[level-membership-for-cardiovascular-category]
Chapter 1 Molecular Mechanisms of Cardiac Electrical Activity
Ionic Equilibrium
The lipid bilayer of the cell membrane is hydrophobic and impermeable to water-soluble substances such as ions. Hence, for the hydrophilic ions to be able to cross the membrane, they need hydrophilic paths that span the membrane (i.e., pores), which are provided by transmembrane proteins called ion channels. Once a hydrophilic pore is available, ions move passively across the membrane driven by two forces: the electrical gradient (voltage difference) and the chemical gradient (concentration difference). The chemical gradient forces the ions to move from a compartment of a higher concentration to one of lower concentration. The electrical gradient forces ions to move in the direction of their inverse sign (i.e., cations [positively charged ions] move toward a negatively charged compartment, whereas anions [negatively charged ions] move toward a positively charged compartment). Because the chemical and electrical gradients can oppose each other, the direction of net ion movement will depend on the relative contributions of chemical gradient and electrical potential (i.e., the net electrochemical gradient), so that ions tend to move spontaneously from a higher to a lower electrochemical potential.1–3
The movement of an ion down its chemical gradient in one direction across the cell membrane results in build-up of excess charge carried by the ion on one side of the membrane, which generates an electrical gradient that impedes continuing ionic movement in the same direction. When the driving force of the electrical gradient across the membrane becomes equal and opposite to the force generated by the chemical gradient, the ion is said to be in electrochemical equilibrium, and the net transmembrane flux (or current) of that particular ion is zero. In this setting, the electrical potential is called the equilibrium potential (Eion) (reversal potential or Nernst potential) of that individual ion. The Eion for a given ion depends on its concentration on either side of the membrane and the temperature, and it measures the voltage that the ion concentration gradient generates when it acts as a battery. At membrane voltages more positive to the reversal potential of the ion, passive ion movement is outward, whereas it is inward at a membrane potential (also known as transmembrane potential; Em) more negative to the Nernst potential of that channel.1,3
When multiple ions across a membrane are removed from their electrochemical equilibrium, each ion will tend to force the Em toward its own Eion. The contribution of each ion type to the overall Em at any given moment is determined by the instantaneous permeability of the plasma membrane to that ion. The larger the membrane conductance to a particular ion, the greater is the ability of that ion to bring the Em toward its own Eion. Hence, the Em is the average of the Eion of all the ions to which the membrane is permeable, weighed according to the membrane conductance of each individual ion relative to the total ionic conductance of the membrane.1,2
Transmembrane Potentials
All living cells, including cardiomyocytes, maintain a difference in the concentration of ions across their membranes. There is a slight excess of positive ions on the outside of the membrane and a slight excess of negative ions on the inside of the membrane, resulting in a difference in the electrical charge (i.e., voltage difference) across the cell membrane, called the membrane potential (Em). A membrane that exhibits an Em is said to be polarized.2
In nonexcitable cells, and in excitable cells in their baseline states (i.e., not conducting electrical signals), the Em is held at a relatively stable value, called the resting potential. All cells have a negative resting Em (i.e., the cytoplasm is electrically negative relative to the extracellular fluid), which arises from the interaction of ion channels and ion pumps embedded in the membrane that maintain different ion concentrations on the intracellular and extracellular sides of the membrane.2
When an ion channel opens, it allows ion flux across the membrane that generates an electrical current (I). This current affects the Em, depending on the membrane resistance (R), which refers to the ratio between the Em and electrical current, as shown in Ohm’s law: E = I × R or R = E/I. Resistance arises from the fact that the membrane impedes the movement of charges across it; hence, the cell membrane functions as a resistor. Conductance describes the ability of a membrane to allow the flux of charged ions in one direction across the membrane. The more permeable the membrane is to a particular ion, the greater is the conductance of the membrane to that ion. Membrane conductance (g) is the reciprocal of R: g = 1/R.1
Because the lipid bilayer of the cell membrane is very thin, accumulation of charged ions on one side gives rise to an electrical force (potential) that pulls oppositely charged particles toward the other side. Hence, the cell membrane functions as a capacitor. Although the absolute potential differences across the cell membrane are small, they give rise to enormous electrical potential gradients because they occur across a very thin surface. As a consequence, apparently small changes in Em can produce large changes in potential gradient and powerful forces that are able to induce molecular rearrangement in membrane proteins, such as those required for opening and closing ion channels embedded in the cell membrane. The capacitance of the membrane is generally fixed and unaffected by the molecules that are embedded in it. In contrast, membrane resistance is highly variable and depends on the conductance of ion channels embedded in the membrane.2,3
The sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (Cl−) ions are the major charge carriers, and their movement across the cell membrane creates a flow of current that generates excitation and signals in cardiac myocytes. The electrical current generated by the flux of an ion across the membrane is determined by the membrane conductance to that ion (gion) and the potential (voltage) difference across the membrane. The potential difference represents the potential at which there is no net ion flux (i.e., the Eion) and the actual Em: current = gion × (Em − Eion).1,4
By convention, an inward current increases the electropositivity within the cell (i.e., causes depolarization of the Em [to be less negative]) and can result from either the movement of positively charged ions (most commonly Na+ or Ca2+) into the cell or the efflux of negatively charged ions (e.g., Cl−) out of the cell. An outward current increases the electronegativity within the cell (i.e., causes hyperpolarization of the Em [to become more negative]) and can result from either the movement of anions into the cell or the efflux of cations (most commonly K+) out of the cell.3
Opening and closing of ion channels can induce a departure from the relatively static resting Em, called a depolarization if the interior voltage rises (becomes less negative) or a hyperpolarization if the interior voltage becomes more negative. The most important ion fluxes that depolarize or repolarize the membrane are passive (i.e., the ions move down their electrochemical gradient without requiring the expenditure of energy), occurring through transmembrane ion channels. In excitable cells, a sufficiently large depolarization can evoke a short-lasting all-or-none event called an action potential, in which the Em very rapidly undergoes specific and large dynamic voltage changes.1
Both resting Em and dynamic voltage changes such as the action potential are caused by specific changes in membrane permeabilities for Na+, K+, Ca2+, and Cl−, which, in turn, result from concerted changes in functional activity of various ion channels, ion transporters, and exchangers.3
The Cardiac Action Potential
During physiological electrical activity, the Em is a continuous function of time. The current flowing through the cell membrane is, at each instant, provided by multiple channels and transporters carrying charge in opposite directions because of their different ion selectivity. The algebraic summation of these contributions is referred to as net transmembrane current.1
