Chapter 13 Multiple organ dysfunction syndrome
The incidence of multiple organ dysfunction syndrome (MODS) is increasing and accounts for up to one-half of deaths in intensive care. Some 50 years ago, multiple organ failure did not exist as a clinical entity. Patients could not be kept alive long enough for the sequential disturbances in the function of distant organs to occur. In the 1960s, acute respiratory failure characterised by bilateral infiltrates on chest radiograph, now termed acute respiratory distress syndrome (ARDS), was described following a variety of non-pulmonary insults. Finally, in 1973, the first description of multiple organ failure appeared in the surgical literature, describing the course of three patients who subsequently died following surgery for ruptured aortic aneurysm.1
Critical illness is often associated with a downward spiral through a systemic inflammatory response syndrome (SIRS) towards frank organ failure and death. Since organ failure is not an all-or-nothing phenomenon, and because dysfunction usually precedes and may progress to gross organ failure, the old term ‘multiple organ failure’ was deemed unsatisfactory. The American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) consensus conference in 1992 proposed that the term ‘dysfunction’ identifies a phenomenon in which organ function is not capable of maintaining homeostasis (Table 13.1).2 However, it should be remembered that it is only a descriptive definition and does not provide any insight into the aetiology or pathogenesis of MODS. In fact, it is not clear whether MODS is a single pathological process with variable clinical expression, or a limited phenotypic expression of a large number of pathological processes. As yet no therapeutic interventions specifically directed at MODS have significantly altered outcome.
Table 13.1 Definition of systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS)3
| Systemic inflammatory response syndrome |
| The systemic inflammatory response to a variety of severe clinical insults is manifest by two or more of the following conditions: |
| Temperature > 38°C or < 36°C |
| Heart rate > 90 beats/min |
| Respiratory rate > 20 breaths/min or PaCO2 < 32 mmHg (or ventilator dependence) |
| White blood cell count > 12 000 cells/mm3, < 4000 cells/mm3 or > 10% band forms |
| Multiple organ dysfunction syndrome |
| The presence of altered function involving at least two or more organ systems in an acutely ill patient such that homeostasis cannot be maintained without intervention |
AETIOLOGY
Uncontrolled infection was classically thought to be the main precedent leading to SIRS and subsequently to MODS. In an Australian epidemiological study, infection was not identified in 912 of a total of 1803 SIRS episodes.4 Therefore it is thought that a substantial proportion of cases of MODS are not initiated by infection. Table 13.2 lists many of the causes of MODS, but is not exhaustive. It must be noted that any number of these causes may in fact act as a secondary insult precipitating MODS. The common features of non-infective aetiologies of MODS are ischaemia, hypoxia, cytokine release, mechanical injury or any combination of these. Thus MODS may occur after all forms of shock and compartment syndromes. Other important causes include trauma, major surgery, burns, pancreatitis, hepatic failure, pulmonary aspiration syndromes and mechanical ventilation.5 Less commonly, cardiopulmonary bypass, blood product and cytokine infusions, and some drug reactions have been associated with MODS.6
Table 13.2 Causes of multiple organ dysfunction syndrome (MODS)
| Infectious agents | Non-infectious insults | Mechanism |
|---|---|---|
| Bacteria | Mechanical ventilation | Ischaemia |
| Viruses | Aspiration | Hypoxia |
| Fungi | Surgery | Cytokine release |
| Protozoa | Burns | |
| Reperfusion syndromes | ||
| Visceral ischaemia | ||
| Pancreatitis | ||
| Hepatic failure | ||
| Cardiopulmonary bypass | ||
| Massive transfusion | ||
| Transfusion reactions | ||
| Hyperthermia | ||
| Malignancy |
PATHOPHYSIOLOGY
INFLAMMATION
The current explanation for the development of SIRS and MODS is local inflammation with activation of the innate immune system and subsequent unbridled systemic inflammation.7 There is a spectrum of clinical sequelae, ranging from the mildest SIRS that just meets the definition, through organ dysfunction that resolves within a few days, to overwhelming SIRS and MODS. The balance between the inflammatory and regulatory arms of the host response may in part explain these varied responses.
