Miscellaneous Airway Diseases
Bronchiectasis
Etiology and Pathogenesis
Another factor that plays a role in some patients is a defect in the ability of the airway to clear itself of, or protect itself against, bacterial pathogens (see Chapter 22). Such a defect predisposes a person to recurrent infections and eventually to airway dilation and bronchiectasis. The abnormality may involve inadequate humoral immunity and insufficient antibody production (hypogammaglobulinemia) or defective leukocyte function. Another problem that has received significant attention is dyskinetic cilia syndrome, in which ciliary dysfunction affects the ability of the ciliary blanket that lines the airway to clear bacteria and protect the airway against infection. The ciliary dysfunction is not limited to the lower airways; it also affects the nasal mucosa and, in males, may affect sperm motility and hence fertility. Pathologically, the dynein arms that are a characteristic feature of the ultrastructure of cilia are frequently absent in this disorder. One specific syndrome associated with bronchiectasis and ciliary dysfunction is Kartagener syndrome, which includes a triad of sinusitis, bronchiectasis, and situs inversus (usually discovered because of the presence of dextrocardia).
Pathology
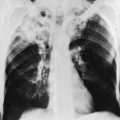
The primary pathologic feature of bronchiectasis is evident on gross inspection of the airways, which are markedly dilated in the involved region (Fig. 7-1). Three specific patterns of dilation have been described: cylindrical (appearing as uniform widening of the involved airways), varicose (having irregularly widened airways resembling varicose veins), and saccular bronchiectasis (characterized by widening of peripheral airways in a balloonlike fashion). These terms are still used when describing radiographic patterns but are much less relevant clinically. The dilated airways are generally filled with a considerable amount of secretions that may be grossly purulent. Microscopic changes of the bronchial wall epithelium, consisting of ulceration and squamous metaplasia, are seen.
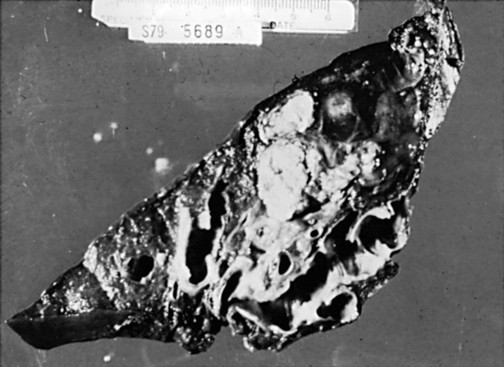
Coexisting disease in the remainder of the tracheobronchial tree is common. Other areas of bronchiectasis may be present, or generalized changes of chronic bronchitis may be seen (see Chapter 6).
Diagnostic Approach
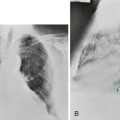
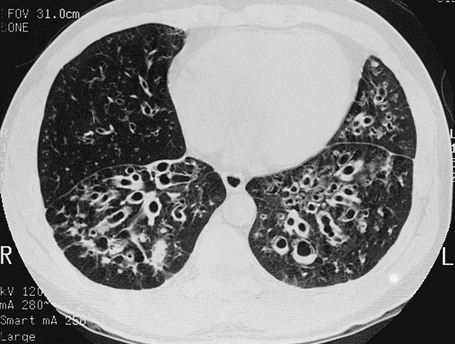
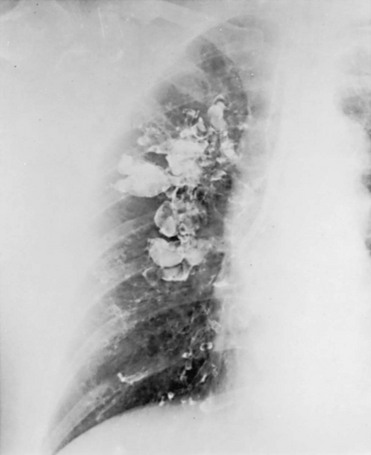
High-resolution computed tomography (HRCT) provides a definitive diagnosis and is the initial procedure used to define the presence, location, and extent of bronchiectasis (Fig. 7-2). HRCT (with sections 1-2 mm thick) provides excellent detail and is particularly useful for detecting subtle bronchiectasis. In the past, the definitive diagnosis depended on bronchography, a radiographic procedure in which an inhaled opaque contrast material was used to outline part of the tracheobronchial tree (Fig. 7-3). This procedure is uncomfortable, can induce bronchospasm, and is not performed today.