The cardiac action potential reflects a balance between inward and outward currents. When a depolarizing stimulus (typically from an electric current from an adjacent cell) abruptly changes the Em of a resting cardiomyocyte to a critical value (the threshold level), the properties of the cell membrane and ion conductances change dramatically, precipitating a sequence of events involving the influx and efflux of multiple ions that together produce the action potential of the cell. In this fashion, an electrical stimulus is conducted from one cell to all the cells that are adjacent to it.2
Unlike skeletal muscle, cardiac muscle is electrically coupled so that the wave of depolarization propagates from one cell to the next, independent of neuronal input. The heart is activated by capacitive currents generated when a wave of depolarization approaches a region of the heart that is at its resting potential. Unlike ionic currents, which are generated by the flux of charged ions across the cell membrane, capacitive currents are generated by the movement of electrons toward and away from the surfaces of the membrane.2,3 The resulting decrease in positive charge at the outer side of the cell membrane reduces the negative charge on the intracellular surface of the membrane. These charge movements, which are carried by electrons, generate a capacitive current. When an excitatory stimulus causes the Em to become less negative and beyond a threshold level (approximately −65 mV for working atrial and ventricular cardiomyocytes), Na+ channels activate (open) and permit an inward Na+ current (INa), resulting in a rapid shift of the Em to a positive voltage range. This event triggers a series of successive opening and closure of selectively permeable ion channels. The direction and magnitude of passive ion movement (and the resulting current) at any given transmembrane voltage are determined by the ratio of the intracellular and extracellular concentrations and the reversal potential of that ion, with the net flux being larger when ions move from the more concentrated side.3
The threshold is the lowest Em at which opening of enough Na+ channels (or Ca2+ channels in the setting of nodal cells) is able to initiate the sequence of channel openings needed to generate a propagated action potential. Small (subthreshold) depolarizing stimuli depolarize the membrane in proportion to the strength of the stimulus and cause only local responses because they do not open enough Na+ channels to generate depolarizing currents large enough to activate nearby resting cells (i.e., insufficient to initiate a regenerative action potential). On the other hand, when the stimulus is sufficiently intense to reduce the Em to a threshold value, regenerative action potential results, whereby intracellular movement of Na+ depolarizes the membrane more, a process that increases conductance to Na+ more, which allows more Na+ to enter, and so on. In this fashion, the extent of subsequent depolarization becomes independent of the initial depolarizing stimulus, and more intense stimuli do not produce larger action potential responses; rather, an all-or-none response results.2
During the action potential, membrane voltages fluctuate in the range of −94 to +30 mV (Fig. 1-1). With physiological external K+, the reversal potential of K+ (EK) is approximately −94 mV, and passive K+ movement during an action potential is out of the cell. On the other hand, because the calculated reversal potential of a cardiac Ca2+ channel (ECa) is +64 mV, passive Ca2+ flux is into the cell.5
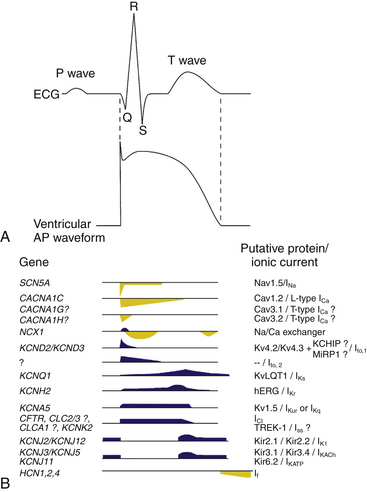
In normal atrial and ventricular myocytes and in His-Purkinje fibers, action potentials have very rapid upstrokes, mediated by the fast inward INa. These potentials are called fast response potentials. In contrast, action potentials in the normal sinus and atrioventricular (AV) nodal cells and many types of diseased tissues have very slow upstrokes, mediated by a slow inward, predominantly L-type voltage-gated Ca2+ current (ICaL), rather than by the fast inward INa (Fig. 1-2). These potentials have been termed slow response potentials.2,5
The Fast Response Action Potential
Phase 4: The Resting Membrane Potential
The Em of resting atrial and ventricular cardiomyocytes remains steady throughout diastole. The resting Em is caused by the differences in ionic concentrations across the membrane and the selective membrane permeability (conductance) to various ions. Large concentration gradients of Na+, K+, Ca2+, and Cl− across the cell membrane are maintained by the ion pumps and exchangers (Table 1-1).
TABLE 1-1 Intracellular and Extracellular Ion Concentrations and Equilibrium Potentials in Cardiomyocytes
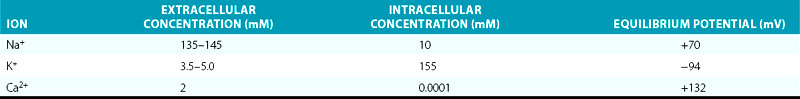
Under normal conditions, the resting membrane is most permeable to K+ and relatively impermeable to other ions. K+ has the largest resting membrane conductance (gK is 100 times greater than gNa) because of the abundance of open K+ channels at rest, whereas Na+ and Ca2+ channels are generally closed. Thus, K+ exerts the largest influence on the resting Em. As a consequence, the resulting Em is almost always close to the K+ reversal potential (Em approximates EK). The actual resting Em is slightly less negative than EK because the cell membrane is slightly permeable to other ions.2
The inwardly rectifying K+ (Kir) channels underlie an outward K+ current (IK1) responsible for maintaining the resting potential near the EK in atrial, His-Purkinje, and ventricular cells, under normal conditions.6,7 Kir channels preferentially allow currents of K+ ions to flow into the cell with a strongly voltage-dependent decline of K+ efflux (i.e., reduction of outward current) on membrane depolarization. As such, IK1 is a strong rectifier that passes K+ currents over a limited range of Em (see Chap. 2 for detailed discussion on the concept of rectification); at a negative Em, IK1 conductance is much larger than that of any other current, thus, it clamps the resting Em close to the reversal potential for K+ (EK). IK1 density is much higher in ventricular than in atrial myocytes, a feature that protects the ventricular cell from pacemaker activity. By contrast, IK1 is almost absent in sinus and AV nodal cells, thus allowing for relatively more depolarized resting diastolic potentials compared with atrial and ventricular myocytes (Table 1-2).2

A unique property of Kir currents is the unusual dependence of rectification on extracellular K+ concentration. Specifically, on increase in extracellular K+, the IK1 current-voltage relationship shifts nearly in parallel with the EK and leads to a crossover phenomenon. One important consequence of such behavior is that at potentials positive to the crossover, K+ conductance increases rather than decreases, against an expectation based on a reduced driving force for K+ ions as a result of elevated extracellular K+ concentration.6–8
Although Ca2+ does not contribute directly to the resting Em (since the voltage-activated Ca2+ channels are closed at the hyperpolarized resting Em), changes in intracellular free Ca2+ concentration can affect other membrane conductance values. Increases in intracellular Ca2+ levels can stimulate the Na+-Ca2+ exchanger (INa-Ca), which exchanges three Na+ ions for one Ca2+ ion; the direction depends on the Na+ and Ca2+ concentrations on the two sides of the membrane and the Em difference. At resting Em and during a spontaneous sarcoplasmic reticulum Ca2+ release event, this exchanger would generate a net Na+ influx, possibly causing transient membrane depolarizations.3
Phase 0: The Upstroke—Rapid Depolarization
On excitation of the cardiomyocyte by electrical stimuli from adjacent cells, its resting Em (approximately −85 mV) depolarizes, leading to opening (activation) of Na+ channels from its resting (closed) state and enabling a large and rapid influx of Na+ ions (inward INa) into the cell down their electrochemical gradient. As a consequence of increased Na+ conductance, the excited membrane no longer behaves like a K+ electrode (i.e., exclusively permeable to K+), but more closely approximates an Na+ electrode, and the membrane potential moves toward the Na+ Eion (ENa; see Table 1-1). Once an excitatory stimulus depolarizes the Em beyond the threshold for activation of Na+ channels (approximately −65 mV), the activated INa is regenerative and no longer depends on the initial depolarizing stimulus; the influx of Na+ ions further depolarizes the membrane and thereby increases conductance to Na+ more, which allows more Na+ to enter.2,9