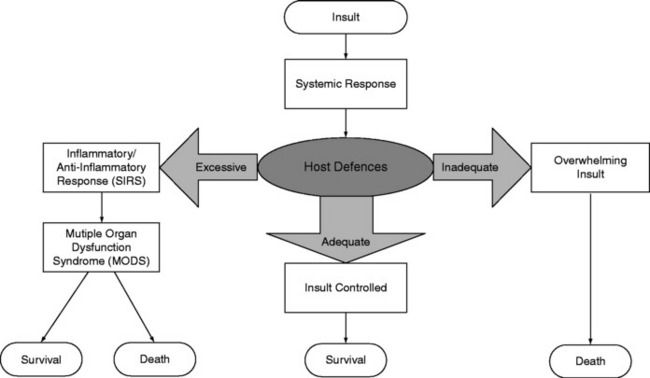
Inflammation involves the activation of circulating immune cells (in particular natural killer (NK), T and B cells and macrophages), the endothelium, and multiple mediator cascades balanced by an anti-inflammatory system. Following injury, proinflammatory mediators are released locally to combat foreign antigens and promote wound healing. Concurrently, anti-inflammatory mediators are released to downregulate this process. If the regulatory mechanisms are unable to contain the response, then inflammatory mediators enter the systemic circulation, additional leukocytes are recruited and activated, and a whole-body response ensues. Homeostasis may be lost and so-called ‘immunological dissonance’ occurs, when the host’s inflammatory or anti-inflammatory response to injury (or both) is excessive or inadequate (Figure 13.1).8 Thus, MODS is not necessarily due to the primary insult, but more likely to be related to an uncontrolled, aggressive systemic response of the host to that primary insult. Without intervention, SIRS may lead to MODS and death.
MOLECULAR MECHANISMS
In sepsis, activated cell/antigen products interact with a member of the Toll-like receptor family to signal gene expression of inflammatory mediators. Toll-like receptors are evolutionarily-conserved receptors expressed by monocytes and macrophages, which recognise whole subsets of pathogens, and represent the phylogenetically ancient innate immune system. Toll-like receptors mediate cell signalling through the same intracellular transcription pathways as cytokines themselves, most notably nuclear factor κB (NF-κB).10 Non-infective causes of MODS seem to have this final common pathway, resulting in mediator release. For example, in ventilator-associated lung injury, the signalling cascade converting the physical stimulus into mediator release operates via NF-κB, but the upstream events remain unclear.11 Large numbers of mediators are involved in inflammation, with chemotactic factors attracting, adhesion molecules focusing, and cytotoxic agents assisting this process. They include cytokines, leukotrienes, prostaglandins, platelet factors and the coagulation and complement systems.
Cytokines are the main mediators of inflammation, with several actions, including directing the cellular response, inducing enzyme production such as inducible nitric oxide synthase (iNOS) and altering adhesion molecules. Their actions are pleiotropic, acting on multiple target cells in different ways depending on timing and local tissue concentration. Several cytokines have been implicated in the development of SIRS and MODS, including tumour necrosis factor (TNF)-α, and interleukins (IL) 1β, 6, and 8. Cytokine and NF-κB concentrations appear to be linked to morbidity and mortality.12,13 In response to proinflammatory mediators, there is endogenous production of anti-inflammatory cytokines such as IL-4, IL-10 and IL-13.
However, it seems that the prevailing internal milieu is likely to be more important than any absolute levels of one mediator or another. Patients at increased risk of SIRS and MODS, such as the elderly and those with pre-existing illnesses, seem to have abnormal cytokine levels.14 The ability of a cell to synthesise pro- or anti-inflammatory mediators is influenced by many factors, both genetic and environmental in origin, but also by its previous state of activation. It may be that an initial insult, insufficient to cause MODS, nevertheless pre-primes a susceptible individual such that a subsequent or secondary insult generates a response that overwhelms homeostasis.
