Chapter 28 Ventricular Arrhythmias in Hypertrophic Cardiomyopathy
Pathophysiology
Changes in myocyte architecture lead to ventricular hypertrophy (Fig. 28-1). The degree and distribution of LV hypertrophy vary markedly. LV hypertrophy can be asymmetrical or symmetrical. The symmetrical form of hypertrophic CMP accounts for more than one-third of cases and is characterized by concentric thickening of the LV with a small ventricular cavity dimension. Asymmetrical septal hypertrophy is the most common variant, which is associated with thickening of the basal anterior septum, which bulges beneath the aortic valve and causes narrowing of the LV outflow region. However, isolated segmental hypertrophy may affect the LV apex (apical hypertrophic CMP) or any portion of the LV. The morphological pattern of LV hypertrophy is not closely predictive of the severity of symptoms or prognosis. Although LV apical aneurysms are observed only rarely (2%), they are associated with a higher rate of adverse disease consequences (10.5% per year) as compared with the general hypertrophic CMP population.1
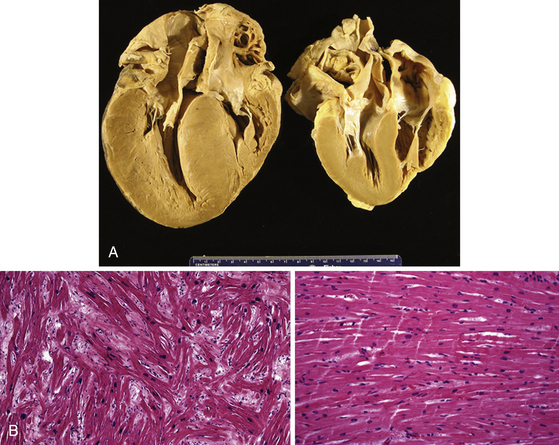
The arrhythmogenic substrate responsible for ventricular tachycardia (VT) occurrence in hypertrophic CMP has not been completely defined. Myofibrillar disarray as well as diffuse interstitial myocardial fibrosis or extensive scarring (likely caused by abnormalities of intramural coronary arteries and focal ischemia), which potentially predispose to disordered conduction patterns and increased dispersion of electrical depolarization and repolarization, have been suggested as factors contributing to ventricular arrhythmogenesis.2
Molecular Genetics
Hypertrophic CMP is familial in approximately half the cases and sporadic in the other half. The disease is transmitted as a Mendelian trait with an autosomal dominant pattern of inheritance and variable clinical penetrance. There is substantial diversity in the genetic causes of hypertrophic CMP. To date, nearly 900 different mutations have been reported in at least 24 genes encoding eight sarcomere proteins, including cardiac alpha- and beta-myosin heavy chains; cardiac troponins T, I, and C; cardiac myosin-binding protein C; alpha-tropomyosin; actin; titin; and essential and regulatory myosin light chains. Among these genes, mutations in MYH7, encoding beta-myosin heavy chain, and MYBPC3, encoding cardiac myosin binding protein-C, are the most common, each accounting for one-quarter to one-third of all cases.3,4 For each gene, several different mutations have been identified, and specific mutations are associated with different disease severity and prognosis.5
In addition, nonsarcomeric protein mutations in genes involved in cardiac metabolism (e.g., the gamma subunit of adenosine monophosphate [AMP]–activated protein kinase, PRKAG2, and lysosome-associated membrane protein 2 [LAMP-2], as in Danon disease), which are responsible for primary cardiac glycogen storage cardiomyopathies in older children and young adults, can be associated with a clinical presentation mimicking or indistinguishable from sarcomeric hypertrophic CMP. A high prevalence of conduction system dysfunction (with the requirement of permanent pacing in 30% of patients) characterizes PRKAG2 mutations. These diseases are often associated with ventricular preexcitation (Wolff-Parkinson-White syndrome). LAMP2 mutations are associated with early-onset LV hypertrophy (often in childhood) with rapid progression of heart failure and poor prognosis. These clinical entities are distinct from hypertrophic CMP caused by sarcomere protein mutations, despite the shared feature of LV hypertrophy. In addition, there can be genetic overlap between hypertrophic CMP and LV noncompaction.4
