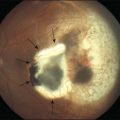
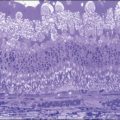
Chapter 141 Molecular Genetics of Choroidal Melanoma
Introduction
While uveal melanoma represents only 5% of all melanomas, it is the most common primary intraocular malignancy of the adult eye, affecting approximately 5–11 individuals per million per year.1 Uveal melanomas arise from melanocytes within the uveal tract, which consists of the iris, ciliary body, and choroid of the eye. Iris melanomas are relatively benign; however, ciliary body and choroidal melanomas still present significant diagnostic and therapeutic challenges.2 Indeed, choroidal melanomas – the most common ocular melanoma – result not only in vision loss, but also in metastasis, which is uniformly fatal. Metastases most commonly target the liver, and the detection of hepatic metastatic lesions predicts a dismal outcome, with a median survival of only a few months.3 Unfortunately, despite advances in the diagnosis and treatment of the primary tumor, we have not witnessed a corresponding improvement in patient survival.
Current treatment for local disease, including eye-sparing approaches (e.g., radioactive plaque therapy, external beam radiation, laser therapy), often leads to profound loss of vision4; thus, the identification of novel targets could provide gene product-targeted therapeutic options, which may avoid local tissue destruction. To this end, it is essential that we determine the molecular mechanism(s) that promote the initiation and progression of uveal melanoma. In the past few years, emerging evidence suggests that a complex series of molecular steps occurs in which uveal melanocytes elude their anti-proliferative and pro-apoptotic harnesses to form a melanoma, and in up to half of patients, metastasize hematogenously to the liver and other organs.5 The high rate of metastasis in patients diagnosed with uveal melanoma despite local treatment is thought to be due to the presence of micrometastases that occur prior to diagnosis and treatment. These micrometastases can remain dormant, often for years or decades, before manifesting clinically as metastatic disease. It is therefore essential that we identify patients at risk for metastasis early, so that we may provide adjuvant systemic therapy in an effort to delay or perhaps prevent the progression to clinical metastatic disease. It is equally important that patients not at risk for metastasis are spared from unnecessary treatment with systemic chemotherapy with their attendant risks and side effects.
Cutaneous melanoma, uveal melanoma, and the ras/raf/mek pathway
Early work exploring the molecular genetics of uveal melanoma was based upon the genetic studies in cutaneous melanoma. Given their common melanocytic origin, observations made in cutaneous melanoma served to guide investigation into the molecular biology of uveal melanoma. Specifically, activating mutations in Ras and B-Raf had been shown to play a fundamental role in the development of cutaneous melanoma, occurring in more than 80% of these tumors.6 Mutations of Ras and B-Raf promote the activation of MEK1/ERK (or the mitogen-activated protein kinase (MAPK) pathway), thereby promoting cell proliferation and survival.7 Uncontrolled proliferation is a key feature of malignant transformation, and activation of the MAPK pathway is a common target for malignant progression. Early work demonstrated that the vast majority of primary uveal melanoma tissue exhibits immunohistochemical evidence of activation of the MAPK pathway.8,9 Based upon these promising findings, several groups have investigated the mutational status of Ras, B-Raf, and MEK1 in primary uveal melanomas, as well as in liver metastases from patients with uveal melanoma. Surprisingly, these studies have been overwhelmingly negative.10,11 Indeed, despite arising from the same cell type, there appear to be more dissimilarities between the molecular genetics of uveal melanoma and cutaneous melanoma than there are similarities.
