Chapter 2 Motor control
Physiology of voluntary and involuntary movements
Movement, whether voluntary or involuntary, is produced by the contraction of muscle. Muscle, in turn, is normally controlled entirely by the anterior horn cells or alpha motoneurons. Some involuntary movement disorders arise from muscle, the alpha motoneuron axon, or the alpha motoneuron itself. While this territory might be considered neuromuscular disease, the border can be fuzzy and patients may well appear in the office of the movement disorder specialist. Examples of involuntary movement arising from neuromuscular disorders that will be discussed in subsequent chapters are listed in Table 2.1.
Table 2.1 Examples of involuntary movements arising from neuromuscular conditions
| Muscle |
| Alpha motoneuron axon |
| Anterior horn cell |
As the sole controller of muscle, the alpha motoneuron is clearly important in understanding the genesis of movement. The influences upon the alpha motoneuron are many and complex, but have been extensively studied. Here only the basics will be reviewed (Hallett, 2003b). Inputs onto the alpha motoneuron can be divided into the segmental inputs and the supraspinal inputs.
Segmental inputs onto the alpha motoneuron
Figure 2.1 depicts the reflex connections onto the alpha motoneuron.
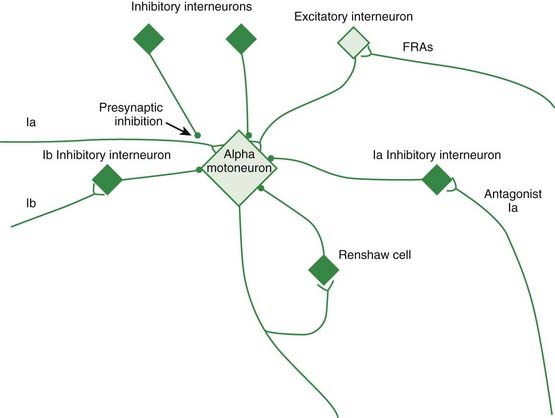
Presynaptic inhibition
Examples of movement disorders arising from segmental dysfunction that will be discussed in subsequent chapters are listed in Table 2.2.
Table 2.2 Examples of movement disorders arising from segmental dysfunction
| Disorder | Mechanism |
|---|---|
| Tetanus | Tetanus toxin blocks the release of GABA and glycine at spinal synapses |
| Stiff-person syndrome | Mainly a disorder of GABA and presynaptic inhibition in the spinal cord |
| Hereditary hyperekplexia | A disorder of glycine receptors with deficient inhibition at multiple synapses including that from the Ia inhibitory interneuron |
Supraspinal control of the alpha motoneuron
The main supraspinal control comes from the corticospinal tract. Approximately 30% of the corticospinal tract arises from the primary motor cortex, and other significant contributions come from the premotor and sensory cortices. The fibers largely cross in the pyramid, but some remain uncrossed. Some terminate as monosynaptic projections onto alpha motoneurons, and others terminate on interneurons including those in the dorsal horn. Other cortical neurons project to basal ganglia, cerebellum, and brainstem, and these structures can also originate spinal projections. Particularly important is the reticular formation that originates several reticulospinal tracts with different functions (Nathan et al., 1996) The nucleus reticularis gigantocellularis mediates some long loop reflexes and is hyperactive in a form of myoclonus. The nucleus reticularis pontis oralis mediates the startle reflex. The inhibitory dorsal reticulospinal tract may have particular relevance for spasticity (Takakusaki et al., 2001). In thinking about the cortical innervation of the reticular formation, it is possible to speak of a corticoreticulospinal tract. The rubrospinal tract, originating in the magnocellular division of the red nucleus, while important in lower primates, is virtually absent in humans.
The basal ganglia
The basal ganglia are of critical importance to many movement disorders, and details of their anatomy are presented in Chapter 3.
The basal ganglia loop anatomy is complex with many connections, but a simplification has become popular that has some heuristic value (Bar-Gad et al., 2003; Wichmann and DeLong, 2003a, 2003b; DeLong and Wichmann, 2007) (Fig. 2.2). In this model there are two pathways that go from the cortex and then back to the cortex. The direct pathway is the putamen, internal division of the globus pallidus (GPi), and thalamus (mainly the Vop nucleus). The indirect pathway is the putamen, external division of the globus pallidus (GPe), subthalamic nucleus (STN), GPi, substantia nigra pars reticulata (SNr), and thalamus. The substantia nigra pars compacta (SNc) is the source of the important nigrostriatal dopamine pathway and appears to modulate the loop, although not being in the loop itself. The putaminal neurons of the direct pathway have dopamine D2 receptors and are facilitated by dopamine, while the putaminal neurons of the indirect pathway have dopamine D1 receptors and are inhibited.
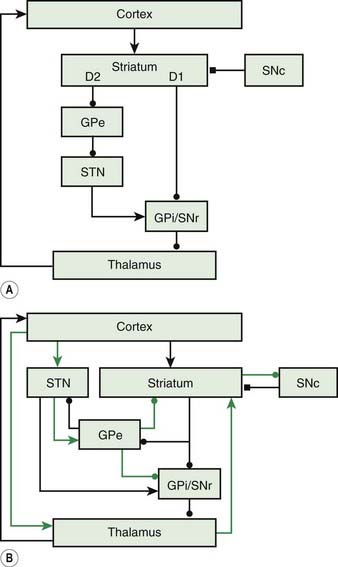
Figure 2.2 also has a more complete diagram indicating more connections and some of the complexity. It is now recognized that even this diagram is too simple, and there is also a hyperdirect pathway directly from the cortex to the STN. Additionally, new importance is given to the pedunculopontine nucleus (PPN), an elongated nucleus in the lateral mesencephalon and pons (Aravamuthan et al., 2007; Hamani et al., 2007; Muthusamy et al., 2007; Shimamoto et al., 2010). This nucleus receives output from the STN and GPi and may be important in balance and gait.
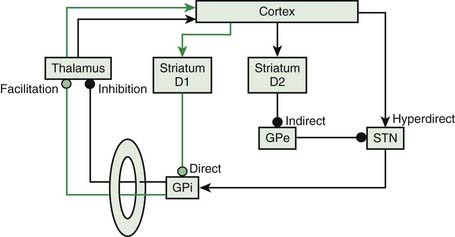
The basal ganglia are anatomically organized to work in a center-surround mechanism. This idea of center-surround organization was one of the possible functions of the basal ganglia circuitry suggested by Alexander and Crutcher (1990). This was followed up nicely by Mink, who detailed the possible anatomy (Fig. 2.3) (Mink, 1996, 2003, 2006). The direct pathway has a focused inhibition in the globus pallidus while the subthalamic nucleus has divergent excitation. The direct pathway (with two inhibitory synapses) is a net excitatory pathway and the indirect pathway (with three inhibitory synapses) is a net inhibitory pathway. Hence, the direct pathway can be the center and the indirect pathway the surround of a center-surround mechanism.
Basal ganglia disorders are characterized by a wide variety of movement signs and symptoms. Often they are divided into hypokinetic and hyperkinetic varieties, implying too little movement on the one hand and too much movement on the other. A full listing of these disorders is in Chapter 1. Here, the pathophysiology of Parkinson disease and dystonia will be emphasized.
Parkinson disease
Parkinson disease (PD) is classically characterized by bradykinesia, rigidity, and tremor-at-rest. All features seem due to the degeneration of the nigrostriatal pathway, but it has not been possible to define a single underlying pathophysiologic mechanism that explains everything. Nevertheless, there are considerable data that give separate understanding to each of the three classic features (Hallett, 2003a; Rodriguez-Oroz et al., 2009).
Bradykinesia
The most important functional disturbance in patients with PD is a disorder of voluntary movement prominently characterized by slowness. This phenomenon is generally called bradykinesia, although it has at least two components, which can be designated as bradykinesia and akinesia (Berardelli et al., 2001). Bradykinesia refers to slowness of movement that is ongoing. Akinesia refers to failure of willed movement to occur. There are two possible reasons for the absence of expected movement. One is that the movement is so slow (and small) that it cannot be seen. A second is that the time needed to initiate the movement becomes excessively long.
While self-paced movements can give information about bradykinesia, the study of reaction time movements can yield information about both akinesia and bradykinesia. In the reaction time situation, a stimulus is presented to a subject, and the subject must make a movement as rapidly as possible. The time between the stimulus and the start of movement is the reaction time; the time from initiation to completion of movement is the movement time. Using this logic, prolongation of reaction time is akinesia, and prolongation of movement time is bradykinesia. Studies of PD patients confirm that both reaction time and movement time are prolonged. However, the extent of abnormality of one does not necessarily correlate with the extent of abnormality of the other (Evarts et al., 1981). This suggests that they may be impaired by separable physiologic mechanisms. In general, prolongation of movement time (bradykinesia) is better correlated with the clinical impression of slowness than is prolongation of reaction time (akinesia).
Some contributing features of bradykinesia are established. One is that there is a failure to energize muscles up to the level necessary to complete a movement in a standard amount of time. This has been demonstrated clearly with attempted rapid, monophasic movements at a single joint (Hallett and Khoshbin, 1980). In this circumstance, movements of different angular distances are accomplished in approximately the same time by making longer movements faster. The electromyographic (EMG) activity underlying the movement begins with a burst of activity in the agonist muscle of 50–100 ms, followed by a burst of activity in the antagonist muscle of 50–100 ms, followed variably by a third burst of activity in the agonist. This “triphasic” pattern has relatively fixed timing with movements of different distance, correlating with the fact of similar total time for movements of different distance. Different distances are accomplished by altering the magnitude of the EMG within the fixed duration burst. The pattern is correct in patients with PD, but there is insufficient EMG activity in the burst to accomplish the movement. These patients often must go thorough two or more cycles of the triphasic pattern to accomplish the movement. Interestingly, such activity looks virtually identical to the tremor-at-rest seen in these patients. The longer the desired movement, the more likely it is to require additional cycles. These findings were reproduced by Baroni et al. (1984), who also showed that levodopa normalized the pattern and reduced the number of bursts.
Berardelli and colleagues (1986) showed that PD patients could vary the size and duration of the first agonist EMG burst with movement size and added load in the normal way. However, there was a failure to match these parameters appropriately to the size of movement required. This suggests an additional problem in scaling of actual movement to the required movement. A problem in sensory scaling of kinesthesia was demonstrated by Demirci et al. (1997). PD patients used kinesthetic perception to estimate the amplitude of passive angular displacements of the index finger about the metacarpophalangeal joint and to scale them as a percentage of a reference stimulus. The reference stimulus was either a standard kinesthetic stimulus preceding each test stimulus (task K) or a visual representation of the standard kinesthetic stimulus (task V). The PD patients’ underestimation of the amplitudes of finger perturbations was significantly greater in task V than in task K. Thus, when kinesthesia is used to match a visual target, distances are perceived to be shorter by the PD patients. Assuming that visual perception is normal, kinesthesia must be “reduced” in PD patients. This reduced kinesthesia, when combined with the well-known reduced motor output and probably reduced corollary discharges, implies that the sensorimotor apparatus is “set” smaller in PD patients than in normal subjects.
In a slower, multijoint movement task, PD patients show a reduced rate of rise of muscle activity that also implies deficient activation (Godaux et al., 1992). On the other hand, Jordan and colleagues (1992) showed that release of force was just as slowed as increase of force, suggesting that slowness to change and not deficient energization was the main problem. If termination of activity is an active process, then this finding really does not argue against deficient energization.
A second physiologic mechanism of bradykinesia is that there is difficulty with simultaneous and sequential movements (Benecke et al., 1987). That PD patients have more difficulty with simultaneous movements than with isolated movements was first pointed out by Schwab and colleagues (1954). Quantitative studies show that slowness in accomplishing simultaneous or sequential movements is more than would be predicted from the slowness of each individual movement. With sequential movements, there is another parameter of interest, the time between the two movements designated the inter-onset latency (IOL) by Benecke and colleagues (1987). The IOL is also prolonged in patients with PD. This problem, similar to the problem with simple movements, can also be interpreted as insufficient motor energy.
Akinesia would seem to be multifactorial, and a number of contributing factors are already known. As noted above, one type of akinesia is the limit of bradykinesia from the point of view of energizing muscles. If the muscle is selected but not energized, then there will be no movement. Such phenomena can be recognized on some occasions with EMG studies where EMG activity will be initiated but will be insufficient to move the body part. Another type of akinesia, again as noted above, is prolongation of reaction time; the patient is preparing to move, but the movement has not yet occurred. Considerable attention has been paid to mechanisms of prolongation of reaction time. One factor is easily demonstrable in patients with rest tremor, who appear to have to wait to initiate the movement together with a beat of tremor in the agonist muscle of the willed movement (Hallett et al., 1977; Staude et al., 1995).
Another mechanism of prolongation of reaction time can be seen in those circumstances where eye movement must be coordinated with limb movement (Warabi et al., 1988). In this situation, there is a visual target that moves into the periphery of the visual field. Normally, there is a coordinated movement of eyes and limb, the eyes beginning slightly earlier. In PD, some patients do not begin to move the limb until the eye movement is completed. This might be due to a problem with simultaneous movements, as noted above. Alternatively, it might be that PD patients need to foveate a target before they are able to move to it.
