CHAPTER 207 Moyamoya Disease
An increasingly recognized cause of stroke, including 6% of all strokes in children, moyamoya syndrome is a cerebrovascular disorder characterized by chronic progressive stenosis of the intracranial internal carotid arteries (ICAs), including the proximal anterior cerebral arteries (ACAs) and middle cerebral arteries (MCAs).1,2 This inexorable occlusion of the anterior circulation occurs simultaneously as characteristic arterial collateral vessels develop at the base of the brain in response to the resultant ischemia. Rarely, in advanced cases, this process can extend to involve the posterior circulation, including the basilar artery and posterior cerebral artery (PCA).
Takeuchi and Shimizu first reported a case of hypoplasia of the bilateral ICAs in 1957, although the term moyamoya (“something hazy, like a puff of smoke”), which describes the characteristic angiographic appearance of abnormally dilated collateral vessels in this condition, was not used until 1969 by Suzuki and Takaku (Fig. 207-1).3,4
Individuals with a well-recognized associated condition (see later) are categorized as having moyamoya syndrome, whereas idiopathic cases with no known risk factors have moyamoya disease. To have moyamoya disease, patients must have bilateral stenosis—patients with only unilateral findings have moyamoya syndrome.5 The term moyamoya, when used alone without the modifier of disease or syndrome, refers to the distinctive findings on arteriography, independent of etiology.
Epidemiology
First described in Japan, moyamoya has now been identified in patients worldwide.6 Although historically considered more prevalent in the Asian population, it affects individuals of many ethnic backgrounds, and there is increasing awareness of this disease in Europe and North America.7 In Japan, it is the most common pediatric cerebrovascular disease.1 In Europe, a recent study cited an incidence of 0.3 patients per center per year, which is approximately a 10th of the incidence in Japan.8 In the United States and Korea, reports have corroborated historical claims of a bimodal age distribution of moyamoya, one group in the pediatric age range (around the first decade of life) and a second group of adults in the 30- to 40-year-old range. Females are affected nearly twice as often as males.9–11 In the United States, moyamoya affects individuals differently, depending on ethnicity; when compared with white individuals, Asian Americans are more than four times as likely, African Americans are twice as likely, and Hispanic Americans are half as likely to have moyamoya.12
Associated Conditions
Particularly strong associations exist between moyamoya and radiotherapy of the head or neck (especially for optic gliomas, craniopharyngiomas, and pituitary tumors), Down syndrome, neurofibromatosis type 1 (with or without hypothalamic–optic pathway tumors), and sickle cell anemia.11,13–15 More tenuous links may exist between moyamoya and other disorders (Table 207-1).11,16
TABLE 207-1 Associated Conditions, Risk Factors, or Syndromes
| RISK FACTOR | NUMBER |
|---|---|
| No associated conditions (idiopathic) | 66 |
| Neurofibromatosis type 1 | 16 |
| Asian | 16 |
| Cranial therapeutic radiation | 15 |
| Hypothalamic–optic system glioma: 8 | |
| Craniopharyngioma: 4 | |
| Medulloblastoma with Gorlin’s syndrome: 1 | |
| Acute lymphocytic leukemia, intrathecal chemotherapy: 2 | |
| Down syndrome | 10 |
| Congenital cardiac anomaly, previously operated | 7 |
| Renal artery stenosis | 4 |
| Hemoglobinopathy (2 sickle cell, 1 “Bryn Mawr”) | 3 |
| Other hematologic: 1 spherocytosis, 1 ITP | 2 |
| Giant cervicofacial hemangiomas | 3 |
| Shunted hydrocephalus | 3 |
| Idiopathic hypertension requiring medication | 3 |
| Hyperthyroidism (one with Graves’ syndrome) | 2 |
Other syndromes, one patient each: Reyes’ (remote), Williams’, Alagille’s, cloacal exstrophy, renal artery fibromuscular dysplasia, and infection with congenital cytomegalic inclusion virus (remote). Two patients had unclassified syndromic manifestations. There were four African Americans, two of whom had sickle cell disease.
ITP, idiopathic thrombocytopenic purpura.
Pathophysiology
Pathologic analysis has demonstrated that affected vessels generally do not exhibit arteriosclerotic or inflammatory changes.17 Rather, vessel occlusion results from a combination of both hyperplasia of smooth muscle cells and luminal thrombosis (Fig. 207-2). The moyamoya collaterals are dilated perforating arteries believed to be a combination of preexisting and newly developed vessels.18,19 A number of growth factors, enzymes, and other peptides have been reported in association with moyamoya, including basic fibroblast growth factor, transforming growth factor-β1, hepatocyte growth factor, vascular endothelial growth factor, matrix metalloproteinases, intracellular adhesion molecules, and hypoxia-inducing factor-1α, among others.19–26 At present, it is difficult to discern which of these peptides are pathologic primary causal agents of disease and which are present merely as part of a normal response to ischemia.
