[level-membership-for-pathology-category]23
Lysosomal and peroxisomal disorders
This chapter deals first with the lysosomal disorders that principally affect gray matter and then with those involving white matter (leukodystrophies). Lastly, the chapter covers the peroxisomal disorders, which include adrenoleukodystrophy. Other leukodystrophies are considered in Chapter 5. Chapter 5 also covers comparative aspects of all of the leukodystrophies, and an approach to their differential diagnosis.
LYSOSOMAL DISORDERS
 GM2 GANGLIOSIDOSIS
GM2 GANGLIOSIDOSIS







GM2 GANGLIOSIDOSIS
MACROSCOPIC APPEARANCES
 ETIOLOGY OF GM2 GANGLIOSIDOSIS
ETIOLOGY OF GM2 GANGLIOSIDOSIS









MICROSCOPIC APPEARANCES
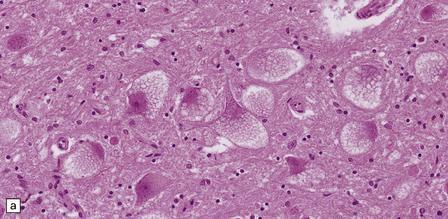
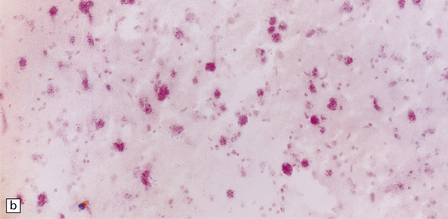
In the older patients neuronal storage of excessive lipofuscin is confined to the basal ganglia, brain stem, cerebellum, and spinal cord. In infantile GM2 gangliosidosis, ballooned neurons are found throughout the CNS and the peripheral nervous system. The foamy nerve cells stain strongly with Luxol fast blue and Sudan black, and in frozen sections the soluble ganglioside is periodic acid–Schiff (PAS)-positive (Fig. 23.1). Microglia are also PAS-positive, retaining stored material even in paraffin sections. Ultrastructural studies show membranous cytoplasmic bodies (MCBs) within neuronal somata.
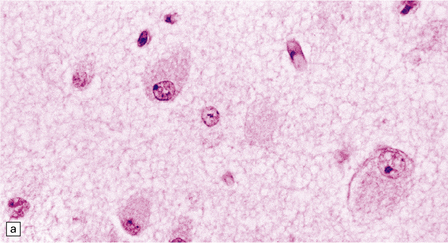
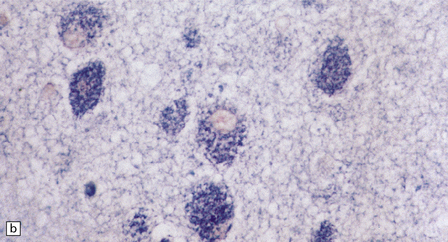
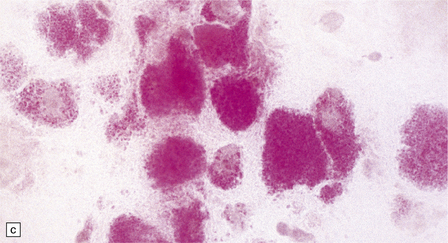
23.1 GM2 gangliosidosis.
(a) Ballooned cortical neurons with foamy cytoplasm and marginated nuclei are negative with PAS in routine paraffin sections. (b) These neurons are, however, strongly stained with Luxol fast blue. (c) The stored ganglioside can be demonstrated in cryostat sections stained with PAS, in this example protected by celloidinization prior to staining.
GM1 GANGLIOSIDOSIS
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Mild gyral atrophy is present in all three subtypes (Fig. 23.2a,b). Ballooned neurons with staining characteristics virtually identical to those seen in GM2 gangliosidosis are widespread in the cerebrum (Fig. 23.2c,d), brain stem, and spinal cord, and in autonomic ganglia (in subtypes 1 and 2). Ballooned neurons are confined to the striatum and pallidum in the adult form. Ultrastructural studies show membranous cytoplasmic bodies.
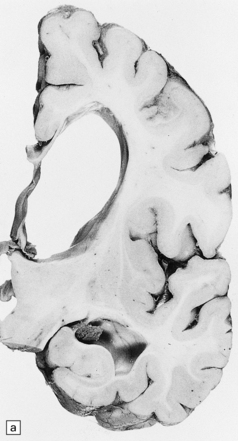
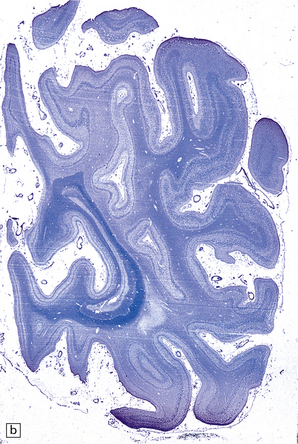
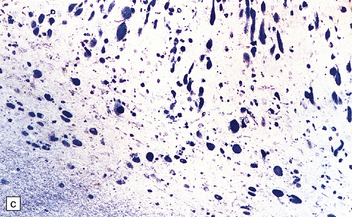
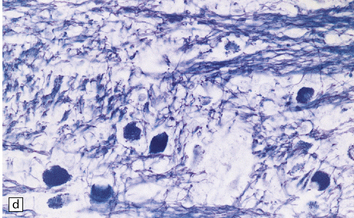
23.2 GM1 gangliosidosis.
(a) Coronal section of cerebral hemisphere from a patient with the type 2 (late-infantile and juvenile) form of GM1 gangliosidosis, who died aged 10 years. There is mild cortical atrophy and ventricular dilatation. (b) Cortical atrophy and poverty of myelination are more evident in a stained section from the occipital lobe. (c) In the cerebral cortex there is extensive neuronal and glial storage. The granular storage bodies fill the neuronal cytoplasm and distend the proximal dendrites. Note also the myelin pallor. (d) Heavy neuronal storage is also present in the pontine nuclei.
 GM1 GANGLIOSIDOSIS
GM1 GANGLIOSIDOSIS




 ETIOLOGY OF GM1 GANGLIOSIDOSIS
ETIOLOGY OF GM1 GANGLIOSIDOSIS

BATTEN’S DISEASE, NEURONAL CEROID LIPOFUSCINOSIS (NCL, CLN)
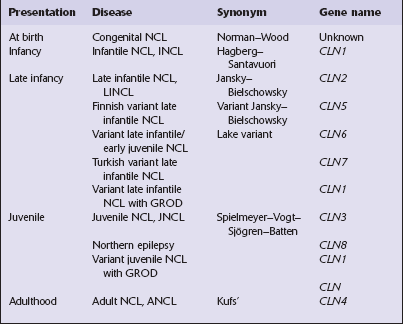
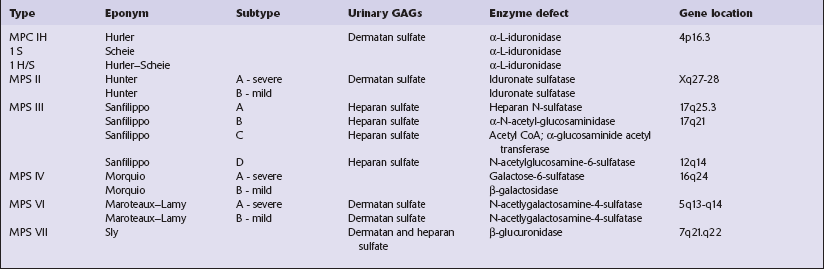
This group of disorders has a confusing set of eponyms (Table 23.1). Classification is further complicated by newer terminology related to the molecular genetics of the disorders. The classification used here combines clinical presentation, age of onset, pathology, and electrophysiology. The diagnosis is most readily obtainable for all forms except Kufs’ disease by cutting cryostat sections of a suction rectal biopsy to examine neurons and other cell types (i.e. smooth muscle, histiocytes, vascular endothelium) for the characteristic accumulations of autofluorescent ceroid lipofuscin, while ultrastructural examination for the various specific types of inclusions is most easily accomplished using buffy coat preparations of lymphocytes. In JNCL (CLN3) numerous vacuolated lymphocytes are demonstrated in the trails of a routine peripheral blood film, and in the right clinical setting this is virtually confirmatory. Skin biopsy is also routinely used: examination of eccrine but not apocrine glands is informative (if the biopsy is from the axilla note that many of the sweat glands will be of apocrine type).
 ETIOLOGY OF BATTEN’S DISEASE
ETIOLOGY OF BATTEN’S DISEASE
 BATTEN’S DISEASE
BATTEN’S DISEASE