Many studies have evaluated reaction time quantitatively with neuropsychological methods (Hallett, 1990). The goal of these studies is to determine the abnormalities in the motor processes that must occur before a movement can be initiated. In order to understand reaction time studies, it is useful to consider from a theoretical point of view the tasks that the brain must accomplish. The starting point is the “set” for the movement. This includes the environmental conditions, initial positions of body parts, understanding the nature of the experiment and, in particular, some understanding of the expected movement. In some circumstances, the expected movement is described completely, without ambiguity. This is the “simple reaction time” condition. The movement can be fully planned. It then needs to be held in store until the stimulus comes to initiate the execution of the movement. In other circumstances, the set does not include a complete description of the required movement. It is intended that the description be completed at the time of the stimulus that calls for the movement initiation. This is the “choice reaction time” condition. In this circumstance, the programming of the movement occurs between the stimulus and the response. Choice reaction time is always longer than simple reaction, and the time difference is due to this movement programming.
In most studies, simple reaction time is significantly prolonged in patients compared with normal subjects (Hallett, 1990). On the other hand, patients appear to have normal choice reaction times or the increase of choice reaction time over simple reaction time is the same in patients and normal subjects. Many studies in which cognitive activity was required for a decision on the correct motor response have shown that PD patients do not have apparent slowing of thinking, called bradyphrenia. The study of choice reaction times was extended by considering three different choice reaction time tasks that required the same simple movement, but differed in the difficulty of the decision of which movement to make (Brown et al., 1993). Comparing PD patients to normal subjects, the patients had a longer reaction time in all three conditions, but the difference was largest when the task was the easiest and smallest when the task was the most difficult. Thus, the greater the proportion of time there is in the reaction time devoted to motor program selection, the closer to normal are the PD results. Labutta et al. (1994) have shown that PD patients have no difficulty holding a motor program in store. Hence, the difficulty must be in executing the motor program. Execution of the movement, however, lies at the end of choice reaction time, just as it does for simple reaction time. How then can it be abnormal and choice reaction time be normal? The answer may be that in the choice reaction time situation both motor programming and motor execution can proceed in parallel.
Transcranial magnetic stimulation (TMS) can be used to study the initiation of execution. With low levels of TMS, it is possible to find a level that will not produce any motor evoked potentials (MEPs) at rest, but will produce an MEP when there is voluntary activation. Using such a stimulus in a reaction time situation between the stimulus to move and the response, Starr et al. (1988) showed that stimulation close to movement onset would produce a response even though there was still no voluntary EMG activity. A small response first appeared about 80 ms before EMG onset and grew in magnitude closer to onset. This method divides the reaction time into two periods. In the first period, the motor cortex remains “unexcitable”; in the second, the cortex becomes increasingly “excitable” as it prepares to trigger the movement. Most of the prolongation of the reaction time appeared due to prolongation of the later period of rising excitability (Pascual-Leone et al., 1994a). This result has been confirmed (Chen et al., 2001). The finding of prolonged initiation time in PD patients is supported by studies of motor cortex neuronal activity in reaction time movements in monkeys rendered parkinsonian with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Watts and Mandir, 1992). In these investigations, there was a prolonged time between initial activation of motor cortex neurons and movement onset.
Another factor that should be kept in mind is that patients appear to have much more difficulty initiating internally triggered movements than externally triggered movements. This is clear clinically in that external cues are often helpful in movement initiation. Examples include improving walking by providing an object to step over or playing march music. This can also be demonstrated in the laboratory with a variety of paradigms (Curra et al., 1997; Majsak et al., 1998).
How does bradykinesia arise from dysfunction of the nigrostriatal pathway? Thinking about the simple basal ganglia diagram, dopamine facilitates the direct pathway and inhibits the indirect pathway. Loss of dopamine will lead to lack of facilitation of movement in both pathways. This could certainly be represented by bradykinesia. This has been referred to as a loss of “motor motivation” (Mazzoni et al., 2007).The origin of rigidity and tremor is less understandable, but also less directly linked to dopamine deficiency clinically (Rodriguez-Oroz et al., 2009).
Rigidity
Tone is defined as the resistance to passive stretch. Rigidity is one form of increased tone that is seen in disorders of the basal ganglia (“extrapyramidal disorders”), and is particularly prominent in PD. Increased tone can result from changes in (1) muscle properties or joint characteristics, (2) amount of background contraction of the muscle, and (3) magnitude of stretch reflexes. There is evidence for all three of these aspects contributing to rigidity. For quantitative purposes, responses can be measured to controlled stretches delivered by devices that contain torque motors. The stretch can be produced by altering the torque of the motor or by altering the position of the shaft of the motor. The perturbation can be a single step or more complex, such as a sinusoid. The mechanical response of the limb can be measured: the positional change if the motor alters force or the force change if the motor alters position. Such mechanical measurements can directly mimic and quantify the clinical impression (Hallett et al., 1994; Hallett, 1999).
Long-latency reflexes are best brought out with controlled stretches with a device such as a torque motor. While long-latency reflexes are normally absent at rest, they are prominent in PD patients (Rothwell et al., 1983; Tatton et al., 1984; Hallett et al., 1994; Hallett, 1999). Long-latency reflexes are also enhanced in PD with background contraction. Since some long-latency stretch reflexes appear to be mediated by a loop through the sensory and motor cortices, the enhancement of long-latency reflexes has been generally believed to indicate increased excitability of this central loop.
There is some evidence that at least one component of the increased long-latency stretch reflex in PD is a group II mediated reflex. This suggestion was first made by Berardelli et al. (1983) on the basis of physiologic features, including insensitivity to vibration. It was subsequently supported by the observation that an enhanced late stretch reflex response could not be duplicated with a vibration stimulus (Cody et al., 1986). Some studies show a correlation between clinically measured increased tone and the magnitude of long-latency reflexes (Berardelli et al., 1983), while others do not (Bergui et al., 1992; Meara and Cody, 1993). Long-latency reflexes contribute significantly to rigidity, but are apparently not completely responsible for it.
Tremor-at-rest
The so called “tremor-at-rest” is the classic tremor of PD and other parkinsonian states such as those produced by neuroleptics or other dopamine-blocking agents such as prochlorperazine and metoclopramide (Elble and Koller, 1990; Hallett, 1991, 1999; Elble, 1997). It is present at rest, disappears with action, but may resume with static posture. That the tremor may also be present during postural maintenance is a significant point of confusion in regard to naming this tremor “tremor-at-rest.” It can involve all parts of the body and can be markedly asymmetrical, but it is most typical with a flexion–extension movement at the elbow, pronation and supination of the forearm, and movements of the thumb across the fingers (“pill-rolling”). Its frequency is 3–7 Hz, but is most commonly 4 or 5 Hz; EMG studies show alternating activity in antagonist muscles.
The anatomical basis of the tremor-at-rest may well differ from the classic neuropathology of PD, that of degeneration of the nigrostriatal pathway. For example, 18F-dopa uptake in the caudate and putamen declines with bradykinesia and rigidity, but is unassociated with degree of tremor (Otsuka et al., 1996). Evidence from a PET study suggests that tremor is associated with a serotonergic deficiency (Doder et al., 2003). Another point in favor of this idea is that the tremor may be successfully treated with a stereotactic lesion or deep brain stimulation of the ventral intermediate (VIM) nucleus of the thalamus, a cerebellar relay nucleus (Jankovic et al., 1995; Benabid et al., 1996).
In parkinsonian tremor-at-rest, there may be some mechanical-reflex component and some 8–12 Hz component, but the most significant component comes from a pathologic central oscillator at 3–5 Hz. This tremor component is unaffected by loading. Evidence for the central oscillator includes the facts that the accelerometric record and the EMG are not affected by weighting, and small mechanical perturbations do not affect it. On the other hand, it can be reset by strong peripheral stimuli such as an electrical stimulus that produces a movement of the body part five times more than the amplitude of the tremor itself (Britton et al., 1993a). Where this strong stimulus acts is not clear, but it does not have to be on the peripheral loop. Additionally, the tremor can be reset by TMS (Britton et al., 1993b; Pascual-Leone et al., 1994b), presumably indicating a role of the motor cortex in the central processes that generate the tremor. In the studies of Pascual-Leone et al. (1994b), using a relatively small stimulus, the tremor was reset with TMS, but not with transcranial electrical stimulation. Since TMS affects the intracortical circuitry more, this seems to be further evidence for a role of the motor cortex.
While cells in the globus pallidus may have oscillatory activity, they are not as well related to the tremor as the cells in the VIM of the thalamus (Hayase et al., 1998; Hurtado et al., 1999). Lenz and colleagues have studied the physiologic properties of cells in the VIM in relation to tremor production (Zirh et al., 1998). They have tried to see if the pattern of spike activity is consistent with specific hypotheses. They examined whether parkinsonian tremor might be produced by the activity of an intrinsic thalamic pacemaker or by the oscillation of an unstable long loop reflex arc. In one study of 42 cells, they found 11 with a sensory feedback pattern, 1 with a pacemaker pattern, 21 with a completely random pattern, and 9 that did not fit any pattern (Zirh et al., 1998). In another study of thalamic neuron activity, some cells with a pacemaker pattern were seen, but these did not participate in the rhythmic activity correlating with tremor (Magnin et al., 2000). These results confirm those of Lenz et al. suggesting that the thalamic cells are not the pacemaker. Using sophisticated analytical techniques, it can be demonstrated that oscillations both in the VIM and in the STN play an efferent role in tremor generation, but that the tremor itself feeds back to these same structures to influence the oscillation (Tass et al., 2010). This does suggest that in some sense the whole loop is responsible for the tremor. The basal ganglia loop may well trigger the cerebellar loop to produce the tremor (Helmich et al., 2011).
Wherever the pacemaker for the tremor, it is important to note that while the tremor is synchronous within a limb, it is not synchronous between limbs (Hurtado et al., 2000). Hence a single pacemaker does not influence the whole body.
Dystonia
Dystonia is characterized by abnormal muscle spasms producing distorted motor control and undesired postures (Defazio et al., 2007; Breakefield et al., 2008). Early on, dystonia is produced only by action, but then it can occur spontaneously. There are presently three general lines of work that may indicate the physiologic substrate for dystonia.
Loss of inhibition
A principal finding in focal dystonia is that of loss of inhibition (Hallett, 2004, 2006a, 2006b, 2011). Loss of inhibition is likely responsible for the excessive movement seen in dystonia patients. Excessive movement includes abnormally long bursts of EMG activity, co-contraction of antagonist muscles, and overflow of activity into muscles not intended for the task (Cohen and Hallett, 1988). Loss of inhibition can be demonstrated in spinal and brainstem reflexes. Examples are the loss of reciprocal inhibition in the arm in patients with focal hand dystonia (Nakashima et al., 1989; Panizza et al., 1990) and abnormalities of blink reflex recovery in blepharospasm (Berardelli et al., 1985). Loss of reciprocal inhibition can be partly responsible for the presence of co-contraction of antagonist muscles that characterizes voluntary movement in dystonia.
Loss of inhibition can also be demonstrated for motor cortical function including the transcranial magnetic stimulation techniques of short intracortical inhibition, long intracortical inhibition, and the silent period (Hallett, 2007a, 2011).
Short intracortical inhibition (SICI) is obtained with paired pulse methods and reflects interneuron influences in the cortex (Ziemann et al., 1996). In such studies, an initial conditioning stimulus is given, enough to activate cortical neurons, but small enough that no descending influence on the spinal cord can be detected. A second test stimulus, at suprathreshold level, follows at a short interval. Intracortical influences initiated by the conditioning stimulus modulate the amplitude of the MEP produced by the test stimulus. At short intervals, less than 5 ms, there is inhibition that is likely largely a GABAergic effect, specifically GABA-A (Di Lazzaro et al., 2000). (At intervals between 8 and 30 ms, there is facilitation, called intracortical facilitation, ICF). There is a loss of intracortical inhibition in patients with focal hand dystonia (Ridding et al., 1995). Inhibition was less in both hemispheres of patients with focal hand dystonia, and this indicates that this abnormality is more consistent as a substrate for dystonia.
The silent period (SP) is a pause in ongoing voluntary EMG activity produced by TMS. While the first part of the SP is due in part to spinal cord refractoriness, the latter part is entirely due to cortical inhibition (Fuhr et al., 1991). This type of inhibition is likely mediated by GABA-B receptors (Werhahn et al., 1999). SICI and the SP show different modulation in different circumstances and clearly reflect different aspects of cortical inhibition. The SP is shortened in focal dystonia.
Intracortical inhibition can also be assessed with paired suprathreshold TMS pulses at intervals from 50 to 200 ms (Valls-Solé et al., 1992). This is called long intracortical inhibition, or LICI, to differentiate it from SICI as noted above. LICI and SICI differ as demonstrated by the facts that with increasing test pulse strength, LICI decreases but SICI tends to increase, and that there is no correlation between the degree of SICI and LICI in different individuals (Sanger et al., 2001). The mechanisms of LICI and the SP may be similar in that both seem to depend on GABA-B receptors. Chen et al. (1997) investigated long intracortical inhibition in patients with writer’s cramp and found a deficiency only in the symptomatic hand and only with background contraction. This abnormality is particularly interesting since it is restricted to the symptomatic setting, and therefore might be a correlate of the development of the dystonia.
There is also neuroimaging evidence consistent with a loss of inhibition. Dopamine D2 receptors are deficient in focal dystonias (Perlmutter et al., 1997). There is weak evidence for reduced GABA concentration both in basal ganglia and motor cortex utilizing magnetic resonance spectroscopy (Levy and Hallett, 2002; Herath et al., 2010).
