Chapter 36 Lysosomal Storage Diseases
Overview and General Concepts
The lysosomal storage diseases are a clinically heterogeneous group of inborn errors of metabolism that have traditionally been classified according to the biochemical nature of the incompletely degraded macromolecules that accumulate in various tissues. The lysosomal storage diseases are represented by about 50 different clinical entities (Table 36-1). Most of the individual conditions also have an eponymous designation in recognition of the investigator who provided seminal descriptions of the typical manifestations and clinical course of specific variants, often before elucidation of their biochemical or molecular bases.
As a central compartment in the endosomal-lysosomal pathway, the lysosome maintains an acidified milieu enriched with catabolic enzymes to facilitate the degradation of various byproducts of cellular turnover, which are mainly delivered to the lysosome through endocytosis. Phagocytosis and autophagy provide alternative points of substrate entry into the lysosome [Falguières et al., 2009]. These complex substrates may include sphingolipids, glycoproteins, and glycosaminoglycans (mucopolysaccharides). The particular metabolite that builds up (and the ensuing pattern and severity of the associated diseases) is primarily dependent on the substrate tissue-source and the relevant metabolic pathway and transport systems that are compromised. For instance, keratan sulfate is a major constituent of cartilage and the cornea; abnormalities in the turnover of keratan sulfate partly explain the characteristic features of skeletal dysplasia and the corneal opacities associated with the mucopolysaccharidoses [Wegrzyn et al., 2004]. Analysis (through a combination of immune quantification assays and tandem mass spectrometry) of the glycosphingolipid and oligosaccharide profiles in the blood and urine of affected patients may prove useful; as surrogate markers of disease activity, they may enable early diagnosis and the monitoring of clinical responses with directed therapies (such as enzyme replacement or substrate synthesis inhibition) [Parkinson-Lawrence et al., 2006].
The enzymes and transport or integral membrane proteins that are responsible for facilitating the intralysosomal metabolism of these substrates are formed within the endoplasmic reticulum and subsequently refined (through distinct post-translational mechanisms) by the Golgi apparatus [Eskelinen et al., 2003]. The uptake of newly synthesized enzymes by the lysosome, as opposed to other cellular organelles such as the peroxisome or mitochondria, is achieved largely through the mannose 6-phosphate (M6P) receptor pathway. Thus, phosphorylation of the mannose residues of the complex carbohydrate side-chains of the various relevant proteins is an essential step in its maturational processing [Urayama et al., 2004]. Elucidation of this pathway of lysosomal protein delivery, including uptake by the mannose receptors, was critical in the development of enzyme therapy for the lysosomal storage diseases [Grabowski and Hopkin, 2003].
Absent or defective hydrolytic activity within the lysosome, which can come about as a consequence of mutations within the specific encoding gene (for the functional enzyme or its activator or co-factor), is the most common cause of disease (Table 36-2) [Ballabio and Gieselmann, 2009]. For example, deficient glucocerebrosidase activity, as in Gaucher’s disease, results in the accumulation of its substrate, the glycosphingolipid glucosylceramide. Sphingolipid co-factors called saposins facilitate the interaction between the water-soluble glycoprotein enzymes and their lipid-soluble substrates [Matsuda et al., 2007]. Mutation of the saposin C gene is associated with the incomplete metabolism of glucosylceramide in vivo and a rare condition that has clinical features that overlap with the chronic neuropathic form of Gaucher’s disease (type 3 disease). Unlike other lysosomal hydrolases, which rely on the M6P receptor pathway, glucocerebrosidase is targeted to the lysosome via LIMP-2 [Reczek et al., 2007]; mutations of the encoding gene for LIMP-2 have been associated with a progressive myoclonic epilepsy and nephrotic syndrome [Balreira et al., 2008].
| Etiology | Disease (Examples) |
|---|---|
| Single hydrolytic enzyme deficiency | Gaucher’s disease Tay–Sachs disease Niemann–Pick disease |
| Single protease enzyme deficiency | Late infantile neuronal ceroid lipofuscinosis (pepinase) |
| Co-factor/activator protein deficiency | Gaucher’s disease variant (saposin) Tay–Sachs disease (GM2 activator) |
| Multiple enzyme deficiencies | Galactosialidosis (protective protein/cathepsin A) Multiple sulfatase deficiency (SUMF-1) I-cell disease (mucolipidosis II) |
| Membrane protein defect | Juvenile neuronal ceroid lipofuscinosis (battenin) |
| Small-molecule transport protein | Cystinosis Sialic acid storage disorders |
| Endocytosis, membrane–vesicle trafficking defect | Mucolipidosis IV Danon’s disease (LAMP-2) Niemann–Pick disease type C |
Other lysosomal disorders result from a defective protein involved in substrate transport (e.g., Niemann–Pick disease type C) or vesicle fusion (e.g., Danon’s disease) [Dierks et al., 2009; Ruivo et al., 2009]. Additional defects of lysosomal transport (e.g., cystinosis, sialic acid storage disorders) involve a dysfunction of intracellular membrane transporters that mediate the movements of the hydrolyzed products outside of the lysosome for their final excretion [Ruivo et al., 2009]. With respect to pathogenesis, cellular toxicity is hypothesized to occur when the lysosomal substrate burden has reached a critical threshold and presumptively triggers a cascade of downstream events that ultimately lead to cell death and the development of disease-specific complications. These consequent mechanisms of disease for most of the individual lysosomal storage diseases remain to be fully clarified.
There is increasing recognition that, besides mechanical forces, other pathologic factors (e.g., inflammation and apoptosis) may play contributory roles in the evolution of the disease phenotype. For instance, in the GM2-gangliosidoses, several markers of an inflammatory response have been found to be elevated and may partly explain the neurodegenerative features encountered in this group of diseases [Jeyakumar et al., 2002, 2003]. Studies have also suggested that certain lysosomal proteases may play active roles in the apoptotic execution process and can act as mediators of programmed cell death [Tardy et al., 2004]. Defects of autophagy are also being recognized as having a role in the pathogenesis of several lysosomal storage diseases [Settembre et al., 2008]. Oxidative damage and cytotoxic cell involvement has been suggested as a factor in the neuronal pathogenesis of mucopolysaccharidosis (MPS) type III [Villani et al., 2009]. Recent studies involving the animal model of mucopolysaccharidosis type I suggest that the absence of recycled precursors results in major shifts in the energy utilization of the cells [Woloszynek et al., 2009]. This finding susggests that lysosomal storage disease may be characterized as diseases of deficiency as well as overabundance (lysosomal storage).
The majority of genes that encode lysosomal proteins are ubiquitously expressed, and each of the various lysosomal storage disease subtypes is characterized by multisystemic involvement. There is wide heterogeneity in the clinical presentation of each entity among individually affected patients, and the concordance between genotype and phenotype is often imperfect even among siblings [Wilcox, 2004]. Disease manifestations may be evident prenatally or at any time from birth to adulthood. Certain lysosomal storage diseases present with nonimmune hydrops fetalis, and the failure to establish causality before the child’s death can lead to potential recurrence in future pregnancies among couples at risk [Staretz-Chacham et al., 2009]. On the other hand, Gaucher’s and Fabry’s diseases may not be associated with prominent symptoms during childhood, and the diagnosis can be missed because the patients have either a mild or an atypical course. This is true especially when the family history is uninformative. To some extent, the variable disease severity encountered between the individual patients has been attributed to different mutations that produce a protein with either no functional activity (in severe cases) or some residual enzyme activity (as observed in those with later onset of clinical expression). Because most lysosomal storage diseases are autosomal-recessive disorders, heterozygotes (or carrier individuals with a single defective gene copy) often have sufficient enzyme activity generated by their other (normal) allele. Carriers for these conditions do not exhibit evidence of tissue storage or suffer from the relevant disease that segregates in the family.
There are at least three disorders, Fabry’s and Danon’s diseases and Hunter’s syndrome (MPS II), that are transmitted as X-linked traits. Although females who are carriers of the trait for Hunter’s syndrome do not appear to develop clinical problems related to the presence of a mutant protein, a proportion of females who are carriers of Fabry’s disease or Danon’s disease may experience disease-related complications, which in a few cases can be as severe as those found in classically affected males. The variable expression in these carrier females has been partly attributed to lyonization (i.e., the random inactivation during early embryogenesis of one of the two X chromosomes). Lyonization results in varying proportions of the mutant gene product in different organ systems [Orstavik, 2009]. Skewed lyonization may be seen in females with an X:autosome translocation. In these cases, there is inactivation of the intact X chromosome. If the X chromosome that is attached to an autosome remains functional and if it happens to bear a gene mutation, disease expression may be unmasked. In addition, rare cases have been reported of females with Hunter’s syndrome who also happen to have Turner’s syndrome (45,X), wherein the single X chromosome that is present bears a mutation of the gene leading to the enzyme (iduronate-2-sulfatase) deficiency.
Although the individual disorders are infrequent to rare and considered orphan disorders by drug regulatory agencies, the lysosomal storage diseases have a combined prevalence of about 1 in 5000 to 8000 [Pinto et al., 2004]. This relatively high occurrence has prompted investigations of the feasibility of targeted screening [Meikle et al., 2004; Zhang et al., 2008]. The rationales given for these programs include:
Table 36-3 Diagnosis and Therapeutic Options for the Lysosomal Storage Disorders
| Disease* | Diagnostic Method† | Treatment‡ |
|---|---|---|
| Tay–Sachs disease (B variant) | Enz; Mol; Prenat (Enz/Mol) | SRT (in clinical trials) |
| GM2 gangliosidosis (three types) | ||
| Sandhoff’s disease (O variant) | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| GM2 gangliosidosis | ||
| GM2 gangliosidosis, AB variant | Func; Mol; Prenat (Mol) | Palliative |
| GM1 gangliosidosis (three types) | Enz; Mol; Prenat (Enz) | Palliative |
| Metachromatic leukodystrophy | Enz; Mol; Prenat (Enz) | HSCT/BMT |
| Metachromatic leukodystrophy variant (saposin B deficiency) | Func/Subs; Mol; Prenat (Mol) | Palliative |
| Krabbe’s disease | Enz; Mol; Prenat (Enz) | HSCT/BMT |
| Fabry’s disease | Enz/Subs; Mol; Prenat (Enz/Mol) | ERT |
| Gaucher’s disease types 1, 2, and 3 | Enz; Mol; Prenat (Enz) | ERT, primarily for types 1 and 3; SRT |
| Gaucher’s disease (saposin C variant) | Func/Subs; Mol; Prenat (Mol) | Palliative |
| Farber’s disease (seven types) | Enz; Mol; Prenat (Enz) | Palliative |
| Niemann–Pick disease types A and B | Enz; Mol; Prenat (Enz) | ERT (in clinical trials primarily for type B) |
| MPS I, Hurler–Scheie | Urine GAG; Enz; Mol; Prenat (Enz) | ERT; HSCT/BMT |
| MPS II, Hunter | Urine GAG; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| MPS IIIA-D, Sanfilippo A to D | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IVA, Morquio A | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IVB, Morquio B | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS VI, Maroteaux–Lamy | Urine GAG; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| MPS VII, Sly | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IX | Enz; Mol; Prenat (none reported) | Palliative |
| Pompe’s disease, glycogen storage disease IIA | Urine oligo; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| Danon’s disease | Mol; Prenat (Mol) | Palliative |
| α-Mannosidosis | Urine oligo; Enz; Mol; Prenat (Subs) | HSCT/BMT |
| β-Mannosidosis | Urine oligo; Enz; Mol; Prenat (Enz) | HSCT/BMT |
| α-Fucosidosis | Mol; Prenat (Mol) | HSCT/BMT |
| Schindler–Kanzaki disease | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| Sialidosis (mucolipidosis I) | Enz; Mol; Prenat (Enz) | Palliative |
| Aspartylglucosaminuria | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| Mucolipidosis II (I-cell disease) | Urine oligo; Mul Enz; Mol; Prenat (Enz) | Palliative; BMT (1 case) |
| Mucolipidosis IV | Mol; Prenat (Mol) | Palliative |
| Galactosialidosis (protective protein/cathepsin A deficiency) | Urine oligo; Mul Enz; Mol; Prenat (Mul Enz) | Palliative |
| Multiple sulfatases | Urine GAG; Mul Enz; Mol; Prenat (Enz) | Palliative |
| Wolman’s disease, cholesteryl ester storage disease (acid lipase deficiency) | Enz; Mol; Prenat (Enz) | Palliative |
| Niemann–Pick disease type C | Func/Subs; Mol; Prenat (Func/Subs) | SRT (in clinical trials) |
| Salla disease, infantile free sialic acid storage disease | Subs; Mol; Prenat (Subs) | Palliative |
| Cystinosis | Func/Subs; Mol; Prenat (Func/Subs) | Cysteamine |
| Pyknodysostosis | Enz; Mol; Prenat (none reported) | Palliative |
| Neuronal ceroid lipofuscinosis | Enz; Mol; Prenat (Enz/Mol for certain subtypes) | Palliative |
† Enz, enzyme assay; Func, functional assay (see text); GAG, glycosaminoglycans; Mol, molecular/DNA/gene defect analysis; Mul Enz, multiple enzyme in either cultured cells/amniotic fluid or serum media; Oligo, oligosaccharides; Prenat, prenatal diagnosis available; Subs, substrate analysis (see text).
‡ BMT, bone marrow transplantation; ERT, enzyme replacement therapy; HSCT, hematopoietic stem cell transplantation; SRT, substrate reduction therapy.
Several advances in the understanding of lysosomal storage diseases have resulted from investigations of animal disease models, which either have arisen spontaneously or have been generated by the application of recombinant genetic techniques (primarily involving mice) [Suzuki et al., 2003]. Numerous insights into the role of certain metabolites in embryonic and fetal development and their functions in various organs have been obtained from experiments involving these different animal models, which have also been useful in the development and preclinical testing of new putative therapies [Desmaris et al., 2004; Haskins, 2009]. Although most mouse disease models are genetically authentic and mimic the human phenotype, several reproduce only some aspects or may have no clinical manifestations at all (e.g., the Tay–Sachs disease knockout mouse) [Elsea and Lucas, 2002].
On the therapeutic front, industrial-scale production of recombinant human enzymes (through manipulation of mammalian cells in culture) has enabled the introduction of enzyme replacement therapy for several disorders, including Gaucher’s, Fabry’s, and Pompe’s (glycogen storage disease II) diseases, and Hurler–Scheie (MPS I H/S), Hunter’s (MPS II) and Maroteaux–Lamy (MPS VI) syndrome [Pastores, 2003; Pastores and Barnett, 2005]. In diseases primarily associated with central nervous system (CNS) degeneration (e.g., Tay–Sachs disease and Niemann–Pick disease type C) and for which protein delivery across the blood–brain barrier represents a major obstacle, trials with substrate reduction therapy have been undertaken [Platt and Lachmann, 2009]. The rationale for this approach is based on the following: in cases wherein mutant cells express residual enzyme activity, metabolic homeostasis may be restored or maintained by limiting the amount of intralysosomal substrate accumulation through inhibition of the amount of precursor material that is synthesized [Pastores and Barnett, 2003; Platt and Jeyakumar, 2008]. In Gaucher’s disease, substrate reduction therapy is feasible with the use of the imino sugar miglustat (which inhibits the activity of ceramide-specific glucosyltransferase, the rate-limiting enzyme involved in the biosynthesis of glycosphingolipids).
Before enzyme replacement therapy and substrate reduction therapy, bone marrow transplantation was undertaken in selected cases (see Table 36-3) [Prasad and Kurtzberg, 2008]. The high procedure-related morbidity and mortality risks associated with bone marrow transplantation (e.g., from graft-versus-host disease or infections) preclude its general application, even in cases in which clear therapeutic gains could be established in the successfully engrafted patients. The use of hematopoietic or mesenchymal stem cells and less intensive (i.e., nonmyeloablative) conditioning regimens is being examined as a means to improve clinical outcome and to minimize procedural risks [Prasad and Kurtzberg, 2008].
Another novel approach, termed enzyme enhancement therapy, is also under investigation. Enzyme enhancement therapy is a potential treatment strategy for diseases resulting from a defective enzyme associated with a mutation that leads to its misfolding and rapid degradation [Desnick, 2004]. In these instances, the use of an agent that acts as a chaperone to stabilize the mutant enzyme may help restore its function. Proof of principle for this strategy has been demonstrated in Fabry’s disease, for which the regular intravenous administration of galactose has led to the resolution of cardiomyopathy and improvement in the associated hemodynamic changes in one patient [Frustaci et al., 2001]. Currently, clinical trials with a pharmacologic chaperone are being undertaken in Gaucher’s and Fabry’s diseases [Pastores and Sathe, 2006].
Gene therapy is also being explored as an option, although most studies to date have only been with animal (primarily mouse) models, except for Gaucher’s disease, for which clinical trials have been performed [Caillaud and Poenaru, 2000; Ellinwood et al., 2004; Eto et al., 2004; Gieselmann et al., 2003b; Shen et al., 2004; Sly and Vogler, 2002]. Unfortunately, gene therapy performed in the patients with Gaucher’s disease resulted in only transient expression of the functional enzyme [Cabrera-Salazar et al., 2002]. Although experiments in animal models provide a foundation for translational studies in humans, the increased size and complexity of the human brain present particular challenges that may not be clarified by studies in the mouse models. Thus, studies in large animal models are increasingly preferred [Haskins, 2009]. Additionally, immunological rejection of the vector and viral products may lead to adverse outcome; ways to address these problems will be needed, to reduce procedure-related risks and promote long-term gene expression. Furthermore, the CNS and synovial joints are sites that can be challenging to treat with systemic gene therapy, and may require direct approaches for delivery of the relevant gene. For most lysosomal storage diseases, the mainstay of treatment has been palliative or supportive care. Treatment of the secondary disabilities (e.g., seizures, sensory impairment, and behavioral, sleep–wake cycle, or communication problems) can have a positive impact on the patient’s quality of life and may help address some sources of parental frustration. Genetic counseling of affected individuals and their relatives is also an important component of comprehensive care of the patient and family.
Sphingolipidoses
The sphingolipidoses are several disorders characterized by abnormalities in the metabolism of multiple glycolipid substrates that form a component of myelin or of lipid rafts within membranes (Figure 36-1). Most of the clinical variants in this group are associated with neurodegenerative features.
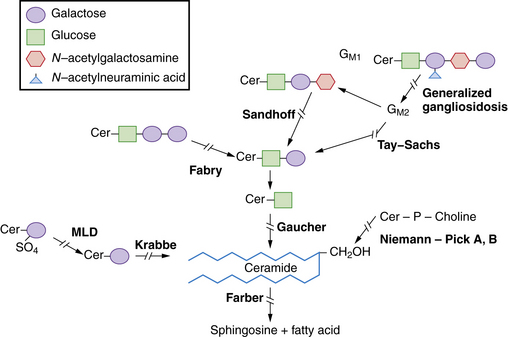
The myelin sheath is an extended, modified plasma membrane that, in the CNS, represents an extension of oligodendroglial cell processes. Examination of normal myelin with polarized microscopy and radiographic diffraction has exhibited a protein-lipid-protein-lipid-protein structure. Chemical investigations have determined that the lipid layers are composed of a bimolecular layer of hydrocarbon chains, cholesterol, phospholipids, and glycolipids (primarily galactocerebroside and sulfatide). The glycolipids are amphiphilic and have a hydrophobic ceramide moiety that acts as a membrane anchor and a hydrophilic, extracellularly oriented oligosaccharide chain. Rather than diffuse distribution in the cell membrane, current studies support the localization of glycosphingolipids, cholesterol, and certain proteins in specialized patches of membranes referred to as lipid rafts. Lipid rafts play key roles in intracellular transport, protein-sorting, and signal-transduction processes. Disturbed distribution of raft lipids and consequently protein subcellular localization due to a gridlock in the endocytic pathway, as seen in certain lysosomal storage diseases, is believed to lead to cell and organ functional impairments [Saravanan et al., 2004; Walkley and Vanier, 2009]. In addition, abnormalities of myelin membrane lipid composition result in the loss of membrane stability and its degeneration.
