CHAPTER 3 Molecules: Structures and Dynamics
This chapter describes the properties of water, proteins, nucleic acids, and carbohydrates as they pertain to cell biology. Chapter 7 covers lipids in the context of biological membranes.
Water
Water is so familiar that its role in cell biology and its fascinating properties tend to be neglected. Water is the most abundant and important molecule in cells and tissues. Humans are about two thirds water. Water is not only the solvent for virtually all cellular compounds but also a reactant or product in thousands of biochemical reactions catalyzed by enzymes, including the synthesis and degradation of proteins and nucleic acids and the synthesis and hydrolysis of adenosine triphosphate (ATP), to name a few examples. Water is also an important determinant of biological structure, as lipid bilayers, folded proteins, and macromolecular assemblies are all stabilized by the hydrophobic effect derived from the exclusion of water from nonpolar surfaces (see Fig. 4-5). Additionally, water forms hydrogen bonds with polar groups of many cellular constituents ranging in size from small metabolites to large proteins. It also associates with small inorganic ions.
Physical chemists are still trying to understand water, one of the most complex liquids. The molecule is roughly tetrahedral in shape (Fig. 3-1A), with two hydrogen bond donors and two hydrogen bond acceptors. The electronegative oxygen withdraws the electrons from the O—H covalent bonds, leaving a partial positive charge on the hydrogens and a partial negative charge on the oxygen. Hydrogen bonds between water molecules are partly electrostatic because of the charge separation (induced dipole) but also have some covalent character, owing to overlap of the electron orbitals. The strength of hydrogen bonds depends on their orientation, being strongest along the lines of tetrahedral orbitals. One can think of oxygens of two water molecules sharing a hydrogen-bonded hydrogen. Given two hydrogen bond donors and acceptors, water can be fully hydrogen-bonded, as it is in ice (Fig. 3-1C). Crystalline water in ice has a well-defined structure with a complete set of tetragonal hydrogen bonds and a remarkable amount (35%) of unoccupied space (Fig. 3-1D).
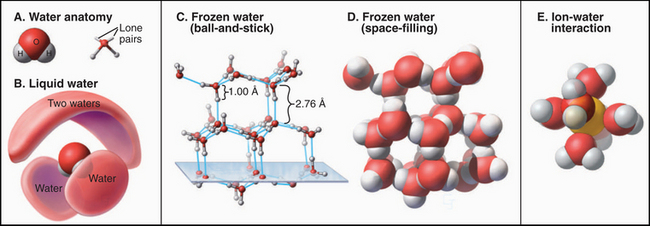
(D–E, From www.nyu.edu/pages/mathmol/library/water, Project MathMol Scientific Visualization Lab, New York University. See “ice.pdb” and “waterbox.pdb.”)
Neither theoretical calculations nor physical observations of liquid water have revealed a consistent picture of its organization. When ice melts, the volume decreases by only about 10%, so liquid water has considerable empty space too. The heat required to melt ice is a small fraction (15%) of the heat required to convert ice to a gas, in which all the hydrogen bonds are lost. Because the heat of melting reflects the number of bonds broken, liquid water must retain most of the hydrogen bonds that stabilize ice. These hydrogen bonds create a continuous, three-dimensional network of water molecules connected at their tetrahedral vertices, allowing water to remain a liquid at a higher temperature than is the case for a similar molecule, ammonia. On the other hand, because liquid water does not have a well-defined, long-range structure, it must be very heterogeneous and dynamic, with rapidly fluctuating regions of local order and disorder. This incomplete picture of water structure limits our ability to understand macromolecular interactions in an aqueous environment.
The properties of water have profound effects on all other molecules in the cell. For example, ions organize shells of water around themselves that compete effectively with other ions with which they might interact electrostatically (Fig. 3-1E). This shell of water travels with the ions, governing the size of pores that they can penetrate. Similarly, hydrogen bonding with water strongly competes with the hydrogen bonding that occurs between solutes, including macromolecules. By contrast, water does not interact as favorably with nonpolar molecules as it does with itself, so the solubility of nonpolar molecules in water is low, and they tend to aggregate to reduce their surface area in contact with water. Such nonpolar interactions are energeti-cally favorable because they reduce unfavorable interactions of nonpolar groups with water and increase favorable interactions of water molecules with each other. This is called the hydrophobic effect (see Fig. 4-5). These interactions of water dominate the behavior of solute molecules in an aqueous environment, where they influence the assembly of proteins, lipids, and nucleic acids into the structures that they assume in the cell. On the other hand, strategically placed water molecules can bridge two macromolecules in functional assemblies.
Proteins
Proteins consist of one or more linear polymers called polypeptides, which consist of various combinations of 20 different amino acids (Figs. 3-2 and 3-3) linked together by peptide bonds (Fig. 3-4). When linked in polypeptides, amino acids are referred to as residues. The sequence of amino acids in each type of polypeptide is unique. It is specified by the gene encoding the protein and is read out precisely during protein synthesis (see Fig. 18-8). The polypeptides of proteins with more than one chain are usually synthesized separately. However, in some cases, a single chain is divided into pieces by cleavage after synthesis.
Polypeptides range widely in length. Small peptide hormones, such as oxytocin, consist of as few as nine residues, while the giant structural protein titin (see Fig. 39-7) has more than 25,000 residues. Most cellular proteins fall in the range of 100 to 1000 residues. Without stabilization by disulfide bonds or bound metal ions, about 40 residues are required for a polypeptide to adopt a stable three-dimensional structure in water.
The sequence of amino acids in a polypeptide can be determined chemically by removing one amino acid at a time from the amino terminus and identifying the product. This procedure, called Edman degradation, can be repeated about 50 times before declining yields limit progress. Longer polypeptides can be divided into fragments of fewer than 50 amino acids by chemical or enzymatic cleavage, after which they are purified and sequenced separately. Even easier, one can sequence the gene or a complementary DNA (cDNA) copy of the messenger RNA for the protein (Fig. 3-16) and use the genetic code to infer the amino acid sequence. This approach misses posttranslational modifications (Fig. 3-3). Analysis of protein fragments by mass spectrometry can be used to sequence even tiny quantities of proteins.
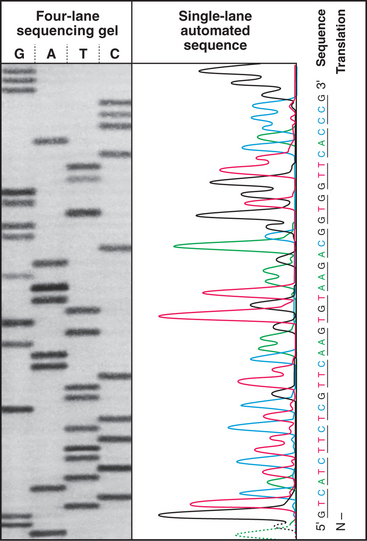
Figure 3-16 The sequence of a purified fragment of DNA is rapidly determined by in vitro synthesis (see Fig. 42-1) using the four deoxynucleoside triphosphates plus a small fraction of one dideoxynucleoside triphosphate. The random incorporation of the dideoxy residue terminates a few of the growing DNA molecules every time that base appears in the sequence. The reaction is run separately with each dideoxynucleotide, and fragments are separated according to size by gel electrophoresis (see Fig. 6-5), with the shortest fragments at the bottom. A radioactive label makes the fragments visible when exposed to an X-ray film. The sequence is read from the bottom as indicated. An automated method uses four different fluorescent dideoxynucleotides to mark the end of the fragments and electronic detectors to read the sequence.
(Based on original data from W-L. Lee, Salk Institute for Biological Studies, San Diego, California.)