Loss of cortical inhibition in motor cortex can give rise to dystonic-like movements in primates. Matsumura et al. showed that local application of bicuculline, a GABA antagonist, onto the motor cortex led to disordered movement and changed the movement pattern from reciprocal inhibition of antagonist muscles to co-contraction (Matsumura et al., 1991). In a second study, they showed that bicuculline caused cells to lose their crisp directionality, converted unidirectional cells to bidirectional cells, and increased firing rates of most cells including making silent cells into active ones (Matsumura et al., 1992).
There is a valuable animal model for blepharospasm that supports the idea of a combination of genetics and environment, and, specifically, that the background for the development of dystonia could be a loss of inhibition (Schicatano et al., 1997). In this model, rats were lesioned to cause a depletion of dopamine; this reduces inhibition. Then the orbicularis oculi muscle was weakened. This causes an increase in the blink reflex drive in order to produce an adequate blink. Together, but not separately, these two interventions produced spasms of eyelid closure, similar to blepharospasm. Shortly after the animal model was presented, several patients with blepharospasm after a Bell’s palsy were reported (Chuke et al., 1996; Baker et al., 1997). This could be a human analog of the animal experiments. The idea is that those patients who developed blepharospasm were in some way predisposed. A gold weight implanted into the weak lid of one patient, aiding lid closure, improved the condition, suggesting that when the abnormal increase in reflex drive was removed, the dystonia could be ameliorated (Chuke et al., 1996).
Loss of surround inhibition, a functional consequence of loss of inhibition
A principle for function of the motor system may be “surround inhibition” (Hallett, 2006a, 2006b; Beck and Hallett, 2011). Surround inhibition is a concept well accepted in sensory physiology (Angelucci et al., 2002). Surround inhibition is poorly known in the motor system, but it is a logical concept. When making a movement, the brain must activate the motor system. It is possible that the brain just activates the specific movement. On the other hand, it is more likely that the one specific movement is generated, and, simultaneously, other possible movements are suppressed. The suppression of unwanted movements would be surround inhibition, and this should produce a more precise movement, just as surround inhibition in sensory systems produces more precise perceptions. For dystonia, a failure of “surround inhibition” may be particularly important since overflow movement is often seen and is a principal abnormality.
There is now good evidence for surround inhibition in human movement. Sohn et al. (2003) have shown that with movement of one finger there is widespread inhibition of muscles in the contralateral limb. Significant suppression of MEP amplitudes was observed when TMS was applied between 35 and 70 ms after EMG onset. Sohn and colleagues have also shown that there is some inhibition of muscles in the ipsilateral limb when those muscles are not involved in any way in the movement (Sohn and Hallett, 2004b). TMS was delivered to the left motor cortex from 3 ms to 1000 ms after EMG onset in the flexor digitorum superficialis muscle. MEPs from abductor digiti minimi were slightly suppressed during the movement of the index finger in the face of increased F-wave amplitude and persistence, indicating that cortical excitability is reduced.
Surround inhibition was studied similarly in patients with focal hand dystonia (Sohn and Hallett, 2004a). The MEPs were enhanced similarly in the flexor digitorum superficialis and abductor digiti minimi indicating a failure of surround inhibition. Using other experimental paradigms, a similar loss of surround inhibition in the hand has been found (Stinear and Byblow, 2004; Beck et al., 2008).
Abnormal plasticity
There is abnormal plasticity of the motor cortex in patients with focal hand dystonia (Quartarone et al., 2006; Weise et al., 2006). This has been demonstrated using the technique of paired associative stimulation (PAS) (Stefan et al., 2000). In PAS, a median nerve shock is paired with a TMS pulse to the sensorimotor cortex timed to be immediately after the arrival of the sensory volley. This intervention increases the amplitude of the MEP produced by TMS to the motor cortex. It has been demonstrated that the process of PAS produces motor learning similar to long-term potentiation (LTP). In patients with dystonia, PAS produces a larger increase in the MEP than that seen in normal subjects. There is also an abnormality in homeostatic plasticity. Homeostatic plasticity is the phenomenon whereby plasticity remains within limits; this can be exceeded in dystonia (Quartarone et al., 2006).
Increased plasticity may arise from decreased inhibition so the inhibitory problem may well be more fundamental. This abnormality may be an important link in demonstrating how environmental influences can trigger dystonia. Abnormal plasticity can arise, at least in part, from abnormal synaptic processes in the basal ganglia (Peterson et al., 2010).
The possibility of increased plasticity in dystonia had been suspected for some time given that repetitive activity over long periods seems to be a trigger for its development. An animal model supported this idea (Byl et al., 1996). Monkeys were trained to hold a vibrating manipulandum for long periods. After some time, they became unable to do so, and this motor control abnormality was interpreted as a possible dystonia. The sensory cortex of these animals was studied, and sensory receptive fields were found to be large. The interpretation of these results was that the synchronous sensory input caused the receptive field enlargement, and that the abnormal sensory function led to abnormal motor function. The results suggested that the same thing might be happening in human focal dystonia: repetitive activity caused sensory receptive field changes and led to the motor disorder.
Abnormal sensory function
Stimulated by the findings of sensory dysfunction in the primate model, investigators began examining sensory function in patients with focal hand dystonia and found it to be abnormal. Although there is no apparent sensory loss on a clinical level, detailed testing of spatial and temporal discrimination revealed subtle impairments (Molloy et al., 2003). The abnormality is present on both hands of patients with unilateral hand dystonia and also on hands of patients with cervical dystonia and blepharospasm. The identification of abnormality of sensation beyond the symptomatic body parts indicated that the sensory abnormality could not be a consequence of abnormal learning, but is more likely a pre-existing physiologic state.
Sensory dysfunction can also be demonstrated with somatosensory evoked potential (SEP) testing (Bara-Jimenez et al., 1998). The dipoles of the N20 from stimulation of individual fingers show disordered representation in the primary sensory cortex (Bara-Jimenez et al., 1998) and these abnormalities are present on both hands of patients with focal hand dystonia (Meunier et al., 2001). The bilateral SEP abnormality was the first indication in the literature that the sensory abnormality was more likely endophenotypic than a consequence of repetitive activity. PET studies show that the sensory cortex is more activated than normal with writing and is more activated when patients are experiencing more dystonia (Lerner et al., 2004). Voxel-based morphometry studies in patients with focal hand dystonia show an increase in gray matter in the primary sensory cortex (Garraux et al., 2004). Such observations indicate that dystonia is a sensory disorder as well as a motor disorder.
There are data from sensory function that are compatible with loss of surround inhibition. Tinazzi and colleagues (2000) studied median and ulnar nerve somatosensory evoked potentials (SEPs) in patients who had dystonia involving at least one upper limb. They compared the amplitude of SEP components obtained by stimulating the median and ulnar nerves simultaneously (MU) with the amplitude value being obtained from the arithmetic sum of the SEPs elicited by stimulating the same nerves separately (M + U). The ratio of MU to (M + U) indicates the interaction between afferent inputs from the two peripheral nerves. No significant difference was found between SEP amplitudes and latencies for individually stimulated median and ulnar nerves in dystonic patients and normal subjects, but recordings in patients yielded a significantly higher percentage ratio for spinal N13, brainstem P14, and cortical N20, P27 and N30 components. The authors state that “these findings suggest that the inhibitory integration of afferent inputs, mainly proprioceptive inputs, coming from adjacent body parts is abnormal in dystonia. This inefficient integration, which is probably due to altered surrounding inhibition, could give rise to an abnormal motor output and might therefore contribute to the motor impairment present in dystonia.”
Another demonstration of loss of surround inhibition in sensory function is in the temporal domain. Patients have difficulty recognizing two stimuli when they are close together. This abnormality seems due to a loss of a short latency inhibition, identified using SEP recovery curves (Tamura et al., 2008).
Cerebellum
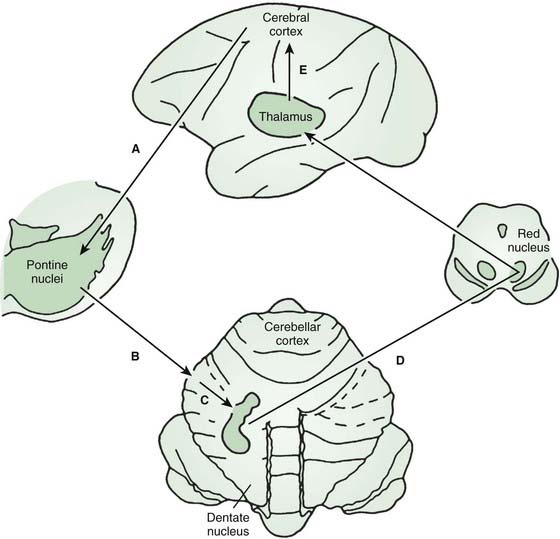
The anatomy of the cerebellar pathways, like the basal ganglia pathways, is complex, but there are simplified models that aid thinking (Schmahmann, 1994; Schmahmann and Pandya, 1997) (Fig. 2.4). The main cortico-cerebellar-cortical loop is frontal lobe, pontine nuclei, cerebellar cortex (via middle cerebellar peduncle), deep cerebellar nuclei, red nucleus and ventral lateral nucleus of thalamus (via superior cerebellar peduncle), and motor cortex. The input fibers to the cerebellar cortex are the mossy fibers that synapse onto granule cells which in turn synapse onto the Purkinje cells. There is also extensive sensory input via spinocerebellar tracts, largely carried in the inferior cerebellar peduncle. A critical modulatory loop involves the inferior olivary nucleus. The inferior olive innervates both the cerebellar cortex and deep nuclei via the inferior cerebellar peduncle and the climbing fibers that synapse directly onto Purkinje cells. Feedback returns to the inferior olive by a dentate-olivary pathway that travels in the superior cerebellar peduncle, goes around the red nucleus, and descends in the central tegmental tract.
Ataxia
The term ataxia, literally meaning without order, refers to disorganized, poorly coordinated or clumsy movement (Massaquoi and Hallett, 2002). Since the time of Holmes (1939), it has been applied more specifically to clumsiness that is due to lesions of the cerebellum and its immediate connecting pathways, of proprioceptive sensory pathways or sometimes of the vestibular system. In order to identify the presence of ataxia, it should not be explained by any abnormality in (maximal isometric) strength, segmental reflexes, muscular tone, ability to isolate movement of individual body parts, or gross motor sequencing or spatial planning. The clumsiness is also not due to spontaneous involuntary movements. Ataxia may be associated with any voluntary movement and with many reflex movements. It commonly affects upright balance, gait, manual coordination and speech, yielding stagger, clumsy manipulation and slurring dysarthria which appear drunken. Indeed, the motor coordination-impairing effects of ethanol are attributed to its specific interference with cerebellar function.
Tonic force control abnormalities: hypotonia and asthenia
Normal individuals have very low, barely perceptible muscle tone when fully relaxed. Holmes noted, however, that acutely injured soldiers with penetrating wounds to the cerebellum had further reduced resistance to passive movement. He viewed hypotonia as a fundamental abnormality which underlies many cerebellar motor deficits (Holmes, 1939). This hypotonia tended to be characteristic especially of the upper extremities and to normalize gradually over weeks to months depending on the severity of the injury. Gilman et al. (1981) have shown that in primates this change parallels the recovery of muscle spindle sensitivity which was acutely depressed by loss of cerebellar fusimotor facilitation.
Large-scale surgical cerebellar ablation in monkeys is generally reported to produce acute weakness especially of the extensor muscles (Gilman et al., 1981). In humans, Holmes clearly distinguished the weakness that followed acute massive damage to the cerebellar hemispheres, asthenia, from that associated with corticospinal tract lesions, paresis. The former did not affect specific muscle groups more than others and was not necessarily associated with changes in tendon reflex sensitivity. Interestingly, asthenia was noted particularly when strength was tested during movement. Static resistance to the examiner was most often normal. Indeed, weakness per se is a very inconstant complaint in cerebellar patients. Further questioning often reveals the problem to be one of easy fatigability and/or a lack of coordination or stability, not of peak strength. Holmes also drew attention to the inability of some patients to maintain steady force levels (astasia, after Luciani). Patients sometimes complain of sudden losses of strength, such as a leg “giving out” or the tendency of an item to drop suddenly from the hand. Holmes attributed these episodes to hypotonia, but their nature remains unclear.
Even in the presence of normal strength and muscular tone, easy fatigability (a second aspect of asthenia) is a prominent complaint of many patients with cerebellar ataxia (Holmes, 1939). The fatigue may affect an individual body part, but may also be sensed more globally. Most patients report that all of the aspects of their ataxia worsen when they are fatigued. A poor night’s sleep, or a particularly busy previous day, predisposes to a day of especially poor motor control. Patients frequently take naps during the day which provide considerable benefit. Fatigue in cerebellar patients appears to be central, and not muscular in origin. Electrophysiologic studies of fatigue in non-depressed patients with cerebellar ataxia by Samii et al. (1997) showed decreased post-exercise facilitation of motor potentials evoked from transcranial magnetic stimulation. This is a central activation defect, similar to that seen in patients with depression and chronic fatigue syndrome. As in these disorders, patients sometimes complain of decreased concentration and mild difficulties with thinking. In this regard, the fatigue is also qualitatively similar to that seen in Parkinson disease. Ultimately, the general fatigue in cerebellar disease and other movement disorders may be related to the increased mental concentration needed to compensate for degraded automatic motor control.