If a relative excess or deficiency of mediator expression can upset inflammatory homeostasis, then it is not surprising to find a strong genetic correlation. Families characterised by low TNF-α production have a 10-fold increased risk of fatal outcome in meningococcal disease, whereas high IL-10 production increases the risk 20-fold.15 TNF-α and IL-10 receptor antagonist polymorphisms are associated with greater susceptibility and worse outcomes to severe sepsis.16 Unfortunately genetic determinants are likely to be more complicated than simple quantitative expression of one mediator or another.
TISSUE INJURY
The final common pathway leading to MODS is often tissue hypoxia. Many factors can compromise oxygen delivery to tissues. Among these factors are arterial hypoxaemia due to acute lung injury, and reduced cardiac output from reduced left ventricular preload and/or impaired ventricular performance. Over and above oxygen delivery abnormalities, evidence is rapidly building that microcirculatory and mitochondrial dysfunction with an abnormal distribution of blood flow and defective oxygen utilisation (‘tissue dysoxia’) is central to the pathogenesis of MODS. Indiscriminate injury by mediators of inflammation leads to deranged endothelial function with altered vascular relaxation and abnormal modulation of coagulation, and mitochondrial and cellular damage. This is compounded by an ongoing initial or subsequent insult. Unchecked, cellular dysfunction leads to loss of ion gradients, leakage of lysosomal enzymes, proteolysis and cell death. If enough tissue injury occurs, organ dysfunction and ultimately failure ensue.
ENDOTHELIAL DYSFUNCTION
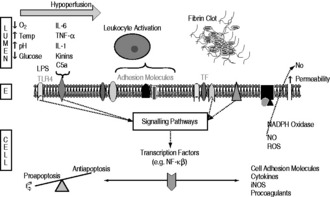
The endothelium is activated by the engagement of antigen, for example lipopolysaccharides from bacterial cell walls, with Toll-like receptors or inflammatory mediators with various receptors. At any stage other environmental factors such as hypoxia, hypoperfusion, increased temperature, acidosis and glycaemic perturbations may also affect endothelial function. The interaction of these extracellular factors with their receptors activates downstream signalling pathways, influencing transcription factors and altering cellular function and/or gene expression. Cell surface adhesion molecules are upregulated, leading to increased leukocyte rolling, adhesion and transmigration, with cytokine-mediated positive feedback and cellular recruitment enhancing this process.17,18 Upregulation of iNOS leads to excessive amounts of NO production.
In addition, inappropriate intravascular coagulation is an important cause of tissue injury. Activation and amplification of the tissue factor pathway, and possibly downregulation of anticoagulatory pathways, lead to the generation of thrombin and hence fibrin formation. Coagulation may result in focal areas of ischaemia, and concurrent depletion of counterregulatory networks. The mechanisms that regulate inflammation, coagulation and the various cell types are intimately linked and cannot be thought of as discrete entities. A schematic diagram of the processes involved in endothelial dysfunction is shown in Figure 13.2.
Relatively new non-invasive techniques such as orthogonal polarisation (Figure 13.3) have demonstrated these abnormalities in microcirculatory blood flow distribution in septic states compared with normal controls. As a consequence of the derecruitment of capillaries, the increased diffusion distances will in turn contribute to the development of hypoxia.
