathies represent 1–5 percent of recessively inherited LGMD and about 33 percent of distal LGMD. Although the distribution of weakness in Miyoshi’s myopathy and LGMD2B is different (distal and proximal, respectively), both syndromes manifest in the late teens, progress slowly, and are associated with high serum creatine kinase levels. Moreover, identical mutations can give rise to both phenotypes in the same family; therefore, environment or differences in genetic background might contribute to the differential expression of muscle weakness [Kawabe et al., 2004].
Molecular Pathogenesis
Molecular pathogenesis is an autosomal-recessive LGMD that is caused by mutations in the dysferlin gene on chromosome 2p13 [Bashir et al., 1996; Passos-Bueno et al., 1996a, 1996b]. In humans, the dysferlin gene encompasses 55 exons spanning more than 150 kilobases of genomic DNA [Aoki et al., 2001]. The dysferlin gene encodes a 230-kDa protein with widespread expression in tissues such as skeletal muscle, cardiac muscle, kidneys, placenta, lungs, and brain. Although dysferlin is expressed most abundantly in skeletal and cardiac muscles, there is little evidence for clinically significant cardiac muscle dysfunction in dysferlin-deficient patients [Choi et al., 2010]. Immunohistochemical studies revealed that, in skeletal muscle, dysferlin is located at the plasma membrane, as well as in cytoplasmic vesicles [Bansal et al., 2003].
The pathogenesis of this class of muscular dystrophy is distinct in that the defect lies in the maintenance, not the structure, of the plasma membrane. The mechanism of defective membrane repair has been comprehensively reviewed [Han and Campbell, 2007]. Histopathologic and immunohistochemical studies in these patients reveal absence of dysferlin [Matsuda et al., 1999]. In addition, a very active inflammatory and degenerative process is characteristic in this disease and often leads to a misdiagnosis of polymyositis. However, the immune response is distinct, with macrophages being more common than T cells; the presence of perivascular and interstitial infiltrate consisting of CD8+ and CD4+ cells, but no B cells; and over-expression of major histocompatibility complex class I on muscle fibers [Gallardo et al., 2001]. These findings differ from those seen in polymyositis but are closely similar to those in SJL/J mice (murine model of dysferlin deficiency); this emphasizes a direct relationship between absence of dysferlin and immune system abnormalities in muscle [Confalonieri et al., 2003], perhaps in response to an inefficient muscle membrane repair and regenerative system [Cenacchi et al., 2005].
Ultrastructural studies provide evidence for an abnormal membrane repair mechanism when dysferlin is absent. Electron microscopy studies demonstrate prominent aggregations of small vacuoles, likely derived from the Golgi apparatus in the subsarcolemmal region, consisting of empty, swollen cisternae. The sarcolemma has multiple small gaps and microvillus-like projections. The basal lamina is thick and has focal duplications, thus having a multilayered appearance [Cenacchi et al., 2005; Selcen et al., 2001]. These alterations are seen mostly over plasmalemmal defects. Characteristic of this disorder is the presence of one to several layers of small vesicles just beneath the sarcolemma consistent with a defective resealing mechanism that fails to repair the sarcolemma [Cenacchi et al., 2005].
Clinical Presentation
There are two common phenotypes, LGMD2B and Miyoshi myopathy, both manifesting in the second decade of life [Takahashi et al., 2003]. Two other phenotypes, distal anterior compartment type and scapuloperoneal type, are seen especially early in the clinical course [Ueyama et al., 2002]. Patients undergo normal early developmental milestones without weakness in childhood; many dysferlin-deficient teenagers are competent athletes. The onset of slowly progressive muscle weakness and wasting usually begins in late adolescence and young adulthood [Mahjneh et al., 2001]. The LGMD2B phenotype is a predominantly proximal muscular dystrophy. Affected individuals have onset of pelvic girdle muscle weakness, with lower limbs abducted and externally rotated, and hyperlordosis as a result of hip muscle weakness. The anterior muscles of the distal legs and distal arms are relatively normal in these patients, even at the later stages of the disease. In contrast, the Miyoshi’s myopathy phenotype is a predominantly distal muscular dystrophy, with early involvement of the posterior compartments of the lower limbs; a common early symptom is the inability to stand on tiptoes. These patients manifest hyperextension of the lower limbs, caused by weakness of the quadriceps and gastrocnemius muscle. Physical examination reveals extensive wasting of the distal posterior muscles of the leg and, later, of the thigh [Ueyama et al., 2002]. Although previously described mainly in Japan, Miyoshi myopathy has been increasingly recognized in Western countries. Ten years after the onset of the disease, pronounced gait abnormalities are present in both the LGMD2B and the Miyoshi myopathy phenotypes; the foot leaves the ground with a double flexion of the knee and the hip. At this stage, upper-extremity weakness is also evident and at times has a distal pattern. Laboratory evaluation demonstrates markedly increased serum creatine kinase levels.
There is no specific treatment for this disorder, and management is in the realm of rehabilitation medicine. Ankle-foot orthosis can improve gait when footdrop is pronounced. Despite the pronounced inflammatory response in muscle biopsy samples, steroids are not beneficial and might worsen the disorder. Murine models of dysferlin deficiency have shown benefit from dual adeno-associated vector-mediated gene transport [Lostal et al., 2010].
Diagnosis
The diagnosis of dysferlinopathy is based on the absence of dysferlin expression on immunoblots or cryostat sections, and on exclusion of dystrophinopathy, sarcoglycanopathy, calpainopathy, and calveolinopathy, because dysferlin deficiency is rarely secondary in other myopathies. However, severe reduction of dysferlin expression is seen only in Miyoshi myopathy or LGMD2B [Anderson et al., 2000]. The diagnosis of dysferlinopathy can also be made by measuring dysferlin expression in peripheral blood mononuclear cells by immunoblot analysis, which has excellent correlation with muscle biopsy findings [Ho et al., 2002]. DNA testing is available commercially.
Limb-Girdle Muscular Dystrophies 2C, 2D, 2E, and 2F: Sarcoglycanopathies
The sarcoglycanopathies are a family of autosomal-recessive disorders that often manifest with an early and severe phenotype. The four sarcoglycans constitute a tetrameric complex of membrane proteins that contributes to the stability of the plasma membrane cytoskeleton and facilitates the association of dystrophin with the dystroglycans. Four sarcoglycan genes (α, β, γ, and δ) are related to each other structurally and functionally, but each has a discrete chromosomal location. Mutations in each gene may produce partial or complete loss of the entire complex [Nigro et al., 1996b]. Patients with sarcoglycanopathies were initially distinguished from children with a severe Duchenne-like presentation who proved dystrophin-positive and in whom autosomal-recessive inheritance was likely [Zatz et al., 1989]. The term severe childhood autosomal-recessive muscular dystrophy (SCARMD) was used to describe the condition of these children; some of them were later found to have a deficiency of a 50-kDa dystrophin-associated protein, the gene for which was localized to chromosome 13q12, the locus for γ-sarcoglycan [Azibi et al., 1993; Matsumura et al., 1992]. Sister proteins were identified [Mizuno et al., 1994], and the phenotypes associated with sarcoglycanopathies broadened to include less severely involved children. Mutations in each of the four sarcoglycans are found worldwide. In outbred populations, such as those in Europe, North America, and Brazil, the relative frequencies of symptomatic mutations of the four genes are, from highest to lowest, α, β, γ, and δ, in an 8:4:2:1 ratio [Duggan et al., 1997]. In other populations, this ratio may vary. A founder mutation, Thr151Arg, in β-sarcoglycan, has been identified in the Indiana Amish community [Duclos et al., 1998]. Specific mutations of γ-sarcoglycan cause the majority of cases of LGMD among Gypsies and persons in northern Africa [Merlini et al., 2000; Noguchi et al., 1995], and a specific mutation of δ-sarcoglycan is seen in a relatively higher frequency in African Brazilians [Nigro et al., 1996a; Vainzof et al., 1999]. Although the worldwide epidemiology of sarcoglycanopathies has not been extensively studied, data obtained in northern Italy suggest an incidence of 5.6 per million [Fanin et al., 1997].
In every child evaluated for sarcoglycanopathy, a primary dystrophinopathy must be excluded. Fewer than 5 percent of males with a Duchenne/Becker phenotype have a sarcoglycanopathy or other LGMD. Young females with a Duchenne/Becker phenotype are nearly as likely to be carriers of a dystrophinopathy as to have an alternative diagnosis [Hoffman and Clemens, 1996]. The majority of dystrophin-positive children presenting with an early-onset Duchenne-like phenotype have a sarcoglycanopathy [Duggan et al., 1996; Vainzof et al., 1999]. In contrast, only about 10 percent of patients with a more benign phenotype evolving in adolescence or young adulthood manifest a sarcoglycanopathy.