Force-rate and movement amplitude scaling deficits: dysmetria, impaired check, and past-pointing
Classic descriptions of cerebellar ataxia include various clinical signs such as dysmetria, dyssynergia (asynergia, decomposition of movement), dysdiadochokinesia, dysrhythmia, and kinetic (intention) and postural tremors (Holmes, 1939; Gilman et al., 1981). Characteristically, ataxic individuals have particular difficulty in properly generating, guiding, and terminating high-speed movements. Movements accelerate somewhat slowly and are relatively late in onset if executed in reaction to a cue. Movements may then either partially arrest prior to reaching their targets or gradually accelerate to excessive speed and overshoot their targets to an abnormal degree. These two types of errors are examples of dysmetria, hypometria and hypermetria, respectively. Two distinct motor control abnormalities appear to underlie dysmetria: force-rate inadequacy and step amplitude mis-scaling. The former causes brief, more consistent velocity-sensitive inaccuracies and the latter, more variable protracted errors.
At a fundamental level, the patient with cerebellar ataxia has difficulty changing voluntary force levels abruptly (Mai et al., 1988). Both acceleration and braking are impaired. In point-to-point movements, for example, this voluntary force-rate deficit is generally corroborated by a slowness in the build-up of agonist EMG and a prolonged agonist action with delayed onset of antagonist EMG (Hallett et al., 1991; Hallett and Massaquoi, 1993). In patients attempting rapid, single-joint movement, the first agonist burst is frequently prolonged regardless of the distance and speed of the movement, and the most striking kinematic abnormality is prolonged acceleration time. The pattern of acceleration time exceeding deceleration time is common in patients but uncommon in normal subjects. Duration of the first agonist burst correlates with, and is largely responsible for, acceleration time. Altered production of appropriate acceleration for rapid voluntary movements may therefore be the primary abnormality in cerebellar dysfunction for attempted rapid voluntary movements. Hypermetria would be the expected resultant movement error unless there is compensation. Hypometria has been attributed to over-compensation, to asthenia in the acute setting, to tremor, or to failure of timely relaxation of the antagonist during movement initiation (Manto et al., 1998). Any of these mechanisms may be contributory in a given movement, and the topography of the lesion might correlate with the type of deficit (Manto et al., 1998).
For point-to-point movements of any given duration, ataxic movements exhibit greater overshoot than normal. In their study of rapid point-to-point elbow flexions, Hore et al. (1991) noted in normal subjects a transient overshoot of about 5–10% of the movement distance. Ataxic patients overshot the target by more than 20% and as much as 35% of the movement distance. On the other hand, whenever there is no observable overshoot, the movements of ataxic individuals are usually abnormally slow or are hypometric. From the point of view of Fitts’ speed–accuracy tradeoff, ataxic patients display decreased motor control bandwidth. Generally, therefore, in the assessment of ataxia it is important to note both the degree of overshoot and the movement time. Appropriate abnormality of either may be consistent with ataxia. Because, however, there may be alternative explanations for increased movement time, slowness is a much less specific finding than overshoot. Due to the inherent tradeoff between speed and accuracy, patients often slow down intentionally in order to maintain error levels that are acceptable to them. Therefore, if it is important to observe maximal speed in a motor task, the examiner must explain that large errors are acceptable and may be, in fact, unavoidable. Even with this encouragement, the examiner is sometimes uncertain that the maximum achievable speed has been elicited.
In spinocerebellar atrophy type 6 (SCA6), there is an abnormality of a voltage-sensitive calcium channel. Hyperventilation enhances the defective function of the channel and increases the behavioral dysfunction. In addition to modifying nystagmus, hypermetria in single-joint movements is exaggerated with hyperventilation (Manto, 2001). This may be a useful clinical provocative test.
Patients with cerebellar deficits also have abnormalities in termination of movement. This problem has been explicitly studied in a task where subjects were asked to make a rapid elbow flexion on the background of tonic elbow extension needed to hold a position against a background force (Hallett et al., 1975). In this circumstance, the tonic triceps activity typically stops before the phasic biceps activity occurs (the “Hufschmidt phenomenon”). Patients with cerebellar dysfunction have a delay in terminating the triceps activity so that it overlaps the beginning of the biceps activity. This delay in stopping leads to overlap of the end of one movement with the beginning of the next.
The practical consequence of sluggishness in termination can be seen at the bedside with the sign called impaired check. If a patient’s elbow which was flexed strongly against the grasp of the examiner is suddenly released, it is difficult for the patient to avoid striking himself/herself with the hand. Impaired check can also be attributed to delay in the triggering of the antagonist muscle (Terzuolo et al., 1973). The distinction between sluggish reduction and delayed changes in force is partially artificial.
In addition to transient overshoot, some patients may show movements that come to rest briefly, or nearly come to rest, at locations that are different from that of the target, most often beyond it (past-pointing). Unlike dynamic overshoot which is always speed-dependent, this effective mis-scaling of the overall movement amplitude is less consistently related to the movement velocity, and often improves with repetition. The sign can be elicited using the Barany pointing test, in which the patient is asked to extend an arm forward, holding it parallel to the floor, and to note its position carefully (Gilman et al., 1981). Next, the patient closes the eyes and points the arm toward the ceiling. The arm is then rapidly brought down to a level as close to its original horizontal position as possible. The ataxic patient without demonstrable proprioceptive deficits may return at least briefly to a steady position beyond (lower than) the original, as if there is an error in the calculation of the distance moved or to have been moved. Among ataxic patients, past-pointing is less consistently observed than is dynamic overshoot, and it is not known whether past-pointing is as closely linked to movement acceleration as is dynamic overshoot. If the patient is allowed to practice and view the error a few times, he or she may become able correct the final position using a second movement while maintaining the eyes closed. It is as if a more precise proprioceptive measurement system can be employed after movement completion. Eventually, the patient may learn to produce a normally scaled movement. That the initial mis-scaling is often correctable may be related to residual cerebellar function, to a retained ability to increase dependence on proprioceptive information, or to rescaling movement at extracerebellar sites.
Exaggerated postural reactions: rebound
The same phenomenon is seen as persistently excessive postural responses to platform perturbations observed by Horak and Diener (1994) in patients with injury to the anterior lobe of the cerebellum. As with rebound in the upper extremity, the excessive initial component of the platform postural response does not attenuate with repetition. Consistent with a cerebellar mechanism, attenuation of initial platform responses in normal subjects appears to be subconscious. Because of secondary, long-latency stabilizing responses, cerebellar patients were still able to avoid falling during the experiments.
Abnormal control of simple multijoint movements: dyssynergia
In ataxic simple multijoint movements, such as intended straight point-to-point hand movements, there is a breakdown in the normal coordination of joint rotations. This has been termed dyssynergia or asynergia and is described as a type of movement decomposition. This typically causes abnormal movement path deviations. As the movements of normal subjects usually display some natural deviation from perfect linearity, abnormality in path is a matter of degree of curvature and of specific pattern. Evidence is now accumulating that ataxic multijoint movements exhibit characteristic trajectory abnormalities (Massaquoi and Hallett, 1996). Thach et al. (1993) have attributed the pronounced ataxia seen in multijoint movements to a hypothetically preferential role for the cerebellum in the coordination of multijoint movement. While the neuroanatomical organization of the cerebellum makes it particularly well suited for coordinating muscle actions of different body parts, the function of the cerebellum in both single and multijoint control may be fundamentally similar.
Analysis of simple, horizontal planar two-joint arm movements suggests that the deficits in acceleration and braking observed at single joints may account, at least in part, for the dyssynergia observed (Hallett and Massaquoi, 1993; Massaquoi and Hallett, 1996). It appears that the force-rate deficit may be accentuated at the joint having the greatest torque-rate demand, which causes an imbalance in the joint accelerations leading to hand movement curvature. This suspected mechanism is consistent with the marked worsening of dyssynergia with increases in intended acceleration. Massaquoi and Slotine (1996) have proposed a theoretical model of intermediate cerebellar function that relates the force production deficit in both single and multijoint limb movements to a common failure of a long-loop feedback control system. The model accounts for the underdamped quality of ataxic motions and reproduces the characteristic curvature of cerebellar patients’ hand trajectories in horizontal planar movements.
As noted by Holmes, ataxia may be especially apparent in multijoint movements because the control problem is more demanding. In addition to the need for forces to launch and stop, there is a requirement for rapid compensation for the disturbing effects of multiple interaction torques between body segments, as well as the need to coordinate more muscles having different individual actions. Because of the additional degrees of force and motion freedom available, failure to compensate for muscular forces and body interaction torques may lead to multidirectional path errors in addition to overall dysmetria. Indeed, several groups have specifically related multijoint trajectory errors in cerebellar ataxia to deficits in interaction torque compensation (Bastian et al., 1996, 2000; Topka et al., 1998). Moreover, Sainburg et al. (1993) have shown deficits in interaction torque control in subjects with sensory ataxia due to peripheral neuropathy. These findings appear to point to similar mechanisms underlying both sensory and cerebellar ataxia.
Several investigators have suggested other mechanisms for asynergia that may be also or alternatively operative. Multijoint movements may be sometimes decomposed into multistep single-joint movement components as a voluntary strategy to simplify programming by minimizing interaction torques between the joints (Bastian et al., 1996). Dyssynergy may be due to the general difficulties cerebellar patients have with timing tasks (Keele and Ivry, 1990). These might yield problems with coordinating the actions of the different joints or muscles within the synergy as suggested by Thach et al. (1992). From this perspective, dysmetria and dyssynergy within simple (single intended velocity peak) single and multijoint movements may have a mechanism similar to that which underlies dysdiadochokinesia, a disruption of compound movements involving more than one intended velocity peak (see below). On the other hand, a servo-control model, such as Massaquoi’s, predicts that timing derangements within single movements occur as secondary effects of muscular activation (force) rate deficits (Massaquoi and Slotine, 1996). The latter view is supported by the prediction, based on dynamics, of a preferred direction for the interjoint timing abnormality (i.e., lag or lead) for a given intended planar hand movement, and therefore of a certain preferred trajectory pattern, rather than random path aberrations.
Abnormalities in timing and coupling movements and other processes: dysrhythmia, dysdiadochokinesia, delayed reaction time and impaired time interval assessment
In addition to timing aberrations that are associated with, and in fact may result from, clumsy movement execution, there appears to be a separate timing abnormality due to failure of a cerebellum-dependent “central clock” (Keele and Ivry, 1990). Theoretically, this clock assists in the timely launching of movements with respect to preceding movements (Diener et al., 1993; Grill et al., 1997). The same system may generally help to launch movements with respect to other events, both external and internal. In all types of reaction tasks, not only does agonist EMG build up more slowly in cerebellar patients, but the EMG onset itself is significantly delayed with respect to the time of the stimulus, as if a triggering system was defective (Grill et al., 1997).
Much physiologic evidence has been accumulated to suggest that the lateral cerebellar hemispheres and dentate nucleus are preferentially involved in context-dependent triggering of movements, while the intermediate and medial regions of the cerebellum control the evolution of ongoing movement of single or multiple body parts. Supporting the existence of an internal triggering/timing system that is separate from that for movement execution control, is the finding by Wing and Kristofferson that timing errors in a simple rhythmic finger tapping task could be partitioned into “implementation” (executional) mistiming and internal clock mistiming, according to a two-component statistical model (Wing et al., 1984). Subsequently, Ivry and Keele found that in cerebellar patients, increased implementation errors were associated with lesions of the medial cerebellum, while clock errors occurred in those having lesions of the lateral hemispheres (Keele and Ivry, 1990). Moreover, cerebellar patients with lateral hemisphere lesions also had difficulty in accurately assessing the difference in the lengths of time intervals between two pairs of tones, while those with medial cerebellar lesions did not. Also noteworthy is their observation that patients with clumsy movements due to either sensory neuropathy or deafferentation showed only executional mistiming. It is not clear, however, whether their abnormal movements appeared clinically identical to cerebellar ataxia.
Abnormalities in motion assessment and prediction: impairment of tracking and mass estimation
Probably closely related to their problems with assessment of time intervals and movement amplitude scaling is cerebellar patients’ basic difficulty in using sensory information to assess and predict motion characteristics. This applies both to body parts, as shown by Grill et al. (1994) and as exhibited in past-pointing tests, and to external objects. Especially in rapid multicomponent movements, a certain amount of motion prediction ability is important for effective performance. Because of the delays in the transmission of neural signals, initiation of movement subcomponents may need to take place well in advance of completion of the preceding subcomponent (Grill et al., 1997). Often, however, details of the plan for the second motion may depend upon the progress of the first motion. For example, in throwing a ball, timing of release must be coordinated with the movement of the arm to produce a properly directed trajectory (e.g., Becker et al., 1990). Similarly, for any control system having nontrivial feedback delays, high-precision tracking of a moving target requires a certain amount of predictive control. This may take the form of additional open-loop (feed-forward) predictive signals or the processing of higher derivatives of error information (e.g., velocity error information for position control) which inherently include some predictive information.