Normally, activation of Na+ channels is transient; fast inactivation (closing of the pore) starts simultaneously with activation, but because inactivation is slightly delayed relative to activation, the channels remain transiently (less than 1 millisecond) open to conduct INa during phase 0 of the action potential before it closes. Additionally, the influx of Na+ into the cell increases the positive intracellular charges and reduces the driving force for Na+. When the ENa is reached, no further Na+ ions enter the cell.9
The rate at which depolarization occurs during phase 0, that is, the maximum rate of change of voltage over time, is indicated by the expression dV/dtmax, which is a reasonable approximation of the rate and magnitude of Na+ entry into the cell and a determinant of conduction velocity for the propagated action potential.2
The threshold for activation of ICaL is approximately −30 to −40 mV. Although ICaL is normally activated during phase 0 by the regenerative depolarization caused by the fast INa, ICaL is much smaller than the peak INa. The amplitude of ICaL is not maximal near the action potential peak because of the time-dependent nature of ICaL activation, as well as the low driving force (Em − ECa) for ICaL. Therefore, ICaL contributes little to the action potential until the fast INa is inactivated, after completion of phase 0. As a result, ICaL affects mainly the plateau of action potentials recorded in atrial and ventricular muscle and His-Purkinje fibers. On the other hand, ICaL can play a prominent role in the upstroke of slow response action potentials in partially depolarized cells in which the fast Na+ channels have been inactivated.2
Phase 1: Early Repolarization
Phase 0 is followed by phase 1 (early repolarization) during which the membrane repolarizes rapidly and transiently to almost 0 mV (early notch), partly because of the inactivation of INa and concomitant activation of several outward currents. The transient outward K+ current (Ito) is mainly responsible for phase 1 of the action potential. Ito rapidly activates (with time constants less than 10 milliseconds) by depolarization and then rapidly inactivates (25 to 80 milliseconds for the fast component of Ito [Ito,f], and 80 to 200 milliseconds for the slow component of Ito [Ito,s]). The influx of K+ ions via Ito channels partially repolarizes the membrane, thus shaping the rapid (phase 1) repolarization of the action potential and setting the height of the initial plateau (phase 2; see Fig. 1-1). Additionally, an Na+ outward current through the Na+-Ca2+ exchanger operating in reverse mode likely contributes to early repolarization.3,6,10
Phase 2: The Plateau
Phase 2 (plateau) represents a delicate balance between the depolarizing inward currents (ICaL and a small residual component of inward INaL) and the repolarizing outward currents (ultrarapidly [IKur], rapidly [IKr], and slowly [IKs] activating delayed outward rectifying currents; see Fig. 1-1). Phase 2 is the longest phase of the action potential, lasting tens (atrium) to hundreds of milliseconds (His-Purkinje system and ventricle). The plateau phase is unique among excitable cells and marks the phase of Ca2+ entry into the cell. It is the phase that most clearly distinguishes the cardiac action potential from the brief action potentials of skeletal muscle and nerve.2
ICaL is activated by membrane depolarization, is largely responsible for the action potential plateau, and is a major determinant of the duration of the plateau phase. ICaL also links membrane depolarization to myocardial contraction. L-type Ca2+ channels activate on membrane depolarization to potentials positive to –40 mV. ICaL peaks at an Em of 0 to +10 mV and tends to reverse at +60 to +70 mV, following a bell-shaped current-voltage relationship.3
Na+ channels also make a contribution, although minor, to the plateau phase. After phase 0 of the action potential, some Na+ channels occasionally fail to inactivate or exhibit prolonged opening or reopening repetitively for hundreds of milliseconds after variable and prolonged latencies, resulting in a small inward INa (less than 1% of the peak INa). This persistent or “late” INa (INaL), along with ICaL, helps maintain the action potential plateau.3,11,12
IKr and IKs are activated at depolarized potentials. IKr activates relatively fast (in the order of tens of milliseconds) on membrane depolarization, thus allowing outward diffusion of K+ ions in accordance with its electrochemical gradient, but voltage-dependent inactivation thereafter is very fast. Hence, only limited numbers of channels remain in the open state, whereas a considerable fraction resides in the nonconducting inactivated state. The fast voltage-dependent inactivation limits outward current through the channel at positive voltages and thus helps maintain the action potential plateau phase that controls contraction and prevents premature excitation. However, as the voltage becomes less positive at the end of the plateau phase of repolarization, the channels recover rapidly from inactivation; this process leads to a progressive increase in IKr amplitudes during action potential phases 2 and 3, with maximal outward current occurring before the final rapid declining phase of the action potential.6,12,13
IKs, which is approximately 10 times larger than IKr, also contributes to the plateau phase. IKs activates in response to membrane depolarization to potentials greater than –30 mV and gradually increases during the plateau phase because its time course of activation is extremely slow, slower than any other known K+ current, and steady-state amplitude is achieved only with extremely long membrane depolarization. Hence, the contribution of IKs to the net repolarizing current is greatest late in the plateau phase, particularly during action potentials of long duration. Importantly, although IKs activates slowly compared with action potential duration, it is also slowly inactivated. As heart rate increases, IKs increases because channel deactivation is slow and incomplete during the shortened diastole. This allows IKs channels to accumulate in the open state during rapid successive depolarizations and mediate the faster rate of repolarization. Hence, IKs plays an important role in determining the rate-dependent shortening of the cardiac action potential.6,12–14
IKur is detected only in human atria but not in the ventricles, so that it is the predominant delayed rectifier current responsible for human atrial repolarization.3,12 The Na+-Ca2+ exchanger operating in forward mode and the Na+-K+ pump provide minor current components during phase 2.
Importantly, during the plateau phase, membrane conductance to all ions falls to rather low values. Thus, less change in current is required near plateau levels than near resting potential levels to produce the same changes in Em. In particular, K+ conductance falls during the plateau phase as a result of inward rectification of IKr and IK1 (that is, voltage-dependent decline of K+ efflux and hence reduction of outward current) on membrane depolarization, in spite of the large electrochemical driving force on K+ ions during the positive phase of the action potential (phases 0, 1, and 2). This property allows membrane depolarization following Na+ channel activation, slows membrane repolarization, and helps maintain a more prolonged cardiac action potential. This also confers energetic efficiency in the generation of the action potential.6,7,14
Phase 3: Final Rapid Repolarization
Phase 3 is the phase of rapid repolarization that restores the Em to its resting value. Phase 3 is mediated by the increasing conductance of the delayed outward rectifying currents (IKr and IKs), the inwardly rectifying K+ currents (IK1 and acetylcholine-activated K+ current [IKACh]), and time-dependent inactivation of ICaL (see Fig. 1-1). Final repolarization during phase 3 results from K+ efflux through the IK1 channels, which open at potentials negative to –20 mV.6,12–14
Phase 4: Restoration of Resting Membrane Potential
Reduction of cytosolic Ca2+ concentration during diastole is achieved by the reuptake Ca2+ by the sarcoplasmic reticulum via activation of the sarco/endoplasmic reticulum Ca2+-ATPase Ca2+ pump (SERCA), in addition to extrusion across the sarcolemma via the Na+-Ca2+ exchanger. In the human heart under resting conditions, the time required for cardiac myocyte depolarization, contraction, relaxation, and recovery is approximately 600 milliseconds.5,15,16
Regional Heterogeneity of the Action Potential
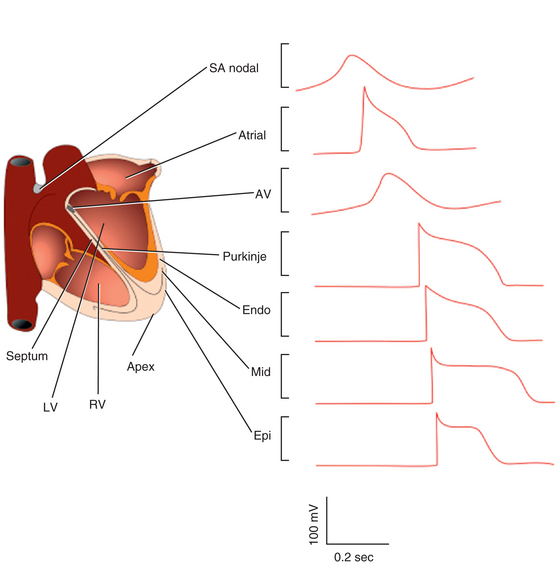
