Schematic representation of the course of lung function for persons with asthma of different severities as compared with persons without asthma. Both lung function at birth and deficits occurring after birth play a role in determining the course of the disease for a lifetime.
Newborn cohorts have also allowed for a detailed study of the natural history of asthma and asthma-related syndromes during childhood. The picture that emerges is one of marked heterogeneity. During the first years of life, many children have recurrent asthma-like symptoms (wheeze, cough, shortness of breath), especially during viral infections, but not all go on to develop the chronic, persistent form of the disease that that is more often associated with the label of asthma. These children have been dubbed “transient early wheezers,” and they are clinically undistinguishable in terms of severity and frequency of symptoms, from “persistent wheezers,” that is, those whose symptoms start before the age of three but are still present in the early school years. Interestingly, transient wheezers were found to have significantly lower levels of lung function measured shortly after birth than children who did not wheeze during the first years of life (12). These children’s lung function also improved concomitantly with the remission of their symptoms, but was still significantly diminished up to early adult life. It is thus plausible to surmise that the congenital airflow limitation present in these children may be causally involved in the pathogenesis of their disease. Because resistance to flow in a tubular structure is a function of the fourth power of the radius and normal infants have narrower airways, relative to the size of their lungs, than older children (13), any further narrowing of the airway would have significant functional consequences for the affected child. The improvement in lung function that these children show with aging could also explain why their symptoms are transient and do not persist beyond the early years.
In summary, the factors that control the level of lung function with which a child is born may play a critical role in determining that child’s susceptibility for the development of both transient and persistent airway obstruction, starting in early life.
Tracking of Lung Function with Age
The availability of cohorts in which lung function has been followed from birth and up to the adult years has provided further support to the concept that factors active in utero may play a critical role in determining the potential level of lung function that a subject may attain into adult life. It has been known for years that lung function tracks markedly during the school years (14), but the role of prenatal versus postnatal influences had not been elucidated. Using data from the Tucson Children’s Respiratory Study, Stern et al. (15) showed that up to 14% of the variance in measurements of airway function in young adults, including FEV1 and FEV1/FVC ratio, was explained by the maximal flows at functional residual capacity, an index of airway function from partial expiratory flow-volume curves, measured in the same subjects at a mean age of two months. The study excluded premature children who required medical attention as newborns. A more detailed analysis of the data suggested that most of the association between infant airway function and adult lung function was attributable to subjects who had already diminished airway function shortly after birth. Individuals who were in the lowest quartile for lung function shortly after birth had mean predicted FEV1/FVC ratios of 75.1% at age twenty-two years, a level that was 5.2% lower than mean values for subjects in the other three quartiles. The authors suggested that this could indicate that subjects with airflow obstruction at birth are those more likely to remain in the lowest end of the distribution until early adult life, whereas tracking of airway function may be less evident in children with normal airways. Thus, combinations of genetic and environmental factors that strongly affect lung growth in utero may have deleterious effects that last into adult life.
COPD and Development of Lung Function
COPD is a disease mostly affecting older adults that is both defined and classified into different stages of severity predominantly based the level of lung function after the administration of a bronchodilator (3). Specifically, for example, subjects who have an FEV1/FVC ratio <70% and an FEV1 that is less than 80% of the value predicted for their sex, size, and age are classified as having stage 2 COPD, stage 1 being mostly a preclinical phase in which only FEV1/FVC is abnormal. Until recently, COPD was considered mostly a disease due to smoking, based on the fact that the great majority of affected individuals in the developed world are indeed smokers. Because only 15–50% of smokers develop COPD, the most accepted hypothesis about the pathogenesis of the disease was that certain subjects were more sensitive than others to the direct toxic effects of cigarettes. Thus, the assumption was that patients with COPD reached early adult life with essentially normal lung function, and that most of their airflow limitation was the result of subsequent accelerated decline in lung function (curves (c) and (d), Figure 16-2). However, recent longitudinal studies of COPD have challenged the idea that this is the only course that the disease can take. In the ECLIPSE study, for example, more than half of patients with COPD followed for a period of three years showed no more decline in FEV1 than that which is observed in subjects without lung disease (16). If COPD is not invariably a progressive disease, the origins of the airflow limitation present in those who do not show excessive lung function decline remained undefined.