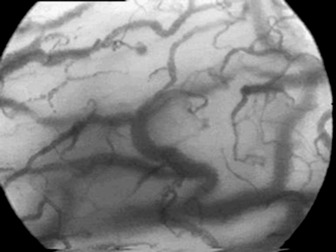
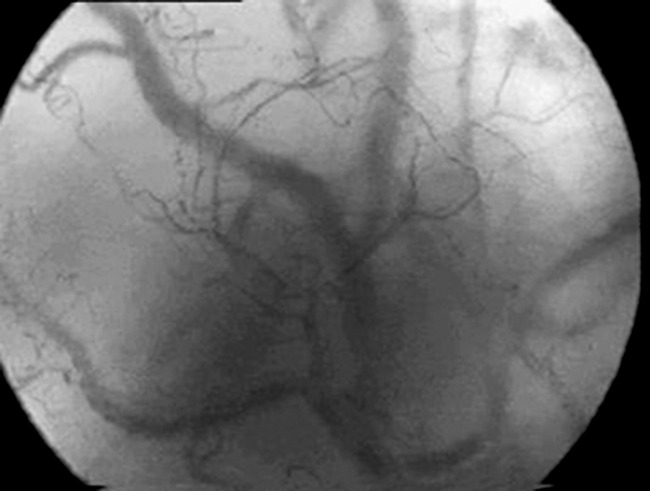
Figure 13.3 Orthogonal polarisation images of the sublingual mucosa in a normal (top) and a septic (bottom) patient. Note, firstly, paucity of smaller blood vessels in the septic sublingual mucosa with increased diffusion distances between blood vessels and, secondly, that the remaining blood vessels are of smaller calibre.
(Courtesy of Cytometrics.)
APOPTOSIS
Changes in the dynamics and regulation of apoptosis in critical illness contribute to organ dysfunction. Apoptosis is the fundamental physiological process by which cells activate an endogenous programme that leads to controlled death and clearance of cells without evoking an inflammatory response. In contrast, necrosis is uncontrolled cellular death with release of intracellular contents and subsequent inflammation. Alterations in the control of apoptosis are caused by changes in the expression of proapoptotic and antiapoptotic genes. Excessive apoptosis has been particularly implicated in gastrointestinal, liver, kidney and cardiac dysfunction. Interestingly, the apoptosis of lymphocytes is delayed, which could lead to prolonged injurious function.20,21
MITOCHONDRIAL DYSFUNCTION
Normally more than 85% of oxygen extracted by tissues is used in the mitochondrial respiratory chain to produce adenosine triphosphate (ATP), the main intracellular energy source. It is widely accepted that oxygen consumption, and hence extraction, is reduced in many disease states leading to MODS. This decrease in oxygen consumption may be out of proportion to any reduction in oxygen delivery caused by microcirculatory dysfunction. Abnormal mitochondrial respiration with markedly reduced ATP production has been implicated in some cases.22 The strong inherited component to the heterogeneity seen in MODS outcome may be influenced by the natural genetic variation in mitochondrial DNA.23
Several mutually compatible mechanisms have been postulated to account for this tissue dysoxia (previously confused with ‘cytopathic hypoxia’). There is diminished delivery of pyruvic acid to the tricarboxylic acid (TCA) cycle by inhibition of pyruvate dehydrogenase leading to increased lactate production. NO and reactive oxygen species (ROS) have been demonstrated to inhibit the mitochondrial respiratory chain significantly. ROS can also induce DNA damage, which activates poly (ADP-ribose) polymerase-1 (PARP-1), consuming NAD+/NADH for repair as a consequence. Since NADH is the main reducing equivalent in oxidative phosphorylation, energy production is compromised. Animal studies suggest that tissue dysoxia occurs after the disease process is well established, which suggests that early intervention may prevent the evolution of MODS.24,25
The mechanisms of tissue injury described above are not mutually exclusive, but the result of interactions between multiple predisposing patient factors, a series of physiologic insults and an endogenous response effected via multiple cell types and hundreds of biochemical mediators. Figure 13.4 attempts to simplify and illustrate the pathogenesis and mechanisms of tissue injury.