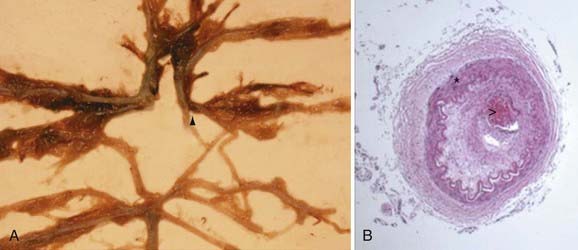
Clues helping to distinguish between these possibilities have come from genetic studies. Associations between moyamoya and loci on chromosomes 3, 6, 8, and 17 (MYMY1, MYMY2, MYMY3), as well as specific HLA haplotypes, have been described.27–32 The familial incidence of affected first-degree relatives in Japan is 7% to 12%, similar to a rate of 6% reported in a recent U.S. series.11,33–38 However, despite evidence supporting a genetic basis of moyamoya, important caveats remain. Reports exist of identical twins with only one affected sibling.11,39 These data support the premise that some type of environmental factor must precipitate the syndrome’s clinical emergence in susceptible patients and suggest that the angiographic changes of moyamoya are the result of a complex interplay between genetic predisposition and external stimuli.
Clinical Findings
Accounting for approximately 6% of childhood strokes in Western countries, moyamoya affects young children in particular, with 50% of patients identified by 10 years of age.1,2 Some patients have rare, intermittent ischemic events or even extended periods of clinical stability, whereas other individuals exhibit rapid neurological decline.11,40 Children are usually initially seen with TIAs or strokes, as evidenced in the largest current report of pediatric moyamoya patients (Table 207-2).11 The much higher rate of stroke in children may be related to less developed verbal skills in this age group, thereby leading to delayed recognition of the underlying moyamoya.41
TABLE 207-2 Symptoms at Initial Evaluation in 143 Patients (Percentage of Patients with Symptoms)
| Stroke | 97 | (67.8%) |
| Transient ischemic attacks (including drop attacks) | 62 | (43.4%) |
| Seizures | 9 | (6.3%) |
| Headache | 9 | (6.3%) |
| Choreiform movements | 6 | (4.2%) |
| Incidental | 6 | (4.2%) |
| Intraventricular or intracerebral bleeding | 4 | (2.8%) |
Symptom totals are greater than patient numbers because some patients had multiple symptoms at initial evaluation.
Ischemic Symptoms
The symptoms of cerebral ischemia in moyamoya are generally related to the territory supplied by the ICAs, including the frontal and temporal lobes. Hemiparesis, dysarthria, aphasia, and cognitive impairment are common.11 Seizures occur frequently, and other, more subtle deficits may be present—an issue particularly problematic in younger patients because they may not be able to articulate their experiences as well as adults. These symptoms may be mistaken for psychiatric illness or developmental delay and include visual deficits, syncope, or personality changes.42–44
Once present, the ischemic symptoms may be transient or fixed. TIAs may be precipitated by events particularly common in children, such as hyperventilation with crying or exertion. The cerebral vessels, already maximally dilated in the setting of chronic ischemia, constrict in response to the decrease in PCO2 and can thus lead to a TIA or stroke.45 Dehydration, a problem in children after colds or fevers, may also give rise to ischemic symptoms.
Hemorrhage
Typically, hemorrhage is a hallmark of adult moyamoya, although children can also have this finding initially.11,46 Hemorrhage can be intraventricular, intraparenchymal (frequently in the region of the basal ganglia), or subarachnoid. Bleeding has historically been attributed to rupture of fragile collateral vessels unable to contain the increased flow shunted from progressive ICA stenosis.47,48 In addition to these friable vessels, studies have revealed that aneurysms in the circle of Willis may form in response to altered circulatory patterns, thus providing another potential cause of hemorrhage.49,50
Headache and Other Symptoms
Children with moyamoya will often complain of headache. Although the etiology is unclear, a recent review has speculated that dilation of meningeal and leptomeningeal collateral vessels may stimulate dural nociceptors.51 Typically, the headache is migraine-like in quality and refractory to medical therapies; although it may improve after surgical treatment of the moyamoya, often concordant with regression of the collateral vessels, it often persists as a troublesome symptom years after other symptoms remit.
Collateral vessels in the basal ganglia have also been implicated in the development of choreiform movements in individuals with advanced moyamoya.11,52 Similar to headache, these symptoms may regress after revascularization of the affected hemisphere.
Natural History and Prognosis
It is difficult to predict the natural history of moyamoya in a given patient. As discussed in the section “Clinical Findings,” patients can have isolated problems with lengthy periods of relative health or can exhibit fulminant deterioration in a very short time.11,40 However, moyamoya syndrome, in terms of both arteriopathy and clinical symptoms, inevitably progresses in untreated patients.4,53 A recent study revealed that the rate of disease progression is high even in asymptomatic patients and that medical therapy alone is of limited use.54 It has been estimated that two thirds of patients with moyamoya have symptomatic progression with poor outcomes if left untreated.55–57 This number contrasts strikingly with an estimated rate of symptomatic progression of just 2.6% after surgical treatment in a recent meta-analysis of more than 1000 patients.58 In general, neurological status at time of treatment, more so than age of the patient, predicts long-term outcome.11,59,60 The unpredictable but relentless course of this disease, coupled with the irreversible nature of deficits once present, dictates a need for early diagnosis whenever possible and prompt treatment once moyamoya is identified.
