CHAPTER 21 Molecular studies in myeloproliferative and myelodysplastic/myeloproliferative neoplasms
Introduction
Almost 60 years after William Dameshek published his description of myeloproliferative disorders,1 clinicians, pathologists, and basic scientists have noticed tremendous progress in the field of these diseases. Due to discovery of molecular markers, it is now possible to discriminate between myeloproliferative and reactive states. Targeted therapies for certain subtypes of myeloproliferative diseases have become available.
Discovery of the so-called Philadelphia (Ph+) chromosome in 1960 by Nowell and Hungerford,2 the subsequent dissection of its molecular structure and of pathways involved in t(9;22) translocation have led to the introduction of imatinib mesylate as the first molecularly targeted therapy in a human malignancy.3 Until 2005, knowledge of molecular aberrations and genetic defects in the Philadelphia-chromosome negative (Ph−) chronic myeloproliferative disorders (CMPD) was rather sparse. In a minority of cases, chromosomal changes such as trisomies 8 and 9, aberrations of chromosome 1, del(20q) or del(13q) in primary myelofibrosis (PMF) or loss of heterozygosity (LOH) at 9p in polycythemia vera (PV) have been demonstrated.4,5
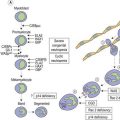
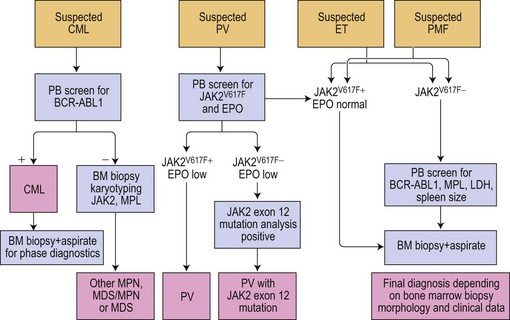
The discovery of the gain-of-function mutation V617F in the tyrosine kinase (TK) Janus kinase 2 (JAK2V617F)6 and other molecular defects in a considerable number of patients with so-called classical Ph− CMPD resulted in a revision of the WHO classification.7 To underline the neoplastic nature of these diseases, the WHO classification published in 2008 replaced the term Ph− CMPD with ‘myeloproliferative neoplasm (MPN)’. This new classification has also introduced new standards for diagnostic algorithms including molecular diagnostics (Fig. 21.1). In this chapter, the current knowledge on molecular changes in MPN and myelodysplastic/myeloproliferative neoplasms (MDS/MPN) is summarized. Molecular aspects of MPN with eosinophilia and of systemic mastocytosis are described in Chapters 25 and 26, respectively.
The Philadelphia chromosome and BCR-ABL in chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) is a clonal stem cell disorder characterized by increased autonomous proliferation of myeloid lineages in the bone marrow (BM). The underlying molecular defect, the balanced reciprocal translocation t(9;22)(q34;q11), was the first chromosomal anomaly consistently present in a human cancer.8 Translocation t(9;22)(q34;q11.2) results in the Philadelphia chromosome found in 90–95 % of patients.9,10 This translocation causes a fusion of the BCR gene from chromosome 22 with the ABL1 gene from chromosome 9.11,12 In 5–10% of patients, a cryptic translocation of 9q34 and 22q11.2 or a variant translocation involving other chromosomes not detectable by conventional karyotype analysis occur. In these patients, the BCR-ABL1 fusion can be demonstrated by molecular methods such as fluorescent in situ hybridization (FISH), reverse transcriptase (RT)-PCR, and Southern blot.13
The BCR-ABL1 fusion results in a deregulated function of the ABL1 gene.14 BCR-ABL1 mRNA encodes for p210 or (rarely) p190 or p230 oncoproteins.15–19 The binding of ATP to the pockets of p210, p190 or p230 allows phosphorylation of selected tyrosine residues on its substrates. The oncogenic properties of the p210, p190, and p230 proteins rely primarily on the constitutively activated TK.
ABL1 plays a central role in the regulation of the cell cycle, cellular response to genotoxic events, and integrin-signaling pathways.20 The fusion of BCR with the SH3 domain of ABL1 interrupts the physiologic suppression of the kinase activity, produces the neoplastic phenotype via deregulation of the impact of ABL1 on apoptosis, cell cycle, and cellular adhesion pathways.21,22 Activation of the RAS pathway results in an increase of mitotic activity and up-regulation of bcl-2, while activation of ‘signal transducers and activators of transcription’ (STAT) 1 and STAT5 proteins results in the inhibition of apoptosis.23–25 Apoptosis inhibition counteracts the protective effect of ABL1 when DNA damage occurs. This results in the accumulation of neoplastic cells and additional genetic changes. Furthermore, BCR-ABL1 negatively influences DNA repair and drives centrosomal hypertrophy, thus increasing the risk of clonal evolution.26,27 BCR-ABL1 induces abnormalities of cytoskeletal cellular function, phosphorylation of adhesion proteins, the production of an abnormal variant of the β1-integrin, and the phosphorylation of Cdc2-related kinase (Crk)-l resulting in reduced adhesion of the CML cells to BM stroma.28–30 Interrupted by BCR-ABL1 interaction between hematopoietic progenitor cells and the BM stroma appears to be important for the control of cellular proliferation.
Nowadays, it is generally accepted that CML originates from a single BCR-ABL1 transformed pluripotent hematopoietic stem cell.31 This immature precursor and its immature daughter cells appear to be rather resistant to therapy utilizing BCR-ABL1 TK inhibitors. Therefore, this type of anti-neoplastic treatment does not seem to cure the disease.32
BCR-ABL1 positive cells or small positive clones have been reported in asymptomatic persons without any signs of CML.33 Moreover, there are differences in the risk of CML between patients with different HLA-B phenotype.34 Thus, immunologic factors may also play a role in the evolution of CML, probably restricting the occurrence of CML to subjects with an immunologic failure of resistance against the BCR-ABL1 positive cell population.
