[level-membership-for-pathology-category]
CHAPTER 29 Lymphoma
Introduction
Peripheral blood (PB) and bone marrow (BM) are relatively frequently involved by lymphomas, particularly those with low-grade clinical behavior.1 BM aspiration and bone marrow trephine biopsy (BMTB) are currently performed at diagnosis as part of the staging of most patients with non-Hodgkin lymphomas (NHL) and in selected patients with Hodgkin lymphoma (HL). Criteria for patient selection for these procedures vary between treatment centers and are undergoing revision as techniques such as magnetic resonance imaging and positron emission tomography are refined to permit non-invasive assessment of disease spread. Evidence that BM involvement influences clinical outcome varies for different disease entities within the spectrum of lymphoproliferative disorders.2–9 BM involvement, and its extent, can be important factors in making clinical decisions concerning choice of treatment.
Lymphoproliferative diseases that present as leukemias have been considered elsewhere (see Chapter 28), as has plasma cell neoplasia (see Chapter 30). In this chapter, blood and BM involvement by lymphomas presenting primarily with lymph node or other solid organ involvement is discussed.
General comments on bone marrow (BM) examination in lymphoproliferative diseases
The full blood count, blood film, BM aspirate and BMTB have complementary roles in the investigation of BM involvement by lymphoma. In different lymphoproliferative diseases, each of these types of specimen may be of greater or lesser value.10–14 In lymphomas that do not readily display leukemic behavior, BMTB sections are frequently found to contain lymphoma when none is evident in the blood or BM aspirate.
Where circulating lymphoma cells are available in sufficient number in the peripheral blood (PB), these give the most consistent morphology and phenotyping by flow cytometry (FCM). BM aspiration without accompanying BMTB may be valuable if representation of disease in the PB is inadequate for diagnosis, permitting cytologic and immunocytochemical assessment of larger numbers of neoplastic cells. However, aspiration for the evaluation of BM involvement by lymphoma has a high false negative rate.13 Deposits of disease frequently occupy sites within the BM microenvironment that are suboptimally sampled by aspiration (e.g. paratrabecular zones). They may also be adherent to stromal components inducing focal fibrosis and less readily aspirated than BM hemopoietic elements. For these reasons BMTB in addition to aspiration is always recommended for the evaluation of BM involvement by lymphoma. Exceptionally, if BM aspiration alone is possible in some patients for whom BMTB cannot be performed, the aspirate films can still provide valuable information about hemopoietic reserve and iron stores. If aspiration proves technically difficult or a poor sample is obtained, BMTB should definitely be performed and an imprint or roll preparation made for cytologic assessment.
BMTB sections are used primarily for morphologic assessment, including analysis of the spatial distribution and extent of lymphomatous deposits.4,8–10,12,14–25 Spatial and cytologic assessment often provide clues to lymphoma subtype (e.g. the pattern of paratrabecular infiltration typical of follicular lymphoma) and can, as a minimum, be used to assess whether disease involvement represents indolent or agressive lymphoma. Histologic sections can also be used for immunohistochemistry (IHC), FISH and PCR; these additional investigations are particularly useful if PB or aspirated BM cells do not provide adequate representation of lymphoma. Where decalcification is employed in association with paraffin wax embedding of trephine cores, use of ethylene diamine tetra-acetic acid (EDTA) to decalcify by chelation, rather than acid exposure, offers excellent antigen and nucleic acid preservation.26–28 For immunophenotyping of lymphoid cell infiltrates in BMTB sections a similar, marginally more restricted, range of antibodies is employed to that used for FCM. Antibodies reactive with additional antigens such as CD79a, cyclin D1, IRF4/MUM1, PAX5 and Ki67 are also available which have been developed specifically for IHC use; these are referred to individually in the text that follows, where appropriate.
Differential diagnosis of reactive lymphoid aggregates vs lymphoma
Nodular aggregates of small lymphoid cells may be found in BMTB sections as a reactive phenomenon, unrelated to any neoplastic lymphoid proliferation. Criteria for distinguishing such aggregates from neoplastic lymphoid infiltrates remain imperfect and controversial.29–31 Immunostaining is helpful in only a minority of examples. Morphologic features remain the best guide, supported by application of molecular genetic techniques, such as PCR amplification and IGH/TCR rearrangement studies, in appropriately processed specimens. However, the sensitivity and specificity of the PCR methods employed may vary and the finding of a monoclonal IGH/TCR rearrangement cannot be assumed to equate with a diagnosis of lymphoma without other supportive evidence.32 In limited circumstances FISH performed using intact sections may be informative (consideration of t(11;14) and t(14;18), for example). For any techniques performed in addition to histological assessment of the original sections, there is a significant likelihood that small or few lymphoid aggregates will not be represented in the material available for testing.
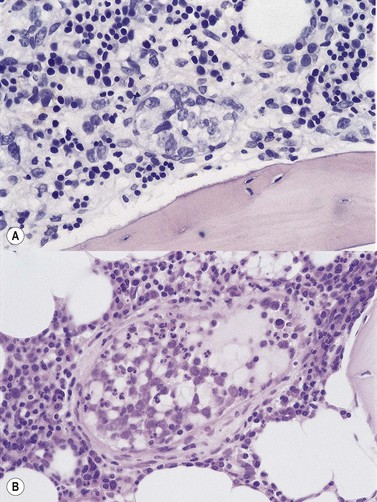
To be accepted as reactive, lymphoid nodules should be few in number, centrally placed within intertrabecular spaces, small and round in profile with well-demarcated margins. A small capillary may be present, running from the periphery into the center of the nodule (Fig. 29.1) and there may be reactive changes such as aggregation of eosinophils in the adjacent hemopoietic tissue. An underlying meshwork of reticulin or CD23+ follicular dendritic cells may be present or absent, probably more dependent on the size of a particular nodule than on its reactive or neoplastic nature. The subjective nature of these criteria will be obvious to the reader but, to date, an objective gold standard for assessment of BM lymphoid nodules remains elusive.
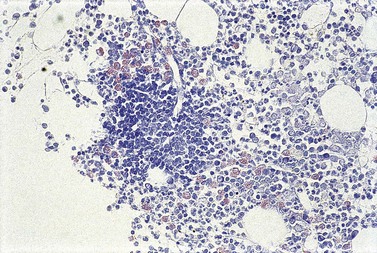
The cytologic composition of lymphoid infiltrates is also critical to their interpretation. Most non-neoplastic aggregates consist of small lymphocytes with only occasional large blast cells; they show little evidence of plasma cell differentiation. Reactive germinal center formation is distinctly uncommon but, when it occurs, the composition of the lymphoid follicle recapitulates that found in lymph nodes and other organized lymphoid tissues. Formation of reactive germinal centers within BM lymphoid aggregates is said to be increased in patients with rheumatoid arthritis and other systemic chronic inflammatory disorders.32 Reactive lymphocytes, predominantly T-cells, also form a significant component of many granulomas in the BM, and the compact infiltrates of systemic mastocytosis; these lesions can be confirmed by IHC, if suspected, and should not be mistaken either for lymphoma or incidental reactive lymphoid nodules.
The laboratory investigation of blood and BM specimens suspected or known to have involvement by lymphoma
Blood
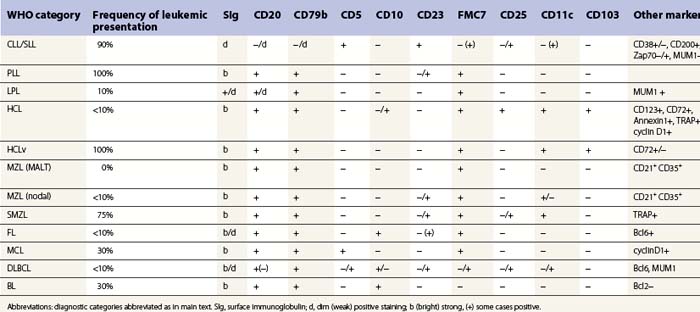
A full blood count and differential white cell count should always be performed. The blood film should be stained using May–Grünwald–Giemsa (MGG) or Wright’s method to assess cytologic features. When circulating abnormal cells are known or suspected to be present immunophenotyping by FCM should be performed; a useful basic antibody panel is shown in Table 29.1. PB cells should also be analyzed by classical cytogenetic and/or molecular genetic methods when the differential diagnosis includes lymphomas known to be associated consistently with abnormal genetic features.
Bone marrow aspirate
A minimum of three films should be prepared and air-dried, using MGG stain for cytologic assessment and differential cell count.33 A Perls stain is also desirable as a routine, for assessment of iron stores. Aspirated BM cells should also be sent in suspension for FCM immunophenotyping and genetic analysis.
Bone marrow trephine biopsy (BMTB)
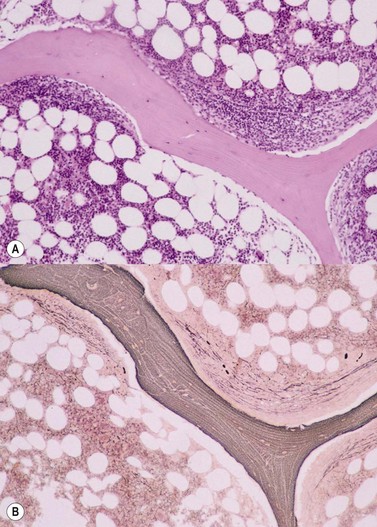
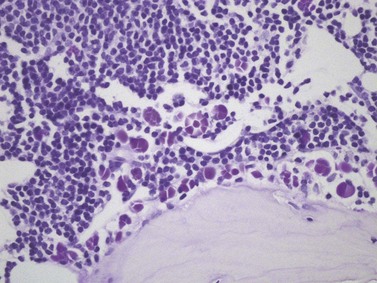
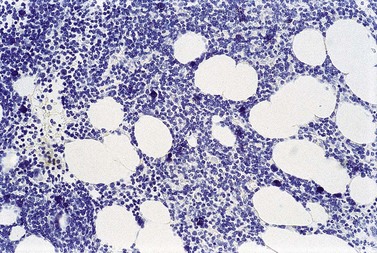
Cores of tissue should be collected that will contain at least 1 cm of interpretable, uncrushed BM after histologic processing. In practice, because of likely inclusion of cortex at the outer end and crushed tissue at the inner end, plus shrinkage that occurs during processing, unfixed cores at the time of collection should be at least 1.5 cm long10,34–36 If aspiration has been unsuccessful, touch preparations should be made using the trephine biopsy core by rolling it gently between two slides and then staining both air-dried slides with MGG. A detailed discussion of technical matters is beyond the scope of this chapter and only brief comments are made here. Further guidance can be found in references.26,37 Plastic-embedded specimens (usually in methyl or glycol methacrylate resin) require modifications to be made to standard tinctorial and IHC methods that may be difficult to incorporate into automated staining schedules but a wide range of immunostains can none the less be performed in laboratories specialized to handle these specimens. After processing the core for histology, thin sections (1–2 µm for plastic-embedded specimens; 3–4 µm for decalcified, paraffin wax-embedded ones) should be cut and stained with H&E from a minimum of three levels through the core. Levels are usually cut 25–50 µm apart, depending on local practice and the diameter of the specimen. On sections from the middle or deepest level, reticulin, PAS and Giemsa stains should be performed routinely. H&E provides a basic and familiar stain with which to assess cell morphology (Fig. 29.2A). Lymphoid infiltrates are often associated with increased reticulin deposition and a disturbance of the normal pattern of reticulin fiber distribution (Fig. 29.2B). PAS stain highlights carbohydrate-rich molecules and consequently stains immunoglobulin if it is of the IgM class and sufficiently abundant. IgM is richly glycosylated; consequently, intracellular and, occasionally, extracellular accumulation of this Ig can be detected in some cases of lymphoplasmacytic lymphoma (Fig. 29.3). The key importance of high-quality Giemsa staining in trephine biopsy sections cannot be over-emphasized (see Chapter 3). Any collection of lymphoid cells will stand out as being turquoise/blue in color, against a generally mauve/pink background (Fig. 29.1). IHC can be useful in confirming the precise diagnosis of lymphoma and in assessing its grade but is of limited value in differentiating reactive lymphoid aggregates in the BM from small deposits of low-grade lymphoma. For the latter task, molecular genetic analysis is gaining importance.30,32
The World Health Organization (WHO) classification of lymphomas
At the end of 1999, an outline version of a new lymphoma classification was published as a result of the WHO lymphoma classification project.38 This embodied the principles of the revised European–American lymphoma (REAL) classification,39 which was published in 1994 and has since become widely accepted in lymphoma diagnostic practice. A revision of the WHO classification was published in 2008 (Box 29.1).40
Box 29.1
Summary of the revised World Health Organization classification of lymphomas (2008)40
B-cell neoplasms
T- and NK-cell neoplasms
Immunodeficiency-associated lymphoproliferative disorders
(Italic text denotes entities regarded as provisional in the WHO 2008 Classification)
A novel and fundamental principle of the REAL classification was the definition of lymphomas according to their distinctive clinical, as well as pathologic, features. Much of the REAL classification remains little changed by the WHO, except for alterations in terminology, but considerable advances in the immunophenotypic and genetic contributions to defining lymphoma types have been incorporated. Some lymphomas considered as provisional entities in the REAL system became accepted as definite entities within the initial WHO classification, and this process, including the addition of new provisional entities, has continued with the 2008 revision. Box 29.1 summarizes the lymphoma categories recognized by the WHO 2008 scheme. Areas of uncertainty in lymphoma classification remain, however, and an important feature of the WHO classification is that it has flexibility to allow for further evolution in the understanding of hematological malignancies. An important advance in the WHO system over previous classifications is the recognition of subtypes of lymphoma that tend to exhibit leukemic behavior (see Chapter 28). Emerging information about the mutational status of immunoglobulin variable region genes in B-cell lymphomas41–46 and gene expresssion micro-array patterns across a wider spectrum of lymphomas,47–59 are likely to permit refinement of the classification as the biological significance of such data becomes clearer. However, while immunophenotypic, cytogenetic and molecular genetic features are central to the WHO classification, with expanded importance in the 2008 revision, little reference is made to the interpretation of patterns of BM involvement as a contribution to the definition of lymphoma entities.
Blood and BM involvement of lymphoma entities according to WHO classification
Description of entities primarily presenting as leukemias is provided in Chapter 28. Features of blood and BM involvement of lymphomas primarily presenting in lymph nodes or as extranodal infiltrates are described below.
Mature B-cell neoplasms
Lymphoplasmacytic lymphoma (LPL)
Clinical features, blood and bone marrow aspiration
Lymphoplasmacytic lymphoma is an indolent lymphoid neoplasm comprising 1–2% of all NHL.40,60–62 Studies in southern Europe have suggested an association between hepatitis C (HCV) infection and LPL,63–65 with treatment of the former being effective in controlling the lymphoma, but no similar association with HCV has been found elsewhere.
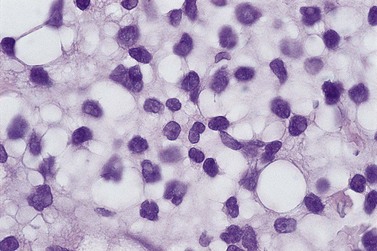
BM involvement is common at presentation and 30% of patients have splenomegaly and/or lymphadenopathy. The clinical presentation usually reflects the presence of a circulating paraprotein (IgM in almost all cases) with or without hyperviscosity symptoms. BM involvement by LPL with an IgM paraprotein underlies the clinical syndrome of Waldenström macrogobulinemia (see Chapter 30). There may be an associated peripheral neuropathy that is believed to be a paraneoplastic phenomenon.62 Pancytopenia may be present as a consequence of tumor burden such as BM failure or fibrosis. Blood involvement is rare, except in advanced disease; it is characterized by the presence in the circulation of plasmacytoid cells with an eccentric nucleus and basophilic cytoplasm. BM aspirate films show a mixture of small lymphocytes, lymphoplasmacytoid cells and mature plasma cells (Fig. 29.4). There is frequently an accompanying increase in mast cells. Typical immunophenotype findings are summarized in Table 29.1.
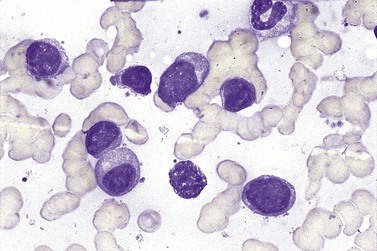
Bone marrow trephine biopsy
Bone marrow is frequently the predominant site of disease involvement in LPL. BMTB sections show infiltrates in most patients, often with more extensive involvement than suggested by aspirate films.10 The cells are predominantly small lymphocytes with varying numbers of plasma cells and cells having intermediate (plasmacytoid) features. The plasma cells may contain Dutcher bodies, which are inclusions of immunoglobulin that invaginate the nuclear membrane and appear intranuclear in histologic sections (Fig. 29.5). Less commonly, single or multiple intracytoplasmic immunoglobulin inclusions (Russell bodies) are found. Scattered lymphoid blast cells may be seen but true para-immunoblasts are absent and no proliferation centers are formed; finding the latter would indicate a diagnosis of B-cell chronic lymphocytic leukemia (B-CLL) with plasmacytic differentiation, rather than LPL. The presence of increased numbers of reactive mast cells in the marrow interstitium, sometimes located preferentially in the periphery of lymphoid infiltrates, may be helpful in supporting a diagnosis of LPL (Fig. 29.6). This phenomenon, however, is also seen in a minority of cases of CLL and is possibly related to IgM expression rather than to other properties of either disease.62,66 It has been suggested that mast cells contribute to B cell proliferation in LPL.67,68 The pattern of infiltration is usually irregular, paratrabecular or diffuse throughout the interstitium; mixtures of these patterns are common in individual cases. Well-defined nodular infiltrates are unusual and when nodules occur in LPL they are usually small and more elliptical or irregular than those seen in other small B-cell lymphomas. The paratrabecular infiltrates of LPL are not usually as extensive or regular as those found in follicular lymphoma. In some patients who have an IgM paraprotein, sinusoids contain PAS-positive proteinaceous material that represents plasma rich in IgM; there may also be interstitial and, rarely, intracytoplasmic deposition of crystalline PAS-positive IgM (Fig. 29.3). IHC shows that the small B-lymphocytes of LPL express CD19, CD20 (which may be weak or absent in cases with prominent plasma cell differentiation), CD79a and PAX5 but lack expression of CD5, CD10, BCL6 and cyclin D1. In occasional cases, a proportion of the neoplastic cells expresses CD23. Plasma cell differentiation is often evident with staining for CD79a and can be demonstrated more clearly using antibodies reactive with CD138 or IRF4/MUM1, or antibody VS38c that reacts with rough endoplasmic reticulum-associated p63 protein.69 Expression of monotypic immunoglobulin in the cytoplasm of cells showing plasmacytic differentiation is usually easily demonstrated, most commonly IgM kappa,40,70 and light chain mRNA production by such cells can also be shown by in situ hybridization (Fig. 29.7). Transformation to large cell lymphoma may occur but is not very common.