Treatment
The three major aspects of treatment of bronchiectasis are antibiotics, bronchopulmonary drainage (clearance of airway secretions), and bronchodilators. Antibiotics are used in various ways. Some patients are treated only when the quantity or appearance of the sputum clearly changes. Other patients, especially those who have severe disease and frequent exacerbations, are given a regimen of intermittent or even continuous antibiotics in an attempt to control chronic infection. Oral agents such as amoxicillin and trimethoprim-sulfamethoxazole, which are effective against many strains of Streptococcus pneumoniae and Haemophilus influenzae, are often used in patients with bronchiectasis. Inhaled tobramycin is sometimes used prophylactically to diminish the growth of gram-negative organisms. When these patients are infected with Pseudomonas organisms, treatment is generally more difficult. Oral fluoroquinolones such as ciprofloxacin have become useful therapy for Pseudomonas infection as an alternative to parenteral antibiotics, but secondary development of resistance to this class of antibiotics is common. Infection with M. avium complex requires prolonged therapy with multiple drugs (see Chapter 24). Chest physical therapy and positioning to allow better drainage of secretions (postural drainage) are frequently used for patients with copious sputum. Alternatively, inflatable vests or mechanical vibrators on the chest are increasingly being used to facilitate clearance of secretions. Bronchodilators may be useful in patients with coexisting airway obstruction that is at least partially reversible. Inhaled DNase has been used to decrease the viscosity of pulmonary secretions in patients with cystic fibrosis (see section on cystic fibrosis) but has not proven effective in bronchiectasis resulting from other causes.
Cystic Fibrosis
Etiology and Pathogenesis
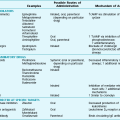
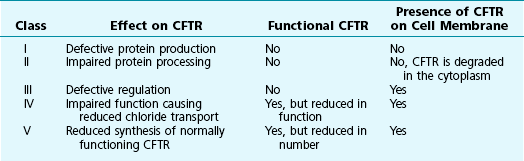
Cystic fibrosis is caused by mutations in the gene that codes for the 1480 amino acid protein cystic fibrosis transmembrane conductance regulator (CFTR), which resides on the long arm of chromosome 7. A member of the adenosine triphosphate (ATP)-binding cassette transporter protein superfamily, CFTR is an epithelial ion channel critically important in regulation of chloride and water absorption and secretion. CFTR mutations are categorized into five different classes according to the resultant functional or processing abnormality in the protein (Table 7-1). The specific defects in each class are potential targets for different approaches to therapy (see Treatment).
Diagnostic Approach
The chest radiograph often shows an increase in markings and the findings of bronchiectasis described in the previous section (Fig. 7-4). Evidence of focal pneumonitis may be seen during the course of the disease.
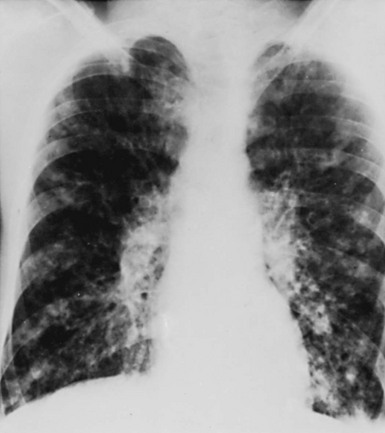
Treatment
In a recent conceptual breakthrough, a new drug, ivacaftor, has been shown to improve respiratory symptoms and lung function in patients with a less common CFTR mutation (G551D-CFTR). This mutation results in a protein that is present in typical amounts on the cell surface but does not function normally. Ivacaftor acts as a “potentiator” to increase the time activated CFTR channels at the cell surface remain open, thus improving the function of the abnormal channel. Unfortunately, the drug does not appear to be effective treatment for patients with the most common defect, ΔF508. However, the success of ivacaftor provides hope that an understanding of the different classes of CFTR mutations (see Table 7-1) may lead to other allele-specific therapies.