GNAQ and GNA11 mutations in uveal melanoma
A recent breakthrough in our understanding of the development of uveal melanoma came from the discovery in uveal melanoma tissue of activating mutations in genes encoding for members of the GαQ family of large G proteins. Genetic screens have shown that approximately half of uveal melanomas exhibit mutations in the gene encoding for GαQ (GNAQ).12 Interestingly, over half of uveal melanomas lacking a mutation in GNAQ exhibit a mutation in the gene encoding for the related protein, Gα11 (GNA11).13 Of note, mutations in genes encoding other heterotrimeric G-protein alpha subunits have been reported in a variety of cancers.14 G proteins are a family of heterotrimeric proteins which signal from cell surface, 7-transmembrane spanning (G protein-coupled) receptors (GPCRs).15 GαQ and Gα11 have 90% sequence homology with each other but may have differing functional roles. Upon ligand binding to its GPCR, the GDP bound to the GαQ/11 subunit is exchanged for GTP, resulting in a conformational change and the subsequent dissociation of the GαQ/11 from the Gβγ subunits. These subunits are then able to regulate various second messengers. The GαQ/11 family mediates its activity through stimulation of phospholipase C-β (PLCβ), which leads to activation of protein kinase C (PKC), and ultimately activates downstream intracellular signaling pathways, including the MAPK signaling pathway. Thus, activating mutations in either GNAQ or GNA11 (GNAQ/11) may provide the link between uveal melanomas and MAPK activation.16
However, mutations in GNAQ/11 have not been found to correlate with clinical, pathologic, immunohistochemical, or genetic factors associated with advanced uveal melanoma.17 Mutations in GNAQ/11 have also been identified in blue nevi (benign intradermal melanocytic proliferations affecting the conjunctiva and periorbital skin).12 Although patients with blue nevi are at higher risk for uveal melanoma, blue nevi are not malignant. Moreover, activating mutations in GNAQ/11 have not been detected in cutaneous melanomas. Indeed, although activating mutations in GNAQ/11 can promote transformation of immortalized melanocytes, mutation of either gene is not sufficient for malignant transformation. Collectively, these findings suggest that mutations in GNAQ/11 may occur early during the development of uveal melanomas. Thus, although helpful in understanding early molecular events in the pathogenesis of this enigmatic tumor, GNAQ/11 is not likely to help identify patients who are at higher risk for later developing metastases. Nonetheless, as GPCRs are the target – directly or indirectly – of 50–60% of all present pharmacological agents,15 GαQ/11 may still provide a novel gene-product specific therapeutic target for the treatment of uveal melanoma.
Chromosomal abnormalities in uveal melanoma
Although several clinical and histological features of uveal melanoma have been associated with a poor prognosis, their ability to predict metastasis in an individual patient is limited. Conversely, specific chromosomal abnormalities appear to provide a more accurate predictor for metastasis and survival.18 Emerging evidence suggests that these specific chromosomal alterations occur either in metastasizing or non-metastasizing tumors relatively late in the progression of uveal melanoma. Several cytogenetic or chromosomal abnormalities (e.g., loss of 1p, loss of chromosome 3, gain of 6p, loss of 6q, loss of 8p, gain of 8q), have been linked statistically to metastatic death in uveal melanoma patients.19–22 However, among these markers, gain of chromosome 6p and loss of one copy of chromosome 3 (monosomy 3) have proved to be the most reliable predictors of metastasis and survival. Gain of chromosome 6p occurs mainly in nonmetastasizing tumors, and carries a better prognosis. Conversely, loss of one copy of chromosome 3 (monosomy 3) occurs most frequently in metastasizing tumors, and predicts a poor outcome.23 Of note, isodisomy 3, in which one copy of chromosome 3 is