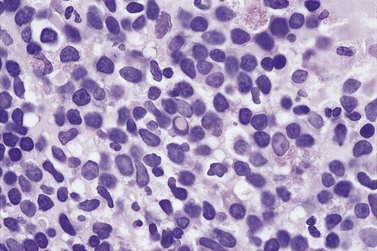
Genetic studies
Initial studies reporting a t(9;14)(p13;q32) IGH/PAX5 translocation in association with LPL70,70 have not been confirmed and no other consistent genetic associations have been identified. A variety of trisomies has been found, of unknown significance, and del(6q) may be associated with an adverse prognosis.71 Immunoglobulin heavy chain genes typically show hypermutation without evidence of ongoing acquisition of further mutations.72
Hairy cell leukemia (HCL)
Clinical features and pathology in the spleen
Hairy cell leukemia is a rare disease accounting for 2% of leukemias and predominantly affecting middle-aged men. The clinical presentation is with anemia, bleeding or infection (often with opportunistic organisms), reflecting PB cytopenias caused by hypersplenism and/or BM failure due to fibrosis associating tumor infiltrates.40 At presentation 60% of patients have splenomegaly and 40% hepatomegaly. In the spleen, HCL is recognized by the presence of a diffuse infiltrate of typical hairy cells (see below), causing effacement of normal red and white pulp architecture. As in the BM, splenic infiltration is accompanied by reticulin deposition, interstitial hemorrhage, sinusoidal vascular ectasia and peliosis-like disruption.73
Blood and bone marrow aspiration
Most patients with HCL have circulating hairy cells although numbers are typically low. These are medium-sized to large lymphoid cells which have abundant, weakly basophilic cytoplasm and hair-like projections from the cell surface.74 The nucleus is frequently indented and has a smooth chromatin pattern with indistinct nucleoli. Typical immunophenotyping results are summarized in Table 29.1. Peripheral cytopenias are common in HCL, particularly neutropenia and monocytopenia. BM aspiration is often unsuccessful due to increased marrow reticulin associated with HCL infiltration; when neoplastic cells are obtained, they are essentially identical in morphology to those found in PB. They may be accompanied by reactive mast cells and plasma cells. In touch preparations from BMTB, HCL cells may lack typical ‘hairy cell’ cytology but present as lymphatic cells with abundant cytoplasm.
Bone marrow trephine biopsy
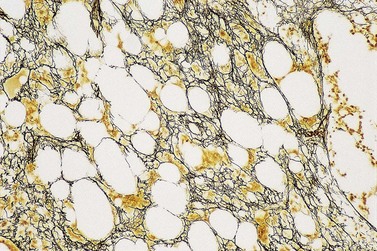
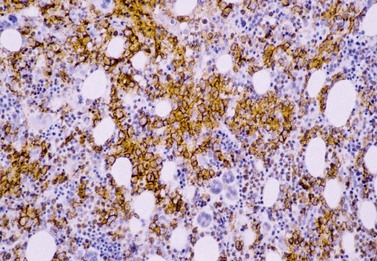
The degree of BM involvement is usually extensive at presentation, with large areas showing almost complete replacement of normal hemopoiesis by infiltrating hairy cells and partial or complete loss of fat spaces. Occasionally, the BM appears hypoplastic, with a subtle pattern of diffuse interstitial infiltration and partial preservation of hemopoiesis; granulopoiesis is often disproportionately reduced and this picture should not be confused with hypoplastic/aplastic anemia or a hypoplastic myelodysplastic syndrome (Fig. 29.8A). Subtle intrasinusoidal infiltration may also occur.25 CD20 staining is essential to determine the extent of infiltrates (Fig. 29.8B). In almost all cases of HCL, interstitial reticulin is greatly increased (Fig. 29.9), which is rarely the case in other hypoplastic states. The infiltrating cells are of medium size with round, oval or bilobed nuclei and abundant, empty-looking cytoplasm (Fig. 29.10). Occasionally, they appear spindle-shaped. The abundant cytoplasm gives an appearance of the cells being widely spaced from one another. Extravasation of red cells into the interstitium is common and sinusoids appear prominent, with gaping lumens, as a result of the background of increased reticulin fibers (Fig. 29.11). Collagen fibrosis is rare. Osteosclerosis may also occur rarely in association with HCL and may regress with treatment.75,76 Reactive mast cells are typically abundant throughout BM infiltrates and polytypic plasma cells may also be increased. Transformation of HCL to a more aggressive, large B-cell lymphoma occurs infrequently.77
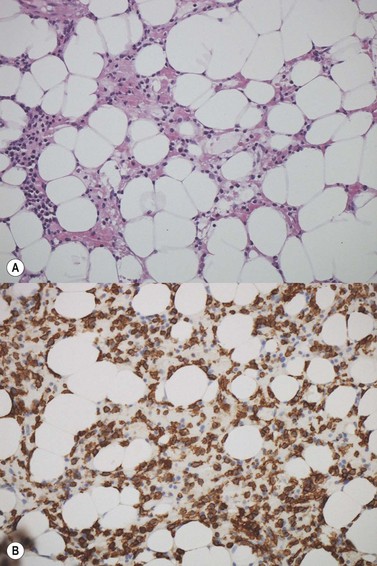
In BMTB sections, hairy cells can be shown by IHC to express CD20, CD79a and CD45RA but not CD5 or CD23. Expression of tartrate-resistant acid phosphatase (TRAP) can usually be demonstrated in extensive infiltrates78,79 although technical performance of anti-TRAP monoclonal antibodies varies and subtle HCL involvement may not be visible against background weak staining of hemopoietic cells. In many cases, hairy cells show heterogeneous, usually weaker than in mantle cell lymphoma, nuclear expression of cyclin D1 and, in a minority of cases, they are positive for CD10.80 Dot-like cytoplasmic expression of CD68 may also be seen. Hairy cells are strongly positive for CD25 and CD123. Hairy cells react well with the monoclonal antibody DBA44,79,81 which recognizes CD7282 a B-cell surface antigen strongly associated with hairy cells but not entirely specific. Annexin A1 and T-cell associated transcription factor T-bet offer further and, to date, highly specific additional markers for HCL.83,84
After treatment, HCL infiltration usually appears dramatically reduced and the increased reticulin resolves rapidly in most patients.75 Assessment of residual disease during and after therapy can be difficult in HCL, in which small interstitial clusters of scattered neoplastic cells may be all that remain; IHC usually reveals more disease than is readily apparent from standard tinctorial stains.
Genetic studies
Knowledge of genetic abnormalities associated with HCL is extremely limited and no consistent genetic associations have been found that appear causative. Immunoglobulin heavy chain genes are hypermutated but stable, without evidence of ongoing mutation. Partial understanding has been gained of genetic abnormalities underpinning the unusual properties of hairy cells;85 over-expression of the activator protein-1 (AP-1) transcription factor drives the expression of CD11c, resulting in some of the unusual stromal interactions and adhesive properties of HCL cells. Up-regulation of AP-1 is secondary to RAS activation in HCL which, in turn, may be due to reduced expression of RhoH that normally competes with Ras proteins at GTP binding sites. Reconstitution of RhoH expression in a mouse xenograft model of HCL reduced proliferation of neoplastic cells and prolonged survival.86
HCL variant (HCLv)
Clinical features and pathology in the spleen
HCLv is a rare disease with incidence of 3 cases per 1 000 000. It may be more common in Asian countries. It has been recognized as a provisional entity in the WHO 2008 classification;40 despite its name, it is clinically, immunophenotypically and genetically distinct from HCL.87,88 Patients typically present with splenomegaly; anemia is common but neutropenia and monocytopenia are rare. HCLv shows overlap between HCL and B-cell prolymphocytic leukemia (B-PLL) in their pattern of splenic involvement, with predominant diffuse red pulp involvement.40
Blood and bone marrow aspiration
There are usually abundant circulating neoplastic cells in HCLv with WBC usually in the range of 20–40 × 109/l. Although HCLv cells have cytoplasmic projections as in HCL, they are larger and have nucleoli more akin to those seen in B-PLL. BM aspiration is usually successful because HCLv is not associated with significantly increased reticulin fiber production in the marrow stroma. The immunophenotype of cells in HCLv is distinct from that found in HCL (see Table 29.1); they are generally negative for CD25, CD123 and annexin A1, with weak or absent TRAP expression. They show CD11c, CD103, FMC7 and strong sIg reactivity as in HCL.
Splenic marginal zone B-cell lymphoma (SMZL)
Clinical features and pathology in the spleen
This disease accounts for fewer than 2% of lymphoid neoplasms and occurs mainly in older individuals, affecting men and women equally.40 An association with hepatitis C has been noted in southern Europe.63
Within the spleen, there is widespread, generally uniform, involvement of the white pulp, giving a miliary appearance to the cut surfaces of splenectomy specimens.90 The germinal centers and mantles of white pulp nodules are atrophic and replaced by densely clustered small lymphoid cells surrounded by expanded marginal zones of more mixed composition. Cells in the marginal zones are predominantly slightly larger, lymphoplasmacytoid or monocytoid cells with more cytoplasm than the lymphocytes present centrally within involved nodules. Within the marginal zones there are also scattered immunoblast-like cells in varying proportions. Small satellite collections of marginal zone-type cells are frequently also present surrounding red pulp capillaries, and are often accompanied by small collections of epithelioid macrophages.73 There may be diffuse infiltration of red pulp cords and sinusoids by the small lymphoid cells. Appearances in splenunculi, when present, are identical to those in the main spleen; hilar lymph nodes show vaguely nodular replacement of follicles by small lymphocytes, usually without morphological evidence of marginal zone differentiation.
Blood and bone marrow aspiration
In PB, neoplastic cells in SMZL are slightly larger than normal lymphocytes, with a round nucleus and mature chromatin pattern. They have a relatively large volume of weakly basophilic cytoplasm and typically exhibit polar villi. BM involvement is almost always present and similar cells are usually readily apparent in aspirate. The immunophenotype (Table 29.1) shows usually positivity for CD19, CD20, surface IgM and in most cases IgD, no CD5 or CD10. CD23 and CD11c are positive in a fraction of cases. Cyclin D1 and Annexin A1 are negative.
Bone marrow trephine biopsy
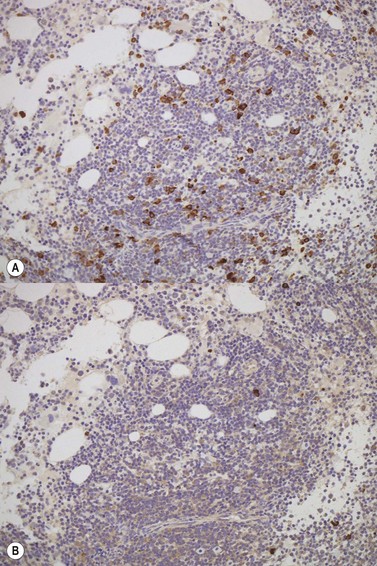
There is usually nodular, interstitial and, less regularly, paratrabecular infiltration by neoplastic cells.10,91 Intrasinusoidal infiltration is often an additional finding, demonstration of which may require IHC (Fig. 29.12). In occasional patients, a subtle and purely intrasinudoidal infiltrate may be present, requiring careful distinction from persistent polyclonal B-cell lymphocytosis (see below). A diffusely infiltrated, packed marrow is found only in rare patients.
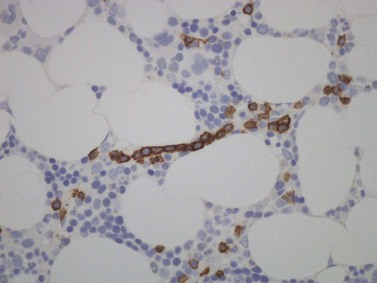
Genetic studies
These neoplasms have clonally rearranged IgH and approximately 50% show hypermutation but evidence of ongoing acquisition of mutations is rare.92 Trisomy 3q has been found in a high proportion of cases, regarded as a late or secondary event in the neoplasm development and of uncertain biological relevance. The occurrence of translocations or allelic losses involving 7q31-32 and 7q21 is believed to be more significant. The latter result in dysregulation of CDK6.93 Microarray and CGH analysis suggests a distinctive profile involving up-regulation of gene expression within the AKT1 and B-cell receptor signaling pathways.94,95 Of note, t(11;18), found in a high proportion of MALT-type marginal zone lymphomas, is absent from SMZL.
Splenic diffuse red pulp small B-cell lymphoma
Clinical features and pathology in the spleen
This lymphoma has been introduced as a provisional entity in the 2008 revision of the WHO classification40 and has been described in patients predominantly of middle age and older, with men and women equally affected. Its clinical behavior is indolent.
Blood and bone marrow aspiration
Circulating lymphocytes often have villous processes and unusually basophilic cytoplasm.96 They may be present only in low numbers. Similar cells are present in BM aspirate. Their imunophenotype resembles that of SMZL but sometimes overlaps with HCLv and increasing experience of these two provisional entities may in future reveal genuine biological overlap between them.
Genetic studies
Hypermutation patterns of IGH and the spectrum of variable region gene usage differ from those found in SMZL and resemble HCL in some cases. A translocation, t(9;14), involving PAX5 and IGH has been found in some patients and genetic alterations associated with splenic, nodal and extranodal marginal zone lymphomas are absent.40,97
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT)-type
Clinical features and pathology at presenting sites
Extranodal marginal zone lymphomas of MALT-type are indolent lymphoproliferative disorders with a 10-year survival of more than 80%.60,61 The most common primary sites of MALT-type extranodal marginal zone lymphoma are within the gastrointestinal tract. Salivary glands, skin, orbit, lung and urogenital organs are the sites of origin in smaller numbers of patients. At presentation, 30% of patients have involvement of more than one mucosal site but lymph node spread is usually absent or localized to nodes close to the mucosal site(s) of involvement. As defined in the WHO classification, these lymphomas are low grade and arise in the context of normal or induced lymphoid tissue in the organs involved (e.g. that caused by infection with Helicobacter pylori in the stomach or Chlamydia psittaci in the orbit).40 The neoplastic cells are typically centrocyte-like, with a tendency to infiltrate epithelial structures and form lympho-epithelial lesions; they show monocytoid or plasmacytic differentiation to varying extents in different individuals.
Blood and bone marrow aspiration
Bone marrow involvement has been reported in 15–40% of cases.98 Fifteen per cent of patients are anemic at presentation but circulating lymphoma cells are not seen. Bone marrow aspiration rarely reveals the presence of lymphoma cells, even when BMTB shows histologic evidence of involvement. Immunophenotype is similar to other marginal zone lymphomas, as summarized in Table 29.1.
Bone marrow trephine biopsy
When present, infiltrates of extranodal marginal zone lymphomas of MALT-type are usually nodular but paratrabecular and interstitial involvement has been described.99
Genetic studies
The translocation t(11;18)(q21;q21) has been found in some cases, particularly of gastric or pulmonary origin, associated with the formation of an API2-MALT1 fusion gene.100 This fusion gene is of uncertain functional significance at present but is absent from node-based and splenic forms of MZL. A t(14;18) translocation, distinct from that associated with FL, has been described mainly in orbital and salivary MALT lymphomas and t(3;14) in thyroid, orbital and skin lymphomas. These are associated with an abnormal API2-MALT1 fusion protein, in the case of t(11;18), or transcriptional deregulation of BCL10, FOXP1 and MALT1. Trisomy 3 has been detected in more than 60% of cases using FISH and comparative genomic hybridization;101,102 this (and other reported associated trisomies, of chromosome 8 and other chromosomes) is not specific for MALT lymphomas. Immunoglobulin genes show a hypermutated pattern indicative of a post-germinal center B cell103,104 with ongoing mutations.
Nodal marginal zone lymphoma (+/− monocytoid B-cells)
Clinical features and lymph node pathology
Nodal marginal zone lymphoma (MZL) is rare, comprising less than 2% of all NHL. Presentation is usually with advanced disease characterized by peripheral and para-aortic lymphadenopathy; 5-year survival is 50–60%.40
The histologic appearances in lymph nodes can be similar to those of nodal involvement secondary to extranodal lymphoma of MALT-type and may also require distinction from reactive monocytoid B-cell hyperplasia. Monocytoid B-cells are medium-sized cells that are quite similar to hairy cells in histologic sections, having oval or bilobed nuclei and abundant, clear cytoplasm. However, infiltrates in nodal MZL are usually heterogeneous, including centrocyte-like cells and others showing marginal zone cell or plasmacytoid differentiation. Distinction from variants of FL with down-regulation of CD10 and, less consistently, of BCL6 can be difficult in some patients in whom nodular architecture is prominent within the lymphoma.105,106
Blood and bone marrow aspiration
The blood is involved in 10% of patients but up to 30% have BM involvement. The appearance of circulating cells varies between patients; they may resemble small lymphocytes or be larger, with monocytoid appearances. The immunophenotype is indistinguishable from extranodal, MALT-type marginal zone lymphoma in many cases (see Table 29.1) although with CD43 and/or weak CD23 expression in some.
Bone marrow trephine biopsy
Few descriptions have been published recording histologic features of BM involvement in nodal MZL. Nodular and paratrabecular patterns of infiltration have been described.107 The infiltrating cells are a mixture of lymphocytes, centrocyte-like cells, monocytoid cells and plasma cells. Clinical features, knowledge of the lymph node or splenic histology and immunophenotype usually exclude the other lymphoma categories. In histologic sections, the neoplastic cells can be shown to express CD20, CD79a but not CD5, CD10, CD23 or cyclin D1.