Immunostaining of muscle with antisarcoglycan antibodies is the standard screening for new cases of sarcoglycanopathy. Muscle biopsy samples from patients with primary dystrophinopathy reveal reduced sarcoglycan immunoreactivity. Therefore, both dystrophin and sarcoglycan immunostaining must be performed on the same muscle tissue. The finding of normal dystrophin staining and deficiency in immunostaining of any of the sarcoglycans suggests that a sarcoglycan mutation is present. Many laboratories perform immunostaining only for α-sarcoglycan, relying on a reduction or absence of immunoreactivity to reflect a mutation in any of the four sarcoglycans. Because the entire sarcoglycan complex tends to be affected as a unit in response to mutations in any of the four components, this is usually adequate. However, staining with antibodies to all four sarcoglycans can provide clues regarding the protein that reflects the primary mutation. This information helps direct DNA testing for definitive diagnosis of the specific sarcoglycan mutation. On occasion, immunoreactivity of components within the sarcoglycan complex appears normal in the presence of a documented reduction in immunostaining of another component [Higuchi et al., 1998; Vainzof et al., 2000b]. Gamma-sarcoglycanopathy, in particular, may produce a marked reduction in γ-sarcoglycan immunoreactivity with relatively good preservation of α, β, and δ-sarcoglycan staining [Nowak et al., 2000; Vainzof et al., 1996]. The pathophysiology of sarcoglycanopathies relates to that of dystrophinopathies, discussed in detail earlier in this chapter. DNA testing for mutations of the four major sarcoglycan species is commercially available and should be performed for confirmation of a specific sarcoglycanopathy, and as a primary diagnostic investigation for a child with a Duchenne phenotype whose muscle biopsy reveals normal dystrophin expression.
Gamma-Sarcoglycanopathy (Limb-Girdle Muscular Dystrophy 2C)
The gene for γ-sarcoglycan is found on chromosome 13q12. It codes for a 35-kDa protein expressed predominantly in skeletal muscle. In addition to its role as a stabilizer of the dystrophin-associated complex, γ-sarcoglycan binds γ-filamin/filamin C [Noguchi et al., 1995]. Minor expression in smooth muscle occurs, as well [Barresi et al., 2000]. The most common γ-sarcoglycan mutation is the deletion of a single thymidine residue, del52IT [Noguchi et al., 1995], which originated in northern Africa as a founder mutation. This mutation produces a truncated protein. However, the clinical severity is variable, even within families, ranging from a Duchenne-like to a mild Becker phenotype [McNally et al., 1996a, 1996b]. The mean age at symptom onset is 5 years. The distribution of weakness is most notable in the glutei and thigh adductors. Upper-extremity weakness usually begins in the periscapular muscle and later involves the deltoid and biceps muscles. In contrast to DMD, the quadriceps tends to be spared and is usually as powerful as the hamstrings. Calf and tongue muscle hypertrophy is common. Cardiac problems occur late in the disease, with evidence of right ventricular hypertrophy on electrocardiograms and asymptomatic ventricular dysfunction [Melacini et al., 1999]. Cognition is normal. Mild sensorineural hearing loss may evolve. Loss of independent ambulation occurs at a mean age of 16 years (range, 10–37 years). Orthopedic problems include progressive lordosis. Surgical stabilization of the spine is rarely needed.
Laboratory studies reveal gross elevations of creatine kinase level, usually more than tenfold normal. Muscle biopsy samples yield dystrophic findings of degeneration and regeneration, and occasional foci of inflammation. Immunostaining of sarcoglycans with a dystrophin-immunostaining control sample is the screening procedure of choice, complemented by DNA studies. Patients with γ-sarcoglycan can have near-normal α-sarcoglycan immunostaining, which yields a false-negative muscle biopsy result [Nowak et al., 2000].
Alpha-Sarcoglycanopathy (Limb-Girdle Muscular Dystrophy 2D)
Alpha-sarcoglycan is encoded on chromosome 17q12–q21. The 50-kDa protein, originally called adhalin, is expressed only in skeletal and cardiac muscle. Most mutations are missense in the extracellular domain [Piccolo et al., 1995]. Worldwide, α-sarcoglycanopathy is the most common of the sarcoglycan disorders, accounting for over 50 percent of patients [Hayashi and Arahata, 1997]. The Arg787Cys missense mutation is the most common, accounting for one-third of all cases of α-sarcoglycanopathy [Carrie et al., 1997]. Null mutations are associated with total absence of α-sarcoglycan and often confer a severe phenotype, with onset of symptoms in the first 3 years of life [Duggan et al., 1997].
Onset of motor problems occurs between 2 and 15 years; earlier onset is predictive of more rapid deterioration and low or absent α-sarcoglycan. In patients with late onset, ambulation may be preserved for decades. Occasional patients manifest a high creatine kinase level in the absence of weakness. The distribution of weakness, atrophy, and hypertrophy is similar to that described for γ-sarcoglycan, but the quadriceps tends to be involved in patients with severe, early-onset disease. The phenotype varies from a severe Duchenne phenotype to a mild Becker phenotype. Early scapular weakness is common. Symptomatic cardiomyopathy is rare, although evidence of cardiac dysfunction is present in about 30 percent of patients [Melacini et al., 1999]. Cognition is normal. Serum creatine kinase levels are high, usually over 5000 IU.
Small therapeutic trials of corticosteroids have been conducted, with reports of benefits similar to those seen in steroid-treated DMD patients [Connolly et al., 1998]. Anecdotes of a decrease in exercise-induced myalgia with prednisone therapy have been shared (Robert Leshner-personal observation).
Beta-Sarcoglycanopathy (Limb-Girdle Muscular Dystrophy 2E)
Beta-sarcoglycan is coded on chromosome 4q12. It is a 43-kDa protein that is expressed not only in skeletal and cardiac muscle but also in brain and kidney tissue. Worldwide, it accounts for about 25 percent of reported cases of sarcoglycanopathy. However, certain populations have a much higher incidence of β-sarcoglycan mutations, including the Indiana Amish [Duclos et al., 1998].
Creatine kinase levels are approximately 5000 IU. Muscle biopsy specimens usually reveal absence of all sarcoglycan immunostaining, and there is occasional reduction of dystrophin immunostaining as well. A knockout mouse model of β-sarcoglycanopathy has been developed, and attempts at gene transfer have been initiated [Dressman et al., 2002]. Corticosteroid treatment produced clinical improvement in two siblings with β-sarcoglycanopathy [Wong-Kisiel and Kuntz, 2010].
Delta-Sarcoglycanopathy (Limb-Girdle Dystrophy 2F)
The gene for δ-sarcoglycanopathy in chromosome region 5q33–q34 codes for a 35-kDa protein localized to the subsarcolemmal region of skeletal and cardiac muscle. It is the rarest of the sarcoglycanopathies. Missense and nonsense mutations are reported, as is a frameshift mutation that produces a premature truncation of protein found in affected Brazilian families [Nigro et al., 1996a]. The clinical phenotype is universally severe, with onset of symptoms occurring between 2 and 10 years of age, and progression to full-time wheelchair use by the mid-teens. Most patients do not survive their second decade. A dilated cardiomyopathy may occur with or without skeletal myopathy [Politano et al., 2001; Tsubata et al., 2000]. Corticosteroid therapy has not been systematically investigated; steroid treatment of a δ-sarcoglycan-deficient mouse model produced worsening myocardial function [Bauer et al., 2008] and needs further evaluation before human trials are considered.
Disorders of α-Dystroglycan Glycosylation
Limb Girdle Muscular Dystrophy 2I: Fukutin-Related Protein Deficiency
LGMD2I is a clinically heterogeneous muscular dystrophy whose causative gene was discovered in 2001 [Brockington et al., 2001b]. Since then, it has become apparent that this is one of the most common forms of LGMD, representing from 11 percent [Zatz et al., 2003] to 19 percent [Krasnianski et al., 2004; Walter et al., 2004] of all cases of LGMD in different series.
Molecular pathogenesis
The disease is caused by a mutation in the fukutin-related protein (FKRP) gene in chromosome 19q13.3 [Brockington et al., 2001a]. The most common mutation is a single point (826C>A) missense mutation in one allele, seen in 90 percent of patients, producing a Leu276Ileu substitution [Brockington et al., 2001b]. Disease severity correlates with a mutation on the second allele; consequently, patients with homozygous mutations causing a Leu276Ileu exhibit a less severe phenotype [Brockington et al., 2002; Mercuri et al., 2003]. Patients heterozygous for this mutation usually have a second heteroallelic mutation on the other allele [Walter et al., 2004]. This disorder is allelic to congenital muscular dystrophy with muscle hypertrophy and a normal central nervous system (merosin-positive congenital muscular dystrophy type [MDC1C]) [Brockington et al., 2001a].
FKRP is a ubiquitous protein with highest levels in skeletal muscle, heart, and placenta tissue. FKRP localizes to rough endoplasmic reticulum and has glycosyltransferase activity [Matsumoto et al., 2004]. FKRP and fukutin are targeted to the medial Golgi apparatus through their N-termini and transmembrane domains [Esapa et al., 2002]. Together with fukutin, FKRP appears to be involved in the initial steps of O-mannosylglycan synthesis of α-dystroglycan [Matsumoto et al., 2004].