The cerebellum appears to be involved in both predictive feedforward and velocity feedback control. Motion prediction deficits can often be identified at the bedside by asking the patient to track, with his or her finger, the examiner’s finger as it moves slowly back and forth in a smooth motion. A motion should be used which would be normally easy to predict and at a speed that would not engender overshoot in a simple point-to-point movement. Cerebellar patients will nevertheless frequently lag the examiner during the motion and/or overshoot at the direction reversals, presumably because they fail to assess properly the examiner’s rate of acceleration and deceleration or the rhythm of the examiner’s overall movement (Morrice et al., 1990). Very slow manual tracking in cerebellar patients also shows breakdown into a sequence of small movements in “staircase” pattern which has been attributed to loss of velocity feedback control (Beppu et al., 1984, 1987).
Holmes (1939) and, more recently Angel (1980), have noted in hemiataxic patients a tendency to overestimate the weight of objects in the affected hand. However, Holmes found no difference between the sides in being able to discriminate accurately between two different weights placed successively in the same hand. Keele and Ivry (1990) also did not find an abnormality in the perception of static force in cerebellar patients. Thus, the cerebellar patient appears compromised in terms of absolute but not relative weight determination. The explanation favored by Holmes and Angel is that individuals tend to assess weight (or mass, as opposed to force per se) by moving an object up and down with their hands, presumably attempting to relate the applied force, or perhaps more accurately, the applied effort, to the rate of acceleration or oscillation frequency. Given patients’ difficulties with the kinesthetic assessment of motion characteristics (Grill et al., 1994), and possibly an element of asthenia, it would not be surprising if a patient’s ability to relate movement effort to hand acceleration is compromised, thus disturbing the assessment of mass as a secondary effect.
Sensory information acquisition and analysis, and motor control
The critical role of sensory information in successful motor control has been long recognized. Based on the deficits that have been noted in cerebellar patients that were described earlier and on the afferent neuroanatomical connections of the cerebellum, it is evident that it plays an important role in processing sensory information to influence motor performance. However, the nature of this influence has been debated. Although it would appear that improved stability and accuracy of body motion are principal purposes of cerebellar function, Bower has put forward the controversial suggestion that the cerebellum is primarily concerned with fine control of the acquisition of sensory information rather than control of movement per se (Gao et al., 1996; Bower, 1997). In particular, it may be chiefly designed to coordinate the positioning and movement of tactile sensory surfaces to optimize the information received.
Increased movement variability
Ataxic motor performance is frequently described as being more variable than normal (Hallett and Massaquoi, 1993; Palliyath et al., 1998). However, aside from the presence of involuntary movements, ataxic variability may arise more as a consequence of enhanced susceptibility to perturbation and of the sequential compounding of errors, than of an inherent noise as might result from the presence of an unstable autonomous generator. This is suggested by the fact that when tasks are constrained sufficiently, ataxic movements become much less variable (Massaquoi and Hallett, 1996). Thus, especially for experimentally conducted single- and two-joint movements for which there is a single attempted movement speed and direction, and where head, eyes, and trunk are fixed, and in the absence of external contacts and forces (i.e., not against gravity), ataxic movements though inaccurate, are much more consistent in their inaccuracy. Because of the loosened control over executive action in the cerebellar patient, movements and perhaps certain cognitive processes are more vulnerable to both internal and external environmental disturbances. Most natural tasks involve multijoint movements which inherently have many degrees of movement freedom, as well as ongoing efforts to guide motion. Elemental trajectory errors may therefore interact, propagate, and become compounded; a process that effectively produces motor control noise.
Cerebellar tremors
Two types of action cerebellar tremor are commonly identified: kinetic and postural tremor. Lesions of the dentate, of the interpositus, and of the cerebellar outflow via the brachium conjunctivum appear to be the most frequently associated with action tremors. Both manifest alternating EMG bursting in agonist and antagonist muscles (Hallett, 1987). All types of cerebellar action tremors may be exacerbated near the point of attempted fixation if greater effort is made to maintain position precisely. Tremor frequency may differ between limbs and the oscillations are generally not synchronous in non-adjacent body parts. However, as are most tremors, cerebellar action tremors are worsened by fatigue. Cerebellar action tremors are often improved and sometimes eliminated by eye closure (Sanes et al., 1988). Propranolol has no substantial effect and alcohol tends to worsen cerebellar action tremors.
Basic mechanisms that have been suggested to underlie cerebellar tremor have included (1) serial voluntary corrections for positioning error (serial dysmetria) (Hallett, 1987), (2) abnormality of transcortical and segmental proprioceptive feedback loops (Hore and Flament, 1986), and (3) action of central oscillators (Ito, 1984). Sufficient evidence has accumulated to indicate that each of these mechanisms is likely to be important to some component of body oscillations in ataxic patients under various circumstances. It is apparent clinically from the slowness of cerebellar voluntary reactions and in performance of rapid alternating movements that serial dysmetria is unlikely to be operative at frequencies greater than around 1–2 Hz at proximal joints or perhaps 3 Hz at the fingers. Thus, only the irregular, low-frequency, ataxic movements exhibited by patients’ limbs as they approach a target are a manifestation of serial dysmetria. Because of the voluntary nature and gross irregularity of these movements, however, serial dysmetria is not really tremor.
Holmes (1939) also drew attention to the intermittent recoveries of posture that patients exhibit when fatigued. These movements consist of slow drifting downward from the intended posture followed by faster upward corrections that appeared voluntary. While these movements can be viewed as a coarse, asymmetric tremor, their nystagmoid character distinguishes them from the more regular, higher-frequency, involuntary oscillations around the intended posture or trajectory that would be characteristic of “true” cerebellar tremors.
The modification of cerebellar tremor by external perturbations and mechanical state (Sanes et al., 1988) indicates at least a partial dependence on peripheral factors, while the persistence of these cerebellar tremors during deafferentation indicates the presence of some central neural instability (Gilman et al., 1976). Several experimental results and models of cerebellar function include the interaction between central and peripheral feedback loops that could be consistent with these observations (Massaquoi and Slotine, 1996).
Increased postural sway and titubation
Ataxic patients exhibit increased irregular sway when standing and sometimes a more regular tremor (titubation). The characteristics of these involuntary movements vary according to the site of the cerebellar system lesion. Diener and Dichgans have performed extensive studies of postural balance in patients with cerebellar system disease (Diener and Dichgans, 1992; Diener et al., 1984). Common to all ataxic patients, except those with lesions restricted to the hemispheres, is the tendency to have abnormally large amplitude sway when the eyes are closed. Patients with anterior lobe atrophy due to chronic alcohol intake and malnutrition and patients with Friedreich ataxia have a high “Romberg quotient,” meaning that they sway considerably more with eyes closed. In general, the eyes-closed instability is greater in Friedreich ataxia patients, who typically have significant proprioceptive loss and may fall without vision. By contrast, the anterior lobe lesion patients tend to oscillate markedly without falling when their eyes are closed. Patients with anterior lobe damage also tend to move much more in an anteroposterior direction while those with Friedreich ataxia have an abnormal degree of lateral sway.
In addition to low-frequency (~1 Hz) sway, a characteristic 2–3 Hz body tremor is seen exclusively in patients with anterior lobe dysfunction when the eyes are closed. Unlike the irregular head and trunk titubation that may be seen with various other cerebellar lesions, the anterior lobe tremor consists of regular anteroposterior oscillation at the head, hip, and ankle. The hip is 180° out of phase with the head so that the center of gravity moves little and balance is maintained despite marked titubation. This tremor has been attributed to increased duration and amplitude of long-latency stretch responses. These long-latency responses are likely to be the scalable, secondary responses observed by Horak and Diener (1994) following exaggerated postural responses. Although abnormally large, these responses eventually stabilize the body after a few decaying oscillations at about 2–3 Hz when the eyes are open. Presumably, persistent titubation occurs especially with eyes closed because the gain of these scalable responses is increased in an effort to compensate for the loss of visual input. This is consistent with the view of titubation as a postural tremor.
Dysarthria
When ataxia affects speech, it is manifested as a clumsy, slurring, poorly modulated dysarthria. No disruption of language usage, structure, or content is attributable to the cerebellar dysfunction. As noted by Gilman et al. (1981), adjectives commonly used to describe cerebellar speech include scanning, slurring, staccato, explosive, hesitant, slow, altered accent, and garbled. These investigators have identified 10 elemental speech abnormalities which are present to varying degrees in different cerebellar patients: (1) imprecise consonants, a feature basic to all dysarthrias; (2) excess and equal stress, the inappropriate allocation of emphasis and accent; (3) irregular articulatory breakdown, the elision of syllables or phonemes; (4) distorted vowels; (5) harshness; (6) prolonged phonemes; (7) prolonged intervals; (8) monopitch; (9) monoloudness; and (10) slow rate. Cerebellar speech may also be tremulous and may trail off to a whisper. However, it should again be noted that many patients are intentionally slow and perhaps regularize their speech, i.e., voluntarily generate “scanning” speech to increase its intelligibility.
Perhaps analogous to the two types of timing deficits in finger movements described by Keele and Ivry (1990), at least two levels of speech control may be abnormal in cerebellar dysarthria. First, it is evident that on simple repetition of syllables, the peak repetition rate is considerably reduced in cerebellar patients and the sounds are not crisp. This could easily be attributed to a difficulty with rapid production and termination of force in the musculature of the vocal tract and respiration. In addition, however, there seems to be a poor regulation of the normal speech prosody or rhythm that is not simply due to decreased ability to speak quickly. Correspondingly, at least two locations for cerebellar control of speech have been suggested. Holmes described dysarthria in gunshot wound patients with damage to the cerebellar hemispheres that was more pronounced when the vermis was also damaged, suggesting important roles for both the vermis and hemispheres.
Dysarthria has been reported in cases where lesions were apparently confined to the vermis (Kadota et al., 1994) and Chiu et al. (1996) have stressed the importance of the vermis and fastigial nuclei in speech integration. On the other hand, Lechtenberg and Gilman (1978) have identified a paravermal site in the left cerebellar hemisphere that is specifically related to cerebellar dysarthria. They speculate that this cerebellar region functions in association with prosody areas in the right cerebral hemisphere to help regulate the timing of speech. The left hemisphere site is probably the more important of at least two cerebellar regions involved in normal speech production.
Gait ataxia
Despite the variability in ataxic locomotion there are still consistent kinematic patterns (Palliyath et al., 1998). As in upper extremity multijoint movement tasks, lower extremity multijoint coordination is characteristically abnormal. In particular, when walking, patients show a relatively greater delay of plantar flexion at the ankle than in flexion at the knee, as well as a relatively sluggish dorsiflexion of the ankle at the onset of swing. In walking, the largest and fastest required force transients are the forceful ankle plantar flexion at the end of stance and the rapid ankle dorsiflexion that follows immediately at the onset of swing. Therefore, as argued with respect to the shoulder in upper extremity multijoint coordination failure, each of these two lower extremity coordination abnormalities is consistent with a force-rate deficit (or perhaps force-delay) at the joint, in this case the ankle, that has the greatest force-rate demand. The situation is not completely clear, however, because a similar ankle–knee relationship may be seen in elderly subjects without ataxia. Further quantitative studies are needed. In any case, owing at least in part to the sluggishness of dorsiflexion, there is a tendency for ataxic patients to trip as their toes fail to clear the ground during swing. That no significant abnormality was noted in the height of toe lift during swing phase by Palliyath et al. (1998) may well be because trials in which stumbles occurred were excluded from analysis.
Ataxic gait, when under control, tends to be slower than normal and to have shortened strides. As argued by Palliyath et al. (1998), this is at least partially a voluntary compensation for the loss of control that occurs at higher speeds. Because walking involves controlled falling, both forward and laterally onto the next foot placement, walking with too slow a cadence demands prolonged balancing on each leg, which is difficult for the ataxic patient. Therefore, as patients slow down, they will tend to adopt a much shorter stride to maintain their cadence, or a wider base to stabilize themselves laterally. Possibly because of the resulting waddle, patients sometimes report that they walk “better” when they move at a moderate speed rather than very slowly, even though they may become more prone to veer or to trip than when they waddle.
Impaired motor learning
The consideration that ataxic patients should have difficulty with motor learning follows from the apparent logic that if they could learn, then why would they still be clumsy. Motor learning itself is a complex phenomenon with a number of different components (Hallett et al., 1996; Hallett and Grafman, 1997). One aspect can be defined as a change in motor performance with practice. Other aspects would include increasing the repertoire of motor behavior and maintenance of a new behavior over a period of time. Even considering only a change in motor performance, there are likely to be several different phenomena. Adaptation and skill learning can be distinguished. Adaptation is simply a change in the nature of the motor output while skill learning is the development of a new capability.
Adaptation learning clearly involves the cerebellum. Adaptation to lateral displacement of vision as produced by prism glasses is a method for assessing learning of a visual-motor task. When prism glasses are used, there is at first a mismatch between where an object is seen and where the pointing is directed. With experience, normal human subjects adjust to this and begin to point correctly. This correct pointing can be a product of both a true change in the visual-motor coordination or an intellectual decision to point in a different direction than where the object appears to be so that the correct movement is made. When the glasses are removed, typically the subject initially points in the opposite direction to that when the glasses were put on. In the naive subject, this is an excellent measure of true change in the visual-motor task since there is no reason for making any intellectual decision to point other than in the direction that the object appears to be. With additional experience, the subjects return to correct performance again. Patients with cerebellar damage show poor or no adaptation (Weiner et al., 1983; Martin et al., 1996). Another paradigm that can test adaptation learning is a task with a change in the visual-motor gain. An example is making movements of the elbow by matching targets on a computer screen. If the gain of the elbow with respect to the display on the computer screen is changed, then the amount of movement to match the targets will change. In the normal circumstance after a gain change, there would be an error that gradually would be reduced with continued practice. Deuschl et al. (1996) found that ataxic patients showed much slower learning than the normal controls.