Diagnosis
Computed Tomography
In most patients with suspected stroke or intracranial hemorrhage, the evaluation typically begins with computed tomography (CT) of the head. In patients with moyamoya, hemorrhage or small areas of hypodensity suggestive of stroke are commonly observed in the cortical watershed zones, basal ganglia, deep white matter, or periventricular regions.61,62
Magnetic Resonance Imaging
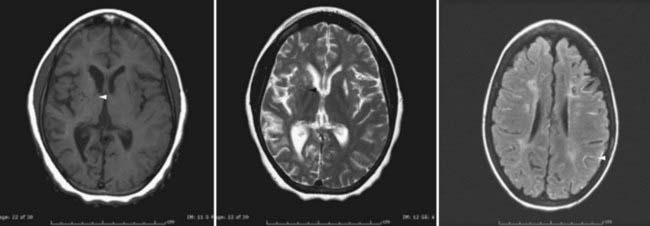
In recent years, magnetic resonance imaging (MRI) has become more widely available, which has led to substantial increases in its use as primary diagnostic modality for patients suspected of having a stroke—including patients undergoing evaluation for moyamoya.63–68 Acute infarction is best seen with diffusion-weighted imaging, whereas chronic infarction is better demonstrated on T1- and T2-weighted images (Fig. 207-3). Diminished cortical blood flow secondary to moyamoya can be inferred from fluid-attenuated inversion recovery (FLAIR) sequences, which demonstrate linear high signal following a sulcal pattern, the so-called ivy sign (Fig. 207-3).69–72 Reduced flow voids in the ICA, MCA, and ACA, coupled with prominent flow voids from the basal ganglia and thalamic collateral vessels, are considered by many to be essentially diagnostic of moyamoya.62,63,73–76 However, it is important to note that although MRI can often establish a diagnosis of moyamoya, it remains limited with regard to helping identify specific collateral networks and areas of marked stenosis—data frequently critical for successful surgical planning.
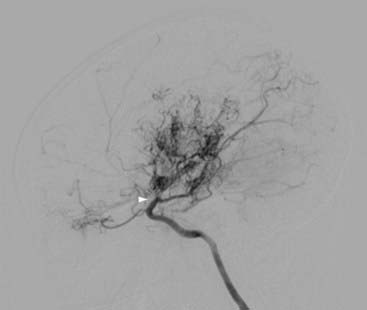
Angiography
Angiography provides crucial surgical planning data and should be performed in all moyamoya patients if possible. In a study of 190 angiograms, the complication rate from angiography in patients with moyamoya syndrome was no higher than that in non–moyamoya-affected populations with other forms of cerebrovascular disease.77 Studies should include all four vessels and external carotid artery (ECA) injections.
The diagnosis of moyamoya is defined by angiography and is characterized by bilateral stenosis of the distal intracranial ICA extending to the proximal ACA, MCA, and PCA, with disease severity classified into six progressive stages by Suzuki and Takaku (see Fig. 207-1).4
Other Diagnostic Techniques
Electroencephalography (EEG) and cerebral blood flow studies may be helpful in the evaluation of patients with moyamoya. Specific findings on EEG, usually in pediatric patients, include posterior or centrotemporal slowing, a hyperventilation-induced diffuse pattern of monophasic slow waves (i.e., build-up), and a characteristic “re–build-up” phenomenon78 (maneuver not recommended in a patient with known moyamoya; see later).
Cerebral blood flow studies include techniques such as transcranial Doppler, perfusion CT, xenon-enhanced CT, positron emission tomography, MR perfusion imaging,79–81 and single-photon emission CT with acetazolamide challenge. These studies can help quantify blood flow, information that some clinicians incorporate into treatment algorithms for children with moyamoya.82–90
Treatment
Medical Therapy
When an individual is considered a poor operative risk or has relatively mild disease, medical therapy has occasionally been used for the treatment of moyamoya. However, there are few data demonstrating either short-term or long-term efficacy of this approach. Antiplatelet agents have been used in individuals whose ischemic symptoms seem to arise as a result of emboli from the formation of microthrombi at sites of arterial stenosis and have been administered routinely in all patients in many operative series.11,40,91,92 Anticoagulants such as warfarin are rarely used, although there has been some experience with low-molecular-weight heparin.93 The other type of medication helpful in the management of moyamoya is calcium channel blockers.91 This class of drugs is particularly useful in ameliorating the symptoms of intractable headache or migraine, commonly seen in moyamoya patients, and also seems to be effective in reducing both the frequency and severity of refractory TIA. The use of calcium channel blockers is generally limited to patients with these symptoms, but caution needs to be exercised to avoid hypotension.