Clinical manifestations
Age at onset ranges from 0.5 to 27 years; 61 percent of patients present before age 5 years. Patients present with either a DMD phenotype (heterozygous mutations) or, with a later onset, a milder limb-girdle syndrome (homozygous mutations). The most severely affected patients present in the first year of life with profound weakness and a congenital muscular dystrophy phenotype. The clinical course varies, usually involving weakness and wasting of the shoulder girdle muscles and proximal extremities, with significant calf hypertrophy and elevated serum creatine kinase levels, increases of the latter ranging from 5- to 70-fold. There is a characteristic predilection for muscles of the lower extremities, with weakness of hip flexion and adduction, knee flexion, and ankle dorsiflexion being more predominant [Fischer et al., 2005]. Other clinical manifestations have been described, including exercise-induced myalgia, in addition to weakness and isolated myalgia, cramps, elevated serum creatine kinase levels, and dilated cardiomyopathy without muscle weakness [Krasnianski et al., 2004]. In some series, most patients presented with muscle pain and myoglobinuria as the earliest symptoms [Walter et al., 2004]. Cardiac and respiratory involvement are relatively common in children with earlier symptom onset, occurring in about 30 percent of patients, and (in contrast to DMD) occasionally while they are still ambulatory [Mercuri et al., 2003; Poppe et al., 2003].
Diagnosis
Methods for genetic sequencing of the FKRP gene from peripheral blood are commercially available, allowing rapid confirmation of the diagnosis. Muscle biopsy samples reveal characteristic dystrophic changes, with muscle fiber size variation, muscle fiber necrosis and regeneration, and mild increase in connective tissue. An important finding is secondary abnormal laminin-2 immunostaining. In evaluating children with a congenital muscular dystrophy phenotype, distinction between FKRP deficiency and laminin α2 deficiency may require DNA testing. Most patients also have a marked decrease in immunostaining of muscle α-dystroglycan and a reduction in its molecular weight on Western blot analysis [Brockington et al., 2001a, 2001b].
Muscle MRI demonstrates a distinct and consistent pattern of muscular involvement in LGMD2I that is correlated with the pattern of muscle weakness in these patients. Predominant weakness of shoulder adduction and internal rotation, elbow flexion, hip flexion and adduction, knee flexion, and ankle dorsiflexion, often independent of the disease duration and the level of clinical severity, is corroborated by the findings on muscle MRI analysis [Fischer et al., 2005]. This pattern is very similar to that of LGMD2A (calpain-3 deficiency), but distinct from dysferlinopathies, sarcoglycanopathies, and dystrophinopathies. In LGMD2I, marked hyperintense signal changes on T1-weighted images can be seen in the adductor and posterior thigh muscles that exceed the changes in the abductor and anterior thigh muscles. Hypertrophy of the gracilis and sartorius muscle is a frequent finding. Furthermore, the degree of signal abnormalities in affected muscles mirrors the degree of muscle weakness in individual patients [Fischer et al., 2005].
Limb Girdle Muscular Dystrophy 2K (POMT1 Deficiency), Limb Girdle Muscular Dystrophy 2M (Fukutin Deficiency), and Limb Girdle Muscular Dystrophy 2N (POMT2 Deficiency)
Autosomal-recessively inherited limb girdle muscular dystrophies K, M, and N are due to mutations in genes that previously have been recognized in more severe clinical phenotypes that will be discussed in the section on congenital muscular dystrophies. All three genes are involved in the O-mannosyl-glycosylation of α-dystroglycan. The severe congenital dystrophy phenotypes are associated with brain malformation and cognitive impairment, giving rise to the classification of these disorders as “syndromic congenital muscular dystrophies.” Creatine kinase levels are grossly increased in all three disorders. Milder phenotypes have been reported, conforming to an LGMD phenotype without structural brain abnormalities; this expands the spectrum of clinical phenotypes and prompts the designation of LGMD2K, M, and N. LGMD2M (Fukutin deficiency) warrants special mention because of significant improvement in motor deficits with corticosteroid therapy [Godfrey et al., 2006].
Sarcomeric Protein Deficiencies
Limb-Girdle Muscular Dystrophy 1A, 2G, and 2J
Limb-girdle muscular dystrophy 1a (myotilinopathy)
LGMD1A was the first autosomal-dominant limb-girdle dystrophy to be linked to a specific gene locus. Initial clinical observations of the co-segregating of two rare diseases, muscular dystrophy and Pelger–Huët syndrome [Schneiderman et al., 1969], in a kindred were followed years later by linkage studies localizing the gene to chromosome region 5q31–q33 [Yamaoka et al., 1994], and the discovery of the first of several missense mutations in the gene coding for the sarcomeric protein myotilin [Hauser et al., 2002].
Myotilin localizes to the Z-disk and interacts with α-actinin and F-actin, efficiently cross-linking thin filaments. This protein also interacts with other filaments, including filamin C. in vitro studies suggest that the expression of myotilin in muscle cells is tightly regulated to become active in the later stages of in vitro myofibrillogenesis, when preassembled myofibrils begin to align. Thus, myotilin appears to have an indispensable role in stabilization and anchorage of the thin filaments that may be needed for correct Z-disk orientation [Salmikangas et al., 2003]. The mutations observed in LGMD1A do not disrupt binding of myotilin to α-actinin, but result in Z-disk streaming.
Myotilin has been implicated in other diseases, including a causal role in some cases of myofibrillar myopathy [Selcen and Engel, 2004; Selcen et al., 2004], and a secondary role in central core disease and nemaline myopathy [Schroder et al., 2003].
Missense mutations underlie the pathophysiology in the initially reported family and the seven additional families expressing this disorder. Patients in the initial kindred had symptoms of proximal weakness in young adulthood, with a mean age at onset of 27 [Hauser et al., 2000]. A suggestion of anticipation has been reported, with a reduction in age at onset by 13 years from one generation to the next, an observation uncommon in diseases not caused by nucleotide repeat expansions. About half of patients with LGMD1A develop nasal dysarthria. Ankle contractures are common. Progression of the disease is slow, and ambulation is maintained for decades. Creatine kinase level elevations from 2- to 10-fold are common. Electrodiagnostic study findings are consistent with a myonecrotic myopathy. Cardiac involvement occurs in about half of the patients but is rarely debilitating. Muscle biopsies reveal Z-line streaming, muscle fiber degeneration, fiber splitting, and central nucleation. Rimmed autophagic vacuoles are reported [Hauser et al., 2002].
Limb-girdle muscular dystrophy 2g (telethoninopathy)
Telethonin is a small 19-kDa sarcomeric protein expressed in skeletal and cardiac muscle [Valle et al., 1997]. Its gene maps to chromosome region 17q11–q12, the candidate site to which samples from members of a Brazilian family with an early-onset LGMD phenotype had mapped [Moreira et al., 1997]. Subsequent studies of this and three other Brazilian families confirmed mutations of the telethonin gene [Moreira et al., 2000]. Telethonin has been implicated in myofibrillar assembly [Mason et al., 1999]. It is phosphorylated by the serine kinase domain of titin and may serve to cap titin in the Z-disk [Knoll et al., 2002]; telethonin is also known as T-CAP.
Patients with telethonin mutations have not been recognized outside Brazil. The onset of symptoms has occurred between 9 and 15 years of age. Weakness is mainly proximal, but of the 13 patients described, 7 had prominent distal weakness with lower-extremity atrophy and footdrop. Paradoxically, six patients had evidence of calf hypertrophy. Only one patient has remained independently ambulatory after age 43. Five of 9 of these original 13 patients studied for cardiac abnormality had mild cardiomyopathy. Creatine kinase levels were elevated 3- to 30-fold in all patients early in their disease. Muscle biopsy revealed degenerating and regenerating muscle fibers and rimmed vacuoles (an unusual finding for LGMD but not for distal myopathy syndromes) [Moreira et al., 1997]. Patients can be screened with immunohistochemical techniques through the use of antitelethonin antibodies. Mutation analysis is simplified because the telethonin gene contains only two exons. DNA testing is commercially available.
Limb-girdle muscular dystrophy 2j (titin)
Titin is a giant structural sarcomere protein with a molecular weight of over 3800 kDa. It is the largest human protein identified and forms the third filament system in striated muscle, along with actin and myosin. Single titin molecules span half sarcomeres from Z-disks to M-lines in skeletal and cardiac muscle. Titin contributes to sarcomere assembly and passive tension of myofibrils, and serves sensor and signaling functions [Granzier and Labeit, 2004; McElhinny et al., 2004]. Titin serine kinase phosphorylates telethonin, another sarcomeric protein that is implicated in LGMD2G. Mutations in the titin gene on chromosome 2q31 most often produce autosomal-dominant tibial muscular dystrophy, a distal muscular dystrophy of mid-adult life, with prominent involvement of the tibialis anterior and toe extensor muscles [Hackman et al., 2002]. This disorder is most commonly seen in persons of Finnish descent. Patients with mutations causing truncated titins have autosomal-dominant familial dilated cardiomyopathy without skeletal muscle disease [Gerull et al., 2002]. Homozygous mutations have produced an adult-onset limb-girdle disorder, LGMD2J [Hackman et al., 2003]. Severe calpain-3 deficiency is seen in patients with LGMD2J and may be an important downstream pathway [Haravuori et al., 2001]. A review of 207 patients heterozygous for the specific C-terminal M-line titin mutation illustrated variability of clinical expression, including one child with infantile-onset generalized weakness and one with onset of an LGMD phenotype at age 6 [Udd et al., 2005]. DNA testing is commercially available.