Substantial differences in the expression levels of ion channels underlie the substantial heterogeneity in action potential duration and configuration between cardiomyocytes located in different cardiac regions. The characteristics of the action potential differ in atrial versus ventricular myocardium, as well as across the ventricular myocardial wall from endocardium, midmyocardium (putative M cells), to epicardium (see Fig. 1-2).3,12
The density of Ito varies across the myocardial wall and in different regions of the heart. The markedly higher densities of Ito, together with the expression of IKur, accelerate the early phase of repolarization and lead to lower plateau potentials and shorter action potentials in atrial cells. In human ventricles, Ito densities are much higher in the epicardium and midmyocardium than in the endocardium. These regional differences are responsible for the shorter duration and the prominent phase 1 notch and the spike and dome morphology of epicardial and midmyocardial action potentials compared with endocardium. A prominent Ito-mediated action potential notch in ventricular epicardium but not endocardium produces a transmural voltage gradient during early ventricular repolarization that registers as a J wave or J point elevation on the ECG.17 Ito densities are also reportedly higher in right than in left (midmyocardial and epicardial) ventricular myocytes, consistent with the more pronounced spike and dome morphology of right, compared with left, ventricular action potentials.3,8,10,12
Experimental studies in wedge preparations strongly suggest the presence of a subpopulation of cells in the midmyocardium (referred to as the M cells) that exhibits distinct electrophysiological (EP) properties, although the presence of M cells has not been consistently confirmed by intact heart experiments.18–21 The putative midmyocardial cells appear to have the longest action potential duration across the myocardial wall, largely attributed to their weaker IKs current but stronger late INa and Na+-Ca2+ exchanger currents. Hence, the M cells have been proposed to underlie the EP basis for transmural ventricular dispersion of repolarization and the T wave on the surface ECG, with the peak of the T wave (in wedge preparations) coinciding with the end of epicardial repolarization and the end of the T wave coinciding with the end of repolarization of the M cells. Although the role of M cells under physiological conditions remains controversial, these cells appear to have a significant role in arrhythmogenesis under a variety of pathological conditions, such as the long QT and Brugada syndromes, secondary to exaggeration of transmural repolarization gradients.18,19
As noted, IKur is detected only in human atria and not in the ventricles, so that it is the predominant delayed rectifier current responsible for human atrial repolarization and is a basis for the much shorter duration of the action potential in the atrium (see Table 1-2).3,12
IK1 density is much higher in ventricular than in atrial myocytes, and this explains the steep repolarization phase in the ventricles (where the more abundant IK1 plays a larger role in accelerating the terminal portion of repolarization) and the more shallow phase in the atria. The higher IK1 channel expression in the ventricle protects the ventricular cell from pacemaker activity.6,7
Changes in expression levels or gating properties of ion channels in pathological conditions can aggravate the regional heterogeneities in action potential duration and configuration that can be arrhythmogenic.3,12
The Slow Response Action Potential
In normal atrial and ventricular myocytes and in the His-Purkinje fibers, action potentials have very rapid upstrokes mediated by the fast inward INa. These potentials are called fast response potentials. In contrast, action potentials in the normal sinus and AV nodal cells and many types of diseased tissue have very slow upstrokes, mediated predominantly by the slow inward ICaL, rather than by the fast inward INa (see Fig. 1-2). These potentials have been termed slow response potentials.5
As noted, the action potentials of pacemaker cells in the sinus and AV nodes are significantly different from those in working atrial and ventricular myocardium. The slow response action potentials are characterized by a more depolarized Em at the onset of phase 4 (−50 to −65 mV), slow diastolic depolarization during phase 4, reduced action potential amplitude, and a much slower rate of depolarization in phase 0 than that in the working myocardial cells, thus resulting in slow conduction velocity of the cardiac impulse in the nodal regions (see Table 1-2). Cells in the His-Purkinje system can also exhibit phase 4 depolarization under special circumstances.6,7
Phase 4: Diastolic Depolarization
In contrast to working atrial and ventricular myocytes and fibers in the His-Purkinje system, which maintain a steady diastolic Em level of approximately –85 mV, sinus and AV nodal excitable cells exhibit a spontaneous, slow, progressive decline in the Em during diastole (spontaneous diastolic depolarization or phase 4 depolarization) that underlies normal automaticity and pacemaking function. Once this spontaneous depolarization reaches threshold (approximately −40 mV), a new action potential is generated.2,22,23
Ic is a hyperpolarization-activated inward current (often referred to as the funny current because, unlike the majority of voltage-sensitive currents, it is activated by hyperpolarization rather than depolarization) that is carried largely by Na+ and, to a lesser extent, K+ ions. The If channels are deactivated during the action potential upstroke and the initial plateau phase of repolarization. However, they begin to activate at the end of the action potential as repolarization brings the Em to levels more negative than approximately −40 to −50 mV, and they are fully activated at approximately −100 mV. Once activated, If depolarizes the membrane to a level where the Ca2+ current activates to initiate an action potential.3 At the end of the repolarization phase of the action potential, because If activation occurs in the background of a decaying outward time-dependent K+ current, the current flow quickly shifts from outward to inward, thus giving rise to a sudden reversal of voltage change (from repolarizing to depolarizing) at the maximum diastolic potential.22–26
The sarcoplasmic reticulum generates spontaneous, rhythmic, local Ca2+ releases (via ryanodine receptors, RyR2) beneath the surface of the membrane, in the absence of Ca2+ overload. Activation of the local oscillatory Ca2+ releases is independent of membrane depolarization and is driven by a high level of basal state phosphorylation of Ca2+ cycling proteins. Critically timed Ca2+ releases occur during the later phase of diastolic depolarization in the form of multiple locally propagating wavelets beneath the cell membrane, and they activate the forward mode of the Na+-Ca2+ exchanger, thus resulting in an inward membrane current (INa-Ca). The INa-Ca causes the late diastolic depolarization to increase exponentially, driving the Em to the threshold to activate a sufficient number of L-type Ca2+ channels and leading to generation of the upstroke of the next action potential. Although regulated by the Em and submembrane Ca2+, the Na+-Ca2+ exchanger does not have time-dependent gating, as do ion channels, but generates an inward current almost instantaneously when submembrane Ca2+ concentration increases.24,25,27,28
Ca2+ influx via ICaL during the action potential triggers Ca2+-induced Ca2+ release from the sarcoplasmic reticulum. The resulting global sarcoplasmic reticular Ca2+ depletion synchronizes the sarcoplasmic reticulum throughout the cell in a Ca2+-depleted state. Refilling of the sarcoplasmic reticulum ensures that the threshold of Ca2+ load required for spontaneous release is achieved at about the time when RyR2 inactivation is removed following prior activation; then the spontaneous local Ca2+ releases occur, thus activating INa-Ca to ignite the next action potential.5,24,28
Such rhythmic, spontaneous intracellular Ca2+ cycling has been referred to as the intracellular Ca2+ clock. Phosphorylation-dependent gradation of speed at which Ca2+ clock cycles is the essential regulatory mechanism of normal pacemaker rate and rhythm. The robust regulation of pacemaker function is ensured by tight integration of the Ca2+ clock and the classic sarcolemmal ion channel clock (formed by voltage-dependent membrane ion channels) to form the overall pacemaker clock. The action potential shape and ion fluxes are tuned by membrane clocks to sustain operation of the Ca2+ clock, which produces timely and powerful ignition of the membrane clocks to effect action potentials.24,25,28