MACROSCOPIC APPEARANCES
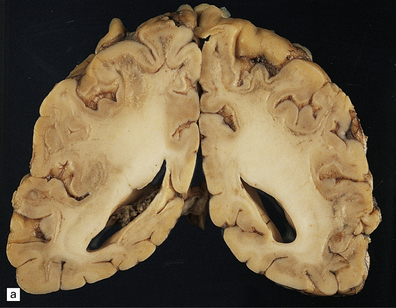
Cerebral atrophy is always present, but is at its most severe in the infantile form (Fig. 23.3a–c), manifesting as a walnut brain with shriveled cortex and rubbery white matter encased in a markedly thickened skull. Atrophy may also be considerable in infantile and juvenile Batten’s disease, and increases with the length of survival. In adult cases (Kufs’ disease), atrophy is more limited, and predominantly in frontal and cerebellar regions.
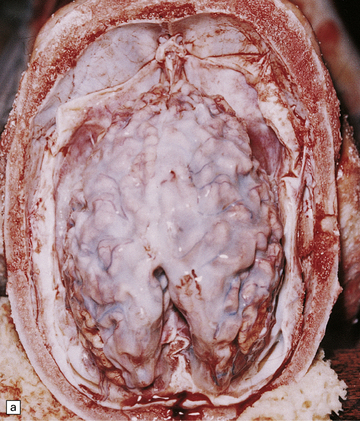
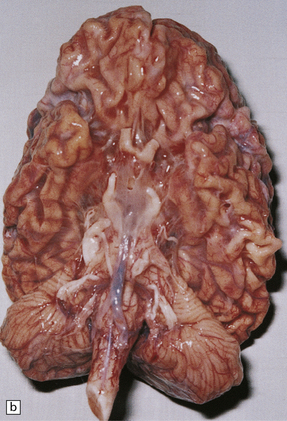
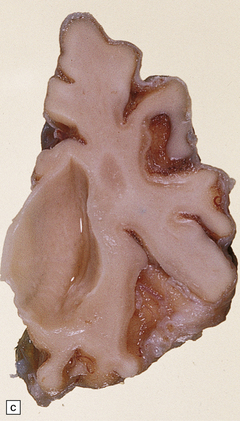
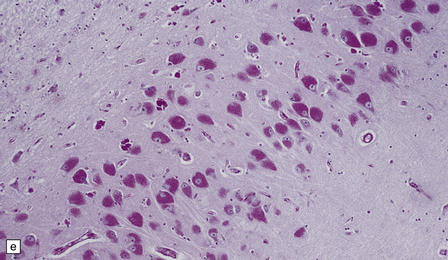
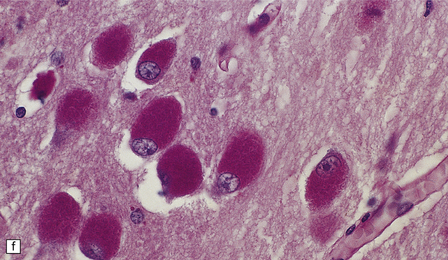
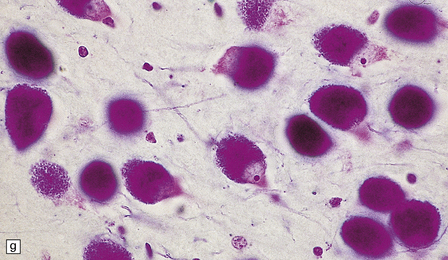
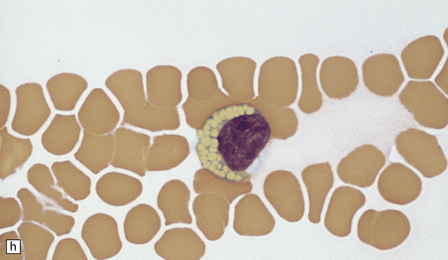
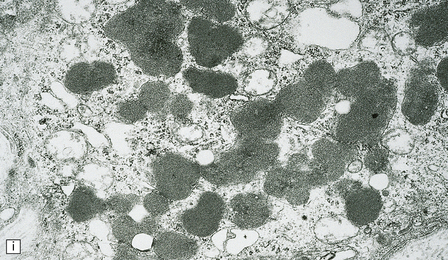
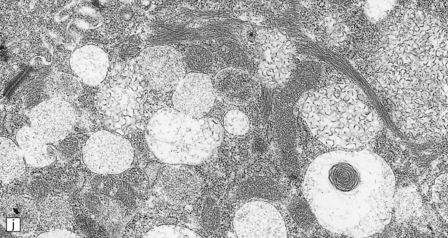
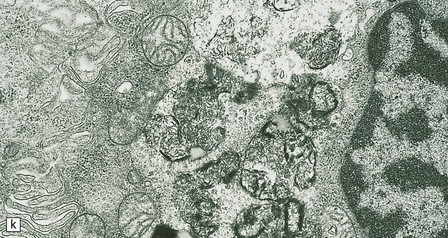
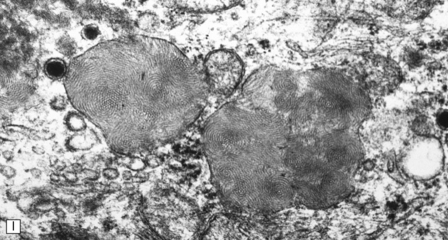
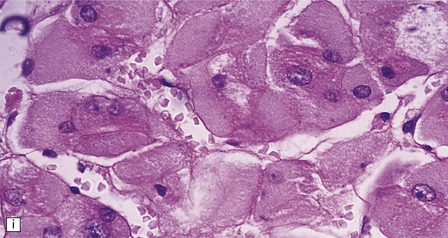
23.3 Batten’s disease.
(a) Infantile Batten’s disease in a boy aged 9 years. The extremely atrophied brain (300 g weight) is covered by gelatinous leptomeninges and markedly thickened dura and surrounded by greatly thickened calvaria. (b) Viewed from below, the cerebral convolutional atrophy is marked and widespread, but the cerebellum and brain stem are relatively spared. (c) A coronal slice of frontal lobe shows a very thin cortical ribbon and tough rubbery white matter. (d) Juvenile Batten’s disease. Neuronal storage material reacts strongly with Sudan black in the cortex. (e) Juvenile Batten’s disease. Neuronal storage material reacts strongly with PAS in hippocampal pyramidal cells. (f) and (g) Kufs’ disease. In the rare adult form of NCL there is similar widespread neuronal storage material, which stains strongly with PAS. (h) Blood film of a patient with juvenile Batten’s disease showing a vacuolated lymphocyte with characteristic large uniform ‘bold’ vacuoles. A similar appearance is seen in GM1 gangliosidosis. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (i) Ultrastructural appearance in infantile Batten’s disease showing granular osmiophilic deposits in a neuron. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (j) Ultrastructural appearance in late-infantile Batten’s disease showing curvilinear bodies within a sweat gland epithelial cell. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (k) Ultrastructural appearance of a sweat gland epithelial cell containing mixed curvilinear and fingerprint bodies in juvenile Batten’s disease (similar in early juvenile and Finnish variant late-infantile Batten’s disease). (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (l) Fingerprint bodies in juvenile Batten’s disease.
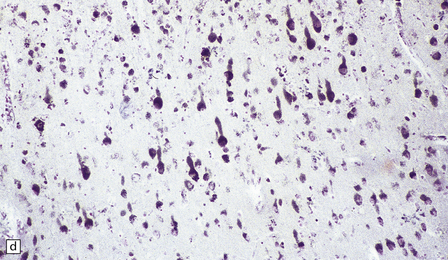
MICROSCOPIC APPEARANCES
The stored material, which is insoluble and therefore readily detectable in paraffin as well as frozen sections, is widespread in the nervous system and in many other tissues. Its tinctorial properties vary slightly between the various subtypes of the disease (Table 23.2). In infantile Batten’s disease, storage is evident in CNS neurons, astrocytes, and macrophages, and in autonomic ganglia from an early stage, but neuronal loss is relatively subtle to begin with, becoming obvious after 2 years. By 4 years of age virtually all cortical neurons have disappeared, and there is dense astrocytic gliosis, and myelin loss. Some astrocytes contain storage material.