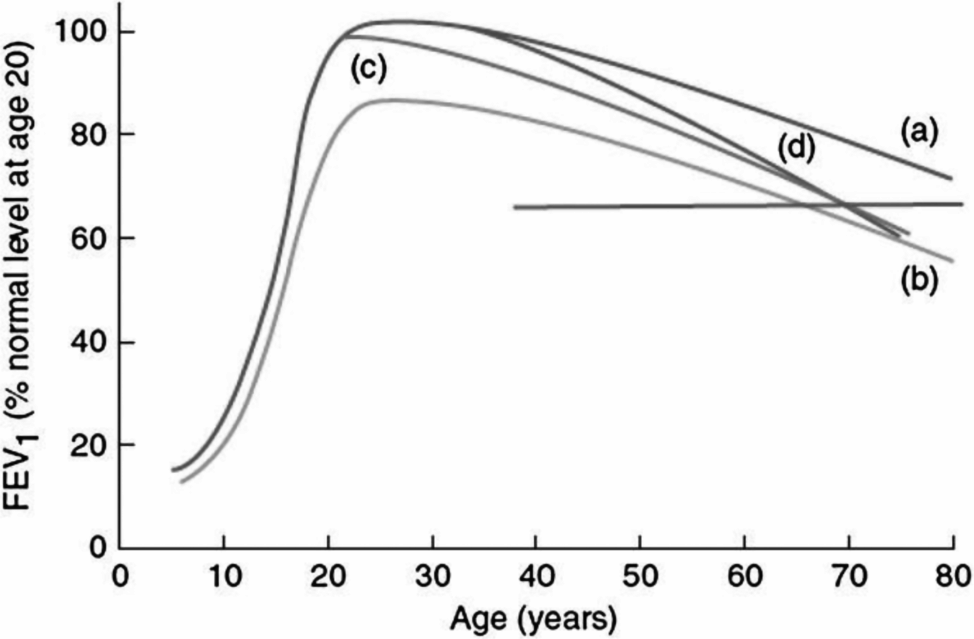
Different pathways to chronic obstructive pulmonary disease (COPD). (a) Normal course of lung function growth and decline in subjects without COPD. (c, d) Premature and late accelerated decline in lung function, mainly associated with active cigarette smoking in subjects who reach early adult life with normal levels of lung function. (b) The early origins of COPD hypothesis: a critically low level of lung function (horizontal line) is achieved by subjects who show normal decline in lung function with aging, but who reached only a fraction of the peak of lung function at age twenty years
One potential explanation could be that genetic and environmental factors that control the development of lung function in utero and during childhood and, therefore, determine the maximum level of lung function attained in adulthood, play an important role in the pathogenesis of COPD. This “early origins” hypothesis (represented in curve (b), Figure 16-2) postulates that many smokers with COPD develop the disease not because of excessive decline in lung function as adults, but because they reach early adult life with a certain degree of irreversible airflow limitation, which in most cases is still asymptomatic. When the normal process of lung function decline takes place, these subjects are at increased risk of reaching the critical level of airway obstruction that defines the different stages of COPD and are also more likely to develop respiratory symptoms associated with smoking, thus fitting the typical clinical presentation of the disease. All this is without the need to show any additional decline of lung function with aging beyond what is characteristic of this age group.
The data from ECLIPSE and other recent longitudinal studies suggest that up to half of all patients with COPD may follow this “early origins” trajectory to the inception of the disease. It is thus critical to understand what determines the level of lung function reached in early adult life.