Genetic studies
Little is known about underlying genetic abnormalities in nodal MZL; trisomies 3, 7 and 18 have been found in a high proportion of cases108 but, like most numerical chromosomal abnormalities in lymphomas, they are probably a secondary phenomenon. The t(11;18) and t(14;18) translocations associated with MALT-type extranodal MZL are not found.
Follicular lymphoma (FL)
Clinical features and lymph node pathology
Follicular lymphoma comprises approximately 35% of NHL. Disease is frequently widespread at diagnosis involving lymph nodes, spleen, and BM. Blood is involved in 10% of cases. The median survival is around 7–9 years and there is approximately 20% risk of transformation to diffuse large B-cell lymphoma within 10 years from diagnosis108 although it is anticipated that intervention with anti-CD20 treatment will improve these outcomes.
In lymph nodes, FL typically replaces normal structures with well defined, uniformly sized germinal centers containing neoplastic centrocytes and centroblasts. The WHO classification divides FL into three grades, depending on the relative proportions of centrocytes and centroblasts present; precise details of how this grading is achieved are available elsewhere.40 Grade 3 disease is further subdivided into grades 3A and 3B. Assessment of grade in FL is of prognostic value; grades 1 and 2 behave as low-grade lymphomas with a chronic course but long survival, while grade 3B disease is more aggressive and has an outcome equivalent to diffuse large B-cell lymphoma. The precise status of grade 3A disease remains controversial but it is generally regarded as more aggressive than grades 1 and 2.109
Blood and bone marrow aspiration
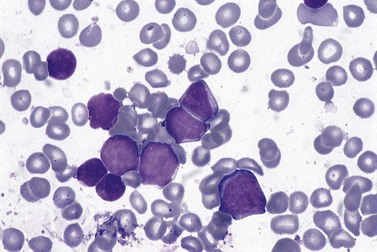
Follicular lymphoma cells, when present in the circulation, are usually centrocytes that appear smaller than those seen in BM or lymph nodes and have little or no cytoplasm. Nuclear chromatin is condensed and uniformly distributed; in typical cases a deep nuclear cleft can be seen (Fig. 29.13). Circulating centroblasts, which are larger cells with prominent nucleoli, are usually only seen in advanced disease. BM aspirate contains detectable centrocytes and/or centroblasts only infrequently, even when BMTB shows clear evidence of histological involvement. Typical immunophenotype findings in FL are summarized in Table 29.1.
Bone marrow trephine biopsy
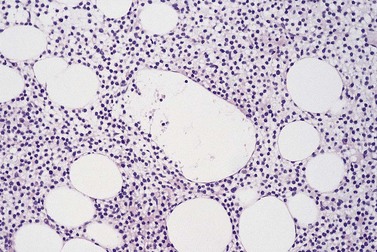
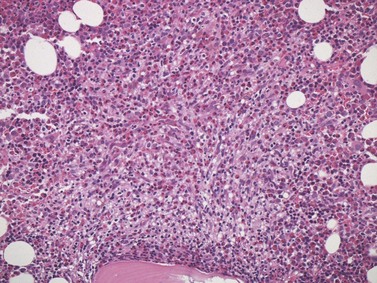
BMTB sections from patients with FL show involvement in the majority of cases but deposits of disease may be small and focal. If no lymphomatous infiltration is detected in initial sections, immunostaining and examination of further sections representing deeper parts of the tissue core are mandatory to avoid missing focal deposits.110 Small or crushed specimens, in which only limited histological assessment is possible, should be regarded as inadequate if no lymphoma is evident, and the biopsy repeated, since there is a high chance of false negative results from such specimens. At least three intact intertrabecular spaces, free from traumatic and other artifacts, are needed for adequate assessment.
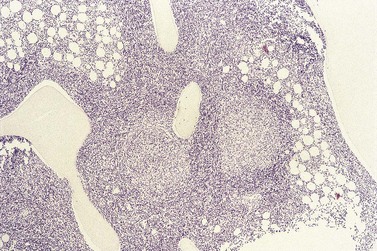
The classical pattern of BM infiltration by FL is paratrabecular. Well-developed paratrabecular infiltrates form bands or ‘crescent moon’ shapes with the longest axis abutting and lying parallel to the trabecular surface (Figs 29.14 and 29.15). Nodular infiltrates are found less often and diffuse interstitial involvement is distinctly uncommon; in either case, typical areas of paratrabecular infiltration are usually also seen. In contrast with the lymph node features, neoplastic germinal centers are rarely formed in BM deposits of FL. If they are present (Fig. 29.16), care must be taken not to mistake them for focal transformation to large cell lymphoma, since they may contain prominent centroblasts. In paratrabecular infiltrates, the cells present are predominantly non-neoplastic small T-lymphocytes, with only small numbers of centrocytes and even fewer centroblasts usually being present. Consequently, immunostaining may be misleading and greater reliance should be placed on recognition of the distinctive paratrabecular distribution of FL infiltrates. It may be very difficult to distinguish minimal non-paratrabecular infiltrates of FL from non-neoplastic lymphoid infiltrates.
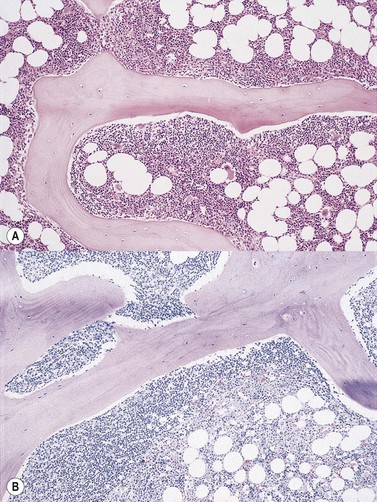
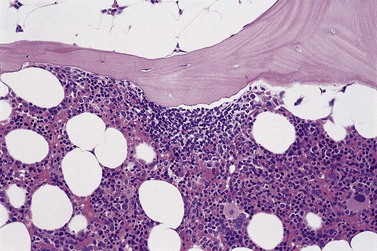
Fig. 29.15 Subtle involvement of bone marrow by FL. In contrast to the extensive linear paratrabecular infiltrates shown in Fig. 29.14, lymphoma in this example is represented by a small ‘crescent moon’ deposit of lymphoma. Although tiny, this deposit can still be seen clearly to be associated with the surface of the adjacent bony trabecula, having its longest axis along the trabecular margin. Decalcified, wax-embedded bone marrow trephine biopsy section; H&E stain, original magnification × 20.
Genetic studies
Immunoglobulin genes are rearranged and hypermutated with evidence of ongoing somatic mutation.45,103 Most cases of FL have a t(14;18)(q32;q21) translocation which dysregulates the BCL2 oncogene by placing it under the influence of the immunoglobulin heavy chain gene promoter. Variant translocations involving kappa and lambda light chain genes occur in a few patients; t(2;18)(p12;q21) and t(18;22)(q21;q11), respectively. High grade examples of FL may have additional BCL6 rearrangements, as found in diffuse large B-cell lymphoma, and very aggressive variants have additional abnormalities of TP53, p16-INK4a and/or MYC.
Mantle cell lymphoma (MCL)
Clinical features and lymph node pathology
MCL comprises approximately 6% of NHL. The median age at diagnosis is 60 years and most patients present with widespread disease involving lymph nodes, spleen and, sometimes, the gastrointestinal (GI) tract (‘lymphomatous polyposis’). Median survival is only 3–5 years.40,111
Blood and bone marrow aspiration
Sixty per cent of patients have BM involvement at diagnosis and circulating lymphoma cells are present in the blood in 30%, particularly those with advanced disease.98 However, there is a subgroup of patients who present with lymphocytosis ± splenomegaly but with no lymphadenopathy and in whom the disease pursues a more indolent course.
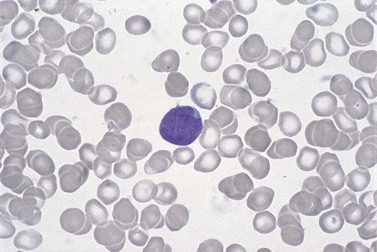
Circulating cells, when present, are pleomorphic; the predominant cell type is medium-sized with moderately abundant chromatin, an irregular nuclear outline and dispersed chromatin. In the blastoid form of MCL, nucleoli are seen within tumor cell nuclei. Similar features characterize the cells in BM aspirate films (Fig. 29.17). By FCM, they have a distinctive immunophenotype (see Table 29.1).
Bone marrow trephine biopsy
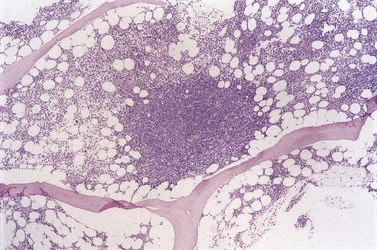
Histologic evidence of BM involvement is found in more than 70% of patients with MCL. The pattern of infiltration varies widely: nodular, interstitial, diffuse and paratrabecular patterns have been described. Nodular infiltration (Fig. 29.18), with or without an additional interstitial component is probably the most commonly seen pattern. Paratrabecular infiltration occurs fairly frequently but, in contrast with FL, is less extensive and is often overshadowed by other patterns of involvement.10,107 The cells are small and, as in lymph nodes, may be lymphocyte-like, may resemble centrocytes or, in a minority of cases, have lymphoblast-like or pleomorphic features. The blastoid variant requires distinction from Burkitt lymphoma and acute lymphoblastic leukemia.
IHC enables demonstration of cyclin D1 expression in the nuclei of neoplastic cells in a great majority of cases of MCL.112 This cell cycle regulatory protein is not detectable in normal lymphoid cells but endothelial cells are positive and may serve as internal positive control for cyclin D1 IHC. The neoplastic cells of CLL, SMZL and FL do not express this molecule. Rare cases of B-PLL have been reported as positive but these may in fact represent examples of leukemic presentation of MCL.113 The cells of MCL also express CD20, CD79a and CD5 but not CD10, CD23 or terminal deoxynucleotidyl transferase (TdT). Expression of Ki67, as a marker of proliferative activity, is of prognostic importance, with high levels (40% or more of cells positive) indicating more aggressive clinical behavior. Accompanying CD23-positive follicular dendritic cell meshworks are less commonly found in bone marrow infiltrates than in involved lymph nodes and other tissue deposits. Occasional cases that appear cyclin D1 negative by IHC can usually be shown by FISH to have t(11;14), using aspirated marrow cells or BMTB sections.
Genetic studies
The translocation t(11;14)(q13;q32) is strongly associated with MCL and places the BCL1 oncogene, which encodes cyclin D1, under regulation of the immunoglobulin heavy chain gene promoter. This leads to inappropriate expression of cyclin D1, as described above. A variant translocation, t(11;22)(q11;q13), juxtaposes BCL1 with the lambda light chain gene in a minority of patients with MCL. Cells of MCL uncommonly show evidence of immunoglobulin variable region gene hypermutation; when present, only low levels of somatic mutation are evident41 and, unlike CLL, the level in an individual patient does not appear to be of prognostic importance with current treatment regimes.
Diffuse large B-cell lymphoma not otherwise specified (DLBCL, NOS)
Clinical features and pathology at presenting sites
Diffuse large B cell lymphoma, not otherwise specified (DLBCL, NOS) comprises approximately 30% of NHL. Presentation may be with node-based or extranodal disease.40 Thirty per cent of patients have B symptoms (fever, weight loss, night sweats) at the time of diagnosis. Assessment of prognostic factors is important for therapy; such factors include age, stage of disease, performance status and serum lactate dehydrogenase (LDH) concentration. Patients with low-risk factors have an 83% 5-year survival while 5-year survival is only 32% in those with high-risk disease.40 A majority of cases of DLBCL, NOS arise de novo but a significant proportion occur following or accompanied by variants of small B-cell lymphoma (particularly B-cell CLL and FL).
Blood and bone marrow aspiration
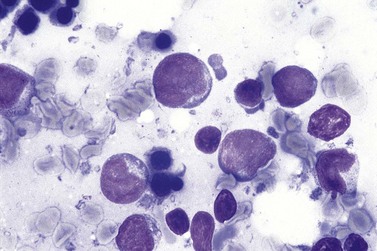
Blood involvement in DLBCL, NOS is rare and, when present, circulating lymphoma cells are usually large, with a large nucleus, prominent multiple nucleoli and abundant cytoplasm. BM aspirate involvement can be recognized by the presence of cells with these features (Fig. 29.19); immunophenotypic findings do not permit their distinction from some other NHL subtypes but remain important in supporting a putative diagnosis of DLCBL, NOS and excluding non-lymphoid blast cell proliferations, as well as contributing to assessment of prognosis.
Bone marrow trephine biopsy
Primary involvement of the BM by DLBCL, NOS is uncommon. When present, it shows no characteristic pattern of distribution within intertrabecular spaces; infiltrates usually form random, solid patches or are dispersed in the interstitium10 (Fig. 29.20). The cellular morphology of large blast cells in BM infiltrates of DLBCL, NOS generally matches that seen at the primary site. In some cases, BM infiltration may be discordant and show low-grade histologic features, even when no accompanying low-grade lymphoma has been found in sections from a lymph node or other diagnostic specimen.114–116 This is occasionally helpful in providing evidence to support origin of an apparently de novo DLBCL, NOS from FL, if typical paratrabecular infiltrates of FL are found in the BMTB sections. The presence of discordant low-grade lymphoma in the BM of patients with DLBCL, NOS does not appear to influence prognosis significantly, whereas the concordant presence of DLBCL, NOS in the BM is an adverse factor.114 It is not always appropriate to assume that the discordant elements represent a single lymphoma; some studies have shown a clonal relationship between IGH rearrangements in only approximately 50% of examples.116 Examples of discordance involving low-grade lymphoma elsewhere and DLBCL, NOS in the BM are very rare. Primary DLCB of bone without any signs of other organ involvement is rare but has been reported to have rather good prognosis.117
The differential diagnosis of DLBCL, NOS in BM includes other subtypes of DLCBL, other large B-cell lymphomas, Burkitt lymphoma, myeloma, acute lymphoblastic leukemia and acute myeloid leukemia. Attention to clinical features, cytologic detail of the infiltrating cells and the appearances of hemopoietic tissue in the background usually permits discrimination between these alternatives. IHC can be used to confirm the B-cell phenotype of neoplastic cells, which express CD79a and in most cases CD20. Less consistently, there may be expression of PAX5, CD5, CD10, BCL2, BLC6, and/or IRF4/MUM1 but staining for myeloid markers and TdT will be negative. Occasionally, the neoplastic cells will be found to express CD30; in the context of a B-cell immunophenotype. Germinal center (GC)-like and non-GC-like immunophenotypes can be deduced from combinations of CD10, BCL6 and IRF4/MUM1 expression,52 providing prognostic information similar to that available from microarray studies (see below). However, genetic and immunophenotypic results do not correlate completely and expression of combinations of different antigens has also been found to be of prognostic value.48 Proliferative activity, conveniently demonstrated by Ki67 immunostaining, is variable but should always be investigated to ensure the differential diagnosis of BL is not missed. Immunocompromised patients, in whom primary presentation of their lymphoma may be in the BM rather than at a nodal or extranodal soft tissue site, are likely to have neoplastic cells harboring latent Epstein–Barr virus (EBV) infection, demonstrable by EBV-EBER in situ hybridization.
Genetic studies
Genetic findings in DLCBL are complex; aneuploidy is common and complex aberrant clones are often found. Cells may show t(14;18)(q32;q21), with BCL2 dysregulation, as in FL. The translocation t(3;14)(q27;q32), and others involving 3q27 breakpoints, dysregulating BCL6 are also relatively common in DLBCL of all morphologic varieties.40 Immunoglobulin variable region genes show evidence of hypermutation with ongoing changes, implying continued exposure to somatic mutator mechanisms.45 Microarray studies have defined germinal center (GC)-like and activated B-cell (ABC)-like molecular expression profiles56,58,118 that are of prognostic significance for patients, even when treated with chemotherapy combinations incorporating anti-CD20. Alternative separation of patients into prognostic groups according to gene expression patterns reflecting immune/stromal response, proliferation characteristics and oxidative phosphorylation is under investigation.54,55
Other diffuse large B-cell lymphoma subtypes
As recognized in the WHO 2008 classification, these are T-cell/histiocyte-rich large B-cell lymphoma, primary DLBCL of the CNS, primary cutaneous DLBCL, leg type, and EBV-positive DLBCL of the elderly. Of these, only T-cell/histiocyte-rich large B cell lymphoma40,119 involves BM with any frequency. Neoplastic cells are rarely represented in blood or aspirated marrow. The histological picture is one of random patchy or diffuse BM replacement by infiltrates that are, as the name implies, rich in reactive small T lymphocytes and macrophages. Large neoplastic blast cells variously resemble large centroblasts, mononuclear Hodgkin and Reed–Sternberg (HRS) cells and the so-called ‘LH’ cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Extensive immunophenotyping is often needed to highlight neoplastic cells, demonstrate their B cell phenotype and confirm absence of HRS features. Excluding an alternative diagnosis of NLPHL can be difficult and is controversial; NLPHL may have a growth pattern closely resembling T-cell/histiocyte-rich large B cell lymphoma and this may develop as a progression from a more typical pattern with repeated relapse over time.120 The genetic basis of T-cell/histiocyte-rich large B cell lymphoma is poorly understood.
Other lymphomas of large B cells
An exception is intravascular large B cell lymphoma, which is a rare and highly aggressive neoplasm that usually presents as a result of CNS or subcutaneous vascular infiltration but may have a primary presentation in BM.6,22,40 Neoplastic cells are rarely recognized in blood or BM aspirate. They are large and anaplastic, resembling cells of Hodgkin or anaplastic large cell lymphoma. They show varying combinations of CD5, CD10 and IRF4/MUM1 expression in addition to expressing CD20 and other pan-B cell markers. Their pattern of BM infiltration is predominantly intrasinusoidal (Fig. 29.21); the marrow interstitium and lumens of larger blood vessels are involved less conspicuously and solid infiltrates are rare. The genetic basis of this rare lymphoma is unknown.