Limb Girdle Muscular Dystrophy 2H (Tripartite Motif-Containing Gene 32)
This rare disorder was described in the Hutterites of Manitoba, Canada, and genetic linkage was established to chromosome 9q31–34, with later definition of a homozygous missense mutation (Asp487Asn) in the E3 ubiquitin ligase TRIM32 gene. TRIM32 is responsible for protein–protein interaction and the labeling of proteins with ubiquitin for proteosome-mediated degradation [Saccone et al., 2008]. The disorder is allelic to sarcotubular myopathy [Schoser et al., 2005; Borg et al., 2009]. Clinical symptoms may begin as early as 8 years, but may not appear until the late 20s. Progression is slow and most patients remain ambulatory for decades. Weakness begins in the pelvic girdle, and the patient has a waddling gait and, occasionally, back pain and fatigue. Serum creatine kinase values are increased 2–30 times normal. Muscle biopsy reveals nonspecific dystrophic changes. Electrocardiographic abnormalities may be seen but clinical cardiomyopathy has not been reported. DNA testing is commercially available.
Limb Girdle Muscular Dystrophy 2L (Anoctamin 5 Deficiency)
Anoctamin (ANO) proteins are a family of ten species, several of which are associated with calcium-activated chloride channels. Mutations in the ANO5 gene, located on chromosome 11.p14.3, have been identified very recently as being responsible for producing a proximal LGMD in three French Canadian families, and a distal non-dysferlin Miyoshi-like myopathy in Dutch and Finnish families [Bolduc et al., 2010]. The mutations identified were splice-site mutations and a single-base pair duplications, all of which resulted in frameshift and introduction of premature stop codons. Missense mutations were identified less commonly. The limb girdle phenotype is characterized by proximal weakness, with prominent asymmetric weakness and atrophy of the quadriceps femoris and weakness of the biceps brachii. The distal myopathy presentation is similar to that seen with dysferlin deficiency, with prominent weakness of calf muscles. The mean age of onset of symptoms is 33, but may be as early as 11 years; males seem to be more affected than females. DNA testing is commercially available.
X-linked Syndromes
McLeod’s Cardiomyopathy
McLeod’s cardiomyopathy is a dilated cardiomyopathy with limited findings for myopathy, linked to chromosome Xp21.1 as a recessive condition involving Kx membrane transport protein. Muscle weakness is mild. Central nervous system involvement includes chorea, seizures, and cerebral and caudate nucleus atrophy [Malandrini et al., 1994]. Acanthocytosis is present, and erythrocytes have abnormal expression of Kell blood group and Kx surface antigens. Creatine kinase levels tend to be high.
Emery–Dreifuss Muscular Dystrophy
Overview
Emery–Dreifuss muscular dystrophy (EDMD) was recognized as a “distinct” phenotype in the mid-1960s [Emery and Dreifuss, 1966]. EDMD is characterized by a clinical triad of early contractures, muscle wasting and weakness in a humeroperoneal distribution, and cardiomyopathy. Considerable phenotypic variability exists regarding age at onset and rate of progression. X-linked transmission and autosomal-dominant transmission are recognized. Three genes have been identified to date and more are likely to be defined.
Molecular Biology
Both X-linked and autosomal-dominant EDMD result from mutations in genes coding for nuclear envelope proteins. The X-linked form of EDMD is most often caused by mutations of the STA gene, located at Xq28, which encodes a 34-kDa ubiquitously expressed nuclear envelope protein, emerin [Bione et al., 1994]. Of mutations producing X-linked emerin-deficient EDMD, 95 percent are null mutations associated with a complete absence of emerin in skeletal muscle, as well as in smooth muscle, skin fibroblasts, leukocytes, and exfoliative buccal cells. A mosaic pattern is seen in female carriers, and the diagnosis of X-linked EDMD in males and identification of female heterozygotes are therefore possible with immunohistochemical studies of muscle and more easily accessed tissue [Emery, 2000; Manilal et al., 1997]. A second X-linked disorder producing EDMD has recently been identified: four-and-a-half LIM domains protein 1 (FHL1), located on Xq27.2 [Gueneau et al., 2009]. The FHL1 variant of EDMD has been studied in three families who proved negative for mutations in the emerin and lamin A/C genes. The clinical picture is a scapuloperoneal distribution of weakness, accompanied by joint contractures and cardiomyopathy, and characterized by conduction defects, arrhythmias, and hypertrophic cardiomyopathy. FHL1 protein deficiency appears to impair myotube formation.
Autosomal-dominant EDMD and, very rarely, autosomal-recessive EDMD [Raffaele Di Barletta et al., 2000] result from mutations of the LMNA gene, located on 1q21, which encodes two nuclear envelope proteins, lamins A and C [Bonne et al., 1999]. Lamins A and C are alternatively spliced products of the same gene and constitute part of the nuclear lamina; they interact with chromatin and other proteins of the inner nuclear membrane, including emerin [Gruenbaum et al., 2003]. Mutations of the lamins A and C genes that produce the autosomal-dominant EDMD phenotype are missense mutations that allow translation of full-length lamins A and C. Immunostaining of lamins A and C is therefore not reliable for confirmation of the diagnosis of autosomal-dominant EDMD [Emery, 2000]. The diagnosis of lamin A or C disorders requires DNA sequencing or mutation scanning [Bonne et al., 2000, 2003; Brown et al., 2001]. The EDMD phenotype and its variants have focused attention on the importance of nuclear envelope proteins [Nagano and Arahata, 2000], but the pathophysiologic mechanisms of the diseases remain conjectural.
Clinical Features
Although X-linked EDMD and autosomal-dominant EDMD usually share a similar phenotype, wide clinical variation with poor genotype–phenotype correlation has been documented in both X-linked EDMD and autosomal-dominant EDMD [Bonne et al., 2000; Fujimoto et al., 1999; Muntoni et al., 1998]. Inter- and intrafamilial phenotypic variability may exist in patients sharing identical mutations.
The onset of contractures occurs early in the disease, in the first or second decade, and often precedes clinically significant weakness. Contractures are most prominent at the elbows, Achilles tendons, and posterior cervical muscles [Emery, 1989] (Figure 92-10 and Figure 92-11). Upper-extremity contractures often precede axial and lower extremity deformities. The arms are held in a semiflexed position (Figure 92-12). The feet are set in talipes equinus, often in association with toe walking. Posterior cervical contractures preclude full neck flexion (Figure 92-13; see also Figure 92-11). Contractures usually remain disproportionate to the degree of weakness [Emery, 2000] and may be the major factor in functional impairment. Muscle weakness is relatively mild and slowly progressive. The distribution of motor deficits is humeroperoneal, with upper-extremity weakness (in biceps, triceps, and spinatus muscles) occurring earlier than leg weakness (in tibialis anterior and peroneal muscles). Pseudohypertrophy is not seen, and its absence helps differentiate X-linked EDMD from dystrophinopathy (BMD) clinically.
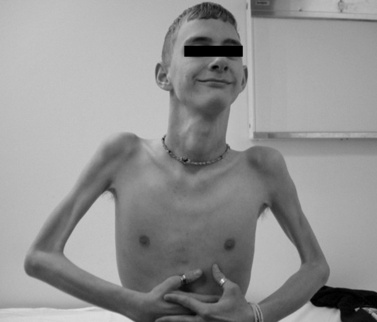
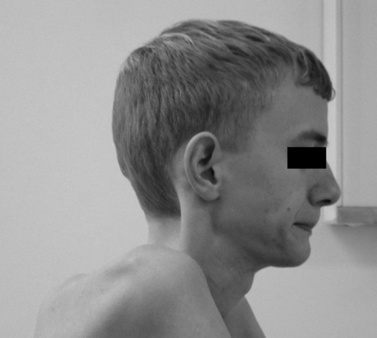
Fig. 92-11 The same male with X-linked Emery–Dreifuss muscular dystrophy.
Significant neck extensor contractures severely limit neck flexion.
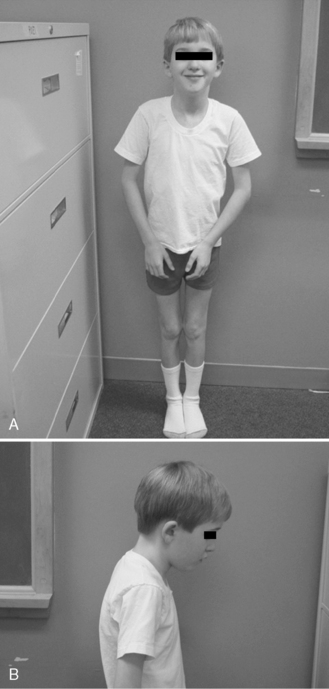

Cardiac symptoms may include palpitations, syncope, and diminished exercise tolerance. Supraventricular dysrhythmias, atrioventricular conduction block, ventricular dysrhythmias, and restrictive or dilated cardiomyopathy may evolve. The risk of ventricular dysfunction and dysrhythmias is greater in autosomal-dominant than in X-linked EDMD [Becane et al., 2000; Bonne et al., 2003], but symptomatic cardiomyopathy may occur in women heterozygous for null emerin mutations. Cardiac symptoms usually evolve after the second decade.