In late-infantile and juvenile Batten’s disease (Fig. 23.3d,e) neuronal loss is less severe and myelin loss, if present, is slight. The rarity of adult cases and the accumulation of lipofuscin during normal aging have impeded the formulation of a consensus view of the histology of Kufs’ disease, although widespread storage is the principal element (Fig. 23.3f,g).
NIEMANN–PICK DISEASE

 Not sphingomyelinase deficient (Table 23.3).
Not sphingomyelinase deficient (Table 23.3).
Table 23.3
Classification of Niemann–Pick disease
| Group I: sphingomyelinase deficient | Group II: not sphingomyelinase deficient |
| Type A: neurovisceral (infantile, juvenile, and adult) | Types C and D (Nova Scotia): neurovisceral |
| Type B: visceral only (infantile, juvenile, and adult) | Possible pure visceral form |
This classification incorporates the earlier four alphabetically defined groups.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Niemann–Pick disease group I
While hepatosplenomegaly is striking in both types, CNS abnormalities are not found in type B. In type A, cerebral atrophy may be slight or absent. Microscopically (Fig. 23.4), there is generalized enlargement of neurons and glia, and storage extends to white matter, which is demyelinated and gliotic. Gastrointestinal tract neuronal plexuses are also affected. Sudanophilic foamy histiocytes containing cholesterol esters, but not ballooned neurons, are numerous in the globus pallidus, substantia nigra, and dentate nucleus. Niemann–Pick cells (Fig. 23.4b) are present throughout the mononuclear phagocyte system, and can fill the alveolar spaces of the lungs. The lymphocytes of patients with type A Niemann–Pick disease contain cytoplasmic vacuoles, which are small and discrete. In contrast, in patients with type B there is minimal or no lymphocytic vacuolation. Bone marrow aspirates show collections of Niemann–Pick cells in patients with type A and in younger patients with type B. In older patients with type B disease there are fewer foamy Niemann–Pick cells, and more prominent ’sea-blue histiocytes’, in which the cytoplasm is filled with small granules that stain intensely blue with the Giemsa or Wright histochemical method.
Niemann–Pick disease group II
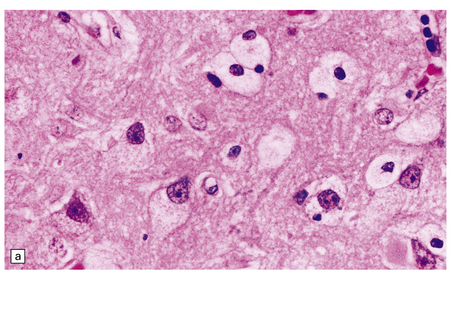
Despite the diverse chemical abnormalities, the morphologic features are fairly uniform. Cerebral atrophy and sclerotic firm white matter are evident. Microscopically, widespread neuronal ballooning is particularly noticeable in the basal ganglia, brain stem, and spinal cord. In addition to finely granular storage material, neuroaxonal dystrophy and Alzheimer-type neurofibrillary tangles are also observed (Fig. 23.5). Neurons, including those of the gastrointestinal tract, store a substance that is lost during routine processing. In frozen sections the substance is only weakly sudanophilic, but includes phospholipid and a PAS-positive sugar-containing compound.
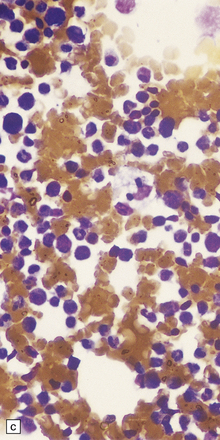
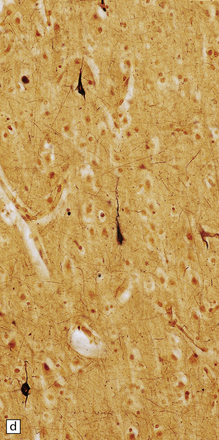
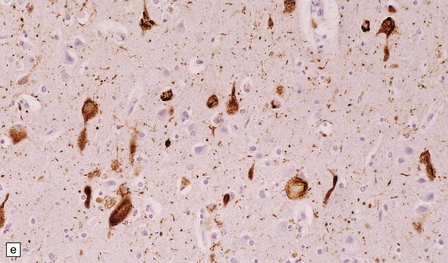
23.5 Niemann–Pick disease type C.
(a) Ballooned neurons in the oculomotor nucleus. (b) Finely granular storage bodies can be demonstrated in neuronal cell bodies and proximal processes using the protected (celloidinized) PAS method in a cryostat section. (c) Foam cells in the bone marrow contain vacuoles of varying size and densely staining fragments of nuclear debris. In most cases, argyrophilic (d), tau-immunoreactive (e) neurofibrillary tangles are present. The tau-immunoreactive material extends into apical dendrites and distends the proximal part of some axons.
 NIEMANN–PICK DISEASE
NIEMANN–PICK DISEASE The onset of type A is in the first year, with hepatosplenomegaly and failure to thrive, followed by mental retardation. Other common findings are cherry red macular spots and diffuse pulmonary infiltration. Survival is uncommon beyond 4 years of age, but in longer survivors neurologic symptoms of dementia, spasticity, and epilepsy may be delayed beyond 5 years of age.
The onset of type A is in the first year, with hepatosplenomegaly and failure to thrive, followed by mental retardation. Other common findings are cherry red macular spots and diffuse pulmonary infiltration. Survival is uncommon beyond 4 years of age, but in longer survivors neurologic symptoms of dementia, spasticity, and epilepsy may be delayed beyond 5 years of age.

 Type C has a variable presentation, and features include congenital ascites, neonatal failure to thrive, hepatosplenomegaly, and prolonged neonatal obstructive jaundice, insidious onset of dementia and ataxia in childhood, various forms of psychosis in adolescence and early adulthood, and dystonia and loss of vertical eye movements in early adulthood or middle age.
Type C has a variable presentation, and features include congenital ascites, neonatal failure to thrive, hepatosplenomegaly, and prolonged neonatal obstructive jaundice, insidious onset of dementia and ataxia in childhood, various forms of psychosis in adolescence and early adulthood, and dystonia and loss of vertical eye movements in early adulthood or middle age.
 ETIOLOGY OF NIEMANN–PICK DISEASE
ETIOLOGY OF NIEMANN–PICK DISEASE
 Group I patients (A & B forms) have a profound deficiency of lysosomal sphingomyelinase activity. Sphingomyelin levels are increased 5–10-fold in the brains of type A patients, while the lipid level in type B patients is normal.
Group I patients (A & B forms) have a profound deficiency of lysosomal sphingomyelinase activity. Sphingomyelin levels are increased 5–10-fold in the brains of type A patients, while the lipid level in type B patients is normal.
 The gene for lysosomal sphingomyelinase is located at 11p15.1–15.4.
The gene for lysosomal sphingomyelinase is located at 11p15.1–15.4.

 The defect in type C and D forms of disease has been linked to chromosome 18q11–12.
The defect in type C and D forms of disease has been linked to chromosome 18q11–12.
GAUCHER’s DISEASE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The characteristic Gaucher cell (Fig. 23.6) is present in many tissues, is large (20–100 mm), and has one or more nuclei. Its cytoplasm is filled with finely or coarsely fibrillar material. The appearance of the cytoplasm in histologic sections has been likened to crumpled tissue paper. Gaucher cells are PAS-positive, negative with lipid stains, and sometimes contain iron pigment. Ultrastructurally, the fibrillar inclusions are membrane-bound elongated bodies containing a tubular arrangement of the glucocerebroside.
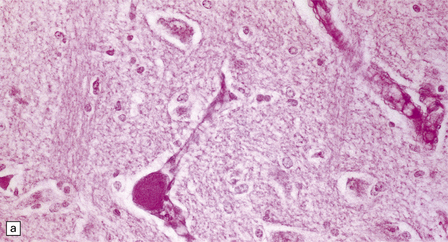
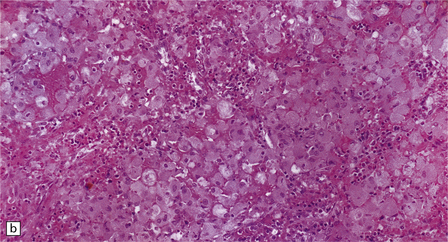
23.6 Gaucher’s disease.
(a) A PAS-positive Gaucher cell close to a capillary in the striatum. (b) Groups of Gaucher cells are present in the spleen. The cytoplasm of these cells may have a vacuolar or fibrillar appearance.
 ETIOLOGY OF GAUCHER’S DISEASE
ETIOLOGY OF GAUCHER’S DISEASE Deficient activity of glucocerebrosidase produces a marked increase in tissue glucocerebroside, especially in the spleen, where its concentration is 100 times normal.
Deficient activity of glucocerebrosidase produces a marked increase in tissue glucocerebroside, especially in the spleen, where its concentration is 100 times normal.
 In some patients, deficiency of a cofactor, saposin C, is responsible for Gaucher’s disease.
In some patients, deficiency of a cofactor, saposin C, is responsible for Gaucher’s disease.
 The gene for glucocerebrosidase, GBA, is located at 1q21.
The gene for glucocerebrosidase, GBA, is located at 1q21.
 Prosaposin, the precursor of saposin C, is encoded on chromosome 10q21.
Prosaposin, the precursor of saposin C, is encoded on chromosome 10q21.
 Note that heterozygous mutations in GBA increase the risk of Parkinson’s disease and dementia with Lewy bodies (see Chapters 28 and 31).
Note that heterozygous mutations in GBA increase the risk of Parkinson’s disease and dementia with Lewy bodies (see Chapters 28 and 31).
 GAUCHER’S DISEASE
GAUCHER’S DISEASE Type 1: chronic non-neuronopathic Gaucher’s disease can begin at any age from birth to adulthood. An early onset is associated with massive splenomegaly and some hepatomegaly.
Type 1: chronic non-neuronopathic Gaucher’s disease can begin at any age from birth to adulthood. An early onset is associated with massive splenomegaly and some hepatomegaly.