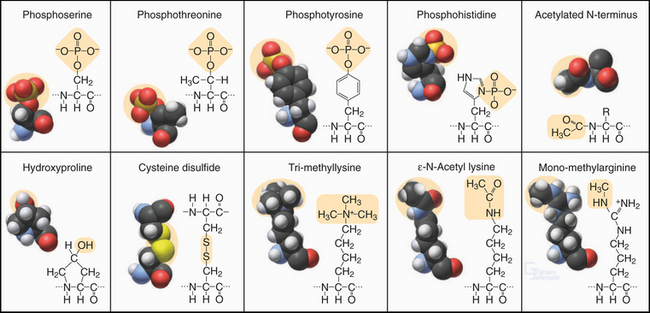
Figure 3-3 modified amino acids. Protein kinases add a phosphate group to serine, threonine, tyrosine, histidine, and aspartic acid (not shown). Other enzymes add one or more methyl groups to lysine, arginine, or histidine (not shown); a hydroxyl group to proline; or an acetate to the N-terminus of many proteins. The reducing environment of the cytoplasm minimizes the formation of disulfide bonds, but under oxidizing conditions within the membrane compartments of the secretory pathway (see Chapter 21), intramolecular or intermolecular disulfide (S—S) bonds form between adjacent cysteine residues.
Properties of Amino Acids
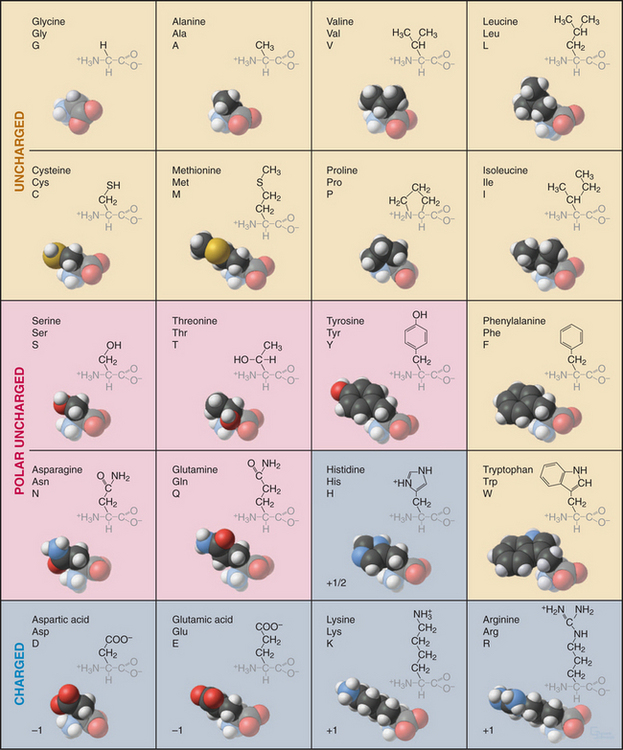
Every student of cell biology should know the chemical structures of the amino acids used in proteins (Fig. 3-2). Without these structures in mind, reading the literature and this book is like spelling without knowledge of the alphabet. In addition to their full names, amino acids are frequently designated by three-letter or single-letter abbreviations.
All but one of the 20 amino acids commonly used in proteins consist of an amino group, bonded to the α-carbon, bonded to a carboxyl group. Proline is a variation on this theme with a cyclic side chain bonded back to the nitrogen to form an imino group. Both the amino group (pK > 9) and carboxyl group (pK = ∼4) are partially ionized under physiological conditions. With the exception of glycine, all amino acids have a β-carbon and a proton bonded to the α-carbon. (Glycine has a second proton instead.) This makes the α-carbon an asymmetrical center with two possible configurations. The l-isomers are used almost exclusively in living systems. Compared with natural proteins, proteins constructed artificially from d-amino acids have mirror-image structures and properties.
Enzymes modify many amino acids after their incorporation into polypeptides. These posttranslational modifications have both structural and regulatory functions (Fig. 3-3). These modifications are referred to many times in this book, especially reversible phosphorylation of amino acid side chains, the most common regulatory reaction in biochemistry (see Fig. 25-1). Methylated and acetylated lysines are important for chromatin regulation in the nucleus (see Fig. 13-3). Whole proteins such as ubiquitin or SUMO can be attached through isopeptide bonds to lysine e-amino groups to act as signals for degradation (see Fig. 23-8) or endocytosis (see Fig. 22-16).
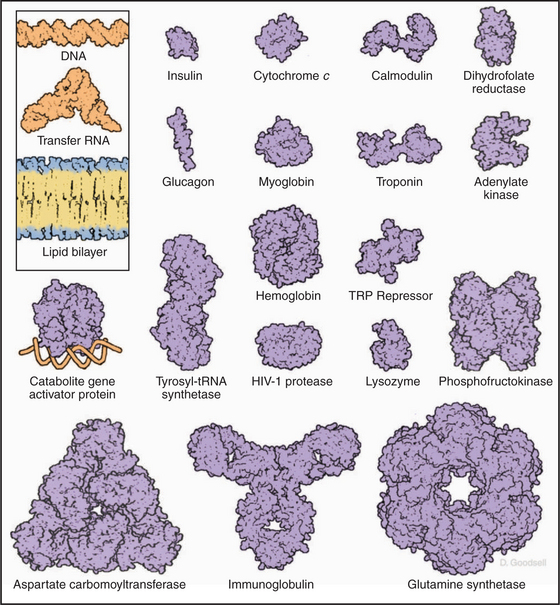
This repertoire of amino acids is sufficient to construct millions of different proteins, each with different capacities for interacting with other cellular constituents. This is possible because each protein has a unique three-dimensional structure (Fig. 3-5), each displaying the relatively modest variety of functional groups in a different way on its surface.
Architecture of Proteins
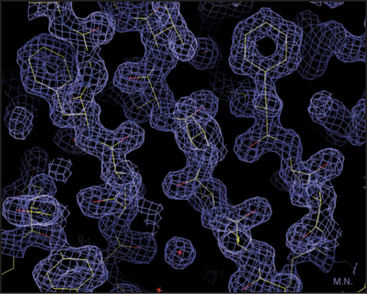
Our knowledge of protein structure is based largely on X-ray diffraction studies of protein crystals or nuclear magnetic resonance (NMR) spectroscopy studies of small proteins in solution. These methods provide pictures showing the arrangement of the atoms in space. X-ray diffraction requires three-dimensional crystals of the protein and yields a three-dimensional contour map showing the density of electrons in the molecule (Fig. 3-6). In favorable cases, all the atoms except hydrogens are clearly resolved, along with water molecules occupying fixed positions in and around the protein. NMR requires concentrated solutions of protein and reveals distances between particular protons. Given enough distance constraints, it is possible to calculate the unique protein fold that is consistent with these spacings. In a few cases, electron microscopy of two-dimensional crystals has revealed atomic structures (see Figs. 7-8B and 34-5).
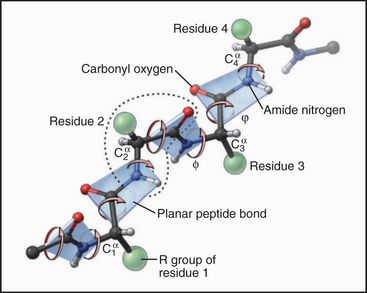
Each amino acid residue contributes three atoms to the polypeptide backbone: the nitrogen from the amino group, the α-carbon, and the carbonyl carbon from the carboxyl group. The peptide bond linking the amino acids together is formed by dehydration synthesis (see Fig. 17-10), a common chemical reaction in biological systems. Water is removed in the form of a hydroxyl from the carboxyl group of one amino acid and a proton from the amino group of the next amino acid in the polymer. Ribosomes catalyze this reaction in cells. Chemical synthesis can achieve the same result in the laboratory. The peptide bond nitrogen has an (amide) proton, and the carbon has a double-bonded (carbonyl) oxygen. The amide proton is an excellent hydrogen bond donor, whereas the carbonyl oxygen is an excellent hydrogen bond acceptor.