lost and the remaining (faulty copy) is duplicated, occurs in 5–10% of cases of patients with uveal melanoma and is prognostically equivalent to monosomy 3.24 Tumors with monosomy 3 have also been found to contain greater numbers of additional chromosomal abnormalities (aneuploidy) than tumors with disomy 3, suggesting that loss of chromosome 3 may promote the genomic instability observed in more aggressive tumors. This has complicated the interpretation of the prognostic significance of some chromosomal abnormalities in uveal melanoma. For example, although gain of copies of chromosome 8q has been reported in uveal melanoma and has been associated with a poor outcome, it may not be an independent risk factor for metastasis, but rather more common in tumors with monosomy 3, which is a very strong metastatic risk factor.25 Conversely, a late chromosomal alteration that does have independent prognostic significance is loss of chromosome 8p, which (in the setting of monosomy 3) predicts earlier metastasis and poorer survival.26
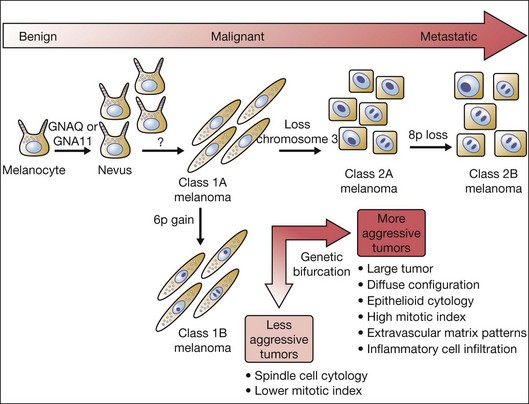
Gain of 6p, loss of 3: the genetic bifurcation in uveal melanoma
Interestingly, gain of chromosome 6p and monosomy 3 are almost completely mutually exclusive,21 representing a genetic bifurcation in uveal melanoma progression (Fig. 141.1). This observation has prompted efforts to use this unique molecular distinction to predict which patients are at higher risk for later developing metastases.
To distinguish between these two populations of uveal melanoma patients, initial efforts focused on cytogenetic analysis: standard karyotyping in which metaphase spreads are directly visualized and chromosomal abnormalities are identified by morphologic changes in chromosome size and banding pattern. However, this technique requires highly trained cytogeneticists, and the accuracy of the results is limited by sampling error attributable to analysis of only a few tumor cells, and to the inability to detect small genetic changes. Moreover, standard karyotyping is unable to identify isodisomy 3, resulting in a missed identification in up to 10% of uveal melanomas. Other techniques that also rely on direct analysis of intact chromosomes include fluorescence in situ hybridization (FISH), spectral karyotyping (SKY), and earlier forms of comparative genomic hybridization (CGH).27 Although these techniques provide some advantages over karyotyping, they are still prone to sampling error attributable to analysis of a small subset of tumor cells, and they are also unable to detect isodisomy.
More recent automated techniques (e.g., array-based CGH, microsatellite analysis (MSA), single-nucleotide polymorphism (SNP) analysis, multiplex ligation-dependent probe amplification (MLPA)), evaluate multiple regions across individual chromosomes using specific molecular probes. By allowing larger numbers of tumor cells to be examined, these techniques reduce the risk of sampling error, and increase the ability to detect smaller abnormalities compared with karyotyping and FISH. Additionally, MSA and SNP can detect the presence of isodisomy 3. However, the prognostic accuracy of these techniques compared with standard karyotyping is unclear.28
Gene expression profiling
The gene-expression data are then analyzed using statistical methods, with the aim of identifying gene clusters that correlate with the patients’ clinical outcome. Specifically, using “supervised” clustering methods for tumor classification guided by knowledge of clinical outcomes, gene clusters are identified. A subset of gene markers can then be identified and used as a “prognosis classifier” that can help predict the later appearance (“poor prognosis”) or absence (“good prognosis”) of clinical metastasis. This approach has been previously applied to other cancers to more accurately classify tumors,29,30 and has recently been tested with uveal melanoma (Fig. 141.2).33
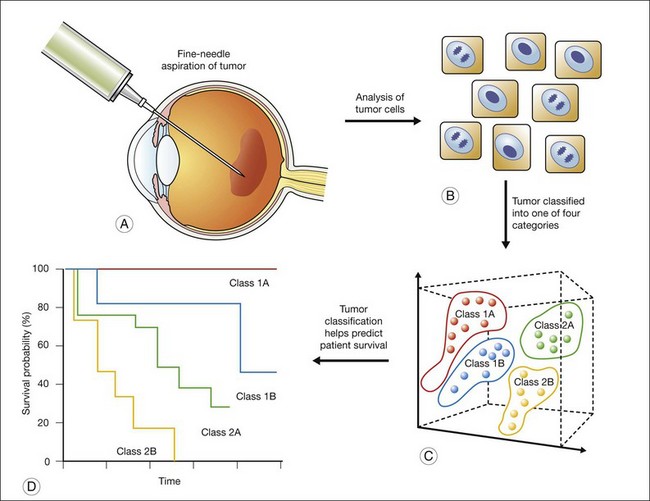