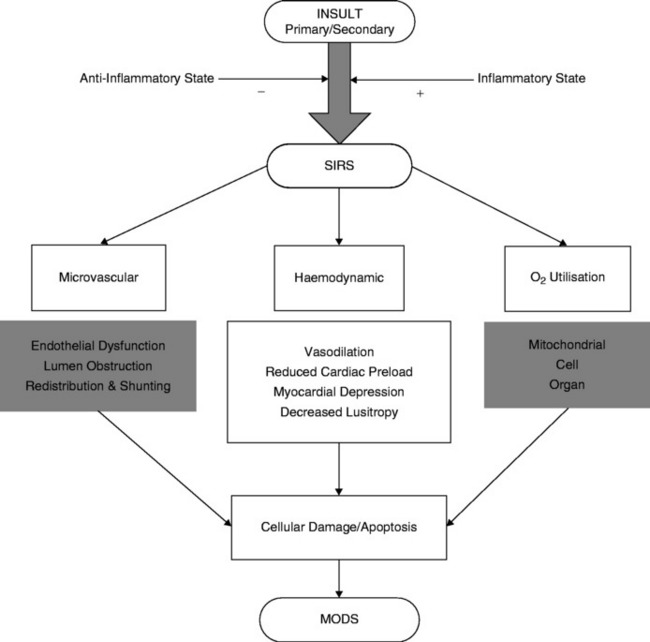
CLINICAL FEATURES
As SIRS progresses, an often unpredictable but nonetheless sequential pattern of organ dysfunction ensues. Once two or more organs are involved, the term ‘MODS’ can be used. Table 13.3 lists the commoner organ systems affected and their associated clinical features.
Table 13.3 Commonly affected organs, with associated clinical features
| Organ system | Associated clinical features | Physiological and biochemical changes |
|---|---|---|
| Neurological | Decreased level of consciousness/encephalopathy (confusion, agitation and/or drowsiness) | Abnormal EEG suggestive of metabolic encephalopathy |
| Cardiovascular | SBP < 90 mmHg or a reduction of > 40 mmHg from baseline | Reduced systemic vascular resistance |
| Tachycardia > 90 beats/min, dysrhythmia | Myocardial depression and reduced lusitropy | |
| Oedema formation | Increased capillary permeability | |
| Respiratory | Tachypnoea > 20 breaths/min | PaCO2 < 32 mmHg |
| Desaturation | Hypoxaemia (reduced PaO2/FiO2 ratio) | |
| Central and peripheral cyanosis | Increased work of breathing | |
| Requirement for mechanical ventilation | Increased lung water | |
| Renal | Urinary output < 0.5 ml/kg per h despite adequate fluid resuscitation | Increasing urea and creatinine |
| Gastrointestinal | Abdominal discomfort and distension | Increased intestinal permeability |
| Large nasogastric aspirates | Splanchnic ischaemia | |
| Failure to absorb enteral nutrition | Ileus | |
| Haemorrhage | Acalculous cholecystitis | |
| Pancreatitis | ||
| Stress ulceration | ||
| Hepatic | Jaundice | Increasing bilirubin |
| Encephalopathy | Increasing lactate | |
| Hyperglycaemia (increased gluconeogenesis, impaired clearance) | ||
| Hypoglycaemia (failing gluconeogenesis) | ||
| Haematological | Haemorrhage, petechial rash Peripheral cyanosis | White blood cell count > 12 000 cells/mm3, < 4000 cells/mm3 or > 10% band forms |
| DIC with coagulopathy and platelets reduced by 50% over 3 days, or < 80 000 cells/mm3 | ||
| Anaemia |
EEG, electroencephalogram; SBP, systolic blood pressure; DIC, disseminated intravascular coagulation.
Encephalopathy is very common and correlates with mortality in sepsis.26 As many as 30% of patients have evidence of myocardial dysfunction (reduced ejection fraction) and ARDS complicates 60% of cases of septic shock.3,27 There is a close relationship between increased intestinal permeability on intensive care unit (ICU) admission and the subsequent development of MODS.28 Increased intestinal permeability may lead to the translocation of endotoxin, bacteria and other mediators. It can therefore be seen that many of these physiological changes can also act as secondary insults in the development of MODS.