The peptide bond has some characteristics of a double bond, owing to resonance of the electrons, and is relatively rigid and planar. The bonds on either side of the α-carbon can rotate through 360 degrees, although a relatively narrow range of bond angles is highly favored. Steric hindrance between the β-carbon (on all the amino acids but glycine) and the α-carbon of the adjacent residue favors a trans configuration in which the side chains alternate from one side of the polymer to the other (Fig. 3-4). Folded proteins generally use a limited range of rotational angles to avoid steric collisions of atoms along the backbone. Glycine without a β-carbon is free to assume a wider range of configurations and is useful for making tight turns in folded proteins.
Folding of Polypeptides
The three-dimensional structure of a protein is determined solely by the sequence of amino acids in the polypeptide chain. This was established by reversibly unfolding and refolding proteins in a test tube. Many, but not all, proteins that are unfolded by harsh treatments (high concentrations of urea or extremes of pH) will refold to regain full activity when returned to physiological conditions. Although many proteins are flexible enough to undergo conformational changes (see later discussion), polypeptides rarely fold into more than one final stable structure. Exceptions with medical importance are prions and amyloid (Box 3-1).
BOX 3-1 Protein Misfolding in Amyloid Diseases
Given that amyloid fibrils form spontaneously and are exceptionally stable, it is not surprising that functional amyloids exist in organisms ranging from bacteria to humans. For example, formation of the pigment granules responsible for skin color depends on a proteolytic fragment of a lysosomal membrane protein that forms amyloid fibrils as a scaffold from melanin pigments. Budding yeast has a number of proteins that can either assume their “native” fold or assemble into amyloid fibrils. The native fold of the protein Sup35p serves as a translation termination factor that stops protein synthesis at the stop codon (see Fig. 17-8). Rarely, Sup35p misfolds and assembles into an amyloid fibril. These fibrils sequester all the Sup35p in fibrils, where it is inactive. The faulty translation termination that occurs in its absence has diverse consequences that are inherited like prions from one generation of yeast to the next.
The following factors influence protein folding:
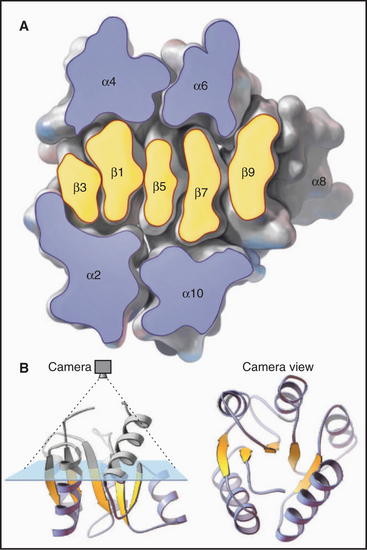
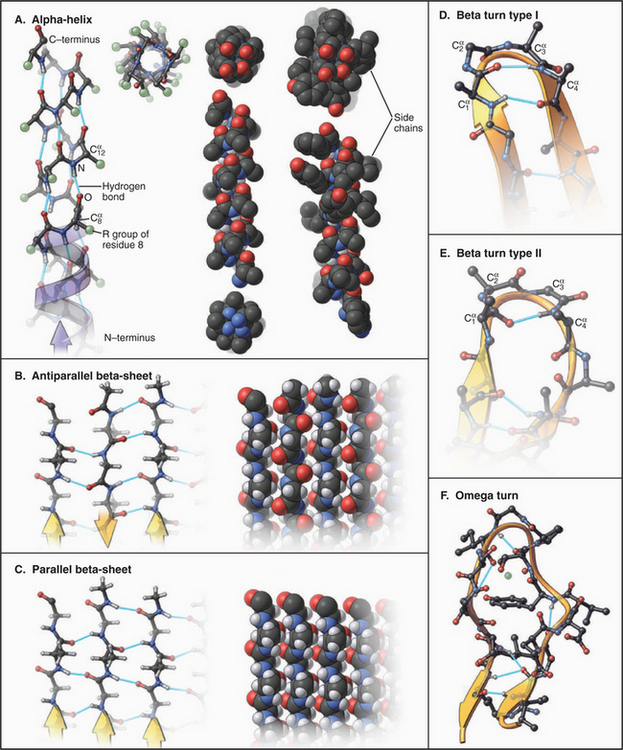
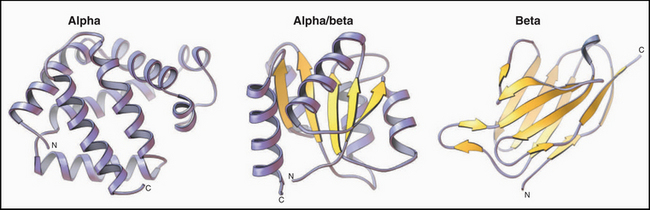
Figure 3-9 ribbon diagrams of protein backbones showing β-strands as flattened arrows, α-helices as coils, and other parts of the polypeptide chains as ropes. Left, The β-subunit of hemoglobin consists entirely of tightly packed α-helices. (PDB file: 1 MBA.) Middle, CheY is a mixed a/b structure, with a central parallel β-sheet flanked by α-helices. Note the right-handed twist of the sheet (defined by the sheet turning away from the viewer at the upper right) and right-handed pattern of helices (defined by the helices angled toward the upper right corner of the sheet) looping across the β-strands. (Compare the cross section in Figure 3-7). (PDB file: 2CHF.) Right, The immunoglobulin VL domain consists of a sandwich of two antiparallel β-sheets. (PDB file: 2IMM.)
These factors tend to maximize the stability of folded proteins in one particular “native” conformation, but the native state of folded proteins is relatively unstable. The standard free energy difference (see Chapter 4) between a folded and globally unfolded protein is only about 40 kJ mol−1, much less than that of a single covalent bond! Even the substitution of a single crucial amino acid can destabilize certain proteins, causing a loss of function. In other cases, misfolding results in noncovalent polymerization of a protein into amyloid fibrils associated with serious diseases (Box 3-1).
The amino acid sequence of each polypeptide contains all the information required to specify folding into the native protein structure, just one of a near infinity of possible conformations. Chapter 17 explains how many conformations of the unfolded polypeptide are rapidly sampled through trial and error to select stable intermediates leading to the native structure. Cells use molecular chaperones to guide and control the quality of folding.
Secondary Structure
Much of the polypeptide backbone of proteins folds into stereotyped elements of secondary structure, especially α-helices and β-sheets (Fig. 3-8). They are shown as spirals and polarized ribbons in “ribbon diagrams” of protein organization used throughout this book. Both α-helices and β-strands are linear, so globular proteins can be thought of as compact bundles of straight or gently curving rods, laced together by surface turns.
a-Helices allow polypeptides to maximize hydrogen bonding of backbone polar groups while using highly favored rotational angles around the α-carbons and tight packing of atoms in the core of the helix (Fig. 3-8). All of these features stabilize the α-helix. Viewed with the amino terminus at the bottom, the amide protons all point downward and the carbonyl oxygens all point upward. The side chains project radially around the helix, tilted toward its N-terminus. Given 3.6 residues in each turn of the right-handed helix, the carbonyl oxygen of residue 1 is positioned perfectly to form a linear hydrogen bond with the amide proton of residue 5. This n to n + 4 pattern of hydrogen bonds repeats along the whole α-helix.
A second strategy used to stabilize the backbone structure of polypeptides is hydrogen bonding of β-strands laterally to form β-sheets (Figs. 3-8 and 3-9). In individual β-strands, the peptide chain is extended in a configuration close to all-trans with side chains alternating top and bottom and amide protons and carbonyl oxygens alternating right and left. β-Strands can form a complete set of hydrogen bonds, with neighboring strands running in the same or opposite directions in any combination. However, the orientation of hydrogen bond donors and acceptors is more favorable in a β-sheet with antiparallel strands than in sheets with parallel strands. Largely parallel β-sheets are usually extensive and completely buried in proteins. β-Sheets have a natural right-handed twist in the direction along the strands. Antiparallel β-sheets are stable even if the strands are short and extensively distorted by twisting. Antiparallel sheets can wrap around completely to form a β-barrel with as few as five strands, but the natural twist of the strands and the need to fill the core of the barrel with hydrophobic residues favors barrels with eight strands.