MUCOPOLYSACCHARIDOSES (MPS)
The classification of the MPSs incorporates historic eponyms, specific enzyme defects, and the analysis of urinary excretion of glycosaminoglycans (GAGs) (Table 23.4). Inheritance is autosomal recessive, except for MPS II (Hunter syndrome), which is transmitted as an X-linked recessive trait.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Macroscopic appearances are usually nondescript, but there may be considerable widening of the skull, dural and meningeal thickening, and sometimes cerebral atrophy. Hydrocephalus may occur (see Chapter 4). A characteristic finding in sectioned brain is the presence of small perivascular cavities in the white matter (Fig. 23.7a–d), which are shown microscopically to contain foamy macrophages.
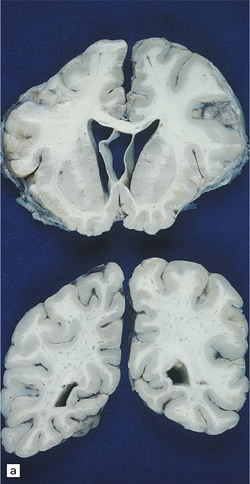
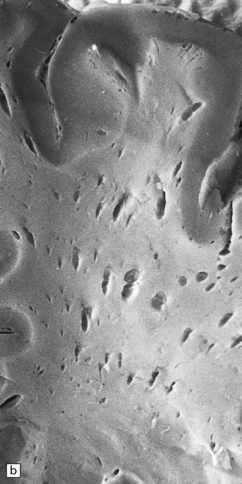
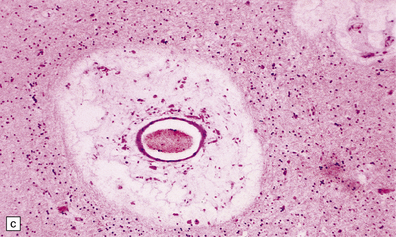
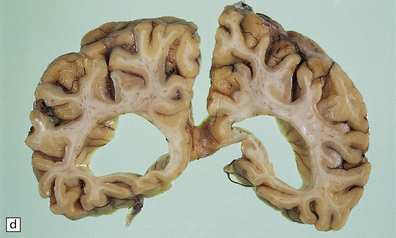
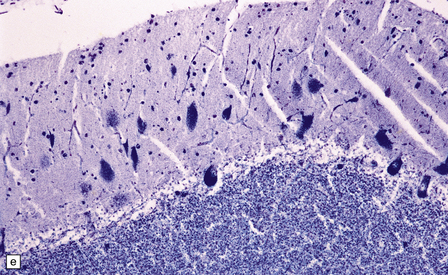
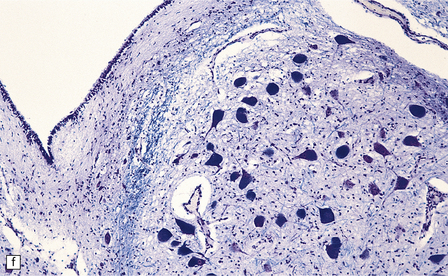
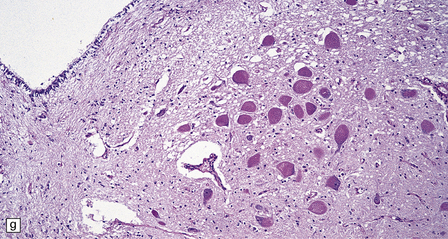
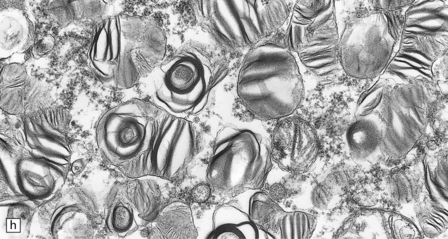
23.7 Mucopolysaccharidoses (MPS).
(a) MPS VI (Maroteaux–Lamy): the white matter is studded with fine pits. (b) On closer inspection the fine pits are perivascular. (c) These perivascular pits are filled with foamy macrophages containing water-soluble mucopolysaccharide, which accounts for their empty appearance in routinely processed sections. (d) MPS IIIa (Sanfilippo A) in an 11-year-old boy: a coronal section of the brain shows severe cerebral atrophy. Neuronal storage of ganglioside is present throughout the brain. (e) In the cerebellar cortex of the patient shown in (d), Purkinje cells and their dendrites are filled with granules. (f) MPS IH: storage of ganglioside in neurons of the 12th cranial nerve nucleus stained with Luxol fast blue. (g) Same as (f), but stained with PAS. (h) Ultrastructurally, the stored material forms membrane stacks or ’zebra’ bodies.
Neuronal storage (Fig. 23.7e–g) is very variable, but usually parallels the severity of mental retardation. The stored material in neurons is ganglioside, and therefore PAS-positive, sudanophilic, and strongly positive with Luxol fast blue. However, neuronal storage of mucopolysaccharide cannot be demonstrated.
 MUCOPOLYSACCHARIDOSES
MUCOPOLYSACCHARIDOSES There are many clinical similarities between the different types of MPS, including coarse facial features, mild to severe skeletal changes (occasionally causing cord compression), hepatosplenomegaly, and mild to severe mental retardation.
There are many clinical similarities between the different types of MPS, including coarse facial features, mild to severe skeletal changes (occasionally causing cord compression), hepatosplenomegaly, and mild to severe mental retardation.
 Carpal tunnel syndrome, cardiomyopathy, and aortic and mitral valve incompetence are common.
Carpal tunnel syndrome, cardiomyopathy, and aortic and mitral valve incompetence are common.


Ultrastructurally, large membrane-bound vacuoles in many tissues are usually empty, but some fine granular material and lamellae are occasionally noted. Various neuronal storage bodies are seen including MCBs, and loosely arranged parallel lamellae juxtaposed with spaces to form ’zebra’ bodies (Fig. 23.7 h).
MANNOSIDOSIS
 MANNOSIDOSIS
MANNOSIDOSIS



FUCOSIDOSIS
Neurons in many areas of the CNS are vacuolated and enlarged. The olives and thalamus are particularly affected. Purkinje cells may be depleted. In some cases, the white matter is extensively demyelinated and gliotic, and the presence of many Rosenthal fibers (Fig. 23.8) produces an appearance reminiscent of Alexander’s disease.
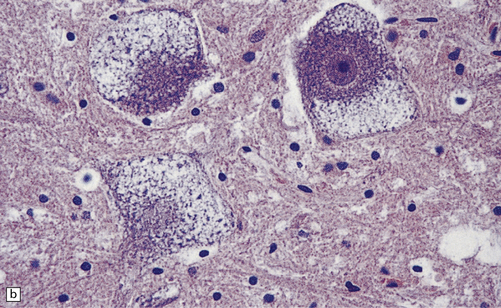
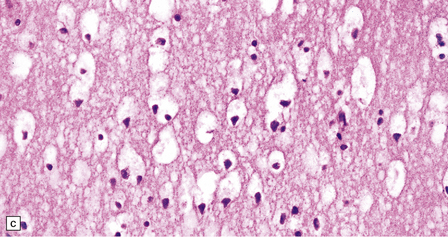
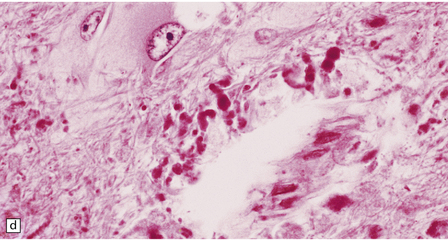
23.8 Fucosidosis.
(a) In this brain from a child aged 6 years, the white matter is strikingly firm and white. (b) Ballooned anterior horn cells. (c) Cortical neurons are distended, but empty of the highly soluble stored fucose compounds. (d) In some patients, the white matter is extremely rich in Rosenthal fibers, which as in Alexander’s disease cluster around blood vessels. (e) The stored material is so soluble that only empty vacuoles can be demonstrated ultrastructurally. (Courtesy of Professor B Lake.)
 FUCOSIDOSIS
FUCOSIDOSIS

FABRY’s DISEASE
Similar storage is present in renal glomerular and tubular epithelia, which are vacuolated (Fig. 23.9a). Urinary deposit derived from desquamated distal tubules contains intensely PAS-positive, mulberry-like cells (Fig. 23.9b,c), which can be analysed biochemically. Glomerular sclerosis supervenes in the later stages of the disease. Cardiac muscle, cells of the mononuclear/phagocyte series (Fig. 23.9d), and peripheral nervous system ganglion cells also exhibit storage.
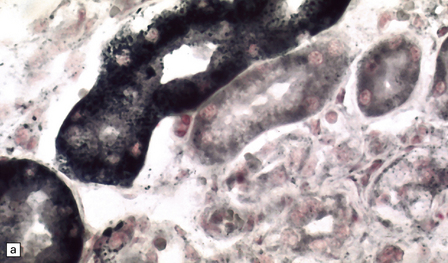
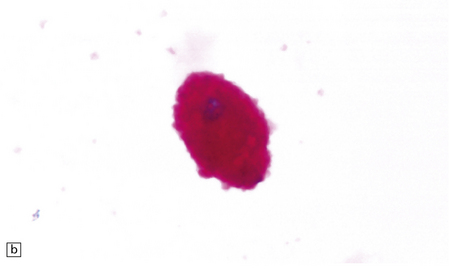
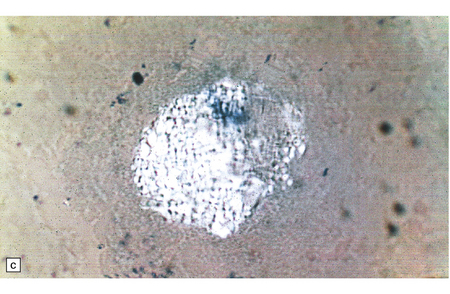
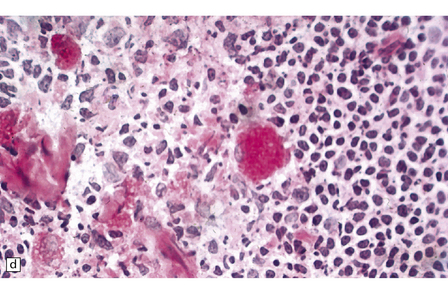
23.9 Fabry’s disease.
(a) Sudan black deposits in renal tubular epithelium; urinary deposit derived from desquamated tubules is a PAS positive mulberry-like cell (b), which is birefringent (c). (d) PAS-positive storage in the spleen.
 ETIOLOGY OF FABRY’S DISEASE
ETIOLOGY OF FABRY’S DISEASE