MANAGEMENT
The cornerstones of the management of patients with SIRS and impending MODS are firstly, the prevention of secondary insults such as nosocomial infection; secondly, early identification of dysfunctional organs with regular clinical assessment and appropriate monitoring, and thirdly, the timely treatment of both primary and secondary insults. Good supportive care, including recognition of the requirement and instigation as necessary of fluid resuscitation, vasopressors/inotropes, mechanical ventilation (with lung protective strategies) and renal replacement therapies, reduce mortality from MODS. In addition, infection control, avoiding oversedation, pressure area care, nursing the patient head-up, early enteral nutrition, and stress ulcer and deep-vein thrombosis thromboprophylaxis are all paramount in reducing secondary insults.
INNOVATIVE THERAPIES
GLYCAEMIC CONTROL
Hyperglycaemia has been reported to impair the immune system, increase endothelial cell apoptosis and cause mitochondrial dysfunction.29 Moreover, insulin promotes endothelial cell survival.30 Hence it may be that tight glucose control could have a protective effect on the endothelium.
In a single-centre study, an absolute mortality risk reduction of 3.4% from MODS and sepsis has been demonstrated in critically ill surgical intensive care patients treated with intensive insulin therapy (aiming for a blood glucose in the range of 4.1–6.1 mmol/l).31
LOW-DOSE GLUCOCORTICOIDS
There has been renewed interest in the therapeutic role of ‘replacement doses’ of steroids in SIRS and MODS, with some evidence suggesting improved survival in patients with septic shock.32 Specifically, steroids inhibit NF-κB and the induction of iNOS by cytokines. Hence the mechanisms by which physiological, and perhaps beneficial, doses of corticosteroids are thought to act include:
MANIPULATION OF THE COAGULATION CASCADE
A 28-day absolute reduction in the risk of death of 6.1% (P = 0.005) has been reported with the administration of human recombinant activated protein C (APC) to patients with severe sepsis and evidence of organ dysfunction (PROWESS study group).33 APC has several potentially beneficial effects:
However, there is a small but significant increased risk of serious bleeding complications. Certain patient groups, notably those with low Acute Physiology and Chronic Health Evaluation (APACHE) II scores (less than 25) and children appear not to benefit from such treatment.34,35 A confirmatory study in patients with APACHE II scores greater than 25 is planned by the original PROWESS investigators.33
Other research into the modulation of the coagulation system has proven disappointing. In contrast to the initial promising preclinical data on antithrombin III, a phase III trial failed to show any benefit, although a recent subgroup analysis of the trial hinted at improved survival in those who also received heparin.36,37 Furthermore, treatment with recombinant tissue factor pathway inhibitor had no effect on all-cause mortality in patients with severe sepsis and high international normalised ratio (INR). Moreover, there was an increased risk of bleeding, irrespective of baseline INR.38
NUTRITION
Studies in ARDS and sepsis patients have suggested that ω-3 fatty acid administration may reduce the release of proinflammatory mediators with decreased cytokine production, NF-κB inhibition and macrophage-mediated systemic dysfunction.39,40 Whilst some studies suggest a reduction in the severity of MODS in humans, conclusive clinical data are lacking. This raises the interesting possibility that not only may nutritional factors influence the course of MODS, but an individual may be susceptible to developing the syndrome in the first instance.
BLOOD PURIFICATION
High-volume haemofiltration (2–6 l/hour) with highly permeable biocompatible membranes has been postulated to remove large quantities of mediators either by filtration or by membrane absorption. While improved cardiovascular parameters have been observed, this may also be due to control of hyperthermia, correction of fluid overload, metabolic acidosis and electrolyte abnormalities.41
NITRIC OXIDE INHIBITORS
Indiscriminate NOS inhibition with NG-monomethyl-L-arginine hydrochloride (L-NMMA), whilst improving vasomotor tone, also leads to significantly increased myocardial and renal complications and mortality. Selective NOS inhibitors may have a role in the treatment of MODS in the future.42
OUTCOMES
The mortality from MODS is related to the number and duration of organ systems in failure,43 and remains the leading cause of death in non-coronary ICUs.44 A simple count of organs affected and the duration of dysfunction will stratify mortality in broad ranges, for example 60–98% depending on age, with three organs affected for more than 1 week.45 Although the incidence and overall outcome of MODS did not seem to change very much in the 1980s, an analysis of the APACHE II and III databases suggests that patients with three or more organ systems in failure did indeed have a better outcome in the later APACHE III database.46 Since the results of research into specific interventions have on the whole been disappointing, any improvement in outcome must be related to better (or earlier) resuscitation and supportive therapies. Survival rates do seem to be improving in this very sick population of patients, but whether MODS can be prevented by any new specific therapy remains to be seen.