Upper Airway Disease
Etiology
On a chronic basis, the upper airway may be partially obstructed by hypertrophy of the tonsils, by tumors (particularly of the trachea), by strictures of the trachea (often resulting from prior instrumentation of the trachea), or by vocal cord paralysis. Tracheomalacia, another chronic condition that may be congenital or acquired, is characterized by flaccidity of supporting airway cartilage and results in upper airway narrowing, especially on forced exhalation. Some patients are subject to recurrent episodes of upper airway obstruction during sleep; this entity is one variety of what is termed sleep apnea syndrome, which is considered further in Chapter 18.
Pathophysiology
On the other hand, if airway diameter changes during the respiratory cycle, the greatest impairment to airflow occurs when the airway diameter is smallest. This type of obstruction is termed a variable obstruction. If the obstruction is located within the thorax, changes in pleural pressure during the respiratory cycle affect the size of the airway and therefore the magnitude of the obstruction. During a forced expiration, the positive pleural pressure causes airway narrowing, making the obstructing lesion more critical. In contrast, during inspiration, the airways increase their diameter, and the effects of a partial obstruction are less pronounced (see Fig. 3-20).
If the obstruction is located above the level of the thorax (i.e., outside the thorax), changes in pleural pressure are not directly transmitted to the airway in question. Rather, the negative airway pressure during inspiration tends to create a vacuum-like effect on extrathoracic upper airways, narrowing them and augmenting the effect of any partial obstruction. During expiration, the pressure generated by the flow of air from the intrathoracic airways tends to widen the extrathoracic airways and decrease the net effect of a partially obstructing lesion (see Fig. 3-20).
Diagnostic Approach
When a fixed lesion is causing a relatively critical obstruction, maximal flow rates generated during inspiration and expiration are approximately equal, and a “plateau” marks both the inspiratory and expiratory parts of the flow-volume curve. When the lesion is variable, the effect of the obstruction depends on whether the lesion is intrathoracic or extrathoracic. With an intrathoracic obstruction, critical narrowing occurs during expiration, and the expiratory part of the flow-volume curve displays a plateau. With an extrathoracic obstruction, the expiratory part of the loop is preserved, and the inspiratory portion displays the plateau. A schematic diagram of the flow-volume loops observed in these types of upper airway obstruction is shown in Figure 3-21.
Barbato, A, Frischer, T, Kuehni, CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009;34:1264–1276.
Barker, AF. Bronchiectasis. N Engl J Med. 2002;346:1383–1393.
Feldman, C. Bronchiectasis: new approaches to diagnosis and management. Clin Chest Med. 2011;32:535–546.
Feldman, C. The use of antiinflammatory therapy and macrolides in bronchiectasis. Clin Chest Med. 2012;33:371–380.
Fujimoto, T, Hillejan, L, Stamatis, G. Current strategy for surgical management of bronchiectasis. Ann Thorac Surg. 2001;72:1711–1715.
Fuschillo, S, De Felice, A, Balzano, G. Mucosal inflammation in idiopathic bronchiectasis: cellular and molecular mechanisms. Eur Respir J. 2008;31:396–406.
Gould, CM, Freeman, AF, Olivier, KN. Genetic causes of bronchiectasis. Clin Chest Med. 2012;33:249–263.
Lillington, GA. Dyskinetic cilia and Kartagener’s syndrome. Bronchiectasis with a twist. Clin Rev Allergy Immunol. 2001;21:65–69.
Metersky, ML. The initial evaluation of adults with bronchiectasis. Clin Chest Med. 2012;33:219–231.
Moulton, BC, Barker, AF. Pathogenesis of bronchiectasis. Clin Chest Med. 2012;33:211–217.
O’Donnell, AE. Bronchiectasis. Chest. 2008;134:815–823.