 FABRY’S DISEASE
FABRY’S DISEASE Presenting features, usually in childhood, are clusters of punctate red–black telangiectases in the bathing trunk area and on mucous membranes (angiokeratoma corporis diffusum), and proteinuria.
Presenting features, usually in childhood, are clusters of punctate red–black telangiectases in the bathing trunk area and on mucous membranes (angiokeratoma corporis diffusum), and proteinuria.
 Renal failure eventually follows.
Renal failure eventually follows.

 Isolated hypertrophic cardiomyopathy is a rare but important presentation.
Isolated hypertrophic cardiomyopathy is a rare but important presentation.
TYPE II GLYCOGENOSIS (POMPE’S DISEASE)
MICROSCOPIC APPEARANCES
The hallmark of the disease is a vacuolar degeneration of skeletal myofibers: extreme in infants, moderate in juveniles, and modest or mild in adults. The vacuoles contain excessive soluble β-particle glycogen and strong acid phosphatase activity. Excess glycogen is also present in capillary endothelium and smooth muscle, and in the infantile form in cardiac muscle fibers in association with cardiomegaly. Neuronal glycogen storage is a feature of the infantile and juvenile forms, but not the adult-onset form. It is found in anterior horn cells (Fig. 23.10) and motor cranial nerve nuclei, basal ganglia, and gastrointestinal tract plexuses. Glycogen is also abundant in astrocytes in the cerebral cortex.
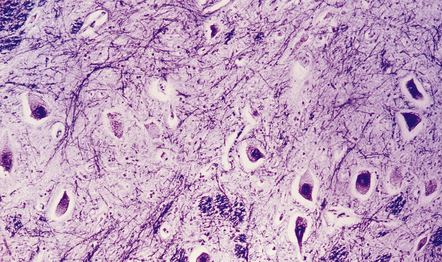
In blood films, lymphocytes have small discrete cytoplasmic glycogen-containing vacuoles.
 TYPE II GLYCOGENOSIS
TYPE II GLYCOGENOSIS



FARBER’s DISEASE
 FARBER’S DISEASE
FARBER’S DISEASE

MICROSCOPIC APPEARANCES
Neuronal loss and gliosis are associated with marked neuronal storage in the basal ganglia, brain stem, and anterior horns, and to a lesser degree in the cerebral cortex and gastrointestinal tract plexuses (Fig. 23.11a–c). The stored material is strongly PAS-positive, weakly sudanophilic, and birefringent in polarized light.
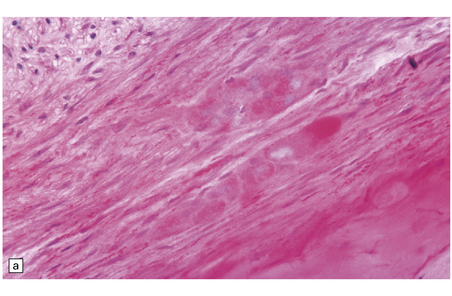
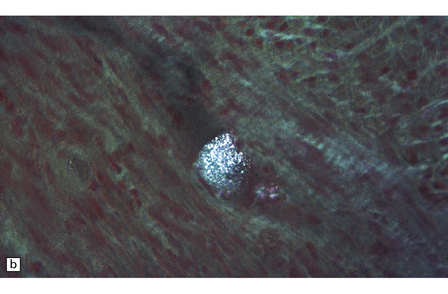
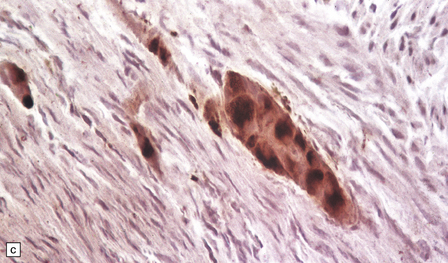
23.11 Farber’s disease:
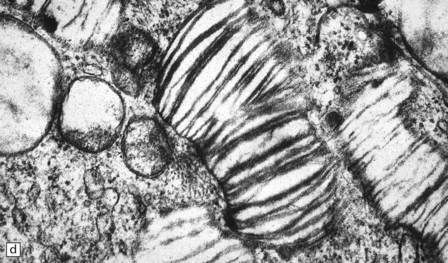
Neuronal storage within ganglion cells of the appendix is PAS positive (a); birefringent in polarized light (b), and shows increased lysosomal activity (c) by acid phosphatase histochemistry; while electron-microscopy (d) demonstrates Zebra bodies. (e) Farber bodies, pathognomic curvilinear tubular or banana-shaped bodies are here demonstrated within a Schwann cell in a sural nerve biopsy. (Courtesy of Drs Jean Jacobs and Michael Groves, Institute of Neurology, London.)
Neurons and endothelial cells contain zebra bodies (Fig. 23.11d). Hepatocytes contain membrane-bound collections of lipid lamellae. Subcutaneous foam cells contain small curvilinear tubular structures (Farber bodies or banana bodies) (Fig. 23.11e).
KRABBE’S LEUKODYSTROPHY
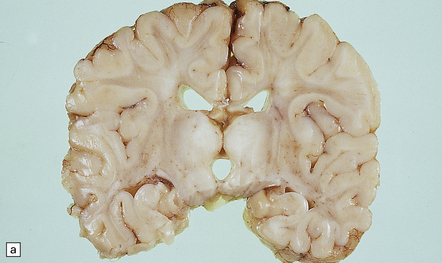
The changes are similar in both the typical infantile form and in the rarer examples with late or adult onset. Externally, there is moderate to severe atrophy, widened sulci and marked weight reduction, while on palpation the firm white matter surrounded by normal cortex gives the impression of an ‘iron fist in a velvet glove’. On cut section the white matter is extensively discolored and grayish, though subcortical white fibers are spared, and the ventricles are dilated (Fig. 23.12a). Cerebellar white matter is similarly affected.
23.12 Krabbe’s globoid cell leukodystrophy.
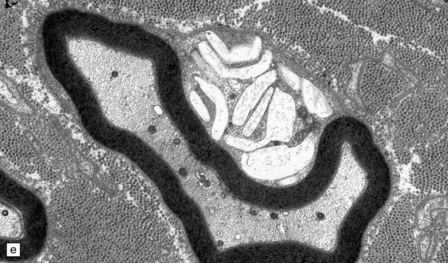
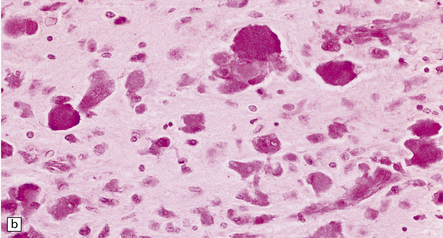
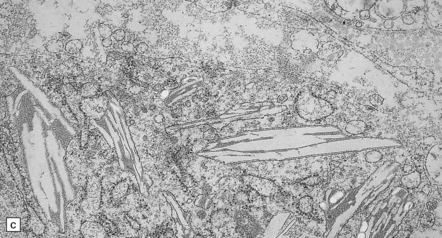
(a) Coronal section through cerebrum. The subcortical U-fibers are generally spared, but the centrum semi-ovale is gray and firm, so the intact brain has the texture of an ’iron fist in a velvet glove’. (b) The demyelinated matter contains both mononuclear and multinucleated macrophages (globoid cells) which are strongly PAS-positive. (c) Ultrastructure of a globoid cell/macrophage. There are numerous curved or straight tubular inclusions with a crystalloid appearance.
MICROSCOPIC APPEARANCES
Principal changes are extensive myelin and oligodendrocyte loss, astrocytic gliosis, and the pathognomonic presence of globoid macrophages which early in the course of the disease are mononuclear but later form perivascular clusters of multinucleated cells with as many as 20 peripheral nuclei. Eventually myelin, axons, and even globoid cells may disappear, leaving only an intense gliosis. Globoid cells are PAS-positive, faintly sudanophilic, but not metachromatic (Fig. 23.12b). They show strong acid phosphatase activity, and ultrastructurally contain straight or curved tubular profiles which are irregularly crystalloid on cross-section (Fig. 23.12c). Demyelination affects most of the cerebral white matter, usually the optic tracts, and the brain stem and cord variably with preservation of cranial and spinal roots. There is no neuronal storage but there may be severe neuronal loss from the dentate and olivary nuclei, and more moderate involvement of other brain stem nuclei and Purkinje cells. Peripheral nerves also show demyelination, fibrosis, and the presence of macrophages containing tubular inclusions.
 KRABBE’S DISEASE AND METACHROMATIC LEUKODYSTROPHY (MLD)
KRABBE’S DISEASE AND METACHROMATIC LEUKODYSTROPHY (MLD)