In infants and children, LV hypertrophy mimicking typical hypertrophic CMP caused by sarcomere protein mutations is often associated with congenital malformations and syndromes, inherited disorders of metabolism, and neuromuscular diseases, such as Fabry disease, Pompe disease, amyloidosis, carnitine deficiency, mitochondrial diseases, Friedreich ataxia, Noonan syndrome, and LEOPARD syndrome. Hypertrophic CMP can be distinguished from these disorders by dysmorphological examination, neuromuscular examination, metabolic screening, family information, and genetic testing.3,6
Clinical Considerations
Clinical Presentation
The clinical presentation of hypertrophic CMP is characterized by extreme variability in disease course, age of onset, severity of symptoms, and risk for SCD. Many patients are either asymptomatic or mildly symptomatic. The majority of patients present during adolescence or young adulthood, but symptoms can develop at any age. Symptomatic patients typically present with dyspnea, chest pain, palpitations, fatigue, orthostatic lightheadedness, presyncope, and syncope. Other complications include atrial and ventricular arrhythmias, infective endocarditis, and congestive heart failure. As noted, SCD can be the first manifestation of the disease.7
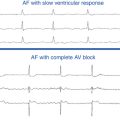
Shortness of breath, particularly exertional, is the most common symptom of hypertrophic CMP, occurring in up to 90% of patients, typically secondary to diastolic LV dysfunction. Syncope occurs in approximately 15% to 25% of patients, typically secondary to abnormal hemodynamic function (i.e., dynamic LV outflow obstruction) or, infrequently, secondary to cardiac arrhythmias.7,8 Atrial fibrillation (AF) is the most common arrhythmia observed in hypertrophic CMP, occurring in approximately 20% to 25% of patients, and is associated with an increased risk of stroke and thromboembolic complications.
Ventricular Arrhythmias
The predominant arrhythmia syndrome associated with hypertrophic CMP is sudden cardiac arrest, presumably due to polymorphic VT or VF. SCD occurs with an annual mortality rate of approximately 6% in referral-based populations and 1% in community-based studies. However, certain patient subgroups can have much higher rates, surpassing the American College of Cardiology/American Heart Association (ACC/AHA) guideline document definition of high risk for SCD (≥2% annual risk).9
Stable sustained monomorphic VT (SMVT) is rare, but can occur in patients with midventricular obstruction or apical LV aneurysms. The recurrence rate of VT is relatively high (56%) in hypertrophic CMP patients, and electrical storm can occur. During electrophysiological (EP) testing, induction of SMVT has a low reproducibility, and polymorphic VT and VF are often induced. The VT is commonly associated with a superior axis on the frontal plane, suggesting LV apical origins. An arrhythmic substrate, such as an aneurysm, can be important for the occurrence of stable SMVT.1,10
Appropriate implantable cardioverter-defibrillator (ICD) discharges in hypertrophic CMP patients occur annually in 10.6% of ICD recipients for secondary prevention after cardiac arrest (5-year cumulative probability, 39%), and in 3.6% of ICD recipients for primary prevention (5-year probability, 17%).11 Of note, arrhythmogenic events do not necessarily portend other adverse clinical outcomes. Specifically, appropriate ICD discharges do not appear to predict the occurrence of heart failure or the need for other invasive therapies (e.g., surgical myectomy, septal ablation).12
Initial Evaluation
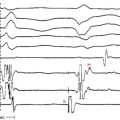
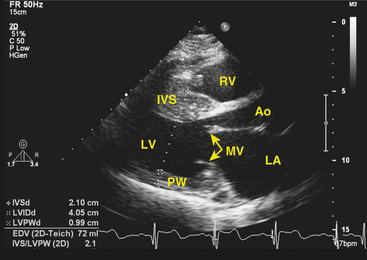
Clinical diagnosis is generally made with transthoracic echocardiography, demonstrating LV hypertrophy in a nondilated ventricle (Fig. 28-2). Cardiac magnetic resonance (MR) imaging also can provide very valuable additional information in the diagnosis of hypertrophic CMP and in differentiating it from other disorders. Marked ECG abnormalities are typically present in the majority of patients.