The degradation of glycosphingolipids takes place in the lysosomes through a stepwise action of specific acid hydrolases. Several of these enzymes (such as β-glucosidase, β-galactocerebrosidase, arylsulfatase A, and acid ceramidase) also require the assistance of nonenzymatic glycoprotein co-factors, the so-called sphingolipid activator proteins (or saposins). Examination of tissue sections obtained from affected individuals with glycosphingolipid storage disease typically reveals membranous cytoplasmic bodies, which represent lipid material enclosed by the lysosomal membranes [Abramovich et al., 2001].
Studies in animal models of Sandhoff’s disease and Niemann–Pick disease type A revealed elevated levels of phosphatidylcholine surfactant in lung tissue [Buccoliero et al., 2004]. Subsequent studies have confirmed these findings in human patients with Sandhoff disease, Gaucher disease type I, and sialidosis type I [Buccoliero et al., 2007]. These observations suggest that changes in phospholipid levels and composition in lung surfactant might be a general feature of sphingolipid storage diseases and may in part be responsible for the increased susceptibility of these patients to respiratory infections and lung disease.
Gangliosidoses
Ganglioside (primarily GM2 and GM3) accumulation has also been found to occur secondarily in other disorders, such as the mucopolysaccharidoses [Walkley and Vanier, 2009]. In the mucopolysaccharidoses, the primary storage of glycosaminoglycans is believed to lead to inhibition of the activity of several ganglioside-degrading lysosomal enzymes. In other disorders (e.g., Niemann–Pick disease type C), secondary ganglioside accumulation is believed to result from a disruption in retroendocytic movement of the substrate and does not appear to reflect simply the nonspecific changes in tissue cellularity (arising from neuronal loss and gliosis) [Walkley and Vanier, 2009].
Neuronal cells that have GM2 ganglioside storage exhibit meganeurite formation (i.e., axonal hillock enlargement) and ectopic dendritogenesis (i.e., the sprouting of new synapse-covered dendritic neurites at the axon hillock) [Walkley and Vanier, 2009]. Axonal spheroid formation (or neuroaxonal dystrophy) is also often noted. The spheroids occur as focal enlargements of various sizes scattered along myelinated and unmyelinated axons in the gray and white matter. In contrast to neuronal cell bodies, which contain characteristic storage material, spheroids consist of collections of multivesicular and dense bodies, mitochondria, and other organelles that would normally be found being transported along axons [Walkley and Vanier, 2009]. These observations suggest that the development of spheroids may involve defective endocytic trafficking within axons.
GM1 Gangliosidoses
Deficient β-galactosidase activity leads to the incomplete metabolism of several other substrates, including galactose-containing glycoproteins, N-acetylgalactosamine, lactose, and keratan sulfate (a glycosaminoglycan). Phenotypic differences likely result from varying activities of the mutant β-galactosidase enzyme against its various substrates, which may explain the dysmorphic facial features reminiscent of the mucopolysaccharidoses in the early infantile form of GM1 gangliosidosis and the predominance of skeletal disease in Morquio’s syndrome type B (due to allelic mutations) [Brunetti-Pierri and Scaglia, 2008]. Allelic mutations involve distinct sequence alterations of the same gene; the phenotype encountered may be different because the specific underlying gene defects involve distinct domains of the protein that subserve different functions. Studies in the mouse model of GM1 gangliosidosis have found that GM1 storage leads to several cellular changes (including activation of the unfolded protein response pathway), the upregulation of BiP and CHOP (C/EBP homologous transcription factor), and the activation of JNK2 and caspase 12, which lead to neuronal apoptosis [Tessitore et al., 2004].
Histologic examination of the brain of two infants with GM1 gangliosidosis revealed a marked decrease in the number of oligodendrocytes and myelin sheaths [van der Voorn et al., 2004]. An immunohistochemical decrease in proteolipid protein and a more profound deficiency of myelin basic protein were also found; these observations indicate that the brain lesions may not simply result from a delay or arrest in myelination, but are due to a “dying-back” oligopathy. In addition, amyloid precursor protein–immunoreactive aggregates were observed in proximal axons and meganeurites, as well as in white matter axons. These data suggest that the myelin deficit results from a loss of oligodendrocytes and abnormal axoplasmic transport, consequent to the massive neuronal storage of GM1 gangliosides.
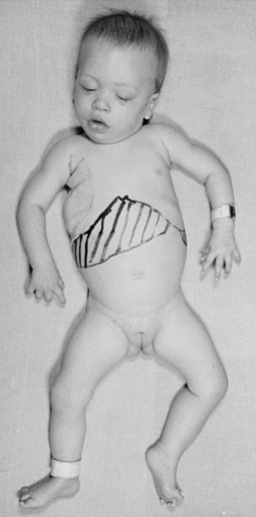
The late infantile form (with onset usually between 12 and 18 months of life) often manifests with gait disturbances and frequent falls. There is usually no distinctive facial dysmorphism, and the liver and spleen are not enlarged. Skeletal deformities, such as hypoplasia of the acetabula and proximal deformity of the metacarpal bones, may be found on radiographic examination but are usually milder than those noted in the early infantile form of GM1 gangliosidosis. Patients develop seizures and spastic quadriparesis, with prominent pseudobulbar signs (i.e., drooling and dysphagia) (Figure 36-2). Death is usual between the ages of 3 and 10 years. The absence of peripheral nerve involvement (i.e., normal conduction velocities) and normal cerebrospinal fluid findings help distinguish this condition from metachromatic leukodystrophy and Krabbe’s disease.
In the juvenile or late-onset form, disease symptoms usually develop in late childhood or adolescence but may also occur as late as the third or fourth decade of life. Delayed onset is partly related to the presence of residual enzyme activity, which in one patient was reported to be associated with homozygosity for R521C mutation in the β-galactosidase gene [Silva et al., 1999]. This clinical variant has been observed in different ethnic groups but appears to be prevalent among the Japanese. Affected individuals have a protracted clinical course characterized by dysarthria and extrapyramidal signs (especially dystonia). Pathologic studies have revealed marked GM1 ganglioside storage in the basal ganglia. Intellectual impairment is slight to moderate.
Mutation analyses have revealed broad heterogeneity, with 102 β-galactosidase gene defects described to date [Brunetti-Pierri and Scaglia, 2008]. Certain gene defects tend to be common in particular subtypes; for instance, the R482H and R208C are often seen in infantile cases, R201C in the late infantile/juvenile patients, and I51T among rare adult/chronic cases. In contrast to the first two mutations, the last two defects are associated with moderate catalytic activity, consistent with expectations for the resultant phenotype. The R59H has been reported to be prevalent among Brazilians, Iberian, and Roma patients with GM1 gangliosidosis.
Only symptomatic treatment is available. In preclinical studies, substrate reduction therapy using two related imino sugars revealed that NB-DGJ led to an optimal response (in terms of survival and fewer side effects in a mouse model of GM1 gangliosidosis), when compared to treatment with NB-DNJ (miglustat). However, functional improvement was greater with NB-DNJ, probably due to the greater impact of NB-DNJ on CNS inflammation [Elliot-Smith et al., 2008].
GM2 Gangliosidoses
The GM2 gangliosidoses include several variants, all of which are associated with the neuronal storage of the monosialoganglioside GM2, resulting in the formation of meganeurites (i.e., distorted and ballooned neurons) and ectopic dendritogenesis. Primary accumulation of GM2 gangliosides occurs with mutations involving the genes encoding the α (Tay–Sachs disease) or β (Sandhoff’s disease) subunit of hexosaminidase A or the GM2 activator protein (in the AB variant) [Fernandes Filho and Shapiro, 2004]. The hexosaminidase A enzyme (with a molecular mass of approximately 100 kDa) is a trimer consisting of one α and two β subunits, encoded by structural genes on different chromosomes (see Table 36-1). Mutations of the β subunit also lead to deficiency of hexosaminidase B (a tetrameric homopolymer of β subunits).
GM2 ganglioside storage is also seen in the so-called B1 variant, resulting from an altered substrate specificity of hexosaminidase A, wherein the mutated enzyme retains the ability to degrade the artificial substrate (used in most diagnostic assays), but not the sulfated or natural substrate in vivo. Clinical and imaging findings are congruent with those reported for Tay–Sachs and Sandhoff’s diseases [Grosso et al., 2003]. The highest incidence (approximately 3.1 per 100,000 live births) for the B1 variant of GM2 gangliosidoses has been described in Portugal, which has been suggested as the point of origin of a founder mutation (R178H) seen in Brazilian patients [Tutor, 2004].
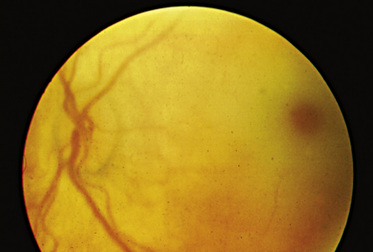
The “classic” infantile form of Tay–Sachs disease is named after a British ophthalmologist (Warren Tay) and an American neurologist (Bernard Sachs); both provided initial descriptions (in the 1880s) of the typical disease manifestations. Affected infants with Tay–Sachs disease have psychomotor deterioration and hyperacusis together with axial hypotonia, bilateral pyramidal signs, and blindness (associated with persistent pupillary responses) (Figure 36-3). The macular cherry-red spot is a characteristic hallmark of the disease (Figure 36-4). Head control is poor, and the size of the head increases markedly in the second year of life from enlargement of the brain (megalencephaly) and not because of tension hydrocephalus. Ultimately, with loss of the neurons and further gliosis, the ventricles may appear enlarged. Affected children become spastic and cachectic. Death usually occurs between 3 and 5 years of age. As patients approach the terminal stages, generalized tonic-clonic and minor motor seizures can occur. The patients do not have any of the following signs: dysmorphic features, visceromegaly, skeletal deformities, and signs of peripheral nerve involvement. These observations are useful in the delineation of Tay–Sachs disease from other lysosomal storage diseases associated with neurodegenerative features (Box 36-1).

Box 36-1 Clinical Manifestations Reported in Patients with Lysosomal Storage Disorders




























































































































* Conductive, sensorineural, or a combination, with involvement of cochlear and CNS dysfunction.
† Describes the normal red macula surrounded by a pale retina, reflecting storage material in the perifoveal ganglion cells.
‡ Refers to constellation of radiologic findings, including macrocephaly, thickened calvaria, J-shaped sella turcica, spatulate ribs, bullet-shaped phalanges that are shortened, rounding and anterior beaking of the vertebral bodies (especially in the lumbar region), and flaring of the iliac wings.
In the general population, the incidence of Tay–Sachs disease has been estimated at 1 in 112,000 live births. Tay–Sachs disease used to be prevalent (1 in 3900 live births) among Ashkenazi Jews, but this situation has been addressed successfully through targeted screening programs. The success of targeted screening has prompted extension of the program to other disorders that are also common in Ashkenazi Jewish individuals, such as Gaucher’s disease, Niemann–Pick disease, and mucolipidosis IV. These particular disorders were chosen because they are found with increased carrier frequency among the Ashkenazi Jews, in whom causality has been attributed to a limited number of “common” mutations [Zhang et al., 2004].
The later-onset forms of GM2 gangliosidosis (referred to as LOTS, which stands for late-onset Tay–Sachs disease) can manifest in childhood, adolescence, or even adulthood. In contrast to the infantile-onset form, patients with juvenile- or adult-onset disease follow a more protracted course and show no ethnic predilection. Affected individuals develop dysarthria and walking problems, primarily resulting from spastic paraparesis (in the childhood-onset form, between the ages of 3 and 6 years) or proximal muscle weakness (in the adult form, with symptom onset in the teens). Differences in the age at onset and disease course have led to distinction of the childhood-onset form (usually referred to as chronic GM2 gangliosidosis) from the adult-onset variant (or late-onset Tay–Sachs disease) [Maegawa et al., 2006]. The phenotype associated with the B1 variant is similar to that encountered in the childhood-onset form.
Ataxia with cerebellar atrophy is also a prominent disease sign in the later-onset forms, and peripheral neuropathy has been described in a subset of patients [Shapiro et al., 2008]. Tonic-clonic or myoclonic seizures are encountered in some children; psychiatric disturbances (i.e., bouts of psychosis and episodes of depression) are present and may be the initial manifestation of disease, particularly among adult-onset patients [Zaroff et al., 2004]. Consideration of late-onset GM2 gangliosidosis should be made in the differential diagnosis of patients with signs of lower motor neuron and spinocerebellar dysfunction. Vision is not impaired and optic fundi are normal, although intellectual deterioration is frequent. The rate of progression and disease severity usually correlate with the age at onset (presumably determined by the severity of the underlying mutation). Childhood-onset disease often leads to death by 15 years of age, preceded by a period characterized by progressive spasticity, rigidity, and dementia, and ending in the vegetative state. Adult-onset patients may live into their 50s or early 60s. Mutation analyses performed in patients with late-onset Tay–Sachs disease have identified a high prevalence of the G269S gene defect, which is associated with residual hexosaminidase A activity, usually in combination with a null allele typical of classic Tay–Sachs disease [Neudorfer et al., 2005].
The AB variant due to a deficiency of the GM2 activator, which is necessary for the hydrolysis of GM2 gangliosides by hexosaminidase A in vivo, has a clinical phenotype that is indistinguishable from the infantile form of Tay–Sachs disease [Li et al., 2003; Sakuraba et al., 1999]. It is a rare condition associated with normal laboratory test results in assays of hexosaminidase A and B enzyme activity with use of the artificial substrates. Diagnostic confirmation requires highly specialized methods, available only through a few laboratories.
Sandhoff’s disease (O variant), due to mutations of the β subunit of hexosaminidase A, has an age at onset and clinical course characterized by neurologic and ophthalmologic findings that are similar to Tay–Sachs disease. Sandhoff’s disease is a pan-ethnic disorder. A distinguishing feature is hepatosplenomegaly and the presence of N-acetylglucosamine–containing oligosaccharides in urine (as only hexosaminidase B, which is also deficient in Sandhoff’s disease but not in Tay–Sachs disease, is responsible for cleaving the N-acetylglucosamine moiety of globosides) [Sakuraba et al., 2002]. Cardiomyopathy has also been noted occasionally. Death usually occurs between 2 and 4 years of age. There are extremely rare cases of patients with juvenile- or late-onset Sandhoff’s disease [Hendriksz et al., 2004].
The enzymatic basis of GM2 gangliosidosis can be ascertained by assays of total hexosaminidase and hexosaminidase A activity, which can be measured in leukocytes and cultured skin fibroblasts. The biochemical tests can be complemented by analysis of the underlying hexosaminidase A gene mutations. Among the Ashkenazi Jewish population, two null hexosaminidase A mutations (a four-base pair insertion [TATC1278] in exon 11 and a splice site gene defect [IVS12]) account for 96 percent of disease alleles among cases of Tay–Sachs disease [Frisch et al., 2004]. In this same population of patients, an allelic glycine to serine substitution at position 269 of the hexosaminidase A gene (G269S) represents the most common mutation among the adult-onset cases of GM2 gangliosidosis. Among non-Jewish patients, one mutation (IVS9) accounts for 14 percent of disease alleles [Cordeiro et al., 2000]. Pseudodeficiency for hexosaminidase A activity associated with decreased enzyme activity in vitro exists and can be clarified by DNA testing or analysis of enzyme activity with use of the natural rather than the artificial substrate. Two mutations (i.e., R247W and R249W) have been associated with pseudodeficient hexosaminidase A activity [Cordeiro et al., 2000].
Investigations have proved that the infantile cases of Tay–Sachs disease are usually due to a combination of severe deleterious mutations, whereas patients with the later-onset forms often have a combination of one deleterious mutation and a second “milder” mutation associated with residual enzyme activity (based on in vitro mutagenesis and protein expression studies). The most frequent mutation responsible for the B1 variant of GM2 gangliosidosis is R178H [Matsuzawa et al., 2003].
Pathologic brain studies reveal widespread neuronal GM2 ganglioside storage throughout the cortex and central nuclear structures, the spinal cord, and the autonomic ganglia. Evidence of neuronal storage has been established in fetuses as early as 12–22 weeks of gestation. GM2 gangliosides are implicated in synaptogenesis, and it has been speculated that an altered pattern of cellular gangliosides may interfere with normal synaptic transmission [Zhao et al., 2004].
Lyso-derivatives of GM2 gangliosides, which are cytotoxic and may be responsible for inducing the neuronal degeneration, are found to be increased in tissues of patients with GM2 gangliosidoses. When they were initially described, the lysolipids were believed to exert their toxicity through inhibition of protein kinase C. More recently, studies in animal models have found that progressive CNS inflammation (as evident from the expression pattern of several inflammatory markers and cytokines) and an alteration in the integrity of the blood–brain barrier occurred coincidentally with the onset of clinical disease signs [Jeyakumar et al., 2003].
Definitive therapy for GM2 gangliosidoses is not available. Substrate reduction therapy is currently under investigation. This approach is based on the inhibition of glucosylceramide synthesis by the imino sugar miglustat (NB-DNJ), and more recently by the galacto-derivative [Andersson et al., 2004]. Preclinical studies have found that animal models of GM2 gangliosidosis (primarily the Sandhoff mouse) given miglustat had a reduction in tissue storage of GM2 ganglioside, with delayed symptom onset and extended survival [Jeyakumar et al., 1999]. Treatment with NB-DGJ demonstrated greater therapeutic efficacy (specifically, extended life expectancy and increased delay in symptom onset) than NB-DNJ, with none of the side effects, such as weight loss and gastrointestinal tract distress, reported with NB-DNJ [Andersson et al., 2004]. Unfortunately, clinical trials using miglustat did not alter ultimate neurologic prognosis in pediatric and adult patients with GM2 gangliosidosis [Maegawa et al., 2009; Shapiro et al., 2009]. Salutary changes noted in the animal models subjected to bone marrow transplantation have generated renewed interest in cellular approaches to treatment of the GM2 gangliosidoses; potentially in combination with substrate reduction therapy [Jeyakumar et al., 2001].
Fabry’s Disease
Fabry’s disease is an X-linked disorder, first described independently in 1898 by Johanes Fabry (from Germany) and William Anderson (from the UK). It is caused by a deficiency of the lysosomal hydrolase α-galactosidase A and the accumulation of glycosphingolipids (predominantly globotriaosylceramide [ceramide trihexoside] and galabiosylceramide) [Schiffmann, 2009]. As a consequence, lipid deposits can be found in the epithelial cells of the cornea, glomeruli and tubules of the kidneys, cardiac myocytes, and endothelial and smooth muscle cells of blood vessels. In the nervous system, lipid deposits are evident in ganglion cells of the dorsal root and autonomic nervous system, and in specific cortical and brainstem structures. The distribution pattern of the storage material determines the associated clinical findings, including corneal and lenticular opacities, acroparesthesias, angiokeratomas, and anhidrosis or hypohidrosis (abnormal sweating) [MacDermot et al., 2001a]. Major morbidity in Fabry’s disease derives from renal, cardiac, and brain involvement.