Phenotypic variability in emerin deficiency relates to the age at onset and disease progression in X-linked EDMD. Cardiomyopathy in FHL1 mutations may be due to deficits in biomechanical function and impaired mediation of cardiac myocyte hypertrophic biomechnical stress [Sheikh et al. 2008].
A broad spectrum of phenotypes has been discovered for mutations of the lamin A/C gene. Phenotype–genotype correlation of LMNA mutations is highly variable [Mercuri et al. 2004; Rankin and Ellard, 2006]. The LMNA gene encoding two nuclear envelope proteins (lamins A and C [lamin A/C]) maps to chromosome 1q21 and has been associated with multiple distinct pathologies. Skeletal and cardiac myopathy without early contractures characterizes autosomal-dominant LGMD1B (see Figure 92-13). Disorders of peripheral nerve, including an autosomal-recessive axonal form of Charcot–Marie–Tooth syndrome (type 2B1) and an autosomal-dominant peripheral neuropathy, are seen in other lamin A or C mutations [De Sandre-Giovannoli et al., 2002]. Dunnigan-type familial partial lipodystrophy [Shackleton et al., 2000] and generalized lipoatrophy with insulin resistance [Caux et al., 2003] have been reported. Autosomal-recessive mandibuloacral dysplasia, characterized by postnatal growth retardation, craniofacial abnormalities, and skeletal malformation, are “non-neuromuscular” syndromes due to alternate LMNA mutations. Premature aging syndromes, including Hutchinson–Gilford progeria [De Sandre-Giovannoli et al., 2003] and atypical Werner’s syndrome [Chen et al., 2003], widen the spectrum of laminopathies. De novo LMNA mutations have been identified in a newly recognized form of congenital muscular dystrophy characterized by severe, early-onset generalized weakness and profound cervicoaxial muscle weakness, producing a “dropped head” phenotype [Quijano-Roy et al., 2008].
Facioscapulohumeral Muscular Dystrophy
Overview
Facioscapulohumeral muscular dystrophy (FSHD) was described by Landouzy and Dejerine in 1885. This disease is inherited as an autosomal-dominant disorder with high penetrance and variable expression. Between 10 and 30 percent of cases represent new mutations [Zatz et al., 1995]. Prevalence is estimated at 1 per 20,000. Clinical signs and symptoms are usually present before age 20 years and are manifested by weakness of the facial muscles, fixators of the scapula, and dorsiflexors of the ankle. Severity is highly variable. Although skeletal muscle symptoms dominate the clinical picture, other systems are often involved, which implies a more widespread developmental/degenerative process.
Molecular Genetics
FSHD maps to chromosome 4q35 [Wijmenga et al., 1993]. More than 95 percent of patients with FSHD have a reduced number of 3.3-kilobase tandem repeat sequences termed D4Z4. The D4Z4 repeat region lies within a region of telomeric heterochromatin. There is no known gene within or telomeric to the D4Z4 repeat region, and the possible molecular biology underlining skeletal muscle and systemic abnormalities has been defined very recently [Lemmers et al., 2010]. An inverse correlation exists between the D4Z4 size and disease severity [Zatz et al., 1995]. Normally, there are 11–100 D4Z4 repeats at 4q35. Patients with FSHD have 1–10 repeats, as confirmed by restriction enzyme digestion. EcoRI-produced fragments are 34 kilobases or smaller (i.e., ten or fewer repeats) [Upadhyaya et al., 1997]. The largest deletions have been associated with severe, congenital-onset FSHD [Brouwer et al., 1994, 1995]. A large deletion, with a 10- to 13-kilobase remaining DNA fragment, usually produces symptoms by 16 years of age, and these may be severe. In some instances, this reflects a new mutation. An intermediate deletion with a remaining DNA fragment greater than 16 kilobases is associated with large families expressing the dominantly inherited condition beginning between ages 8 and 22 years. Small deletions are usually found in kindreds with disease of later onset, usually from 15 to 23 years. In these families, a few individuals carrying the small deletion appear phenotypically normal, which is suggestive of nonpenetrance. To express FSHD, the truncated D4Z4 repeat sequence must be coupled with a permissive chromosomal background: a specific 4qA haplotype. If the truncated repeat sequence is paired with a 4qB haplotype, FSHD will not evolve. In addition, at least one 4q35 D4Z4 microsatellite is required for disease expression. Investigations have shown that FSHD patients “carry specific single-nucleotide polymorphisms in the chromosomal region distal to the last D4Z4 repeat. This FSHD-predisposing configuration creates a canonical polyadenylation signal for transcripts derived from DUX4, a double homeobox gene of unknown function that straddles the last repeat unit and the adjacent sequence. Transfection studies revealed that DUX4 transcripts are efficiently polyadenylated and are more stable when expressed from permissive chromosomes. These findings suggest that FSHD arises through a toxic gain of function attributable to the stabilized distal DUX4 transcript” [Lemmers et al., 2010].
Commercially available testing of D4Z4 tandem repeats on chromosome 4q35 is the most specific and sensitive diagnostic test for FSHD. Subtelomeric translocations between chromosomes 4q35 and 10q26 occur relatively frequently in the general population and may complicate molecular diagnosis. A D4Z4 deletion results in disease if the deletion occurs on chromosome 4, but deletions on chromosome 10 have no clinical significance. Detection of a deletion in an individual with a 4–10 translocation must be interpreted with caution [van der Maarel et al., 1999; van Deutekom et al., 1996]. In 5 percent of FSHD patients, a 4q35 deletion is not identified, which suggests that at least one additional genetic disorder, designated facioscapulohumeral muscular dystrophy 2, produces a FSHD phenotype that is clinically indistinguishable from classic 4q35 FSHD [Yamanaka et al., 2004; de Greef et al., 2010].
Clinical Features
Clinical diagnostic criteria have been established by the Facioscapulohumeral Muscular Dystrophy Consortium [Tawil et al., 1998]. Inclusion criteria specify autosomal-dominant inheritance; bifacial weakness; and weakness of the scapular stabilizer muscles, ankle dorsiflexor muscles, or both. Supporting criteria include asymmetry of motor deficits (a finding far more common in FSHD than any other muscular dystrophy); sparing of deltoid, neck flexor, and calf muscles; and involvement of wrist extensors and abdominal muscles. Hearing loss involving high frequencies and retinal vasculopathy (Coats disease) are additional supportive findings. Exclusion criteria include eyelid ptosis, extraocular muscle weakness, skin rash, elbow contractures, cardiomyopathy, sensory loss, neurogenic changes documented in muscle biopsy, and myotonia or neurogenic motor unit potentials documented on needle electromyography.
Weakness is usually appreciated first in the facial muscles. Patients have difficulty with puckering the lips, whistling, sipping through a straw, or blowing up balloons because of weakness of the orbicularis oris muscle. Most patients have a transverse smile and limited facial expression. Weakness of the orbicularis oculi is usually asymptomatic, but the examiner can recognize facial muscle weakness by observing incomplete burying of the eyelashes with forced eyelid closure and the ease with which the closed eyelids can be pried apart. A history of sleeping with the eyes open may be offered by a parent or spouse. Facial weakness may be asymmetric (Figure 92-14 and Figure 92-15).
Weakness of scapular fixation is evidenced by scapular winging, accentuated by arm extension (Figure 92-16). The scapulae ride high on the back, producing the illusion of hypertrophied trapezius muscles (Figure 92-17). Arm abduction is impaired in the presence of normal power and bulk of the deltoid muscles. Wasting of the biceps and triceps with preservation of deltoid and forearm muscles yields a “Popeye” configuration to the arms. Wasting of the clavicular head of the pectoral muscles produces a reversal of the axillary folds with a deep upward slope. Weakness of lower abdominal muscles may result in a “pot belly” when standing and a positive Beevor sign (cephalad movement of the umbilicus with neck flexion) when the patient lies supine.
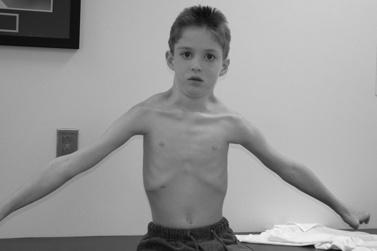
Motor deficits are slowly progressive. Some patients report long periods of clinical stability, punctuated by abrupt stepwise loss of power in restricted muscle groups. Asymmetry of weakness and muscle atrophy is the rule and may be striking. Most patients remain functionally independent throughout life. About 20 percent of FSHD patients (usually those with early disease onset) eventually require a wheelchair. Life expectancy is normal. Men afflicted with FSHD 1 appear to be more severely affected than women, and greater penetrance of the disorder has been documented in male patients [Zatz et al., 1998]. Sensorineural hearing loss is frequently documented, especially in children presenting before 10 years of age [Takeya et al., 1990; Voit et al., 1986], but can often be asymptomatic. Retinal vasculopathy with telangiectasia and retinal detachment (Coats disease) may develop, especially in patients with early childhood symptom onset [Fitzsimons et al., 1987; Gurwin et al., 1985; Padberg et al., 1995]; regular ophthalmology evaluations are warranted.