Clinical genetic testing for hypertrophic CMP comprising the eight most common disease-causing genes is now commercially available. Overall, the yield of genetic testing in probands with hypertrophic CMP is approximately 60%, but it depends on patient selection, falling to approximately 30% in sporadic disease.13,14 Although the knowledge of the underlying gene and mutation has a limited role in risk stratification and management of the individual patient, genetic testing is recommended for patients with an established clinical diagnosis of hypertrophic CMP in whom mutation-specific confirmatory testing would benefit, confirm, or exclude the diagnosis in at-risk family members. Once a pathogenic mutation has been detected, cascade screening of the relatives is indicated. On the other hand, genetic testing is not recommended for diagnosis of hypertrophic CMP in patients with nondiagnostic clinical features because the absence of a sarcomere mutation cannot rule out familial hypertrophic CMP.Furthermore, variants of uncertain significance can pose a problem in subjects with lower clinical pretest probability of a pathogenic sarcomere mutation.4
Risk Stratification
The traditionally acknowledged noninvasive risk stratification strategy for primary prevention uses five clinical markers that have been defined in several retrospective and observational studies. These primary prevention risk factors (applicable to hypertrophic CMP patients without prior cardiac arrest) are: (1) premature hypertrophic CMP-related sudden death in one or more relatives; (2) unexplained syncope (especially in the young and related to exertion); (3) multiple, repetitive (or prolonged) nonsustained VT on ambulatory (Holter) monitoring; (4) severe LV hypertrophy (maximal wall thickness of ≥30 mm); and (5) an abnormal blood pressure response during upright exercise (i.e., failure of blood pressure to rise appropriately by more than 20 to 30 mm Hg from baseline).8,15,16
The relative weight that can be assigned to each of the traditional risk markers has not been defined. A consensus document on hypertrophic CMP from the ACC and European Society of Cardiology (ESC) categorized known risk factors for SCD as “major” and “possible in individual patients” (Table 28-1). Although the absence of risk factors identifies a low-risk group, the positive predictive value of any single risk factor is limited. Risk stratification based on incorporation of multiple risk factors would likely improve positive predictive accuracy.9
TABLE 28-1 Risk Factors for Sudden Cardiac Death in Hypertrophic Cardiomyopathy
| MAJOR RISK FACTORS | POSSIBLE IN INDIVIDUAL PATIENTS |
|---|---|
From Zipes DP, Camm AJ, Borggrefe M, et al: ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines, J Am Coll Cardiol 48:e247-e346, 2006.
It is also important to note that the traditional risk stratification strategy in hypertrophic CMP remains imprecise and a few SCDs have been reported in young hypertrophic CMP patients judged to be at low risk without any of the acknowledged risk markers.17
In addition to the five traditional risk factors, other features of hypertrophic CMP may increase SCD risk for patients in selected subgroups. LV apical aneurysm (which is observed only in 2% of hypertrophic CMP patients) has been associated with a substantial (10%) annual event rate, largely because of the arrhythmogenic substrate created by the fibrotic thin-walled aneurysm.1,18 Similarly, patients in the end-stage phase of the disease (characterized by LV systolic dysfunction, wall thinning, and chamber enlargement) awaiting heart transplantation have a substantial arrhythmia-related event rate of 10% per year.17