Published reports directly comparing outcomes between medical and surgical therapy for moyamoya are limited in number. A large survey from Japan in 1997 found no significant differences in outcome between medically and surgically treated moyamoya patients in the short term, although a more recent review revealed that 38% of 651 moyamoya patients who were initially treated medically subsequently required surgery as a result of progressive symptoms.5,94
Surgery
Traditionally, direct procedures have been used in adults, with immediate restoration of blood supply being cited as a major benefit. Protection from ischemia is delayed for several weeks with indirect techniques while new vessel ingrowth is established. However, direct bypass is often technically difficult to perform in children because of the small size of donor and recipient vessels, thus making indirect techniques appealing in pediatric populations. Nonetheless, direct operations have been successful in children, as have indirect procedures in adults.95–97 Considerable debate exists regarding the relative merits and shortcomings of the two approaches, with some centers advocating combinations of both.94,97–100
Numerous variations of indirect revascularization procedures exist, including encephaloduroarteriosynangiosis, encephalomyoarteriosynangiosis, and simply drilling bur holes without vessel synangiosis.99,101–105 A modification termed pial synangiosis has been used with encouraging results in both adults and children (Fig. 207-4).11 A review of 143 patients treated by pial synangiosis demonstrated marked reductions in stroke frequency after surgery. Sixty-seven percent had strokes before treatment, 7.7% had strokes in the perioperative period, and just 3.2% had strokes after at least 1 year of follow-up. The long-term stroke rate was 4.3% in individuals with a minimum of 5 years of follow-up.11
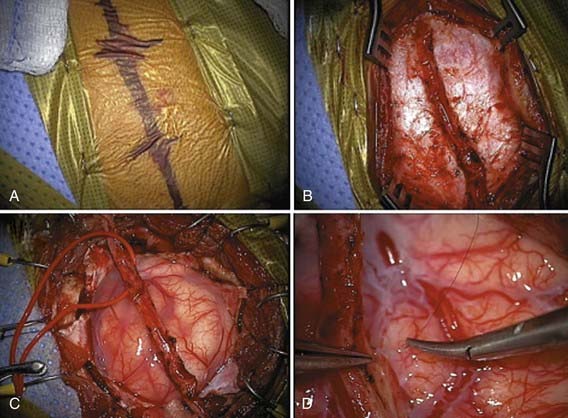
Given the poor response to medical therapy, the relentless course of the disease, and the excellent response to surgery, operative indications for revascularization in moyamoya are broad.58 Two large studies with long-term follow-up demonstrated a good safety profile for surgical treatment of moyamoya (approximately 4% risk for stroke within 30 days of surgery per hemisphere), with a 96% probability of remaining stroke free over a 5-year follow-up period.11,55 A recent meta-analysis of 1156 moyamoya patients treated surgically concluded that 87% derived symptomatic benefit from surgical revascularization, with indirect and direct/combined techniques demonstrating equal effectiveness.58 In general, all patients with documented moyamoya should be considered operative candidates.
Perioperative Care
Moyamoya patients are at additional risk for ischemic events during the perioperative period. Potential complications of surgery for moyamoya include stroke, infection, and intracranial hemorrhage. Crying and hyperventilation can lower PaCO2 and induce ischemia secondary to cerebral vasoconstriction. Any techniques to reduce pain—including the use of perioperative sedation, painless wound-dressing techniques, and closure of the wound with absorbable suture—may reduce the likelihood of stroke and shorten the hospitalization.106 Similarly, it is important to avoid hypotension, hypovolemia, hyperthermia, and hypocapnia both intraoperatively and perioperatively.11 Postoperatively, patients should be given intravenous fluids at 1.25 to 1.5 times the normal maintenance rate for 48 to 72 hours.106
Acute Symptoms
In addition to providing surgical therapy for patients with moyamoya, there is often a need to ameliorate acute symptoms. Rapid institution of measures capable of increasing cerebral blood flow and oxygenation can reduce the likelihood of a TIA progressing to a completed stroke. The initial steps are similar to perioperative management and should include intravenous hydration with isotonic fluids (usually at 1 to 1.5 times maintenance), avoidance of hypotension, and administration of supplemental oxygen.11,106,107 Hyperventilation is to be avoided. Serum electrolyte and glucose levels should be normalized. Seizure activity, if present, should be treated with appropriate pharmacologic agents.
Emergency imaging can ascertain whether ischemia or hemorrhage is the cause of the new symptoms. Although commonly evaluated with CT, MRI with diffusion-weighted images can be helpful in the identification of completed stroke. In the absence of hemorrhage, antiplatelet agents have been used on the assumption that emboli from microthrombus formation at sites of arterial stenosis may contribute to the symptoms.11,40,91,92 Aspirin is used at many institutions: 325 mg for adults and 81 mg (or less) for preteen children.
Follow-up
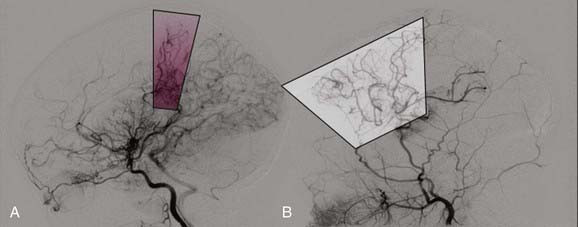
Several reports suggest that periodic clinical and radiographic re-examination of patients with moyamoya disease may be helpful in some clinical settings.5,58 Recent studies have demonstrated that bilateral involvement eventually develops in approximately a third of patients with unilateral moyamoya.108,109 There is also evidence that progression of the disease is more likely to occur rapidly and more frequently in younger patients.108,109 Of patients who underwent surgery (for either bilateral or unilateral disease), reoperation because of refractory disease was necessary in 1.8% to 18% of patients.94 Many institutions perform angiography 1 year postoperatively, followed by annual MRI studies for several years thereafter (Fig. 207-5).