Burkitt lymphoma (BL)
Clinical features and pathology at presenting sites
The WHO classification recognizes three clinical subtypes of BL (endemic, sporadic and immunodeficiency-associated).40 Aggressive lymphomas previously regarded as ‘Burkitt-like’ or ‘atypical Burkitt’s lymphoma’ are now classified within the WHO 2008 revision as ‘B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma’.121 Endemic BL is a disease of childhood in sub-Saharan Africa and is frequently extranodal at presentation, with a high incidence of jaw tumors. Sporadic BL occurs in all parts of the world and has a wide age distribution; ileocecal tumor formation in young males is a common presentation. Burkitt lymphoma in the context of immunodeficiency is most commonly seen in association with human immunodeficiency virus (HIV) infection but may also occur as a form of post-transplant lymphoproliferative disease and in other immunodeficiency states; its histologic features resemble those of the sporadic disease. All subtypes of BL are characterized histologically by diffuse proliferation of medium-sized cells that typically have a round nucleus, small or inconspicuous nucleoli and a rim of basophilic cytoplasm that may contain lipid vacuoles. Cells in BL associated with immunosuppression may show more evidence of plasmablastic differentiation than those in the other subtypes. Much variation in cellular features occurs between patients, however, and BL cannot be diagnosed reliably on the basis of cell morphology. High cell turnover in all subtypes is reflected by the presence of abundant tingible body macrophages, responding to the high rate of apoptotic cell death and providing the well-known ‘starry sky’ appearance. It should be noted that this feature is not specific to BL but may also be found in other aggressive lymphomas with high rates of apoptotic cell death.
Blood and bone marrow aspiration
Circulating tumor cells are present in a minority of patients with BL; in effect, these patients have a mature B-cell acute leukemia, of ALL-L3 subtype as defined historically by the French-American-British (FAB) group.122 The cells are fairly large, with cytoplasmic basophilia and vacuolation. Nuclear chromatin is dispersed and nucleoli are indistinct. The same morphology is represented in BM aspirate films, when involved, and the immunophenotype is distinguished from B-ALL by absence of nuclear TdT expression. Within the WHO classification, this presentation is categorized as Burkitt leukemia.40
Bone marrow trephine biopsy
The BM is not commonly involved by BL at presentation, except those patients who present with Burkitt leukemia. When present, BL cells in BMTB sections resemble those seen at other sites of disease involvement, being medium-sized with round nuclei, inconspicuous nucleoli and basophilic cytoplasm. Vacuolation of the cytoplasm is difficult to appreciate in histologic sections, even when prominent in cytology preparations. Mitotic figures are usually numerous. Infiltration may be interstitial, nodular or diffuse10 and may be accompanied by tingible body macrophages, giving a similar ‘starry sky’ appearance to that seen at other sites of involvement. The differential diagnosis includes ALL, blastoid variants of MCL and aggressive morphologic variants of DLCBL; clinical context and immunophenotype are usually discriminatory. The cells of BL express CD20 (sometimes only weakly), CD79a and CD10 but not CD5, cyclin D1, TdT or IRF4/MUM1. BCL2 is absent in most cases. Demonstration of Ki67 expression provides indirect evidence that cell cycle control has been dysregulated to leave all BL cells active in the cell cycle. Essentially 100% of tumor cells in BL express Ki67, with uniformly strong intensity, a picture that is very uncommon in all other lymphomas. The presence of EBV in tumor cells can be demonstrated in BMTB sections by IHC for latent membrane protein-1 (LMP-1) or EBV nuclear antigen-2 (EBNA-2), or by in situ hybridization to demonstrate EBV early RNA species (EBER). As mentioned above, however, EBV is less commonly associated with sporadic BL than with endemic and HIV-associated cases.
Genetic studies
At a genetic level, BL cells characteristically have translocations involving the MYC oncogene and immunoglobulin heavy or light chain genes: t(8;14)(q24;q32), t(2;8)(p12;q24), t(8;22)(q24;q11). The precise breakpoints vary between endemic, sporadic and immunodeficiency-associated subtypes and, in a substantial proportion of patients, the partner genes involved in MYC translocations are unknown. The subtypes also vary in their association with clonal latent infection by EBV; endemic BL is highly associated with EBV latency in tumor cells, sporadic BL less so and, of the immunodeficiency-associated cases, EBV latency is most frequently encountered in those arising in HIV-positive patients. Immunoglobulin variable region genes are hypermutated in BL. Gene profiling with microarray techniques shows an expression signature clearly distinct from that of DLCBL, although intermediate patterns are also found.50
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL
This category within the WHO 2008 classification includes aggressive lymphomas showing different combinations of the morphological, immunophenotypic and genetic features of both BL and DLCBL but not fully meeting all clinical and pathological criteria for either.40,121 The lymphomas encompassed are heterogeneous. Most have cytomorphology that is intermediate between the two; others have typical BL morphology but an atypical immunophenotype. The presence of MYC rearrangement may be found in association with a complex karyotype or an additional t(14;18), the latter suggesting origin by transformation from FL and being associated with particularly aggressive clinical behavior. Additional BCL6 rearrangement is also commonly found. Features in blood and BM have not been described specifically although patients may present with leukemia. When BM is involved, the infiltrates share the characteristics shown at other sites of involvement. This category should be used with caution in patients presenting with BM as the only diagnostic tissue unless extensive immunophenotyping and cytogenetic or FISH analysis is employed to provide the wide range of information needed.
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma
This category has been created within the 2008 revision of the WHO classification of lymphomas to recognize rare large cell lymphomas which, as the name indicates, share characteristics of large B cell lymphoma (particularly primary mediastinal B cell lymphoma) and classical Hodgkin lymphoma (HL) without clearly meeting diagnostic criteria for either.40,121 Typical presentation is in young men, with a large anterior mediastinal mass; unlike classical HL, EBV is present only in a relatively small minority of patients. Blood and BM features have not been described specifically although spread to BM is noted. Marrow infiltration, when present, would be expected to have similar features to those found at other sites. A high content of very pleomorphic large blast cells is usual, accompanied by fibrosis but relatively low numbers of inflammatory cells such as macrophages and eosinophils. There is more evidence of B cell-associated antigen expression than in classical HL, with both CD20 and CD79a being strongly expressed. There is retention of CD45 positivity by neoplastic cells, expression of CD30 and, in most cases, also of CD15. This category should be used with caution in patients for whom BM is the sole diagnostic tissue unless clinical features are strongly indicative and extensive immunophenotyping is employed to characterize infiltrates.
Mature T-cell and NK-cell neoplasms
Extranodal NK/T lymphoma, nasal type; enteropathy-associated T-cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma
Clinical features and pathology at presenting sites
Lymphomas of NK/T-cell, nasal type are much more common in Asia than anywhere else and are associated with latent EBV infection in most cases. It is important to note that, although the classical presentation is with a necrotizing mid-facial neoplasm, these lymphomas also occur at other body sites.24,40 Inflammatory features frequently mask the presence of neoplastic cells and the diagnosis may be difficult to establish.
Enteropathy-associated T-cell lymphoma (EATL)40 usually presents in adults with small bowel obstruction due to constricting tumor or with perforation due to tumor ulceration; multiple sites of small bowel tumor formation may be found at laparotomy. The neoplastic cells are medium-sized or large, with a cytotoxic T-cell phenotype, often associated with extensive tissue necrosis underlying the formation of ulcers. Most cases show background histologic features of gluten-sensitive enteropathy in non-neoplastic small bowel tissue and a proportion of patients have clinically overt celiac disease, usually of adult onset.
As its name implies, subcutaneous panniculitis-like T-cell lymphoma presents with clinical features of panniculitis and also has striking inflammatory histologic features accompanying dispersed malignant cells in subcutaneous tissue.40 The neoplastic T/NK-cells in these lymphoma subtypes are large and frequently pleomorphic. They typically have cytotoxic features, with granules containing perforin, granzymes and TIA-1 contributing to their necrotizing/inflammatory behavior.
Genetic studies
TCR-beta and -gamma genes are in germline configuration in most examples of nasal type NK/T-cell lymphoma but are often clonally rearranged in rare cases presenting in the lymph nodes as well as in subcutaneous panniculitis-like T-cell lymphoma and EATL. Patients with EATL have a high frequency of an underlying constitutional HLA-DQA1*0501,DQB1*0201 genotype, which is present in a very high proportion of patients with celiac disease.123 In contrast with other T cell lymphomas, complex abnormalities of 9q or deletions involving 16q are present in most cases of EATL.124
Hepatosplenic T-cell lymphoma
Clinical features, pathology, blood and bone marrow aspirate findings
This is a rare lymphoma with a median age at presentation of approximately 30 years. Most patients present with hepatosplenomegaly and have systemic symptoms.40 Most are pancytopenic at presentation and approximately half have circulating lymphoma cells. The presence of the latter heralds aggressive, end-stage disease in some patients, although prognosis in all cases is poor.11,17,125
Bone marrow trephine biopsy
Histologic sections show erythroid and megakaryocytic hyperplasia, with interstitial and intrasinusoidal infiltration by neoplastic cells,21,40 which may be subtle and almost invisible without IHC. The cells are of variable size, as described above, and are often pleomorphic; IHC stains reveal that they express CD2, CD3 and CD56 but usually neither CD4 nor CD8. They lack CD5 expression and show selective expression of the cytotoxic granule proteins TIA-1 and granzyme M without perforin or granzyme B. If involvement is slight, IHC staining of sinusoidal endothelium for von Willebrand factor, CD31 or CD34 may be useful to highlight the location of infiltrating cells. The presence of prominent intrasinusoidal infiltration by relatively large neoplastic cells permits distinction of this disease from other T-cell lymphomas. The marrow reaction and T-cell phenotype of the neoplastic cells distinguish it from intravascular large B-cell lymphoma. T-cell phenotype, cellular pleomorphism and absence of nodular infiltrates also distinguish hepatosplenic T-cell lymphoma from SMZL, which may show similar predominance of intrasinusoidal infiltration.
Genetic studies
Molecular genetic studies have shown monoclonal rearrangements of either gamma or beta or both sets of TCR genes in individual patients. TCR-beta rearrangement is generally unproductive but, while most cases express only alpha-beta T-cell receptor proteins, neoplastic cells in a minority of patients do express the alpha-beta T-cell receptor.126–128 This does not alter clinical behavior compared with cases showing gamma-delta T-cell receptor expression. Most cases have an isochromosome 7q in the neoplastic cell clone and progression is associated with increased copies of this or with abnormalities involving the second chromosome 7.129 EBV latent genes are not expressed in this lymphoma.
Anaplastic large cell lymphoma (ALCL), ALK-positive, and ALCL, ALK-negative
Clinical and pathologic features
Anaplastic large cell lymphoma (ALCL) accounts for 2% of adult and 13% of pediatric non-Hodgkin lymphoma.61,130 Approximately 50% express the anaplastic lymphoma-associated kinase (ALK; CD246 – see below). Patients with ALK-positive disease have different clinical features from those with ALK-negative lymphoma. This has been recognized by creation of a new entity of ALK-negative ALCL in the 2008 revised WHO classification, distinct from ALK-positive ALCL.40 ALK expression is associated with younger age, male sex, combinations of nodal and extranodal involvement and the occurrence of ‘B’ symptoms. Patients with ALK-positive disease have significantly better survival following intensive chemotherapy than do patients with ALK-negative disease.131–133
In ALK-positive ALCL, variation in the precise nature of the underlying chromosomal translocation pairs ALK with one of several alternative partner genes. The nature of the fusion partner influences subcellular localization of the expressed ALK enzyme but this is not known to influence clinical behavior of the disease.134 IHC can reveal the distribution of ALK within neoplastic cells, correlating well in most patients with the cytogenetic and molecular genetic findings.135
Blood and bone marrow aspiration
PB cytopenias occur and correlate with the presence of BM involvement but circulating tumor cells have been reported in only rare cases of ALCL with small cell cytology. Neoplastic cells are occasionally represented in aspirated BM; they can be recognized by their large size, abundant moderately basophilic cytoplasm and large irregular, sometimes multiple, nuclei with prominent nucleoli. Reactive hemophagocytosis may be found.23
Bone marrow trephine biopsy
BMTB sections reveal evidence of BM infiltration in 10–30% of patients, with the higher end of this range reflecting use of immunostains in addition to standard H&E staining. Few studies have attempted to assess ALK-positive and ALK-negative ALCL as distinct entities.14 BM involvement is possibly more common in the elderly, who are more likely to have ALK-negative disease. Infiltration is usually interstitial or focal and may be subtle, with only small clusters or single cells present.7,14 Morphology of the neoplastic cells resembles that at the primary site, with Reed–Sternberg cell-like features in most cases but small cell variants and admixed inflammatory cells may confuse the picture. IHC staining for T-cell associated antigens plus CD30 and ALK (CD246) is helpful, although expression of several T-cell associated antigens may be down-regulated. The major differential diagnosis is with Hodgkin lymphoma, but the characteristic background inflammatory infiltrate is missing. Alternative diagnoses of metastatic carcinoma, melanoma, Langerhans cell histiocytosis and large B cell lymphomas require consideration in some cases. Further IHC studies to demonstrate cytokeratins and other epithelial markers, melanoma markers (HMB45, MelanA, S100 protein) and antigens expressed by Langerhans cells (S100 protein, CD1a and CD2) and B-cells may be needed to establish the correct diagnosis.
Genetic studies
Most cases have clonally arranged TCR genes. The variation in ALK translocation fusion partners, the resultant subcellular localization of ALK protein expression and, where known, the molecular basis of this variation is summarized in the WHO 2008 classification.40 Secondary structural and numerical chromosomal changes occur frequently, involving numerous targets that differ between ALK-positive and ALK-negative ALCL. Gene expression profiling by microarray techniques has also shown patterns which differ between the two,53 further justifying their consideration as distinct entities.
Angioimmunoblastic T-cell lymphoma (AIL-T NHL)
Clinical features and lymph node pathology
Patients are usually adults and present with lymphadenopathy, often disseminated although not bulky. Additional features of fever, autoimmune phenomena, drug hypersensitivities and hypergammaglobulinemia (usually polyclonal) are common.136
Lymph nodes involved by AIL-T NHL usually show T-zone expansion with inactive B-cell follicles although greater histological variation has been recognized in recent years, including examples accompanied by marked follicular hyperplasia.137,138 The expanded paracortex may contain vaguely nodular areas of infiltration. High endothelial venules and other arborizing small blood vessels are usually prominent and are seen well with PAS staining. The latter may also show deposits of extravascular PAS-positive material, of uncertain origin. Cells within infiltrated areas are mixed, with scattered blast cells, macrophages, plasma cells and single or clustered medium-sized lymphoid cells that have abundant clear cytoplasm. The clear cells may appear to be clustered around small blood vessels; immunostaining reveals these cells to be of T-cell phenotype, usually of CD4 subtype and expressing PD-1 (programmed death-1), indicative of a follicular helper T-cell phenotype.139,140 The scattered blasts are a mixture of T- and B-cells. In most cases an irregular meshwork of dendritic cells expressing CD21 and CD23 is present underlying expanded areas of paracortex. Evidence of latent EBV infection can be demonstrated in most cases, with EBV-EBER expression in varying proportions of the B-cell blasts. Transformation to diffuse large B-cell lymphoma occurs occasionally via overgrowth of a monoclonal large B-cell population.136
Blood and bone marrow aspiration
Direct involvement of PB is very rare in AIL-T NHL but there may be cytopenias involving erythroid cells, platelets, granulocytes and lymphocytes in various combinations. Reactive increases in plasma cells, plasmacytoid cells and atypical lymphocytes may be seen141 and there may be a polyclonal increase in plasma immunoglobulins. The presence of a leukoerythroblastic blood picture may reflect BM infiltration. BM aspirate films usually reflect similar nonspecific findings but neoplastic T-cells can occasionally be found; FACS and/or molecular genetic analysis are required for confirmation if cells are seen that raise suspicion of neoplasia.
Bone marrow trephine biopsy
BMTB sections show evidence of infiltration relatively frequently in AIL-T NHL, although the reported incidence varies widely between different studies.17,19,20 Involvement is usually focal, with infiltrates distributed randomly within marrow spaces (Fig. 29.22).20 Infiltrates have a similar mixed composition to those found in affected lymph nodes and the underlying stroma usually has increased reticulin fibers, sometimes with a locally increased number of capillaries. Extracellular PAS-positive material is rarely seen. The background hemopoietic tissue frequently shows dysplastic features, with abnormalities of cell distribution within marrow spaces. Megakaryocytes and erythroid cells may show cytological atypia and granulopoiesis may be left-shifted, with relatively increased numbers of promyelocytes and myelocytes.
Genetic studies
Clonal rearrangements of TCR genes are present in almost all cases and additional clonal or oligoclonal IGH rearrangements are also found in a substantial minority.142 The latter correlate with the presence of EBV-positive B-cell blasts. Trisomies of chromosomes 3 and 5 are relatively frequent, as is an additional X chromosome. Gains of chromosomes 19, 22q and 11q plus losses of 13q have been found by comparative genomic hybridization studies.143 Gene expression profiling confirms a molecular signature of follicular helper T-cells.51
Peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS)
Clinical features and lymph node pathology
These lymphomas account for approximately 6% of NHL and occur in a wide age range of patients (median 61 years; range 17–90 years).61 There is usually extensive nodal involvement at presentation, with 65% of patients having stage IV disease.98 Extranodal involvement occurs more frequently than in B-cell NHL and systemic features, such as fever, are also common. Peripheral T-cell lymphomas in this category are probably a mixture of different entities that cannot currently be separated into defined prognostic groups;40 overall, they are aggressive neoplasms with worse prognoses than DLBCL.
Bone marrow trephine biopsy
While PTCL, NOS accounts for only a minority of non-Hodgkin lymphomas, BM involvement in this category of lymphoma is frequent, with the majority of reported cases in several series having positive BM staging biopsies.17,19 Some patients with PTCL, NOS present with BM as the sole or predominant site of disease. Infiltration is usually focal and patchy or diffuse throughout the interstitium. Reactive cells are frequently admixed with the infiltrates, as in AIL-T NHL, but the neoplastic cells in PTCL, NOS are usually larger and more obviously atypical, with marked pleomorphism. There is usually a patchy or diffuse increase in reticulin, sometimes with increased capillaries in areas of focal infiltration. Dysplastic hemopoietic features may be seen in non-infiltrated areas of marrow,16 as in AIL-T NHL. Lymph node histology is required to distinguish PTCL, NOS from AIL-T NHL. IHC may be necessary to exclude possible alternative diagnoses of BM involvement by Hodgkin lymphoma, DLBCL or ALCL. The BM histology may also mimic a post-transplant lymphoproliferative disorder or the polymorphous lymphoid aggregates found in some patients with HIV infection; clinical context usually suggests the likelihood of one or other of these latter types of infiltration.