Although the disease primarily affects males, a significant proportion of females may also develop disease-related complications; however, onset among carrier females may be delayed [MacDermot et al., 2001b]. Disease incidence has been estimated at 1 in 117,000 males; however, this is likely an underestimate as certain patients may have atypical features (including later onset of cardiac complications) and not be properly identified. Most estimates also do not include females who may be missed because they remain asymptomatic. Recent studies have led to identification of cases of Fabry’s disease among patients with cryptogenic stroke, and those with hypertrophic cardiomyopathy or on dialysis or post-kidney transplant [Rolfs et al., 2005; Monserrat et al., 2007; Kleinert et al., 2009]. In the absence of a positive family history, the diagnosis of Fabry’s disease is often delayed (by as much as a decade).
In late childhood or adolescence, affected individuals may experience recurrent attacks of burning or lancinating pains in the distal extremities (acroparesthesias), often associated temporally with physical activity and fever [MacDermot and MacDermot, 2001]. The pains may last for days or weeks, and edema of the hands and feet may occur. Physical examination often reveals angiokeratomas (telangiectatic skin lesions) distributed over the “bathing trunk” area between the nipple line and above the knees, in the umbilical region, and, in males, over the penis and scrotum (Figure 36-5). Corneal opacities, noted as a diffuse haze or in a verticillate or whorl-like pattern on slit-lamp examination, are typical; they are usually also seen among females and used to identify potential carriers before the availability of genetic test results. These eye findings do not interfere with vision, although central retinal artery occlusion can rarely occur and may lead to acute blindness [Orssaud et al., 2003]. Unilateral or bilateral hearing loss may develop [Germain et al., 2002]. Most patients also experience abdominal pains and bouts of diarrhea. The vasomotor, sweating, and gastrointestinal problems may reflect a dysfunction of the autonomic nervous system [Kolodny and Pastores, 2002].
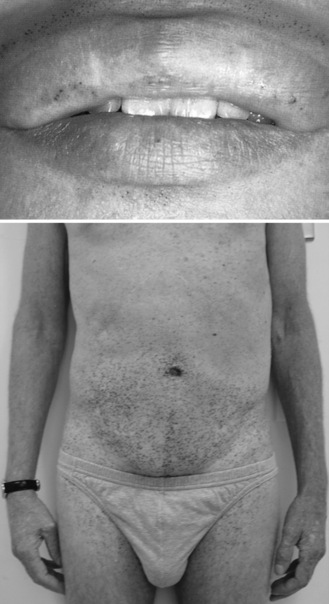
In classically affected males, the condition leads to chronic renal insufficiency and eventually renal failure (usually between the ages of 35 and 45 years), often preceded by proteinuria (mostly in the non-nephrotic range) and hyposthenuria [Thadhani et al., 2002]. In early or middle adulthood, patients may develop cardiac conduction problems or arrhythmia together with hypertrophic cardiomyopathy and ultimately heart failure [Glass et al., 2004]. Obstructive airway disease may contribute to limitations in exercise tolerance. Patients are also at risk for transient ischemic events or stroke, related to ischemic or thrombotic events [Kolodny and Pastores, 2002]. Cerebral imaging and blood flow studies have paradoxically revealed hyperperfusion attributed to altered vascular reactivity rather than arterial occlusive disease [Moore et al., 2003]. There is increased tortuosity of the blood vessels (predominantly in the posterior circulation) and white matter signal abnormalities. Studies have also found that patients with Fabry’s disease have evidence of endothelial dysfunction and leukocyte activation consistent with a prothrombotic state, which may explain the risk for focal cerebral signs such as aphasia and hemiplegia [Altarescu et al., 2001]. Median cumulative survival in untreated males (50 years) and females (70 years) is reduced [MacDermot et al., 2001a, b].
The diagnosis of Fabry’s disease can be established in males (hemizygotes) on the basis of reduced α-galactosidase A activity in plasma, or more reliably in leukocytes and cultured skin fibroblasts. A significant proportion of carrier females (heterozygotes) may have residual enzyme activity that overlaps with measurements obtained for the general population. Mutation analysis is a more reliable means for assignment of female carrier status, although this approach necessitates sequence characterization to identify the causal gene defect because most affected individuals tend to have a “private” mutation (i.e., unique or limited to few affected families). Lyonization may partly explain the wide variability in clinical presentation among carrier females. In addition, certain mutations may be associated with significant residual enzyme activity and may explain the delayed onset of symptoms or atypical disease course observed in some patients [Garman and Garboczi, 2002]. Affected individuals have elevated urinary excretion of ceramide trihexoside, although this requires demonstration with specialized testing (by high-performance liquid chromatography or tandem mass spectrometry). Biochemical or molecular genetic testing can be performed for prenatal diagnosis.
Before the introduction of enzyme therapy with agalsidase alfa (Replagal) or agalsidase beta (Fabrazyme), treatment was primarily symptomatic. Analgesics and often opiate-type medications are given for acroparesthesia. The frequency and severity of the pain episodes may be reduced or eliminated by chronic low-dose phenytoin (Dilantin) or carbamazepine (Tegretol) treatment. Patients are usually also given low-dose aspirin or antiplatelet aggregating agents for stroke prophylaxis and angiotensin-converting enzyme inhibitors (e.g., enalapril [Vasotec]) for renal protection in those with proteinuria. Dialysis or kidney transplantation is introduced after renal failure, and patients may require a pacemaker or defibrillator for cardiac conduction abnormalities or arrhythmias. Patients with severe heart valve problems may need a mechanical or tissue heart valve replacement, and cardiac transplantation has been performed in patients with cardiomyopathy and heart failure [Karras et al., 2008].
Enzyme therapy for Fabry’s disease is safe and effective in reducing the amount of tissue lipid storage. Associated benefits include the reduction of both pain episodes and the use of chronic pain medications, stabilization or improvement of renal function in patients with normal or mildly reduced kidney function (glomerular filtration rate of 60 mL/min or higher), and a reduction of excess cardiac mass among those so affected [Pastores and Thadhani, 2002; Lidove et al., 2007]. Gastrointestinal complaints and abnormal sweating have also improved with enzyme therapy [Hoffmann et al., 2004; Schiffmann et al., 2003]. Cerebrovascular studies have demonstrated reversibility of the functional abnormalities with treatment, although patients remain at risk for stroke [Moore et al., 2002; Schiffmann, 2009]. Long-term studies will be required to ascertain the effectiveness of therapy in altering the natural history of the disease and improving survival. Additional studies will also be necessary to ascertain the determinants of therapeutic response, which is likely influenced by the presence of preexisting pathologic processes (e.g., glomerulosclerosis, calcified and stenotic heart valves). A significant proportion of enzyme-treated patients form antibodies against the administered protein, which results in neutralization of enzyme activity [Linthorst et al., 2004]. The long-term implications of antibody formation will need to be carefully assessed, although serial measurements indicate immunotolerance (i.e., a decrease in antibody titers over time) [Pastores, 2004]. Substrate reduction therapy and the use of pharmacologic chaperones, which remain investigational, are potential alternative or adjunctive strategies in the treatment of Fabry disease [Heare et al., 2007; Benjamin et al., 2009]. Gene therapy experiments currently being undertaken in the mouse model of Fabry’s disease appear to suggest promising results [Ziegler et al., 2004].
Gaucher’s Disease
The disease described by PCE Gaucher (in 1882) as an epithelioma of the spleen is known to be a lipid storage disorder resulting from a deficiency of the lysosomal enzyme glucocerebrosidase (acid β-glucosidase) [Butters, 2007]. The accumulation of its incompletely metabolized substrate, glucocerebroside (glucosylceramide), is confined to cells of monocyte-macrophage lineage. Glucocerebroside in the peripheral system is derived primarily from the breakdown of senescent red and white blood cell membranes. In the CNS, glucocerebroside is a byproduct of the catabolism of globosides and gangliosides.
There are three major clinical subtypes, delineated on the basis of the presence or absence of neurologic involvement. Assignment of clinical subtype is based on observations of the age at onset, evolution of the disease, and, when it is present, the rate and severity of psychomotor deterioration [Goker-Alpan et al., 2003].
Type 1 Gaucher’s disease encompasses the non-neuropathic type associated with hepatosplenomegaly, anemia and thrombocytopenia, pulmonary involvement, and bone disease. Although type 1 Gaucher’s disease has been referred to as the adult type, this designation is misleading as onset of disease may be evident from childhood. There is wide heterogeneity in clinical presentation. A small proportion of patients remain asymptomatic for most of their adult lives; others may develop significant disease progression earlier. Although the patients do not have a primary neurodegenerative disease, there can be secondary neurologic complications (e.g., local nerve injury due to hemorrhage and entrapment, or from spinal cord compression with collapse of a vertebral body) [Pastores et al., 2003].
There are several reports of adult patients with type 1 Gaucher’s disease who have developed Parkinson’s disease [Lwin et al., 2004]. Neuropathologic studies of brains obtained from patients with type 1 Gaucher’s disease have revealed focal regions of gliosis (especially in the hippocampal CA2–CA4 region and calcarine cortex) and the presence of α-synuclein–immunoreactive cortical and brainstem-type Lewy bodies [Wong et al., 2004]. These observations suggest a potential causal relationship that requires further investigation [Mata et al., 2008].
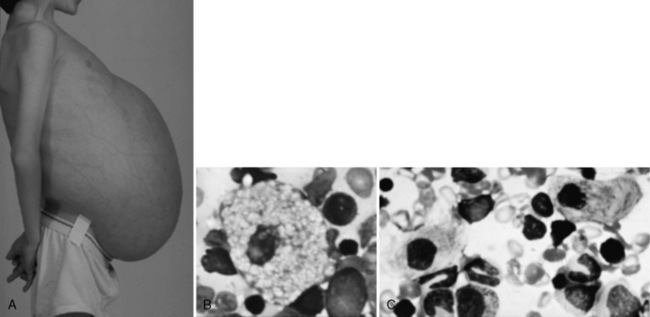
Type 1 Gaucher’s disease is a pan-ethnic disorder that has been noted as prevalent among individuals of Ashkenazi Jewish descent (carrier frequency of approximately 1 in 20) [Colombo, 2000]. In most populations, type 1 Gaucher’s disease represents the most common lysosomal storage disorder with an estimated frequency of about 1 in 50,000 [Applegarth et al., 2000].The hematologic and visceral findings that typify type 1 Gaucher’s disease are also encountered in the neuropathic subtypes (Figure 36-6).
Type 2 Gaucher’s disease represents the acute neuropathic form, associated with disease onset before the age of 12 months. Patients develop spasticity with head retraction, dysphagia, and a rapidly fatal course (with death usually between 2 and 3 years of age). Laryngeal stridor and trismus (due to bulbar spasticity) are added problems, and aspiration pneumonia is a frequent complication. Patients do not have dysmorphic features or the cherry-red spot that is typical of infantile Tay–Sachs disease and Niemann–Pick disease. However, the most severely affected patients can present with congenital ichthyosiform-collodion skin abnormalities or nonimmune hydrops fetalis [Staretz-Chacham et al., 2009]. The latter phenotype is characterized by an aggressive course, with death in the first few weeks of life. Similar findings were noted in the animal (mouse) model for Gaucher’s disease in association with an alteration of the glucocerebroside to ceramide ratio and a disruption in the lipid membranes at the level of the stratum corneum [Eblan et al., 2005]. Bone involvement, characteristically found in patients with types 1 and 3 Gaucher’s disease, is not seen in type 2 Gaucher’s disease, perhaps because there is insufficient time for the skeletal disease process to evolve.
Type 3 Gaucher’s disease, which may manifest before the age of 2 years, is associated with chronic neurologic problems including tonic-clonic and myoclonic seizures, ataxia, and extrapyramidal rigidity, often complicated by severe pulmonary involvement (including interstitial lung disease) [Campbell et al., 2003]. Paralytic strabismus with oculomotor apraxia (i.e., saccadic initiation failure) is a characteristic finding in patients with primary neuropathic Gaucher’s disease. Brainstem auditory-evoked response testing often reveals diverse results, including prolonged peak and interpeak latencies for waves I–V and generally dysmorphic wave formation [Bamiou et al., 2001].
The clinical expression of type 3 Gaucher’s disease can be complex, but neurologic problems can occasionally be restricted to supranuclear horizontal gaze palsy despite significant extraneurologic systemic problems. An increased number of cases have been identified in the Norrbottnian region of Sweden, attributed to a founder effect (for the L444P mutation) [Dahl et al., 1993]. The founder effect refers to the phenomenon wherein the increased frequency of a certain gene defect within a defined population is explained by common ancestry (i.e., a shared identity by descent) [Slatkin, 2004].
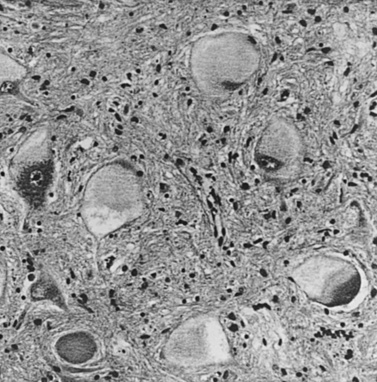
The basis of primary neurologic involvement in certain Gaucher’s disease subtypes has not been fully elucidated. In contrast to the amount of lipid storage material measured in the liver and spleen, levels of glucocerebroside are not significantly elevated in brain tissue. Glucosylsphingosine (psychosine), an alternative metabolic byproduct of severely deficient glucocerebrosidase deficiency, is believed to play a contributory role [Schueler et al., 2003]. Evidence of neuronal apoptosis has been demonstrated in type 2 Gaucher’s disease, and lipid-filled cells can be found in the perivascular Virchow–Robin spaces. Neuronophagic microglial nodules can also be found in several regions of the brain (e.g., cortex, thalamus, basal ganglia, brainstem, and cerebellum) and in the spinal cord (Figure 36-7). Elevation of glucocerebroside in neurons, possibly involving endoplasmic reticulum membranes, leads to increased calcium stores (possibly secondary to modulation of the ryanodine receptors) [Lloyd-Evans et al., 2003]. These cellular changes may be added factors that promote neuropathic forms of Gaucher’s disease. Analysis of gene expression profiles in brains of the Gaucher’s disease mouse model revealed downregulation of the bcl-2 gene in the brainstem and cerebellum, but not in cortex [Hong et al., 2004a]. In situ labeling of DNA fragmentation by the terminal transferase-mediated dUTP nick-end labeling (TUNEL) assay confirmed that apoptosis occurred at these sites. These observations suggest that the accumulation of either glucocerebroside or glucosylsphingosine affects the expression of apoptosis mediators and could be responsible for neuronal cell death. Reduced expression of brain-derived neurotrophic factor and nerve growth factor has also been observed in the cerebral cortex, brainstem, and cerebellum of the Gaucher’s mouse, compared with that in wild-type mouse, and extracellular signal-regulated kinase (ERK) 1/2 expression was downregulated in neurons and correlated with a decreased number of neurons [Kim et al., 2006].
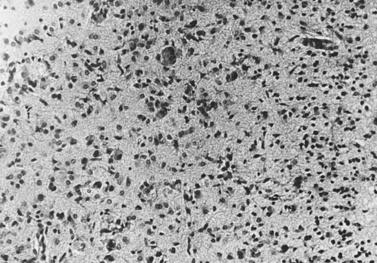
Fig. 36-7 Section of occipital cortex in an infant with Gaucher’s disease.
(Courtesy Joo Ho Sung, Minneapolis, MN.)
An atypical rare variant characterized predominantly by cardiac problems, such as aortic and mitral valve calcification, appears to be restricted to patients with a unique genotype (i.e., homozygosity for the D409H mutation) [George et al., 2001]. These findings were first reported among Arab patients with Gaucher’s disease from the Jenin area, but this phenotype has since been reported among other ethnic groups.
Rare patients with evidence of glucocerebroside storage and normal glucocerebrosidase activity in vitro have been described; careful investigations have revealed a deficiency of saposin [Elleder et al., 2005]. Patients with saposin C or prosaposin deficiency usually present with symptoms characteristic of subacute neuropathic Gaucher’s disease (i.e., progressive horizontal ophthalmoplegia, pyramidal and cerebellar signs, myoclonic jerks, and generalized seizures). Saposin C, which is derived from the proteolytic cleavage of a precursor molecule (prosaposin), is a co-factor required by glucocerebrosidase in the hydrolysis of glucocerebroside. In biochemical assays to measure glucocerebrosidase activity, the role of saposin C is replaced by the use of detergents. This replacement explains the normal results obtained when the activity of glucocerebrosidase is measured in vitro. Prosaposin, through activation of mitogen-activated protein kinases, may act as an apoptosis suppressor [Misasi et al., 2004]. Thus, a prosaposin deficiency could promote neuronal cell loss.
Several markers, readily measured in the plasma or serum of patients with Gaucher’s disease, are often elevated. Testing often reveals elevated measures of tartrate-resistant acid phosphatase, angiotensin-converting enzyme, ferritin, CCL18/PARC, and chitotriosidase activity [Wajner et al., 2004; Aerts et al., 2005]. Although these findings have not been associated with a specific pathologic mechanism, the alterations have been taken to reflect the systemic burden of disease. Polyclonal hypergammaglobulinemia is also a frequent finding, particularly in the elderly, and the risk for multiple myeloma in these patients is increased [Taddei et al., 2009].
Bone involvement (e.g., severe osteopenia and osteonecrosis of major joints including the humeral and femoral heads) can be a major source of morbidity for patients with types 1 and 3 Gaucher’s disease. The indicators of bone turnover (e.g., osteocalcin, bone-specific alkaline phosphatase, N-telopeptides) have not been consistently abnormal, even when significant bone disease exists, suggesting the dominance of local (possibly vascular) over systemic factors [Ciana et al., 2003; Pastores and Meere, 2005].
Cardiopulmonary complications can occur, and severe interstitial lung diseases and alveolar consolidation have been described. Rare cases of patients with pulmonary hypertension, which has been found primarily among splenectomized adult patients, have also been reported [Mistry et al., 2002].
Biochemical testing is often complemented by analysis of the common mutations (specifically, N370S, L444P, IVS2+1, 84insG, and R496H). Carrier assignment can also be performed more reliably through mutation analysis. The five gene defects noted account for approximately 96 percent of disease alleles among Ashkenazi Jewish individuals, but for only about 40–60 percent of mutations among the non-Jewish patients (wherein the 55-base pair deletion is frequent) [Wallerstein et al., 2001]. Correlation studies have revealed genotype–phenotype concordance to be imperfect, with isolated exceptions. For instance, the presence of at least one N370S (1226G) allele precludes development of primary CNS involvement. In general, patients homozygous for the N370S mutation tend to have milder disease compared with other patients who are compound heterozygotes for the other more deleterious alleles. Patients homozygous for the L444P mutation tend to have severe disease, often (but not invariably) associated with primary neurologic manifestations.
Examination of bone marrow tissue (not the preferred method of diagnosis) reveals clumps of lipid-engorged macrophages (Gaucher cells). Magnetic resonance imaging (MRI) of the spine (sagittal views) and femurs (coronal views) has been useful in assessment of the degree of bone marrow infiltrative disease in Gaucher’s disease [Maas et al., 2003]. On occasion, Gaucher-like cells (or pseudo-Gaucher cells) in isolation may be detected in other disorders, such as chronic myelocytic leukemia, and in disease states associated with increased cell turnover [Saito et al., 2007]. True Gaucher cells are periodic acid–Schiff-positive, and their cytoplasm has an irregular wrinkled appearance (see Figure 36-6B). Studies have revealed that Gaucher cells are alternative activated macrophage-like cells that express acid phosphatase, CD68, CD14, and HLA class II, but not CD11b, CD40, or dendritic cell markers [Boven et al., 2004]. Furthermore, Gaucher cells exhibited infrequent immunoreactivity for mannose receptor and did not express proinflammatory cytokines such as tumor necrosis factor-α and monocyte chemoattractant protein 1, but they did express the markers CD163, CCL18, and interleukin-1 receptor antagonist.