Imatinib mesylate and second generation formulas such as nilotinib and dasatinib represent TK inhibitors that have been designed to work against the constitutively activated chimeric BCR-ABL1 protein, which is responsible for the aberrant phenotype of affected cells in CML patients.35,36
Karyotyping and conventional cytogenetics accompany the inevitable histopathological evaluation of BM biopsy in the initial diagnostic algorithm in order to demonstrate the presence of the Ph+ chromosome. Detection of the BCR-ABL1 fusion products by reverse transcriptase (RT)-PCR analysis in patients with CML (and also Ph+ ALL) has become the golden standard in the diagnostic setting. Standardized real-time quantitative (RQ)-PCR has become the methodology of choice for monitoring molecular response to treatment with TK inhibitors and/or interferon.37 The achievement of the major molecular response (MMR) is the key issue in evaluating therapy effects. MMR can only be determined by quantitative RT-PCR methodology and was originally defined as the reduction of the BCR-ABL1 fusion transcripts by three logs below a standardized baseline level. The latter was initially introduced as a part of the International Randomized Study of Interferon versus STI571 (IRIS) trial.38 Since most patients treated with imatinib or other TK inhibitors achieve a complete cytogenetic response (CCyR), the monitoring of minimal residual disease (MRD) by quantification of the BCR-ABL1 transcripts has become increasingly important. It has been shown that CCyR and MMR after 12 months of treatment indicates an improved progression-free survival at 24 months in 100% of the patients treated with imatinib as compared to 95% in the group showing no MMR and 85% in patients who were not in CCyR at 12 months.38 All patients who achieved CCyR and MMR at 18 months had a 5-year progression-free survival. Moreover, studies in patients followed for several years showed that those achieving MMR early in the first year of treatment had significantly longer CCyR39.
Despite the improved prognosis of CML patients since start of the imatinib era, a response failure is observed in 25–30% of patients.40 The individual risk of developing resistance to imatinib is hard to predict and underlying mechanisms are largely unknown. The majority of patients develop de novo mutations in the ABL kinase domain during therapy. Some may even have pre-existing mutations hindering a good response to initial treatment.
A study of lymphoid blast crisis of CML has recently shown that lymphoid blasts but not CML cells express a B-cell specific mutator named AID (antibody diversification enzyme activation-induced deaminase), which promotes genetic instability and somatic hypermutation of tumor suppressor and DNA repair genes.41 AID may also be responsible for the acquisition of imatinib resistance in CML cells, which justifies testing for the potential targeting of AID and further stresses the need for BCR-ABL1 transcript level monitoring during TK inhibitor treatment.
Due to the clinical importance of MRD, efforts to standardize the methodologies for quantification of BCR-ABL1 transcripts in CML patients are now in progress. Most studies refer to the IRIS trial and the associated International Scale (IS) for BCR-ABL1 transcript quantification,38 but significant inter-laboratory differences concerning definition of patient molecular status still exist. The increase of BCR-ABL1 transcripts to a level >0.1% on the IS scale can be interpreted as a potential response failure and should lead to a close-meshed monitoring of BCR-ABL1 transcript levels.42 Successful efforts to standardize methodologies have already been reported.43 The goal of a recently initiated study is to introduce a conversion factor (CF) allowing the harmonization and comparison of results obtained during molecular monitoring of CML patients before and during therapy.44 In brief, every participating laboratory generates its own CF, which will allow comparison of BCL-ABL1 transcript levels for upcoming multicenter studies.
Interestingly, there is no concordance between BCR-ABL1 transcript levels in either sorted or unsorted BM aspirate cells and the levels determined in peripheral blood cells, both mononuclear cells and granulocytes. Since drawing blood is less invasive as compared to BM aspiration, it should be considered the standard choice to determine the BCR-ABL1 transcript levels. Apart from BCR-ABL1 levels, other prognostic parameters such as the presence of BM fibrosis45 or increasing numbers of BM blasts are of importance during clinical follow-up (see Chapter 24 for details).
The Janus kinase 2 in MPN
JAK2V617F
The molecular nature of the gain-of-function mutation in JAK2 is a hotspot in exon 14 where the wild-type (WT) guanine (G) is changed to a mutant thymine (T) with consecutive replacement of valine by phenylalanine at position 617 (V617F) in the JH2 pseudokinase.6,46–48 Even though functional studies are not yet available, it is well accepted that the bulky amino acid phenylalanine changes the conformation of the protein thereby blocking the interaction of the auto-inhibitory JH2 pseudokinase domain with the catalytic domain JH1. This hampers control of catalytic activity and leads to a constitutively activated JAK2 kinase and subsequently, the activation of downstream targets in affected cells.49
The effects of mutated JAK2V617F show a considerable diversity in various cellular lineages and MPN subtypes affected by this mutation. The JAK2V617F clone is autonomous and highly proliferative in vitro, even in the absence of growth factors. All MPN subtypes harboring the JAK2V617F show high numbers of endogenous erythroid colonies (EEC) in BM cultures. In addition, the affected lineages are additionally hypersensitive to growth factors such as erythropoietin (EPO), granulocyte-colony stimulating factor (G-CSF) or interleukins. In mouse models, the mutation is powerful enough to mediate a PV-like phenotype and leads to development of myelofibrosis.47,50,51 Similar findings were demonstrated in MPN patients harboring the JAK2V617F mutation, but some of the features observed in the animal model might be due to toxic effects of the JAK2V617F overdose. It is of note that strain-specific differences were found in two different mice models showing the JAK2V617F mutation. Whereas both the Balb/C and the C57Bl/6 mice showed an increase in hemoglobin and hematocrit, the number of leukocytes and the degree of myelofibrosis were strikingly higher in the Balb/C mice.50 No thrombocytosis was seen in these models, even though an increase of megakaryocytes in the BM was noted. Direct transfer of knowledge concerning JAK2V617F mutation derived from mice models to human MPN appears to be difficult and different phenotypes observed in humans may also develop as a consequence of individual host modifiers.