Up to 25% of the residues in globular proteins are present in bends at the surface (Fig. 3-8D-F). Residues constituting bends are generally hydrophilic. The presence of glycine or proline in a turn allows the backbone to deviate from the usual geometry in tight turns, but the composition of bends is highly variable and not a strong determinant of folding or stability. Turns between linear elements of secondary structure are called reverse turns, as they reverse the direction of the polypeptide. Those between β-strands have a few characteristic conformations and are called β-bends.
Many parts of polypeptide chains in proteins do not have a regular structure. At one extreme, small segments of polypeptide, frequently at the N- or C-terminus, are truly disordered in the sense that they are mobile. Many other irregular segments of polypeptide are tightly packed into the protein structure. Omega loops are compact structures consisting of 6 to 16 residues, generally on the protein surface, that connect adjacent elements of secondary structure (Fig. 3-8F). They lack regular structure but typically have the side chains packed in the middle of the loop. Some are mobile, but many are rigid. Omega loops form the antigen-binding sites of antibodies. In other proteins, they bind metal ions or participate in the active sites of enzymes.
Packing of Secondary Structure in Proteins
Elements of secondary structure can pack together in almost any way (Fig. 3-9), but a few themes are favored enough to be found in many proteins. For example, two β-sheets tend to pack face to face at an angle of about 40 degrees with nonpolar residues packed tightly, knobs into holes, in between. α-Helices tend to pack at an angle of about 30 degrees across β-sheets, always in a right-handed arrangement. Adjacent α-helices tend to pack together at an angle of either +20 degrees or −50 degrees, owing to packing of side chains from one helix into grooves between side chains on the other helix.
Coiled-coils are a common example of regular superstructure (Fig. 3-10). Two α-helices pair to form a fibrous structure that is widely used to create stable polypeptide dimers in transcription factors (see Fig. 15-18) and structural proteins (see Fig. 39-4). Typically, two identical α-helices wrap around each other in register in a left-handed super helix that is stabilized by hydrophobic interactions of leucines and valines at the interface of the two helices. Intermolecular ionic bonds between the side chains of the two polypeptides also stabilize coiled-coils. Given 3.6 residues per turn, the sequence of a coiled-coil has hydrophobic residues regularly spaced at positions 1 and 4 of a “heptad repeat.” This pattern allows one to predict the tendency of a polypeptide to form coiled-coils from its amino acid sequence.
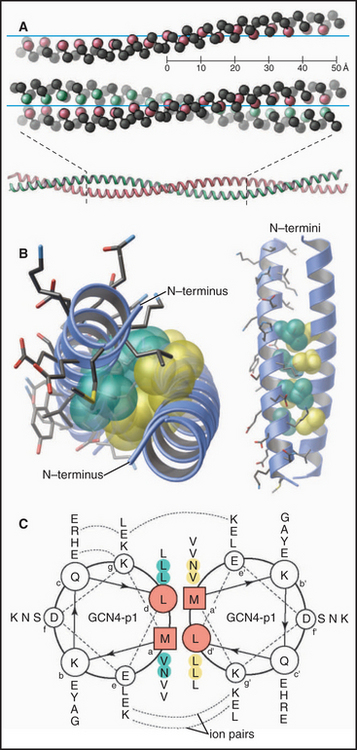
Figure 3-10 coiled-coils. A, Comparison of a single α-helix, represented by spheres centered on the α-carbons, and a two-stranded, left-handed coiled-coil. Two identical α-helices make continuous contact along their lengths by the interaction of the first and fourth residue in every two turns (seven residues) of the helix. (PDB file: 2TMA.) B, Atomic structure of the GCN4 coiled-coil, viewed end-on. The coiled-coil holds together two identical peptides of this transcription factor dimer (see Fig. 15-17 for information on its function). Hydrophobic side chains fit together like knobs into holes along the interface between the two helices. (PDB file: GCN4.) C, Helical wheel representation of the GCN4 coiled-coil. Following the arrows around the backbone of the polypeptides, one can read the sequences from the single-letter code, starting with the boxed residues and proceeding to the most distal residue. Note that hydrophobic residues in the first (a) and fourth (d) positions of each two turns of the helices make hydrophobic contacts that hold the two chains together. Electrostatic interactions (dashed lines) between side chains at positions e and g stabilize the interaction. Other coiled-coils consist of two different polypeptides (see Fig. 15-18), and some are antiparallel (see Fig. 13-19).
(C, Redrawn from O’Shea E, Klemm JD, Kim PS, Alber T: X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled-coil. Science 254:539–544, 1991.)
b-Sheets can also form extended structures. One called a β-helix consists of a continuous polypeptide strand folded into a series of short β-sheets that form a three-sided helix. Fig. 24-4 shows end-on and side views of two β-helices of a growth factor receptor.
Interaction of Proteins with Solvent
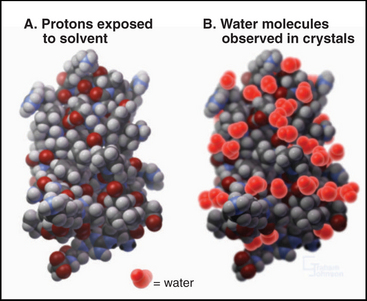
The surface of proteins is almost entirely covered with protons (Fig. 3-11). Some protons are potential hydrogen bond donors, but many are inert, being bonded to backbone or side chain aliphatic carbons. Although most of the charged side chains are exposed on the surface, so are many nonpolar side chains. Many water molecules are ordered on the surface of proteins by virtue of hydrogen bonds to polar groups. These water molecules appear in electron density maps of crystalline proteins but exchange rapidly, on a picosecond (10−12 second) time scale. Waters that are in contact with nonpolar atoms maximize hydrogen bonding with each other, forming a dynamic layer of water with reduced translational diffusion compared with bulk water. This lowers the entropy of the water by increasing its order and provides a thermodynamic impetus to protein folding pathways that minimize the number of hydrophobic atoms displayed on the surface (see Fig. 4-5).
Protein Dynamics
In addition to relatively small, local variations in structure, many proteins undergo large conformational changes (Fig. 3-12). These changes in structure often reflect a change of activity or physical properties. Conformational changes play roles in many biological processes ranging from opening and closing ion channels (see Fig. 10-5) to cell motility (see Fig. 36-5). Many conformational changes have been observed indirectly by spectroscopy or hydrodynamic methods or directly by crystallography or NMR. For example, when glucose binds the enzyme hexokinase, the two halves of the protein clamp around this substrate by rotating 12 degrees about a hinge consisting of two polypeptides. Guanosine triphosphate (GTP) binding to elongation factor EF-Tu causes a domain to rotate 90 degrees about two glycine residues (see Fig. 25-7)! Similarly, phosphorylation of glycogen phosphorylase causes a local rearrangement of the N-terminus that transmits a structural change over a distance of more than 2nm to the active site (see Fig. 27-3). The Ca2+ binding regulatory protein calmodulin undergoes a dramatic conformational change (Fig. 3-12) when wrapping tightly around a helical peptide of a target protein (also see Chapter 26).