METACHROMATIC LEUKODYSTROPHY (MLD)
In the late infantile form the brain may appear externally normal, slightly enlarged, or atrophic (changes are slight in juvenile and adult cases). On section the white matter is a dull chalky white, firm to touch, and sharply demarcated from the cortex (Fig. 23.13a). Cerebellar atrophy may also occur.
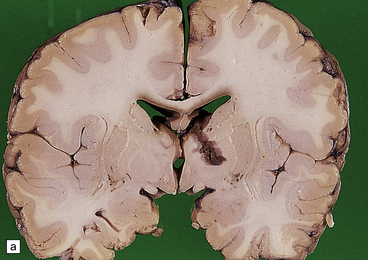
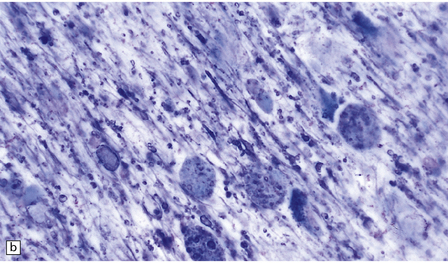
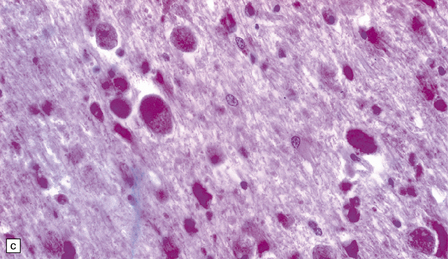
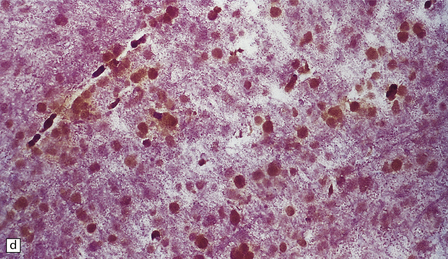
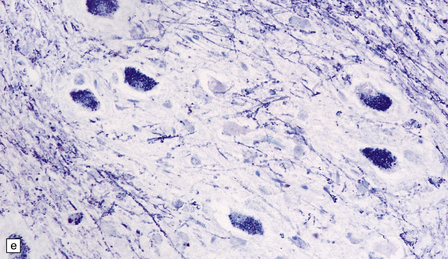
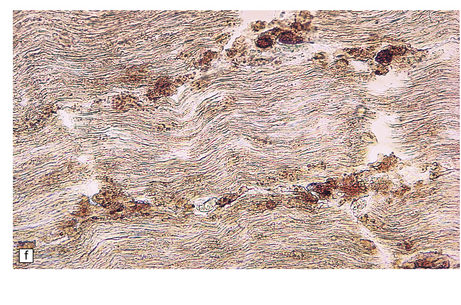
23.13 Metachromatic leukodystrophy.
(a) Coronal section through cerebrum. The hemispheric white matter has a dull chalky white appearance and is firm to touch. (b) In the white matter there is marked destruction and loss of myelin accompanying accumulation within uninucleated macrophages. (c) As for (b) (PAS). (d) The stored material in cerebral white matter is sulfatide, which is metachromatic in frozen sections stained with thionin or acidified cresyl violet. (e) Neuronal storage of sulfatide in the cerebellar dentate nucleus. (f) Metachromatic sulfatide in peripheral nerve. (g) Sulfatide in renal tubular epithelium.
MICROSCOPIC APPEARANCES
Demyelination and considerable axon loss are extensive in cerebral and cerebellar white matter and corticospinal tracts. There is oligodendrocyte loss, intense astrogliosis, and accumulation of PAS and Luxol fast blue positive macrophages which, in frozen sections, demonstrate brown metachromasia with acidified cresyl violet, toluidine blue or thionine (Fig. 23.13c,d). This sulfatide deposition also occurs in neurons in the basal ganglia, dentate nucleus, some brain stem nuclei and dorsal root ganglia, as well as within macrophages and Schwann cells, in peripheral nerves, within renal tubular epithelium, macrophages in lymph nodes and spleen, liver, Kupffer cells, biliary duct epithelium, adrenal medulla, islets of Langerhans, and many other organs. Ultrastructurally, there are three types of inclusion: prismatic, tuffstone, and laminated.
PEROXISOMAL DISORDERS
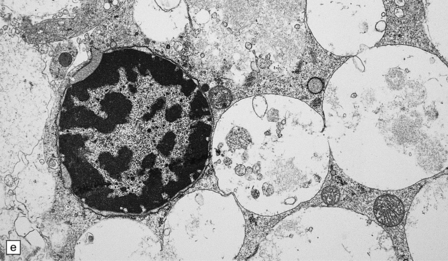
Peroxisomes are tiny organelles, 0.05–0.2 μm in diameter in the cytoplasm of all nucleated cells (in the liver and kidney they are ~10 times larger). They are identified by their structure, positive histochemical reaction for catalase (Fig. 23.14a), or immunohistochemistry. New peroxisomes form by budding from existing peroxisomes. Peroxisomal proteins, both enzymes and membrane proteins, are encoded by nuclear genes and imported via special receptors into the organelle. The many functions of peroxisomes are listed in Table 23.5.
Table 23.5
Processes integral to normal metabolism of the nervous system, adrenals, and liver
Plasmalogen biosynthesis (important components in cell membranes and myelin)
Cholesterol biosynthesis
Bile acid biosynthesis
β-oxidation of fatty acids (including very long chain fatty acids)
Other functions
Dolichol synthesis (through action of mevalonate kinase)
Glyoxalate transamination
Peroxide-based respiration/peroxidatic oxidation
Pipecolic acid oxidation
Glutaric acid oxidation
Phytanic acid α-oxidation
Alcohol dehydrogenase (medium chain)
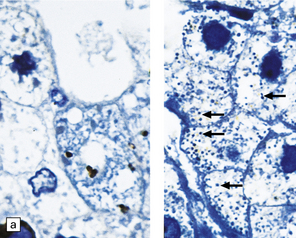
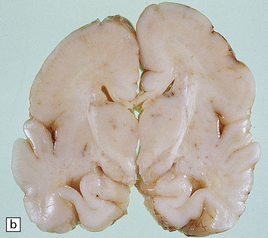
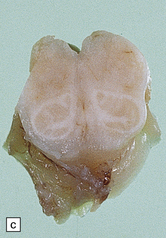
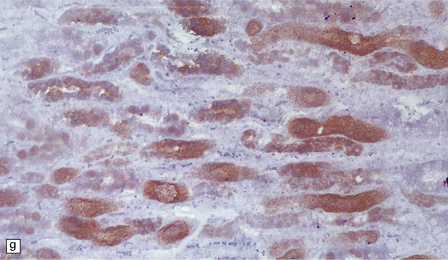
23.14 Zellweger cerebrohepatorenal syndrome.
(a) Peroxisomes are readily demonstrated in liver by catalase histochemistry. They appear as darkly stained bodies (arrows) in a normal control (right panel), but are absent in Zellweger syndrome (left panel). (Courtesy of Professor B Lake.) (b) Macrogyric convolutions and thickened cortical ribbon in a neonate with Zellweger syndrome. (c) Horizontal slice through the medulla showing the dysplastic inferior olivary nuclei, which are coarse, thickened dorsally, and can be partially fragmented (see Chapter 3, Fig 3.100b).
A classification of peroxisomal disorders is given in Table 23.6. Although this is a long list and probably still far from complete, morphologic data remain scanty for many of these conditions. Disorders with significant neurologic disturbance and well-documented neuropathology are fewer: in particular, the adrenoleukodystrophies and Zellweger syndrome and its variants.
Table 23.6
Classification of peroxisomal disorders
1. Peroxisomes absent or severely reduced (defective peroxisomal membrane synthesis or import of matrix protein results in defective peroxisomal assembly and generalized enzyme defects)
Zellweger cerebrohepatorenal syndrome
Neonatal adrenoleukodystrophy
Infantile Refsum’s disease
Zellweger-like syndrome
Rhizomelic chondroplasia punctata (classic form)
Pseudo-infantile Refsum’s disease
2. Peroxisomes present, but may be structurally abnormal (single peroxisomal enzyme defect)
X-linked adrenoleukodystrophy
Pseudo-neonatal adrenoleukodystrophy
Rhizomelic chondroplasia punctata
Bifunctional enzyme deficiency
Pseudo-Zellweger syndrome
Trihydroxycholestanoic acidemia
Pipecolic acidemia (isolated)
Refsum’s disease
Atypical Refsum’s disease
Glutaric aciduria type III
Primary hypoxaluria type I
Acatalasemia
Mevalonic aciduria
Sjögren–Larsson syndrome
ZELLWEGER CEREBROHEPATORENAL SYNDROME
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The weight of the brain is often increased and there are widespread gyral abnormalities with both widened pachygyric convolutions and polymicrogyria (Fig. 23.14b).
 LYSOSOMAL ENZYME DEFECTS CAUSING LEUKODYSTROPHY
LYSOSOMAL ENZYME DEFECTS CAUSING LEUKODYSTROPHY