Congenital onset of FSHD has been associated with mental retardation and epilepsy in children with the largest D4Z4 deletions (EcoRI fragment size of 10 kilobases: i.e., fewer than 4 repeats). The congenital-onset, or infantile, phenotype is often severe and rapidly progressive, accompanied by sensorineural hearing loss and Coats disease [Fitzsimons et al., 1987; Small, 1968; Taylor et al., 1982]. Marked shoulder and pelvic girdle weakness is present before 6 years of age, with loss of ambulation by 15 years. The condition may be mistaken for Möbius’ syndrome. Profound weakness of facial muscles is typical (Figure 92-18).

Laboratory Findings
Creatine kinase determinations are either normal or slightly elevated; creatine kinase levels exceeding five times the laboratory norms are suggestive of an alternative diagnosis. Genetic testing has largely replaced electrodiagnostic and muscle biopsy evaluations in persons with suspected FSHD. Electromyographic studies of moderately weak muscles reveal brief, low-amplitude motor unit potentials consistent with myopathy; sparse fibrillations reflecting muscle fiber necrosis are also present. However, the needle electromyographic examination may be normal in clinically powerful muscles. Neurogenic motor units are virtually never seen and, if present, should raise the possibility of another disorder, such as X-linked spinal and bulbar muscular atrophy. Muscle biopsy findings are often nonspecific, with occasional small angular fibers, necrotic fibers, and regenerating fibers. A modest increase in the percentage of hypertrophic type II muscle fibers may be seen. Foci of inflammatory cells in a perivascular or endomysial distribution are commonly encountered and, on occasion, may lead to a pathologic misdiagnosis of polymyositis [Rothstein et al., 1971].
Treatment
There is no definitive treatment for FSHD. Results of therapeutic trials of corticosteroids [Rose and Tawil, 2004] and the β-adrenergic agonist albuterol [Kissel et al., 2001] have been disappointing. Trials of the calcium blocker diltiazem [Elsheikh and Bollman, 2007] and an industry-sponsored trial of monoclonal antibody directed against human myostatin [Wagner et al., 2008] failed to increase strength or function.
Footdrop is best managed with ankle-foot orthoses. Orthopedic scapular fixation may improve upper-extremity function [Bunch and Siegel, 1993; Twyman et al., 1996] but the gain may be short-lived. Muscle-strengthening exercises may be of some benefit for preserving power in relatively unaffected muscle groups. Severe weakness of the orbicularis oculi may predispose to corneal injury; artificial tears can be helpful.
Oculopharyngeal Muscular Dystrophy
The earliest description of oculopharyngeal muscular dystrophy (OPGMD) is credited to Taylor [1915], who described the combination of ptosis and pharyngeal palsy, key findings of the disorder, in related family members. OPGMD is an adult-onset, autosomal-dominant disorder, typically manifesting after 50 years of age [Brais et al., 1995]. The disease maps to chromosome 14q11.2 and is caused by a small trinucleotide GCG repeat expansion in the coding region of the polyalanine binding protein nuclear 1 (PABPN1) gene. Many affected individuals of French Canadian descent are traced to a single French ancestor [Barbeau, 1966]. OPGMD is recognized in the fourth to sixth decades by onset of ptosis, partial extraocular muscle paresis, dysphagia, and tongue weakness. Weakness of the proximal upper- and lower-extremity muscles slowly develops. The creatine kinase level may be normal or mildly elevated. Progressive oculopharyngeal dysfunction is described in Greek siblings, aged 11 and 14 years, both of whom had rimmed vacuoles; one was found to have cytoplasmic and intranuclear tubulofilamentous inclusions 25 nm in diameter on muscle biopsy [Rose et al., 1997]. Another childhood-onset oculopharyngeal syndrome manifested with intestinal pseudo-obstruction [Amato et al., 1995]. These may be examples of recessively inherited conditions clinically similar to but genetically distinct from autosomal-dominant OPGMD. These patients do not have the PABPN1 repeat expansion. Progressive ophthalmoplegia in a child should be interpreted with caution. Often, this symptom is secondary to a mitochondrial disorder or variant of centronuclear myopathy rather than a muscular dystrophy [Rowland et al., 1997].
Congenital Muscular Dystrophies
The congenital muscular dystrophies (CMDs) are a heterogeneous group of disorders characterized by hypotonia, weakness, and variable degrees of muscle contractures. Signs and symptoms are evident in the neonatal period or become evident in first few months of life. Serum creatine kinase levels are usually elevated. Muscle biopsy abnormalities suggestive of a myopathic or dystrophic process, including variation in muscle fiber size, necrotic and regenerating fibers, and an increase in endomysial connective tissue, establish a working diagnosis of CMD (Figure 92-19). Muscle biopsy is also important to exclude alternative diagnoses, such as a morphologically definable congenital myopathy. With the exceptions of a recently defined syndrome due to de novo mutations in the lamin A/C gene, and some cases of Ullrich disease, the CMDs are inherited as autosomal-recessive disorders.
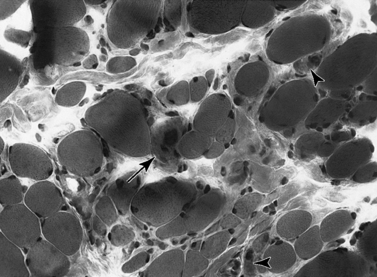
Delineation of a precise diagnosis requires integration of clinical muscle and systemic findings; muscle biopsy, including immunostaining or Western blot of extracellular proteins; and DNA testing. The CMD syndromes have traditionally been classified into those associated with brain and eye malformations (syndromic CMDs, Table 92-3) and those without (nonsyndromic or “classic” CMDs, Table 92-4). Within this broad classification, considerable phenotypic variation exists, and clinical differences may be more quantitative than qualitative. Although “pseudospecific syndromes,” such as Walker–Warburg syndrome and muscle-eye-brain disease, will be described in this section, the clinician is faced with great variation in genotype–phenotype correlation. A more useful classification of the CMDs is based on a combination of clinical features and primary biochemical defects [Muntoni and Voit, 2004]. Clinical leaders have suggested a CMD classification into one of four major groups, based on genes involved and the predicted localization and function of their protein products [Bonnemann and Bertini, 2010].
The summed incidence of all forms of CMD was reported as approximately 1 per 21,500 [Mostacciuolo et al., 1996]. Increasing familiarity with the spectrum of clinical phenotypes and more widespread availability of protein immunostaining and DNA testing will undoubtedly result in a greater identification of CMD patients and facilitate more precise diagnoses [Peat et al., 2008]. The recognition of clinical findings and symptoms shared by many patients afflicted with the various CMDs has led to standard of care guidelines for this vulnerable group of children [Wang et al., 2010].
Abnormalities of α-Dystroglycan Glycosylation
Fukuyama Congenital Muscular Dystrophy
Fukuyama congenital muscular dystrophy is one of the most common autosomal-recessive diseases and is the second most common form of muscular dystrophy in Japan, where the carrier frequency is estimated at 1 in 90 [Toda et al., 2000]. The disease is uncommon outside of Japan. Fukuyama CMD is caused by mutations of chromosome 9q31 on the gene coding for fukutin, a protein localized to the basement membrane of muscle. The protein is also found in fetal, but not in postnatal, neurons. The most common mutation is a 3-kilobase retrotransposon, which is suggestive of a founder effect [Kobayashi et al., 1998]. Fukutin deficiency is associated with decreased immunostaining for α-dystroglycan and merosin, with preservation of the dystrophin-related complex [Hayashi et al., 2001].
Laboratory studies reveal consistent elevation of creatine kinase levels. Electrodiagnostic studies reveal myopathic potentials on needle electromyography; nerve conduction velocities are normal. Brain imaging studies and postmortem studies reveal brain malformations, including polymicrogyria, pachygyria, and agyria of the cerebrum and cerebellum (type II cobblestone lissencephaly; see Chapter 26), with a lack of normal lamination of the cerebral cortex. There is a paucity of cortical gyrations in the temporal and occipital regions.
MDC 1C (Fukutin-Related Protein Deficiency)
MDC 1C is allelic to LGMD2I. The genetic defect maps to chromosome 19q13.3 and the gene coding for fukutin-related protein (FKRP). Symptoms usually manifest at birth or in the first few weeks of life, with hypotonia and leg muscle hypertrophy. Creatine kinase values are greatly increased (1000–10,000 IU/L). Children follow a regressive course that may include cardiomyopathy. Routine histochemical analysis of muscle biopsy specimens does not differentiate these patients from children with merosin-negative CMD, but immunohistochemical staining reveals partial merosin deficiency and severely reduced glycosylated α-dystroglycan. In addition, reduced molecular weight of α-dystroglycan on Western blot indicates defective glycosylation, central to the pathophysiology of MDC 1C. Thus, the spectrum of this protein deficiency includes the milder and more commonly encountered limb-girdle phenotype LGMD2I, discussed earlier in this chapter, and a CMD phenotype [Brockington et al., 2001a, 2002; Mercuri et al., 2003].