The information derived from GEP in uveal melanoma has proven to be a useful tool in predicting a patient’s clinical outcome.31,32 In patients with uveal melanoma, GEP has identified two major subgroups referred to as class 1 and class 2; approximately half of uveal melanomas fall into each subgroup.33 Patients with class 1 tumors have a lower risk of metastasis and patients with class 2 tumors have a higher risk of metastasis.34 Interestingly, the class 2 gene expression profile is closely associated with previously identified prognostic features, such as larger tumor size, epithelioid cytology, extravascular looping matrix patterns, and monosomy 3.35 The prognostic accuracy of the class 2 expression profile has been reported to be greater than for any of these other features (individually or in combination).33 The identification of a subset of genes has enabled a polymerase chain reaction (PCR)-based platform that further increased the prognostic accuracy of GEP for uveal melanoma.36 This PCR-based method requires less tissue and has further proven useful for the analysis of archival (formalin-fixed, paraffin-embedded) samples.
The two classes have further been subdivided into four prognostically significant subclasses (1A, 1B, 2A and 2B), based on gene-expression profiling.33 The metastasis-free survival is longest for class 1A and shortest for class 2B. The subclass 1B signature corresponds closely to gain of chromosome 6p, and the subclass 2B signature corresponds closely to loss of chromosome 8p. If molecular forecasting of the outcome of uveal melanoma using GEP proves reliable, it would certainly be a significant advance on existing prognostic methods, to be sure.
Clinical implications of genetic prognostication
Ironically, the additional information provided by our greater understanding of the molecular genetics of uveal melanoma has ultimately led to new questions about both the disease and how and when we should treat patients with uveal melanoma. Central to these questions is the debate as to whether or not we should offer (or perform) genetic testing for every patient with uveal melanoma. Alternatively, it has been argued that we should offer this test to only a subgroup of uveal melanoma patients. However, identifying which patients should be offered genetic testing remains unclear (Fig. 141.3). Should testing be limited to patients who undergo enucleation or local tumor resection or should it include patients who undergo brachytherapy, which would require fine-needle biopsy as an extra procedure? Does tumor location or size influence the decision? What (if any) is the role for genetic testing on patients with a suspicious nevus?
Tissue procurement
Which test(s) should we use?
Until alternative (non-invasive) approaches (e.g., circulating tumor cells, radioimmunoscintigraphy, serum biomarkers) are available, many clinicians agree that transscleral or transvitreous FNAB is the most safe and effective means to obtain tissue for genetic testing. However, the best test to perform for genetic testing remains unresolved. Moreover, it has further been argued that as current tests are not FDA-approved for use in diagnostic procedures, they should be considered investigational, and require institutional review board approval for their use for prognostication.28 Not surprisingly, there is significant confusion and controversy in the ocular oncology field regarding the use of these prognostic tests in the clinical management of uveal melanoma. The future of uveal melanoma diagnostic/prognostic tests may rest in the use of microarray technologies, either for the evaluation of changes in copy number or for gene expression profiling. Although these techniques have historically been technically demanding and expensive, emerging PCR-based platforms may provide less expensive, and more accurate assays for genetic prognostication. Ultimately, studies looking at molecular prognostic testing in uveal melanoma have been retrospective in nature; prospective validation of these tests is essential before they can be considered routine for patients in clinic.