Genetic studies
This is a heterogeneous group of T-cell lymphomas with complex karyotypes. A wide variety of recurring chromosomal gains and losses have been described that differ from those seen in AIL-T NHL and ALCL.143 Gene expression profiles also differ from other peripheral T cell lymphomas.59 Accompanying EBV latent infection is relatively infrequent.
Hodgkin lymphoma (HL)
Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL)
Clinical features and lymph node pathology
Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) comprises approximately 5% of all HL. The median age at presentation is mid-30s and males are affected significantly more often than females. The disease usually involves peripheral lymph nodes (neck, axillary or inguinal groups) and approximately 80% of patients have stage I or II disease at diagnosis. The prognosis is good; 90% of patients remain alive at 10 years from diagnosis, mostly having had removal of involved lymph nodes with or without adjunctive local radiotherapy as their sole treatment.120 There is, however, a higher risk of late disease relapse than in classical Hodgkin lymphoma and relapse may involve disease transformation into large B cell lymphoma.
In lymph nodes, NLPHL is characterized histologically by replacement of normal structures with expanded, B-cell-rich nodules enclosing scattered CD30-negative large blast cells.40 These large cells have distinctive ‘popcorn cell’ appearances, express CD45 and are positive for B-cell markers such as CD20 and CD79a; they are typically accompanied by scattered or loosely clustered macrophages and rosettes of T-lymphocytes. T-cells within rosettes show up-regulation of IRF4/MUM1 and express PD-1, indicating a follicular helper T-cell phenotype. Scattered as well as rosetted T-cells include a high proportion of CD57-positive cells, unlike the reactive T-cells accompanying classical Hodgkin lymphoma. True Reed–Sternberg cells are not present and the features of NLPHL may be accompanied by progressive transformation of germinal centers, a histologically distinctive reactive process; differential diagnosis between NLPHL and this reactive process is sometimes difficult when only few large cells are present. In other cases, diffuse architecture may lead to overlapping features with T-cell/histiocyte-rich large B cell lymphoma and the distinction of NLPHL from, or its possible relationship to, this variant of large B cell lymphoma is controversial.
Bone marrow trephine biopsy
Involvement of the BM is distinctly uncommon in NLPHL and, like aspiration, BMTB for staging is performed relatively infrequently. The finding of lymphoid or lymphohistiocytic infiltrates in BMTB sections from a patient diagnosed as having NLPHL should prompt review of the original diagnosis, since stage IV disease occurs rarely. Most such cases, upon review, are found to represent lymphocyte-rich classical Hodgkin lymphoma or occur in patients with a history of multiply-relapsed NLPHL in whom a high stage, T cell/histiocyte-rich pattern of disease, overlapping with T-cell/histiocyte-rich large B cell lymphoma may emerge over time.9 Histological features in the BM in such patients resemble those at other sites. In typical NLPHL at first presentation, BM histology is usually entirely normal.
Genetic studies
The relative paucity of neoplastic cells in NLPHL renders clonal IGH rearrangement undetectable in most cases unless specialized techniques such as single-cell PCR are employed. Isolated neoplastic cells, however, have been shown to have clonally rearranged IGH with a high degree of somatic hypermutation. They demonstrate ongoing acquisition of further mutations144 and have functional immunoglobulin production. Translocations and other abnormalities of BCL6 are relatively common.145 Latent EBV infection is absent.
Classical Hodgkin lymphoma
Clinical features and lymph node pathology
All variants of classical Hodgkin lymphoma are characterized histologically by the presence of Reed–Sternberg (RS) cells.40 These cells frequently have distinctive lacunar morphology in nodular sclerosing classical Hodgkin lymphoma (NSCHL) but, in the other subtypes, classical binucleate and multinucleate RS cells are found. They express CD30 and, in a majority of cases, CD15; they generally lack expression of CD45 and EMA. PAX5 and IRF4/MUM1 are consistently expressed but there is usually no demonstrable production of immunoglobulin or J-chain. CD20 and CD79a are only rarely positive (usually one or the other, but not both). The transcription factor OCT2 and its coactivator BOB1 are negative in a great majority of cases. Latent EBV infection in RS cells, and in occasional bystander small B cells, can be demonstrated in approximately 50% of cases; this is most frequent in MCCHL and occurs to a lesser extent in NSCHL. The pattern of latency is such that there is consistent expression of LMP1, which can be shown conveniently by IHC. Alternatively, in situ hybridization can be employed to demonstrate EBV-EBER. Mononuclear Hodgkin cells sharing these phenotypic properties are present in most cases and predominate in some. In all subtypes of classical Hodgkin lymphoma, RS cells are accompanied by reactive lymphoid cells, macrophages and eosinophils. Bands of fibrosis separating nodular areas of mixed, cellular infiltration are present in NSCHL. Lymphocyte-depleted Hodgkin lymphoma (LDCHL) is much less frequently diagnosed now than in pre-immunohistochemistry days, since many cases that would previously have been categorized as LDCHL can now be shown to be variants of anaplastic large cell lymphoma. Lymphocyte-rich classical Hodgkin lymphoma (LRCHL) is characterized by appearances superficially resembling NLPHL but with true RS cells and clinical behavior more in keeping with mixed cellularity Hodgkin lymphoma (MCCHL).40
Bone marrow trephine biopsy
BMTB is much more sensitive than aspiration for detection of BM involvement by classical HL, at least in part because the infiltrates cause stromal fibrosis and are difficult to aspirate. Depending on patient selection criteria, different studies have reported up to 15% of BMTB performed for staging in classical HL to be positive.3 This figure is undoubtedly an over-estimate, since many patients with clinical stage IA or IIA disease do not undergo BM examination. Bilateral biopsies or single long-core biopsy increase the positive detection rate in classical HL and in non-Hodgkin lymphomas. However, the clinical value of BM staging in classical HL has been questioned,1 since patient management is rarely influenced directly by the outcome. Occasionally, BM is the primary or sole site of involvement by classical HL; this is particularly the case in patients with HIV-associated disease.
Involvement of BM by classical HL is rarely subtle; the infiltrates usually form sizeable, irregular patches with dense underlying fibrosis. Occasionally, all or most of the biopsy core is replaced. Extensively involved BM may contain areas of necrosis, even in previously untreated patients. Classical RS cells may or may not be found and mononuclear Hodgkin cells are usually few, scattered widely within irregular areas of macrophage and lymphocyte infiltration (Fig. 29.23). Plasma cells and eosinophils are usually also present in these infiltrates but the number of these cells varies considerably between patients. Sometimes, the infiltrates appear densely fibrotic, or loose and edematous, with few cells of any type other than fibroblasts evident, and remodeling of trabecular bone may be seen; this pattern is relatively common in specimens taken for re-staging after treatment.
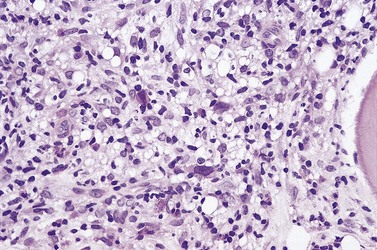
Genetic studies
Reed–Sternberg cells in cases of classical HL studied by single-cell PCR techniques have been shown to have clonally rearranged and hypermutated IGH without intraclonal heterogeneity (i.e., with no evidence of ongoing somatic mutation).146 However, immunoglobulins are not transcribed, due either to crippling mutations or defective regulatory elements such as OCT2 and BOB1. Integrated, clonal, EBV is present in RS and bystander B-cells in a high proportion of cases of classical HL, particularly in MCCHL and in classical HL arising in immunocompromised patients, with a distinctive pattern of latent gene expression including LMP1 and EBNA2 expression.147 Intracellular signaling pathways involving NFkappaB are constitutively activated, and regulation of the JAK/STAT pathway is disrupted, promoting proliferation and inhibiting apoptosis.148 Gene expression studies using microarray techniques show a molecular signal reflecting these disturbances and closely resembling that found in primary mediastinal large B cell lymphoma;149 the WHO 2008 classification recognizes overlap between large B cell lymphoma, predominantly of mediastinal type, and classical HL.40
Post-transplant lymphoproliferative disorders (PTLD) and other lymphoid proliferations associated with impaired immunity
Clinical and pathologic features
The clinical presentation of PTLD is very variable. Patients presenting in the early post-transplant period often have an infectious mononucleosis-like disease characterized by constitutional symptoms and rapid enlargement of tonsils and cervical lymph nodes. Patients with PTLD of late onset, usually a year or more after transplantation, have less severe constitutional symptoms and frequently have extranodal disease similar to HIV-related lymphomas. The cumulative incidence of PTLD at 10 years is in the order of 5% following heart transplantation and 1% following renal or BM transplantation.150,151 The incidence of PTLD is higher in patients who are seronegative for EBV at the time of transplantation, compared with those who are seropositive. In solid organ recipients, PTLD is almost always of host origin while, in recipients of allogeneic BM grafts, not surprisingly it arises from donor cells.
Lymphoproliferative disorders arising after solid organ or BM transplantation also vary greatly in histologic appearance and immunophenotype between individuals.40,152 Although they may appear to arise from either T- or B-cells, EBV infection is associated with the majority of cases, through reactivation of latent infection in the graft recipient or as a result of primary infection acquired from graft tissue. Similar EBV-associated lymphoid proliferations occur in other acquired and inherited immune deficiency states and in association with age-related decline in immunity in some elderly patients.153 Morphologically, examples of PTLD have been reported that are equivalent to most subtypes of large B-cell, Burkitt, peripheral T-cell and Hodgkin lymphomas, plus polyclonal and monoclonal plasmacytic proliferations. Also well described in the spectrum of PTLD are polymorphous lymphoid infiltrates composed of complex mixtures of inflammatory cells and atypical lymphoid cells, including B-cells at all stages of maturation.
Bone marrow trephine biopsy
Approximately 50% of patients with polymorphous PTLD have BM involvement, seen in trephine biopsy sections as poorly defined irregular or nodular infiltrates of mixed cells (Fig. 29.24). BM involvement by PTLD in patients who have variants equivalent to B-cell, T-cell or Hodgkin lymphomas shares the morphology of the primary tumor and has features as described above for the equivalent lymphomas seen in immunocompetent individuals. The presence of EBV can be demonstrated in a majority of patients by in situ hybridization for EBER but LMP1 is often not expressed and immunostaining for the latter should not be relied upon in this context. BM involvement has been associated with poor prognosis.154
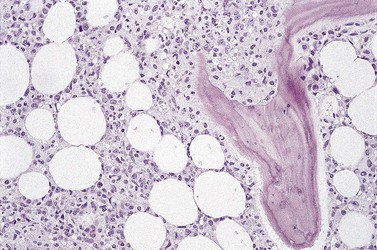
Appearances of lymphoma infiltrates following therapy
Apart from quantitative changes in the degree of BM infiltration by lymphoma following chemotherapy, there may also be changes in the morphology of cell infiltrates. At present, it remains controversial whether nodular lymphoid infiltrates (a common finding in this context), not occupying paratrabecular or perisinusoidal locations, are reactive phenomena or residual deposits of low-grade lymphoma. A prominent, hypercellular rim of granulocytes, particularly eosinophils, often surrounds such nodules and they are usually composed predominantly of small T-lymphocytes, with few B-cells, regardless of the original lymphoma diagnosis (Fig. 29.25).155 It is relatively common to see such nodules following anti-CD20 immunotherapy of low-grade lymphoma. This treatment leads to down-regulation of CD20 expression by B-cells, an effect which may persist for long periods, so that use of CD20 as a B-cell marker for IHC in this context is unreliable. Typical findings are normal expression of antigens such as CD19 and CD79a in the almost complete absence of CD20. Absence of demonstrable clonal IGH rearrangement in microdissected nodules of this type has led some authors to conclude that they are not neoplastic,156,157 but they may represent ‘tombstone’ lesions rather than newly formed reactive lymphoid nodules; whether or not they are truly inert in terms of potential for re-growth of lymphoma is unproven.
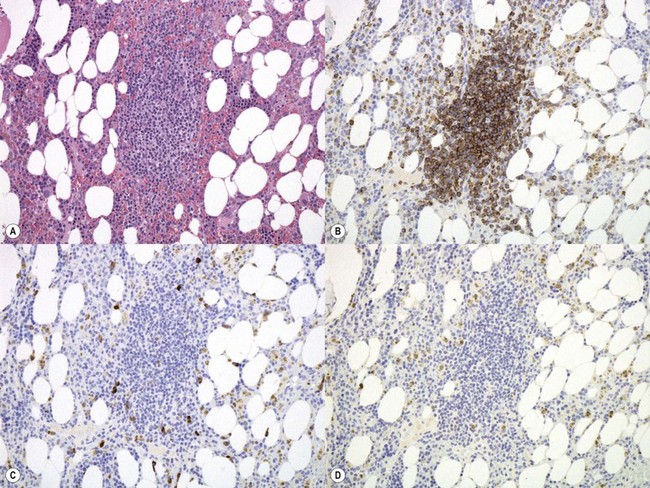
BM hyperplastic, dysplastic and stromal reactions to lymphoma
Hemopoiesis often appears remarkably unaffected by significant levels of BM infiltration by lymphoma. However, certain types of lymphoma are regularly associated with spatial and cytologic abnormalities of hemopoiesis, often with increased stromal reticulin. Such effects are presumably secondary to cytokine production by the neoplastic cells themselves or by inflammatory cells stimulated in consequence of their presence. Infiltration of the BM by angioimmunoblastic T-cell lymphoma or peripheral T-cell lymphoma, not otherwise specified, has particularly been reported in association with such phenomena.4,16,19,158 However, in occasional patients, B-cell lymphomas of various subtypes are also accompanied by marked hemopoietic cell hyperplasia and/or by stromal edema and fibrosis. In these cases, the presence of lymphomatous infiltrates is usually obvious; attention to megakaryocyte morphology usually excludes a myeloproliferative neoplasm but it can be difficult to determine whether or not a myelodysplastic syndrome is represented. Immunostaining for CD42b or CD61 can assist by revealing the full spectrum of megakaryocytes, including any true micromegakaryocytes. The latter would not be expected in a reactive myelopathy. Lack of an increase in CD34-positive early hemopoietic cells may be additionally reassuring. Hematological follow-up, with BM aspiration and cytogenetic studies ± repeat trephine biopsy may be helpful.
Differential diagnosis and classification of lymphoma when bone marrow trephine biopsy provides the sole source of tissue for assessment
Occasionally, lymphoma presents with BM as the sole site of disease or as the most accessible site for diagnostic biopsy. As an additional problem, BM aspiration frequently yields no lymphoma cells in patients in whom BMTB sections show definite evidence of lymphoma infiltration, so that FCM data cannot be obtained to support the diagnosis. In these circumstances, BM histology is crucial to diagnosis. The spatial distribution and cellular composition of lymphoma infiltrates, supplemented by judicious use of IHC, permits accurate WHO classification in most cases. The various spatial patterns of lymphoma in different diagnostic categories have been described in earlier sections of this chapter. No single pattern is unique to any lymphoma entity but it is possible to make some helpful generalizations. Paratrabecular infiltration is highly suggestive of FL although it may also be seen in LPL, MCL and SMZL; in the latter conditions, paratrabecular infiltrates are rarely as extensive or linear as in FL and are usually accompanied by other patterns (varying combinations of nodular and interstitial). Paratrabecular infiltration occurs with exceptional rarity in CLL; stromal alterations in treated patients with residual disease at follow-up, or in patients with coincidental marrow pathologies, may occasionally result in formation of focal paratrabecular infiltrates but the dominant pattern remains typical. The typical pattern in CLL is nodular infiltration with greater or lesser degrees of interstitial spread. The presence of scattered or clustered para-immunoblasts in nodular areas of small lymphoid cell infiltration strongly suggests CLL. The presence of scattered blast cells in LPL and SMZL or the rare formation of neoplastic follicles in FL can mimic para-immunoblasts but FL will generally show at least some areas of paratrabecular infiltration while infiltrates of LPL are usually more irregular in shape and less well defined than those of CLL. In SMZL, nodularity is distinctively centered on well preserved nodular clusters of follicular dendritic cells, possibly representing colonized non-neoplastic follicles and readily demonstrated by CD21 or CD23 staining. There is often an additional component of intrasinusoidal infiltration, but a pure intrasinusoidal pattern should raise suspicion of persistent polyclonal B lymphocytosis mimicking SMZL.159 The presence of numerous plasma cells and formation of Dutcher bodies supports a diagnosis of LPL, both features being rare in CLL; an increased number reactive mast cells typically accompany LPL infiltrates and this feature is less common in CLL. Nodular infiltration is also a frequent pattern in MCL but the nodules in this disease appear monotonous cytologically, without blast cells or plasma cells.
IHC for CD5, CD10, CD23, BCL6 and cyclin D1 (plus CD20 or CD79a and CD3 to assess overall T- and B-cell numbers) usually distinguishes between these various small B-cell lymphomas (see Table 29.1). The ‘spaced out’ infiltrates of HCL are histologically distinctive and are rarely confused with other small cell subtypes of lymphoma; reticulin staining and IHC for TRAP and annexin A1 provide confirmation, if needed.
Infiltrates of large lymphoid cells require distinction from increased numbers of immature myelomonocytic cells, poorly differentiated plasma cell neoplasms and, rarely, metastatic solid cancers. Diffuse large B cell lymphoma occasionally arises primarily within BM160 and IHC is highly valuable in this context, as described earlier in this chapter in relation to DLBCL presenting at more usual sites.
1 Bairey O, Shpilberg O. Is bone marrow biopsy obligatory in all patients with non-Hodgkin’s lymphoma? Acta Haematol. 2007;118:61-64.
2 Bartl R, Frisch B, Burkhardt R, et al. Assessment of bone marrow histology in the malignant lymphomas (non-Hodgkin’s): correlation with clinical factors for diagnosis, prognosis, classification and staging. Br J Haematol. 1982;51:511-530.
3 Bartl R, Frisch B, Burkhardt R, et al. Assessment of bone marrow histology in Hodgkin’s disease: correlation with clinical factors. Br J Haematol. 1982;51:345-360.