Pasteur, MC, Bilton, D, Hill, AT, British Thoracic Society Bronchiectasis non-CF Guideline Group. British Thoracic Society guideline for non-CF bronchiectasis. Thorax. 2010;65:i1–58.
Rosen, MJ. Chronic cough due to bronchiectasis: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:122s–131s.
Smith, MP. Non-cystic fibrosis bronchiectasis. J R Coll Physicians Edinb. 2011;41:132–139.
Strippoli, MP, Frischer, T, Barbato, A, et al. for the ERS Task Force on primary ciliary dyskinesia in children: Management of primary ciliary dyskinesia in European children: recommendations and clinical practice. Eur Respir J. 2012;39:1482–1491.
Todd, JL, Palmer, SM. Bronchiolitis obliterans syndrome: the final frontier for lung transplantation. Chest. 2011;140:502–508.
Boucher, RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146.
Boyle, MP. Adult cystic fibrosis. JAMA. 2007;298:1787–1793.
Chmiel, JF, Konstan, MW. Inflammation and anti-inflammatory therapies for cystic fibrosis. Clin Chest Med. 2007;28:331.
Cohen-Cymberknoh, M, Shoseyov, D, Kerem, E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;183:1463–1471.
Corbyn, Z. Promising new era dawns for cystic fibrosis treatment. Lancet. 2012;379:1475–1476. 21
Davis, PB, Yasothan, U, Kirkpatrick, P. Ivacaftor. Nat Rev Drug Discov. 2012;30(11):349–350.
Donaldson, SH, Boucher, RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631–1636.
Drumm, ML, Konstan, MW, Schluchter, MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443.
Elizur, A, Cannon, CL, Ferkol, TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489–495.
Farrell, PM, Rosenstein, BJ, White, TB, et al. Cystic Fibrosis Foundation: Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153:S4–S14.
Flume, PA, Liou, TG, Borowitz, DS, et al. for the VX08–770–104 Study Group: Ivacaftor in Subjects with Cystic Fibrosis who are Homozygous for the F508del-CFTR Mutation. Chest. 2012. Mar 1. [Epub ahead of print]
Flume, PA, Mogayzel, PJ, Jr., Robinson, KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Clinical Practice Guidelines for Pulmonary Therapies Committee. Am J Respir Crit Care Med. 2009;180:802–808.
Freedman, SD, Blanco, PG, Zaman, MM, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med. 2004;350:560.
Kerem, E. Pharmacological induction of CFTR function in patients with cystic fibrosis: mutation-specific therapy. Pediatr Pulmonol. 2005;40:183.
Mehta, A. CFTR: more than just a chloride channel. Pediatr Pulmonol. 2005;39:292–298.
Pier, GB. The challenges and promises of new therapies for cystic fibrosis. J Exp Med. 2012;209:1235–1239.
Ramsey, BW, Davies, J, McElvaney, NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. VX08–770–102 Study Group. N Engl J Med. 2011;365:1663–1672.
Rogan, MP, Stoltz, DA, Hornick, DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139:1480–1490.
Yarden, J, Radojkovic, D, De Boeck, K, et al. Association of tumour necrosis factor alpha variants with the CF pulmonary phenotype. Thorax. 2005;60:320.
Ernst, A, Feller-Kopman, D, Becker, HD, et al. Central airway obstruction. Am J Respir Crit Care Med. 2004;169:1278–1297.
Gaissert, HA, Burns, J. The compromised airway: tumors, strictures, and tracheomalacia. Surg Clin North Am. 2010;90:1065–1089.
Grenier, PA, Beigelman-Aubry, C, Brillet, PY. Nonneoplastic tracheal and bronchial stenoses. Radiol Clin North Am. 2009;47:243–260.
Hsia, D, Musani, AI. Interventional pulmonology. Med Clin North Am. 2011;95(6):1095–1114.
Morris, MJ, Christopher, KL. Diagnostic criteria for the classification of vocal cord dysfunction. Chest. 2010;138:1213–1223.
Weinberger, M, Abu-Hasan, M. Pseudo-asthma: when cough, wheezing, and dyspnea are not asthma. Pediatrics. 2007;120:855–864.