Although LV outflow obstruction (subaortic gradient ≥30 mm Hg at rest) is a determinant of progressive heart failure and cardiovascular death (particularly from stroke), the specific relationship to SCD is weak and insufficient to regard LV outflow obstruction as a primary SCD risk marker.19 Reduction of obstruction by myotomy/myectomy (or alcohol ablation) is not considered a primary strategy for mitigating SCD risk. Similarly, the severity of clinical symptoms such as dyspnea, chest pain, and effort intolerance has not been correlated with increased risk of SCD.9
Currently, genetic testing appears to have little clinical application to the assessment of prognosis. Although certain mutations (e.g., some beta-myosin heavy chain and troponin T mutations) responsible for hypertrophic CMP can be associated with a higher risk for SCD compared with other mutations, there is substantial overlap between different disease gene groups, and exceptions are common.4 Hence, the prognostic value of disease-causing mutations in hypertrophic CMP for risk stratification is questionable, and the clinical course of individual patients cannot be reliably predicted based solely on particular genetic substrates and disease-causing mutations.17
Similarly, there is insufficient evidence to regard specific 12-lead ECG patterns, T wave alternans, heart rate variability, QT interval prolongation and dispersion, or coronary arterial bridging as risk markers in hypertrophic CMP patients.17,20
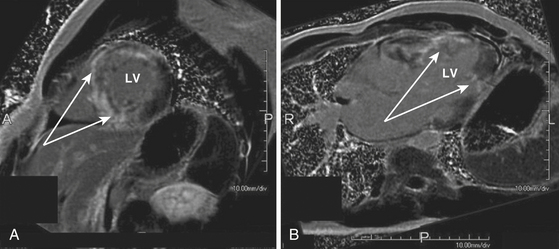
The role of cardiac MR imaging in risk stratification in patients with hypertrophic CMP is being evaluated. Cardiac MR imaging can help determine the severity and distribution of LV hypertrophy. Detection of myocardial fibrosis by gadolinium cardiac MR imaging (i.e., delayed enhancement) may have a role as a clinical predictor for increased risk of SCD. Up to 80% of patients with hypertrophic CMP have some degree of myocardial gadolinium hyperenhancement (the presumptive imaging correlate of increased myocardial fibrosis or scar) on cardiac MR (Fig. 28-3). However, the extent of hyperenhancement is greater in patients with progressive disease (28.5% versus 8.7%) and in patients with two or more risk factors for SCD (15.7% versus 8.6%).21 Additionally, patients with diffuse rather than confluent enhancement had two or more risk factors for SCD (87% versus 33%).17
Electrocardiographic Features
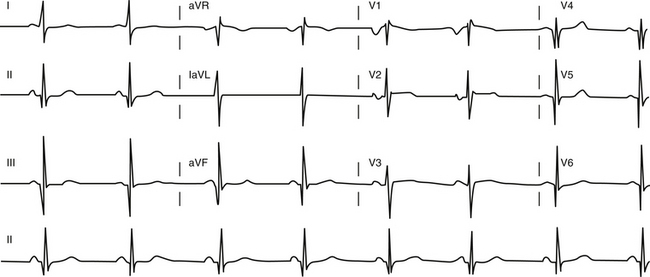
The ECG is abnormal in more than 90% of patients with hypertrophic CMP and in 75% of asymptomatic relatives. An abnormal ECG in the young is a sensitive marker of early disease expression. ECGs show a wide variety of abnormal patterns; however, no particular ECG pattern is characteristic or predictive of future events. Left atrial enlargement, repolarization abnormalities (ST-T changes including marked T wave inversion), and deep and narrow Q waves (most commonly in the inferolateral leads) are the most frequent ECG findings (Fig. 28-4). Voltage criteria for LV hypertrophy alone are nonspecific and are often seen in normal young adults. Giant negative T waves in the midprecordial leads are characteristic of hypertrophy confined to the LV apex.
Principles of Management
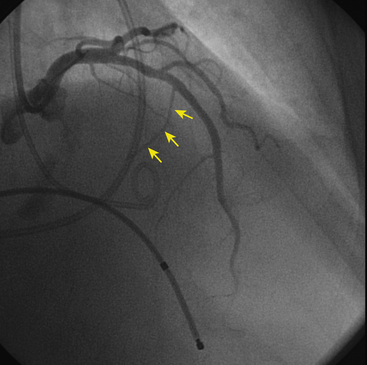
No treatments to prevent or modify disease progression currently exist. Therefore current treatment focuses on symptom management, assessment for risk of SCD, and family screening. Patients with LV outflow obstructive symptoms or heart failure are treated with beta blockers, verapamil, and disopyramide. Reduced heart rate and decreased contractility resulting from their action can potentially alleviate symptoms related to LV outflow obstruction. In cases that are unresponsive to drugs, septal surgical myotomy/myectomy or percutaneous alcohol septal ablation (Fig. 28-5) should be considered.