Before the introduction of enzyme therapy, the care of affected individuals was primarily palliative and included analgesics for bone pain, blood transfusions for severe anemia and thrombocytopenia, and splenectomy for severe hypersplenism. In addition, supplemental agents (such as bisphosphonates, calcium, and vitamin D) have been administered to patients (primarily adults) with Gaucher’s disease and severe osteopenia. Pregnancy in affected women requires careful monitoring and attention to risk reduction for bleeding and other problems, especially around labor and delivery in those who are severely thrombocytopenic [Elstein et al., 2004].
Enzyme replacement therapy is based on the regular intravenous administration of glucocerebrosidase, initially purified from human placenta (alglucerase [Ceredase]), and more recently by use of the recombinant enzyme (imiglucerase [Cerezyme]) and generated from overexpression in cultured mammalian cells [Charrow, 2009]. The therapeutic protein is modified from its native form by sequential deglycosylation to expose the mannose residues necessary for targeted cellular uptake through the mannose receptor.
Enzyme therapy reverses the hematologic and visceral manifestations of Gaucher’s disease and leads to the stabilization or improvement of bone involvement [Charrow et al., 2004; Pastores et al., 2004]. Enzyme therapy does not influence the ultimate progression of neurologic complications in patients with type 2 Gaucher’s disease, and thus its use for this subgroup of patients is deemed inappropriate. Although intravenously administered enzyme does not lead to delivery of significant amounts of the protein across the blood–brain barrier, its use in patients with type 3 Gaucher’s disease leads to improved quality of life (as a consequence of the reversal of the extraneurologic disease manifestations). Abnormal brainstem auditory-evoked response findings noted at baseline in eight children with type 3 Gaucher’s disease continued to deteriorate with high-dose enzyme replacement therapy [Campbell et al., 2004]. Thus, patients with type 3 Gaucher’s disease and their parents should be counseled appropriately about long-term outcome and the possibility that certain aspects of the disease may continue to progress. Enzyme therapy alone appears to improve bone density in patients with significant osteopenia, but only after years of treatment; thus, it is not uncommon for these patients to be prescribed supplemental treatment with bisphosphonates [Wenstrup et al., 2004; Sims et al., 2008].
Substrate reduction therapy (with miglustat [Zavesca]) has been shown to lead to improvements in several key clinical features of Gaucher’s disease. This approach, when it is used alone (i.e., monotherapy), is based on the partial reduction of the synthesis of the substrate (to meet the metabolic capacity of the mutant enzyme) [Cox et al., 2000; Pastores and Barnett, 2003]. Studies have proved that a significant proportion of the orally administered agent (miglustat) can be found in cerebrospinal fluid [Platt and Butters, 2000]. Unfortunately, clinical trials with this drug in patients with type 3 Gaucher’s disease on concomittant enzyme therapy has not been shown to alter ultimate neurologic prognosis [Schiffmann et al., 2008].
A minority of patients (about 10–15 percent) receiving enzyme replacement therapy have developed antibodies (primarily immunoglobulin G) against the infused glucocerebrosidase, although only a few have developed overt adverse events (mainly pruritus and hives), which respond readily to oral premedications (such as antihistamines) [Charrow, 2009]. This safety profile has led to consideration of enzyme therapy as the preferred treatment for patients with Gaucher’s disease, including those with type 3 disease. In patients with type 3 Gaucher’s disease and progressive CNS manifestations, bone marrow transplantation has been performed with favorable outcomes. However, the procedural risks associated with bone marrow transplantation and the prospect of graft-versus-host disease have limited its indication to carefully selected cases. The feasibility of gene therapy for Gaucher’s disease remains to be re-examined [Hong et al., 2004b]. Meanwhile, alternative treatment options, including the use of a pharmacologic chaperone (isofagomine), are being explored [Yu et al., 2007].
Niemann–Pick Disease, Including Types A and B
Niemann–Pick disease is the eponymous designation for the storage disorders characterized by a primary deficiency of acid sphingomyelinase (ASM), associated with the progressive storage of sphingomyelin (phosphorylcholine) in the reticuloendothelial system. Impaired formation of ceramide within the lysosome or in the plasma membrane and its role as an apoptosis inducer are being actively examined as a potential added mechanism of disease [Jenkins et al., 2009]. In the acid sphingomyelinase “knockout” mouse, which develops a phenotype largely mimicking that of the neuronopathic form of Niemann–Pick disease (type A), the observed myelin deficiency has been attributed to reduced oligodendrocyte metabolic activity [Buccinnà et al., 2009].
Currently, Niemann–Pick disease types A and B are the terms used to refer to the infantile neuropathic and later-onset non-neuropathic forms of the disease, respectively. In actuality, both Niemann–Pick disease types A and B are allelic disorders (i.e., involving different mutations of the same gene) that result in primary deficiency of acid sphingomyelinase and represent the spectrum of phenotypes associated with mutations of the acid sphingomyelinase gene. Additional subtypes, referred to as Niemann–Pick disease types C and D in a classification scheme proposed by A. Crocker (1961), have clinical features that overlap with the classic Niemann–Pick disease types A and B. Eventually, Niemann–Pick disease types C and D were recognized as being associated with secondary and not primary acid sphingomyelinase deficiency. Indeed, a different cause for Niemann–Pick disease type C had been suggested by studies involving the fusion of cells (or formation of heterokaryons) from type A or B disease with type C cells. More recent investigations have found that Niemann–Pick disease types C and D are also allelic disorders due to mutations of one of two different genes (designated NPC1 and NPC2) leading to disruption in the trafficking or metabolism of cholesterol and sphingolipids [Walkley and Suzuki, 2004]. These various Niemann–Pick disease subtypes are all inherited as autosomal-recessive traits.
Patients with early infantile Niemann–Pick disease (type A) present with failure to thrive and hepatomegaly in the first few weeks or months of life, usually before evident neurologic regression and the appearance of the macular cherry-red spot. The disorder is prevalent among individuals of Ashkenazi Jewish ancestry (carrier frequency of 1 in 90). In contrast to Gaucher’s disease, there is marked abdominal distention due to hepatosplenomegaly and liver dysfunction. Serum levels of acid phosphatase are normal. Affected infants do not have dysmorphic facial features or skeletal deformities. Respiratory problems develop, and chest radiographs often reveal a fine miliary infiltration of the lungs [Minai et al., 2000]. There is developmental delay, with head lag and the inability to sit, and eventual regression with reduction in spontaneous movements and loss of interest in the surroundings. Unlike in Tay–Sachs disease, an exaggerated acousticomotor response cannot be elicited, and head circumference is either normal or moderately reduced. With disease progression, there is rigidity and opisthotonos. The findings on cerebrospinal fluid examination are normal. Death occurs in the second or third year of life.
Niemann–Pick disease type B is associated with visceral involvement (and by convention no neurologic problems) [Kolodny, 2000; Wasserstein et al., 2004]. The absence of significant brain involvement may be a consequence of the underlying mutation’s association with residual enzyme activity. Hepatosplenomegaly and pulmonary infiltrative disease may be detected in late infancy or early childhood. Progressive pulmonary infiltration is generally the major disease complication. On high-resolution chest computed tomography (CT), the main abnormalities consist of thickening of the interlobular septa and patchy areas of ground-glass attenuation [Minai et al., 2000]. These changes correspond to the reticular or reticulonodular pattern of abnormalities, involving mainly the lower lung zones, that can be seen on chest radiographs. Abnormal linear growth and delayed skeletal maturation are also common in children and adolescents with Niemann–Pick disease type B [Wasserstein et al., 2003].
An intermediate phenotype may be encountered, with retinal degeneration and other neurologic signs (including slowed nerve conduction velocities) appearing in late childhood, adolescence, or adulthood (Figure 36-8). A study involving 45 patients (23 males and 22 females, ranging in age from 3 to 65 years) with Niemann–Pick disease type B revealed the presence of macular halos or cherry-red spots that incidentally were found not to be an absolute predictor of neurodegeneration [McGovern et al., 2004a]. The course of disease in patients with a phenotype intermediate between type A and type B is similar to that observed in type 3 Gaucher’s disease, although the latter condition is often characterized by oculomotor apraxia and the absence of the cherry-red spot. In addition, there are differences found in the levels of chitotriosidase activity between patients with Gaucher’s disease (600-fold increase) and those with Niemann–Pick disease (30-fold increase) [Wajner et al., 2004].
Investigations of lipid abnormalities in ten patients with Niemann–Pick disease type A and 30 additional patients with Niemann–Pick disease type B revealed that all had low (<35 mg/dL) high-density lipoprotein cholesterol [McGovern et al., 2004b]. Also noted were hypertriglyceridemia and increased low-density lipoprotein cholesterol in 25 of 40 (62 percent) and 27 of 40 (67 percent) of the patients, respectively. Coronary artery calcium scores were positive (>1.0) in 10 of 18 type B patients studied, which suggests that Niemann–Pick disease may be associated with a risk for early atherosclerotic heart disease.
The diagnosis of Niemann–Pick disease types A and B can be established on the basis of deficient acid sphingomyelinase activity, demonstrable in leukocytes or cultured skin fibroblasts. It has been reported that the biochemical assay to measure acid sphingomyelinase activity by use of the artificial substrate 2-N-(hexadecanoyl)-amino-4-nitrophenyl phosphorylcholine can occasionally lead to a missed diagnosis [Harzer et al., 2003]. In this particular study, four patients had the Q292K mutation on at least one allele. Thus, when the diagnosis of Niemann–Pick disease remains strongly suspected and a negative result is obtained on enzyme testing, it is important to determine whether the assay was performed with the artificial substrate (as is the custom in most laboratories). Another approach in these cases may entail examination of bone marrow (see Figure 36-6B).
Bone marrow examination (not the preferred initial means of diagnosis) reveals lipid-laden “foamy” histiocytes (with a soap bubble appearance), also evident in tissue sections obtained from the liver. The accumulated material is sudanophilic and periodic acid–Schiff-negative. On occasion, the atypical storage cells in bone marrow have been designated sea-blue histiocytes [Candoni et al., 2001].
Three acid sphingomyelinase gene mutations (specifically, two missense defects, R496L and L302P, and one frameshift mutation, fsP330) represent the most common Niemann–Pick disease gene defects, accounting for approximately 95 percent of disease alleles among Ashkenazi Jewish patients with Niemann–Pick disease type A [Schuchman and Miranda, 1997]. Among Niemann–Pick disease type B cases from North Africa, a three-base in-frame deletion of exon 6 (R608del) is frequent (87 percent of mutant alleles). This mutation was also found to be prevalent among patients with Niemann–Pick disease type B from the Canary Islands of Spain, located on the northwest coast of Africa [Fernandez-Burriel et al., 2003]. Among patients with Niemann–Pick disease type B, homozygosity for the R608del mutation tends to be associated with a relatively milder phenotype compared with other combinations of acid sphingomyelinase gene mutations [Simonaro et al., 2002]. Several mutations in patients (primarily of Italian ancestry) with Niemann–Pick disease type B have been identified; in these studies, no correlation was found between onset of pulmonary symptoms and genotypes [Pittis et al., 2004]. Information about the causal mutation for specific families enables accurate carrier assignment. Niemann–Pick disease types A and B are pan-ethnic disorders, but the mutations that lead to disease outside of the populations just described tend to be private and require analysis of the acid sphingomyelinase gene sequence for identification. Prenatal diagnosis can be made by assay of acid sphingomyelinase activity in cultured chorionic villi or amniocytes [Vanier, 2002]. Preimplantation genetic diagnosis for Niemann–Pick disease type B has been reported [Hellani et al., 2004].
Only symptomatic treatment is available for Niemann–Pick disease types A and B, although enzyme therapy is under consideration for subtype B. In the murine model of Niemann–Pick disease, pulmonary delivery of recombinant acid sphingomyelinase improved clearance of lysosomal sphingomyelin from the lungs [Ziegler et al., 2009]. Repeated subcutaneous implantation of amniotic epithelial cells has not been shown to influence the ultimate disease course in prior therapeutic studies [Bembi et al., 1992]. Niemann–Pick disease type A is not amenable to hematopoietic stem cell transplantation because of its rapid progression, but limited experience suggests that hematopoietic stem cell transplantation may have some beneficial effects for Niemann–Pick disease type B. Report of a 16-year follow-up of allogeneic bone marrow transplantation carried out on a 3-year-old girl with Niemann–Pick disease type B revealed severe neurologic involvement (including bilateral cherry-red spots), despite normal findings on neurologic examination at the time of transplantation [Victor et al., 2003]. However, baseline pulmonary infiltration regressed after bone marrow transplantation, although there was no clinical evidence of pulmonary insufficiency. Her post-transplant course was complicated by severe graft-versus-host disease and respiratory arrest, factors that may have had an adverse influence on the neurologic outcome.
Niemann–Pick Disease, Including Types C and D
Niemann–Pick disease type C is a neurovisceral lipid storage disorder associated with a wide spectrum of clinical phenotypes. It is characterized neuropathologically by the widespread appearance of axonal spheroids, an accumulation of intraneuronal cytoplasmic inclusions, and neuronal loss [Vanier and Millat, 2003]. The disease incidence has been estimated at 1 in 150,000. Approximately 95 percent of cases have mutations in a gene (designated NPC1) that encodes a large transmembrane glycoprotein, localized primarily in the endosomes [Sturley et al., 2004]. A smaller group of patients who are clinically and biochemically indistinguishable from individuals with NPC1 mutations have a defect of the NPC2 gene, which encodes a small soluble lysosomal protein with cholesterol-binding properties (previously identified as epididymal secretory glycoprotein 1 [HE1]) [Vanier and Millat, 2004]. It is likely that both protein products (i.e., NPC1 and NPC2) function in a coordinated fashion, as studies performed on skin fibroblasts obtained from patients with either mutation reveal the accumulation of unesterified cholesterol and glycolipids in the lysosome–late endosome. Furthermore, the mating of mice genetically deficient for each protein (i.e., NPC1 or NPC2) results in the generation of double-deficient mice that resemble the single-disease mutants [Pentchev, 2004]. A consequence of cholesterol sequestration is a reduction in its availability, and an impairment of cholesterol-mediated homeostatic responses in the endoplasmic reticulum [Karten et al., 2009]. An abnormality of autophagy has also been demonstrated in cells obtained from the Niemann–Pick disease type C mouse model [Ishibashi et al., 2009], and other studies have shown an impairment of neurosteroidogenesis [Griffin et al., 2004].
Immunohistochemical studies involving the HE1/NPC2 protein revealed its localization in pyramidal or projection neurons in the cerebral cortex and amygdala, and in the Purkinje neurons of the cerebellum [Ong et al., 2004]. This regional pattern of expression is similar to the findings with NPC1, with a low level of expression of both proteins in regions derived from the diencephalon (such as the thalamus and hypothalamus). In contrast to NPC1, which is found predominantly in astrocytes, HE1/NPC2 has been observed mainly in neurons. The HE1/NPC2 protein has also been found in the cytosol of dendrites and on postsynaptic densities. These results suggest that NPC1 and HE1/NPC2 are differentially enriched in astrocytes and neurons, respectively, and that HE1/NPC2 may function in supporting the integrity of the postsynaptic densities of neurons [Ong et al., 2004].
The classically affected patients (accounting for 50–60 percent of cases) often have a benign, self-limited jaundice in early infancy and an unremarkable childhood [Vanier and Millat, 2003]. Eventually, between the ages of 3 and 8 years (less often after the 10th year of life), they are noted to be clumsy, have a tendency to fall, and develop ataxia. Hepatosplenomegaly may be noted, and supranuclear (vertical) gaze palsy, which may be accompanied by blinking or head thrusting, is evident on examination. With disease progression, patients experience dysarthria, dysphagia, and dementia. Additional neurologic manifestations include dystonia, choreoathetosis, and seizures (of the generalized tonic-clonic and myoclonic type). A proportion of patients with Niemann–Pick disease type C may present at birth with ascites or develop neonatal (cholestatic) jaundice and a rapidly fatal disease.
Cataplexy also occurs late in the disease but rarely may be the presenting feature of Niemann–Pick disease type C [Vankova et al., 2003]. Typically, the loss of postural tone is evoked by a humorous stimulus (gelastic cataplexy). Examination of cerebrospinal fluid has revealed reduced hypocretin-1 levels in two patients with Niemann–Pick disease type C, which suggests that the storage abnormalities may have an impact on the hypothalamus and, more specifically, hypocretin-containing cells [Kanbayashi et al., 2003]. These changes may be partially responsible for the sleep abnormalities and cataplexy seen in these patients.
Psychiatric disturbances, including psychosis, have been reported to occur in patients with Niemann–Pick disease type C, usually coinciding with the onset of puberty [Josephs et al., 2003]. Most patients develop spasticity and rigidity, and die of pulmonary complications in their teens or early adulthood. Pertinent negative findings include the absence of retinal changes, visual failure, and signs of lower motor neuron disease; cerebrospinal fluid examination findings are also normal.
A clinical severity scale has been developed to characterize and quantify disease progression in patients with Niemann–Pick disease type C. A linear increase in severity scores over time has been observed, independent of age of onset [Yanjanin et al., 2009]. In addition to neuronal storage, studies involving the mouse model of Niemann–Pick disease type C have revealed conspicuous hypomyelination in the cerebral white matter and corpus callosum. In brains from patients with Niemann–Pick disease type C, studies have found neuropathologic abnormalities (e.g., formation of tauopathies and neurofibrillary tangles) similar to those found in Alzheimer’s disease [Nixon, 2004]. Furthermore, the aggregated amyloid precursor protein cleavage product amyloid beta accumulates in late endosomes. In addition, presenilins (normally localized in the endoplasmic reticulum and endoplasmic reticulum–Golgi intermediate compartment) were distributed to late endosomes. These observations suggest that endosome dysfunction and the disruption of cholesterol homeostasis may promote mechanisms common to both Niemann–Pick disease type C and Alzheimer’s disease. In the mouse model, intracerebroventricular administration of two potent inhibitors (roscovitine and olomoucine) of the cyclin-dependent kinases attenuated the hyperphosphorylation of the neurofilament tau and mitotic proteins [Zhang et al., 2004]. These observations were associated with a reduction in the number of spheroids, the modulation of Purkinje neuron death, and the amelioration of the motor defects in these mice. The diagnosis of Niemann–Pick disease type C is based on demonstration in cultured skin fibroblasts of impaired cholesterol esterification and intralysosomal storage of unesterified cholesterol (which gives off an intense perinuclear fluorescence with filipin staining) (Figure 36-9). Identification of the causal NPC1 or NPC2 mutations requires specialized studies because of the existence of genetic heterogeneity (i.e., similar clinical disorder or phenotype resulting from two different gene defects) and private mutations in most of the patients (outside defined populations). In one study involving 143 unrelated patients with Niemann–Pick disease type C, mutation analysis led to identification of 251 of 286 (88 percent) disease alleles [Park et al., 2003]. The most common NPC1 mutation, I1061T, was detected in 18 percent of NPC alleles.
The designation Niemann–Pick disease type D was used to describe affected individuals among the French Acadians in Nova Scotia (Canada) [Greer et al., 1999]. In these patients, disease onset is usually between the ages of 2 and 4 years. After characterization of the NPC1 gene, studies revealed a common founder mutation (G309T) in this population of patients. Similarly, a specific gene defect (I106T) has been identified among Spanish American cases of Niemann–Pick disease type C in the Upper Rio Grande Valley (Colorado) attributed to a founder effect [Millat et al., 1999].