4 Caulet S, Delmer A, Audouin J, et al. Histopathological study of bone marrow biopsies in 30 cases of T-cell lymphoma with clinical, biological and survival correlations. Hematol Oncol. 1990;8:155-168.
5 Chung R, Lai R, Wei P, et al. Concordant but not discordant bone marrow involvement in diffuse large B-cell lymphoma predicts a poor clinical outcome independent of the International Prognostic Index. Blood. 2007;110:1278-1282.
6 Dufau JP, Le TA, Molina T, et al. Intravascular large B-cell lymphoma with bone marrow involvement at presentation and haemophagocytic syndrome: two Western cases in favour of a specific variant. Histopathology. 2000;37:509-512.
7 Fraga M, Brousset P, Schlaifer D, et al. Bone marrow involvement in anaplastic large cell lymphoma. Immunohistochemical detection of minimal disease and its prognostic significance. Am J Clin Pathol. 1995;103:82-89.
8 Khoury JD, Jones D, Yared MA, et al. Bone marrow involvement in patients with nodular lymphocyte predominant Hodgkin lymphoma. Am J Surg Pathol. 2004;28:489-495.
9 Skinnider BF, Connors JM, Gascoyne RD. Bone marrow involvement in T-cell-rich B-cell lymphoma. Am J Clin Pathol. 1997;108:570-578.
10 Bain BJ, Clark DM, Wilkins B. Bone marrow pathology. Oxford: Blackwell Wiley; 2010.
11 Sallah S, Smith SV, Lony LC, et al. Gamma/delta T-cell hepatosplenic lymphoma: review of the literature, diagnosis by flow cytometry and concomitant autoimmune hemolytic anemia. Ann Hematol. 1997;74:139-142.
12 Schmid C, Isaacson PG. Bone marrow trephine biopsy in lymphoproliferative disease. J Clin Pathol. 1992;45:745-750.
13 Schmidt B, Kremer M, Gotze K, et al. Bone marrow involvement in follicular lymphoma: comparison of histology and flow cytometry as staging procedures. Leuk Lymphoma. 2006;47:1857-1862.
14 Weinberg OK, Seo K, Arber DA. Prevalence of bone marrow involvement in systemic anaplastic large cell lymphoma: are immunohistochemical studies necessary? Hum Pathol. 2008;39:1331-1340.
15 Audouin J, Le TA, Molina T, et al. Patterns of bone marrow involvement in 58 patients presenting primary splenic marginal zone lymphoma with or without circulating villous lymphocytes. Br J Haematol. 2003;122:404-412.
16 Auger MJ, Nash JR, Mackie MJ. Marrow involvement with T cell lymphoma initially presenting as abnormal myelopoiesis. J Clin Pathol. 1986;39:134-137.
17 Dogan A, Morice WG. Bone marrow histopathology in peripheral T-cell lymphomas. Br J Haematol. 2004;127:140-154.
18 Franco V, Florena AM, Ascani S, et al. CD27 distinguishes two phases in bone marrow infiltration of splenic marginal zone lymphoma. Histopathology. 2004;44:381-386.
19 Gaulard P, Kanavaros P, Farcet JP, et al. Bone marrow histologic and immunohistochemical findings in peripheral. T-cell lymphoma: a study of 38 cases. Hum Pathol. 1991;22:331-338.
20 Grogg KL, Morice WG, Macon WR. Spectrum of bone marrow findings in patients with angioimmunoblastic T-cell lymphoma. Br J Haematol. 2007;137:416-422.
21 Vega F, Medeiros LJ, Bueso-Ramos C, et al. Hepatosplenic gamma/delta T-cell lymphoma in bone marrow. A sinusoidal neoplasm with blastic cytologic features. Am J Clin Pathol. 2001;116:410-419.
22 Parrens M, Dubus P, Agape P, et al. Intrasinusoidal bone marrow infiltration revealing intravascular lymphomatosis. Leuk Lymphoma. 2000;37:219-223.
23 Wong KF, Chan JK, Ng CS, et al. Anaplastic large cell Ki-1 lymphoma involving bone marrow: marrow findings and association with reactive hemophagocytosis. Am J Hematol. 1991;37:112-119.
24 Wong KF, Chan JK, Cheung MM, So JC. Bone marrow involvement by nasal NK cell lymphoma at diagnosis is uncommon. Am J Clin Pathol. 2001;115:266-270.
25 Ya-In C, Brandwein J, Pantalony D, Chang H. Hairy cell leukemia variant with features of intrasinusoidal bone marrow involvement. Arch Pathol Lab Med. 2005;129:395-398.
26 Wilkins BS, Clark DM. Making the most of bone marrow trephine biopsy. Histopathology. 2009;55:631-640.
27 Wickham CL, Sarsfield P, Joyner MV, et al. Formic acid decalcification of bone marrow trephines degrades DNA: alternative use of EDTA allows the amplification and sequencing of relatively long PCR products. Mol Pathol. 2000;53:336.
28 Wickham CL, Boyce M, Joyner MV, et al. Amplification of PCR products in excess of 600 base pairs using DNA extracted from decalcified, paraffin wax embedded bone marrow trephine biopsies. Mol Pathol. 2000;53:19-23.
29 Faulkner-Jones BE, Howie AJ, Boughton BJ, Franklin IM. Lymphoid aggregates in bone marrow: study of eventual outcome. J Clin Pathol. 1988;41:768-775.
30 Krober SM, Horny HP, Greschniok A, Kaiserling E. Reactive and neoplastic lymphocytes in human bone marrow: morphological, immunohistological, and molecular biological investigations on biopsy specimens. J Clin Pathol. 1999;52:521-526.
31 Thiele J, Zirbes TK, Kvasnicka HM, Fischer R. Focal lymphoid aggregates (nodules) in bone marrow biopsies: differentiation between benign hyperplasia and malignant lymphoma – a practical guideline. J Clin Pathol. 1999;52:294-300.
32 Brinckmann R, Kaufmann O, Reinartz B, Dietel M. Specificity of PCR-based clonality analysis of immunoglobulin heavy chain gene rearrangements for the detection of bone marrow involvement by low-grade B-cell lymphomas. J Pathol. 2000;190:55-60.
33 Bain BJ. Bone marrow aspiration. J Clin Pathol. 2001;54:657-663.
34 Bishop PW, McNally K, Harris M. Audit of bone marrow trephines. J Clin Pathol. 1992;45:1105-1108.
35 Campbell JK, Matthews JP, Seymour JF, et al. Optimum trephine length in the assessment of bone marrow involvement in patients with diffuse large cell lymphoma. Ann Oncol. 2003;14:273-276.
36 Lee SH, Erber WN, Porwit A, et al. ICSH guidelines for the standardization of bone marrow specimens and reports. Int J Lab Hematol. 2008;30:349-364.
37 Bain BJ, Clark DM, Wilkins B. Bone marrow pathology. Oxford: Blackwell Wiley; 2010.
38 Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting – Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835-3849.
39 Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361-1392.
40 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphatic Tissues. Lyon: IARC; 2008.
41 Camacho FI, Algara P, Rodriguez A, et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood. 2003;101:4042-4046.
42 Dunn-Walters D, Thiede C, Alpen B, Spencer J. Somatic hypermutation and B-cell lymphoma. Philos Trans R Soc Lond B Biol Sci. 2001;356:73-82.
43 Klein U, Goossens T, Fischer M, et al. Somatic hypermutation in normal and transformed human B cells. Immunol Rev. 1998;162:261-280.
44 Lossos IS, Alizadeh AA, Eisen MB, et al. Ongoing immunoglobulin somatic mutation in germinal center B cell-like but not in activated B cell-like diffuse large cell lymphomas. Proc Natl Acad Sci USA. 2000;97:10209-10213.
45 Ottensmeier CH, Thompsett AR, Zhu D, et al. Analysis of VH genes in follicular and diffuse lymphoma shows ongoing somatic mutation and multiple isotype transcripts in early disease with changes during disease progression. Blood. 1998;91:4292-4299.
46 Zhu D, Orchard J, Oscier DG, et al. V(H) gene analysis of splenic marginal zone lymphomas reveals diversity in mutational status and initiation of somatic mutation in vivo. Blood. 2002;100:2659-2661.
47 Bain B, Gilliland DG, Vardiman J, Horny HP. Chronic eosinophilic leukaemia, not otherwise specified. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. Lyon: IARC Press; 2008. p. 51–3
48 Amen F, Horncastle D, Elderfield K, et al. Absence of cyclin-D2 and Bcl-2 expression within the germinal centre type of diffuse large B-cell lymphoma identifies a very good prognostic subgroup of patients. Histopathology. 2007;51:70-79.
49 Martinez N, Camacho FI, Algara P, et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res. 2003;63:8226-8232.
50 Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431-2442.
51 de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952-4963.
52 Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275-282.
53 Lamant L, de RA, Duplantier MM, et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007;109:2156-2164.
54 Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313-2323.
55 Monti S, Savage KJ, Kutok JL, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105:1851-1861.
56 Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937-1947.
57 Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185-197.
58 Wright G, Tan B, Rosenwald A, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991-9996.
59 Zettl A, Rudiger T, Konrad MA, et al. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am J Pathol. 2004;164:1837-1848.
60 Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107:265-276.
61 Muller AM, Ihorst G, Mertelsmann R, Engelhardt M. Epidemiology of non-Hodgkin’s lymphoma (NHL): trends, geographic distribution, and etiology. Ann Hematol. 2005;84:1-12.
62 Vijay A, Gertz MA. Waldenstrom macroglobulinemia. Blood. 2007;109:5096-5103.
63 de Sanjose S, Benavente Y, Vajdic CM, et al. Hepatitis C and non-Hodgkin lymphoma among 4784 cases and 6269 controls from the International Lymphoma Epidemiology Consortium. Clin Gastroenterol Hepatol. 2008;6:451-458.
64 Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol. 2007;60:1378-1383.
65 Martyak LA, Yeganeh M, Saab S. Hepatitis C and lymphoproliferative disorders: from mixed cryoglobulinemia to non-Hodgkin’s lymphoma. Clin Gastroenterol Hepatol. 2009;7:900-905.
66 Wilkins BS, Buchan SL, Webster J, Jones DB. Tryptase-positive mast cells accompany lymphocytic as well as lymphoplasmacytic lymphoma infiltrates in bone marrow trephine biopsies. Histopathology. 2001;39:150-155.
67 Ho AW, Hatjiharissi E, Ciccarelli BT, et al. CD27-CD70 interactions in the pathogenesis of Waldenstrom macroglobulinemia. Blood. 2008;112:4683-4689.
68 Tournilhac O, Santos DD, Xu L, et al. Mast cells in Waldenstrom’s macroglobulinemia support lymphoplasmacytic cell growth through CD154/CD40 signaling. Ann Oncol. 2006;17:1275-1282.
69 Banham AH, Turley H, Pulford K, et al. The plasma cell associated antigen detectable by antibody VS38 is the p63 rough endoplasmic reticulum protein. J Clin Pathol. 1997;50:485-489.
70 Iida S, Rao PH, Nallasivam P, et al. The t(9;14)(p13;q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood. 1996;88:4110-4117.
71 Ocio EM, Schop RF, Gonzalez B, et al. 6q deletion in Waldenstrom macroglobulinemia is associated with features of adverse prognosis. Br J Haematol. 2007;136:80-86.
72 Walsh SH, Laurell A, Sundstrom G, et al. Lymphoplasmacytic lymphoma/Waldenstrom’s macroglobulinemia derives from an extensively hypermutated B cell that lacks ongoing somatic hypermutation. Leuk Res. 2005;29:729-734.
73 Wilkins B, Wright DH. Illustrated pathology of the spleen. Cambridge, UK: Cambridge University Press; 2000.
74 Bain B. Blood cells. A practical guide. Oxford: Blackwell Publishing; 2006.
75 Verhoef GE, de Wolf-Peeters C, Zachee P, Boogaerts MA. Regression of diffuse osteosclerosis in hairy cell leukaemia after treatment with interferon. Br J Haematol. 1990;76:150-151.
76 VanderMolen LA, Urba WJ, Longo DL, et al. Diffuse osteosclerosis in hairy cell leukemia. Blood. 1989;74:2066-2069.
77 Sun T, Grupka N, Klein C. Transformation of hairy cell leukemia to high-grade lymphoma: a case report and review of the literature. Hum Pathol. 2004;35:1423-1426.
78 Hoyer JD, Li CY, Yam LT, et al. Immunohistochemical demonstration of acid phosphatase isoenzyme 5 (tartrate-resistant) in paraffin sections of hairy cell leukemia and other hematologic disorders. Am J Clin Pathol. 1997;108:308-315.
79 Went PT, Zimpfer A, Pehrs AC, et al. High specificity of combined TRAP and DBA.44 expression for hairy cell leukemia. Am J Surg Pathol. 2005;29:474-478.
80 Chen YH, Tallman MS, Goolsby C, Peterson L. Immunophenotypic variations in hairy cell leukemia. Am J Clin Pathol. 2006;125:251-259.
81 Hounieu H, Chittal SM, al Saati T, et al. Hairy cell leukemia. Diagnosis of bone marrow involvement in paraffin-embedded sections with monoclonal antibody DBA.44. Am J Clin Pathol. 1992;98:26-33.
82 Parnes JR, Pan C. CD72, a negative regulator of B-cell responsiveness. Immunol Rev. 2000;176:75-85.
83 Falini B, Tiacci E, Liso A, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet. 2004;363:1869-1870.
84 Johrens K, Stein H, Anagnostopoulos I. T-bet transcription factor detection facilitates the diagnosis of minimal hairy cell leukemia infiltrates in bone marrow trephines. Am J Surg Pathol. 2007;31:1181-1185.
85 Cawley JC. The pathophysiology of the hairy cell. Hematol Oncol Clin North Am. 2006;20:1011-1021.
86 Galiegue-Zouitina S, Delestre L, Dupont C, et al. Underexpression of RhoH in hairy cell leukemia. Cancer Res. 2008;68:4531-4540.
87 Cessna MH, Hartung L, Tripp S, et al. Hairy cell leukemia variant: fact or fiction. Am J Clin Pathol. 2005;123:132-138.
88 Matutes E, Wotherspoon A, Catovsky D. The variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16:41-56.
89 Brito-Babapulle V, Matutes E, Oscier D, et al. Chromosome abnormalities in hairy cell leukaemia variant. Genes Chromosomes Cancer. 1994;10:197-202.
90 Mollejo M, Menarguez J, Lloret E, et al. Splenic marginal zone lymphoma: a distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am J Surg Pathol. 1995;19:1146-1157.
91 Isaacson PG, Matutes E, Burke M, Catovsky D. The histopathology of splenic lymphoma with villous lymphocytes. Blood. 1994;84:3828-3834.
92 Algara P, Mateo MS, Sanchez-Beato M, et al. Analysis of the IgV(H) somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002;99:1299-1304.
93 Corcoran MM, Mould SJ, Orchard JA, et al. Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene. 1999;18:6271-6277.
94 Novara F, Arcaini L, Merli M, et al. High-resolution genome-wide array comparative genomic hybridization in splenic marginal zone B-cell lymphoma. Hum Pathol. 2009;40:1628-1637.
95 Ruiz-Ballesteros E, Mollejo M, Rodriguez A, et al. Splenic marginal zone lymphoma: proposal of new diagnostic and prognostic markers identified after tissue and cDNA microarray analysis. Blood. 2005;106:1831-1838.
96 Traverse-Glehen A, Baseggio L, Bauchu EC, et al. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: a distinct clinicopathologic and molecular entity? Blood. 2008;111:2253-2260.
97 Baro C, Salido M, Domingo A, et al. Translocation t(9;14)(p13;q32) in cases of splenic marginal zone lymphoma. Haematologica. 2006;91:1289-1291.
98 Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol. 1998;16:2780-2795.
99 Griesser H, Kaiser U, Augener W, et al. B-cell lymphoma of the mucosa-associated lymphatic tissue (MALT) presenting with bone marrow and peripheral blood involvement. Leuk Res. 1990;14:617-622.
100 Rosenwald A, Ott G, Stilgenbauer S, et al. Exclusive detection of the t(11;18)(q21;q21) in extranodal marginal zone B cell lymphomas (MZBL) of MALT type in contrast to other MZBL and extranodal large B cell lymphomas. Am J Pathol. 1999;155:1817-1821.
101 Brynes RK, Almaguer PD, Leathery KE, et al. Numerical cytogenetic abnormalities of chromosomes 3, 7, and 12 in marginal zone B-cell lymphomas. Mod Pathol. 1996;9:995-1000.
102 Dierlamm J, Rosenberg C, Stul M, et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia. 1997;11:747-758.
103 Walsh SH, Rosenquist R. Immunoglobulin gene analysis of mature B-cell malignancies: reconsideration of cellular origin and potential antigen involvement in pathogenesis. Med Oncol. 2005;22:327-341.
104 Du M, Diss TC, Xu C, et al. Ongoing mutation in MALT lymphoma immunoglobulin gene suggests that antigen stimulation plays a role in the clonal expansion. Leukemia. 1996;10:1190-1197.
105 Arcaini L, Lucioni M, Boveri E, Paulli M. Nodal marginal zone lymphoma: current knowledge and future directions of an heterogeneous disease. Eur J Haematol. 2009;83:165-174.
106 Traverse-Glehen A, Felman P, Callet-Bauchu E, et al. A clinicopathological study of nodal marginal zone B-cell lymphoma. A report on 21 cases. Histopathology. 2006;48:162-173.
107 Henrique R, Achten R, Maes B, et al. Guidelines for subtyping small B-cell lymphomas in bone marrow biopsies. Virchows Arch. 1999;435:549-558.
108 Arcaini L, Lucioni M, Boveri E, Paulli M. Nodal marginal zone lymphoma: current knowledge and future directions of an heterogeneous disease. Eur J Haematol. 2009;83:165-174.
109 Ott G, Katzenberger T, Lohr A, et al. Cytomorphologic, immunohistochemical, and cytogenetic profiles of follicular lymphoma: 2 types of follicular lymphoma grade 3. Blood. 2002;99:3806-3812.