Genetic testing in clinical trials
Genetic testing in uveal melanoma has, and will continue to have, a significant impact on our understanding of the molecular pathogenesis of the disease. Certainly, the gene expression profile of class 2 uveal melanomas provides clues as to the molecular events that promote progression and metastasis in this subset of tumors. This, in turn, may help identify novel therapeutic targets for these patients. Moreover, knowledge that there are two classes of uveal melanomas may influence the success of future clinical trials designed to examine the response of these tumors to novel treatments. A limitation of the Collaborative Ocular Melanoma Study (COMS)37 may have been that the consequence of the two classes of uveal melanomas (class 1 and 2) were not previously recognized at the time of the study’s design. The good outcome of half the patients in the trial (class 1) may have been largely independent of treatment, and may have limited our ability to uncover a therapeutic advantage to one of the treatment arms. It should be noted that eligibility of patients in future trials, which includes patients who have received local treatment, may be limited to those patients who either have already had genetic testing or who have tissue available for genetic testing prior to entry into the trial. Recognizing that there are two classes of uveal melanoma patients may further help in the identification of new and/or more accurate (non-invasive) biomarkers for the development of metastasis in high-risk (class 2A/2B) patients. There is little doubt that the additional information provided by genetic studies of uveal melanoma will provide a fund of knowledge that may benefit future patients with this disease.
Diagnosis and treatment of current uveal melanoma patients
A key ethical consideration for patients who undergo genetic testing is informed consent. Most specialists would agree that it is important to inform patients of the investigational nature of the test and that the test is not a routine clinical test and has not yet been proven to be of benefit in either guiding management or prolonging survival. However, reasonable disagreement remains as to whether genetic testing for uveal melanoma should only be performed in a research setting.28 For patients requiring FNAB, the purpose of the procedure, along with its potential complications, must also be explained to the patient.
Nonetheless, the potential impact of genetic testing on the early diagnosis of uveal melanoma patients who are more likely to develop metastases (prognostication) is a compelling argument for its use for these patients. However, as in other settings in medicine, disease prediction continues to outpace our ability to treat or cure metastatic uveal melanoma, resulting in a therapeutic rift between what we know and what we can do. This raises the reasonable, albeit philosophical question, as to whether there is an inherent value to simply knowing that a patient will – or will not – develop metastatic disease. Although emerging evidence suggests that the survival of patients with solitary metastatic nodules can be prolonged with early detection and treatment (e.g., resection, chemoembolization, or intrahepatic arterial infusion),38 the value of genetic testing even in this setting remains a topic of debate.
It has also been suggested that cytogenetic markers can be used to determine which small suspicious uveal melanomas should be treated (class 2 or monosomy 3) and which may be safe to observe (class 1 or gain of 6p).39 However, this approach assumes that class 2 tumors have not already metastasized, in which case local treatment may not benefit patient survival. Conversely, it is possible that treatment of class 1A tumors (rather than observing) may prevent them from converting to class 2 tumors. It has also been suggested that genetic testing may still influence surveillance for metastasis. Patients with class 1 tumors may require less frequent monitoring than patients with class 2 tumors. This is complicated by the fact that genetic prognostication is not an exact science. Thus, despite the paucity of treatments for metastatic disease, the risk of potentially delaying the diagnosis of metastasis by decreasing the frequency of surveillance remains controversial. While some patients may find comfort in knowing that they have a lower risk for metastases based on the genetics of their tumors, there is a real potential for adversely affecting the quality of life of a patient who is told that they have a high risk of metastases given that there is currently no effective adjuvant therapy available for metastatic uveal melanoma.
To date, all studies for genetic testing have focused on prognostication: the ability to predict outcome (e.g., metastases or survival) in the absence of treatment. However, these tests may further play an essential role in predicting treatment response. For example, it is more likely that therapies specifically targeting GNAQ or GNA11 would be effective in primary tumors with activating mutations in GNAQ/11. Similarly, patients with metastases that harbor specific mutations in genes that promote metastasis (e.g., BAP140) may be more responsive to adjuvant systemic chemotherapies that are designed to target these genes. Clearly, as new therapeutic options for metastatic uveal melanoma are introduced, the value of genetic prognostication will continue to evolve.
1 Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18:75–84.