Among MPN, PV showed the highest JAK2V617F frequency (~95%) followed by PMF and ET (~50% JAK2V617F mutated cases).52–55 It is of note that patients with JAK2V617F mutated ET show PV-like features, e.g. elevated hemoglobin and hematocrit levels. Also, it has been suggested that PV patients with a homozygous mutation status (i.e. 50% or more alleles showing the mutant T allele) had a higher risk for development of myelofibrosis.56–58 However, the mutation per se is probably not responsible for myelofibrosis development, since no correlation between the JAK2 status and the clinical course or degree of fibrosis could be demonstrated in PMF.59,60 A retrospective analysis of 490 MPN patients along with evaluation of follow-up biopsies revealed a correlation between JAK2V617F mutant allele burden and MPN subtypes. In this study, a considerable number of ET cases were characterized by lower allele burden.61
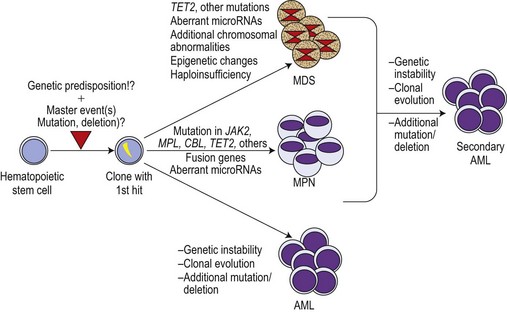
Although the discovery of the JAK2V617F mutation was a breakthrough in the field, it became increasingly clear that this molecular defect is not the initiating event in MPN pathogenesis. This is illustrated by X-chromosome inactivation patterns (XCIP) in female ET patients with the JAK2V617F mutation.62–64 XCIP studies found that the proportion of JAK2V617F positive cells was lower than the total number of clonal cells in a given patient. A higher proportion of cells with the deletion of chromosome 20q in comparison to the percentage of cells with the JAK2V617F mutation was found in patients carrying both anomalies.64 These studies clearly indicate that the neoplastic hematopoietic stem cell (HSC) must have acquired at least one molecular aberration preceding the JAK2V617F mutation. This master event could be an as yet undiscovered small chromosomal deletion or an insertion spanning only small-size region(s) of a gene, thereby impossible to detect by conventional cytogenetic techniques (Fig. 21.2). This event must lead to a selective advantage of the affected cell, allowing autonomous expansion accompanied by ongoing chromosomal instability and additional molecular defects such as mutations inducing entity-typical phenotypes. In MPN, the mimicry of phenotypes between different entities is not uncommon. This might be a result of two or more aberrations existing in parallel. It is therefore now widely accepted that JAK2V617F is a secondary mutation leading towards PV or a PV-like clinical appearance, particularly in ET.
JAK2 (K539L and other aberrations in exon 12)
In the diagnostic setting, some patients show high hemoglobin levels, high hematocrit and low serum EPO levels but do not harbor the JAK2V617F mutation. These patients are usually affected by a rather rare molecular defect in exon 12 of the JAK2 gene. Exon 12 aberrations vary between point mutations leading to the K539L mutation and insertions/deletions leading to various amino acid substitutions.65 Two interesting epidemiologic features characterize JAK2 exon 12 aberrations: 1) more women than men were affected when compared to idiopathic erythrocytosis showing the JAK2WT; 2) patients presented at younger age when compared to those with JAK2V617F mutation.66 However, due to the low frequency of JAK2 exon 12 aberrations and the complexity of testing, this type of molecular analysis should be restricted to patients who are JAK2V617F negative but show clinical parameters strongly suggesting clonal erythropoiesis.
The JAK2V617F mutation in de novo acute leukemia and in MPN transformed to acute leukemia
MPN transformation into acute myeloid leukemia (AML) is the major life-threatening event in the course of the disease. The frequency of transformation varies among the MPN subtypes. The general risk for evolving to AML is much higher for patients with PV or PMF than for those having ET. The impact of a history of prior treatment, i.e. with hydroxyurea, is unclear.67,68 The mechanism of the transforming switch is totally unknown. After discovery of the JAK2V617F mutation, this molecular defect has been suggested to be a powerful co-factor in the process of transformation. A series of studies investigated the occurrence of JAK2V617F in secondary AML developing in patients with a history of MPN and in de novo AML or acute lymphoblastic leukemia (ALL). In one study the JAK2V617F mutation was not detected in de novo AML M0 – M6-subtype (according to the French-American-British (FAB) classification) but some positive cases of AML M7 were found (2/11, 18%).69 Another investigation described one case of AML M6 (1/53) positive for JAK2V617F but no positive cases in AML M5 (0/85) or AML M7 (0/14) FAB categories.70 In one study a frequency of almost 3.0% was reported (3/113 de novo AML) with two AML cases showing both the JAK2V617F and t(8;21)(q22;q22). One case assigned to the ‘AML without maturation’ category had a JAK2 (V607N) mutation.71 In B- and T-ALL, the JAK2 mutation seems to be extremely rare, except for children with Down syndrome.72,73
To investigate the role of JAK2V617F in the transformation of MPN, the mutant allele burden (‘gene dosage’) has been retrospectively studied in individual patients. It was found that patients with a prior history of MPN and detectable JAK2V617F at diagnosis showed no increase of mutant allele burden during transformation to acute leukemia.74 The same study demonstrated that both the development of myelofibrosis and leukemic transformation in PV and PMF was found in patients with the JAK2WT status.
It has been noted that in JAK2V617F positive patients, the populations of blasts in transformed MPN are frequently negative for the JAK2 mutation.63 These findings are in line with the suggestion of the pre-JAK2V617F phase, in which clonality has already been achieved by an as yet undefined mechanism (see Fig. 21.2). Thus, JAK2V617F-negative AML developing during course of MPN might arise from the primary clone and could be the key to discovery of the primary master hit. The other scenario could be that, indeed, two separate clones develop independently in an individual patient. Studies of the molecular signatures of JAK2V617F-negative blasts from the AML transformation of MPN could open up relevant insights into the pathogenesis of early MPN development.