Fukui Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg. 1997;99(suppl 2):S238-S240.
Fung Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst. 2005;21:358-364.
Scott Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 360(12), 2009. 1226-1223
1 Nagaraja D, Verma A, Taly AB, et al. Cerebrovascular disease in children. Acta Neurol Scand. 1994;90:251-255.
2 Soriano SG, Sethna NF, Scott RM. Anesthetic management of children with moyamoya syndrome. Anesth Analg. 1993;77:1066-1070.
3 Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerve. 1957;9:37-43.
4 Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease: Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20:288-299.
5 Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg. 1997;99(suppl 2):S238-S240.
6 Suzuki J, Kodama N. Moyamoya disease—a review. Stroke. 1983;14:104-109.
7 Caldarelli M, Di Rocco C, Gaglini P. Surgical treatment of moyamoya disease in pediatric age. J Neurosurg Sci. 2001;45:83-91.
8 Yonekawa Y, Ogata N, Kaku Y, et al. Moyamoya disease in Europe, past and present status. Clin Neurol Neurosurg. 1997;99(suppl 2):S58-S60.
9 Baba T, Houkin K, Kuroda S. Novel epidemiological features of moyamoya disease. J Neurol Neurosurg Psychiatry. 2008;79:900-904.
10 Wakai K, Tamakoshi A, Ikezaki K, et al. Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey. Clin Neurol Neurosurg. 1997;99(suppl 2):S1-S5.
11 Scott RM, Smith JL, Robertson RL, et al. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg Spine. 2004;100:142-149.
12 Uchino K, Johnston SC, Becker KJ, et al. Moyamoya disease in Washington State and California. Neurology. 2005;65:956-958.
13 Hankinson TC, Bohman LE, Heyer G, et al. Surgical treatment of moyamoya syndrome in patients with sickle cell anemia: outcome following encephaloduroarteriosynangiosis. J Neurosurg Pediatr. 2008;1:211-216.
14 Ullrich NJ, Robertson R, Kinnamon DD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. 2007;68:932-938.
15 Jea A, Smith ER, Robertson R, et al. Moyamoya syndrome associated with Down syndrome: outcome after surgical revascularization. Pediatrics. 2005;116:e694-701.
16 Connor SE, Hewes D, Ball C, et al. Alagille syndrome associated with angiographic moyamoya. Childs Nerv Syst. 2002;18:186-190.
17 Fukui M, Kono S, Sueishi K, et al. Moyamoya disease. Neuropathology. 2000;20(suppl):S61-S64.
18 Kono S, Oka K, Sueishi K. Histopathologic and morphometric studies of leptomeningeal vessels in moyamoya disease. Stroke. 1990;21:1044-1050.
19 Lim M, Cheshier S, Steinberg GK. New vessel formation in the central nervous system during tumor growth, vascular malformations, and moyamoya. Curr Neurovasc Res. 2006;3:237-245.
20 Takagi Y, Kikuta K, Nozaki K, et al. Expression of hypoxia-inducing factor-1 alpha and endoglin in intimal hyperplasia of the middle cerebral artery of patients with moyamoya disease. Neurosurgery. 2007;60:338-345.
21 Malek AM, Connors S, Robertson RL, et al. Elevation of cerebrospinal fluid levels of basic fibroblast growth factor in moyamoya and central nervous system disorders. Pediatr Neurosurg. 1997;27:182-189.
22 Nanba R, Kuroda S, Ishikawa T, et al. Increased expression of hepatocyte growth factor in cerebrospinal fluid and intracranial artery in moyamoya disease. Stroke. 2004;35:2837-2842.
23 Soriano SG, Cowan DB, Proctor MR, et al. Levels of soluble adhesion molecules are elevated in the cerebrospinal fluid of children with moyamoya syndrome. Neurosurgery. 2002;50:544-549.
24 Ueno M, Kira R, Matsushima T, et al. Moyamoya disease and transforming growth factor-beta1. J Neurosurg. 2000;92:907-908.
25 Hojo M, Hoshimaru M, Miyamoto S, et al. Role of transforming growth factor-beta1 in the pathogenesis of moyamoya disease. J Neurosurg. 1998;89:623-629.
26 Yoshimoto T, Houkin K, Takahashi A, Abe H. Angiogenic factors in moyamoya disease. Stroke. 1996;27:2160-2165.
27 Ikeda H, Sasaki T, Yoshimoto T, et al. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am J Hum Genet. 1999;64:533-537.
28 Nanba R, Tada M, Kuroda S, et al. Sequence analysis and bioinformatics analysis of chromosome 17q25 in familial moyamoya disease. Childs Nerv Syst. 2005;21:62-68.