The management of patients with Niemann–Pick disease type C is primarily symptomatic. A therapeutic trial involving dietary restriction of cholesterol and the administration of cholesterol-lowering agents (i.e., statins) has resulted in the reduction of hepatic and plasma cholesterol levels but no effect on neurologic outcome [Patterson and Platt, 2004].
Secondary accumulation of GM2 ganglioside in the brains of patients with Niemann–Pick disease type C, and as also shown in the mouse model, is believed to contribute to the neuronal degeneration. Preclinical studies in the knockout mouse model treated with miglustat have led to delayed onset of symptoms and increased survival. Outcome in the affected mice was enhanced when substrate reduction therapy was given in combination with bone marrow transplantation [Jeyakumar et al., 2001]. Miglustat given to one patient with Niemann–Pick disease type C reduced pathologic lipid storage, improved the endosomal uptake, and returned lipid trafficking to normal in peripheral blood B lymphocytes [Lachmann et al., 2004]. Demonstration that treatment with miglustat, which has no direct effect on cholesterol metabolism, corrects the abnormal lipid trafficking suggests that glycosphingolipid accumulation is the primary pathogenetic event. In a randomized controlled study, miglustat treatment was shown to improve or stabilize several clinically relevant markers, such as horizontal saccadic eye movement (which is impaired in affected individuals), improved swallowing capacity and auditory acuity, and led to a slower deterioration in ambulatory index [Patterson et al., 2010].
Farber’s Disease (Lipogranulomatosis)
Farber’s disease, initially described by S. Farber and colleagues (1957), is characterized by widespread granulomatous tissue infiltration by ceramide-containing foamy macrophages resulting from a deficiency of lysosomal acid ceramidase [Park and Schuchman, 2006]. Examination of the lysosomal degradation of sphingomyelin-derived ceramide in situ with use of the affected patient’s skin fibroblasts has found a significant correlation between the ceramide accumulation and the severity of Farber’s disease [Levade et al., 1995]. Insertional mutagenesis of the mouse acid ceramidase gene has been shown to lead to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes [Li et al., 2002]. Recent studies suggest that acid ceramidase is an essential factor required for embryo survival, and functions by removing ceramide from the newly formed embryos and inhibiting the default apoptosis pathway [Eliyahu et al., 2007].
Clinical features include painful swollen and deformed joints and dysphonia (hoarse cry), with onset usually before the age of 4 months [Mondal et al., 2009; Sana et al., 2009] (Figure 36-10). Dyspnea due to infiltration of the peribronchial and perialveolar spaces is common. Firm subcutaneous nodules may be found on the fingers, wrist, elbows, and spine. Hand radiographs reveal destructive juxta-articular bone lesions [Toppet et al., 1978]. Although lipid storage is present in neurons and glial cells (mainly in the brainstem nuclei, basal ganglia and ganglion cells of the retina, and anterior horn cells), neurologic signs are usually not prominent (Figure 36-11). Formal evaluation of any developmental problem is complicated by the severe inanition and fixed joints [Jameson et al., 1987]. Some patients have progressive mental deterioration and visual loss. Cerebrospinal fluid protein levels are increased. Anorexia, vomiting, and swallowing difficulties lead to cachexia. Cholestatic jaundice and rapidly evolving liver failure have been described in two siblings [Willis et al., 2008]. Severe muscle atrophy, in part a consequence of immobilization of the joints, is also seen. Death generally occurs within the first 2 years of life, although there are rare cases of patients with an attenuated form of the disease and more prolonged clinical course [Pavone et al., 1980].
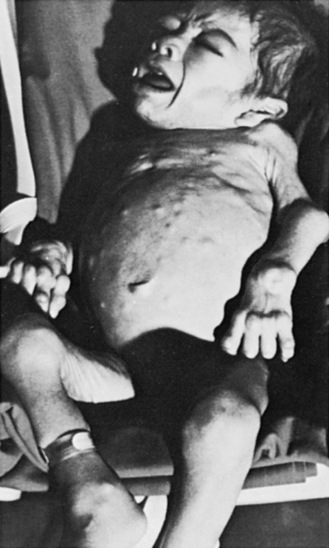
Fig. 36-10 A severely cachectic patient with Farber’s disease.
The patient exhibits multiple subcutaneous lymphogranulomas and periarticular swelling.
(From Gellis S, Feingold M. Contributed by TRC Sisson. Am J Dis Child 1971;122:573.)
Fig. 36-11 Section of spinal cord from a patient with Farber’s disease.
The section exhibits swollen neurons, neuronophagia, and loss of neurons in the anterior horn area.
(Courtesy Hugo W. Moser, Baltimore, MD.)
The diagnosis is often based on quantification of ceramide (a fatty acylsphingosine) accumulation in tissues or cultured skin fibroblasts [Mitsuo et al., 1988]. Analysis of ceramidase activity in vivo can also be performed on the same tissues; however, residual ceramidase activity has not been found to correlate with disease severity. Several mutations in the gene encoding lysosomal acid ceramidase have been identified [Bar et al., 2001].
There is no specific treatment, although affected individuals may obtain relief from joint pains with analgesics. In a 9-year-old girl with occasional painful locking of the metacarpophalangeal (MCP) joints of the middle fingers and severe tenderness of the dorsal aspect of the wrists, resection of several nodules within the MCP joint and of a nodule that was firmly attached to the extensor pollicis longus tendon beneath the extensor retinaculum relieved pain and enabled the patient to perform daily activities [Moritomo et al., 2002]. Supportive care may be required for laryngeal and respiratory symptoms. Gene therapy is under investigation; one study has evaluated in vivo delivery of human acid ceramidase via cord blood transplantation and direct injection of lentivirus as treatment approaches [Ramsubir et al., 2008].
Krabbe’s Disease (Globoid Cell Leukodystrophy)
In 1916, K. Krabbe, a Danish neurologist, described two siblings with an acute infantile diffuse “sclerosis” of the brain (characterized by unusual cells in the white matter termed globoid cells by Collier and Greenfield) (Figure 36-12). The condition has since been attributed to a deficiency of galactocerebroside β-galactosidase (β-galactosidase C, galactosylceramidase), which normally cleaves the bond between ceramide and the galactose moiety of galactosylceramide [Suzuki, 2003]. Disease incidence in the general population is estimated at 1 in 201,000, although an increased prevalence (1 in 25,000) of the early infantile form has been reported in Sweden [Arvidsson et al., 1995]. An authentic animal model (the twitcher mouse, which is galactosylceramidase-deficient) has been described [Yagi et al., 2004].
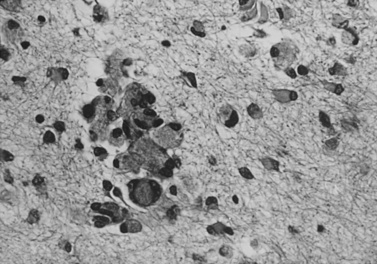
Fig. 36-12 Clumps of globoid and epithelioid cells in cerebral white matter in Krabbe’s disease.
The background area demonstrates demyelination and gliosis.
(Courtesy Joo Ho Sung, Minneapolis, MN.)
The majority of cases are an early infantile form (with onset between 3 and 6 months of life) (Figure 36-13). Patients demonstrate irritability and develop rapidly progressive generalized rigidity and tonic spasms [Korn-Lubetzki and Nevo, 2003]. Affected infants often have a clenched fist and develop myoclonic jerks. Blindness and optic atrophy appear later, along with pendular nystagmus. Abnormalities of the brainstem auditory-evoked responses are among the first objective indications of CNS disease; visual-evoked potential abnormalities also occur. Brain MRI shows signs of leukodystrophy, with symmetric signal abnormalities usually noted in the periventricular region of the posterior cerebral hemispheres. Diffusion tensor imaging has been shown to detect significant differences in the corticospinal tracts of asymptomatic neonates [Escolar et al., 2009]. Fluorine 18-labeled 2-fluoro-2-deoxyglucose positron emission tomography (PET) in one 2½-year-old male revealed a marked decrease in the metabolism of the left cerebral cortex and no uptake in the caudate heads [Al-Essa et al., 2000]. However, normal glucose uptake was observed in the thalami, lentiform nuclei, and cerebellum.
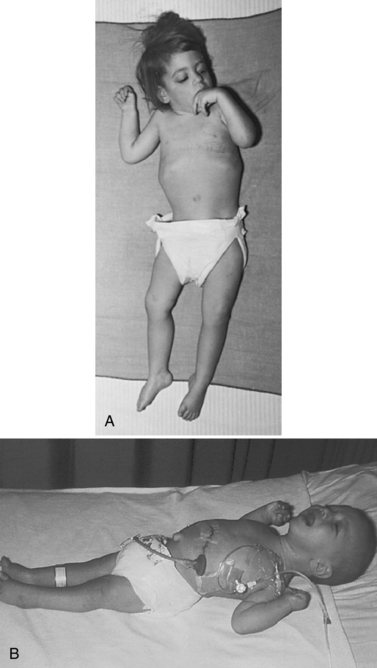
Cerebrospinal fluid protein is elevated in Krabbe’s disease, and examination of the peripheral nervous system reveals markedly reduced nerve conduction velocities [Husain et al., 2004]. There is no visceromegaly or skeletal abnormalities. Death, usually due to respiratory difficulties and bronchopneumonia, occurs between the ages of 1 and 2 years. A clinical disease staging system for affected infants has been developed, which is based solely on signs and symptoms of disease. The index was found to be useful in predicting outcomes after umbilical cord blood transplantation, whereas standard neurophysiological and neuroimaging tests were not useful in the staging algorithm [Escolar et al., 2006].
In a small proportion (about 10–15 percent) of patients, onset is in the late infantile or juvenile period. Several such cases have been identified in southern Italy (and Sicily). These patients exhibit signs of progressive walking problems, spastic paraparesis, and cerebellar ataxia, mostly before the age of 5 years [Lyon et al., 1991]. With disease progression, patients may develop dystonia and visual failure (due to optic atrophy and demyelination of the optic radiation). In one report, a 48-year-old man presented with asymmetric upper and lower motor neuron signs initially attributed to a motor neuron disease [Henderson et al., 2003]. In this patient, nerve conduction studies and neuroimaging provided important signs leading to the correct diagnosis. In another report, which described three affected brothers with disease onset in the fifth decade, the neurologic examination found features of asymmetric peripheral neuropathy associated with pyramidal signs [Sabatelli et al., 2002]. Two of the patients in this family died at 59 and 61 years of age of respiratory failure. Other cases have been described with presentations consistent with spastic paraplegia [Bajaj et al., 2002]. Behavioral changes and intellectual impairment have also been reported, and these problems may be the initial complaints, particularly in juvenile-onset patients before recognition of the other features of Krabbe’s disease.
A study examined the results of testing involving 26 children with Krabbe’s disease, 20 with the early infantile disease (range of ages at onset, 0.5–6 months), and 6 with the late-onset disease (range of ages at onset, 10–60 months) [Aldosari et al., 2004]. The results revealed that patients with early infantile disease had abnormal flash visual-evoked potentials (53 percent), abnormal brainstem auditory-evoked response (88 percent), and abnormal electroencephalograms (65 percent). Nerve conduction studies were abnormal in all of these patients. Among the late-onset patients, none had abnormal visual-evoked potentials, whereas brainstem auditory-evoked responses and electroencephalograms were abnormal in 40 percent and 33 percent of the children. Only 20 percent of the late-onset patients had abnormal nerve conduction studies. Abnormal neurophysiologic studies were found to correlate with more extensive disease as measured by MRI scans [Korn-Lubetzki et al., 2003].
Brain magnetic resonance spectroscopy (MRS) performed in patients with infantile Krabbe’s disease has revealed elevation of myo-inositol and choline-containing compounds in affected white matter, consistent with demyelination and glial proliferation [Brockmann et al., 2003; Zarifi et al., 2001]. The accompanying decrease of N-acetylaspartate points to neuroaxonal loss. In juvenile-onset cases, MRS indicated astrocytosis with minor neuroaxonal damage in white matter [Farina et al., 2000]. Spinal involvement may be evident on MRI as abnormal contrast enhancement of the lumbosacral nerve roots [Given et al., 2001].
Pathologic studies of brains from patients with Krabbe’s disease reveal a rapid disappearance of myelin and myelin-forming cells (i.e., oligodendrocytes in the CNS and Schwann cells in the peripheral nervous system). In addition, there is reactive astrocytic gliosis and tissue infiltration by multinucleated macrophages (globoid cells) filled with materials that are periodic acid–Schiff-positive [Itoh et al., 2002]. These findings are considered to reflect a reaction to psychosine (galactosylsphingosine), a toxic metabolite that accumulates in the tissues of patients with Krabbe’s disease [Haq et al., 2003]. Exposure of an immortalized human oligodendroglial cell line (MO3.13) to exogenous psychosine results in the concurrent upregulation of JNK AP-1 (a pro-apoptotic pathway mediator) and downregulation of the NF-κB pathway (an anti-apoptotic pathway factor) [Haq et al., 2003]. Another study, which examined a mouse-derived oligodendrocyte progenitor cell line (OLP-II), revealed that psychosine caused cytotoxic effects in a dose-dependent manner [Zaka and Wenger, 2004]. Moreover, psychosine treatment resulted in the activation or cleavage of initiator caspases 8 and 9, and effector caspase 3. These results provide further support for a role for psychosine in cell death through an apoptotic mechanism. Recently, psychosine has been shown to accumulate specifically in lipid rafts in the twitcher mouse brain and sciatic nerve, and in samples from brains of human Krabbe’s disease patients. The psychosine accumulation was accompanied by an increase in cholesterol in these domains and changes in the distribution of the lipid raft markers flotillin-2 and caveolin-1, potentially leading to a disruption of multiple signaling pathways [White et al., 2009a, b].
In a separate study that examined brain obtained from three patients with Krabbe’s disease, white matter lesions were found (in the corona radiata, corpus callosum, and cerebellar peduncles) in addition to the neuronal loss in the thalamus, cerebellum, and inferior olivary nucleus [Itoh et al., 2002]. Ramified microglia was immunoreactive for ferritin and HLA-DR α chain, and both the small and large globoid cells exhibited immunoreactivity for ferritin KP-1 and neural cell adhesion molecule. T lymphocytes immunoreactive for leukocyte common antigen, ubiquitin carboxyl-terminal esterase L1, and CD3 were increased around the vessels in the white matter. These data suggest that immunoreactive changes may partly be involved in the myelin breakdown and glial pathologic changes in Krabbe’s disease.
The diagnosis of Krabbe’s disease is based on demonstration of decreased galactosylceramidase activity in peripheral leukocytes or cultured skin fibroblasts [Randell et al., 2000]. Mutation analysis has led to identification of more than 40 gene defects responsible for Krabbe’s disease [Fu et al., 1999]. A deletion mutation (502del) has been reported as common, accounting for up to 40 percent of disease alleles. Homozygosity for this mutation is associated with the severe infantile form of the disease. Mutation analysis in a patient with late-onset disease revealed compound heterozygosity for the 809GÆA (G270D) and 1609GÆA (G537R) mutations [De Gasperi et al., 1999]. The relatively mild clinical phenotype observed in this patient was attributed to the 809GÆA allele, based on the presence of residual galactosylceramidase activity in vitro. Indeed, this mutation has been found in several other patients with late-onset Krabbe’s disease.
There is no specific treatment for the infantile form of Krabbe’s disease. However, the irritability encountered in the infantile form of Krabbe’s disease has been successfully controlled with low-dose morphine [Stewart et al., 2001]. Bone marrow transplantation with appropriate timing in the later-onset forms has returned cerebrospinal fluid protein to normal levels and stabilized the neurologic findings, neuropsychologic function, and extent of demyelination on MRI [Caniglia et al., 2002]. Newborn screening has been recommended for early detection and early intervention by hematopoietic stem cell transplantation to improve outcome [Galvin-Parton, 2003; Li et al., 2004]. Transplantation of umbilical cord blood from unrelated donors in newborns with infantile Krabbe’s disease has been reported to alter the natural history of the disease favorably, but only when initiated before the onset of symptoms [Escolar et al., 2005]. However, a longer period of follow-up has indicated that over 80 percent of the transplanted infants have gross motor problems, and in some instances required assistance with ambulation [Prasad and Kurtzberg, 2008]. These observations and other considerations have raised several concerns regarding the newborn screening program for Krabbe’s disease initiated in New York State and planned to be introduced in the state of Illinois [Steiner, 2009]. Enzyme replacement therapy, which may necessitate intracerebroventricular administration of galactocerebrosidase to bypass the blood–brain barrier, is under investigation [Lee et al., 2005, 2007]. Stem cell-based strategies, using glial progenitor cells and other cell types, are also being explored [Goldman et al., 2008]. Experimental studies in the twitcher mouse model with vascular endothelial growth factor (VEGF), an endothelial cell mitogen and permeability factor, given intravenously at the time of birth, led to increased blood–brain barrier permeability [Young et al., 2004]. Systemic treatment with VEGF before bone marrow transplantation or the administration of recombinant lentivirus resulted in increased numbers of either donor cells or virus-transduced cells in the recipient brain. This approach may prove to be useful in future studies of cellular or enzyme replacement therapies for Krabbe’s disease and other lysosomal storage diseases with primary CNS involvement.
Deficiency of saposin A was demonstrated to cause late-onset, slowly progressive globoid cell leukodystrophy in the mouse [Matsuda et al., 2001b]. The saposin A-null mouse developed slowly progressive hind-limb paralysis, with onset at 2.5 months of age. Tremors and shaking, prominent in other myelin mutants, were not obvious until the terminal stage. Pathologic and analytic biochemistry revealed findings that were qualitatively identical to but generally much milder than those seen in the typical human infantile Krabbe’s disease. Interestingly, in affected female mice that became pregnant, neurologic symptoms greatly improved, compared with affected nulliparous female mice or affected male mice. Immune-related gene (monocyte chemoattractant protein 1, tumor necrosis factor-α) expression was found to be significantly downregulated in the brain of pregnant saposin A-null mice. In addition, there was intense expression of the estrogen receptors (alpha and beta) on the globoid cells, activated astrocytes, and microglia in the demyelinating area [Matsuda et al., 2001a]. When these mice were subcutaneously implanted with timed-release 17β-estradiol pellets, the pathologic process was vastly improved. These findings suggest that the higher levels of estrogen during pregnancy may have a protective effect. Saposin A deficiency has not been described in human patients but might be anticipated as a cause of late-onset chronic leukodystrophy without primary galactosylceramidase deficiency.
Metachromatic Leukodystrophy (Sulfatide Lipidosis)
Deficiency of arylsulfatase A activity leads to the accumulation of its incompletely metabolized substrate, galactosylsulfatide (cerebroside sulfate), in the white matter of the central and peripheral nervous system [Gieselmann, 2003]. The associated clinical condition derives its name from the brownish or reddish birefringent (metachromatic) color of the sulfatide deposits, noted when involved tissues are stained with cresyl violet or toluidine blue [Gieselmann et al., 2003a]. Sulfatides are a component of myelin, and their increased concentration in tissues of the patients with metachromatic leukodystrophy may lead to instability and myelin breakdown (Figure 36-14). It has also been hypothesized that lysosulfatide (a deacylated form of sulfatide) may be cytotoxic and promote the dissolution of myelin. Lysosulfatide has been shown to regulate the motility of a neural precursor cell line (B35 neuroblastoma cells) via calcium-mediated process collapse (by rapidly promoting process retraction and cell rounding) [Hans et al., 2009].
Fig. 36-14 Myelinated fiber obtained from the sural nerve.
(Courtesy Stephen A. Smith, Minneapolis, MN.)