MODS may serve as a useful outcome measure of disease severity for risk adjustment and outcome markers in critical care. The advantage of using MODS is that it may be a less biased measure of the original insult and subsequent care provided. One specific instrument designed to assess organ failure is the Sequential Organ Function Assessment (SOFA) score (see Appendix 8). Using the worst values for six commonly measured parameters (PaO2/FiO2 ratio, platelet count, bilirubin, blood pressure, Glasgow Coma Score and urine output or creatinine), a score is assigned on admission to intensive care and repeated every 48 hours until discharge. The advantage of the SOFA score is in its ability to describe the epidemiology of critical illness both at the time of admission and during ICU care. There is a positive association between total SOFA score on admission and ICU mortality before 7 days, but not after. Interestingly, ICU mortality after 7 days is associated with high SOFA scores on day 6 after adjusting for admission score. Thus, illness on admission becomes less important, and the course during the ICU stay becomes more relevant, with the presence or development of cardiovascular dysfunction having the strongest independent effect on the probability of death compared with other organ system dysfunctions.47
Although mortality is an important outcome measure in ICU, permanent restrictions in daily activities as measured by quality of life (QOL) scores are often also relevant to both the individual and society. MODS has been shown in a prospective observational study using the QOL score RAND-36 to have a statistically significant negative effect on vitality and emotional role limitations. Some 47.2% of all patients were unable to return to work and/or experienced severe limitations in their daily activities and 3.8% were unable to live at home 1 year after hospital discharge.48 Similarly diminished QOL scores have been found in post cardiac surgery and acute lung injury patients.49,50 However, patients with acute renal dysfunction appear to have a less marked deterioration in their QOL.
1 Tilney NL, Bailey GL, Morgan AP. Sequential system failure after rupture of abdominal aortic aneurysms: an unsolved problem in postoperative care. Ann Surg. 1973;178:117-122.
2 American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864-874.
3 Parrillo JE, Parker MM, Natanson C, et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227-242.
4 Finfer S, Bellomo R, Lipman J, et al. Adult-population incidence of severe sepsis in Australian and New Zealand intensive care units. Intens Care Med. 2004;30:589-596.
5 Evans TW, Smithies M. ABC of intensive care: organ dysfunction. Br Med J. 1999;318:1606-1609.
6 Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018-1028.
7 Singer M, De Santis V, Vitale D, et al. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364:545-548.
8 Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann Intern Med. 1996;125:680-687.
9 Johnson D, Mayers I. Multiple organ dysfunction syndrome: a narrative review. Can J Anaesth. 2001;48:502-509.
10 Glauser MP. Pathophysiologic basis of sepsis: considerations for future strategies of intervention. Crit Care Med. 2000;28:S. 8
11 Held HD, Boettcher S, Hamann L, et al. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163:711-716.
12 Pinsky MR, Vincent JL, Deviere J, et al. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest. 1993;103:565-575.
13 Paterson RL, Galley HF, Dhillon JK, et al. Increased nuclear factor kappa B activation in critically ill patients who die. Crit Care Med. 2000;28:1047-1051.
14 Bone RC. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med. 1996;24:163-172.
15 Westendorp RG, Langermans JA, Huizinga TW, et al. Genetic influence on cytokine production and fatal meningococcal disease. Lancet. 1997;349:170-173.
16 Freeman BD, Buchman TG. Gene in a haystack: tumor necrosis factor polymorphisms and outcome in sepsis. Crit Care Med. 2000;28:3090-3091.