Eye blink conditioning is recognized as a form of motor learning and could be argued to fit the definition proposed here for adaptation learning. In nonhuman animal studies, eye blink conditioning seems to require the cerebellum, at least for the expression and timing of the response. A number of groups have studied eye blink conditioning in patients with cerebellar lesions and found them to be markedly deficient (Daum et al., 1993; Topka et al., 1993).
There have been fewer studies of motor skill learning. Topka et al. (1998) evaluated the ability of patients to learn a multijoint two-dimensional trajectory with the upper extremity. While the performance of the patients was clearly impaired, they improved their performance as much as the normal subjects as long as the task was done slowly. With rapid movements, learning did slow down abnormally in the patients. Skill learning has many components including features such as the sequencing of the different components. Other parts of the brain play important roles in these other functions and can be responsible for the learning in the ataxic patients. Functional imaging studies, for example, show increased activation with learning in motor cortex, premotor cortex, and parietal areas (Hallett et al., 1996; Hallett and Grafman, 1997). The cortical network becomes more tightly connected (Wu et al., 2008).
Clinical localization
Virtually any lesion of the cerebellar parenchyma can be associated with ataxia. Presumably due to the considerable redundancy, complex interconnectedness, and plasticity of cerebellar circuits, as well as to inter-individual physiologic differences, attempts at precisely localizing cerebellar function on the basis of experimental and natural lesions have yielded inconsistent results. Similarly, it is usually not possible to predict lesion site within the cerebellum with great accuracy from the clinical examination. The clinicoanatomic correlations described below should therefore be viewed only as predominant patterns. See Timmann et al. (2008) for a review.
In general, signs that involve only the limbs unilaterally are most often due to lesions of the ipsilateral cerebellar hemispheres. However, this is not uniformly the case. In a series of 106 patients having unilateral or predominantly unilateral hemispheric injury (most postsurgical cases) studied by Lechtenberg and Gilman (Gilman et al., 1981), predominantly right limb dysmetria was associated with right hemisphere damage in 22 of 26 (85%) cases and left limb dysmetria with left hemispheric damage in 37 of 42 (88%) cases. For dysdiadochokinesia, ipsilateral hemispheric lesions were seen in 11 of 12 (91%) right predominant cases and 25 of 32 (78%) left predominant cases. The mechanism of strictly contralateral hemispheric effects on limb movement is not known. The handedness of the subjects was not reported. However, assuming the usual prevalence, the findings for dysdiadochokinesia are consistent with a mild additional impairment of the nondominant hand. This is consistent with the apparent importance of cerebrocerebellar interaction with rapid alternating movements (see above). When vermal lesions affected the limbs, the deficits were usually seen bilaterally and when there was asymmetry of deficit, the left limbs tended to be more severely affected. Overall, tremor was found less often than dysmetria or dysdiadochokinesia, but it occurred with similar rates in vermal and unilateral hemispheric disease.
Cortical control mechanisms
As noted earlier, the primary motor cortex provides the principal output to the corticospinal tract. Thus, its inputs determine the brain’s contribution to movement. The main inputs come from the premotor cortices, including the lateral premotor cortex, the supplementary motor area, and the caudal parts of the cingulate motor area. These areas in turn receive their input from wide areas of brain including the presupplementary motor area, rostral parts of the cingulate motor area, dorsolateral prefrontal cortex, and parietal areas. Considerable attention has been given recently to the parietal-premotor connections, which are highly specific and appear to provide important links between sensory and motor function (Rizzolatti et al., 1998; Rizzolatti and Luppino, 2001) (Fig. 2.5).
Apraxia
The apraxias are disorders of motor control, characterized by a loss of the motor program, not explicable by more elemental motor, sensory, coordination, or language impairments (Haaland et al., 2000; Hanna-Pladdy et al., 2001; Zadikoff and Lang, 2005; Gross and Grossman, 2008). Idiomotor apraxia is present when there is knowledge of the task, but there are temporal and spatial errors in performance (Wheaton and Hallett, 2007). It has long been suspected to be due to a disconnection between parietal and premotor areas. Table 2.3 lists the types of apraxias.
| Limb kinetic apraxia | Loss of hand and finger dexterity; significantly affecting manipulative movements |
| Ideomotor apraxia | Deficit in pantomiming tool use and gestures with temporal and spatial errors. Knowledge of tasks is still present |
| Ideational apraxia | Failure to carry out a series of tasks using multiple objects for an intended purpose; problem in the sequencing of actions. Tools are identifiable |
| Conceptual apraxia | Loss of tool knowledge; inappropriate use of tools and objects; inability to solve mechanical problems |
| (Verbal-motor) Dissociation apraxia | Failure to respond to verbal commands, but use of objects is appropriate |
| Conduction apraxia | Problems with imitating, but not with responding to verbal commands |
Modified from Wheaton LA, Hallett M. Ideomotor apraxia: A review. J Neurol Sci 2007;260:1–10.
What is a voluntary movement?
While clearly much is known about the anatomy and physiology of the motor system, there is still considerable difficulty with the concept of voluntariness. Many movements are triggered by sensory stimuli, and the physiology of this mechanism is relatively clear. However, there are certainly movements that appear to be “internally triggered” and humans have the sense that they have willed the movement. The self-initiation of movement and conscious awareness of movement appear to involve mesial motor structures such as the supplementary motor area and the dorsolateral prefrontal cortex (Deiber et al., 1999). As pointed out by Paus (2001), the mesial motor structures including the anterior cingulate cortex, in particular, is a place of convergence for motor control, homeostatic drive, emotion, and cognition. Looked at critically, the sense of voluntariness is clearly a “perception of consciousness” (what can be called a quale). There is very little understanding of how this evolves.
Disorders of willed movement
In neurology, there are many disorders where the issue of will arises (Hallett, 2006c, 2007b, 2009).
Although tics are generally considered involuntary, patients with tics often cannot say whether their movements are voluntary or involuntary. This may not be a relevant distinction in their minds. It is perhaps a better description to say that they can suppress their movements or they just let them happen. Tics look like voluntary movements in all respects from the point of view of EMG and kinesiology (Hallett, 2000). Interestingly, they are often not preceded by the normal brain potential, the Bereitschaftspotential, and hence the brain mechanisms for their production clearly differs from ordinary voluntary movement (Obeso et al., 1981; Karp et al., 1996).
The symptom of loss of voluntary movement is often called abulia or, in the extreme, akinetic mutism (Fisher, 1983). The classic lesion is in the midline frontal region affecting areas including the supplementary motor area and cingulate motor areas. The bradykinesia and akinesia of Parkinson disease are related.
The alien hand phenomenon is characterized by unwanted movements that arise without any sense of their being willed. In addition to simple, unskilled, quasi-reflex movements (such as grasping), there can also be complex, skilled movements such as intermanual conflict or interference (Fisher, 2000). There appears to be difficulty in self-initiating movement and excessive ease in the production of involuntary and triggered movements. In cases with discrete lesions, this seems to have its anatomical correlation in the territory of the anterior cerebral artery.
Conversion psychogenic movements are movements interpreted by the patient as involuntary. Their etiology is actually obscure since the physiology of conversion is really unknown. EEG investigation of these movements shows a normal looking Bereitschaftspotential preceding them (Toro and Torres, 1986; Terada et al., 1995). The normal brain potential, however, indicates that there must be substantial sharing of brain voluntary movement mechanisms (Hallett, 2010).
Acknowledgment
The ataxia section of the chapter is updated from a syllabus written for the 1999 meeting of the American Academy of Neurology, which itself was extensively modified and updated from a chapter by Massaquoi and Hallett (1998). The Parkinson disease section is modified from an earlier chapter (Hallett, 2003a). The dystonia section is modified from earlier chapters (Hallett, 2004).
Alexander G.E., Crutcher M.D. Functional achitecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266-271.
Angel R.W. Barognosis in a patient with hemiataxia. Ann Neurol. 1980;7(1):73-77.
Angelucci A., Levitt J.B., Lund J.S. Anatomical origins of the classical receptive field and modulatory surround field of single neurons in macaque visual cortical area V1. Prog Brain Res. 2002;136:373-388.
Aravamuthan B.R., Muthusamy K.A., Stein J.F., et al. Topography of cortical and subcortical connections of the human pedunculopontine and subthalamic nuclei. Neuroimage. 2007;37(3):694-705.
Baker R.S., Sun W.S., Hasan S.A., et al. Maladaptive neural compensatory mechanisms in Bell’s palsy-induced blepharospasm. Neurology. 1997;49(1):223-229.
Bar-Gad I., Morris G., Bergman H. Information processing, dimensionality reduction and reinforcement learning in the basal ganglia. Prog Neurobiol. 2003;71(6):439-473.
Bara-Jimenez W., Catalan M.J., Hallett M., Gerloff C. Abnormal somatosensory homunculus in dystonia of the hand. Ann Neurol. 1998;44:828-831.
Baroni A., Benvenuti F., Fantini L., et al. Human ballistic arm abduction movements: effects of L-dopa treatment in Parkinson’s disease. Neurology. 1984;34(7):868-876.
Bastian A.J., Martin T.A., Keating J.G., Thach W.T. Cerebellar ataxia. Abnormal control of interaction torques across multiple joints. J Neurophysiol. 1996;76:492-509.
Bastian A.J., Zackowski K.M., Thach W.T. Cerebellar ataxia: torque deficiency or torque mismatch between joints? J Neurophysiol. 2000;83(5):3019-3030.
Beck S., Hallett M. Surround inhibition in the motor system. Exp Brain Res. 2011;210(2):165-172.
Beck S., Richardson S.P., Shamim E.A., et al. Short intracortical and surround inhibition are selectively reduced during movement initiation in focal hand dystonia. J Neurosci. 2008;28(41):10363-10369.
Becker W.J., Kunesch E., Freund H.J. Coordination of a multi-joint movement in normal humans and in patients with cerebellar dysfunction. Can J Neurol Sci. 1990;17(3):264-274.
Benabid A.L., Pollak P., Gao D., et al. Chronic electrical stimulation of the ventralis intermedius nucleus of the thalamus as a treatment of movement disorders. J Neurosurg. 1996;84:203-214.
Benecke R., Rothwell J.C., Dick J.P., et al. Simple and complex movements off and on treatment in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1987;50(3):296-303.
Beppu H., Nagaoka M., Tanaka R. Analysis of cerebellar motor disorders by visually-guided elbow tracking movement. 2. Contribution of the visual cues on slow ramp pursuit. Brain. 1987;110(Pt 1):1-18.
Beppu H., Suda M., Tanaka R. Analysis of cerebellar motor disorders by visually guided elbow tracking movement. Brain. 1984;107(Pt 3):787-809.
Berardelli A., Dick J.P., Rothwell J.C., et al. Scaling of the size of the first agonist EMG burst during rapid wrist movements in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1986;49(11):1273-1279.
Berardelli A., Rothwell J.C., Day B.L., Marsden C.D. Pathophysiology of blepharospasm and oromandibular dystonia. Brain. 1985;108:593-608.
Berardelli A., Rothwell J., Thompson P.D., Hallett M. Pathophysiology of bradykinesia in Parkinson’s disease. Brain. 2001;124(11):2131-2146.
Berardelli A., Sabra A.F., Hallett M. Physiological mechanisms of rigidity in Parkinson’s Disease. J Neurol Neurosurg Psychiatry. 1983;46(1):45-53.
Bergui M., Lopiano L., Paglia G., et al. Stretch reflex of quadriceps femoris and its relation to rigidity in Parkinson’s disease. Acta Neurol Scand. 1992;86(3):226-229.
Bower J.M. Control of sensory data acquisition. Int Rev Neurobiol. 1997;41:489-513.
Breakefield X.O., Blood A.J., Li Y., et al. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9(3):222-234.
Britton T.C., Thompson P.D., Day B.L., et al. Modulation of postural tremors at the wrist by supramaximal electrical median nerve shocks in essential tremor, Parkinson’s disease and normal subjects mimicking tremor. J Neurol Neurosurg Psychiatry. 1993;56:1085-1089.
Britton T.C., Thompson P.D., Day B.L., et al. Modulation of postural wrist tremors by magnetic stimulation of the motor cortex in patients with Parkinson’s disease or essential tremor and in normal subjects mimicking tremor. Ann Neurol. 1993;33:473-479.
Brown V.J., Schwarz U., Bowman E.M., et al. Dopamine dependent reaction time deficits in patients with Parkinson’s disease are task specific. Neuropsychologia. 1993;31(5):459-469.
Byl N., Merzenich M.M., Jenkins W.M. A primate genesis model of focal dystonia and repetitive strain injury: I. Learning-induced dedifferentiation of the representation of the hand in the primary somatosensory cortex in adult monkeys. Neurology. 1996;47:508-520.
Chen R., Kumar S., Garg R.R., Lang A.E. Impairment of motor cortex activation and deactivation in Parkinson’s disease. Clin Neurophysiol. 2001;112(4):600-607.
Chen R., Wassermann E., Caños M., Hallett M. Impaired inhibition in writer’s cramp during voluntary muscle activation. Neurology. 1997;49:1054-1059.