The overall incidence of metachromatic leukodystrophy has been estimated at 1 in 121,000. The relatively high frequency (1 in 2520 live births, with an estimated carrier frequency of 1 in 25 to 1 in 50) in the Navajo Indians of the southwestern United States has been attributed to a “genetic bottleneck” that occurred in the mid-19th century [Holve et al., 2001]. Genetic bottleneck refers to an evolutionary event leading to a drastic reduction in population size. The same phenomenon explains the increased frequency (1 in 75 live births) of metachromatic leukodystrophy in a small Jewish community that lived in Habban (Yemen), isolated from the other Jewish populations [Zlotogora et al., 1995]. There have been rare patients with saposin B deficiency who are unable to catabolize sulfatide and other glycosphingolipids in vivo, leading to their accumulation and a neurodegenerative disease that is viewed as a variant of metachromatic leukodystrophy [Wrobe et al., 2000; Deconinck et al., 2008]. Saposin B (also known as sphingolipid activator protein 1) is a small glycoprotein that acts as an activator of arylsulfatase A and is required for the hydrolysis of galactosylsulfatide [Whitelegge et al., 2003]. Patients with saposin B deficiency have clinical features that overlap with the juvenile-onset form of metachromatic leukodystrophy [Wrobe et al., 2000]. These individuals have evidence of tissue sulfatide storage with normal arylsulfatase A activity in vitro; their diagnosis can be confirmed by tests using specific antibodies against saposin B or identification of the causal saposin B gene mutation.
Late infantile-onset metachromatic leukodystrophy represents the most common clinical form of the disease, accounting for 60–70 percent of all cases (Figure 36-15). Later-onset forms of the disease, developing in childhood or adolescence and even during adulthood, have been described.
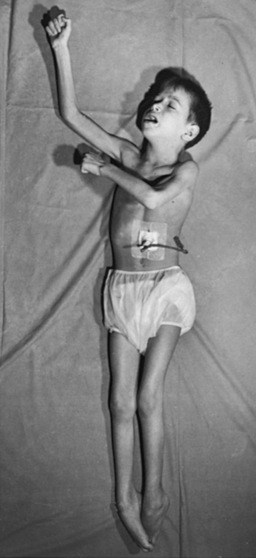
In late infantile metachromatic leukodystrophy, affected children usually present with progressive gait problems, manifesting between the ages of 14 and 16 months. Examination of the lower extremities may initially reveal a mixed picture, with flaccid paralysis or more commonly with pyramidal signs (e.g., spasticity and bilateral Babinski signs) and depressed or hyperactive deep tendon reflexes. Electrophysiologic testing reveals evidence of peripheral nerve involvement, with marked slowing of the motor and sensory nerve conduction velocities. The cerebrospinal fluid protein level is elevated. Brain MRI reveals leukodystrophy, with a hyperintense signal in the periventricular and central white matter on T2-weighted images, which may initially be limited to the parieto-occipital region [Sener, 2003]. In one 17-month-old male with metachromatic leukodystrophy, an echo-planar diffusion magnetic resonance sequence revealed a restricted diffusion pattern in the deep white matter [Sener, 2002]. Proton MRS revealed a marked decrease in choline, a metabolite related to myelin turnover [Sener, 2003]. These observations likely represent dysmyelination.
The disease is progressive, and affected patients with metachromatic leukodystrophy are eventually unable to stand or sit up. Additional findings include dysarthria, drooling, and dysphagia. There may be visual failure. Seizures occur in a small proportion of patients in the terminal stages of their disease, by which time the children are rigid and assume decerebrate or decorticate postures [Cameron et al., 2004]. Deposition of sulfatides in the gallbladder can cause the formation of papillomatous changes and lead to cholecystitis [Vettoretto et al., 2001]. Death in patients with late infantile metachromatic leukodystrophy usually occurs between the ages of 3 and 7 years.
Metachromatic leukodystrophy may manifest in later life, from early childhood (3–8 years of age) to adolescence or even adulthood [Ito et al., 2009]. Behavioral abnormalities, cognitive deficits, and dementia may be more prominent in the later-onset forms [Baumann et al., 2002; Black et al., 2003; Mihaljevic-Peles et al., 2001]. Abnormalities of nerve conduction and cerebrospinal fluid protein elevation may be minimal. Brain white matter alterations on MRI may also be less pronounced. The variability in clinical presentation can often lead to a missed diagnosis, and metachromatic leukodystrophy should be considered in cases characterized by deterioration in behavioral or mental capacities, a progressive disorder of gait or coordination, or a progressive polyneuropathy. It is important to exclude arylsulfatase A pseudodeficiency in these patients. The course of disease in the late-onset forms of metachromatic leukodystrophy can be protracted, with survival of more than 5–10 years not unusual. However, disease may evolve more rapidly in some patients.
Impaired cortical glucose metabolism (especially of the medial temporal and frontal cortices) was observed with PET and fluorine 18-labeled fluorodeoxyglucose in a 41-year-old male patient with late-onset metachromatic leukodystrophy [Johannsen et al., 2001]. In addition, brain MRI displayed confluent hyperintensities of periventricular and subcortical white matter. Neuropsychologic assessment demonstrated that he had mild amnesia, visuospatial dysfunction, and attention deficits with a slow psychomotor speed. The neuropsychologic deficits in this individual were related to the location of deficits in glucose metabolism.
To a certain extent, the clinical course of metachromatic leukodystrophy is associated with the nature of the causal mutation [Biffi et al., 2008]. Patients with a combination of deleterious alleles for this autosomal-recessive condition have a late infantile onset, whereas those with a combination of a severe and mild mutation or two mild mutant alleles have delayed onset and follow a subacute or chronic disease course. Two gene defects – namely, 459+1G to A and 1277C to T (Pro426Leu) – account for almost 50 percent of metachromatic leukodystrophy mutations; the remaining alleles are rare or private [Eng et al., 2003, 2004]. In a study of 12 adult metachromatic leukodystrophy patients with primary motor signs (pyramidal, cerebellar, and less often dystonia) and a peripheral neuropathy, the major adult arylsulfatase A mutation P426L [Lugowska et al., 2002]. In contrast, patients with the psychocognitive forms (associated with modifications of mood, peculiar social reactions, and a progressive mental deterioration) were often noted to be compound heterozygotes, with the I179S mutation on one allele [Lugowska et al., 2002]. In a separate study, screening for the mutations in 34 unrelated patients with metachromatic leukodystrophy from Poland revealed that the I179S mutation accounted for 17 percent of examined alleles (2 of 12) in adults and as much as 42 percent (5 of 12 alleles) in patients with late juvenile metachromatic leukodystrophy [Berger et al., 1997]. The I179S mutation has not been found in the late infantile or early juvenile cases.
The diagnosis of metachromatic leukodystrophy is based on demonstration of deficient arylsulfatase A activity in leukocytes or cultured skin fibroblasts. Peripheral nerve biopsy, which is often unnecessary, reveals the typical prismatic and tuffstone inclusions in Schwann cells. Polymorphisms that give rise to arylsulfatase A pseudodeficiency (associated with decreased arylsulfatase A activity in vitro) are common in the general population (approximately 10 percent) [Rafi et al., 2003]. Biochemical assays for the enzyme with the artificial 4-methylumbelliferyl substrate may lead to an erroneous diagnosis of affected and carrier status in these individuals. Increased urinary sulfatide excretion (detected by thin-layer chromatography) is found only in affected patients with metachromatic leukodystrophy who have a true deficiency of arylsulfatase A activity [Whitfield et al., 2001]. Sulfatide loading tests performed with tissues in culture can help distinguish arylsulfatase A pseudodeficiency from a true carrier or affected status, but these methods require specialized expertise and have generally been replaced by a combination of biochemical and molecular assays.
The sequence variation leading to arylsulfatase A deficiency has been characterized, and most laboratories offering the biochemical test can check for these mutations. In families in which the causal arylsulfatase A gene defects are known, true carrier status can be assigned reliably by mutation analysis. In families at risk for having an affected child, the presence of the arylsulfatase A pseudodeficiency allele should be ascertained in both parents to avoid mistakes in prenatal diagnosis. One study reported a higher incidence of evident or possible microorganic brain damage in metachromatic leukodystrophy–pseudodeficiency and metachromatic leukodystrophy heterozygotes, although none of these carriers had overt clinical problems [Tylki-Szymanska et al., 2002b]. On the other hand, there is a separate report that describes the neuropsychologic profiles and brain MRI findings of eight family members of a 7-year-old female with metachromatic leukodystrophy [Weber Byars et al., 2001]. All eight relatives were heterozygous carriers, including five who were also carriers of the arylsulfatase A pseudodeficiency gene. The patient’s younger sister had features of nonverbal learning disability despite a normal MRI study, whereas two members with minor white matter findings did not [Weber Byars et al., 2001]. Thus, the findings in this family do not support the concept of a syndrome of nonverbal learning disability in heterozygous carriers of the gene for metachromatic leukodystrophy, even in association with the metachromatic leukodystrophy pseudodeficiency gene.
There is no effective treatment for metachromatic leukodystrophy. In a child with metachromatic leukodystrophy who required surgery for gastroesophageal reflux, general anesthesia was performed with a lumbar epidural technique [Hernandez-Palazon, 2003]. In this patient, the epidural catheter enabled the delivery of analgesia postoperatively, avoiding the risk of respiratory depression associated with the use of opioids in these high-risk patients.
Bone marrow transplantation, when it is undertaken early, may slow or halt the progression of the disease, but not when neuropsychologic signs are advanced. Bone marrow transplantation may be a reasonable option for presymptomatic patients, or while neuropsychologic function and independence in activities of daily living remain intact. Recently, the outcome of umbilical cord blood transplantation in three siblings with juvenile metachromatic leukodystrophy at different stages of disease has been described [Pierson et al., 2008]. After transplant, the oldest sibling experienced disease progression, whereas his two siblings had near- or total resolution of signal abnormalities on neuroimaging. Neuropsychological testing has remained stable, and nerve conduction studies have shown improvement.
Investigators are currently examining the role of mesenchymal stem cell infusion. In a study of four patients with metachromatic leukodystrophy, there were significant improvements in nerve conduction velocities after mesenchymal stem cell infusion [Koc et al., 2002]. Bone mineral density was either maintained or slightly improved in all the patients. However, there was no clinically apparent change in the patients’ overall health and mental and physical development after mesenchymal stem cell infusion. Enzyme replacement therapy is also being evaluated as a potential therapeutic option. Repeated intravenous injection of the cognate enzyme in a mouse model of metachromatic leukodystrophy improved ataxic gait and CNS histopathology [Matzner et al., 2009]. Sulfatide accumulation in oligodendroglia and neurons has been described in the arylsulfatase A-null mouse model of metachromatic leukodystrophy [D’Hooge et al., 2001]. Neuronal sulfatide storage was most prominent in many nuclei of the medulla oblongata and pons, and in several nuclei of the midbrain and forebrain. In the mouse studies, sulfatide storage did not affect the overall composition of most myelin proteins but specifically caused a severe reduction of myelin and lymphocyte (MAL) protein, indicating the existence of a regulatory mechanism between lipid and myelin protein synthesis in oligodendrocytes [Wittke et al., 2004]. MAL is a myelin proteolipid protein with four transmembrane domains that tightly binds glycosphingolipids with particular sulfatides. MAL is developmentally regulated in differentiated cultured oligodendrocytes, and its expression level peaks in the nervous system during myelin formation. The specific reduction and mistargeting of MAL protein may contribute to the pathogenic mechanisms in metachromatic leukodystrophy. Interestingly, there was no obvious sign of widespread demyelination in the arylsulfatase A-null mice, except for loss of neurons in two nuclei of the auditory pathway of aged mice, specifically in the ventral cochlear nucleus and nucleus of the trapezoid body. Several observations in the arylsulfatase A-null mice, such as hyperactivity, motor incoordination and slowing, and age-related learning and memory deficits, may relate to the decline of neuromotor and cognitive functions seen in patients with metachromatic leukodystrophy. These findings could be used as correlative or outcome measures in preclinical investigations of prospective therapies before their introduction in human trials.
Multiple Sulfatase Deficiency (Austin’s Disease)
A defect of post-translational modification of several sulfatases leads to a neurovisceral disorder characterized by tissue accumulation of sulfatides, glycosaminoglycans (mucopolysaccharides), and cholesteryl sulfate [Dierks et al., 2009]. The gene mutated in this disease is SUMF1, which encodes a protein, the human C(α)-formylglycine generating enzyme, which activates sulfatases by modifying a key cysteine residue within their catalytic domains [Cosma et al., 2003; Dierks et al., 2003]. The particular clinical picture encountered in the individual patient depends on the pattern of enzyme deficiencies [Macaulay et al., 1998]. An accumulation of autophagosomes resulting from defective autophagosome–lysosome fusion has also been observed in the mouse model, which is hypothesized to play a contributory role in disease resulting from multiple sulfatase deficiency [Settembre et al., 2008]. Activated microglia have been detected in the cerebellum and brain cortex of the mutant mouse model, associated with remarkable astroglyosis and neuronal cell loss. An increase in the expression levels of inflammatory cytokines and of apoptotic markers in both the CNS and liver suggests that inflammation and apoptosis may occur during the late stage of the disease process [Settembre et al., 2007].
The clinical features of multiple sulfatase deficiency represent a combination of the neurologic findings noted in patients with early infantile metachromatic leukodystrophy and the dysmorphic facial features and skeletal deformities (i.e., dysostosis multiplex) seen with mucopolysaccharidosis [Schlotawa et al., 2008]. Patients have ichthyosis (dry, scaly skin), usually evident in the first few weeks of life [Busche et al., 2009; Castano Suarez et al., 1997]. Inclusions (Alder–Reilly granules) may be seen in peripheral blood leukocytes [Soong et al., 1988]. Associated findings include retarded psychomotor development, hepatosplenomegaly, deafness, and peripheral neuropathy [Guerra et al., 1990]. Brain MRI has been reported to demonstrate extensive diffuse symmetrical high signal in the deep white matter of both cerebral hemispheres and brainstem, with enlargement of sulci and subdural spaces and mild brain atrophy [Zafeiriou et al., 2008]. Nerve conduction velocities are decreased, and cerebrospinal fluid protein is elevated. Urine analysis reveals increased excretion of sulfatides, heparan sulfate, and dermatan sulfate.
Two unusual cases of multiple sulfatase deficiency with variable enzymatic deficiency of arylsulfatases A, B, and C have been reported. Both patients had ichthyosis, broad thumbs and index fingers, and an unusually slow progression of their neurologic symptoms [Blanco-Aguirre et al., 2001]. In addition, they lacked the hepatosplenomegaly that is typical of multiple sulfatase deficiency. Olivopontocerebellar atrophy was present, and one of the patients had a large retrocerebellar cyst. Mucopolysaccharides were not detected in the urine from either subject. Another patient has also been reported to develop early, severe visual impairment associated with prominent pigmentary retinopathy.
Analysis of the lipid composition of the brain obtained from two patients with multiple sulfatase deficiency revealed sulfatide concentration in the white matter that was 3–4 times that of normal brains [Eto et al., 1976]. The ganglioside pattern in the gray and white matter was abnormal, with a higher proportion of GM3, GM2, and GD3 gangliosides. Increased amounts of glucocerebroside, ceramide lactoside, and ceramide trihexoside were also present. The fatty acid compositions of myelin sulfatide and sphingomyelin were almost normal, whereas the nonhydroxy fatty acids of cerebroside contained fewer long-chain fatty acids.
A variant of multiple sulfatase deficiency described in Saudi Arabian children presented with corneal clouding, macrocephaly, and dysostosis multiplex [al Aqeel et al., 1992]. Ichthyosis was absent, and steroid sulfatase activity was normal. Mental retardation was reported to be mild. This Saudi variant of multiple sulfatase deficiency accounts for 5 percent of all lysosomal storage disease cases in the Cell Repository Registry of the Inborn Errors of Metabolism Laboratory at the King Faisal Specialist Hospital and Research Centre in Riyadh, Saudi Arabia.
The diagnosis of multiple sulfatase deficiency can be made on the basis of characteristic clinical manifestations in conjunction with variable deficiencies of the arylsulfatases A, B (N-acetylgalactosamine-4-sulfate sulfatase), and C (steroid sulfatase), and four other sulfatases involved in the degradation of specific glycosaminoglycans [Mancini et al., 2001]. The identification of the responsible gene has led to characterization of several different mutations associated with variable clinical expression, including milder disease variants [Cosma et al., 2004].
Wolman’s Disease and Cholesteryl Ester Storage Disorder
The enzyme lysosomal acid lipase is normally involved in the hydrolysis of cholesteryl esters and triglycerides that have been internalized by receptor-mediated endocytosis of lipoprotein particles [Wolman, 1995]. Deficiency of lysosomal acid lipase leads to the massive accumulation of exogenous cholesteryl esters throughout most organ systems, giving rise to one of two phenotypes: the severe Wolman’s disease or the more benign cholesteryl ester storage disease. The observed differences in clinical disease course are likely due to expression of genetic mutations that either abolish lysosomal acid lipase enzyme activity in the more severe Wolman’s disease phenotype or lead to some residual activity as in cholesteryl ester storage disease [Hooper et al., 2008; Zschenker et al., 2001].
Wolman’s disease was first described in 1956 by Abramov and colleagues, who referred to it as a generalized xanthomatosis with calcified adrenals. The condition is clinically characterized by vomiting, diarrhea, malabsorption, and failure to thrive, with onset in infancy [Browne et al., 2003]. Patients are hypotonic and develop anemia and massive hepatosplenomegaly. Liver tissue sections reveal microsteatosis and macrosteatosis, and cholesterol crystals in hepatocyte cytoplasm. The adrenal glands are enlarged and calcified, features detectable on CT scans. Bone marrow aspiration demonstrates a marked increase in foamy macrophages. Death occurs within the first year of life as a consequence of liver failure, coagulopathy, and pancytopenia.
Cholesteryl ester storage disease has a more variable phenotype; it may be benign and remain undetected into adulthood [D’Agostino et al., 1988]. Lipid deposition is widespread, but hepatomegaly is the principal and sometimes the only sign of the disease, even when it is noted from birth or in early childhood. Patients develop early atherosclerosis and hepatic fibrosis, with possible evolution to cirrhosis. Atypical combined hyperlipidaemia in a patient with “fatty liver disease,” in the absence of overweight, should lead to consideration of cholesteryl ester storage disease as a potential diagnosis [Decarlis et al., 2009].
The diagnosis of Wolman’s disease and cholesteryl ester storage disease is based on demonstration of deficient lysosomal acid lipase activity in cultured skin fibroblasts and blood lymphocytes. Bone marrow transplantation has been reported to lead to normalization of peripheral leukocyte lysosomal acid lipase activity in a patient (6.5 months of age) with Wolman’s disease and to result in the resolution of diarrhea and return of liver function to normal [Krivit et al., 2000]. At the age of 4 years, the patient was noted to be gaining developmental milestones. Follow-up data have revealed that the patient has required treatment for seizures, severe short stature, and hyperthyroidism [Tolar et al., 2009]. Treatment of Wolman’s disease with hematopoietic stem cell transplantation is deemed to be extremely challenging, with the majority of attempts resulting in death because of liver failure from progressive disease or sinusoidal obstruction syndrome, as well as other transplant-related complications including infection, graft-versus-host disease, and failure to engraft [Gramatges et al., 2009].
Enzyme therapy and gene therapy experiments have been performed in the mouse model of Wolman’s disease [Du et al., 2002, 2008], establishing preclinical proof of concept for these therapeutic strategies.
Mucopolysaccharidoses
The mucopolysaccharidoses are an etiologically heterogeneous group of disorders resulting from various individual deficiencies of the lysosomal enzymes (i.e., glycosidases and sulfatases) involved in the sequential degradation of glycosaminoglycans [Clarke, 2008; Muenzer, 2004] (Figure 36-16). The glycosaminoglycans consist of polysaccharide chains that are attached to a polypeptide core through a xylose link; their incomplete hydrolysis leads to tissue deposition and increased urinary excretion of the substrates dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate. The pattern of substrate storage and corresponding urinary excretion profile are influenced by the specific underlying enzyme deficiency [Hochuli et al., 2003].