2 Shields CL, Shields JA. Ocular melanoma: relatively rare but requiring respect. Clin Dermatol. 2009;27:122–133.
3 Bell DJ, Wilson MW. Choroidal melanoma: natural history and management options. Cancer Control. 2004;11:296–303.
4 Damato B. Developments in the management of uveal melanoma. Clin Experiment Ophthalmol. 2004;32:639–647.
5 Patel M, Smyth E, Chapman PB, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin Cancer Res. 2011;17:2087–2100.
6 Haluska FG, Ibrahim N. Therapeutic targets in melanoma: map kinase pathway. Curr Oncol Rep. 2006;8:400–405.
7 Poulikakos PI, Rosen N. Mutant BRAF melanomas – dependence and resistance. Cancer Cell. 2011;19:11–15.
8 Weber A, Hengge UR, Urbanik D, et al. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab Invest. 2003;83:1771–1776.
9 Zuidervaart W, van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032–2038.
10 Cohen Y, Goldenberg-Cohen N, Parrella P, et al. Lack of BRAF mutation in primary uveal melanoma. Invest Opthalmol Vis Sci. 2003;44:2876–2878.
11 Wong CW, Fan YS, Chan TL, et al. BRAF and NRAS mutations are uncommon in melanomas arising in diverse internal organs. J Clin Pathol. 2005;58:640–644.
12 Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602.
13 Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199.
14 Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94.
15 Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 40, 2000. re1
16 Sisley K, Doherty R, Cross NA. What hope for the future? GNAQ and uveal melanoma. Br J Ophthalmol. 2011;95:620–623.
17 Bauer J, Kilic E, Vaarwater J, et al. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br J Cancer. 2009;101:813–815.
18 Singh AD, Damato B, Howard P, et al. Uveal melanoma: genetic aspects. Ophthalmol Clin North Am. 2005;18:85–97.
19 Gordon KB, Thompson CT, Char DH, et al. Comparative genomic hybridization in the detection of DNA copy number abnormalities in uveal melanoma. Cancer Res. 1994;54:4764–4768.
20 Prescher G, Bornfeld N, Becher R. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990;82:1765–1769.
21 Speicher MR, Prescher G, du Manoir S, et al. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54:3817–3823.
22 Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59:3032–3037.
23 Prescher G, Bornfeld N, Hirche H, et al. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225.
24 White VA, McNeil BK, Thiberville L, et al. Acquired homozygosity (isodisomy) of chromosome 3 during clonal evolution of a uveal melanoma: association with morphologic heterogeneity. Genes Chromosomes Cancer. 1996;15:138–143.
25 Ehlers JP, Worley L, Onken MD, et al. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008;14:115–122.
26 Onken MD, Worley LA, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008;14:3737–3745.
27 Teixeira MR. Combined classical and molecular cytogenetic analysis of cancer. Eur J Cancer. 2002;38:1580–1584.
28 Harbour JW. Molecular prognostic testing and individualized patient care in uveal melanoma. Am J Ophthalmol. 2009;148:823–829.
29 Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235.
30 Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511.
31 Onken MD, Worley LA, Ehlers JP, et al. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209.
32 Tschentscher F, Hüsing J, Hölter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–2584.
33 Landreville S, Agapova OA, Harbour JW. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008;4:629–636.
34 Onken MD, Worley LA, Harbour JW. Association between gene expression profile, proliferation and metastasis in uveal melanoma. Curr Eye Res. 2010;35:857–863.
35 Worley LA, Onken MD, Person E, et al. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007;13:1466–1471.
36 Onken MD, Worley LA, Tuscan MD, et al. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2007;12:461–468.
37 Singh AD, Kivela T. The collaborative ocular melanoma study. Ophthalmol Clin North Am. 2005;18:129–142.
38 Sato T. Locoregional management of hepatic metastasis from primary uveal melanoma. Semin Oncol. 2010;37:127–138.
39 Shields JA, Shields CL, Materin M, et al. Role of cytogenetics in management of uveal melanoma. Arch Ophthalmol. 2008;126:416–419.
40 Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413.