Myeloproliferative leukemia virus oncogene (MPL) mutations in MPN
Thrombopoietin (TPO) and its receptor MPL (CD110) are important for hematopoietic stem-cell survival, the differentiation of hematopoietic progenitor cells, and are key regulatory factors in megakaryocyte development and platelet formation.75 MPL is expressed by hematopoietic tissues, hematopoietic stem cells, erythroid progenitors, megakaryocytes and platelets.76 Aberrant expression of MPL has been implicated in some hematological malignancies. Earlier studies showed that reduced MPL expression in megakaryocytes from patients with MPN is due to impaired post-translational modifications.77,78 Reduced platelet MPL expression and weakly labeled megakaryocytes by immunohistochemistry were found to be of diagnostic and prognostic value in MPN.79–82
Several studies demonstrated that the megakaryocytic and erythroid lineages co-express MPL and the erythropoietin receptor (EPO-R) as well as transcription factors such as nuclear factor-erythroid derived (NF-E)1 (identical to globin transcription factor (GATA1)) and NF-E2.83–85 Due to structural homologies between MPL, EPO-R and their ligands, it has been suggested that both megakaryocytic and erythroid lineages arise from a common progenitor.85 Therefore, abnormal expression and function of MPL or EPO-R could induce proliferation of both megakaryocytic and erythroid lineages.86 In mice overexpressing the functional MPL receptor, enhanced erythropoiesis and reduced megakaryopoiesis have been demonstrated.87
Since the JAK-STAT pathway is constitutively activated in MPN but only half of PMF and ET patients harbor the JAK2V617F mutation, screening for mutations in other cytokine receptors by high throughput DNA sequencing has been performed.88 Whereas defined regions of the granulocyte-colony stimulating factor receptor (G-CSF-R) and the EPO-R showed no molecular defects, a point mutation W515L in the juxtamembrane domain of MPL (exon 10) was discovered.88 Cell lines transfected with MPLW515L show cytokine-independent growth and hypersensitivity for TPO. Mouse strains carrying the mutation exhibit high levels of thrombocytosis, massive hepatosplenomegaly and infarctions of the spleen. Analysis of a large cohort (1182 patients) detected a frequency of ~5% MPLW515L in patients with PMF and ET.89 The latter study revealed another mutation in the MPL gene: MPLW515K as well as the occurrence of MPL mutations together with JAK2V617F in individual patients.
In a study of 776 samples from ET patients, the overall frequency of MPL exon 10 mutations was 8.5% in patients with JAK2WT.90 Patients with the MPLW515K mutation had a higher mutant allele burden than those with MPLW515L. As compared to patients with JAK2V617F, MPL mutated ET patients showed lower hemoglobin levels, higher EPO levels, higher platelets, and endogenous megakaryocyte colony growth but there was no increase in endogenous erythroid colonies and overall lower bone marrow cellularity. MPL mutations lacked prognostic significance with regard to frequency of hemorrhage, thrombosis, development of myelofibrosis, and survival.
One study investigated in a large series of MPN the potential correlation between reduced platelet MPL protein expression and the mutant JAK2V617F allele load in neutrophils.91 In all MPN subtypes, the higher percentage of mutant JAK2V617F alleles was related to lower MPL expression, which suggests that the presence of JAK2V617F is associated with down-regulation of MPL. In ET cases showing JAK2WT, the MPL expression was significantly higher than in ET with JAK2V617F. PMF patients in this study exhibited the lowest platelet MPL levels followed by PV and ET cases. It has been suggested that JAK2V617F may possibly bind MPL in the endoplasmic reticulum (as shown for JAK2WT and EPO-R) or impair further processing in the Golgi apparatus, directly inducing down-regulation of MPL. This could also be a counteracting mechanism inhibiting autonomous proliferation mediated by negative regulators such as the adaptor protein Lnk (linker), which normally controls MPL expression.92
The axis: growth factor (ligand) – growth factor receptor – intracellular tyrosine kinase seems to involve a large number of players in parallel, making every step in signal transduction rather complicated. Cellular transformation mediated by gain-of-function mutations such as the JAK2V617F is dependent on expression of other components such as functional G-CSF-receptor and EPO-R. As shown for JAK2V617F, a functional EPO-R is necessary not only for JAK2WT signaling but also for the mutant form.93 It is plausible that a composition of molecular aberrations, i.e. concomitant JAK2V617F and MPLW515K/L mutations in an individual patient could either amplify or abrogate the transforming cellular effects. Thus, the clinical and histological phenotype in MPN could be the result of a mixture of molecular aberrations.