17 Vallet B. Bench-to-bedside review: endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Crit Care. 2003;7:130-138.
18 Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765-3777.
19 Drexler H. Nitric oxide synthases in the failing human heart: a doubled-edged sword? Circulation. 1999;99:2972-2975.
20 Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28:N1-N13.
21 Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29:S99-S106.
22 Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219-223.
23 Baudouin SV, Saunders D, Tiangyou W, et al. Mitochondrial DNA and survival after sepsis: a prospective study. Lancet. 2005;366:2118-2121.
24 Fink MP. Bench-to-bedside review: cytopathic hypoxia. Crit Care. 2002;6:491-499.
25 Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:21-237.
26 Eidelman LA, Putterman D, Putterman C, et al. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996;275:470-473.
27 Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med. 1995;332:27-37.
28 Doig CJ, Sutherland LR, Sandham JD, et al. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444-451.
29 Baumgartner-Parzer SM, Wagner L, Pettermann M, et al. High-glucose-triggered apoptosis in cultured endothelial cells. Diabetes. 1995;44:1323-1327.
30 Hermann C, Assmus B, Urbich C, et al. Insulin-mediated stimulation of protein kinase Akt: a potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:402-409.
31 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
32 Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862-871.
33 Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
34 Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332-1341.
35 Nadel S, Goldstein B, Williams MD, et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet. 2007;369:836-843.
36 Warren BL, Eid A, Singer P, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled tria. JAMA. 2001;286:1869-1878.
37 Wiedermann CJ, Hoffmann JN, Juers M, et al. High-dose antithrombin III in the treatment of severe sepsis in patients with a high risk of death: efficacy and safety. Crit Care Med. 2006;34:285-292.
38 Abraham E, Reinhart K, Opal S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238-247.
39 Mayer K, Fegbeutel C, Hattar K, et al. Omega-3 vs. omega-6 lipid emulsions exert differential influence on neutrophils in septic shock patients: impact on plasma fatty acids and lipid mediator generation. Intens Care Med. 2003;29:1472-1481.
40 Pacht ER, DeMichele SJ, Nelson JL, et al. Enteral nutrition with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants reduces alveolar inflammatory mediators and protein influx in patients with acute respiratory distress syndrome. Crit Care Med. 2003;31:491-500.
41 Heering P, Morgera S, Schmitz FJ, et al. Cytokine removal and cardiovascular hemodynamics in septic patients with continuous venovenous hemofiltration. Intens Care Med. 1997;23:288-296.
42 Watson D, Grover R, Anzueto A, et al. Cardiovascular effects of the nitric oxide synthase inhibitor NG-methyl-L-arginine hydrochloride (546C88) in patients with septic shock: results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002). Crit Care Med. 2004;32:1-20.
43 Knaus WA, Draper EA, Wagner DP, et al. Prognosis in acute organ-system failure. Ann Surg. 1985;202:685-693.
44 Marshall JC, Cook DJ, Christou NV, et al. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23:1638-1652.
45 Barriere SL, Lowry SF. An overview of mortality risk prediction in sepsis. Crit Care Med. 1995;23:376-393.
46 Zimmerman JE, Knaus WA, Wagner DP, et al. A comparison of risks and outcomes for patients with organ system failure: 1982–1990. Crit Care Med. 1996;24:1633-1641.
47 Nfor TK, Walsh TS, Prescott RJ. The impact of organ failures and their relationship with outcome in intensive care: analysis of a prospective multicentre database of adult admissions. Anaesthesia. 2006;61:731-778.
48 Pettila V, Kaarlola A, Makelainen A. Health-related quality of life of multiple organ dysfunction patients one year after intensive care. Intens Care Med. 2000;26:1473-1479.
49 Nielsen D, Sellgren J, Ricksten SE. Quality of life after cardiac surgery complicated by multiple organ failure. Crit Care Med. 1997;25:52-57.
50 Weinert CR, Gross CR, Kangas JR, et al. Health-related quality of life after acute lung injury. Am J Respir Crit Care Med. 1997;156:1120-1128.