Chiu M.J., Chen R.C., Tseng C.Y. Clinical correlates of quantitative acoustic analysis in ataxic dysarthria. Eur Neurol. 1996;36(5):310-314.
Chuke J.C., Baker R.S., Porter J.D. Bell’s Palsy-associated blepharospasm relieved by aiding eyelid closure. Ann Neurol. 1996;39:263-268.
Cody F.W., MacDermott N., Matthews P.B., Richardson H.C. Observations on the genesis of the stretch reflex in Parkinson’s disease. Brain. 1986;109(Pt 2):229-249.
Cohen L.G., Hallett M. Hand cramps: clinical features and electromyographic patterns in a focal dystonia. Neurology. 1988;38:1005-1012.
Curra A., Berardelli A., Agostino R., et al. Performance of sequential arm movements with and without advance knowledge of motor pathways in Parkinson’s disease. Mov Disord. 1997;12(5):646-654.
Daum I., Schugens M.M., Ackermann H., et al. Classical conditioning after cerebellar lesions in humans. Behav Neurosci. 1993;107:748-756.
Defazio G., Berardelli A., Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain. 2007;130(Pt 5):1183-1193.
Deiber M.P., Honda M., Ibanez V., et al. Mesial motor areas in self-initiated versus externally triggered movements examined with fMRI: effect of movement type and rate. J Neurophysiol. 1999;81(6):3065-3077.
DeLong M.R., Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64(1):20-24.
Demirci M., Grill S., McShane L., Hallett M. A mismatch between kinesthetic and visual perception in Parkinson’s disease. Ann Neurol. 1997;41(6):781-788.
Deuschl G., Toro C., Zeffiro T., et al. Adaptation motor learning of arm movements in patients with cerebellar diseases. J Neurol Neurosurg Psychiatry. 1996;60:515-519.
Di Lazzaro V., Oliviero A., Meglio M., et al. Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex. Clin Neurophysiol. 2000;111(5):794-799.
Diener H.C., Dichgans J. Pathophysiology of cerebellar ataxia. Mov Disord. 1992;7:95-109.
Diener H.C., Dichgans J., Bacher M., Gompf B. Quantification of postural sway in normals and patients with cerebellar diseases. Electroencephalogr Clin Neurophysiol. 1984;57:134-142.
Diener H.C., Hore J., Ivry R., Dichgans J. Cerebellar dysfunction of movement and perception. Can J Neurol Sc. 1993;20(Suppl 3):S62-S69.
Doder M., Rabiner E.A., Turjanski N., et al. Tremor in Parkinson’s disease and serotonergic dysfunction: an 11C-WAY 100635 PET study. Neurology. 2003;60(4):601-605.
Elble R.J. The pathophysiology of tremor. In: Watts R.L., Koller W.C., editors. Movement Disorders Neurologic Principles and Practice. New York: McGraw-Hill; 1997:405-417.
Elble R.J., Koller W.C. Tremor. Baltimore: Johns Hopkins University Press; 1990.
Evarts E.V., Teravainen H., Calne D.B. Reaction time in Parkinson’s disease. Brain. 1981;104(Pt 1):167-186.
Fisher C.M. Honored guest presentation: abulia minor vs. agitated behavior. Clin Neurosurg. 1983;31:9-31.
Fisher C.M. Alien hand phenomena: a review with the addition of six personal cases. Can J Neurol Sci. 2000;27(3):192-203.
Fuhr P., Agostino R., Hallett M. Spinal motor neuron excitability during the silent period after cortical stimulation. Electroencephalogr Clin Neurophysiol. 1991;81:257-262.
Gao J.H., Parsons L.M., Bower J.M., et al. Cerebellum implicated in sensory acquisition and discrimination rather than motor control. Science. 1996;272(5261):545-547.
Garraux G., Bauer A., Hanakawa T., et al. Changes in brain anatomy in focal hand dystonia. Ann Neurol. 2004;55(5):736-739.
Gilman S., Bloedel J.R., Lechtenberg R. Disorders of the Cerebellum. Philadelphia: F.A. Davis; 1981.
Gilman S., Carr D., Hollenberg J. Kinematic effects of deafferentation and cerebellar ablation. Brain. 1976;99(2):311-330.
Godaux E., Koulischer D., Jacquy J. Parkinsonian bradykinesia is due to depression in the rate of rise of muscle activity. Ann Neurol. 1992;31(1):93-100.
Grill S.E., Hallett M., Marcus C., McShane L. Disturbances of kinesthesia in patients with cerebellar disorders. Brain. 1994;117:1433-1447.
Grill S.E., Hallett M., McShane L.M. Timing of onset of afferent responses and of use of kinesthetic information for control of movement in normal and cerebellar-impaired subjects. Exp Brain Res. 1997;113(1):33-47.
Gross R.G., Grossman M. Update on apraxia. Curr Neurol Neurosci Rep. 2008;8(6):490-496.
Haaland K.Y., Harrington D.L., Knight R.T. Neural representations of skilled movement. Brain. 2000;123(Pt 11):2306-2313.
Hallett M. Differential diagnosis of tremor. In: Vinken P.J., Bruyn G.W., Klawans H.L., editors. Handbook of Clinical Neurology: Extrapyramidal Disorders. Amsterdam: Elsevier Science Publishers; 1987:583-595.
Hallett M. Clinical neurophysiology of akinesia. Rev Neurol (Paris). 1990;146:585-590.
Hallett M. Classification and treatment of tremor. JAMA. 1991;266:1115-1117.
Hallett M. Electrophysiologic evaluation of movement disorders. In: Aminoff M.J., editor. Electrodiagnosis in Clinical Neurology. fourth ed. New York: Churchill Livingstone; 1999:365-380.
Hallett M. Neurophysiology of tics. In: Cohen D.J., Goetz C.G., Jankovic J., editors. Tourette Syndrome. Philadelphia: Lippincott Williams & Wilkins; 2000:237-244.
Hallett M. Parkinson revisited: pathophysiology of motor signs. Adv Neurol. 2003;91:19-28.
Hallett M., editor. Movement Disorders. Amsterdam: Elsevier, 2003.
Hallett M. Dystonia: abnormal movements result from loss of inhibition. Adv Neurol. 2004;94:1-9.
Hallett M. Pathophysiology of dystonia. J Neural Transm Suppl.. 2006;70:485-488.
Hallett M. Pathophysiology of writer’s cramp. Hum Mov Sci.. 2006;25(4–5):454-463.
Hallett M. Voluntary and involuntary movements in humans. In: Hallett M., Fahn S., Jankovic J., Lang A.E., Cloninger C.R., Yudofsky S.C., editors. Psychogenic Movement Disorders Neurology and Psychiatry. Philadelphia: AAN Press, Lippincott Williams & Wilkins; 2006:189-195.
Hallett M. Transcranial magnetic stimulation: a primer. Neuron. 2007;55(2):187-199.
Hallett M. Volitional control of movement: The physiology of free will. Clin Neurophysiol.. 2007;118(6):1179-1192.
Hallett M. Physiology of volition. In: Murphy N., Ellis G.F.R., O’Connor T., editors. Downward Causation and the Neurobiology of Free Will. Berlin: Springer; 2009:127-143.
Hallett M. Physiology of psychogenic movement disorders. J Clin Neurosci. 2010;17(8):959-965.
Hallett M. Neurophysiology of dystonia: The role of inhibition. Neurobiol Dis. 2011;42(2):177-184.
Hallett M., Berardelli A., Delwaide P., et al. Central EMG and tests of motor control. Report of an IFCN Committee. Electroencephalogr Clin Neurophysiol. 1994;90:404-432.
Hallett M., Berardelli A., Matheson J., et al. Physiological analysis of simple rapid movements in patients with cerebellar deficits. J Neurol Neurosurg Psychiatry. 1991;53:124-133.
Hallett M., Grafman J. Executive function and motor skill learning. In: Schmahmann J.D., editor. The Cerebellum and Cognition. San Diego: Academic Press; 1997:297-323.
Hallett M., Khoshbin S. A physiological mechanism of bradykinesia. Brain. 1980;103:301-314.
Hallett M., Massaquoi S. Physiologic studies of dysmetria in patients with cerebellar deficits. Can J Neurol Sci. 1993;20(Suppl 3):S83-S89.
Hallett M., Pascual-Leone A., Topka H. Adaptation and skill learning. Evidence for different neural substrates. In: Bloedel J.R., Ebner T.J., Wise S.P., editors. Acquisition of Motor Behavior in Vertebrates. Cambridge: MIT Press; 1996:289-301.
Hallett M., Shahani B.T., Young R.R. EMG analysis of patients with cerebellar deficits. J Neurol Neurosurg Psychiatry. 1975;38(12):1163-1169.
Hallett M., Shahani B.T., Young R.R. Analysis of stereotyped voluntary movements at the elbow in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1977;40:1129-1135.
Hamani C., Stone S., Laxton A., Lozano A.M. The pedunculopontine nucleus and movement disorders: anatomy and the role for deep brain stimulation. Parkinsonism Relat Disord. 2007;13(Suppl 3):S276-S280.
Hanna-Pladdy B., Heilman K.M., Foundas A.L. Cortical and subcortical contributions to ideomotor apraxia: analysis of task demands and error types. Brain. 2001;124(Pt 12):2513-2527.
Hayase N., Miyashita N., Endo K., Narabayashi H. Neuronal activity in GP and Vim of parkinsonian patients and clinical changes of tremor through surgical interventions. Stereotact Funct Neurosurg. 1998;71(1):20-28.
Helmich R.C., Janssen M.J., Oyen W.J., et al. Pallidal dysfunction drives a cerebellothalamic circuit into Parkinson tremor. Ann Neurol. 2011;69(2):269-281.
Herath P., Gallea C., van der Veen J.W., et al. In vivo neurochemistry of primary focal hand dystonia: a magnetic resonance spectroscopic neurometabolite profiling study at 3T. Mov Disord. 2010;25(16):2800-2808.
Holmes G. The cerebellum of man. Brain. 1939;62:1-30.
Horak F.B., Diener H.C. Cerebellar control of postural scaling and central set in stance. J Neurophysiol. 1994;72(2):479-493.
Hore J., Flament D. Evidence that a disordered servo-like mechanism contributes to tremor in movements during cerebellar dysfunction. J Neurophysiol. 1986;56(1):123-136.
Hore J., Wild B., Diener H.C. Cerebellar dysmetria at the elbow, wrist, and fingers. J Neurophysiol. 1991;65(3):563-571.
Hurtado J.M., Gray C.M., Tamas L.B., Sigvardt K.A. Dynamics of tremor-related oscillations in the human globus pallidus: a single case study. Proc Natl Acad Sci USA. 1999;96(4):1674-1679.
Hurtado J.M., Lachaux J.P., Beckley D.J., et al. Inter- and intralimb oscillator coupling in parkinsonian tremor. Mov Disord. 2000;15(4):683-691.
Ito M. The Cerebellum and Neural Control. New York: Raven Press; 1984.
Jankovic J., Cardoso F., Grossman R.G., Hamilton W.J. Outcome after stereotactic thalamotomy for parkinsonian, essential, and other types of tremor. Neurosurgery. 1995;37:680-686.
Jordan N., Sagar H.J., Cooper J.A. A component analysis of the generation and release of isometric force in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1992;55(7):572-576.
Kadota O., Sakaki S., Kumon Y., et al. Large cystic cavernous angioma of the cerebellum – case report. Neurol Med Chir (Tokyo). 1994;34(11):768-772.
Karp B.I., Porter S., Toro C., Hallett M. Simple motor tics may be preceded by a premotor potential. J Neurol Neurosurg Psychiatry. 1996;61:103-106.
Keele, S.W., Ivry, R., 1990. Does the cerebellum provide a common computation for diverse tasks? A timing hypothesis. Ann NY Acad Sci 608, 179–207.
Labutta R.J., Miles R.B., Sanes J.N., Hallett M. Motor program memory storage in Parkinson’s disease patients tested with a delayed response task. Mov Disord. 1994;9:218-222.
Lechtenberg R., Gilman S. Speech disorders in cerebellar disease. Ann Neurol. 1978;3(4):285-290.
Lerner A., Shill H., Hanakawa T., et al. Regional cerebral blood flow correlates of the severity of writer’s cramp symptoms. NeuroImage. 2004;21:904-913.
Levy L.M., Hallett M. Impaired brain GABA in focal dystonia. Ann Neurol. 2002;51(1):93-101.
Magnin M., Morel A., Jeanmonod D. Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neuroscience. 2000;96(3):549-564.
Mai N., Bolsinger P., Avarello M., et al. Control of isometric finger force in patients with cerebellar disease. Brain. 1988;111(Pt 5):973-998.
Majsak M.J., Kaminski T., Gentile A.M., Flanagan J.R. The reaching movements of patients with Parkinson’s disease under self- determined maximal speed and visually cued conditions. Brain. 1998;121(Pt 4):755-766.
Manto M.U. Effects of hyperventilation on fast goal-directed limb movements in spinocerebellar ataxia type 6. Eur J Neurol. 2001;8(5):401-406.
Manto M.U., Setta F., Jacquy J., et al. Different types of cerebellar hypometria associated with a distinct topography of the lesion in cerebellum. J Neurol Sci. 1998;158(1):88-95.
Martin T.A., Keating J.G., Goodkin H.P., et al. Throwing while looking through prisms. I. Focal olivocerebellar lesions impair adaptation. Brain. 1996;119(Pt 4):1183-1198.