TET2 mutations in MPN and other myeloid malignancies
Soon after the report of Vainchenker et al. at American Society for Hematology Meeting 2008, collaborative studies confirmed that acquired molecular aberrations in the Ten-Eleven Translocation 2 (TET2) gene are frequent in MPN and MDS/MPN.94,95 These aberrations show considerable variation including frameshifts, nonsense or missense mutations in exon 4 and 12 of the TET2 gene.94 The overall TET2 mutation frequency in MPN is 13% (5% in ET, 16% and 17% in PV and PMF, respectively) and 50% in patients with chronic myelomonocytic leukemia (CMML). TET2 mutations occur in both JAK2V617F and JAK2WT cases with frequencies of 17% and 7%, respectively. TET2 mutations significantly increase with age: mutational frequency is 23% in patients >60 years of age compared to only 4% in younger individuals.94 In PV and PMF patients, the occurrence of TET2 mutations did not correlate with any prognostic factor such as survival, rate of transformation to AML or risk of thrombosis. Based on its prevalence in older patients, which often already acquired the JAK2V617F mutation, the usefulness of TET2 as a diagnostic marker appears to be limited. However, another study in patients with PV showed that TET2 mutations may occur in both JAK2V617F and JAK2WT clones of an individual patient.96 An overall frequency of 20% TET2 mutations was found in a cohort of 61 cases of familial MPN with no age correlation. In this study, sequential occurrence of JAK2V617F and subsequently TET2 mutations in individual patients was reported.97 Different TET2 alterations occurred in the same individual suggesting molecular dynamics probably due to genetic instability. TET2 mutations were also found in other myeloid malignancies with considerably higher mutation frequencies: in de novo AML (42%),98 systemic mastocytosis (29%),99 MDS with rearranged 4q24 (19%),100 and in secondary AML (up to 32%).96 Therefore, alterations in the TET2 gene appear to be a common event in myeloid malignancies including those showing dysplastic features, chronic myeloproliferation and acute leukemia (see Fig. 21.2). Future studies will show whether the TET2 gene functions as a tumor suppressor gene or if structurally altered TET2 contributes to malignant transformation due to its relevant role in normal hematopoietic differentiation. Before discovery of TET2 alterations, the function of this gene was unclear. Since TET2 alterations are loss-of-function defects, knock-out and knock-in studies will show the impact of this gene on normal and malignant hematopoiesis.
However, the TET1 gene, which fusion with the histone methyltransferase MLL has been identified in AML associated with t(10;11)(q22;q23) translocation, was shown to catalyze the reaction from 5-methylcytosine to 5-hydroxymethylcytosine in the DNA suggesting a role for TET proteins in epigenetic regulation.101
JAK2V617F, MPLW515L/K, BCR-ABL, KITD816V: molecular markers may be combined in an individual patient
Shortly after the JAK2V617F mutation and other molecular aberrations were described in MPN, numerous studies reported that various molecular markers in MPN may be combined in an individual patient. The first reported patients with concomitant presence of BCR-ABL and JAK2V617F 102 presented typical Philadelphia-positive CML and quickly achieved molecular remission after start of imatinib treatment. However, under imatinib treatment a JAK2V617F clone evolved leading to a Philadelphia-negative MPN with myelofibrosis. A retrospective molecular analysis revealed that the JAK2V617F positive population with low mutant allele burden was already present at time of CML diagnosis. This case clearly illustrated that different clones may co-exist in MPN and that therapy may create an advantage for one clone, after a competing clone has been successfully repressed.
Besides the combination of BCR-ABL and JAK2V617F, other molecular marker combinations have been described. These include: JAK2V617F and MPLW515L/K in MPN and MDS/MPN,103–107 JAK2V617F and KITD816V in systemic mastocytosis with associated non-mast cell hematological disease (SM-AHNMD),103 TET2 mutation and JAK2V617F in MPN and MDS/MPN96,97.
In Philadelphia-negative MPN carrying solely the JAK2V617F mutation, megakaryocytes showed variation in their mutant allele burden when this single defect was quantified.60 Accordingly, various cellular lineages in individuals with combined molecular defects may also show different molecular changes. In a patient with concomitant JAK2V617F and MPLW515L mutations, CD34-positive cells, granulocytes, and monocytes showed the presence of both mutations. However, T and B lymphocytes carried the MPLW515L mutation but were JAK2WT, whereas the erythropoiesis and the megakaryocytic lineage showed JAK2V617F with MPLWT.104 These findings strongly argue against a sequential acquisition of molecular defects in various cellular lineages in MPN and favor the hypothesis of parallel proliferation of unrelated hematopoietic stem-cell clones.
The casitas B-lineage lymphoma gene (CBL) aberrancies in MPN and other myeloid malignancies
The proto-oncogene CBL is a negative regulator of several receptor tyrosine kinase signaling pathways and an adaptor protein in tyrosine-phosphorylation dependent signaling.108 Ubiquitination of receptor protein-tyrosine kinases (PTKs) terminates signaling by marking active receptors for degradation. CBL is an adaptor protein for receptor PTKs that positively regulates receptor PTK ubiquitination. Ubiquitin-protein ligases, also known as E3s, are the components of ubiquitination pathways that recognize target substrates and promote their ligation to ubiquitin. CBL protein acts as an E3 that can recognize tyrosine-phosphorylated substrates, such as the activated platelet-derived growth factor receptor.109 It was concluded that CBL functions as an ubiquitin-protein ligase and thus provides a distinct mechanism for substrate targeting in the ubiquitin system (Table 21.1).
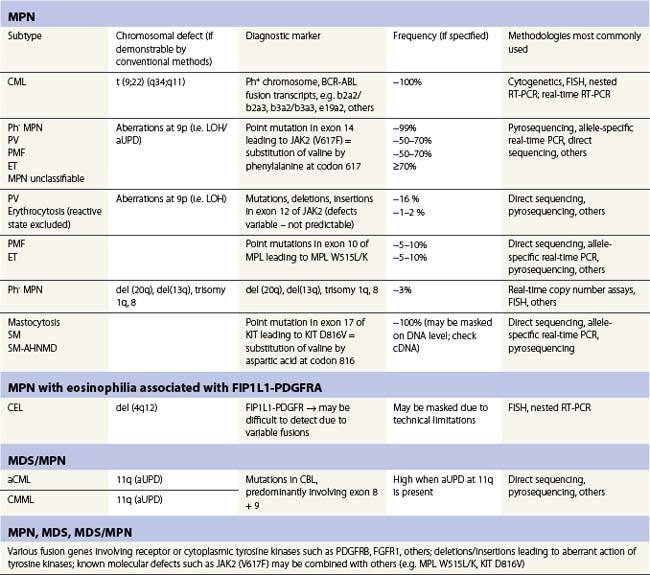
Table 21.1 Selected molecular aberrancies in myeloproliferative (MPN) and myelodysplastic/myeloproliferative (MDS/MPN) neoplasms
The CBL gene is located on the 11q23.3 telomeric of the MLL gene, which is frequently fused to loci on other chromosomes in translocations causing various types of leukemia. Southern blot analysis in conjunction with FISH in a patient with AML (FAB M1) suggested that the CBL/MLL fusion was the result of an interstitial deletion.110 Two other studies showed the frequent occurrence of molecular defects other than fusion involving the CBL gene.111,112 Acquired uniparental disomy (aUPD) that leads to loss-of-heterozygosity and can induce the loss-of-function of tumor suppressors or gain-of-function of proto-oncogenes could be demonstrated in the CBL gene. aUPD induces a gain-of-function leading to inhibition of the wild-type CBL and prolonged action of tyrosine kinases.112 In the study by Grand et al., genome-wide SNP screening detected that atypical CML (aCML), CMML and MPN cases with manifest myelofibrosis were most frequently affected by CBL mutations.111 CBL mutations were often associated with aUPD at 11q and showed involvement of CBL exon 8 and 9 as well as intron regions. These changes span over a region of several hundred base pairs making the diagnostic assay much more difficult by comparison to detection of JAK2V617F. Notably, an underlying molecular defect in the CBL gene seems to correlate with a shorter overall survival and progression-free survival in MPN patients.111 Especially in aCML and CMML, CBL aberrations offer new insights into the molecular pathology of these MPN subtypes and serve as an important clonal marker allowing exclusion of reactive states in borderline cases.