There is some degree of uncertainty about the relative role of If versus that of intracellular Ca2+ cycling in controlling the normal pacemaker cell automaticity. Furthermore, the interactions between the membrane ion channel clock and the intracellular Ca2+ clock and cellular mechanisms underlying this internal Ca2+ clock are not completely elucidated. A further debate has arisen around their individual (or mutual) relevance in mediating the positive and negative chronotropic effects of neurotransmitters. Nevertheless, these interactions are of fundamental importance for understanding the integration of pacemaker mechanisms at the cellular level.24,25,27
Phase 0: The Upstroke—Slow Depolarization
IK1 is almost absent in sinus and AV nodal cells, thus allowing for relatively more depolarized resting diastolic potentials (−50 to −65 mV) compared with atrial and ventricular myocytes and facilitating diastolic depolarization mediated by the inward currents (e.g., If). At the depolarized level of the maximum diastolic potential of pacemaker cells, most Na+ channels are inactivated and unavailable for phase 0 depolarization. Consequently, action potential upstroke is mainly achieved by ICaL.6,7
L-type Ca2+ channels activate on depolarization to potentials positive to –40 mV, and ICaL peaks at 0 to +10 mV. The peak amplitude ICaL is less than 10% that of INa, and the time required for activation and inactivation of ICaL is approximately an order of magnitude slower than that for INa. As a consequence, the rate of depolarization in phase 0 (dV/dt) is much slower and the peak amplitude of the action potential is less than that in the working myocardial cells.5,15,29
Excitability
Excitability of a cardiac cell describes the ease with which the cell responds to a stimulus with a regenerative action potential. A certain minimum charge has to be applied to the cell membrane to elicit a regenerative action potential (i.e., the stimulus should be sufficiently intense to reduce the Em to the threshold value); excitability is inversely related to the charge required for excitation.3
Excitability of a cardiac cell depends on the passive and active properties of the cell membrane. The passive properties include the membrane resistance and capacitance and the intercellular resistance. The more negative the Em, the more Na+ channels are available for activation, the greater the influx of Na+ into the cell during phase 0, and the greater the conduction velocity. In contrast, membrane depolarization to approximately −60 to −70 mV can inactivate half the Na+ channels, and depolarization to −50 mV or less can inactivate all the Na+ channels, thereby rendering Na+ channels unavailable for mediating an action potential upstroke and reducing tissue excitability.5
On the other hand, supernormal excitability can be observed during a brief period at the end of phase 3 of the action potential. During the supernormal period, excitation is possible in response to an otherwise subthreshold stimulus; that same stimulus fails to elicit a response earlier or later than the supernormal period. Two factors are responsible for supernormality: the availability of fast Na+ channels and the proximity of the Em to threshold potential. During the supernormal phase of excitability, the cell has recovered enough to respond to a stimulus (i.e., an adequate number of Na+ channels is available for activation). At the same time, because the Em is still reduced, it requires only a little additional depolarization to bring the fiber to threshold; thus, a smaller stimulus than is normally required elicits an action potential.30,31
Reduced membrane excitability occurs in certain physiological and pathophysiological conditions. Genetic mutations that result in loss of Na+ channel function, Na+ channel blockade with class I antiarrhythmic drugs, and acute myocardial ischemia can cause reduced membrane excitability.32
Refractoriness
During a cardiac cycle, once an action potential is initiated, the cardiomyocyte becomes unexcitable to stimulation (i.e., unable to initiate another action potential in response to a stimulus of threshold intensity) for some duration of time (which is slightly shorter than the “true” action potential duration) until its membrane has repolarized to a certain level. This period of refractoriness to stimulation is physiologically necessary for the mechanical function of the heart; it allows only gradual recovery of excitability, thus permitting relaxation of cardiac muscle before subsequent activation. Additionally, the refractory period acts as a protective mechanism by preventing multiple, compounded action potentials from occurring (i.e., it limits the frequency of depolarization and heart rate). Therefore, this property is a determinant of susceptibility to arrhythmias.33
The refractory period is determined, in part, by the action potential duration and the Em, and the degree of refractoriness primarily reflects the number of Na+ channels that have recovered from their inactive state. With repolarization, the Na+ channel normally recovers rapidly from inactivation (within 10 milliseconds) and is ready to open again. The ERP extends from phase 0 to approximately −60 mV during phase 3 of the action potential, a time during which it is impossible for the myocardium to respond with a propagated action potential, or even to a strong stimulus. The relative refractory period extends from approximately −60 mV during phase 3 to the end of phase 3 of the action potential. During this period, a depressed response is possible to a strong stimulus. Therefore, when premature stimulation occurs during the relative refractory period (i.e., before full recovery and at less negative potentials of the cell membrane), a portion of Na+ channels will still be refractory and unavailable for activation. Consequently, the INa and phase 0 of the next action potential will be reduced, and conduction of the premature stimulus will be slowed.3,32
In pacemaking tissues, INa is predominantly absent, and excitability is mediated by the activation of ICaL. After inactivation, the transition of Ca2+ channels from the inactivated to the closed resting state (i.e., recovery from inactivation) is relatively slow. The time constant for recovery from inactivation depends on both the Em and the intracellular Ca2+ concentration (typically 100 to 200 milliseconds at −80 mV and low intracellular Ca2+ concentration). This means that ICaL must recover from inactivation between action potentials. As a result, excitability in pacemaking cells may not be recovered by the end of phase 3 of the action potential and full restoration of maximum diastolic potential, because L-type Ca2+ channels require longer time to recovery from inactivation to be able to mediate the upstroke of a new action potential. In other words, sinus and AV nodal cells remain refractory for a time interval that is longer than the time it takes for full voltage repolarization to occur, a phenomenon termed postrepolarization refractoriness.5,29 This can also occur during some disease states such as myocardial infarction.
Conduction
Conduction velocity refers to the speed of propagation of the action potential through cardiac tissue and is determined by source-sink relationships, which reflect the interplay between the active membrane properties of cardiac cells (i.e., electrical excitability or refractoriness of the source generating the action potential, or both) and the passive properties determined by cell-to-cell coupling and tissue geometry (sink).3,34
During action potential propagation, an excited cell serves as a source of electrical charge for depolarizing neighboring unexcited cells. The requirements of adjacent resting cells to reach the threshold Em constitute an electrical sink (load) for the excited cell. For propagation to succeed, the excited cell must provide sufficient charge to bring the Em at a site in the sink from its diastolic value to the threshold. Once threshold is reached and action potential is generated, the load on the excited cell is removed, and the newly excited cell switches from being a sink to being a source for the downstream tissue, thus perpetuating the process of action potential propagation. Action potentials are “regenerative” because they can be conducted over large distances without attenuation.