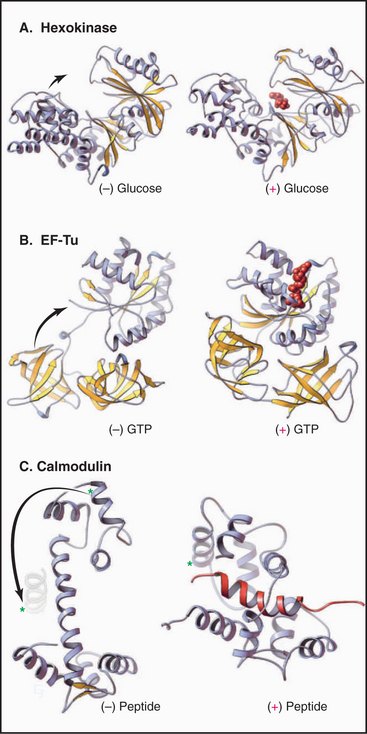
Figure 3-12 conformational changes of proteins. A, The glycolytic enzyme hexokinase. The two domains of the protein hinge together to surround the substrate, glucose. (PDB files: 2YHX and 1HKG.) B, EF-Tu, a cofactor in protein synthesis (see Fig. 17-10), folds more compactly when it binds guanosine triphosphate. (PDB files: 1EFU and 1EFT.) C, Calmodulin (see Chapter 26) binds Ca2+ and wraps itself around an α-helix (red) in target proteins. Note the large change in position of the helix marked with an asterisk. (PDB files: CLN and 2BBM.)
Modular Domains in Proteins
Most polypeptides consist of linear arrays of multiple independently folded, globular regions, or domains, connected in a modular fashion (Fig. 3-13). Most domains consist of 40 to 100 residues, but kinase domains and motor domains (see Figs. 36-3 and 36-9) are much larger. Each of more than 1000 recognized families of domains is thought to have evolved from a different common ancestor. In this sense, the members of a family are said to be homologous. Through the processes of gene duplication, transposition, and divergent evolution, the most widely used domains (e.g., the immunoglobulin domain) have become incorporated into hundreds of different proteins, where they serve unique functions. Homologous domains in different proteins have similar folds but may differ significantly in amino acid sequences. Nevertheless, most related domains can be recognized from characteristic patterns of amino acids along their sequences. For example, cysteine residues of immunoglobulin G (Ig) domains are spaced in a pattern required to make intramolecular disulfide bonds (Fig. 3-3).
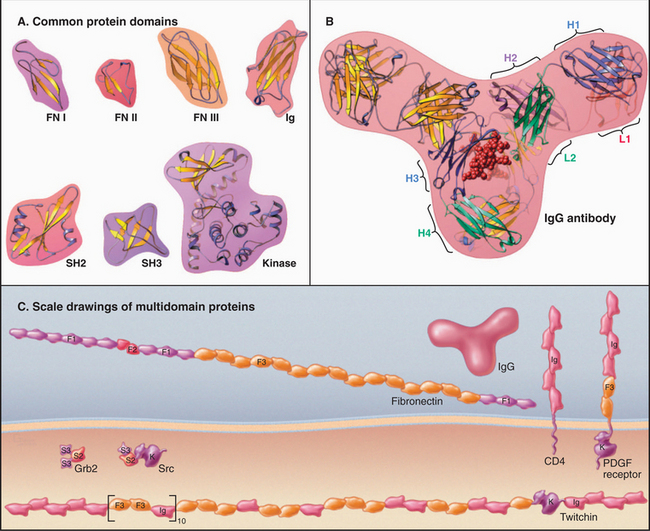
Figure 3-13 modular proteins constructed from evolutionarily homologous, independently folded domains. A, Examples of protein domains used in many proteins: fibronectin 1 (FN I), fibronectin 2 (FN II), fibronectin 3 (FN III), immunoglobulin (Ig), Src homology 2 (SH2), Src homology 3 (SH3), kinase. (PDB files: FN7, 1PDC, 1FNA, 1IG2, 1HCS, 1PRM, and 1CTP.) B, Immunoglobulin G (IgG), a protein composed of 12Ig domains on four polypeptide chains. Two identical heavy chains (H) consist of four Ig domains, and two identical light chains (L) consist of two Ig domains. The sequences of these six Ig domains differ, but all of the domains are folded similarly. The two antigen-binding sites are located at the ends of the two arms of the Y-shaped molecule composed of highly variable loops contributed by domains H1 and L1. (PDB file: 1IG2.) C, Examples of proteins constructed from the domains shown in A: fibronectin (see Fig. 29-15), CD4 (see Figs. 27-8 and 28-9), PDGF-receptor (see Fig. 24-4), Grb2 (see Fig. 27-6), Src (see Fig. 25-3 and Box 27-1), and twitchin (see Chapter 39). Each of the 31 FN3 domains in twitchin has a different sequence. F1 is FI, F2 is FII, and F3 is FIII.
Nucleic Acids
Nucleic acids, polymers of a few simple building blocks called nucleotides, store and transfer all genetic information. This is not the limit of their functions. RNA enzymes, ribozymes, catalyze some biochemical reactions. Other RNAs are receptors (riboswitches) or contribute to the structures and enzyme activities of major cellular components, such as ribosomes (see Fig. 17-7) and spliceosomes (see Fig. 16-5). In addition, nucleotides themselves transfer chemical energy between cellular systems and information in signal transduction pathways. Later chapters elaborate on each of these topics.
Building Blocks of Nucleic Acids
Nucleotides consist of three parts: (1) a base built of one or two cyclic rings of carbon and a few nitrogen atoms, (2) a five-carbon sugar, and (3) one or more phosphate groups (Fig. 3-14). DNA uses four main bases: the purines adenine (A) and guanosine (G) and the pyrimidines cytosine (C) and thymine (T). In RNA, uracil (U) is found in place of thymine. Some RNA bases are chemically modified after synthesis of the polymer. The sugar of RNA is ribose, which has the aldehyde oxygen of carbon 4 cyclized to carbon 1. The DNA sugar is deoxyribose, which is similar to ribose but lacks the hydroxyl on carbon 2. In both RNA and DNA, carbon 1 of the sugar is conjugated with nitrogen 1 of a pyrimidine base or with nitrogen 9 of a purine base. The hydroxyl of sugar carbon 5 can be esterified to a chain of one or more phosphates, forming nucleotides such as adenosine monophosphate (AMP), adenosine diphosphate (ADP), and ATP.
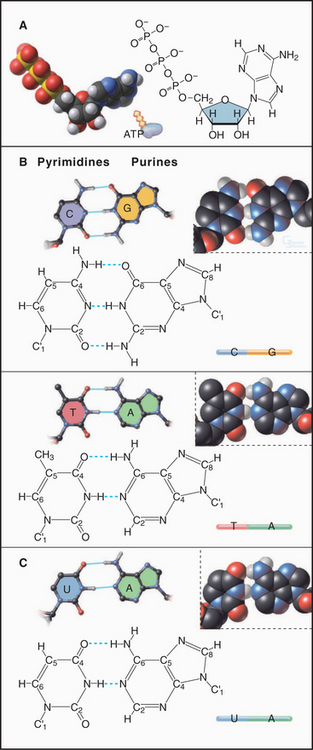
Covalent Structure of Nucleic Acids
DNA and RNA are polymers of nucleotides joined by phosphodiester bonds (Fig. 3-15). The backbone links a chain of five atoms (two oxygens and three carbons) from one phosphorous to the next—a total of six backbone atoms per nucleotide. Unlike the backbone of proteins, in which the planar peptide bond greatly limits rotation, all six bonds along a polynucleotide backbone have some freedom to rotate, even that in the sugar ring. This feature gives nucleic acids much greater conformational flexibility than polypeptides, which have only two variable torsional angles per residue. The backbone phosphate group has a single negative charge at neutral pH. The N—C bond linking the base to the sugar is also free to rotate on a picosecond time scale, but rotation away from the backbone is strongly favored. The bases have a strong tendency to stack upon each other, owing to favorable van der Waals interactions (see Chapter 4) between these planar rings.
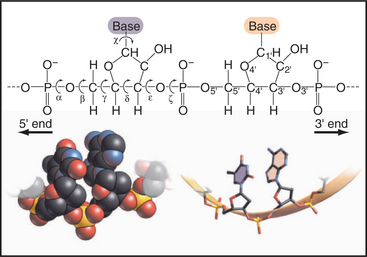
Figure 3-15 rotational freedom of the backbone of a polynucleotide, rna in this case. The stick figure of two residues shows that all six of the backbone bonds are rotatable, even the C4′—C′ bond that is constrained by the ribose ring. This gives polynucleotides more conformational freedom than polypeptides. Note the phosphodiester bonds between the residues and the definition of the 3′ and 5′ ends. Space-filling and stick figures at the bottom show a uridine (U) and adenine (A) from part of Figure 3-17.