Deficiency of fukutin-related protein seen in MDC 1C and LGMD2I usually does not result in pathologic neuronal migration. However, evaluation of 13 patients with FKRP gene mutations and a clinical phenotype of CMD revealed that 8 had a range of structural brain abnormalities, including cerebellar cysts, unilateral periventricular nodular heterotopia, and pontine hypoplasia similar to that seen in patients with the clinical phenotype of muscle-eye-brain disease and Walker–Warburg syndrome. Cognitive impairment was appreciated in 3 patients [Mercuri et al., 2006]. Of the known genes implicated in the genesis of the CMDs, FKRP deficiency has the broadest phenotypic spectrum and underscores the critical role that glycosylated α-dystroglycan plays in neuronal migration. DNA testing is commercially available.
Muscle-Eye-Brain Disease
The first description of patients with the muscle-eye-brain phenotype were from Finland [Santavuori et al., 1989], but the syndrome has now been seen worldwide. Initial reports suggested the muscle-eye-brain disease gene, localized to chromosome region 1p32–p34 [Cormand et al., 2001] and coded for loss-of-function mutations in protein O-mannose β1,2-N-acetylglucosaminyltransferase (POMGnT1), a type II membrane protein similar to other Golgi glycosyltransferases. It is now evident that the muscle-eye-brain phenotype is genetically heterogeneous and may be the result of mutations in other genes along the α-dystroglycan O-mannosyl-glycosylation pathway. The clinical phenotype may be severe, dominated by neonatal hypotonia, profound developmental delay with psychomotor retardation, and ocular abnormalities. Affected children may attain independent ambulation by age 4 years. Some acquire limited language skills. Motor abilities decline late in the first decade, in association with the development of contractures and spasticity. Visual failure is in the form of severe myopia, followed by retinal degeneration, glaucoma, and cataracts. Seizures are common. Central nervous system dysgenesis includes lissencephaly type II (cobblestone complex), frontal pachygyria, cerebellar hypoplasia, and occipital micropolygyria. There is severe disorganization of the cerebral and cerebellar cortices [Haltia et al., 1997]. The associated myopathy may be overshadowed by central nervous system and ocular abnormalities. Creatine kinase levels are increased, especially after the first year of life, and electromyography studies show myopathic abnormalities. DNA testing for at-risk families is available.
Walker–Warburg Syndrome
Walker–Warburg syndrome has no ethnic predominance. The clinical stigmata are dominated by the most severe brain malformations seen in any CMD syndrome, including lissencephaly type II, hydrocephalus, occipital encephalocele, fusion of the cerebral hemispheres, absence of the corpus callosum, and hypoplasia of the cerebellum and brainstem. Ocular findings have included congenital cataracts, microphthalmia, buphthalmus, and Peter’s anomaly. At birth, affected children are blind, markedly hypotonic, and poor feeders. Myopathy is manifest by mild elevations of creatine kinase level and variable abnormalities on muscle biopsy samples. All affected infants manifest profound psychomotor retardation, and the median length of survival is only 4 months. Epilepsy is a common comorbidity. Clinical similarities in muscle-eye-brain disease and Walker–Warburg syndrome caused debate as to whether they are separate entities [Cormand et al., 2001] or a spectrum of single or multiple autosomal-recessive disorders. The initial detection of a distinct gene locus on chromosome 9q34.1, coding for protein O-mannosyl-transferase 1 (POMT1) in patients with Walker–Warburg syndrome have been supplemented by numerous reports of Walker–Warburg syndrome due to other disorders of α-dystroglycan, confirming that Walker–Warburg syndrome and muscle-eye-brain disease represent a clinical spectrum secondary to multiple mutations in multiple genes rather than distinct diseases.
MDC 1D: Large
CMD due to LARGE gene mutations (MDC 1D) is a very rare disorder. The LARGE gene maps to 22q12.3–q13 and codes a glycosyltransferase-like protein: LARGE. Although the precise mechanism of action is not known, the LARGE protein co-localizes to Golgi markers and stimulates the production of highly glycosylated α-dystroglycan [Brockington et al., 2005]. Mutations in the LARGE gene have been associated with a Walker–Warburg syndrome phenotype [van Reeuwijk et al., 2007]. A murine model of MDC 1D has been identified. Over-expression of LARGE improves the murine phenotype and may offer a therapeutic strategy for other muscular dystrophies due to α-dystroglycan glycosylation defects [Barresi et al., 2004].
Congenital Muscular Dystrophy with Integrin α7 Deficiency
Integrins are transmembrane heterodimeric (α/β) receptors crucial in establishing linkages between the extracellular matrix and muscle cytoskeleton. Integrin α7β1D is the major integrin receptor of adult muscle. Integrin α7 and α-dystroglycan are the main muscle receptors for laminin α2. Deficiency of integrin α7 is a very rare disorder, with only four patients reported to date. This entity was discovered during analysis of muscle biopsy specimens from 117 patients with unclassified congenital myopathy and CMD phenotypes. Three patients evidenced integrin α7 deficiency on muscle immunohistochemistry profiles, which was correlated with three different mutations in the integrin α7 deficiency gene on chromosome 12q13 [Hayashi et al., 1998]. Another study of 210 patients with undefined myopathy indicates that secondary deficiencies of integrin α7 also occur, complicating the diagnostic significance of immunostaining of this protein [Pegoraro et al., 2002]. Although cognitive impairment was noted in a single patient, the very small number of proven cases have not evidenced central nervous system involvement, as seen in the severe phenotypes of α-dystroglycan glycosylation disorders.
Abnormalities of Extracellular Matrix Proteins
MDC 1A: Laminin-α2 (Merosin)-Negative Congenital Muscular Dystrophy LAMA2 (“Nonsyndromic Congenital Muscular Dystrophy”)
Laminin α2 (merosin)-negative CMD (MDC 1A) is the most commonly diagnosed of the CMD disorders and accounts for about 40 percent of cases of nonsyndromic autosomal-recessive CMD (see Table 92-4). The extracellular matrix protein is coded on 6922–q23. The merosin (LAMA2) gene contains 64 exons with mutations distributed through a large coding sequence of 9.5 kilobases. Merosin is the heavy α2 chain of laminin 2 (LAMA2). The laminins are heterotrimeric proteins that influence cell adhesion, growth, and migration. LAMA2 is expressed in the basement membrane of muscle fibers, in Schwann cells of intramuscular nerves, and at neuromuscular junctions. LAMA2 promotes myotube stability and inhibits apoptosis [Vachon et al., 1996].
Patients with complete absence of merosin present as hypotonic infants. Limb weakness is usually more prominent than bulbar or respiratory impairment, but feeding difficulties, including aspiration [Philpot et al., 1999] and ventilatory insufficiency necessitating respiratory support, occur frequently [Fardeau et al., 1996]. Most patients achieve independent sitting, but few stand or walk unassisted. Multiple joint contractures may be present at birth or develop in infancy or early childhood. Progressive contractures mandate periodic orthopedic consultation. Weakness tends to be stable or slowly progressive (Figure 92-20). Most patients exhibit normal brain structure and cognitive development. Focal cortical dysplasia of the occipital lobes occurs in some cases with concurrent intellectual impairment [Taratuto et al., 1999], but the extensive neuronal migration abnormalities characteristic of patients with syndromic CMD is not seen. Epilepsy occurs in approximately 30 percent of cases, even in patients with normal intellect [Pegoraro et al., 1998].

Virtually all merosin-negative CMD patients have characteristic white-matter lesions on MRI scans, most prominent on T2-weighted images. These findings may not be evident at birth but invariably develop by age 6 months. White-matter abnormalities involve the centrum semiovale and spare the corpus callosum, internal capsule, cerebellum, and brainstem [Caro et al., 1999]. The MRI findings are consistent with abnormal water distribution in the white matter that results from disruption of the blood–brain barrier secondary to merosin deficiency in cerebral vasculature [Villanova et al., 1997]. Serum creatine kinase levels are usually elevated in the neonatal period in excess of 1000 U/L but tend to decrease over years. Electrodiagnostic studies yield nonspecific myopathic findings on needle electromyography. Nerve conduction studies reveal slowing of conduction velocity, consistent with demyelinating polyneuropathy [Gilhuis et al., 2002]. Muscle biopsy samples demonstrate changes consistent with a dystrophic process, including adipose tissue replacement and connective tissue proliferation. In addition, inflammatory changes may be striking enough to suggest a diagnosis of infantile polymyositis [Pegoraro et al., 1996]. Absence of immunohistochemical staining for the laminin α2 chain on snap frozen muscle biopsy or skin biopsy tissue provides confirmation of the diagnosis of merosin deficiency.