An action potential traveling down a cardiac muscle fiber is propagated by local circuit currents, much as it does in nerve and skeletal muscle. Conduction velocity along the cardiac fiber is directly related to the action potential amplitude (i.e., the voltage difference between the fully depolarized and the fully polarized regions) and the rate of change of potential (i.e., the rate of rise of phase 0 of the action potential [dV/dt]). These factors depend on the amplitude of INa, which, in turn, is directly related to the Em at the time of stimulation, the availability of Na+ channels for stimulation, and the size of the Na+ electrochemical potential gradient across the cell membrane. A reduction in INa, leading to a reduction in the rate or amplitude of depolarization during phase 0 of the action potential, can decrease axial current flow (and therefore capacitive current) and slow conduction or produce conduction block. Tissues with high concentration of Na+ channels, such as Purkinje fibers, which contain up to 1 million Na+ channels per cell, have a large, fast inward current. The large INa spreads quickly within and between cells to support rapid conduction (approximately 4 m/sec). In contrast, at a less negative resting Em, Na+ channel availability is limited because of the inactivation of a portion of the channels; hence, INa amplitude is attenuated, and the upstroke velocity is slowed. With progressive reduction of excitability, less Na+ source current is generated, and conduction velocity and the safety factor decrease monotonically. When the safety factor falls to less than 1, conduction can no longer be sustained, and failure (conduction block) occurs. Action potentials with reduced upstroke velocity resulting from partial inactivation of Na+ channels are called depressed fast responses.35
Excitation-Contraction Coupling
Excitation-contraction coupling describes the physiological process by which electrical stimulation of the cardiomyocytes (the action potential) results in a mechanical response (muscle contraction). The contraction of a cardiac myocyte is governed primarily by intracellular Ca2+ concentration (Fig. 1-3). Ca2+ enters the cell during the plateau phase of the action potential through the L-type Ca2+ channels that line areas of specialized invaginations known as transverse (T) tubules. Although the rise in intracellular Ca2+ is small and not sufficient to induce contraction, the small amount of Ca2+ entering the cell via ICaL triggers a massive release of Ca2+ from the sarcoplasmic reticulum (the major store for Ca2+) into the cytosol by opening the RyR2 channels (present in the membrane of the sarcoplasmic reticulum) in a process known as Ca2+-induced Ca2+ release (CICR). Approximately 75% of Ca2+ present in the cytoplasm during contraction is released from the sarcoplasmic reticulum.
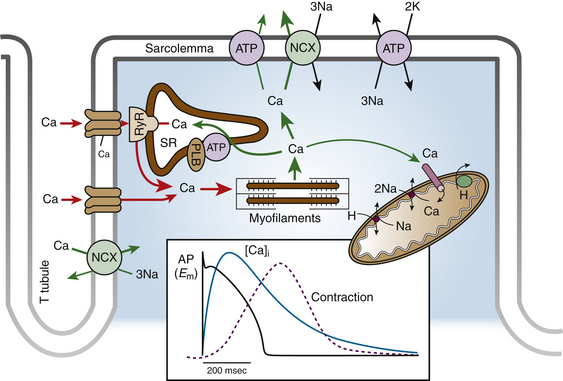
Each junction between the sarcolemma (T tubule) and sarcoplasmic reticulum, where 10 to 25 L-type Ca2+ channels and 100 to 200 RyRs are clustered, constitutes a local Ca2+ signaling complex, or a couplon. When a Ca2+ channel opens, local cytosolic Ca2+ concentration rises in less than 1 millisecond in the junctional cleft to 10 to 20 µM, and this activates RyR2 to release Ca2+ from the sarcoplasmic reticulum. The close proximity of the RyR2 to the T tubule enables each L-type Ca2+ channel to activate 4 to 6 RyR2s and generate a Ca2+ spark. Ca2+ influx via ICaL simultaneously activates approximately 10,000 to 20,000 couplons in each ventricular cardiomyocyte with every action potential.36–38
CICR typically induces release of only approximately 50% to 60% of sarcoplasmic reticulum Ca2+ content. RyR2 channels are inactivated by a feedback mechanism from the rising Ca2+ concentration in the cleft and, more importantly, by the decline of sarcoplasmic reticulum Ca2+ content (a process referred to as luminal Ca2+-dependent deactivation). This process ensures that the sarcoplasmic reticulum never is fully depleted of Ca2+ physiologically.38,39
Relaxation requires the removal of Ca2+ from the cytosol, a process vital for enabling ventricular chamber relaxation and filling, as well as for prevention of arrhythmias. At the end of phase 2 of the action potential, Ca2+ entry into the cell slows, and most of the surplus Ca2+ in the cytosol is resequestered into the sarcoplasmic reticulum by the SERCA, the activity of which is controlled by the phosphoprotein phospholamban. Additionally, some of the Ca2+ is extruded from the cell by the sarcolemmal Na+-Ca2+ exchanger and, to a minor degree, the cell membrane Ca2+ ATPase, to balance the Ca2+ that enters with ICaL. As the cytosolic Ca2+ concentration drops, Ca2+ dissociates rapidly from the myofilaments, thus inducing a conformational change in the troponin complex leading to troponin I inhibition of the actin binding site. At the end of the cycle, a new ATP binds to the myosin head and displaces the adenosine diphosphate, and the initial sarcomere length is restored, thus ending contraction. Recurring Ca2+ release-uptake cycles provide the basis for periodic elevations of cytosolic Ca2+ concentration and contractions of myocytes, hence for the orderly beating of the heart.38,39
1. Zaza A. Control of the cardiac action potential: the role of repolarization dynamics. J Mol Cell Cardiol. 2010;48:106-111.
2. Katz A.M. The cardiac action potential. In: Katz A.M., editor. Physiology of the heart. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2011:369-400.
3. Grant A.O. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2:185-194.
4. Katz A.M. Cardiac ion channels. In: Katz A.M., editor. Physiology of the heart. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2011:343-368.
5. Bodi I., Mikala G., Koch S.E., et al. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306-3317.
6. Tamargo J., Caballero R., Gomez R., et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9-33.
7. Anumonwo J.M., Lopatin A.N. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010;48:45-54.
8. Oudit G.Y., Backx P.H. Voltage-regulated potassium channels. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:29-42.
9. Andavan G.S., Lemmens-Gruber R. Voltage-gated sodium channels: mutations, channelopathies and targets. Curr Med Chem. 2011;18:377-397.
10. Niwa N., Nerbonne J.M. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12-25.
11. Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2-11.
12. Amin A.S., Tan H.L., Wilde A.A. Cardiac ion channels in health and disease. Heart Rhythm. 2010;7:117-126.
13. Charpentier F., Merot J., Loussouarn G., Baro I. Delayed rectifier K(+) currents and cardiac repolarization. J Mol Cell Cardiol. 2010;48:37-44.
14. Tristani-Firouzi M., Sanguinetti M.C. Structural determinants and biophysical properties of HERG and KCNQ1 channel gating. J Mol Cell Cardiol. 2003;35:27-35.
15. Benitah J.P., Alvarez J.L., Gomez A.M. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26-36.
16. van der Heyden M.A., Wijnhoven T.J., Opthof T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc Res. 2005;65:28-39.
17. Antzelevitch C., Yan G.X. J wave syndromes. Heart Rhythm. 2010;7:549-558.
18. Wilson L.D., Jennings M.M., Rosenbaum D.S. Point: M cells are present in the ventricular myocardium. Heart Rhythm. 2011;8:930-933.
19. Glukhov A.V., Fedorov V.V., Lou Q., et al. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981-991.
20. Janse M.J., Coronel R., Opthof T. Counterpoint: M cells do not have a functional role in the ventricular myocardium of the intact heart. Heart Rhythm. 2011;8:934-937.
21. Opthof T., Coronel R., Janse M.J. Is there a significant transmural gradient in repolarization time in the intact heart? Repolarization gradients in the intact heart. Circ Arrhythm Electrophysiol. 2009;2:89-96.
22. Mangoni M.E., Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008;88:919-982.
23. Baruscotti M., Barbuti A., Bucchi A. The cardiac pacemaker current. J Mol Cell Cardiol. 2010;48:55-64.
24. Lakatta E.G. A paradigm shift for the heart’s pacemaker. Heart Rhythm. 2010;7:559-564.
25. Venetucci L.A., Trafford A.W., O’Neill S.C., Eisner D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285-292.
26. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434-446.
27. Maltsev V.A., Lakatta E.G. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res. 2008;77:274-284.
28. Lakatta E.G., Maltsev V. A new functional paradigm for the heart’s pacemaker: mutual entrainment of intracellular calcium clocks and surface membrane ion channel clocks. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:235-247.
29. Ono K., Iijima T. Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol. 2010;48:65-70.
30. Kilborn M.F. Electrocardiographic manifestations of supernormal conduction, concealed conduction, and exit block. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 4. Philadelphia: Saunders; 2004:733-738.