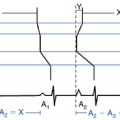
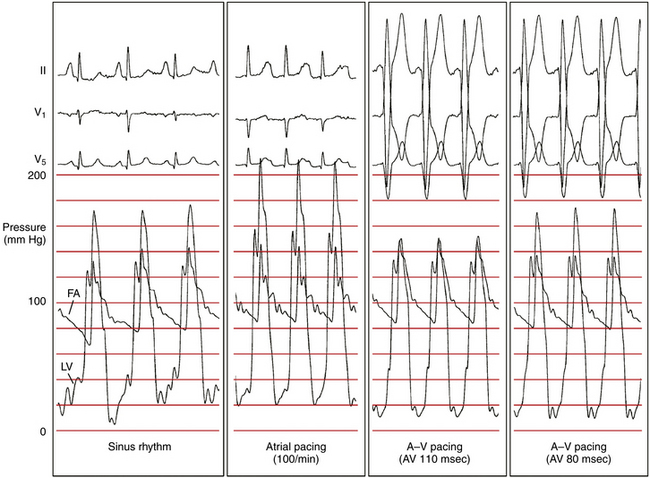
Dual-chamber pacing modifies the LV excitation pattern by changing the depolarization synchrony of the LV contraction, and can potentially be useful in a subset of symptomatic patients who have substantial LV outflow gradients (>30 mm Hg at rest or >50 mm Hg provoked) refractory to medical therapy and are not candidates for surgical myotomy/myectomy or alcohol septal ablation (Fig. 28-6). Although the initial observational studies in patients with hypertrophic CMP showed promising results, subsequent randomized clinical studies failed to show a significant benefit of dual-chamber pacing, and there are currently no data available to support the contention that pacing alters the clinical course of the disease or improves survival or long-term quality of life in hypertrophic CMP. Hence, routine implantation of dual-chamber pacemakers is no longer used, except in rare situations.22
Implantable Cardioverter-Defibrillator
ICD therapy is the most effective and reliable approach for reduction of the risk of SCD in patients with hypertrophic CMP, both for secondary prevention and altering the natural history of this disease. Of note, the interval from ICD implantation to first appropriate device intervention is quite variable, and often considerable in length. Even ICD recipients for secondary prevention after a cardiac arrest can survive for many years with or without the aid of ICD discharge.17 On the other hand, prophylactic pharmacological treatment alone (amiodarone, beta blockers, or verapamil) does not offer hypertrophic CMP patients reliable protection against SCD and is generally not indicated.
It is important to note that the number of risk factors in ICD recipients for primary prevention is unrelated to the likelihood of an appropriate therapy; about 35% of patients with appropriate ICD interventions for VT/VF had undergone implantation for only a single risk factor; and the likelihood of appropriate discharge was similar in patients with one, two, or three or more risk markers.11 Therefore, a single marker of high risk may represent sufficient evidence to justify the recommendation for a prophylactic ICD in selected patients with hypertrophic CMP. Indeed, the most recent ACC/AHA/ESC practice guidelines make primary prevention with the ICD a class IIa indication, based on the presence of one or more risk factors.9,22 Whether hypertrophic CMP patients should have ICDs implanted routinely following alcohol septal ablation is presently unresolved.
Catheter Ablation
In patients with apical aneurysms and SMVT, recurrence is quite high and antiarrhythmic drug therapy or catheter ablation should be considered. Antiarrhythmic medications have limited efficacy. When catheter ablation is undertaken, it should be appreciated that an epicardial approach may be warranted in a significant number of patients; in a recent report, an electrophysiologically identifiable epicardial scar was present in 80% of patients compared with 60% with an endocardial scar.1,2,18,23,24
Participation in Sports
Participation in intense competitive sports can represent a potential risk factor in athletes with hypertrophic CMP, even when conventional markers are absent, and exclusion from most participation in contact and most organized competitive sports is recommended (even in patients with ICDs), with the exception of low-intensity, class 1A sports (e.g., golf, bowling, billiards).
Family Screening
Because penetrance is age dependent, many family members may not express the phenotype at the time of examination and may be falsely considered “unaffected.” Thus, periodic evaluation of the “unaffected” family members is necessary, as some may develop hypertrophic CMP later in life. Annual screening should be considered in adolescents and young adults (ages 12 to 25 years), in athletes, and in those with a family history of early-onset disease. Screening every 3 to 5 years in other individuals may be adequate (Table 28-2).13
TABLE 28-2 Clinical Screening Strategies with Echocardiography and 12-Lead ECG for Detection of Hypertrophic Cardiomyopathy in Families
| AGE OF FAMILY MEMBER | SCREENING STRATEGY |
|---|---|
| <12 yr old | Optional unless: |
CMP = cardiomyopathy.