Summary and future perspectives
Discovery of important mechanisms necessary for proper signal transduction in hematopoiesis, i.e. the induction of signaling by growth factors, receptor activation, recruitment of tyrosine kinases such as JAKs and transmission of signals by effector molecules such as the STATs was indispensable for exploring some of the molecular defects in MPN. However, we have to realize that JAK2V617F, MPLW515L/K, TET2 and other defects might only represent the tip of the iceberg. Even though a constitutively activated kinase is a promising target for up-coming therapies, these therapies will probably not hit the molecular ‘master event’ responsible for the formation of a pre-malignant clone (Fig. 21.2). Also, we have to consider that in the presence of potentially co-existing clones the successful targeting of the JAK2V617F positive clone could lead to a selective growth advantage of another clone (the pre-JAK2V617F clone or a smaller bystander clone).
The reliable discrimination of the myeloproliferative (neoplastic) from the reactive state by detection of JAK2V617F, aberrations in JAK2 (exon 12), MPLW515L/K and TET2 has become a major step in the diagnostic approach. However, we still have to face the diagnostic dilemma that in terms of MPN classification, the detection of the JAK2V617F or other molecular defects per se is of limited value. Apart from PV, in which JAK2V617F in ~99% exerts its effects predominantly on the erythroid lineage, ET and PMF exhibit distinct features that are demonstrated in cases with and without molecular changes. Even though ET showing the JAK2V617F could be interpreted as a modified form of PV, true ET clearly shows another course including rare leukemic transformation and virtually no development of myelofibrosis. In a subset of ET cases, the lower mutant allele burden of JAK2V617F allows discrimination from the hypercellular, prefibrotic stage of PMF.61 This finding can be of clinical value as PMF has a high risk for the development of myelofibrosis and the early stage of PMF is sometimes difficult to distinguish from ET by morphological and clinical parameters.
1 Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372-375.
2 Nowell PC. Discovery of the Philadelphia chromosome: a personal perspective. J Clin Invest. 2007;117:2033-2035.
3 Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of BCR-Abl positive cells. Nat Med. 1996;2:561-566.
4 Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol. 2009;82:329-338.
5 Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30:229-236.
6 Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779-1790.
7 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphatic Tissues. Lyon: IARC; 2008.
8 Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
9 Nowell PC, Hungerford DA. Chromosome studies in human leukemia. II. Chronic granulocytic leukemia. J Natl Cancer Inst. 1961;27:1013-1035.
10 Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290-293.
11 de Klein A, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765-767.
12 Bartram CR, de Klein A, Hagemeijer A, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277-280.
13 Dreazen O, Klisak I, Rassool F, et al. The bcr gene is joined to c-abl in Ph1 chromosome negative chronic myelogenous leukemia. Oncogene Res. 1988;2:167-175.
14 Muller AJ, Young JC, Pendergast AM, et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11:1785-1792.
15 Ben-Neriah Y, Daley GQ, Mes-Masson AM, et al. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212-214.
16 Pane F, Intrieri M, Quintarelli C, et al. BCR/ABL genes and leukemic phenotype: from molecular mechanisms to clinical correlations. Oncogene. 2002;21:8652-8667.
17 Inokuchi K, Dan K, Takatori M, et al. Myeloproliferative disease in transgenic mice expressing P230 Bcr/Abl: longer disease latency, thrombocytosis, and mild leukocytosis. Blood. 2003;102:320-323.
18 Ohsaka A, Shiina S, Kobayashi M, et al. Philadelphia chromosome-positive chronic myeloid leukemia expressing p190(BCR-ABL). Intern Med. 2002;41:1183-1187.
19 Ravandi F, Cortes J, Albitar M, et al. Chronic myelogenous leukaemia with p185(BCR/ABL) expression: characteristics and clinical significance. Br J Haematol. 1999;107:581-586.
20 Kawai H, Nie L, Yuan ZM. Inactivation of NF-kappaB-dependent cell survival, a novel mechanism for the proapoptotic function of c-Abl. Mol Cell Biol. 2002;22:6079-6088.
21 Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247-259.
22 Pendergast AM, Gishizky ML, Havlik MH, Witte ON. SH1 domain autophosphorylation of P210 BCR/ABL is required for transformation but not growth factor independence. Mol Cell Biol. 1993;13:1728-1736.
23 Bedi A, Zehnbauer BA, Barber JP, et al. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood. 1994;83:2038-2044.
24 Spiekermann K, Pau M, Schwab R, et al. Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp Hematol. 2002;30:262-271.