29 Inoue TK, Ikezaki K, Sasazuki T, et al. Linkage analysis of moyamoya disease on chromosome 6. J Child Neurol. 2000;15:179-182.
30 Inoue TK, Ikezaki K, Sasazuki T, et al. Analysis of class II genes of human leukocyte antigen in patients with moyamoya disease. Clin Neurol Neurosurg. 1997;99(suppl 2):S234-S237.
31 Han H, Pyo CW, Yoo DS, et al. Associations of moyamoya patients with HLA class I and class II alleles in the Korean population. J Korean Med Sci. 2003;18:876-880.
32 Sakurai K, Horiuchi Y, Ikeda H, et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004;49:278-281.
33 Kitahara T, Ariga N, Yamaura A, et al. Familial occurrence of moya-moya disease: report of three Japanese families. J Neurol Neurosurg Psychiatry. 1979;42:208-214.
34 Sogaard I, Jorgensen J. Familial occurrence of bilateral intracranial occlusion of the internal carotid arteries (Moya Moya). Acta Neurochir (Wien). 1975;31:245-252.
35 Hamada JI, Yoshioka S, Nakahara T, et al. Clinical features of moyamoya disease in sibling relations under 15 years of age. Acta Neurochir (Wien). 1998;140:455-458.
36 Nanba R, Kuroda S, Tada M, et al. Clinical features of familial moyamoya disease. Childs Nerv Syst. 2006;22:258-262.
37 Nanba R, Kuroda S, Ishikawa T, et al. [Familial moyamoya disease—clinical features and current study.]. No Shinkei Geka. 2004;32:7-16.
38 Kaneko Y, Imamoto N, Mannoji H, et al. Familial occurrence of moyamoya disease in the mother and four daughters including identical twins. Neurol Med Chir (Tokyo). 1998;38:349-354.
39 Tanghetti B, Capra R, Giunta F, et al. Moyamoya syndrome in only one of two identical twins. Case report. J Neurosurg. 1983;59:1092-1094.
40 Ohaegbulam C, Magge S, Scott RM. Moyamoya syndrome. In: McLone D, editor. Pediatric Neurosurgery. Philadelphia: WB Saunders; 2001:1077-1092.
41 Nishimoto A, Ueta K, Onbe H. Cooperative study on moyamoya disease in Tokyo, Japan. Abstracts of the 10th Meeting on Surgery for Stroke. 1981:53-58.
42 Karasawa J, Touho H, Ohnishi H, et al. Cerebral revascularization using omental transplantation for childhood moyamoya disease. J Neurosurg. 1993;79:192-196.
43 Lubman DI, Pantelis C, Desmond P, et al. Moyamoya disease in a patient with schizophrenia. J Int Neuropsychol Soc. 2003;9:806-810.
44 Miyamoto S, Kikuchi H, Karasawa J, et al. Study of the posterior circulation in moyamoya disease. Part 2: Visual disturbances and surgical treatment. J Neurosurg. 1986;65:454-460.
45 Tagawa T, Naritomi H, Mimaki T, et al. Regional cerebral blood flow, clinical manifestations, and age in children with moyamoya disease. Stroke. 1987;18:906-910.
46 Han DH, Nam DH, Oh CW. Moyamoya disease in adults: characteristics of clinical presentation and outcome after encephalo-duro-arterio-synangiosis. Clin Neurol Neurosurg. 1997;99(suppl 2):S151-S155.
47 Iwama T, Morimoto M, Hashimoto N, et al. Mechanism of intracranial rebleeding in moyamoya disease. Clin Neurol Neurosurg. 1997;99(suppl 2):S187-S190.
48 Irikura K, Miyasaka Y, Kurata A, et al. A source of haemorrhage in adult patients with moyamoya disease: the significance of tributaries from the choroidal artery. Acta Neurochir (Wien). 1996;138:1282-1286.
49 Kawaguchi S, Sakaki T, Morimoto T, et al. Characteristics of intracranial aneurysms associated with moyamoya disease. A review of 111 cases. Acta Neurochir (Wien). 1996;138:1287-1294.
50 Kuroda S, Houkin K, Kamiyama H, et al. Effects of surgical revascularization on peripheral artery aneurysms in moyamoya disease: report of three cases. Neurosurgery. 2001;49:463-467.
51 Seol HJ, Wang KC, Kim SK, et al. Headache in pediatric moyamoya disease: review of 204 consecutive cases. J Neurosurg. 2005;103:439-442.
52 Parmar RC, Bavdekar SB, Muranjan MN, et al. Chorea: an unusual presenting feature in pediatric moyamoya disease. Indian Pediatr. 2000;37:1005-1009.
53 Imaizumi T, Hayashi K, Saito K, et al. Long-term outcomes of pediatric moyamoya disease monitored to adulthood. Pediatr Neurol. 1998;18:321-325.
54 Kuroda S, Ishikawa T, Houkin K, et al. Incidence and clinical features of disease progression in adult moyamoya disease. Stroke. 2005;36:2148-2153.