When the causative mutation in the index patient is identified, genetic testing is recommended for all first-degree relatives and can have particular advantages over clinical screening, as ECG or echocardiographic abnormalities may be absent or subtle, or develop late in life. Furthermore, genetic testing affords permanent reassurance to those family members who test gene-negative, obviating the need for clinical investigations or long-term follow-up and screening of their offspring.4,14
1. Furushima H., Chinushi M., Iijima K., et al. Ventricular tachyarrhythmia associated with hypertrophic cardiomyopathy: incidence, prognosis, and relation to type of hypertrophy. J Cardiovasc Electrophysiol. 2010;21:991-999.
2. Santangeli P., Di Biase L., Lakkireddy D., et al. Radiofrequency catheter ablation of ventricular arrhythmias in patients with hypertrophic cardiomyopathy: safety and feasibility. Heart Rhythm. 2010;7:1036-1042.
3. Morita H., Rehm H.L., Menesses A., et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899-1908.
4. Ackerman M.J., Priori S.G., Willems S., et al. HRS/EHRA Expert Consensus Statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 2011;8:1308-1339.
5. Boussy T., Paparella G., de Asmundis C., et al. Genetic basis of ventricular arrhythmias. Heart Fail Clin. 2010;6:249-266.
6. Pinto J.R., Parvatiyar M.S., Jones M.A., et al. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy,. J Biol Chem. 2009;284:19090-19100.
7. Williams L., Frenneaux M. Syncope in hypertrophic cardiomyopathy: mechanisms and consequences for treatment. Europace. 2007;9:817-822.
8. Spirito P., Autore C., Rapezzi C., et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703-1710.
9. Zipes D.P., Camm A.J., Borggrefe M., et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol. 2006;48:e247-e346.
10. Lim K.K., Maron B.J., Knight B.P. Successful catheter ablation of hemodynamically unstable monomorphic ventricular tachycardia in a patient with hypertrophic cardiomyopathy and apical aneurysm. J Cardiovasc Electrophysiol. 2009;20:445-447.
11. Maron B.J., Spirito P., Shen W.K., et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
12. Maron B.J., Haas T.S., Shannon K.M., et al. Long-term survival after cardiac arrest in hypertrophic cardiomyopathy. Heart Rhythm. 2009;6:993-997.
13. Maron B.J., Seidman J.G., Seidman C.E. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2125-2132.
14. Bos J.M., Towbin J.A., Ackerman M.J. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201-211.
15. Miller M.A., Gomes J.A., Fuster V. Risk stratification of sudden cardiac death in hypertrophic cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2007;4:667-676.
16. Christiaans I., van Engelen K., van Langen I.M., et al. Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy: systematic review of clinical risk markers. Europace. 2010;12:313-321.
17. Maron B.J., Spirito P. Implantable defibrillators and prevention of sudden death in hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2008;19:1118-1126.
18. Maron M.S., Finley J.J., Bos J.M., et al. Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. 2008;118:1541-1549.
19. Efthimiadis G.K., Parcharidou D.G., Giannakoulas G., et al. Left ventricular outflow tract obstruction as a risk factor for sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol. 2009;104:695-699.
20. Sherrid M.V., Cotiga D., Hart D., et al. Relation of 12-lead electrocardiogram patterns to implanted defibrillator-terminated ventricular tachyarrhythmias in hypertrophic cardiomyopathy. Am J Cardiol. 2009;104:1722-1726.
21. Adabag A.S., Maron B.J., Appelbaum E., et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51:1369-1374.
22. Epstein A.E., DiMarco J.P., Ellenbogen K.A., et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: executive summary. Heart Rhythm. 2008;5:934-955.
23. Dukkipati S.R., d’Avila A., Soejima K., et al. Long-term outcomes of combined epicardial and endocardial ablation of monomorphic ventricular tachycardia related to hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:185-194.
24. Dukkipati S.R., d’Avila A., Soejima K., et al. Long-term outcomes of combined epicardial and endocardial ablation of monomorphic ventricular tachycardia related to hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:185-194.