25 McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708-722.
26 Deutsch E, Dugray A, AbdulKarim B, et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97:2084-2090.
27 Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319-327.
28 Salgia R, Li JL, Ewaniuk DS, et al. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100:46-57.
29 Salgia R, Quackenbush E, Lin J, et al. The BCR/ABL oncogene alters the chemotactic response to stromal-derived factor-1alpha. Blood. 1999;94:4233-4246.
30 Bhatia R, Munthe HA, Forman SJ. Abnormal growth factor modulation of beta1-integrin-mediated adhesion in chronic myelogenous leukaemia haematopoietic progenitors. Br J Haematol. 2001;115:845-853.
31 Jorgensen HG, Holyoake TL. Characterization of cancer stem cells in chronic myeloid leukaemia. Biochem Soc Trans. 2007;35:1347-1351.
32 Bhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701-4707.
33 Biernaux C, Loos M, Sels A, et al. Detection of major BCR-ABL gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86:3118-3122.
34 Posthuma EF, Falkenburg JH, Apperley JF, et al. HLA-B8 and HLA-A3 coexpressed with HLA-B8 are associated with a reduced risk of the development of chronic myeloid leukemia. The Chronic Leukemia Working Party of the EBMT. Blood. 1999;93:3863-3865.
35 Hochhaus A, Kantarjian HM, Baccarani M, et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109:2303-2309.
36 Jabbour E, Cortes JE, Kantarjian H. Optimizing treatment with BCR-Abl tyrosine kinase inhibitors in Philadelphia chromosome-positive chronic myeloid leukemia: focus on dosing schedules. Clin Lymphoma Myeloma. 2008;8(Suppl. 3):S75-S81.
37 Branford S, Cross NC, Hochhaus A, et al. Rationale for the recommendations for harmonizing current methodology for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia. 2006;20:1925-1930.
38 Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349:1423-1432.
39 Cortes J, Talpaz M, O’Brien S, et al. Molecular responses in patients with chronic myelogenous leukemia in chronic phase treated with imatinib mesylate. Clin Cancer Res. 2005;11:3425-3432.
40 Goldman JM. Initial treatment for patients with CML. Hematology Am Soc Hematol Educ Program. 2009:453-460.
41 Klemm L, Duy C, Iacobucci I, et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16:232-245.
42 Hughes TP, Branford S. Measuring minimal residual disease in chronic myeloid leukemia: fluorescence in situ hybridization and polymerase chain reaction. Clin Lymphoma Myeloma. 2009;9(Suppl. 3):S266-S271.
43 Branford S, Fletcher L, Cross NC, et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood. 2008;112:3330-3338.
44 Muller MC, Cross NC, Erben P, et al. Harmonization of molecular monitoring of CML therapy in Europe. Leukemia. 2009;23:1957-1963.
45 Buesche G, Ganser A, Schlegelberger B, et al. Marrow fibrosis and its relevance during imatinib treatment of chronic myeloid leukemia. Leukemia. 2007;21:2420-2427.
46 Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-1061.
47 James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144-1148.
48 Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387-397.
49 Kaushansky K. On the molecular origins of the chronic myeloproliferative disorders: it all makes sense. Blood. 2005;105:4187-4190.
50 Wernig G, Mercher T, Okabe R, et al. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274-4281.
51 Lacout C, Pisani DF, Tulliez M, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652-1660.
52 Lippert E, Boissinot M, Kralovics R, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865-1867.
53 Tefferi A, Lasho TL, Schwager SM, et al. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer. 2006;106:631-635.
54 Horn T, Kremer M, Dechow T, et al. Detection of the activating JAK2 V617F mutation in paraffin-embedded trephine bone marrow biopsies of patients with chronic myeloproliferative diseases. J Mol Diagn. 2006;8:299-304.
55 Bock O, Busche G, Koop C, et al. Detection of the single hotspot mutation in the JH2 pseudokinase domain of Janus kinase 2 in bone marrow trephine biopsies derived from chronic myeloproliferative disorders. J Mol Diagn. 2006;8:170-177.
56 Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia. 2005;19:1847-1849.
57 Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol. 2005;131:208-213.
58 Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366:1945-1953.
59 Bock O, Neuse J, Hussein K, et al. Aberrant collagenase expression in chronic idiopathic myelofibrosis is related to the stage of disease but not to the JAK2 mutation status. Am J Pathol. 2006;169:471-481.
60 Hussein K, Brakensiek K, Buesche G, et al. Different involvement of the megakaryocytic lineage by the JAK2 V617F mutation in polycythemia vera, essential thrombocythemia and chronic idiopathic myelofibrosis. Ann Hematol. 2007;86:245-253.
61 Hussein K, Bock O, Theophile K, et al. JAK2(V617F) allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp Hematol. 2009;37:1186-1193.
62 Kiladjian JJ, Elkassar N, Cassinat B, et al. Essential thrombocythemias without V617F JAK2 mutation are clonal hematopoietic stem cell disorders. Leukemia. 2006;20:1181-1183.
63 Kralovics R, Teo SS, Li S, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377-1380.
64 Campbell PJ, Baxter EJ, Beer PA, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108:3548-3555.
65 Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459-468.
66 Schnittger S, Bacher U, Haferlach C, et al. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica. 2009;94:414-418.
67 Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008;22:1494-1502.
68 Huang J, Li CY, Mesa RA, et al. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer. 2008;112:2726-2732.
69 Jelinek J, Oki Y, Gharibyan V, et al. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome–negative CML, and megakaryocytic leukemia. Blood. 2005;106:3370-3373.
70 Frohling S, Lipka DB, Kayser S, et al. Rare occurrence of the JAK2 V617F mutation in AML subtypes M5, M6, and M7. Blood. 2006;107:1242-1243.
71 Lee JW, Kim YG, Soung YH, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25:1434-1436.
72 Levine RL, Loriaux M, Huntly BJ, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377-3379.
73 Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484-1492.