31. Fisch C., Knoebel S.B. Supernormal conduction and excitability. In: Fisch C., Knoebel S.B., editors. Electrocardiography of clinical arrhythmias. Armonk, NY: Futura; 2000:237-252.
32. Kleber A.G., Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431-488.
33. Hund T.J., Rudy Y. Determinants of excitability in cardiac myocytes: mechanistic investigation of memory effect. Biophys J. 2000;79:3095-3104.
34. Noorman M., van der Heyden M.A., van Veen T.A., et al. Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23-31.
35. Amin A.S., Asghari-Roodsari A., Tan H.L. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223-237.
36. Kushnir A., Marks A.R. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol. 2010;59:1-30.
37. Mohamed U., Napolitano C., Priori S.G. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007;18:791-797.
38. Bers D.M. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23-49.
39. Gyorke S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2009;6:123-129.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 1 Molecular Mechanisms of Cardiac Electrical Activity
Ionic Equilibrium
The lipid bilayer of the cell membrane is hydrophobic and impermeable to water-soluble substances such as ions. Hence, for the hydrophilic ions to be able to cross the membrane, they need hydrophilic paths that span the membrane (i.e., pores), which are provided by transmembrane proteins called ion channels. Once a hydrophilic pore is available, ions move passively across the membrane driven by two forces: the electrical gradient (voltage difference) and the chemical gradient (concentration difference). The chemical gradient forces the ions to move from a compartment of a higher concentration to one of lower concentration. The electrical gradient forces ions to move in the direction of their inverse sign (i.e., cations [positively charged ions] move toward a negatively charged compartment, whereas anions [negatively charged ions] move toward a positively charged compartment). Because the chemical and electrical gradients can oppose each other, the direction of net ion movement will depend on the relative contributions of chemical gradient and electrical potential (i.e., the net electrochemical gradient), so that ions tend to move spontaneously from a higher to a lower electrochemical potential.1–3
The movement of an ion down its chemical gradient in one direction across the cell membrane results in build-up of excess charge carried by the ion on one side of the membrane, which generates an electrical gradient that impedes continuing ionic movement in the same direction. When the driving force of the electrical gradient across the membrane becomes equal and opposite to the force generated by the chemical gradient, the ion is said to be in electrochemical equilibrium, and the net transmembrane flux (or current) of that particular ion is zero. In this setting, the electrical potential is called the equilibrium potential (Eion) (reversal potential or Nernst potential) of that individual ion. The Eion for a given ion depends on its concentration on either side of the membrane and the temperature, and it measures the voltage that the ion concentration gradient generates when it acts as a battery. At membrane voltages more positive to the reversal potential of the ion, passive ion movement is outward, whereas it is inward at a membrane potential (also known as transmembrane potential; Em) more negative to the Nernst potential of that channel.1,3
When multiple ions across a membrane are removed from their electrochemical equilibrium, each ion will tend to force the Em toward its own Eion. The contribution of each ion type to the overall Em at any given moment is determined by the instantaneous permeability of the plasma membrane to that ion. The larger the membrane conductance to a particular ion, the greater is the ability of that ion to bring the Em toward its own Eion. Hence, the Em is the average of the Eion of all the ions to which the membrane is permeable, weighed according to the membrane conductance of each individual ion relative to the total ionic conductance of the membrane.1,2
Transmembrane Potentials
All living cells, including cardiomyocytes, maintain a difference in the concentration of ions across their membranes. There is a slight excess of positive ions on the outside of the membrane and a slight excess of negative ions on the inside of the membrane, resulting in a difference in the electrical charge (i.e., voltage difference) across the cell membrane, called the membrane potential (Em). A membrane that exhibits an Em is said to be polarized.2
In nonexcitable cells, and in excitable cells in their baseline states (i.e., not conducting electrical signals), the Em is held at a relatively stable value, called the resting potential. All cells have a negative resting Em (i.e., the cytoplasm is electrically negative relative to the extracellular fluid), which arises from the interaction of ion channels and ion pumps embedded in the membrane that maintain different ion concentrations on the intracellular and extracellular sides of the membrane.2
When an ion channel opens, it allows ion flux across the membrane that generates an electrical current (I). This current affects the Em, depending on the membrane resistance (R), which refers to the ratio between the Em and electrical current, as shown in Ohm’s law: E = I × R or R = E/I. Resistance arises from the fact that the membrane impedes the movement of charges across it; hence, the cell membrane functions as a resistor. Conductance describes the ability of a membrane to allow the flux of charged ions in one direction across the membrane. The more permeable the membrane is to a particular ion, the greater is the conductance of the membrane to that ion. Membrane conductance (g) is the reciprocal of R: g = 1/R.1
Because the lipid bilayer of the cell membrane is very thin, accumulation of charged ions on one side gives rise to an electrical force (potential) that pulls oppositely charged particles toward the other side. Hence, the cell membrane functions as a capacitor. Although the absolute potential differences across the cell membrane are small, they give rise to enormous electrical potential gradients because they occur across a very thin surface. As a consequence, apparently small changes in Em can produce large changes in potential gradient and powerful forces that are able to induce molecular rearrangement in membrane proteins, such as those required for opening and closing ion channels embedded in the cell membrane. The capacitance of the membrane is generally fixed and unaffected by the molecules that are embedded in it. In contrast, membrane resistance is highly variable and depends on the conductance of ion channels embedded in the membrane.2,3
The sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (Cl−) ions are the major charge carriers, and their movement across the cell membrane creates a flow of current that generates excitation and signals in cardiac myocytes. The electrical current generated by the flux of an ion across the membrane is determined by the membrane conductance to that ion (gion) and the potential (voltage) difference across the membrane. The potential difference represents the potential at which there is no net ion flux (i.e., the Eion) and the actual Em: current = gion × (Em − Eion).1,4
By convention, an inward current increases the electropositivity within the cell (i.e., causes depolarization of the Em [to be less negative]) and can result from either the movement of positively charged ions (most commonly Na+ or Ca2+) into the cell or the efflux of negatively charged ions (e.g., Cl−) out of the cell. An outward current increases the electronegativity within the cell (i.e., causes hyperpolarization of the Em [to become more negative]) and can result from either the movement of anions into the cell or the efflux of cations (most commonly K+) out of the cell.3
Opening and closing of ion channels can induce a departure from the relatively static resting Em, called a depolarization if the interior voltage rises (becomes less negative) or a hyperpolarization if the interior voltage becomes more negative. The most important ion fluxes that depolarize or repolarize the membrane are passive (i.e., the ions move down their electrochemical gradient without requiring the expenditure of energy), occurring through transmembrane ion channels. In excitable cells, a sufficiently large depolarization can evoke a short-lasting all-or-none event called an action potential, in which the Em very rapidly undergoes specific and large dynamic voltage changes.1
Both resting Em and dynamic voltage changes such as the action potential are caused by specific changes in membrane permeabilities for Na+, K+, Ca2+, and Cl−, which, in turn, result from concerted changes in functional activity of various ion channels, ion transporters, and exchangers.3
The Cardiac Action Potential
During physiological electrical activity, the Em is a continuous function of time. The current flowing through the cell membrane is, at each instant, provided by multiple channels and transporters carrying charge in opposite directions because of their different ion selectivity. The algebraic summation of these contributions is referred to as net transmembrane current.1