(Redrawn from Jaeger JA, SantaLucia J, Tinoco I: Determination of RNA structure and thermodynamics. Annu Rev Biochem 62:255–287, 1993.)
Each type of nucleic acid has a unique sequence of nucleotides. Simple laboratory procedures employing the enzymatic synthesis of DNA allow the sequence to be determined rapidly (Fig. 3-16). All DNA and RNA molecules are synthesized biologically in the same direction (see Figs. 15-11 and 42-1) by adding a nucleoside triphosphate to the 3′ sugar hydroxyl of the growing strand. Cleavage of the two terminal phosphates from the new subunit provides energy for extension of the polymer in the 5′ to 3′ direction. Newly synthesized DNA and RNA molecules have a phosphate at the 5′ end and a 3′ hydroxyl at the other end. In certain types of RNA (e.g., messenger RNA [mRNA]), the 5′ nucleotide is subsequently modified by the addition of a specialized cap structure (see Figs. 16-2 and 17-2).
Secondary Structure of DNA
A few viruses have chromosomes consisting of single-stranded DNA molecules, but most DNA molecules are paired with a complementary strand to form a right-handed double helix, as originally proposed by Watson and Crick (Fig. 3-17). Key features of the double helix are two strands running in opposite directions with the sugar-phosphate backbone on the outside and pairs of bases hydrogen-bonded to each other on the inside (Fig. 3-14). Pairs of bases are stacked 0.34nm apart, nearly perpendicular to the long axis of the polymer. This regular structure is referred to as B-form DNA, but real DNA is not completely regular. On average, in solution, β-form DNA has 10.5 base pairs per turn and a diameter of 1.9nm. Hydrogen bonds between adenine and thymine and between guanine and cytosine span nearly the same distance between the backbones, so the helix has a regular structure that, to a first approximation, is independent of the sequence of bases. One exception is a run of As that tends to bend adjoining parts of the helix. Because the bonds between the bases and the sugars are asymmetrical, the DNA helix is asymmetrical: The major groove on one side of the helix is broader than the other, minor groove. Most cellular DNA is approximately in the β-form conformation, but proteins that regulate gene expression can distort the DNA significantly (see Fig. 15-7).
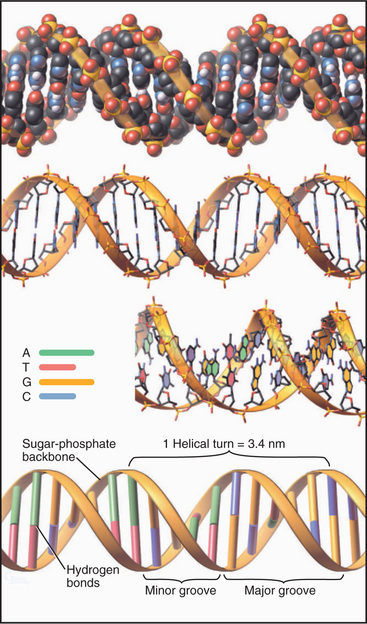
Figure 3-17 models of β-form dna. The molecule consists of two complementary antiparallel strands arranged in a right-handed double helix with the backbone (Fig. 3-15) on the outside and stacked pairs of hydrogen-bonded bases (see Fig. 3-14) on the inside. Top, Space-filling model. Middle, Stick figures, with the lower figure rotated slightly to reveal the faces of the bases. Bottom, Ribbon representation.
(Idealized 24–base pair model built by Robert Tan, University of Alabama, Birmingham.)
DNA molecules are either linear or circular. Human chromosomes are single linear DNA molecules (see Fig. 12-1). Many, but not all, viral and bacterial chromosomes are circular. Eukaryotic mitochondria and chloroplasts also have circular DNA molecules.
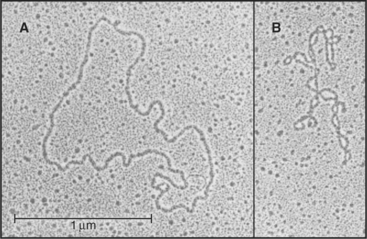
When circular DNAs or linear DNAs with both ends anchored (as in chromosomes; see Chapter 13) are twisted about their long axis, the strain is relieved by the development of long-range bends and twists called supercoils or superhelices (Fig. 3-18). Supercoiling can be either positive or negative depending on whether the DNA helix is wound more tightly or somewhat unwound. Supercoiling is biologically important, as it can influence the expression of genes. Under some circumstances, supercoiling favors unwinding of the double helix. This can promote access of proteins involved in the regulation of transcription from DNA (see Chapter 15).
Secondary and Tertiary Structure of RNAs
RNAs range in size from micro-RNAs of 20 nucleotides (see Fig. 16-12) to messenger RNAs with more than 80,000 nucleotides. Because each nucleotide has about three times the mass of an amino acid, RNAs with a modest number of nucleotides are bigger than most proteins (see Fig. 1-4). The 16S RNA of the small ribosomal subunit of bacteria consists of 1542 nucleotides with a mass of about 460 kD, much larger than any of the 21 proteins with which it interacts (see Fig. 17-7).
Except for the RNA genomes of a few viruses, RNAs generally do not have a complementary strand to pair with each base. Instead they form specific structures by optimizing intramolecular base pairing (Figs. 3-19 and 3-20). Comparison of homologous RNA sequences provides much of what is known about this intramolecular base pairing. The approach is to identify pairs of nucleotides that vary together across the phylogenetic tree. For example, if an A and a U at discontinuous positions in one RNA are changed together to C and a G in homologous RNAs, it is inferred that they are hydrogen-bonded together. This covariant method works remarkably well, because hundreds to thousands of homologous sequences for the major classes of RNA are available from comparative genomics. Conclusions about base pairing from covariant analysis have been confirmed by experimental mutagenesis of RNAs and direct structure determination.
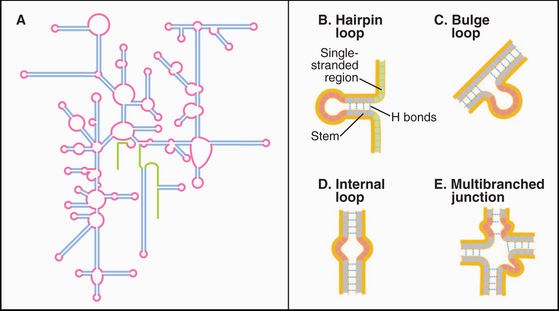
The simplest RNA secondary structure is an antiparallel double helix stabilized by hydrogen bonding of complementary bases (Figs. 3-20 and 3-21). Similarly to DNA, G pairs with C and U pairs with A. Unlike the case in DNA, G also frequently pairs with U in RNA. Helical base pairing occurs between both contiguous and discontiguous sequences. When contiguous sequences form a helix, the strand is often reversed by a tight turn, forming an antiparallel stem-loop structure. These hairpin turns frequently consist of just four bases. A few sequences are highly favored for turns, owing to their compact, stable structures. Bulges due to extra bases or noncomplementary bases frequently interrupt base-paired helices of RNA.