110 Raynaud P, Caulet-Maugendre S, Foussard C, et al. T-cell lymphoid aggregates in bone marrow after rituximab therapy for B-cell follicular lymphoma: a marker of therapeutic efficacy? Hum Pathol. 2008;39:194-200.
111 Landgren O, Tilly H. Epidemiology, pathology and treatment of non-follicular indolent lymphomas. Leuk Lymphoma. 2008;49(Suppl. 1):35-42.
112 Cheuk W, Wong KO, Wong CS, Chan JK. Consistent immunostaining for cyclin D1 can be achieved on a routine basis using a newly available rabbit monoclonal antibody. Am J Surg Pathol. 2004;28:801-807.
113 Ruchlemer R, Parry-Jones N, Brito-Babapulle V, et al. B-prolymphocytic leukaemia with t(11;14) revisited: a splenomegalic form of mantle cell lymphoma evolving with leukaemia. Br J Haematol. 2004;125:330-336.
114 Conlan MG, Bast M, Armitage JO, Weisenburger DD. Bone marrow involvement by non-Hodgkin’s lymphoma: the clinical significance of morphologic discordance between the lymph node and bone marrow. Nebraska Lymphoma Study Group. J Clin Oncol. 1990;8:1163-1172.
115 Kluin PM, van Krieken JH, Kleiverda K, Kluin-Nelemans HC. Discordant morphologic characteristics of B-cell lymphomas in bone marrow and lymph node biopsies. Am J Clin Pathol. 1990;94:59-66.
116 Kremer M, Spitzer M, Mandl-Weber S, et al. Discordant bone marrow involvement in diffuse large B-cell lymphoma: comparative molecular analysis reveals a heterogeneous group of disorders. Lab Invest. 2003;83:107-114.
117 Heyning FH, Hogendoorn PC, Kramer MH, et al. Primary non-Hodgkin’s lymphoma of bone: a clinicopathological investigation of 60 cases. Leukemia. 1999;13:2094-2098.
118 Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-511.
119 El Weshi A, Akhtar S, Mourad WA, et al. T-cell/histiocyte-rich B-cell lymphoma: clinical presentation, management and prognostic factors: report on 61 patients and review of literature. Leuk Lymphoma. 2007;48:1764-1773.
120 Fan Z, Natkunam Y, Bair E, et al. Characterization of variant patterns of nodular lymphocyte predominant Hodgkin lymphoma with immunohistologic and clinical correlation. Am J Surg Pathol. 2003;27:1346-1356.
121 Quintanilla-Martinez L, de Jong D, de Mascarel A, et al. Gray zones around diffuse large B cell lymphoma. Conclusions based on the workshop of the XIV meeting of the European Association for Hematopathology and the Society of Hematopathology in Bordeaux, France. J Hematop. 2009;2:211-236.
122 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451-458.
123 Howell WM, Leung ST, Jones DB, et al. HLA-DRB, -DQA, and -DQB polymorphism in celiac disease and enteropathy-associated T-cell lymphoma. Common features and additional risk factors for malignancy. Hum Immunol. 1995;43:29-37.
124 Obermann EC, Diss TC, Hamoudi RA, et al. Loss of heterozygosity at chromosome 9p21 is a frequent finding in enteropathy-type T-cell lymphoma. J Pathol. 2004;202:252-262.
125 Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14:991-997.
126 Cooke CB, Krenacs L, Stetler-Stevenson M, et al. Hepatosplenic T-cell lymphoma: a distinct clinicopathologic entity of cytotoxic gamma delta T-cell origin. Blood. 1996;88:4265-4274.
127 Macon WR, Levy NB, Kurtin PJ, et al. Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol. 2001;25:285-296.
128 Lai R, Larratt LM, Etches W, et al. Hepatosplenic T-cell lymphoma of alphabeta lineage in a 16-year-old boy presenting with hemolytic anemia and thrombocytopenia. Am J Surg Pathol. 2000;24:459-463.
129 Wlodarska I, Martin-Garcia N, Achten R, et al. Fluorescence in situ hybridization study of chromosome 7 aberrations in hepatosplenic T-cell lymphoma: isochromosome 7q as a common abnormality accumulating in forms with features of cytologic progression. Genes Chromosomes Cancer. 2002;33:243-251.
130 Abouyabis AN, Shenoy PJ, Lechowicz MJ, Flowers CR. Incidence and outcomes of the peripheral T-cell lymphoma subtypes in the United States. Leuk Lymphoma. 2008;49:2099-2107.
131 Benharroch D, Meguerian-Bedoyan Z, Lamant L, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood. 1998;91:2076-2084.
132 Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93:2697-2706.
133 Stein H, Foss HD, Durkop H, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96:3681-3695.
134 Falini B, Pulford K, Pucciarini A, et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood. 1999;94:3509-3515.
135 Pulford K, Lamant L, Morris SW, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. 1997;89:1394-1404.
136 Dogan A, Attygalle AD, Kyriakou C. Angioimmunoblastic T-cell lymphoma. Br J Haematol. 2003;121:681-691.
137 Ree HJ, Kadin ME, Kikuchi M, et al. Angioimmunoblastic lymphoma (AILD-type T-cell lymphoma) with hyperplastic germinal centers. Am J Surg Pathol. 1998;22:643-655.
138 Rodriguez-Justo M, Attygalle AD, Munson P, et al. Angioimmunoblastic T-cell lymphoma with hyperplastic germinal centres: a neoplasia with origin in the outer zone of the germinal centre? Clinicopathological and immunohistochemical study of 10 cases with follicular T-cell markers. Mod Pathol. 2009;22:753-761.
139 Roncador G, Garcia Verdes-Montenegro JF, Tedoldi S, et al. Expression of two markers of germinal center T cells (SAP and PD-1) in angioimmunoblastic T-cell lymphoma. Haematologica. 2007;92:1059-1066.
140 Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ. Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2006;30:802-810.
141 Yamane A, Awaya N, Shimizu T, et al. Angioimmunoblastic T-cell lymphoma with polyclonal proliferation of plasma cells in peripheral blood and marrow. Acta Haematol. 2007;117:74-77.
142 Smith JL, Hodges E, Quin CT, et al. Frequent T and B cell oligoclones in histologically and immunophenotypically characterized angioimmunoblastic lymphadenopathy. Am J Pathol. 2000;156:661-669.
143 Thorns C, Bastian B, Pinkel D, et al. Chromosomal aberrations in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma unspecified: a matrix-based CGH approach. Genes Chromosomes Cancer. 2007;46:37-44.
144 Marafioti T, Hummel M, Anagnostopoulos I, et al. Origin of nodular lymphocyte-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med. 1997;337:453-458.
145 Wlodarska I, Stul M, de Wolf-Peeters C, Hagemeijer A. Heterogeneity of BCL6 rearrangements in nodular lymphocyte predominant Hodgkin’s lymphoma. Haematologica. 2004;89:965-972.
146 Marafioti T, Hummel M, Foss HD, et al. Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood. 2000;95:1443-1450.
147 Deacon EM, Pallesen G, Niedobitek G, et al. Epstein–Barr virus and Hodgkin’s disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med. 1993;177:339-349.
148 Hinz M, Loser P, Mathas S, et al. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood. 2001;97:2798-2807.
149 Calvo KR, Traverse-Glehen A, Pittaluga S, Jaffe ES. Molecular profiling provides evidence of primary mediastinal large B-cell lymphoma as a distinct entity related to classic Hodgkin lymphoma: implications for mediastinal gray zone lymphomas as an intermediate form of B-cell lymphoma. Adv Anat Pathol. 2004;11:227-238.
150 Swinnen LJ. Diagnosis and treatment of transplant-related lymphoma. Ann Oncol. 2000;11(Suppl. 1):45-48.
151 Thomas JA, Crawford DH, Burke M. Clinicopathologic implications of Epstein–Barr virus related B cell lymphoma in immunocompromised patients. J Clin Pathol. 1995;48:287-290.
152 Harris NL, Ferry JA, Swerdlow SH. Posttransplant lymphoproliferative disorders: summary of Society for Hematopathology Workshop. Semin Diagn Pathol. 1997;14:8-14.
153 Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
154 Evens AM, David KA, Helenowski I, et al. Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol. 2010;28:1038-1046.
155 Foran JM, Norton AJ, Micallef IN, et al. Loss of CD20 expression following treatment with rituximab (chimaeric monoclonal anti-CD20): a retrospective cohort analysis. Br J Haematol. 2001;114:881-883.
156 Chetty R, Echezarreta G, Comley M, Gatter K. Immunohistochemistry in apparently normal bone marrow trephine specimens from patients with nodal follicular lymphoma. J Clin Pathol. 1995;48:1035-1038.
157 Douglas VK, Gordon LI, Goolsby CL, et al. Lymphoid aggregates in bone marrow mimic residual lymphoma after rituximab therapy for non-Hodgkin lymphoma. Am J Clin Pathol. 1999;112:844-853.
158 Hanson CA, Brunning RD, Gajl-Peczalska KJ, et al. Bone marrow manifestations of peripheral T-cell lymphoma. A study of 30 cases. Am J Clin Pathol. 1986;86:449-460.
159 Feugier P, De March AK, Lesesve JF, et al. Intravascular bone marrow accumulation in persistent polyclonal lymphocytosis: a misleading feature for B-cell neoplasm. Mod Pathol. 2004;17:1087-1096.
160 Alvares CL, Matutes E, Scully MA, et al. Isolated bone marrow involvement in diffuse large B cell lymphoma: a report of three cases with review of morphological, immunophenotypic and cytogenetic findings. Leuk Lymphoma. 2004;45:769-775.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 29 Lymphoma
Introduction
Peripheral blood (PB) and bone marrow (BM) are relatively frequently involved by lymphomas, particularly those with low-grade clinical behavior.1 BM aspiration and bone marrow trephine biopsy (BMTB) are currently performed at diagnosis as part of the staging of most patients with non-Hodgkin lymphomas (NHL) and in selected patients with Hodgkin lymphoma (HL). Criteria for patient selection for these procedures vary between treatment centers and are undergoing revision as techniques such as magnetic resonance imaging and positron emission tomography are refined to permit non-invasive assessment of disease spread. Evidence that BM involvement influences clinical outcome varies for different disease entities within the spectrum of lymphoproliferative disorders.2–9 BM involvement, and its extent, can be important factors in making clinical decisions concerning choice of treatment.
Lymphoproliferative diseases that present as leukemias have been considered elsewhere (see Chapter 28), as has plasma cell neoplasia (see Chapter 30). In this chapter, blood and BM involvement by lymphomas presenting primarily with lymph node or other solid organ involvement is discussed.
General comments on bone marrow (BM) examination in lymphoproliferative diseases
The full blood count, blood film, BM aspirate and BMTB have complementary roles in the investigation of BM involvement by lymphoma. In different lymphoproliferative diseases, each of these types of specimen may be of greater or lesser value.10–14 In lymphomas that do not readily display leukemic behavior, BMTB sections are frequently found to contain lymphoma when none is evident in the blood or BM aspirate.
Where circulating lymphoma cells are available in sufficient number in the peripheral blood (PB), these give the most consistent morphology and phenotyping by flow cytometry (FCM). BM aspiration without accompanying BMTB may be valuable if representation of disease in the PB is inadequate for diagnosis, permitting cytologic and immunocytochemical assessment of larger numbers of neoplastic cells. However, aspiration for the evaluation of BM involvement by lymphoma has a high false negative rate.13 Deposits of disease frequently occupy sites within the BM microenvironment that are suboptimally sampled by aspiration (e.g. paratrabecular zones). They may also be adherent to stromal components inducing focal fibrosis and less readily aspirated than BM hemopoietic elements. For these reasons BMTB in addition to aspiration is always recommended for the evaluation of BM involvement by lymphoma. Exceptionally, if BM aspiration alone is possible in some patients for whom BMTB cannot be performed, the aspirate films can still provide valuable information about hemopoietic reserve and iron stores. If aspiration proves technically difficult or a poor sample is obtained, BMTB should definitely be performed and an imprint or roll preparation made for cytologic assessment.
BMTB sections are used primarily for morphologic assessment, including analysis of the spatial distribution and extent of lymphomatous deposits.4,8–10,12,14–25 Spatial and cytologic assessment often provide clues to lymphoma subtype (e.g. the pattern of paratrabecular infiltration typical of follicular lymphoma) and can, as a minimum, be used to assess whether disease involvement represents indolent or agressive lymphoma. Histologic sections can also be used for immunohistochemistry (IHC), FISH and PCR; these additional investigations are particularly useful if PB or aspirated BM cells do not provide adequate representation of lymphoma. Where decalcification is employed in association with paraffin wax embedding of trephine cores, use of ethylene diamine tetra-acetic acid (EDTA) to decalcify by chelation, rather than acid exposure, offers excellent antigen and nucleic acid preservation.26–28 For immunophenotyping of lymphoid cell infiltrates in BMTB sections a similar, marginally more restricted, range of antibodies is employed to that used for FCM. Antibodies reactive with additional antigens such as CD79a, cyclin D1, IRF4/MUM1, PAX5 and Ki67 are also available which have been developed specifically for IHC use; these are referred to individually in the text that follows, where appropriate.
Differential diagnosis of reactive lymphoid aggregates vs lymphoma
Nodular aggregates of small lymphoid cells may be found in BMTB sections as a reactive phenomenon, unrelated to any neoplastic lymphoid proliferation. Criteria for distinguishing such aggregates from neoplastic lymphoid infiltrates remain imperfect and controversial.29–31 Immunostaining is helpful in only a minority of examples. Morphologic features remain the best guide, supported by application of molecular genetic techniques, such as PCR amplification and IGH/TCR rearrangement studies, in appropriately processed specimens. However, the sensitivity and specificity of the PCR methods employed may vary and the finding of a monoclonal IGH/TCR rearrangement cannot be assumed to equate with a diagnosis of lymphoma without other supportive evidence.32 In limited circumstances FISH performed using intact sections may be informative (consideration of t(11;14) and t(14;18), for example). For any techniques performed in addition to histological assessment of the original sections, there is a significant likelihood that small or few lymphoid aggregates will not be represented in the material available for testing.
To be accepted as reactive, lymphoid nodules should be few in number, centrally placed within intertrabecular spaces, small and round in profile with well-demarcated margins. A small capillary may be present, running from the periphery into the center of the nodule (Fig. 29.1) and there may be reactive changes such as aggregation of eosinophils in the adjacent hemopoietic tissue. An underlying meshwork of reticulin or CD23+ follicular dendritic cells may be present or absent, probably more dependent on the size of a particular nodule than on its reactive or neoplastic nature. The subjective nature of these criteria will be obvious to the reader but, to date, an objective gold standard for assessment of BM lymphoid nodules remains elusive.
The cytologic composition of lymphoid infiltrates is also critical to their interpretation. Most non-neoplastic aggregates consist of small lymphocytes with only occasional large blast cells; they show little evidence of plasma cell differentiation. Reactive germinal center formation is distinctly uncommon but, when it occurs, the composition of the lymphoid follicle recapitulates that found in lymph nodes and other organized lymphoid tissues. Formation of reactive germinal centers within BM lymphoid aggregates is said to be increased in patients with rheumatoid arthritis and other systemic chronic inflammatory disorders.32 Reactive lymphocytes, predominantly T-cells, also form a significant component of many granulomas in the BM, and the compact infiltrates of systemic mastocytosis; these lesions can be confirmed by IHC, if suspected, and should not be mistaken either for lymphoma or incidental reactive lymphoid nodules.
The laboratory investigation of blood and BM specimens suspected or known to have involvement by lymphoma
Blood
A full blood count and differential white cell count should always be performed. The blood film should be stained using May–Grünwald–Giemsa (MGG) or Wright’s method to assess cytologic features. When circulating abnormal cells are known or suspected to be present immunophenotyping by FCM should be performed; a useful basic antibody panel is shown in Table 29.1. PB cells should also be analyzed by classical cytogenetic and/or molecular genetic methods when the differential diagnosis includes lymphomas known to be associated consistently with abnormal genetic features.
Bone marrow aspirate
A minimum of three films should be prepared and air-dried, using MGG stain for cytologic assessment and differential cell count.33 A Perls stain is also desirable as a routine, for assessment of iron stores. Aspirated BM cells should also be sent in suspension for FCM immunophenotyping and genetic analysis.
Bone marrow trephine biopsy (BMTB)
Cores of tissue should be collected that will contain at least 1 cm of interpretable, uncrushed BM after histologic processing. In practice, because of likely inclusion of cortex at the outer end and crushed tissue at the inner end, plus shrinkage that occurs during processing, unfixed cores at the time of collection should be at least 1.5 cm long10,34–36 If aspiration has been unsuccessful, touch preparations should be made using the trephine biopsy core by rolling it gently between two slides and then staining both air-dried slides with MGG. A detailed discussion of technical matters is beyond the scope of this chapter and only brief comments are made here. Further guidance can be found in references.26,37 Plastic-embedded specimens (usually in methyl or glycol methacrylate resin) require modifications to be made to standard tinctorial and IHC methods that may be difficult to incorporate into automated staining schedules but a wide range of immunostains can none the less be performed in laboratories specialized to handle these specimens. After processing the core for histology, thin sections (1–2 µm for plastic-embedded specimens; 3–4 µm for decalcified, paraffin wax-embedded ones) should be cut and stained with H&E from a minimum of three levels through the core. Levels are usually cut 25–50 µm apart, depending on local practice and the diameter of the specimen. On sections from the middle or deepest level, reticulin, PAS and Giemsa stains should be performed routinely. H&E provides a basic and familiar stain with which to assess cell morphology (Fig. 29.2A). Lymphoid infiltrates are often associated with increased reticulin deposition and a disturbance of the normal pattern of reticulin fiber distribution (Fig. 29.2B). PAS stain highlights carbohydrate-rich molecules and consequently stains immunoglobulin if it is of the IgM class and sufficiently abundant. IgM is richly glycosylated; consequently, intracellular and, occasionally, extracellular accumulation of this Ig can be detected in some cases of lymphoplasmacytic lymphoma (Fig. 29.3). The key importance of high-quality Giemsa staining in trephine biopsy sections cannot be over-emphasized (see Chapter 3). Any collection of lymphoid cells will stand out as being turquoise/blue in color, against a generally mauve/pink background (Fig. 29.1). IHC can be useful in confirming the precise diagnosis of lymphoma and in assessing its grade but is of limited value in differentiating reactive lymphoid aggregates in the BM from small deposits of low-grade lymphoma. For the latter task, molecular genetic analysis is gaining importance.30,32
The World Health Organization (WHO) classification of lymphomas
At the end of 1999, an outline version of a new lymphoma classification was published as a result of the WHO lymphoma classification project.38 This embodied the principles of the revised European–American lymphoma (REAL) classification,39 which was published in 1994 and has since become widely accepted in lymphoma diagnostic practice. A revision of the WHO classification was published in 2008 (Box 29.1).40
Box 29.1
Summary of the revised World Health Organization classification of lymphomas (2008)40
B-cell neoplasms
T- and NK-cell neoplasms
Immunodeficiency-associated lymphoproliferative disorders
(Italic text denotes entities regarded as provisional in the WHO 2008 Classification)
A novel and fundamental principle of the REAL classification was the definition of lymphomas according to their distinctive clinical, as well as pathologic, features. Much of the REAL classification remains little changed by the WHO, except for alterations in terminology, but considerable advances in the immunophenotypic and genetic contributions to defining lymphoma types have been incorporated. Some lymphomas considered as provisional entities in the REAL system became accepted as definite entities within the initial WHO classification, and this process, including the addition of new provisional entities, has continued with the 2008 revision. Box 29.1 summarizes the lymphoma categories recognized by the WHO 2008 scheme. Areas of uncertainty in lymphoma classification remain, however, and an important feature of the WHO classification is that it has flexibility to allow for further evolution in the understanding of hematological malignancies. An important advance in the WHO system over previous classifications is the recognition of subtypes of lymphoma that tend to exhibit leukemic behavior (see Chapter 28). Emerging information about the mutational status of immunoglobulin variable region genes in B-cell lymphomas41–46 and gene expresssion micro-array patterns across a wider spectrum of lymphomas,47–59 are likely to permit refinement of the classification as the biological significance of such data becomes clearer. However, while immunophenotypic, cytogenetic and molecular genetic features are central to the WHO classification, with expanded importance in the 2008 revision, little reference is made to the interpretation of patterns of BM involvement as a contribution to the definition of lymphoma entities.