The cardiac action potential reflects a balance between inward and outward currents. When a depolarizing stimulus (typically from an electric current from an adjacent cell) abruptly changes the Em of a resting cardiomyocyte to a critical value (the threshold level), the properties of the cell membrane and ion conductances change dramatically, precipitating a sequence of events involving the influx and efflux of multiple ions that together produce the action potential of the cell. In this fashion, an electrical stimulus is conducted from one cell to all the cells that are adjacent to it.2
Unlike skeletal muscle, cardiac muscle is electrically coupled so that the wave of depolarization propagates from one cell to the next, independent of neuronal input. The heart is activated by capacitive currents generated when a wave of depolarization approaches a region of the heart that is at its resting potential. Unlike ionic currents, which are generated by the flux of charged ions across the cell membrane, capacitive currents are generated by the movement of electrons toward and away from the surfaces of the membrane.2,3 The resulting decrease in positive charge at the outer side of the cell membrane reduces the negative charge on the intracellular surface of the membrane. These charge movements, which are carried by electrons, generate a capacitive current. When an excitatory stimulus causes the Em to become less negative and beyond a threshold level (approximately −65 mV for working atrial and ventricular cardiomyocytes), Na+ channels activate (open) and permit an inward Na+ current (INa), resulting in a rapid shift of the Em to a positive voltage range. This event triggers a series of successive opening and closure of selectively permeable ion channels. The direction and magnitude of passive ion movement (and the resulting current) at any given transmembrane voltage are determined by the ratio of the intracellular and extracellular concentrations and the reversal potential of that ion, with the net flux being larger when ions move from the more concentrated side.3
The threshold is the lowest Em at which opening of enough Na+ channels (or Ca2+ channels in the setting of nodal cells) is able to initiate the sequence of channel openings needed to generate a propagated action potential. Small (subthreshold) depolarizing stimuli depolarize the membrane in proportion to the strength of the stimulus and cause only local responses because they do not open enough Na+ channels to generate depolarizing currents large enough to activate nearby resting cells (i.e., insufficient to initiate a regenerative action potential). On the other hand, when the stimulus is sufficiently intense to reduce the Em to a threshold value, regenerative action potential results, whereby intracellular movement of Na+ depolarizes the membrane more, a process that increases conductance to Na+ more, which allows more Na+ to enter, and so on. In this fashion, the extent of subsequent depolarization becomes independent of the initial depolarizing stimulus, and more intense stimuli do not produce larger action potential responses; rather, an all-or-none response results.2
During the action potential, membrane voltages fluctuate in the range of −94 to +30 mV (Fig. 1-1). With physiological external K+, the reversal potential of K+ (EK) is approximately −94 mV, and passive K+ movement during an action potential is out of the cell. On the other hand, because the calculated reversal potential of a cardiac Ca2+ channel (ECa) is +64 mV, passive Ca2+ flux is into the cell.5
In normal atrial and ventricular myocytes and in His-Purkinje fibers, action potentials have very rapid upstrokes, mediated by the fast inward INa. These potentials are called fast response potentials. In contrast, action potentials in the normal sinus and atrioventricular (AV) nodal cells and many types of diseased tissues have very slow upstrokes, mediated by a slow inward, predominantly L-type voltage-gated Ca2+ current (ICaL), rather than by the fast inward INa (Fig. 1-2). These potentials have been termed slow response potentials.2,5
The Fast Response Action Potential
Phase 4: The Resting Membrane Potential
The Em of resting atrial and ventricular cardiomyocytes remains steady throughout diastole. The resting Em is caused by the differences in ionic concentrations across the membrane and the selective membrane permeability (conductance) to various ions. Large concentration gradients of Na+, K+, Ca2+, and Cl− across the cell membrane are maintained by the ion pumps and exchangers (Table 1-1).
TABLE 1-1 Intracellular and Extracellular Ion Concentrations and Equilibrium Potentials in Cardiomyocytes
Under normal conditions, the resting membrane is most permeable to K+ and relatively impermeable to other ions. K+ has the largest resting membrane conductance (gK is 100 times greater than gNa) because of the abundance of open K+ channels at rest, whereas Na+ and Ca2+ channels are generally closed. Thus, K+ exerts the largest influence on the resting Em. As a consequence, the resulting Em is almost always close to the K+ reversal potential (Em approximates EK). The actual resting Em is slightly less negative than EK because the cell membrane is slightly permeable to other ions.2
The inwardly rectifying K+ (Kir) channels underlie an outward K+ current (IK1) responsible for maintaining the resting potential near the EK in atrial, His-Purkinje, and ventricular cells, under normal conditions.6,7 Kir channels preferentially allow currents of K+ ions to flow into the cell with a strongly voltage-dependent decline of K+ efflux (i.e., reduction of outward current) on membrane depolarization. As such, IK1 is a strong rectifier that passes K+ currents over a limited range of Em (see Chap. 2 for detailed discussion on the concept of rectification); at a negative Em, IK1 conductance is much larger than that of any other current, thus, it clamps the resting Em close to the reversal potential for K+ (EK). IK1 density is much higher in ventricular than in atrial myocytes, a feature that protects the ventricular cell from pacemaker activity. By contrast, IK1 is almost absent in sinus and AV nodal cells, thus allowing for relatively more depolarized resting diastolic potentials compared with atrial and ventricular myocytes (Table 1-2).2
A unique property of Kir currents is the unusual dependence of rectification on extracellular K+ concentration. Specifically, on increase in extracellular K+, the IK1 current-voltage relationship shifts nearly in parallel with the EK and leads to a crossover phenomenon. One important consequence of such behavior is that at potentials positive to the crossover, K+ conductance increases rather than decreases, against an expectation based on a reduced driving force for K+ ions as a result of elevated extracellular K+ concentration.6–8
Although Ca2+ does not contribute directly to the resting Em (since the voltage-activated Ca2+ channels are closed at the hyperpolarized resting Em), changes in intracellular free Ca2+ concentration can affect other membrane conductance values. Increases in intracellular Ca2+ levels can stimulate the Na+-Ca2+ exchanger (INa-Ca), which exchanges three Na+ ions for one Ca2+ ion; the direction depends on the Na+ and Ca2+ concentrations on the two sides of the membrane and the Em difference. At resting Em and during a spontaneous sarcoplasmic reticulum Ca2+ release event, this exchanger would generate a net Na+ influx, possibly causing transient membrane depolarizations.3
Phase 0: The Upstroke—Rapid Depolarization
On excitation of the cardiomyocyte by electrical stimuli from adjacent cells, its resting Em (approximately −85 mV) depolarizes, leading to opening (activation) of Na+ channels from its resting (closed) state and enabling a large and rapid influx of Na+ ions (inward INa) into the cell down their electrochemical gradient. As a consequence of increased Na+ conductance, the excited membrane no longer behaves like a K+ electrode (i.e., exclusively permeable to K+), but more closely approximates an Na+ electrode, and the membrane potential moves toward the Na+ Eion (ENa; see Table 1-1). Once an excitatory stimulus depolarizes the Em beyond the threshold for activation of Na+ channels (approximately −65 mV), the activated INa is regenerative and no longer depends on the initial depolarizing stimulus; the influx of Na+ ions further depolarizes the membrane and thereby increases conductance to Na+ more, which allows more Na+ to enter.2,9
Normally, activation of Na+ channels is transient; fast inactivation (closing of the pore) starts simultaneously with activation, but because inactivation is slightly delayed relative to activation, the channels remain transiently (less than 1 millisecond) open to conduct INa during phase 0 of the action potential before it closes. Additionally, the influx of Na+ into the cell increases the positive intracellular charges and reduces the driving force for Na+. When the ENa is reached, no further Na+ ions enter the cell.9
The rate at which depolarization occurs during phase 0, that is, the maximum rate of change of voltage over time, is indicated by the expression dV/dtmax, which is a reasonable approximation of the rate and magnitude of Na+ entry into the cell and a determinant of conduction velocity for the propagated action potential.2
[/not-level-membership-for-cardiovascular-category]