Genetics of Lung Function
There is now solid evidence indicating that levels of lung function, adjusted for age, sex, and height, are genetically controlled. Heritability of lung function, that is, the fraction of phenotype variability that can be attributed to genetic variation, can be calculated both from hundreds of thousands of single-nucleotide polymorphisms (SNPs) that span the whole genome and that have been widely used to study genetic association with a large number of human phenotypes, and from segregation of lung function within pedigrees. Similar heritability coefficients (0.50 for FEV1 to 0.66 for FEV1/FVC) were reported for genome-wide association studies (GWAS)- and pedigree-based studies when both methods have been assessed in the same population (17). A meta-analysis of the results for GWAS for spirometry performed in four cohorts for a total of 20,890 individuals of European ancestry (18) identified eight loci associated with FEV1/FVC: the gene for hedgehog interacting protein (HHIP) on chromosome 4q31, which is known to participate in developmental processes; the gene for a G protein-coupled receptor, GPR126 on chromosome at 6q24.1, known to be involved in the genetics of stature (19), and thus possibly resulting from insufficient adjustment of the results for height; a gene for a member of the “a disintegrin and metalloprotease” (ADAM) family of membrane-anchored glycoproteins that control cell-matrix interactions and help regulate growth and morphogenesis, ADAM19 at 5q33.3; a locus between the genes for the advanced glycosylation end product (AGER) and Palmitoyl-Protein Thioesterase 2 (PPT2) at 6p21; a gene of unknown function, FAM13A at 4q22.1; a gene for Protein patched homolog 1, PTCH1, which is also a hedgehog interacting protein, at 9q22.32; and genes for phosphotyrosine interaction domain containing 1 (PID1) at 2q36.3 and 5-hydroxytryptamine (serotonin) receptor 4 (HTR4) at 5q31-33. One locus associated with FEV1 contained several genes and was located at 4q24. A second meta-analysis of GWAS results from the SpiroMeta consortium (20), which included 20,288 individuals of European ancestry, replicated many of the loci described earlier, and described one additional locus associated with FEV1/FVC ratio, thrombospondin type I domain containing 4 (THSD4) at 15q23, which interestingly is also related to the ADAM family.
Subsequent to the meta-analysis described earlier, an even larger GWAS assessed genetic determinants of FEV1/FVC ratio and FEV1 in 48,201 individuals of European ancestry with follow-up of the top hits in up to an additional 46,411 individuals (21). This study identified an additional sixteen loci associated with lung function and replicated many of the loci reported earlier.
Both children and adults were included in these two very large studies, and results were not confounded by smoking, which suggests that the genes identified most likely regulate lung functional growth during fetal life and the growing postnatal years. However, as is true for almost all complex human phenotypes, the loci identified explain only a small fraction of the variability of lung function in the population. In the Soler–Artigas study, which involved almost 100,000 persons, the authors calculated that the twenty-six newly identified and replicated loci explained approximately 3.2% of the additive polygenic variance for FEV1/FVC and 1.5% of the variance for FEV1 (21). Using statistical tools that estimate the number of susceptibility loci and the distribution of their effect sizes for a trait on the basis of discoveries from existing GWAS, the authors calculated that there were an additional seventy-six putative undiscovered variants, which, when added to the other twenty-six loci, collectively explain around 7.5% of the additive polygenic variance for FEV1/FVC and 3.4% of the variance for FEV1. Clearly, even when extremely large numbers of subjects are studied, a large gap remains between the heritability calculations described earlier (i.e., 50–66% of variation in lung function explained by genetic variation) and the low fraction of that variation detected through GWAS.
Several explanations have been proposed for this gap, the so-called hidden heritability. It is quite likely that, much like is the case for stature, lung function is a highly polygenic condition, and, therefore thousands of common and rare genetic variants with very small effects may be at play. If effects of common variants included in GWAS are too small, there would be very little power to detect them with a few thousand cases and controls. In the case of rare variants, which are usually not included in GWAS chips, the only way to thoroughly address their role is to sequence the whole genomes of large numbers of individuals, which until recently was not feasible due to the high costs of such an enterprise. Fortunately, costs of sequencing are rapidly decreasing, and it is thus to be expected that studies of the association between rare variants and lung function will become available in the near future.