55 Choi JU, Kim DS, Kim EY, et al. Natural history of moyamoya disease: comparison of activity of daily living in surgery and non surgery groups. Clin Neurol Neurosurg. 1997;99(suppl 2):S11-S18.
56 Kurokawa T, Chen YJ, Tomita S, et al. Cerebrovascular occlusive disease with and without the moyamoya vascular network in children. Neuropediatrics. 1985;16:29-32.
57 Ezura M, Takahashi A, Yoshimoto T. Successful treatment of an arteriovenous malformation by chemical embolization with estrogen followed by conventional radiotherapy. Neurosurgery. 1992;31:1105-1107.
58 Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst. 2005;21:358-364.
59 Fukuyama Y, Umezu R. Clinical and cerebral angiographic evolutions of idiopathic progressive occlusive disease of the circle of Willis (“moyamoya” disease) in children. Brain Dev. 1985;7:21-37.
60 Maki Y, Enomoto T. Moyamoya disease. Childs Nerv Syst. 1988;4:204-212.
61 Shin IS, Cheng R, Pordell GR. Striking CT scan findings in a case of unilateral moyamoya disease—a case report. Angiology. 1991;42:665-671.
62 Fujita K, Shirakuni T, Kojima N, et al. [Magnetic resonance imaging in moyamoya disease.]. No Shinkei Geka. 1986;14:324-330.
63 Yamada I, Matsushima Y, Suzuki S. Moyamoya disease: diagnosis with three-dimensional time-of-flight MR angiography. Radiology. 1992;184:773-778.
64 Yamada I, Suzuki S, Matsushima Y. Moyamoya disease: comparison of assessment with MR angiography and MR imaging versus conventional angiography. Radiology. 1995;196:211-218.
65 Yamada I, Suzuki S, Matsushima Y. Moyamoya disease: diagnostic accuracy of MRI. Neuroradiology. 1995;37:356-361.
66 Katz DA, Marks MP, Napel SA, et al. Circle of Willis: evaluation with spiral CT angiography, MR angiography, and conventional angiography. Radiology. 1995;195:445-449.
67 Takanashi JI, Sugita K, Niimi H. Evaluation of magnetic resonance angiography with selective maximum intensity projection in patients with childhood moyamoya disease. Eur J Paediatr Neurol. 1998;2:83-89.
68 Chang KH, Yi JG, Han MH, et al. MR imaging findings of moyamoya disease. J Korean Med Sci. 1990;5:85-90.
69 Maeda M, Tsuchida C. “Ivy sign” on fluid-attenuated inversion-recovery images in childhood moyamoya disease. AJNR Am J Neuroradiol. 1999;20:1836-1838.
70 Yoon HK, Shin HJ, Chang YW. “Ivy sign” in childhood moyamoya disease: depiction on FLAIR and contrast-enhanced T1-weighted MR images. Radiology. 2002;223:384-389.
71 Fujiwara H, Momoshima S, Kuribayashi S. Leptomeningeal high signal intensity (ivy sign) on fluid-attenuated inversion-recovery (FLAIR) MR images in moyamoya disease. Eur J Radiol. 2005;55:224-230.
72 Chabbert V, Ranjeva JP, Sevely A, et al. Diffusion- and magnetisation transfer-weighted MRI in childhood moya-moya. Neuroradiology. 1998;40:267-271.
73 Sunaga Y, Fujinaga T, Ohtsuka T. [MRI findings of moyamoya disease in children.]. No To Hattatsu. 1992;24:375-379.
74 Brady AP, Stack JP, Ennis JT. Moyamoya disease—imaging with magnetic resonance. Clin Radiol. 1990;42:138-141.
75 Rolak LA. Magnetic resonance imaging in moyamoya disease. Arch Neurol. 1989;46:14.
76 Bruno A, Yuh WT, Biller J, et al. Magnetic resonance imaging in young adults with cerebral infarction due to moyamoya. Arch Neurol. 1988;45:303-306.
77 Robertson RL, Chavali RV, Robson CD, et al. Neurologic complications of cerebral angiography in childhood moyamoya syndrome. Pediatr Radiol. 1998;28:824-829.
78 Kodama N, Aoki Y, Hiraga H, et al. Electroencephalographic findings in children with moyamoya disease. Arch Neurol. 1979;36:16-19.
79 Kim JH, Shin T, Park JH, et al. Various patterns of perfusion-weighted MR imaging and MR angiographic findings in hyperacute ischemic stroke. AJNR Am J Neuroradiol. 1999;20:613-620.
80 Seitz RJ, Meisel S, Weller P, et al. Initial ischemic event: perfusion-weighted MR imaging and apparent diffusion coefficient for stroke evolution. Radiology. 2005;237:1020-1028.
81 Koga M, Reutens DC, Wright P, et al. The existence and evolution of diffusion-perfusion mismatched tissue in white and gray matter after acute stroke. Stroke. 2005;36:2132-2137.
82 Takase K, Kashihara M, Hashimoto T. Transcranial Doppler ultrasonography in patients with moyamoya disease. Clin Neurol Neurosurg. 1997;99(suppl 2):S101-S105.