Partial merosin deficiency has been reported in rare patients with a benign childhood- or adult-onset LGMD phenotype [Allamand et al., 1997]; routine immunohistochemical staining for LAMA2 is therefore suggested in evaluating patients with both CMD and LGMD phenotypes. Prenatal diagnosis of merosin-deficient CMD is possible by direct mutation analysis of chorionic villus biopsy material. Direct immunostaining of trophoblasts from chorionic villus samples with laminin α2 chain antibodies provides an additional method for prenatal diagnosis. DNA testing is commercially available, and the diagnosis of laminin α2-deficient CMD should be confirmed with direct mutation analysis whenever possible to offer optimal genetic counseling [Vainzof et al., 2005].
Ullrich’s Congenital Muscular Dystrophy and Bethlem’s Myopathy
Ullrich’s CMD and Bethlem’s myopathy are being recognized much more frequently as clinicians have become familiar with the clinical phenotypes. Ullrich CMD is usually caused by autosomal-recessive mutations in any of three collagen 6 genes (COL6A1, COL6A2, and COL6A3) [Vanegas et al., 2002; Zhang et al., 2002]. However, a clinical phenotype of Ullrich CMD is often due to autosmal-dominant mutation [Baker et al., 2005]. Collagen 6 immunostaining of muscle basal lamina and endomysial connective tissue is severely reduced or absent in the Ullrich patient [Higuchi et al., 2003]. In cases demonstrating normal immunostaining for collagen 6, double immunostaining for both collagen 6 and another extracellular matrix protein, such as collagen IV or perlecan, has shown that collagen 6 may be present in the interstitium but absent from the sarcolemma [Okada et al., 2007].
Ullrich CMD is characterized by infantile onset of weakness, hypotonia, and early findings of severe proximal joint contractures in the face of striking hyperlaxity of distal joints [Furukawa and Toyokura, 1977; Lampe et al., 2007]. Other orthopedic findings may include congenital hip dislocation, prominent calcanei, and transient kyphosis. Progressive flexion contractures of the wrists, fingers, and ankles often replace distal limb laxity. The disease is slowly progressive. Although some youngsters acquire independent ambulation, weakness and orthopedic deformities lead to full-time wheelchair use. Involvement of the chest wall, producing restrictive pulmonary disease, often necessitates nocturnal ventilatory support, but cardiac function is usually unaffected. Serum creatine kinase levels are usually normal or slightly elevated. Muscle biopsy samples demonstrate nonspecific myopathic or dystrophic changes of degenerating and regenerating muscle cells, with increased endomysial connective tissue.
Collagen 6 is composed of three different α peptide chains. The α1 and α2 chains are encoded by two genes (COL6A1 and COL6A2), located head to tail at chromosome 21q22.3 [Heiskanen et al., 1995]. The gene for COL6A3 maps to locus 2q37. Collagen 6 is an extracellular matrix protein expressed in nearly all connective tissues. The phenotype of Ullrich CMD merges with that of Bethlem’s myopathy, originally described as a benign myopathy with autosomal-dominant inheritance [Bethlem and Wijngaarden, 1976]. The clinical manifestations are more variable than those of Ullrich CMD. The onset of signs and symptoms of proximal weakness is usually in early childhood, manifesting by difficulty climbing stairs and running. Gowers sign is often present. In more severe cases, findings may be present in the neonatal period, with mild hypotonia, foot deformities, congenital torticollis, or arthrogryposis. Unlike the progressive contractures in Ullrich CMD, the neonatal arthrogryposis seen in patients with Bethlem’s myopathy tends to improve spontaneously. Weakness is usually nonprogressive. However, contractures may develop late in the first decade, often localized to the ankles, elbow flexors, and long finger flexors. In other patients, the clinical presentation is delayed until the fourth decade. Dominant mutations in any of the collagen 6 genes may produce the Bethlem myopathy phenotype or a clinical picture of Ullrich CMD [Jobsis et al., 1999]. The family history is helpful in determining whether the disorder has been transmitted as an autosomal-dominant or recessive trait, but de novo mutations and parental germline mosaicism may complicate this assessment. Immunostaining for collagen 6 in muscle is usually normal in Bethlem’s myopathy. The diagnostic sensitivity of immunostaining of cultured fibroblasts is superior to muscle. Ultimately, DNA studies of the COL6A genes are necessary for definitive diagnosis [Lampe et al., 2005].
Management of Ullrich CMD and Bethlem myopathy is symptomatic. Aggressive physical therapy and surgical intervention address contractures. Severely affected children need active mobilization, and standing frames are often helpful. Nocturnal ventilatory support is often required [Wallgren-Pettersson et al., 2004]. The combination of restrictive lung disease and progressive orthopedic deformity in severely affected children makes the timing of surgical interventions critical. Murine models of COL6 mutations and humans with Ullrich’s CMD demonstrate mitochondrial dysfunction with dysregulation of the mitochondrial permeability transition pore (PTP). Experimental investigations utilizing the PTP inhibitor cyclosporine are in progress [Merlini and Bernardi, 2008; Hicks et al., 2009].
Congenital Muscular Dystrophy with Early Rigid Spine Syndrome
Development of axial paraspinal muscle contractures produces the rigid spine syndrome (SEPN1) in a number of congenital myopathies and muscular dystrophies, including Emery–Dreifuss muscular dystrophy and Ullrich’s CMD. A “distinct” phenotype–genotype correlation was defined in consanguineous families of Moroccan, Turkish, and Iranian descent [Moghadaszadeh et al., 2001]. In another American family presenting a phenotype of infantile hypotonia, prominent axial-cervical muscle weakness, and early spinal rigidity and scoliosis, linkage to the chromosome 1p rigid spine syndrome locus was proved [Flanigan et al., 2000]. The gene for rigid spine syndrome in these patients mapped to chromosome region 1p36, coding for a selenoprotein (SEPN1), known to be expressed in skeletal muscle, as well as brain and lung. The SEPN1 protein localizes to the endoplasmic reticulum, but little is known about its function. The disorder is transmitted as an autosomal-recessive trait. Most children present with mild hypotonia and weakness, recognized in the first year of life. Early evolution of rigid spine due to axial muscle contractures and Achilles tendon contracture are characteristic. A thin body habitus is frequently noted and there is no muscle hypertrophy. Creatine kinase serum levels are normal. Most children achieve independent ambulation. Restrictive lung disease nearly always complicates rigid spine syndrome and mandates close observation for nocturnal hypoventilation and early institution of noninvasive respiratory support [Schara et al., 2008]. SEPN1 deficiency has also been shown in cases of congenital fiber type disproportion myopathy and multi-mini-core diseases.
Lamin A/C-Associated Congenital Muscular Dystrophy
The broad spectrum of neuromuscular and non-neuromuscular disorders due to mutations in the lamin A/C gene have been outlined earlier in the section describing Emery–Dreifuss muscular dystrophy. Special attention is given here to a recently recognized syndrome of CMD associated with de novo mutations of the LMNA gene. Isolated cases of an early-onset, severe CMD phenotype were first reported in 2000 [Bonne et al., 2000]. A recent summary of 15 patients defines the clinical syndrome and its molecular genetic correlate [Quijano-Roy et al., 2008]. All patients presented with weakness and gross motor delays in the first year of life. No family history of neuromuscular disease was obtained. All patients had de novo heterozygous LMNA mutations. Three had severe, early-onset disease with failure to attain motor milestones. Twelve later developed axial cervical muscle weakness, resulting in a “dropped head ” phenotype – the nearly unique clinical feature of this syndrome. Creatine kinase levels were elevated in all patients. Progression of weakness was of variable severity, but all patients demonstrated selective axial weakness and wasting of the cervicoaxial muscles. Proximal muscle weakness dominates motor deficits in the upper limbs and distal weakness predominates in the lower limbs. A rigid spine syndrome with thoracic lordosis evolves rapidly, followed by development of lower-limb flexion contractures at the knees and ankles. Cardiomyopathy is manifest by cardiac arrhythmias. There is rapid progression of respiratory muscle involvement and early ventilatory support is needed in the majority of cases. Muscle biopsy reveals myopathic and/or dystrophic features. Immunohistochemical studies for lamin A/C have been normal, necessitating DNA studies for definitive diagnosis.
Nesprin-Associated Congenital Muscular Dystrophy
A single Sicilian sibship with the clinical phenotype of CMD associated with adducted thumbs is mentioned here because it represents another disorder underlain by a defective nuclear envelope protein. The male and female siblings manifested congenital weakness, hypotonia, contractures of thumbs and toes, and mental retardation [Voit et al., 2002]. Motor deficits were progressive. Muscle biopsy findings were nonspecific. Mid-thigh muscle MRI revealed widespread fatty replacement of muscle, and brain MRI revealed cerebellar hypoplasia. Positional cloning identified that the gene involved was that coding for nesprin-1, a nuclear envelope protein that binds to both emerin and lamin A/C. The role of nesprin and other nuclear envelope proteins in the genesis of Emery–Dreifuss muscular dystrophy, congenital muscular dystrophies, and other myopathic phenotypes will undoubtedly be defined better in the next few years [Zhang et al., 2007].
 The complete list of references for this chapter is available online at
The complete list of references for this chapter is available online at