As explained earlier, adult lung function is determined by a combination of the level of lung function present at birth and the deficits in lung function growth that emerge during the different phases of postnatal development and senescence. GWAS of lung function have included mixed populations of children and adults, and it is thus not possible to know if the genetic variants identified in these studies are determinants of the “basal” lung function that tracks with age from birth or of the deficits in lung function growth (or accelerated decline) occurring in postnatal life. This issue is a complex one to address because it requires the availability of a rather large population in which both newborn and postnatal lung function has been reliably tested. Kreiner-Møller et al. (22) measured lung function at birth and at seven years of age in 284 children of asthmatic mothers and assessed the association between indices from these measurements and a composite score of genetic risk, calculated based on some of the reported SNPs for adult FEV1/FVC ratio and FEV1 described earlier, as the number of risk alleles weighted on the effect size reported in the original adult studies. They reported that the genetic risk scores were not significantly associated with lung function measures at age one month, but the genetic risk score for adult FEV1/FVC was significantly associated with reduced forced expiratory volume at 50% of vital capacity (FEF50) at age seven years and similarly with reduced growth in FEF50 from birth to age seven years. These results thus suggested that a significant proportion of the variance explained by the GWAS studies may reflect deficits of lung functional growth that occur after birth. However, this result may be influenced by the fact that all children in this study had mothers with asthma, and it is thus plausible to surmise that these children may be more likely to have asthma, with associated deficits in lung functional growth associated with the disease. Larger studies involving unselected general populations are needed to further elucidate these complex issues.
It is also still possible that there are common genetic variants with meaningful effects that are not detected by the current GWAS chips. This could be the case for common variants that are not included and are not correlated (in technical terms: not in “linkage disequilibrium”) with the large number of SNPs included in those chips. An interesting example is vascular endothelial growth factor A (VEGFA). VEGFA is known to play a major role in the regulation of airway and alveolar growth in utero. Reduction of VEGF expression during fetal life is known to impair lung microvascular, airway, and airspace maturation in mice (23,24). Simpson et al. reported that genetic variants not present in available GWAS chips, and not correlated with variants included in those chips, were associated with lung function measured shortly after birth, during childhood, and up to adult life (25). The most strongly associated variant, rs305028, appeared to alter splicing and affect the balance between the inhibitory (VEGF-A165b) and active (VEGF-A165a) isoforms of the molecule. In summary, genetics plays a major role in determining the distribution of lung function in the general population, and these effects can be detected even shortly after birth.
Prematurity and Lung Function
There is increasing evidence that premature birth is associated with significant respiratory sequelae, both in terms of respiratory morbidity and long-term alterations in lung function. Data from a small number of adolescents and young adults born before 1973 and who developed bronchopulmonary dysplasia (BPD) showed that 68% had airway obstruction, as demonstrated by an abnormally low FEV1, and 24% had fixed airflow limitation (26). A Dutch study followed forty-two children born in 1983 with a gestational age of less than thirty-two weeks and/or a birth weight under 1,500 g up to the age of nineteen years (27). As compared to controls, former premature subjects had significantly lower FEV1 and a decreased diffusing capacity. Although the authors claimed that there were no differences in lung function between preterms that had or did not have a history of BPD, the numbers of subjects were probably too small to justify definitive conclusions (28). In a study of all singleton infants born in Sweden in 1973–1979 (n = 622,616) and followed up to early adult life, Crump et al. (29) reported that those born extremely premature (twenty-three to twenty-seven weeks gestation) were 2.4 times more likely (adjusted 95% CI: 1.41–4.06) to be prescribed asthma medications in 2005–2007 than those who were born at term. No association was found between later preterm birth (twenty-eight–thirty-two or thirty-three–thirty-six weeks gestation) and asthma medications in young adulthood.