83 Morgenstern C, Griewing B, Muller-Esch G, et al. Transcranial power-mode duplex ultrasound in two patients with moyamoya syndrome. J Neuroimaging. 1997;7:190-192.
84 Ikezaki K, Matsushima T, Kuwabara Y, et al. Cerebral circulation and oxygen metabolism in childhood moyamoya disease: a perioperative positron emission tomography study. J Neurosurg. 1994;81:843-850.
85 Tanaka Y, Nariai T, Nagaoka T, et al. Quantitative evaluation of cerebral hemodynamics in patients with moyamoya disease by dynamic susceptibility contrast magnetic resonance imaging—comparison with positron emission tomography. J Cereb Blood Flow Metab. 2006;26:291-300.
86 Khan N, Yonekawa Y. Moyamoya angiopathy in Europe. Acta Neurochir Suppl. 2005;94:149-152.
87 Shirane R, Yoshida Y, Takahashi T, et al. Assessment of encephalo-galeo-myo-synangiosis with dural pedicle insertion in childhood moyamoya disease: characteristics of cerebral blood flow and oxygen metabolism. Clin Neurol Neurosurg. 1997;99(suppl 2):S79-S85.
88 Liu HM, Peng SS, Li YW. The preoperative and postoperative cerebral blood flow and vasoreactivity in childhood moyamoya disease. Keio J Med. 2000;49(suppl 1):A86-A89.
89 Nambu K, Suzuki R, Hirakawa K. Cerebral blood flow: measurement with xenon-enhanced dynamic helical CT. Radiology. 1995;195:53-57.
90 Takeuchi S, Tanaka R, Ishii R, et al. Cerebral hemodynamics in patients with moyamoya disease. A study of regional cerebral blood flow by the 133Xe inhalation method. Surg Neurol. 1985;23:468-474.
91 Scott RM. Moyamoya syndrome: a surgically treatable cause of stroke in the pediatric patient. Clin Neurosurg. 2000;47:378-384.
92 Scott RM. Surgery for moyamoya syndrome? Yes. Arch Neurol. 2001;58:128-129.
93 Bowen MD, Burak CR, Barron TF. Childhood ischemic stroke in a nonurban population. J Child Neurol. 2005;20:194-197.
94 Ikezaki K. Rational approach to treatment of moyamoya disease in childhood. J Child Neurol. 2000;15:350-356.
95 Isono M, Ishii K, Kobayashi H, et al. Effects of indirect bypass surgery for occlusive cerebrovascular diseases in adults. J Clin Neurosci. 2002;9:644-647.
96 Smith ER, Scott RM. Surgical management of moyamoya syndrome. Skull Base. 2005;15:15-26.
97 Veeravagu A, Guzman R, Patil CG, et al. Moyamoya disease in pediatric patients: outcomes of neurosurgical interventions. Neurosurg Focus. 2008;24(2):E16.
98 Matsushima T, Inoue T, Ikezaki K, et al. Multiple combined indirect procedure for the surgical treatment of children with moyamoya disease. A comparison with single indirect anastomosis with direct anastomosis. Neurosurgical Focus. 1998;5(5):e4.
99 Matsushima T, Inoue T, Katsuta T, et al. An indirect revascularization method in the surgical treatment of moyamoya disease—various kinds of indirect procedures and a multiple combined indirect procedure. Neurol Med Chir (Tokyo). 1998;38(suppl):297-302.
100 Matsushima Y. [Indirect anastomoses for moyamoya disease.]. No Shinkei Geka. 1998;26:769-786.
101 Kawaguchi S, Okuno S, Sakaki T. Effect of direct arterial bypass on the prevention of future stroke in patients with the hemorrhagic variety of moyamoya disease. J Neurosurg. 2000;93:397-401.
102 Houkin K, Kamiyama H, Abe H, et al. Surgical therapy for adult moyamoya disease. Can surgical revascularization prevent the recurrence of intracerebral hemorrhage? Stroke. 1996;27:1342-1346.
103 Sencer S, Poyanli A, Kiris T, et al. Recent experience with moyamoya disease in Turkey. Eur Radiol. 2000;10:569-572.
104 Houkin K, Kuroda S, Nakayama N. Cerebral revascularization for moyamoya disease in children. Neurosurg Clin N Am. 2001;12:575-584. ix
105 Dauser RC, Tuite GF, McCluggage CW. Dural inversion procedure for moyamoya disease. Technical note. J Neurosurg. 1997;86:719-723.
106 Nomura S, Kashiwagi S, Uetsuka S, et al. Perioperative management protocols for children with moyamoya disease. Childs Nerv Syst. 2001;17:270-274.
107 Fujiwara J, Nakahara S, Enomoto T, et al. The effectiveness of O2 administration for transient ischemic attacks in moyamoya disease in children. Childs Nerv Syst. 1996;12:69-75.
108 Kawano T, Fukui M, Hashimoto N, et al. Follow-up study of patients with “unilateral” moyamoya disease. Neurol Med Chir (Tokyo). 1994;34:744-747.
109 Smith ER, Scott RM. Progression of disease in unilateral moyamoya syndrome. Neurosurg Focus. 2008;24(2):E17.