Extensively deranged cortical migration manifests as pachygyria, polymicrogyria, and neuronal heterotopias (see Chapter 3). Focal gray heterotopias are present in cerebellar white matter, the dentate nucleus is dysplastic, and the inferior olivary nuclei show a characteristic malformation (Fig. 23.14c) (see Chapter 3). The white matter shows decreased myelin and evidence of myelin breakdown with lipid deposition (cholesterol esterified to very long chain fatty acids) in astrocytes and macrophages in some cases. A neuronal lipidosis and neuroaxonal dystrophy in the spinal nucleus of Clarke and in the lateral cuneate nucleus have also been reported.
 ZELLWEGER SYNDROME
ZELLWEGER SYNDROME Onset at birth with severe hypotonia and characteristic dysmorphic features (i.e. high forehead, large fontanelles, epicanthic folds, shallow supraorbital ridges, micrognathia, abnormal ears, and high arched palate). There is often stippled calcification of the patella.
Onset at birth with severe hypotonia and characteristic dysmorphic features (i.e. high forehead, large fontanelles, epicanthic folds, shallow supraorbital ridges, micrognathia, abnormal ears, and high arched palate). There is often stippled calcification of the patella.



 ETIOLOGY OF ZELLWEGER SYNDROME (ZWS) AND PSEUDO-ZWS
ETIOLOGY OF ZELLWEGER SYNDROME (ZWS) AND PSEUDO-ZWS




ADRENOLEUKODYSTROPHY
Externally relatively normal, the sliced brain shows an extensive symmetric white matter abnormality which in the fixed state is firm and gray. There is a caudal to rostral gradient of severity with the frontal areas often less severely involved (Fig. 23.15a,b).
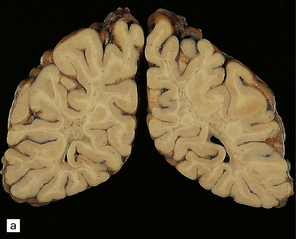
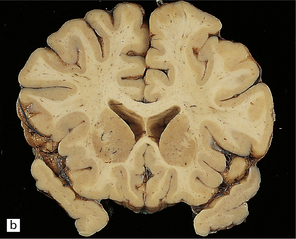
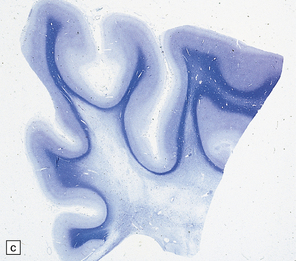
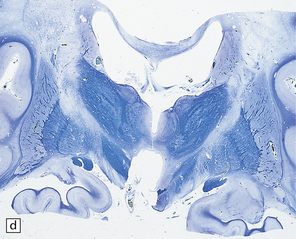
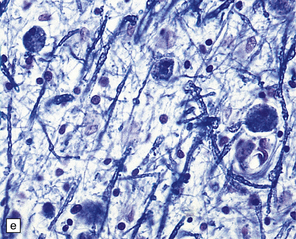
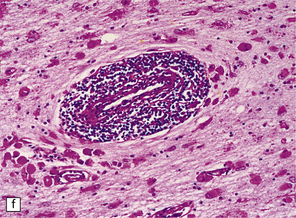
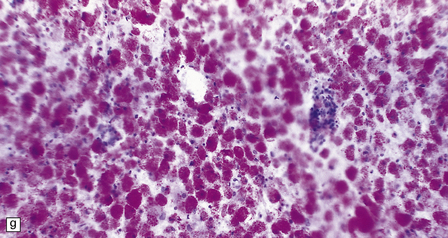
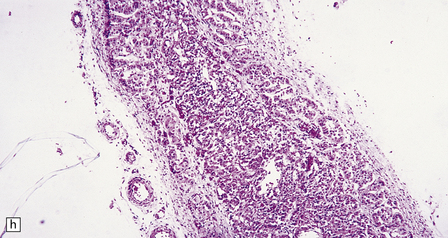

23.15 Adrenoleukodystrophy.
Macroscopically, the affected white matter is firm and gray, and is usually more severely affected posteriorly (a) than anteriorly (b). (c) Section through the frontal lobe showing extensive myelin loss from the centrum semiovale and corpus callosum, but with sparing of the U-fibers. (d) Section through the basal ganglia at the level of the amygdala showing extensive myelin loss from the centra semiovalia, corpus callosum, and internal capsules, but with sparing of the U-fibers. (e) Myelin fragmentation and a sparse infiltrate of macrophages are found at the advancing edge of the demyelinating process. (f) In the center of the demyelinated areas there are prominent perivascular collections of lymphocytes and groups of PAS-positive macrophages. (g) These macrophages may form large aggregates. (h) The adrenal gland is usually severely atrophic and may be difficult to find at necropsy. (i) The remaining adrenocortical cells are enlarged, eosinophilic, and show prominent striations. (j) The lipid deposits contain very long chain fatty acids and appear ultrastructurally as needle-like trilaminar bodies up to 7 nm long and 10 nm wide.
 ADRENOLEUKODYSTROPHY (ALD)
ADRENOLEUKODYSTROPHY (ALD)


 PEROXISOMAL DEFECTS CAUSING ADRENOLEUKODYSTROPHY (ALD)/ADRENOMYELONEUROPATHY
PEROXISOMAL DEFECTS CAUSING ADRENOLEUKODYSTROPHY (ALD)/ADRENOMYELONEUROPATHY