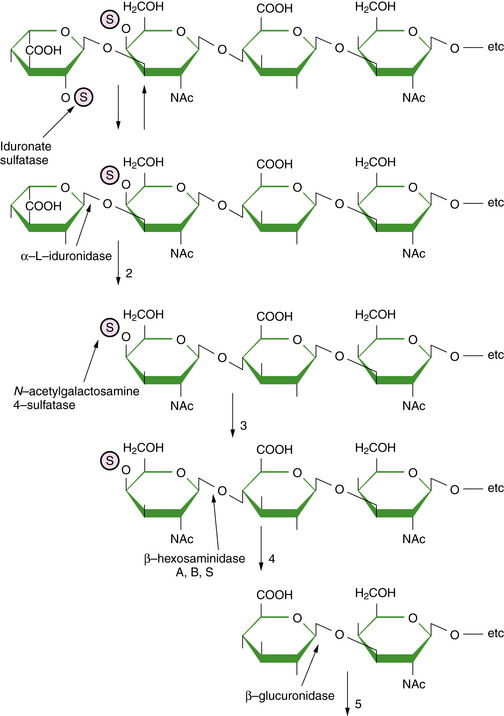
Glycosaminoglycans (GAG) are engaged in the trafficking of molecules through the endosomal network, maintenance of steady state levels of intracellular and extracellular oligosaccharides and proteoglycans, the activity of lysosomal enzyme, and the regulation of GAG-dependent inhibitors (serpins, serine proteinase inhibitors and cathepsins) [Clarke, 2008]. Thus, defects in the lysosomal degradation of substrates likely influence diverse biochemical and physiological processes. Tissue deposits, when examined under the electron microscope, reveal different types of intralysosomal inclusion bodies, the most characteristic of which have been designated zebra bodies. Testing for the presence and pattern of urinary glycosaminoglycan excretion is often used to screen for the diagnosis of mucopolysaccharidosis (MPS) in suspected cases. However, this test often yields a false-negative result [Mabe et al., 2004]. Definitive diagnosis of a particular mucopolysaccharidosis subtype is ultimately based on the specific assay for a particular enzyme with use of plasma, leukocytes, or cultured skin fibroblasts.
The mucopolysaccharidoses are infrequent, with a collective incidence of about 1 in 25,000 to 1 in 50,000 [Nelson et al., 2003]. The individual disorders are assigned a number (based on the sequence of their historical description), and each has an eponymous designation. However, the former classification of MPS V and VIII as separate disorders has been dropped, after the recognition that these entities represent an attenuated form of other previously designated mucopolysaccharidosis subtypes. The relatively milder form of disease expression in certain variants has been partly attributed to residual enzyme activity associated with less deleterious mutations. The absence of perfect concordance between residual enzyme activity and the clinical disease severity is hypothesized to result from the failure to account for the efficiency of substrate synthesis.
Although each MPS subtype is due to a distinct enzyme deficiency, there is significant overlap in the non-neurologic manifestations across the individual diagnostic entities. The two most characteristic manifestations are coarse facial features and dysostosis multiplex. Coarsening of the facial features is a consequence of the storage of glycosaminoglycans in the soft tissues of the orofacial region and the underlying facial bone dysostosis. The term dysostosis multiplex refers to the typical skeletal changes and constellation of radiographic findings (e.g., bullet-shaped phalanges, flattening of the vertebral bodies and their anterior beaking) (Figure 36-17). These changes, in conjunction with deposition and thickening of the skin, lead to joint contractures and limitations in the range of neck and limb movements. Increased apoptosis has been described in chondrocytes from animals affected with MPS VI (Maroteaux–Lamy syndrome), and may be a common pathologic mechanism for this group of disorders [Simonaro et al., 2001]. Recent studies suggest that the accumulation of GAG in murine MPS I bone has an inhibitory effect on cathepsin K activity, resulting in impaired osteoclast activity and decreased cartilage resorption, changes that may contribute to the skeletal dysplasia seen in the mucopolysaccharidoses [Wilson et al., 2009].
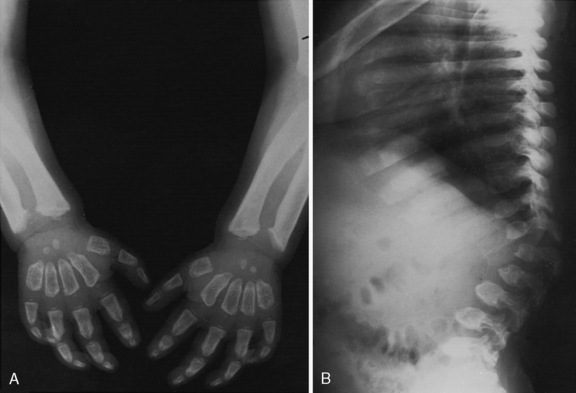
Ophthalmologic complications, such as corneal stromal opacity, pigmentary retinal degeneration, optic nerve atrophy, and glaucoma, are also common in patients with mucopolysaccharidoses. Membrane-bound vacuoles in the nonpigmented epithelium of the ciliary processes have been observed by electron microscopy. Multiple iridociliary cysts have also been reported in patients with MPS I S (Scheie’s syndrome) and MPS VI [Sato et al., 2002]. Other abnormalities of diagnostic importance include deafness, hepatosplenomegaly, obstructive airway problems, and umbilical and inguinal hernia [Shih et al., 2002].
Recurrent upper respiratory tract and ear infections are prominent complaints in infancy, and cardiovascular abnormalities are frequent. In a study of 39 patients (aged 4–22 years) with various mucopolysaccharidoses, valvular lesions, and different forms of cardiac involvement were detected [Mohan et al., 2002]. The most common finding, regardless of the MPS type, was thickening of the mitral valve with regurgitation or stenosis [Rigante and Segni, 2002].
Developmental regression is a feature of most but not all MPS subtypes, each of which constitutes a spectrum of disorders associated with variable expression of the skeletal and somatic anomalies noted before. The accumulation of heparan sulfate and the secondary storage of gangliosides in neurons may be partly responsible for the cerebral dysfunction. In one study, two patients with MPS IIIA with signs of CNS degeneration but only mild somatic features excreted a highly sulfated variant of heparan sulfate, whereas a patient with MPS I S and two patients with MPS II who presented primarily with coarse facial features, joint contractures, and skeletal deformities excreted a different type of heparan sulfate with lower sulfation [Perkins et al., 2001]. In the most severely involved cases, the storage problem can be compounded by tension hydrocephalus due to the deposition of glycosaminoglycans (which leads to impairment of cerebrospinal fluid absorption) and the histiocytic infiltration and collagen proliferation in the meninges (pachymeningitis) [Paulson et al., 1974]. Progressive glycosaminoglycan storage at various sites may lead to compartment syndromes (e.g., carpal tunnel syndrome) and secondary focal neurologic deficits due to spinal cord or nerve root compression. Brain imaging studies involving 20 patients with different forms of MPS (with mental retardation in 11 individuals and normal cognitive function in 9) indicated a significant correlation between white matter alterations and mental retardation [Gabrielli et al., 2004].
The mucopolysaccharidoses are inherited in an autosomal-recessive manner, with one exception (MPS II, or Hunter’s syndrome, which is transmitted as an X-linked trait). Most forms are associated with significant morbidity as a consequence of skeletal and visceral complications; application of palliative care can have a positive influence on the patient’s health-related and functional quality of life [Dumas et al., 2004]. Attention to obstructive airway problems and consideration of continuous positive airway pressure or tracheostomy, when appropriate, can lead to significant relief [Khan and Khan, 2002; Shinhar et al., 2004]. Anterior instrumented correction and fusion of the spine is effective in treating the thoracolumbar kyphosis associated with MPS [Dalvie et al., 2001a].
Bone marrow transplantation has altered the course of disease in certain subtypes of MPS, and enzyme therapy has become available for MPS I, II, and VI. There have also been several studies involving different animal models (of various MPS subtypes) that have examined the potential safety and effectiveness of gene therapy for these disorders [Bosch and Heard, 2003; Haskins et al., 2002]. Substrate reduction therapy may also be a potential therapeutic option. Genistein (4′,5,7-trihydroxyisoflavone), a small-molecule nutripharmaceutical, has been shown to inhibit the synthesis of GAGs (mediated through an epidermal growth factor-dependent pathway) in cultures of fibroblasts of MPS patients, thereby reducing tissue storage [Jakóbkiewicz-Banecka et al., 2009]. The availability of enzyme therapy for MPS I, II, and VI and its consideration in the other mucopolysaccharidoses necessitate a sensitive and reliable method not only for early diagnosis but also for serial quantitative monitoring of therapeutic responses, if any [Kakkis, 2002]. In one study, examination of urine samples from 68 patients with MPS (by electrospray ionization–tandem mass spectrometry) led to identification of an oligosaccharide profile for each group of patients [Fuller et al., 2004; Ramsay et al., 2003]. Each of the urine oligosaccharide profiles enabled the identification of all patients and their subtypes, with the exception of MPS IIIB and IIIC. In three individuals with different MPS disorders (specifically, MPS I, IVA, and VI) who underwent bone marrow transplantation, a substantial reduction in the level of diagnostic oligosaccharides was observed after transplantation. These observations provide a disease-specific fingerprint that can be used in targeted screening and the biochemical monitoring of current and proposed therapies. Two other factors have been recently identified: heparin co-factor II–thrombin complex and dipeptidyl peptidase IV (DPP-IV; CD26), which may be used as surrogate marker of disease stage and in the monitoring of treatment [Beesley et al., 2009; Randall et al., 2008].
Prenatal diagnosis is available for all MPS subtypes.
Mucopolysaccharidosis Type I (MPS I)
This disorder is caused by a deficiency of α-l-iduronidase (IDUA) in association with the accumulation in tissues and increased urinary excretion of dermatan and heparan sulfate. The phenotypic spectrum has been arbitrarily divided into three clinical forms on the basis of the age at onset and severity of disease expression. Hurler’s syndrome (MPS IH) is named after G. Hurler, who (in 1919) described infants with a gibbus deformity, corneal clouding, and mental retardation. This subtype constitutes the most severe end of the clinical spectrum. Scheie’s syndrome (MPS IS), formerly classified under MPS V, includes patients initially described by Scheie and colleagues in 1962. Affected individuals with MPS IS have normal intelligence, but suffer from restricted movements of their joints and develop the clawhand deformity. They may have normal or more often short stature. The intermediate form has been designated the Hurler–Scheie syndrome (MPS I H/S).
Children with Hurler’s syndrome appear normal at birth but gradually develop a distinctive facies typical of the mucopolysaccharidoses (Figure 36-18). Affected infants have a large scaphocephalic head, depressed nasal bridge, frontal bossing, and thickened skin, scalp hair, and eyebrows. These findings, including developmental delay, may not be apparent even to the parents until the age of 12–15 months, particularly when the affected infant is the first-born child. A review of the past medical history often reveals recurrent upper respiratory tract and ear infections. Additional clues to the diagnosis include the presence of clawhands with stubby digits, kyphosis, enlarged tongue, hepatosplenomegaly, umbilical and inguinal hernias, and corneal opacities. The eye findings may initially be mild and detectable only by slit-lamp examination. Body hair is also often increased, and the skin has a leathery (thickened) texture.
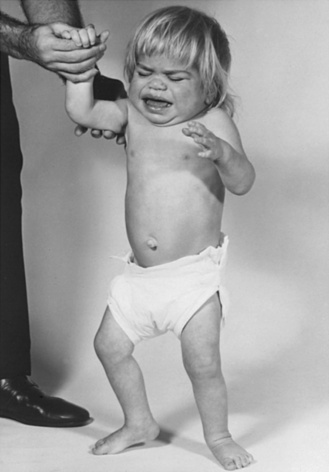
Brain MRI in patients with MPS I reveals several abnormalities, including abnormal signal intensity in the white matter, widening of the cortical sulci and the size of the supratentorial ventricles, dilatation of the perivascular spaces, and enlargement of the subarachnoid spaces. In one study of six patients with MPS I, no relationship was found between the brain MRI findings and clinical manifestations of the disease [Matheus et al., 2004].
Complications that often develop in affected children include upper airway obstruction, coronary artery occlusion, cardiomegaly, and heart valve problems. A study involving 99 patients (aged 1–24 years; median age, 10.3 years) with MPS I found that increasing age and abnormalities of the cardiac ejection fraction were significant risk factors for death [Mohan et al., 2002]. Pulmonary complications, such as sleep apnea, develop because of airway obstruction due to a combination of factors, including enlarged tonsils and adenoids, narrowing of the trachea, and thickening of the mucosal lining of the airways. These airway problems are aggravated by a small chest and an enlarged liver and spleen pushing up against the diaphragm.
Individuals with the attenuated form, Scheie’s syndrome, are often missed because they have normal intelligence and may achieve normal adult stature. Characteristic findings on close inspection may reveal corneal clouding, short neck, stiff joints, and cardiac valvular disease. in vivo confocal microscopy images of the cornea in a 13-year-old male with MPS I S revealed brighter intercellular spaces than those of normal corneas [Grupcheva et al., 2003]. Cicatrization of the anterior stroma was identified, and the keratocytes of the middle and posterior stroma exhibited markedly altered morphologic shape, often round or elliptic, and with clearly demarcated, hyporeflective centers. In addition, the nerve fibers of the sub-basal plexus were somewhat more irregular and difficult to distinguish, possibly because of underlying fibrosis.
Molecular characterization of MPS I patients has resulted in the identification of more than 70 distinct mutations in the IDUA gene [Matte et al., 2003; Yogalingam et al., 2004]. Mutation analyses have indicated that, to a certain extent, the wide variability in clinical presentation of patients with MPS I is a reflection of the high degree of genetic heterogeneity [Terlato and Cox, 2003]. Two mutations, W402X and Q70X, that are prevalent among individuals of European extraction (and account for up to 70 percent of MPS I disease alleles) tend to be associated with the severe MPS I H when they are found in homozygosity or in combination with another deleterious allele. On the other hand, two other less common mutations, R89Q and R89W, tend to be found in patients with the attenuated phenotypes MPS I H/S and MPS I S [Hein et al., 2003]. Additional common mutations in the IDUA gene include P533R (among Moroccan and Sicilian Italian patients with MPS I) and R383H; these two mutations are associated with residual enzyme activity. In a survey of 29 Brazilian patients with MPS I, the frequency of common mutations identified was as follows: W402X, 37 percent; P533R, 11.6 percent; and R383H, 3.3 percent [Matte et al., 2003].
Palliative care, including measures to relieve signs of upper airway obstruction (e.g., continuous positive airway pressure, tracheostomy) and shunting for hydrocephalus (as indicated), is an important component of the overall management [Chan et al., 2003; Muenzer et al., 2009]. The patients also are at increased risk for complications during administration of anesthesia, and appropriate precautions must be taken before any elective surgical procedure [Ard et al., 2005; Walker et al., 2003b].
The routine follow-up care of affected individuals, including those with attenuated disease subtypes, must include a thorough neurologic examination because motor and sensory deficits (e.g., carpal tunnel syndrome, spinal cord compression) may develop, necessitating appropriate intervention to eliminate or to minimize long-term neurologic deficits. Carpal tunnel syndrome may be under-recognized because its onset is insidious and may be associated with few or none of the typical symptoms of numbness and tingling, except for thenar atrophy [Yuen et al., 2007]. The diagnosis of carpal tunnel syndrome often requires testing of nerve conduction velocities. Congenital umbilical hernias in MPS I are generally treated conservatively because complications such as incarceration or perforation are rare [Hulsebos et al., 2004].
Bone marrow transplantation in infants with Hurler’s syndrome, by providing an endogenous source of the enzyme, leads to clinical improvements such as the resolution of hepatosplenomegaly and obstructive airway disease [Souillet et al., 2003]. These positive changes mostly occur in the early years after bone marrow transplantation, and clinical benefit is greatest in the patients who received their transplant before the age of 2 years. Neuropsychologic responses to bone marrow transplantation are variable and related to the patient’s age and intellectual capacity at the time of engraftment [Staba et al., 2004]. Certain disease features, such as skeletal problems, have a much poorer response, likely because of the poor penetration of the enzyme into the relevant tissues [Weisstein et al., 2004]. Unfortunately, most children who successfully undergo transplantation ultimately require major orthopedic surgery for genu valgum, hip dysplasia, kyphoscoliosis, carpal tunnel syndrome, and trigger digits. Intellectual and developmental deterioration may occur, and cardiac valvular deformities persist. A proportion of patients undergoing transplantation also experience graft failure, and life-threatening pulmonary hemorrhage has been encountered after the procedure [Gassas et al., 2003].
Enzyme therapy for MPS I has been approved on the basis of clinical trials primarily involving patients with the Hurler–Scheie phenotype. The studies proved that enzyme replacement therapy leads to improved pulmonary function (based on increased forced expiratory volume in 1 second) and increased exercise capacity (based on the 6-minute walk test, wherein distance traveled over time is measured) [Wraith et al., 2004]. Added observations noted in the treated patients include increased range of joint motion, reduction in sleep apnea or hypopnea, resolution of hepatosplenomegaly, and decreased urinary excretion of glycosaminoglycans. Weekly infusions of the recombinant enzyme (laronidase [Aldurazyme]) given to patients orally premedicated (with acetaminophen and diphenhydramine) were relatively well tolerated, despite the high incidence of antibody formation. Antibody formation was not associated with loss of therapeutic benefit, perhaps because of a decrease in antibody titers with time [Kakavanos et al., 2003]. The recombinant enzyme formulation has also been prescribed to infants not subjected to or just before bone marrow transplantation and until engraftment has been established [Wraith et al., 2007; Wynn et al., 2009]. The benefit of enzyme therapy in these clinical settings remains to be established. Reversal of somatic and visceral problems, commonly encountered in all clinical forms, is anticipated to improve the treated patient’s quality of life.
Intrathecally administered recombinant human α-iduronidase given to MPS I-affected dogs penetrates the brain and reaches tissues in deep brain sections lacking cerebrospinal fluid contact [Kakkis et al., 2004]. These findings suggest that intrathecal enzyme administration may be an effective way to treat the CNS disease in MPS I. Intrathecal enzyme adminstration in a patient with symptomatic spinal cord compression has been reported to be beneficial; however, additional studies are required before this approach can be generally recommended [Munoz-Rojas et al., 2008].
Mucopolysaccharidosis Type II (MPS II)
There are two clinical MPS II variants, which have similar somatic features but vary in degree [Wraith et al., 2008]. The MPS II subtypes are distinguished on the basis of intellectual development and rate of disease progression. In the severe form, there is mental retardation and neurologic involvement (as noted in patients with Hurler’s syndrome) (Figure 36-19). In the attenuated form, intelligence is preserved or mildly impaired. Median age at death was significantly lower in patients with cognitive involvement (11.7 years) compared with those without cognitive involvement (14.1 years) [Jones et al., 2009]. Neurological complications encountered in patients with Hunter syndrome include hydrocephalus, spinal cord compression, cervical myelopathy, optic nerve compression, and hearing impairment [Al Sawaf et al., 2008]. Chronic diarrhea is also a common problem in these patients, whose serial evaluations require careful attention to the potential for pulmonary and cardiac complications. Some patients have whitish macular skin lesions over the back, shoulders, and thighs; this is a distinctive dermatologic feature of Hunter’s syndrome and is useful in its differential diagnosis.