Massaquoi S., Hallett M. Kinematics of initiating a two-joint arm movement in patients with cerebellar ataxia. Can J Neurol Sci. 1996;23:3-14.
Massaquoi S.G., Hallett M. Ataxia and other cerebellar syndromes. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. third ed. Baltimore: Williams & Wilkins; 1998:623-686.
Massaquoi S.G., Hallett M. Ataxia and other cerebellar syndromes. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. fourth ed. Philadelphia: Lippincott, Williams & Wilkins; 2002:393-408.
Massaquoi S.G., Slotine J.J. The intermediate cerebellum may function as a wave-variable processor. Neurosci Lett. 1996;215(1):60-64.
Matsumura M., Sawaguchi T., Kubota K. GABAergic inhibition of neuronal activity in the primate motor and premotor cortex during voluntary movement. J Neurophysiol. 1992;68:692-702.
Matsumura M., Sawaguchi T., Oishi T., et al. Behavioral deficits induced by local injection of bicuculline and muscimol into the primate motor and premotor cortex. J Neurophysiol. 1991;65:1542-1553.
Mazzoni P., Hristova A., Krakauer J.W. Why don’t we move faster? Parkinson’s disease, movement vigor, and implicit motivation. J Neurosci. 2007;27(27):7105-7116.
Meara R.J., Cody F.W. Stretch reflexes of individual parkinsonian patients studied during changes in clinical rigidity following medication. Electroencephalogr Clin Neurophysiol. 1993;89(4):261-268.
Meunier S., Garnero L., Ducorps A., et al. Human brain mapping in dystonia reveals both endophenotypic traits and adaptive reorganization. Ann Neurol. 2001;50(4):521-527.
Mink J.W. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381-425.
Mink J.W. The basal ganglia and involuntary movements: impaired inhibition of competing motor patterns. Arch Neurol. 2003;60(10):1365-1368.
Mink J.W. Neurobiology of basal ganglia and Tourette syndrome: basal ganglia circuits and thalamocortical outputs. Adv Neurol. 2006;99:89-98.
Molloy F.M., Carr T.D., Zeuner K.E., et al. Abnormalities of spatial discrimination in focal and generalized dystonia. Brain. 2003;126(Pt 10):2175-2182.
Morrice B.L., Becker W.J., Hoffer J.A., Lee R.G. Manual tracking performance in patients with cerebellar incoordination: effects of mechanical loading. Can J Neurol Sci. 1990;17(3):275-285.
Muthusamy K.A., Aravamuthan B.R., Kringelbach M.L., et al. Connectivity of the human pedunculopontine nucleus region and diffusion tensor imaging in surgical targeting. J Neurosurg. 2007;107(4):814-820.
Nakashima K., Rothwell J.C., Day B.L., et al. Reciprocal inhibition in writer’s and other occupational cramps and hemiparesis due to stroke. Brain. 1989;112:681-697.
Nathan P.W., Smith M., Deacon P. Vestibulospinal, reticulospinal and descending propriospinal nerve fibres in man. Brain. 1996;119(Pt 6):1809-1833.
Obeso J.A., Rothwell J.C., Marsden C.D. Simple tics in Gilles de la Tourette’s syndrome are not prefaced by a normal premovement potential. J Neurol Neurosurg Psychiatry. 1981;44:735-738.
Otsuka M., Ichiya Y., Kuwabara Y., et al. Differences in the reduced 18F-Dopa uptakes of the caudate and the putamen in Parkinson’s disease: correlations with the three main symptoms. J Neurol Sci. 1996;136:169-173.
Palliyath S., Hallett M., Thomas S.L., Lebiedowska M.K. Gait in patients with cerebellar ataxia. Mov Disord. 1998;13(6):958-964.
Panizza M., Lelli S., Nilsson J., Hallett M. H-reflex recovery curve and reciprocal inhibition of H-reflex in different kinds of dystonia. Neurology. 1990;40:824-828.
Pascual-Leone A., Valls-Solé J., Brasil-Neto J., et al. Akinesia in Parkinson’s disease. I. Shortening of simple reaction time with focal, single-pulse transcranial magnetic stimulation. Neurology. 1994;44:884-891.
Pascual-Leone A., Valls-Solé J., Toro C., et al. Resetting of essential tremor and postural tremor in Parkinson’s disease with transcranial magnetic stimulation. Muscle Nerve. 1994;17:800-807.
Paus T. Primate anterior cingulate cortex: where motor control, drive and cognition interface. Nat Rev Neurosci. 2001;2(6):417-424.
Perlmutter J.S., Stambuk M.K., Markham J., et al. Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J Neurosci. 1997;17:843-850.
Peterson D.A., Sejnowski T.J., Poizner H. Convergent evidence for abnormal striatal synaptic plasticity in dystonia. Neurobiol Dis. 2010;37(3):558-573.
Quartarone A., Siebner H.R., Rothwell J.C. Task-specific hand dystonia: can too much plasticity be bad for you? Trends Neurosci. 2006;29(4):192-199.
Ridding M.C., Sheean G., Rothwell J.C., et al. Changes in the balance between motor cortical excitation and inhibition in focal, task specific dystonia. J Neurol Neurosurg Psychiatry. 1995;59:493-498.
Rizzolatti G., Luppino G. The cortical motor system. Neuron. 2001;31(6):889-901.
Rizzolatti G., Luppino G., Matelli M. The organization of the cortical motor system: new concepts. Electroencephalogr Clin Neurophysiol. 1998;106(4):283-296.
Rodriguez-Oroz M.C., Jahanshahi M., Krack P., et al. Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol. 2009;8(12):1128-1139.
Rothwell J.C., Obeso J.A., Traub M.M., Marsden C.D. The behaviour of the long-latency stretch reflex in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1983;46(1):35-44.
Sainburg R.L., Poizner H., Ghez C. Loss of proprioception produces deficits in interjoint coordination. J Neurophysiol. 1993;70(5):2136-2147.
Samii A., Wassermann E.M., Hallett M. Decreased postexercise facilitation of motor evoked potentials in patients with cerebellar degeneration. Neurology. 1997;49(2):538-542.
Sanes J.N., LeWitt P.A., Mauritz K.H. Visual and mechanical control of postural and kinetic tremor in cerebellar system disorders. J Neurol Neurosurg Psychiatry. 1988;51(7):934-943.
Sanger T.D., Garg R.R., Chen R. Interactions between two different inhibitory systems in the human motor cortex. J Physiol. 2001;530(Pt 2):307-317.
Schicatano E.J., Basso M.A., Evinger C. Animal model explains the origins of the cranial dystonia benign essential blepharospasm. J Neurophysiol. 1997;77(5):2842-2846.
Schmahmann J.D. The cerebellum in autism. Clinical and anatomical perspectives. In: Bauman M.L., Kemper T.L., editors. The Neurobiology of Autism. Baltimore: Johns Hopkins University Press; 1994:195-226.
Schmahmann J.D., Pandya D.N. The cerebrocerebellar system. Int Rev Neurobiol. 1997;41:31-60.
Schwab R.S., Chafetz M.E., Walker S. Control of two simultaneous voluntary motor acts in normals and in parkinsonism. Arch Neurol. 1954;72:591-598.
Shimamoto S.A., Larson P.S., Ostrem J.L., et al. Physiological identification of the human pedunculopontine nucleus. J Neurol Neurosurg Psychiatry. 2010;81(1):80-86.
Sohn Y.H., Hallett M. Disturbed surround inhibition in focal hand dystonia. Ann Neurol.. 2004;56(4):595-599.
Sohn Y.H., Hallett M. Surround inhibition in human motor system. Exp Brain Res.. 2004;158(4):397-404.
Sohn Y.H., Jung H.Y., Kaelin-Lang A., Hallett M. Excitability of the ipsilateral motor cortex during phasic voluntary hand movement. Exp Brain Res. 2003;148(2):176-185.
Starr A., Caramia M., Zarola F., Rossini P.M. Enhancement of motor cortical excitability in humans by non-invasive electrical stimulation appears prior to voluntary movement. Electroencephalogr Clin Neurophysiol. 1988;70:26-32.
Staude G., Wolf W., Ott M., et al. Tremor as a factor in prolonged reaction times of parkinsonian patients. Mov Disord. 1995;10(2):153-162.
Stefan K., Kunesch E., Cohen L.G., et al. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123(Pt 3):572-584.
Stinear C.M., Byblow W.D. Impaired modulation of intracortical inhibition in focal hand dystonia. Cereb Cortex. 2004;14(5):555-561.
Takakusaki K., Kohyama J., Matsuyama K., Mori S. Medullary reticulospinal tract mediating the generalized motor inhibition in cats: parallel inhibitory mechanisms acting on motoneurons and on interneuronal transmission in reflex pathways. Neuroscience. 2001;103(2):511-527.
Tamura Y., Matsuhashi M., Lin P., et al. Impaired intracortical inhibition in the primary somatosensory cortex in focal hand dystonia. Mov Disord. 2008;23(4):558-565.
Tass P., Smirnov D., Karavaev A., et al. The causal relationship between subcortical local field potential oscillations and Parkinsonian resting tremor. J Neural Eng. 2010;7(1):16009.
Tatton W.G., Bedingham W., Verrier M.C., Blair R.D. Characteristic alterations in responses to imposed wrist displacements in parkinsonian rigidity and dystonia musculorum deformans. Can J Neurol Sci. 1984;11:281-287.
Terada K., Ikeda A., Van Ness P.C., et al. Presence of Bereitschaftspotential preceding psychogenic myoclonus: clinical application of jerk-locked back averaging. J Neurol Neurosurg Psychiatry. 1995;58:745-747.
Terzuolo C.A., Soechting J.F., Viviani P. Studies on the control of some simple motor tasks. II. On the cerebellar control of movements in relation to the formulation of intentional commands. Brain Res. 1973;58(1):217-222.
Thach W.T., Goodkin H.P., Keating J.G. The cerebellum and the adaptive coordination of movement. Ann Rev Neurosci. 1992;15:403-442.
Thach W.T., Perry J.G., Kane S.A., Goodkin H.P. Cerebellar nuclei: rapid alternating movement, motor somatotopy, and a mechanism for the control of muscle synergy. Rev Neurol (Paris). 1993;149:607-628.
Timmann D., Brandauer B., Hermsdorfer J., et al. Lesion-symptom mapping of the human cerebellum. Cerebellum. 2008;7(4):602-606.
Tinazzi M., Priori A., Bertolasi L., et al. Abnormal central integration of a dual somatosensory input in dystonia. Evidence for sensory overflow. Brain. 2000;123(Pt 1):42-50.
Topka H., Konczak J., Schneider K., et al. Multijoint arm movements in cerebellar ataxia: abnormal control of movement dynamics. Exp Brain Res. 1998;119(4):493-503.
Topka H., Valls-Solé J., Massaquoi S., Hallett M. Deficit in classical conditioning in patients with cerebellar degeneration. Brain. 1993;116:961-969.
Toro C., Torres F. Electrophysiological correlates of a paroxysmal movement disorder. Ann Neurol. 1986;20(6):731-734.
Valls-Solé J., Pascual-Leone A., Wassermann E.M., Hallett M. Human motor evoked responses to paired transcranial magnetic stimuli. Electroencephalogr Clin Neurophysiol. 1992;85(6):355-364.
Warabi T., Yanagisawa N., Shindo R. Changes in strategy of aiming tasks in Parkinson’s disease. Brain. 1988;111(Pt 3):497-505.
Watts R.L., Mandir A.S. The role of motor cortex in the pathophysiology of voluntary movement deficits associated with parkinsonism. Neurol Clin. 1992;10(2):451-469.
Weiner M.J., Hallett M., Funkenstein H.H. Adaptation to lateral displacement of vision in patients with lesions of the central nervous system. Neurology. 1983;33:766-772.
Weise D., Schramm A., Stefan K., et al. The two sides of associative plasticity in writer’s cramp. Brain. 2006;129(Pt 10):2709-2721.
Werhahn K.J., Kunesch E., Noachtar S., et al. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J Physiol (Lond). 1999;517(Pt 2):591-597.
Wheaton L.A., Hallett M. Ideomotor apraxia: A review. J Neurol Sci. 2007;260:1-10.
Wichmann T., DeLong M.R. Functional neuroanatomy of the basal ganglia in Parkinson’s disease. Adv Neurol.. 2003;91:9-18.
Wichmann T., DeLong M.R. Pathophysiology of Parkinson’s disease: the MPTP primate model of the human disorder. Ann NY Acad Sci.. 2003;991:199-213.
Wing A.M., Keele S., Margolin D.I. Motor disorder and the timing of repetitive movements. Ann NY Acad Sci. 1984;423:183-192.
Wu T., Chan P., Hallett M. Modifications of the interactions in the motor networks when a movement becomes automatic. J Physiol. 2008;586(Pt 17):4295-4304.
Zadikoff C., Lang A.E. Apraxia in movement disorders. Brain. 2005;128(Pt 7):1480-1497.
Ziemann U., Rothwell J.C., Ridding M.C. Interaction between intracortical inhibition and facilitation in human motor cortex. J Physiol (Lond). 1996;496(Pt 3):873-881.
Zirh T.A., Lenz F.A., Reich S.G., Dougherty P.M. Patterns of bursting occurring in thalamic cells during parkinsonian tremor. Neuroscience. 1998;83(1):107-121.