74 Mesa RA, Powell H, Lasho T, et al. A longitudinal study of the JAK2(V617F) mutation in myelofibrosis with myeloid metaplasia: analysis at two time points. Haematologica. 2006;91:415-416.
75 Kaushansky K. Mpl and the hematopoietic stem cell. Leukemia. 2002;16:738-739.
76 Kaushansky K. The role of the MPL receptor in myeloproliferative disorders. Leukemia. 1998;12(Suppl. 1):S47-S50.
77 Moliterno AR, Spivak JL. Posttranslational processing of the thrombopoietin receptor is impaired in polycythemia vera. Blood. 1999;94:2555-2561.
78 Horikawa Y, Matsumura I, Hashimoto K, et al. Markedly reduced expression of platelet c-mpl receptor in essential thrombocythemia. Blood. 1997;90:4031-4038.
79 Mesa RA, Hanson CA, Li CY, et al. Diagnostic and prognostic value of bone marrow angiogenesis and megakaryocyte c-Mpl expression in essential thrombocythemia. Blood. 2002;99:4131-4137.
80 Tefferi A, Yoon SY, Li CY. Immunohistochemical staining for megakaryocyte c-mpl may complement morphologic distinction between polycythemia vera and secondary erythrocytosis. Blood. 2000;96:771-772.
81 Yoon SY, Li CY, Tefferi A. Megakaryocyte c-Mpl expression in chronic myeloproliferative disorders and the myelodysplastic syndrome: immunoperoxidase staining patterns and clinical correlates. Eur J Haematol. 2000;65:170-174.
82 Teofili L, Pierconti F, Di FA, et al. The expression pattern of c-mpl in megakaryocytes correlates with thrombotic risk in essential thrombocythemia. Blood. 2002;100:714-717.
83 Martin DI, Zon LI, Mutter G, Orkin SH. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344:444-447.
84 Vigon I, Mornon JP, Cocault L, et al. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: identification of a member of the hematopoietic growth factor receptor superfamily. Proc Natl Acad Sci USA. 1992;89:5640-5644.
85 McDonald TP, Sullivan PS. Megakaryocytic and erythrocytic cell lines share a common precursor cell. Exp Hematol. 1993;21:1316-1320.
86 Yan XQ, Lacey DL, Saris C, et al. Ectopic overexpression of c-mpl by retroviral-mediated gene transfer suppressed megakaryopoiesis but enhanced erythropoiesis in mice. Exp Hematol. 1999;27:1409-1417.
87 Kieran MW, Perkins AC, Orkin SH, Zon LI. Thrombopoietin rescues in vitro erythroid colony formation from mouse embryos lacking the erythropoietin receptor. Proc Natl Acad Sci USA. 1996;93:9126-9131.
88 Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270.
89 Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472-3476.
90 Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112:141-149.
91 Moliterno AR, Williams DM, Rogers O, Spivak JL. Molecular mimicry in the chronic myeloproliferative disorders: reciprocity between quantitative JAK2 V617F and Mpl expression. Blood. 2006;108:3913-3915.
92 Gery S, Gueller S, Chumakova K, et al. Adaptor protein Lnk negatively regulates the mutant MPL, MPLW515L associated with myeloproliferative disorders. Blood. 2007;110:3360-3364.
93 Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci USA. 2005;102:18962-18967.
94 Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905-911.
95 Kosmider O, Gelsi-Boyer V, Ciudad M, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676-1681.
96 Delhommeau F, Dupont S, Della VV, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289-2301.
97 Saint-Martin C, Leroy G, Delhommeau F, et al. Analysis of the ten-eleven translocation 2 (TET2) gene in familial myeloproliferative neoplasms. Blood. 2009;114:1628-1632.
98 Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144-147.
99 Tefferi A, Levine RL, Lim KH, et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23:900-904.
100 Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113:6403-6410.
101 Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-935.
102 Hussein K, Bock O, Theophile K, et al. Chronic myeloproliferative diseases with concurrent BCR-ABL junction and JAK2V617F mutation. Leukemia. 2008;22:1059-1062.
103 Sotlar K, Bache A, Stellmacher F, et al. Systemic mastocytosis associated with chronic idiopathic myelofibrosis: a distinct subtype of systemic mastocytosis associated with a [corrected] clonal hematological non-mast [corrected] cell lineage disorder carrying the activating point mutations KITD816V and JAK2V617F. J Mol Diagn. 2008;10:58-66.
104 Hussein K, Bock O, Theophile K, et al. Biclonal expansion and heterogeneous lineage involvement in a case of chronic myeloproliferative disease with concurrent MPLW515L/JAK2V617F mutation. Blood. 2009;113:1391-1392.
105 Hussein K, Theophile K, Buhr T, et al. Different lineage involvement in myelodysplastic/myeloproliferative disease with combined MPLW515L and JAK2V617F mutation. Br J Haematol. 2009;145:673-675.
106 Pardanani A, Lasho TL, Finke C, et al. Extending Jak2V617F and MplW515 mutation analysis to single hematopoietic colonies and B and T lymphocytes. Stem Cells. 2007;25:2358-2362.
107 Lasho TL, Pardanani A, McClure RF, et al. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br J Haematol. 2006;135:683-687.
108 Smit L, Borst J. The Cbl family of signal transduction molecules. Crit Rev Oncog. 1997;8:359-379.
109 Joazeiro CA, Wing SS, Huang H, et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309-312.
110 Fu JF, Hsu JJ, Tang TC, Shih LY. Identification of CBL, a proto-oncogene at 11q23.3, as a novel MLL fusion partner in a patient with de novo acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37:214-219.
111 Grand FH, Hidalgo-Curtis CE, Ernst T, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113:6182-6192.
112 Sanada M, Suzuki T, Shih LY, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904-908.
113 Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009;6:627-637.
114 Mullighan CG. TET2 mutations in myelodysplasia and myeloid malignancies. Nat Genet. 2009;41:766-767.