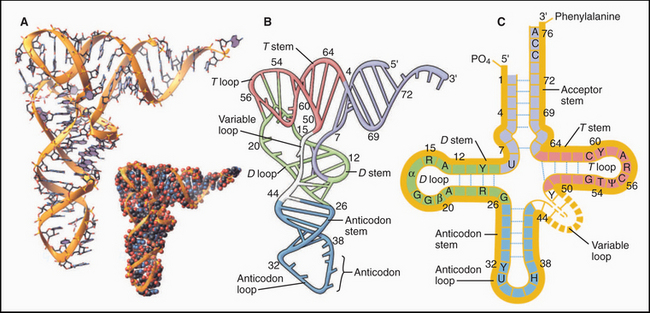
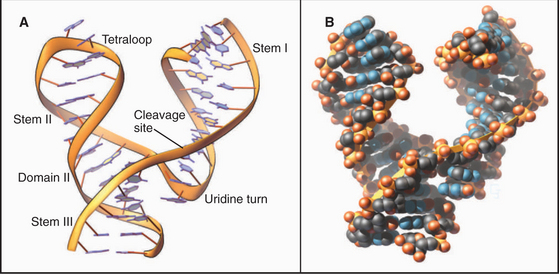
Crystal structures of RNAs such as tRNAs (Fig. 3-20) and a hammerhead ribozyme (Fig. 3-21) established that RNAs have novel, specific, three-dimensional structures. Crystal structures of ribosomes (see Fig. 17-7) showed that larger RNAs fold into specific structures using similar principles. Crystallization of RNAs is challenging, and NMR provides much less information on RNA than on proteins of the same size, so much is yet to be learned about RNA structures.
As in proteins, many residues in RNAs are in conventional secondary structures, especially stems consisting of base-paired double helices; however, RNA backbones make sharp turns that allow unconventional hydrogen bonds between bases, ribose hydroxyls, and backbone phosphates. Generally, the phosphodiester backbone is on the surface with most of the hydrophobic bases stacked internally. Some bases are hydrogen-bonded together in triplets (Fig. 3-22) rather than in pairs. Four or five Mg2+ ions stabilize regions of tRNA with high densities of negative charge.
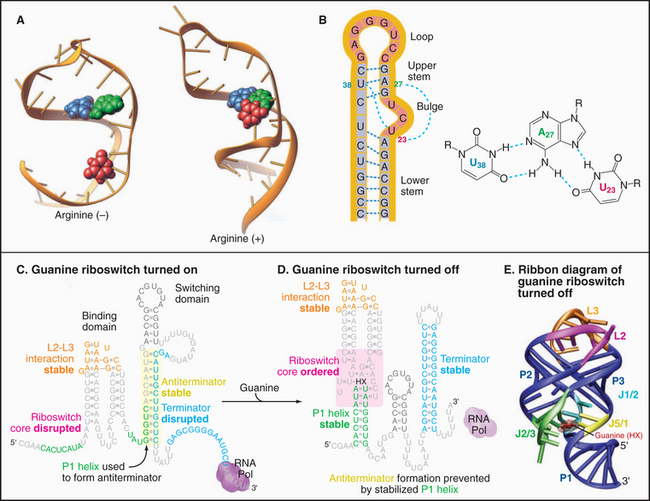
Like proteins, RNAs can change conformation. The TAR RNA is a stem-loop structure with a bulge formed by three unpaired nucleotides (Fig. 3-22). TAR is located at the 5′ end of all RNA transcripts of the human immunodeficiency virus (HIV) that causes AIDS. Bind-ing of a regulatory protein called TAT changes the conformation of TAR and promotes elongation of the RNA. Binding arginine also changes the conformation of TAR.
Like proteins, RNAs can bind ligands. About 2% of the genes in the bacterium Bacillus subtillis are regulated by RNA sequences located in the mRNAs. For example, mRNAs for enzymes used to synthesize purines such as guanine have a guanine-sensitive riboswitch that controls translation (Fig. 3-22C-D). At low guanine levels, the conformation allows transcription. High concentrations of guanine bind the RNA, causing a massive reorganization that blocks transcription. This negative feedback loop optimizes the cellular concentration of guanine.
Carbohydrates
Carbohydrates serve four main functions:
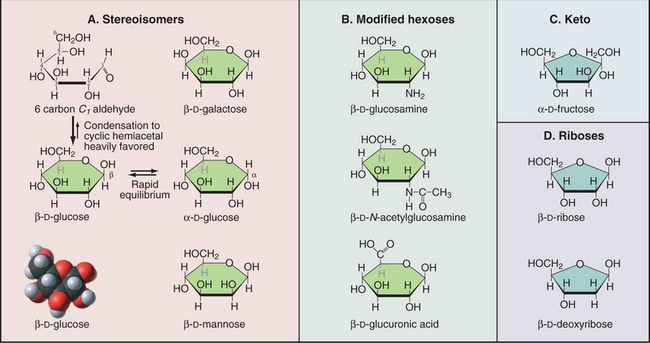
A modest number of simple sugars (Fig. 3-23) form the vast array of different complex carbohydrates found in nature. These sugars consist of three to seven carbons with one aldehyde or ketone group and multiple hydroxyl groups. In water, the common five-carbon (pentose) and six-carbon (hexose) sugars cyclize by reaction of the aldehyde or ketone group with one of the hydroxyl carbons. This forms a compact structure that is used in all the glycoconjugates considered in this book. Given several asymmetrical carbons in each sugar, a great many stereochemical isomers exist. For example, the hydroxyl on carbon 1 can either be above (b-isomer) or below (a-isomer) the plane of the ring. Proteins (enzymes, lectins, and receptors) that interact with sugars distinguish these stereoisomers.
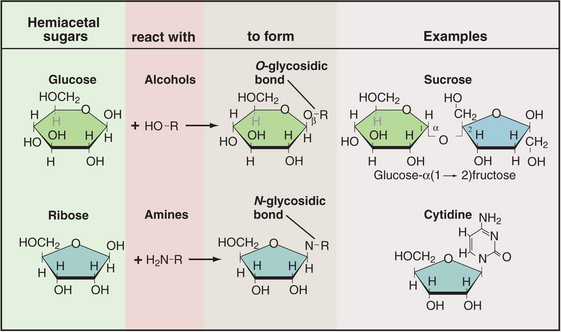
Sugars are coupled to other molecules by highly specific enzymes, using a modest repertoire of intermolecular bonds (Fig. 3-24). The common O-glycosidic (carbon-oxygen-carbon) bond is formed by removal of water from two hydroxyls—the hydroxyl of the carbon bonded to the ring oxygen of a sugar and a hydroxyl oxygen of another sugar or the amino acids serine and threonine. A similar reaction couples a sugar to an amine, as in the bond between a sugar and a nucleoside base. Sugar phosphates with one or more phosphates esterified to a sugar hydroxyl are components of nucleotides as well as of many intermediates in metabolic pathways.
Glycoconjugates—polymers of one or more types of sugar molecules—are present in massive amounts in nature and are used as both energy stores and structural components (Fig. 3-25). Cellulose (unbranched β-1,4 polyglucose), which forms the cell walls of plants, and chitin (unbranched β-1,4 poly N-acetylglucosamine), which forms the exoskeletons of many invertebrates, are the first and second most abundant biological polymers found on the earth. In animals, giant complex carbohydrates are essential components of the extracellular matrix of cartilage and other connective tissues (see Figs. 29-13 and 34-3). Glycogen, a branched α-1,4 polymer of glucose, is the major energy store in animal cells. Starch-polymers of glucose with or without a modest level of branching-performs the same function for plants.
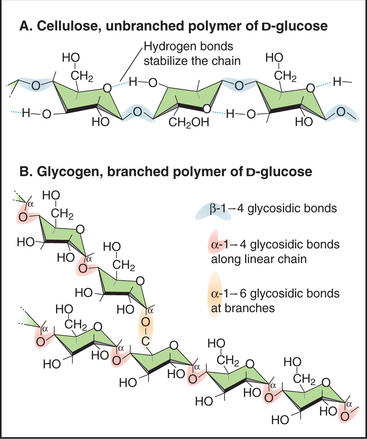
Figure 3-25 examples of simple glycoconjugates. A, Cellulose, an unbranched homopolymer of glucose used to construct plant cell walls. B, Glycogen, a branched homopolymer of glucose used by animal cells to store sugar. Many glycoconjugates consist of several different types of sugar subunits (see Figs. 21-26 and 29-13).