MICROSCOPIC APPEARANCES
Demyelination is severe in the central cerebral white matter, optic nerves, internal capsule, and commissures, while U-fibers and stria of Gennari are relatively spared (Fig. 23.15c,d). There may be some involvement of the descending brain stem tracts (much more evident in the adrenomyeloneuropathy variant). Histologically, three zones of abnormality may be discerned. In the most recent there is demyelination with preservation of axons, and scattered sudanophilic PAS-positive macrophages (Fig. 23.15e). Older lesions also demonstrate a pronounced perivascular mononuclear infiltrate and numerous macrophages (Fig. 23.15f,g). The most ancient lesions are without macrophage or inflammatory activity, depleted of axons and oligodendroglia, and heavily gliotic.
Adrenal atrophy may cause severe difficulty in identifying the adrenals at autopsy: histologically, ballooned cells with striated cytoplasm replace the cortex (Fig. 23.15 h,i). With electron microscopy, adrenal, testis, peripheral nerves, and brain show cytoplasmic inclusions comprising needle-like trilaminar bodies of very long chain fatty acid esters (Fig. 23.15j).
REFERENCES
Fournier, B., Smeitink, J.A., Dorland, L., et al. Peroxisomal disorders. A review. J Inherit Metab Dis.. 1994;17:470–486.
Goebel, H.H. The neuronal ceroid-lipofuscinoses. J Child Neurol.. 1995;10:424–437.
Lake, B. Lysosomal and peroxisomal disorders. In Graham D., Lantos P., eds.: Greenfield’s neuropathology, 6th ed., London: Arnold, 1997.
Love, S., Bridges, L.R., Case, C.P. Neurofibrillary tangles in Niemann–Pick disease type C. Brain.. 1995;118:119–129.
, Mendelian Inheritance in Man, OMIM (TM). Baltimore, MD: McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University; 2000. Bethesda, MD: National Center for Biotechnology Information, National Library of Medicine; Online: http://www.ncbi.nlm.nih.gov/omim/.
Powers, J.M. Adreno-leukodystrophy (adreno-testiculo-leukomyelo-neuropathic-complex). Clin Neuropathol.. 1985;4:181–199.
Scriver, C.R., Beaudet, A.L., Valle, D., et al. The metabolic and molecular basis of inherited disease, 7th ed. New York: McGraw Hill; 1995.
Sharp, J.D., Wheeler, R.B., Lake, B.D., et al. Loci for classical and variant late infantile neuronal ceroid lipofuscinosis map to chromosomes 11p15 and 15q21-23. Hum Mol Genet.. 1997;6:591–595.
Walkley, S.U. Pathogenic cascades in lysosomal disease – Why so complex? J Inherit Metab Dis.. 2009;32:181–189.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]23
Lysosomal and peroxisomal disorders
This chapter deals first with the lysosomal disorders that principally affect gray matter and then with those involving white matter (leukodystrophies). Lastly, the chapter covers the peroxisomal disorders, which include adrenoleukodystrophy. Other leukodystrophies are considered in Chapter 5. Chapter 5 also covers comparative aspects of all of the leukodystrophies, and an approach to their differential diagnosis.
LYSOSOMAL DISORDERS
GM2 GANGLIOSIDOSIS
MACROSCOPIC APPEARANCES
MICROSCOPIC APPEARANCES
In the older patients neuronal storage of excessive lipofuscin is confined to the basal ganglia, brain stem, cerebellum, and spinal cord. In infantile GM2 gangliosidosis, ballooned neurons are found throughout the CNS and the peripheral nervous system. The foamy nerve cells stain strongly with Luxol fast blue and Sudan black, and in frozen sections the soluble ganglioside is periodic acid–Schiff (PAS)-positive (Fig. 23.1). Microglia are also PAS-positive, retaining stored material even in paraffin sections. Ultrastructural studies show membranous cytoplasmic bodies (MCBs) within neuronal somata.
23.1 GM2 gangliosidosis.
(a) Ballooned cortical neurons with foamy cytoplasm and marginated nuclei are negative with PAS in routine paraffin sections. (b) These neurons are, however, strongly stained with Luxol fast blue. (c) The stored ganglioside can be demonstrated in cryostat sections stained with PAS, in this example protected by celloidinization prior to staining.
GM1 GANGLIOSIDOSIS
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Mild gyral atrophy is present in all three subtypes (Fig. 23.2a,b). Ballooned neurons with staining characteristics virtually identical to those seen in GM2 gangliosidosis are widespread in the cerebrum (Fig. 23.2c,d), brain stem, and spinal cord, and in autonomic ganglia (in subtypes 1 and 2). Ballooned neurons are confined to the striatum and pallidum in the adult form. Ultrastructural studies show membranous cytoplasmic bodies.
23.2 GM1 gangliosidosis.
(a) Coronal section of cerebral hemisphere from a patient with the type 2 (late-infantile and juvenile) form of GM1 gangliosidosis, who died aged 10 years. There is mild cortical atrophy and ventricular dilatation. (b) Cortical atrophy and poverty of myelination are more evident in a stained section from the occipital lobe. (c) In the cerebral cortex there is extensive neuronal and glial storage. The granular storage bodies fill the neuronal cytoplasm and distend the proximal dendrites. Note also the myelin pallor. (d) Heavy neuronal storage is also present in the pontine nuclei.
BATTEN’S DISEASE, NEURONAL CEROID LIPOFUSCINOSIS (NCL, CLN)
This group of disorders has a confusing set of eponyms (Table 23.1). Classification is further complicated by newer terminology related to the molecular genetics of the disorders. The classification used here combines clinical presentation, age of onset, pathology, and electrophysiology. The diagnosis is most readily obtainable for all forms except Kufs’ disease by cutting cryostat sections of a suction rectal biopsy to examine neurons and other cell types (i.e. smooth muscle, histiocytes, vascular endothelium) for the characteristic accumulations of autofluorescent ceroid lipofuscin, while ultrastructural examination for the various specific types of inclusions is most easily accomplished using buffy coat preparations of lymphocytes. In JNCL (CLN3) numerous vacuolated lymphocytes are demonstrated in the trails of a routine peripheral blood film, and in the right clinical setting this is virtually confirmatory. Skin biopsy is also routinely used: examination of eccrine but not apocrine glands is informative (if the biopsy is from the axilla note that many of the sweat glands will be of apocrine type).
MACROSCOPIC APPEARANCES
Cerebral atrophy is always present, but is at its most severe in the infantile form (Fig. 23.3a–c), manifesting as a walnut brain with shriveled cortex and rubbery white matter encased in a markedly thickened skull. Atrophy may also be considerable in infantile and juvenile Batten’s disease, and increases with the length of survival. In adult cases (Kufs’ disease), atrophy is more limited, and predominantly in frontal and cerebellar regions.
23.3 Batten’s disease.
(a) Infantile Batten’s disease in a boy aged 9 years. The extremely atrophied brain (300 g weight) is covered by gelatinous leptomeninges and markedly thickened dura and surrounded by greatly thickened calvaria. (b) Viewed from below, the cerebral convolutional atrophy is marked and widespread, but the cerebellum and brain stem are relatively spared. (c) A coronal slice of frontal lobe shows a very thin cortical ribbon and tough rubbery white matter. (d) Juvenile Batten’s disease. Neuronal storage material reacts strongly with Sudan black in the cortex. (e) Juvenile Batten’s disease. Neuronal storage material reacts strongly with PAS in hippocampal pyramidal cells. (f) and (g) Kufs’ disease. In the rare adult form of NCL there is similar widespread neuronal storage material, which stains strongly with PAS. (h) Blood film of a patient with juvenile Batten’s disease showing a vacuolated lymphocyte with characteristic large uniform ‘bold’ vacuoles. A similar appearance is seen in GM1 gangliosidosis. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (i) Ultrastructural appearance in infantile Batten’s disease showing granular osmiophilic deposits in a neuron. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (j) Ultrastructural appearance in late-infantile Batten’s disease showing curvilinear bodies within a sweat gland epithelial cell. (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (k) Ultrastructural appearance of a sweat gland epithelial cell containing mixed curvilinear and fingerprint bodies in juvenile Batten’s disease (similar in early juvenile and Finnish variant late-infantile Batten’s disease). (Courtesy of Professor B Lake, Great Ormond Street Hospital, London.) (l) Fingerprint bodies in juvenile Batten’s disease.
MICROSCOPIC APPEARANCES
The stored material, which is insoluble and therefore readily detectable in paraffin as well as frozen sections, is widespread in the nervous system and in many other tissues. Its tinctorial properties vary slightly between the various subtypes of the disease (Table 23.2). In infantile Batten’s disease, storage is evident in CNS neurons, astrocytes, and macrophages, and in autonomic ganglia from an early stage, but neuronal loss is relatively subtle to begin with, becoming obvious after 2 years. By 4 years of age virtually all cortical neurons have disappeared, and there is dense astrocytic gliosis, and myelin loss. Some astrocytes contain storage material.
In late-infantile and juvenile Batten’s disease (Fig. 23.3d,e) neuronal loss is less severe and myelin loss, if present, is slight. The rarity of adult cases and the accumulation of lipofuscin during normal aging have impeded the formulation of a consensus view of the histology of Kufs’ disease, although widespread storage is the principal element (Fig. 23.3f,g).
NIEMANN–PICK DISEASE
Not sphingomyelinase deficient (Table 23.3).
Table 23.3
Classification of Niemann–Pick disease
| Group I: sphingomyelinase deficient | Group II: not sphingomyelinase deficient |
| Type A: neurovisceral (infantile, juvenile, and adult) | Types C and D (Nova Scotia): neurovisceral |
| Type B: visceral only (infantile, juvenile, and adult) | Possible pure visceral form |
This classification incorporates the earlier four alphabetically defined groups.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Niemann–Pick disease group I
While hepatosplenomegaly is striking in both types, CNS abnormalities are not found in type B. In type A, cerebral atrophy may be slight or absent. Microscopically (Fig. 23.4), there is generalized enlargement of neurons and glia, and storage extends to white matter, which is demyelinated and gliotic. Gastrointestinal tract neuronal plexuses are also affected. Sudanophilic foamy histiocytes containing cholesterol esters, but not ballooned neurons, are numerous in the globus pallidus, substantia nigra, and dentate nucleus. Niemann–Pick cells (Fig. 23.4b) are present throughout the mononuclear phagocyte system, and can fill the alveolar spaces of the lungs. The lymphocytes of patients with type A Niemann–Pick disease contain cytoplasmic vacuoles, which are small and discrete. In contrast, in patients with type B there is minimal or no lymphocytic vacuolation. Bone marrow aspirates show collections of Niemann–Pick cells in patients with type A and in younger patients with type B. In older patients with type B disease there are fewer foamy Niemann–Pick cells, and more prominent ’sea-blue histiocytes’, in which the cytoplasm is filled with small granules that stain intensely blue with the Giemsa or Wright histochemical method.
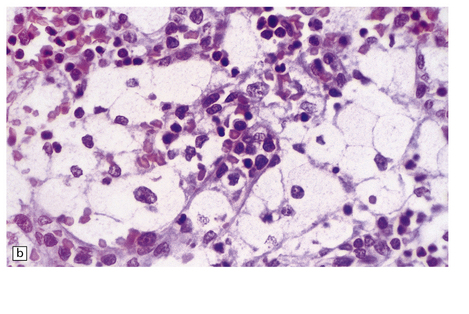
23.4 Niemann–Pick disease type A.
(a) Prominent neuronal and glial ballooning are seen in the cortex. (b) Large mulberry-like storage cells filling the red pulp of the spleen contain a single nucleus, uniform vacuoles, and occasional red cell debris.
[/not-level-membership-for-pathology-category]