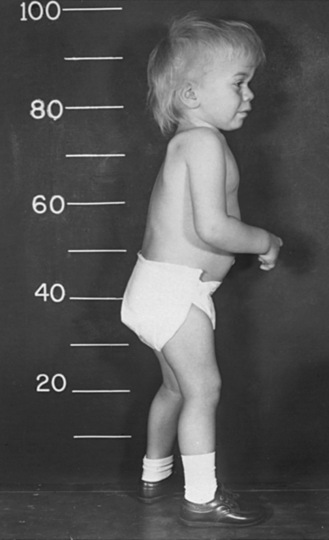
Fig. 36-19 Hunter’s syndrome.
This 16-month-old male with Hunter’s syndrome has many similarities with the patient in Figure 36-18. Flexion contractures of the knees and elbows are prominent. The facial features are also characteristic.
The diagnosis of MPS II is based on demonstration of deficient iduronate-2-sulfatase activity in leukocytes and cultured skin fibroblasts. Prenatal diagnosis is feasible on the basis of enzyme assay. A new method for diagnosis with a novel fluorigenic 4-methylumbelliferyl substrate has been described [Keulemans et al., 2002]. Mutation analyses in patients with MPS II indicate that most have unique gene defects [Froissart et al., 2007]. However, more than 35 percent of independent point mutations at the iduronate-2-sulfatase gene locus are found at CpG sites [Tomatsu et al., 2004b]. Large deletions of the gene encoding iduronate-2-sulfatase tend to correlate with more severe degrees of mental retardation. An Alu-mediated recombination has been implicated in a patient with MPS II who showed the skipping of exon 8 at the cDNA level and the generation of a new large rearrangement [Ricci et al., 2003]. Laboratory studies have revealed that evaluation of residual iduronate-2-sulfatase activity by gene analysis, expression studies, and transcript analysis does not always allow prediction of a patient’s phenotype.
Bone marrow transplantation ameliorates the systemic manifestions of Hunter’s syndrome, but reported neuropsychological outcomes have been variable and appeared to be related to the severity. A recent report describes the outcome in eight boys who received bone marrow transplanted between the ages of 3 and 16 years [Guffon et al., 2009]. On follow-up, from 7 to 17 years post-transplant, cardiovascular abnormalities were stabilized, hepatosplenomegaly was resolved, and joint stiffness improved. Perceptual hearing defects remained stable, while transmission hearing defects improved. Only one child required subsequent surgery to correct kyphosis. Enzyme therapy (idursulfase) for MPS II has been shown to lead to a marked decrease in urinary GAG as well as reduced GAG accumulation in several tissues [Muenzer et al., 2006]. Associated clinical findings in treated patients include improvement in soft-tissue joint contractures, increased physical endurance as measured by the distance that they can achieve in the 6-minute walk test, and stabilization or improvement in pulmonary function tests [Muenzer et al., 2006]. Somatic gene therapy, with viral and nonviral (i.e., encapsulated heterologous cells and muscle electrogene transfer) approaches, is under investigation with use of MPS II primary fibroblasts [Tomanin et al., 2002].
Mucopolysaccharidosis Type III (MPS III)
The Sanfilippo syndrome (or MPS III) includes a group of patients with a similar phenotype resulting from a defect of heparan sulfate degradation on account of four different enzyme deficiencies (see Table 36-1) [Valstar et al., 2008]. Diagnosis of the individual subtypes is based on the specific enzyme deficiency, as measured in biochemical assays performed on leukocytes or cultured skin fibroblasts. Each disorder is further classified by alphabetical letters into MPS III A, B, C, and D. MPS IIIA is the most common variant, and its expression tends to be more severe. The protein that is deficient in patients with MPSIIIC has been characterized as a lysosomal membrane enzyme, heparan sulfate acetyl-CoA (AcCoA): alpha-glucosaminide N-acetyltransferase. This protein catalyzes transmembrane acetylation of the terminal glucosamine residues of heparan sulfate prior to their hydrolysis by alpha-N-acetylglucosaminidase. Several mutations have been reported among patients with MPS IIIA and IIIB [Beesley et al., 2005; Esposito et al., 2000; Yogalingam and Hopwood, 2001]; mutations in MPS IIIC and IIID are just beginning to be identified [Beesley et al., 2003; Feldhammer et al., 2009].
Unlike the other MPS subtypes that are associated with prominent skeletal and somatic features, the Sanfilippo syndrome has clinical features dominated by early and marked behavioral problems (Figure 36-20). Hyperactivity and aggressive behavior, which become apparent between the ages of 2 and 6 years, represent common initial concerns, followed by signs of arrest in intellectual development and speech delay. Altered sleep–wake cycles and insomnia are also frequent sources of parental frustration [Fraser et al., 2002]. Examination of the brain of the mouse model of MPS IIIB (alpha-N-acetylglucosaminidase deficiency) revealed P-tau in the medial entorhinal cortex [Ohmi et al., 2009]. Electron microscopy of dentate gyrus neurons showed cytoplasmic inclusions of paired helical filaments, P-tau aggregates characteristic of tauopathies. Separate studies in the mouse model have also shown that heparan sulfate oligosaccharides, which accumulate in MPS IIB, activate the degradation of synaptophysin by the proteasome with consequences on synaptic vesicle components [Vitry et al., 2009].
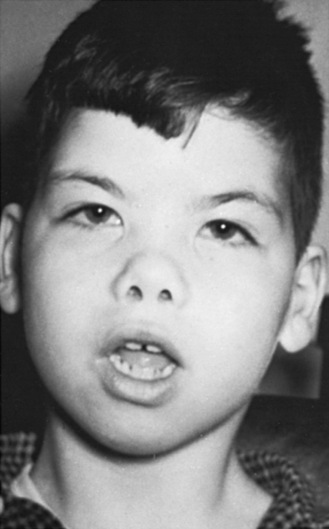
The disease is progressive, and the patients become severely demented and often are bedridden by the end of the first decade. An exceptional report has described two sisters with MPS IIIC with adult-onset dementia and retinitis pigmentosa [Berger-Plantinga et al., 2004]. Another report also provides a description of an adult patient with MPS IIIA who presented at the age of 45 years with hypertension [Van Hove et al., 2003]. At the age of 53 years, she was found to have severe concentric hypertrophic nonobstructive cardiomyopathy. There was no coarsening of facial features. In addition, her neurologic function, skeleton, cornea, liver, and spleen were normal.
As the somatic and skeletal features of MPS III may be subtle and the diagnosis often missed, inclusion of this disorder in the differential diagnosis of children presenting with behavioral problems is important. Studies of bone turnover and bone mineral density in three patients with MPS III revealed low serum vitamin D concentration and greatly reduced bone mineral density at lumbar and femoral sites [Rigante and Caradonna, 2004]. Screening for excessive urinary excretion of heparan sulfate may enable detection of MPS III. As testing of the urine alone may give false-negative results, in circumstances when the diagnosis remains strongly suspected, analysis for the putative enzyme deficiency should be performed [Chih-Kuang et al., 2002].
There is no specific treatment for MPS III. Investigations suggest that the sleep disruption in patients with MPS III consists of an irregular sleep–wake pattern, which at its onset might appear to be a disorder of initiating or maintaining sleep [Mariotti et al., 2003]. This finding could explain why some patients do not respond to conventional hypnotics. Therapies aimed at resynchronization (such as behavioral treatment, light therapy, or melatonin) have been recommended. Behavior modification and medications to control aggressive behavior may be required, although these approaches only work modestly at best. The use of antipsychotic medications has been reported to lead potentially to increased incidence of extrapyramidal syndrome in affected individuals [Tchan and Sillence, 2009]. Several gene therapy experiments conducted in various mouse models for MPS IIIA and IIIB provide a rationale for this approach in human patients [Fu et al., 2007; McIntyre et al., 2008]. Pathologic studies in the caprine (goat) model indicate that prenatal therapeutic intervention is likely to be necessary to prevent or to ameliorate substantive CNS and systemic lesions [Jones et al., 2004].
Mucopolysaccharidosis Type IV (MPS IV)
This subtype, also referred to as Morquio’s syndrome, includes two genetically distinct enzyme deficiency disorders (identified as MPS IV A and B) [Haas, 2002]. Both subtypes are phenotypically similar or have overlapping clinical features. Intellect is usually normal, but secondary neurologic complications are frequent and related to the skeletal abnormalities and increased joint laxity [Ebara et al., 2003]. Affected patients have a high risk for cervical cord compression because of severe hypoplasia of the odontoid process and laxity of the cervical ligaments. The potential for atlantoaxial subluxation during anesthesia and while engaged in sports necessitates appropriate precautions [Dalvie et al., 2001b; Morgan et al., 2002]. Lower leg paresis due to kyphoscoliosis was reported in a 65-year-old woman who developed compression of the spinal cord around T12 to L2 [Ebara et al., 2003]. In these patients, surgery to stabilize the cervical spine is usually necessary as a preventive measure [White et al., 2009a]. With increased survival, there is a risk for respiratory failure secondary to abnormalities of the thoracic cage.
Keratan sulfate is increased in urine among affected individuals with MPS IV from deficiency of either N-acetylgalactosamine-6-sulfate (MPS IVA) or β-galactosidase (MPS IVB). Histological analysis, performed on bone, cartilage, and fibroblasts obtained from two unrelated patients with MPS IVA, showed that the collagen of bone and the collagen deposited by fibroblasts were normal, whereas the extracellular matrix of cartilage was abnormal and prone to degeneration [Bank et al., 2009].
Mutation analysis performed on MPS IVA patients from Latin America revealed 12 different gene defects, with R386C accounting for 32.5 percent of the unrelated mutant alleles [Tomatsu et al., 2004a]. One mutation (R380S) was associated with an attenuated disease form.
The underlying enzyme deficiencies for MPS IVB and GM1 gangliosidosis are identical; the variable disease expression has been attributed to distinct allelic mutations in the gene encoding β-galactosidase. An imbalanced substrate specificity of the mutant β-galactosidase that induces predominant accumulation of keratan sulfate has been demonstrated in skin fibroblasts of patients with MPS IVB and may be useful in differential diagnostic analysis and prognostication [Hinek et al., 2000]. Most mutations in MPS IVB are associated with homozygosity or heterozygosity for the W273L mutation, which expresses residual enzyme activity [Paschke et al., 2001].
There is no specific treatment. The anesthetic care of these patients, before any elective surgical procedures, should take into consideration increased risks resulting from the respiratory, craniofacial, cardiac, skeletal, ocular, and hepatic abnormalities [Walker et al., 2003a]. Because intellect is normal and skeletal deformities associated with the lysosomal storage diseases are not significantly modified by bone marrow transplantation, there is no role for this procedure in the management of patients with MPS IV A or B. Enzyme therapy in the mouse model for MPS IVA resulted in reduction of keratan sulfate storage in visceral organs, sinus lining cells in bone marrow, heart valves, ligaments, and connective tissues, providing a rationale for this therapeutic approach in human patients [Tomatsu et al., 2008].
Mucopolysaccharidosis Type VI (MPS VI)
Maroteaux–Lamy syndrome (MPS VI) results from a deficiency of N-acetylgalactosamine-4-sulfate (arylsulfatase B) in association with the excessive urinary excretion of dermatan sulfate. Investigations in fibroblasts from MPS VI patients has revealed lysosomal storage of dermatan sulfate to be associated with impaired autophagy, accumulation of polyubiquitinated proteins, and mitochondrial dysfunction [Tessitore et al., 2009].
There is a wide variability in clinical expression, including mild and severe phenotypes, with skeletal and somatic features that are typical for the mucopolysaccharidoses. Corneal opacities result from deposition of keratan sulfate and other proteoglycans in lysosomes, causing the death of keratocytes and an abnormal build-up of proteoglycans in the stroma [Akhtar et al., 2002]. These changes are believed to be responsible for the lateral aggregation of collagen fibrils and impaired fibrillogenesis in MPS VI. Degenerate swollen keratocytes, together with gross changes in epithelial, stromal, and endothelial cells, would be expected to increase light scattering significantly in these involved corneas.
Affected individuals with MPS VI usually have normal intelligence. Myelopathy secondary to thickening of the dura in the cervical region has been described. Hydrocephalus is another complication that, if unrecognized, may contribute to mental decline. The recognition of precocious excessive growth in two patients with MPS VI facilitated an early diagnosis [Heron et al., 2004]. A study involving Brazilian patients with MPS VI found no significant correlation between clinical manifestations and the levels of urinary glycosaminoglycans or the residual arylsulfatase B activity [Azevedo et al., 2004]. In this population of patients, a 23-base pair deletion causing a shift in reading frame has been found to be relatively common [Petry et al., 2003]. Molecular characterization of the gene encoding arylsulfatase B among other groups of patients has revealed broad heterogeneity in underlying gene defects [Garrido et al., 2008].
Bone marrow transplantation in patients with MPS VI has resulted in the resolution of hepatomegaly and splenomegaly, stabilization of cardiopulmonary function, and improvement of visual acuity and joint mobility [Wang et al., 2008]. Corneal transplantation performed in a female who underwent bone marrow transplantation at the age of 13 years has been reported to help maintain clear corneal grafts [Ucakhan et al., 2001]. In clinical trials involving 39 patients (ranging in age from 5 to 29 years), enzyme therapy (galsulfase) for MPS VI led to improvements in physical endurance, as measured by the distance walked in 12 minutes and the number of steps taken during the 3-minute stair climb test [Harmatz et al., 2006a]. In treated patients, as opposed to patients receiving placebo, these findings were associated with significant reductions in urinary excretion of glycosaminoglycans. Infusions of the recombinant investigational enzyme (Aryplase [BioMarin Pharmaceutical Inc.] at 1 mg/kg weekly) were generally well tolerated. Longer-term follow-up of treated patients indicated that the clinical improvements were sustained, except in those with advanced skeletal disease who were at high risk for progression of hip disease and cervical spinal cord compression [Harmatz et al., 2008]. Enzyme therapy studies in the feline model indicate overall improvement in the disease (from a physical, neurologic, and skeletal perspective) that was most pronounced for cats treated from birth compared with those treated at a later age. These findings support the arguments for early intervention [Auclair et al., 2003].
Mucopolysaccharidosis Type VII (MPS VII)
Sly’s syndrome (MPS VII) results from a deficiency of β-glucuronidase. There are about 40 cases reported in the medical literature. The disease is characterized by a wide spectrum of clinical manifestations. A severe neonatal form is associated with nonimmune hydrops fetalis [Cheng et al., 2003]. Craniovertebral instability and spinal cord compression have also been reported to occur in MPS VII [Dickerman et al., 2004].
Studies in the mouse model to elucidate the basis of the skeletal disease in MPS VII revealed that the growth plates had reduced tyrosine phosphorylation of STAT3 (a pro-proliferative transcription factor), decreased expression of leukemia inhibitory factor (LIF) and other interleukin-6 family cytokines, and reduced phosphorylated tyrosine kinase 2 (TYK2), Janus kinase 1 (JAK1), and JAK2 (known activators of STAT3 phosphorylation) [Metcalf et al., 2009]. These observations are consistent with reduced chondrocyte proliferation and ultimately shortened bones. Using brain tissue obtained from the mouse model, gene expression profile studies have indicated roles for microglial activation and induction of apoptotic pathways in the neurodegenerative process associated with MPS VII [Richard et al., 2008].
Bone marrow transplantation has been performed in a 12-year-old female homozygous for the A619V mutation in the β-glucuronidase gene [Yamada et al., 1998]. The donor cells were obtained from an HLA-identical unrelated female donor. Reported improvements included almost normal β-glucuronidase activity in circulating lymphocytes and greatly diminished urinary excretion of glycosaminoglycans. These observations were associated with improved motor function (including independent ambulation), and decreased frequency of upper respiratory tract and middle ear infections. Previous problems that the patient had experienced, such as dyspnea on exertion and vertigo, also improved.
The diagnosis of MPS VII is based on demonstration of decreased β-glucuronidase activity. Several mutations have been described in the β-glucuronidase gene [Tomatsu et al., 2009]. In the longest known survivor (a patient who died at the age of 37 years), mutation analysis revealed two novel missense mutations, K350N and R577L [Storch et al., 2003]. Expression studies revealed that the K350N gene defect was associated with residual enzyme activity, whereas the mutant protein resulting from the R577L transcript was rapidly degraded. Prenatal diagnosis of MPS VII can be made reliably by analysis of enzyme activity in amniotic fluid or cultured chorionic villi or amniocytes [Natowicz et al., 2003].
Several studies, including investigations of gene therapy, are taking place in the animal model of MPS VII [Sferra et al., 2004]. A report noted that peripheral administration of a recombinant adeno-associated virus type 2 vector in adult MPS VII mice led to transgene expression levels sufficient for improvements in both the peripheral and central manifestations of this disease [Sakurai et al., 2004]. These results correlate with findings in another study, which revealed that enzyme transport into brain parenchyma in early postnatal life in the mice was mediated by the mannose 6-phosphate/insulin-like growth factor-2 receptor, which was not observed in the adult mice [Urayama et al., 2004]. The transplantation of genetically modified bone marrow stromal cells to the lateral ventricle of newborn MPS VII mice has been found to reduce the brain content of glycosaminoglycans, with marked improvement in cognitive function compared with the untreated mice [Shen et al., 2004]. In a separate study, β-glucuronidase gene transfer led to a reversal of storage lesions in neurons that project to the gene expression site, but not in nearby structures that would have been corrected if the effect had been mediated by diffusion [Frisella et al., 2001]. In addition, transduction of cells in the subventricular zone resulted in the uptake of β-glucuronidase by cells entering the rostral migratory stream. These findings suggest that gene transfer to specific neuronal circuits or cells in the migratory pathways may facilitate delivery to the global brain lesions found in these disorders [Shen et al., 2004]. Interestingly, chemical modification of the human β-glucuronidase enzyme to eliminate its uptake by mannose 6-phosphate and mannose receptors in cultured cells, resulted in slowed plasma clearance and improved effectiveness in clearing CNS storage, compared with changes observed using the native enzyme at the same dose [Grubb et al., 2008].
Mucopolysaccharidosis Type IX (MPS IX)
The first patient subsequently classified as having MPS IX (Natowicz syndrome) had a relatively mild phenotype (compared with patients with other forms of MPS). Clinical characteristics included periarticular soft tissue masses, mild short stature, and acetabular erosions of the joints [Natowicz et al., 1996]. The patient also did not have signs of neurologic or visceral involvement. Histologic examination of affected tissues revealed macrophages with hyaluronan-filled lysosomes. Serum hyaluronidase activity was noted to be deficient, and the concentration of hyaluronan in the serum was elevated 38- to 90-fold above normal [Natowicz et al., 2003]. Hyaluronan is an important structural and functional component of the extracellular matrix. Changes in hyaluronan turnover have been associated with many processes involving cell proliferation, migration, and differentiation, and with embryogenesis, inflammation, wound healing, and metastasis. Histological studies of the knee joint in the mouse model of MPS IX showed a loss of proteoglycans occurring as early as 3 months and which progressed with age [Martin et al., 2008]. An increased number of chondrocytes displaying intense pericellular and/or cytoplasmic hyaluronic acid staining were detected in the epiphyseal and articular cartilage.
Two mutations (as expected for an autosomal-recessive trait) have been identified in the lysosomal hyaluronidase 1 gene. A 1412G to A mutation introduces a nonconservative amino acid substitution [Glu268Lys] in a putative active site residue [Triggs-Raine et al., 1999]. The second mutation is a complex intragenic rearrangement [1361del37ins14] that results in a premature termination codon. Subsequent studies have revealed that additional enzymes (hyaluronidase 2 and 3) are encoded by genes found to cluster on human chromosome 3p21.3 [Csoka et al., 2001]. The three enzymatic forms of hyaluronidase have markedly different tissue expression patterns, consistent with their differing roles in hyaluronan metabolism.