Blood and BM involvement of lymphoma entities according to WHO classification
Description of entities primarily presenting as leukemias is provided in Chapter 28. Features of blood and BM involvement of lymphomas primarily presenting in lymph nodes or as extranodal infiltrates are described below.
Mature B-cell neoplasms
Lymphoplasmacytic lymphoma (LPL)
Clinical features, blood and bone marrow aspiration
Lymphoplasmacytic lymphoma is an indolent lymphoid neoplasm comprising 1–2% of all NHL.40,60–62 Studies in southern Europe have suggested an association between hepatitis C (HCV) infection and LPL,63–65 with treatment of the former being effective in controlling the lymphoma, but no similar association with HCV has been found elsewhere.
BM involvement is common at presentation and 30% of patients have splenomegaly and/or lymphadenopathy. The clinical presentation usually reflects the presence of a circulating paraprotein (IgM in almost all cases) with or without hyperviscosity symptoms. BM involvement by LPL with an IgM paraprotein underlies the clinical syndrome of Waldenström macrogobulinemia (see Chapter 30). There may be an associated peripheral neuropathy that is believed to be a paraneoplastic phenomenon.62 Pancytopenia may be present as a consequence of tumor burden such as BM failure or fibrosis. Blood involvement is rare, except in advanced disease; it is characterized by the presence in the circulation of plasmacytoid cells with an eccentric nucleus and basophilic cytoplasm. BM aspirate films show a mixture of small lymphocytes, lymphoplasmacytoid cells and mature plasma cells (Fig. 29.4). There is frequently an accompanying increase in mast cells. Typical immunophenotype findings are summarized in Table 29.1.
Bone marrow trephine biopsy
Bone marrow is frequently the predominant site of disease involvement in LPL. BMTB sections show infiltrates in most patients, often with more extensive involvement than suggested by aspirate films.10 The cells are predominantly small lymphocytes with varying numbers of plasma cells and cells having intermediate (plasmacytoid) features. The plasma cells may contain Dutcher bodies, which are inclusions of immunoglobulin that invaginate the nuclear membrane and appear intranuclear in histologic sections (Fig. 29.5). Less commonly, single or multiple intracytoplasmic immunoglobulin inclusions (Russell bodies) are found. Scattered lymphoid blast cells may be seen but true para-immunoblasts are absent and no proliferation centers are formed; finding the latter would indicate a diagnosis of B-cell chronic lymphocytic leukemia (B-CLL) with plasmacytic differentiation, rather than LPL. The presence of increased numbers of reactive mast cells in the marrow interstitium, sometimes located preferentially in the periphery of lymphoid infiltrates, may be helpful in supporting a diagnosis of LPL (Fig. 29.6). This phenomenon, however, is also seen in a minority of cases of CLL and is possibly related to IgM expression rather than to other properties of either disease.62,66 It has been suggested that mast cells contribute to B cell proliferation in LPL.67,68 The pattern of infiltration is usually irregular, paratrabecular or diffuse throughout the interstitium; mixtures of these patterns are common in individual cases. Well-defined nodular infiltrates are unusual and when nodules occur in LPL they are usually small and more elliptical or irregular than those seen in other small B-cell lymphomas. The paratrabecular infiltrates of LPL are not usually as extensive or regular as those found in follicular lymphoma. In some patients who have an IgM paraprotein, sinusoids contain PAS-positive proteinaceous material that represents plasma rich in IgM; there may also be interstitial and, rarely, intracytoplasmic deposition of crystalline PAS-positive IgM (Fig. 29.3). IHC shows that the small B-lymphocytes of LPL express CD19, CD20 (which may be weak or absent in cases with prominent plasma cell differentiation), CD79a and PAX5 but lack expression of CD5, CD10, BCL6 and cyclin D1. In occasional cases, a proportion of the neoplastic cells expresses CD23. Plasma cell differentiation is often evident with staining for CD79a and can be demonstrated more clearly using antibodies reactive with CD138 or IRF4/MUM1, or antibody VS38c that reacts with rough endoplasmic reticulum-associated p63 protein.69 Expression of monotypic immunoglobulin in the cytoplasm of cells showing plasmacytic differentiation is usually easily demonstrated, most commonly IgM kappa,40,70 and light chain mRNA production by such cells can also be shown by in situ hybridization (Fig. 29.7). Transformation to large cell lymphoma may occur but is not very common.
Genetic studies
Initial studies reporting a t(9;14)(p13;q32) IGH/PAX5 translocation in association with LPL70,70 have not been confirmed and no other consistent genetic associations have been identified. A variety of trisomies has been found, of unknown significance, and del(6q) may be associated with an adverse prognosis.71 Immunoglobulin heavy chain genes typically show hypermutation without evidence of ongoing acquisition of further mutations.72
Hairy cell leukemia (HCL)
Clinical features and pathology in the spleen
Hairy cell leukemia is a rare disease accounting for 2% of leukemias and predominantly affecting middle-aged men. The clinical presentation is with anemia, bleeding or infection (often with opportunistic organisms), reflecting PB cytopenias caused by hypersplenism and/or BM failure due to fibrosis associating tumor infiltrates.40 At presentation 60% of patients have splenomegaly and 40% hepatomegaly. In the spleen, HCL is recognized by the presence of a diffuse infiltrate of typical hairy cells (see below), causing effacement of normal red and white pulp architecture. As in the BM, splenic infiltration is accompanied by reticulin deposition, interstitial hemorrhage, sinusoidal vascular ectasia and peliosis-like disruption.73
Blood and bone marrow aspiration
Most patients with HCL have circulating hairy cells although numbers are typically low. These are medium-sized to large lymphoid cells which have abundant, weakly basophilic cytoplasm and hair-like projections from the cell surface.74 The nucleus is frequently indented and has a smooth chromatin pattern with indistinct nucleoli. Typical immunophenotyping results are summarized in Table 29.1. Peripheral cytopenias are common in HCL, particularly neutropenia and monocytopenia. BM aspiration is often unsuccessful due to increased marrow reticulin associated with HCL infiltration; when neoplastic cells are obtained, they are essentially identical in morphology to those found in PB. They may be accompanied by reactive mast cells and plasma cells. In touch preparations from BMTB, HCL cells may lack typical ‘hairy cell’ cytology but present as lymphatic cells with abundant cytoplasm.
Bone marrow trephine biopsy
The degree of BM involvement is usually extensive at presentation, with large areas showing almost complete replacement of normal hemopoiesis by infiltrating hairy cells and partial or complete loss of fat spaces. Occasionally, the BM appears hypoplastic, with a subtle pattern of diffuse interstitial infiltration and partial preservation of hemopoiesis; granulopoiesis is often disproportionately reduced and this picture should not be confused with hypoplastic/aplastic anemia or a hypoplastic myelodysplastic syndrome (Fig. 29.8A). Subtle intrasinusoidal infiltration may also occur.25 CD20 staining is essential to determine the extent of infiltrates (Fig. 29.8B). In almost all cases of HCL, interstitial reticulin is greatly increased (Fig. 29.9), which is rarely the case in other hypoplastic states. The infiltrating cells are of medium size with round, oval or bilobed nuclei and abundant, empty-looking cytoplasm (Fig. 29.10). Occasionally, they appear spindle-shaped. The abundant cytoplasm gives an appearance of the cells being widely spaced from one another. Extravasation of red cells into the interstitium is common and sinusoids appear prominent, with gaping lumens, as a result of the background of increased reticulin fibers (Fig. 29.11). Collagen fibrosis is rare. Osteosclerosis may also occur rarely in association with HCL and may regress with treatment.75,76 Reactive mast cells are typically abundant throughout BM infiltrates and polytypic plasma cells may also be increased. Transformation of HCL to a more aggressive, large B-cell lymphoma occurs infrequently.77
In BMTB sections, hairy cells can be shown by IHC to express CD20, CD79a and CD45RA but not CD5 or CD23. Expression of tartrate-resistant acid phosphatase (TRAP) can usually be demonstrated in extensive infiltrates78,79 although technical performance of anti-TRAP monoclonal antibodies varies and subtle HCL involvement may not be visible against background weak staining of hemopoietic cells. In many cases, hairy cells show heterogeneous, usually weaker than in mantle cell lymphoma, nuclear expression of cyclin D1 and, in a minority of cases, they are positive for CD10.80 Dot-like cytoplasmic expression of CD68 may also be seen. Hairy cells are strongly positive for CD25 and CD123. Hairy cells react well with the monoclonal antibody DBA44,79,81 which recognizes CD7282 a B-cell surface antigen strongly associated with hairy cells but not entirely specific. Annexin A1 and T-cell associated transcription factor T-bet offer further and, to date, highly specific additional markers for HCL.83,84
After treatment, HCL infiltration usually appears dramatically reduced and the increased reticulin resolves rapidly in most patients.75 Assessment of residual disease during and after therapy can be difficult in HCL, in which small interstitial clusters of scattered neoplastic cells may be all that remain; IHC usually reveals more disease than is readily apparent from standard tinctorial stains.
Genetic studies
Knowledge of genetic abnormalities associated with HCL is extremely limited and no consistent genetic associations have been found that appear causative. Immunoglobulin heavy chain genes are hypermutated but stable, without evidence of ongoing mutation. Partial understanding has been gained of genetic abnormalities underpinning the unusual properties of hairy cells;85 over-expression of the activator protein-1 (AP-1) transcription factor drives the expression of CD11c, resulting in some of the unusual stromal interactions and adhesive properties of HCL cells. Up-regulation of AP-1 is secondary to RAS activation in HCL which, in turn, may be due to reduced expression of RhoH that normally competes with Ras proteins at GTP binding sites. Reconstitution of RhoH expression in a mouse xenograft model of HCL reduced proliferation of neoplastic cells and prolonged survival.86
HCL variant (HCLv)
Clinical features and pathology in the spleen
HCLv is a rare disease with incidence of 3 cases per 1 000 000. It may be more common in Asian countries. It has been recognized as a provisional entity in the WHO 2008 classification;40 despite its name, it is clinically, immunophenotypically and genetically distinct from HCL.87,88 Patients typically present with splenomegaly; anemia is common but neutropenia and monocytopenia are rare. HCLv shows overlap between HCL and B-cell prolymphocytic leukemia (B-PLL) in their pattern of splenic involvement, with predominant diffuse red pulp involvement.40
Blood and bone marrow aspiration
There are usually abundant circulating neoplastic cells in HCLv with WBC usually in the range of 20–40 × 109/l. Although HCLv cells have cytoplasmic projections as in HCL, they are larger and have nucleoli more akin to those seen in B-PLL. BM aspiration is usually successful because HCLv is not associated with significantly increased reticulin fiber production in the marrow stroma. The immunophenotype of cells in HCLv is distinct from that found in HCL (see Table 29.1); they are generally negative for CD25, CD123 and annexin A1, with weak or absent TRAP expression. They show CD11c, CD103, FMC7 and strong sIg reactivity as in HCL.
Splenic marginal zone B-cell lymphoma (SMZL)
Clinical features and pathology in the spleen
This disease accounts for fewer than 2% of lymphoid neoplasms and occurs mainly in older individuals, affecting men and women equally.40 An association with hepatitis C has been noted in southern Europe.63
Within the spleen, there is widespread, generally uniform, involvement of the white pulp, giving a miliary appearance to the cut surfaces of splenectomy specimens.90 The germinal centers and mantles of white pulp nodules are atrophic and replaced by densely clustered small lymphoid cells surrounded by expanded marginal zones of more mixed composition. Cells in the marginal zones are predominantly slightly larger, lymphoplasmacytoid or monocytoid cells with more cytoplasm than the lymphocytes present centrally within involved nodules. Within the marginal zones there are also scattered immunoblast-like cells in varying proportions. Small satellite collections of marginal zone-type cells are frequently also present surrounding red pulp capillaries, and are often accompanied by small collections of epithelioid macrophages.73 There may be diffuse infiltration of red pulp cords and sinusoids by the small lymphoid cells. Appearances in splenunculi, when present, are identical to those in the main spleen; hilar lymph nodes show vaguely nodular replacement of follicles by small lymphocytes, usually without morphological evidence of marginal zone differentiation.
Blood and bone marrow aspiration
In PB, neoplastic cells in SMZL are slightly larger than normal lymphocytes, with a round nucleus and mature chromatin pattern. They have a relatively large volume of weakly basophilic cytoplasm and typically exhibit polar villi. BM involvement is almost always present and similar cells are usually readily apparent in aspirate. The immunophenotype (Table 29.1) shows usually positivity for CD19, CD20, surface IgM and in most cases IgD, no CD5 or CD10. CD23 and CD11c are positive in a fraction of cases. Cyclin D1 and Annexin A1 are negative.
Bone marrow trephine biopsy
There is usually nodular, interstitial and, less regularly, paratrabecular infiltration by neoplastic cells.10,91 Intrasinusoidal infiltration is often an additional finding, demonstration of which may require IHC (Fig. 29.12). In occasional patients, a subtle and purely intrasinudoidal infiltrate may be present, requiring careful distinction from persistent polyclonal B-cell lymphocytosis (see below). A diffusely infiltrated, packed marrow is found only in rare patients.
Genetic studies
These neoplasms have clonally rearranged IgH and approximately 50% show hypermutation but evidence of ongoing acquisition of mutations is rare.92 Trisomy 3q has been found in a high proportion of cases, regarded as a late or secondary event in the neoplasm development and of uncertain biological relevance. The occurrence of translocations or allelic losses involving 7q31-32 and 7q21 is believed to be more significant. The latter result in dysregulation of CDK6.93 Microarray and CGH analysis suggests a distinctive profile involving up-regulation of gene expression within the AKT1 and B-cell receptor signaling pathways.94,95 Of note, t(11;18), found in a high proportion of MALT-type marginal zone lymphomas, is absent from SMZL.
Splenic diffuse red pulp small B-cell lymphoma
Clinical features and pathology in the spleen
This lymphoma has been introduced as a provisional entity in the 2008 revision of the WHO classification40 and has been described in patients predominantly of middle age and older, with men and women equally affected. Its clinical behavior is indolent.
Blood and bone marrow aspiration
Circulating lymphocytes often have villous processes and unusually basophilic cytoplasm.96 They may be present only in low numbers. Similar cells are present in BM aspirate. Their imunophenotype resembles that of SMZL but sometimes overlaps with HCLv and increasing experience of these two provisional entities may in future reveal genuine biological overlap between them.
Genetic studies
Hypermutation patterns of IGH and the spectrum of variable region gene usage differ from those found in SMZL and resemble HCL in some cases. A translocation, t(9;14), involving PAX5 and IGH has been found in some patients and genetic alterations associated with splenic, nodal and extranodal marginal zone lymphomas are absent.40,97
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT)-type
Clinical features and pathology at presenting sites
Extranodal marginal zone lymphomas of MALT-type are indolent lymphoproliferative disorders with a 10-year survival of more than 80%.60,61 The most common primary sites of MALT-type extranodal marginal zone lymphoma are within the gastrointestinal tract. Salivary glands, skin, orbit, lung and urogenital organs are the sites of origin in smaller numbers of patients. At presentation, 30% of patients have involvement of more than one mucosal site but lymph node spread is usually absent or localized to nodes close to the mucosal site(s) of involvement. As defined in the WHO classification, these lymphomas are low grade and arise in the context of normal or induced lymphoid tissue in the organs involved (e.g. that caused by infection with Helicobacter pylori in the stomach or Chlamydia psittaci in the orbit).40 The neoplastic cells are typically centrocyte-like, with a tendency to infiltrate epithelial structures and form lympho-epithelial lesions; they show monocytoid or plasmacytic differentiation to varying extents in different individuals.
Blood and bone marrow aspiration
Bone marrow involvement has been reported in 15–40% of cases.98 Fifteen per cent of patients are anemic at presentation but circulating lymphoma cells are not seen. Bone marrow aspiration rarely reveals the presence of lymphoma cells, even when BMTB shows histologic evidence of involvement. Immunophenotype is similar to other marginal zone lymphomas, as summarized in Table 29.1.
Bone marrow trephine biopsy
When present, infiltrates of extranodal marginal zone lymphomas of MALT-type are usually nodular but paratrabecular and interstitial involvement has been described.99
[/not-level-membership-for-pathology-category]