Glycoconjugates differ from proteins and nucleic acids in that they have a broader range of conformations owing to the flexible glycosidic linkages between the sugar subunits. Although sugar polymers may be stabilized by extensive intramolecular hydrogen bonds and some glycosidic linkages are relatively rigid, NMR studies have revealed that many glycosidic bonds rotate freely, allowing the polymer to change its conformation on a submillisecond time scale. This dynamic behavior limits efforts to determine glycoconjugate structures. They are reluctant to crystallize, and the multitude of conformations does not lend itself to NMR analysis. Structural details are best revealed by X-ray crystallography of a glycoconjugate bound to a protein, such as a lectin or a glycosidase (a degradative enzyme).
Sugars are linked to proteins in three different ways (Fig. 3-26) by specific enzymes that recognize unique protein conformations. Glycoprotein side chains vary in size from one sugar to polymers of hundreds of sugars. These sugar side chains can exceed the mass of the protein to which they are attached. Chapters 21 and 29 consider glycoprotein biosynthesis.
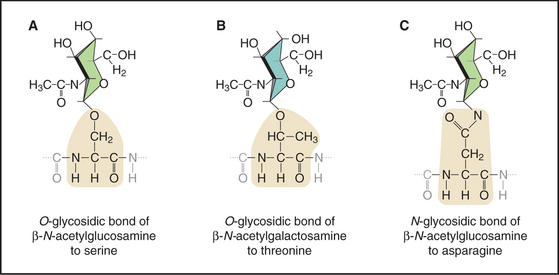
Figure 3-26 three types of glycosidic bonds link glycoconjugates to proteins. A, An O-glycosidic bond links N-acetylglucosamine to serine residues of many intracellular proteins. B, An O-glycosidic bond links N-acetylgalactosamine to serine or threonine residues of core proteins, initiating long glycoconjugate polymers called glycosaminoglycans on extracellular proteoglycans (see Fig. 29-13). C, An N-glycosidic bond links N-acetylglucosamine to asparagine residues of secreted and membrane glycoproteins (see Fig. 21-26). A wide variety of glycoconjugates extend the sugar polymer from the N-acetylglucosamine. These stick figures illustrate the conformations of the sugar rings.
Compared with the nearly invariant sequences of proteins and nucleic acids, glycoconjugates are heterogeneous, because enzymes assemble these sugar polymers without the aid of a genetic template. These glycosyltransferases link high-energy sugar-nucleosides to acceptor sugars. These enzymes are specific for the donor sugar-nucleoside and selective, but not completely specific, for the acceptor sugars. Thus, cells require many different glycosyltransferases to generate the hundreds of types of sugar-sugar bonds found in glycoconjugates. Particular cells consistently produce the same range of specific glycoconjugate structures. This reproducible heterogeneity arises from the repertoire of glycosyltransferases expressed, their localization in specific cellular compartments, and the availability of suitable acceptors. Glycosyltransferases compete with each other for acceptors, yielding a variety of products at many steps in the synthesis of glycoconjugates. For example, the probability of encountering a particular glycosyltransferase depends upon the part of the Golgi apparatus (see Fig. 21-14) in which a particular acceptor finds itself.
The Aqueous Phase of Cytoplasm
The aqueous phase of cells contains a wide variety of solutes, including inorganic ions, building blocks of major organic constituents, intermediates in metabolic pathways, carbohydrate and lipid energy stores, and high concentrations of proteins and RNA. In addition, eukaryotic cells have a dense network of cytoskeletal fibers (Fig. 3-27). Cells control the concentrations of solutes in each cellular compartment, because many (e.g., pH, Na+, K+, Ca2+, and cyclic AMP) have essential regulatory or functional significance in particular compartments.

The high concentration of macromolecules and the network of cytoskeletal polymers make the cytoplasm a very different environment from the dilute salt solutions that are usually employed in biochemical experiments on cellular constituents. The presence of 300 mg/mL of protein and RNA causes the cytoplasm to be crowded. The concentration of bulk water in cytoplasm is less than the 55M in dilute solutions, but the microscopic viscosity of the aqueous phase in live cells is remarkably close to that of pure water. Crowding lowers the diffusion coefficient of the molecules by a factor of about 3, but it also enhances macromolecular associations by raising the chemi-cal potential of the diffusing molecules through an “excluded volume” effect. Macromolecules take up space in the solvent, so the concentration of each molecule is higher in relation to the available solvent. At cellular concentrations of macromolecules, the chemical potential of a molecule (see Chapter 4) may be one or more orders of magnitude higher than its concentration. (The chemical potential, rather than the concentration, determines the rate of reactions.) Therefore, crowding favors protein-protein, protein–nucleic acid, and other macromolecular assembly reactions that depend on the chemical potential of the reactants. Crowding also changes the rates and equilibria of enzymatic reactions, usually increasing the activity as compared with values in dilute solutions.
ACKNOWLEDGMENTS
Thanks go to Tom Steitz and Andrew Miranker for their suggestions on revisions to this chapter.
Brandon C, Tooze J. Introduction to Protein Structure. New York: Garland Publishing, 1999;350.
Bryant RG. The dynamics of water-protein interactions. Annu Rev Biophys Biomol Struct. 1996;25:29-53.
Chothia C, Hubbard T, Brenner S, et al. Protein folds in the all-b and all-a classes. Annu Rev Biophys Biomol Struct. 1997;26:597-627.
Creighton TE. Proteins: Structure and Molecular Principles, 2nd ed., New York: WH Freeman; 1993:507.
Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18-25.
Dobson CM. Protein folding and misfolding. Nature. 2003;426:884-890.
Doherty EA, Doudna JA. Ribozyme structures and mechanisms. Annu Rev Biophys Biomolec Struct. 2001;30:457-475.
Feizi T, Mulloy B. Carbohydrates and glycoconjugates: Glycomics: The new era of carbohydrate biology. Curr Opin Struct Biol. 2003;13:602-604.
Huff ME, Balch WE, Kelly JW. Pathological and functional amyloid formation orchestrated by the secretory pathway. Curr Opin Struct Biol. 2003;13:674-682.
Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355-382.
Kubelka J, James Hofrichter J, Eaton WA. The protein folding “speed limit.”. Curr Opin Struct Biol. 2004;14:76-88.
Kuhlman B, Baker D. Exploring folding free energy landscapes using computational protein design. Curr Opin Struct Biol. 2004;14:89-95.
Lilley DMJ. The origins of RNA catalysis in ribozymes. Trends Biochem Sci. 2003;28:495-501.
Lupas A. Coiled-coils: New structures and new functions. Trends Biochem Sci. 1996;21:375-382.
Murthy VL, Srinivasan R, Draper DE, Rose GD. A complete conformational map for RNA. J Mol Biol. 1999;291:313-327.
Narlikar GJ, Hershlag D. Mechanistic aspects of enzyme catalysis: Lessons from comparisons of RNA and protein enzymes. Annu Rev Biochem. 1997;66:19-60.
Onoa B, Tinoco I. RNA folding and unfolding. Curr Opin Struct Biol. 2004;14:374-379.
Parak FG. Proteins in action: The physics of structural fluctuations and conformational changes. Curr Opin Struct Biol. 2003;13:552-557.
Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-533.
Ponting CP, Russell RR. The natural history of protein domains. Annu Rev Biophys Biomolec Struct. 2002;31:45-71.
Soukup JK, Soukup GA. Riboswitches exert genetic control through metabolite-induced conformational change. Curr Opin Struct Biol. 2004;14:344-349.
Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96-103.
Vogel C, Bashton M, Kerrison ND, et al. Structure, function and evolution of multi-domain proteins. Curr Opin Struct Biol. 2004;14:208-216.
Wedekind JE, McKay DR. Crystallographic structures of the hammerhead ribozyme: Relationship to ribozyme folding and catalysis. Annu Rev Biophys Biomol Struct. 1998;27:475-502.