Subjects included in these three studies were born before the availability of surfactant for therapeutic use. More recent surveys have assessed long-term sequelae of current graduates of neonatal intensive care, in whom BPD is most often observed in extremely premature infants who have been treated with surfactant and also with gentler ventilatory regimens than in the past. Anatomical studies indicate that this so-called “new BPD” is characterized by decreased incidence of airway fibrosis (30), suggesting the possibility that the long-term impairment in airway function in these children may be less severe than that observed in former preterms from the presurfactant era. Friedrich et al. (31) showed that healthy infants born prematurely had decreased forced expiratory flows and normal forced vital capacities during the first and second years of life. Based on their finding that increase in lung function with growth was similar to those observed in full-term children, they speculated that premature birth is associated with altered lung function development that may persist beyond the first years of life. Fawke et al. (32) reported the results of lung function measurements at age eleven in 182 participants enrolled in the EPICure Study, which included all children born at or less than twenty-five full weeks of gestation in the United Kingdom and Ireland between March and December 1995. They found that, as a group and compared with appropriate controls, these children had significantly lower mean ± SE prebronchodilator FEV1 (83 ± 14% predicted vs. 100 ± 12% predicted) and FEV1/FVC ratio (86 ± 0.1% vs. 79 ± 0.1%).
Interestingly, alterations were most severe among participants with prior BPD (71% of entire cohort): mean ± SE values were 80 ± 13% predicted and 78 ± 10% for FEV1 and FEV1/FVC ratio, respectively. Reports of asthma-like symptoms were also higher in all preterms, but especially among those with BPD. These results have been recently confirmed by a longitudinal study of children with BPD followed up to a mean age of 9.5 years (33), which also showed a mild restrictive component (i.e., a significant decrease in FVC) in these children.
A recent meta-analysis (34) assessed all available studies in which FEV1 was assessed in later life in preterm-born subjects, with or without BPD, compared with term-born controls. Fifty-nine studies were included. Former preterm-born subjects without BPD, those with BPD with supplemental oxygen requirement at twenty-eight days, and those with BPD with supplemental oxygen dependency at thirty-six weeks postmenstrual age had deficits in size-adjusted FEV1 of 7.2%, 16.2%, and 18.9%, respectively, when compared with term-born controls. Pooled mean % predicted FEV1 for all subjects included in the different studies was 91% (95% confidence interval 88.8% to 93.1%) for preterm born subjects without BPD, 83.7% (80.2% to 87.2%) for BPD with supplemental oxygen requirement at twenty-eight days, and 79.1% (76.9% to 81.3%) for BPD with supplemental oxygen dependency at thirty-six weeks postmenstrual age.
Few studies have studied lung function longitudinally to determine if there is any improvement during childhood in children with different degrees of prematurity. Filbrun et al. (35) assessed lung function on two occasions in preschool children with a history of BPD. They confirmed that these children had a marked obstructive spirometric pattern with, in addition, a modest restrictive component. In the group as a whole, maximal flows and volumes tracked with age, thus suggesting no apparent catch-up growth in lung function. However, children with above average somatic growth during the interval between the two tests showed improvement in both restrictive and obstructive pattern as compared with their peers with less somatic growth. This finding strongly supports the contention that aggressive postnatal nutritional care may have a favorable effect on the respiratory outcome of children with a history of BPD (36). Using data from the Avon Longitudinal Study of Parents and Children (ALSPAC), spirometric indices were compared at eight to nine years of age (n = 6705) and fourteen to seventeen years of age (n = 4508) between term-born children and those born after twenty-five to thirty-two, thirty-three to thirty-four, and thirty-five to thirty-six weeks gestation, respectively (37). No significant deficits in any lung function measurements were found among children born after thirty-five to thirty-six weeks at either age eight to nine or fourteen to seventeen years. At eight to nine years of age, FEV1, but not FVC, was significantly lower, in the twenty-five- to thirty-two-week gestation group compared with the term group. At this same age, FEV1 and FVC were significantly lower in the children born after thirty-three to thirty-four weeks gestation as compared with the term group. Mean deficits compared with term infants after adjustment for age, gender, and height were 143 mL for FEV1 and 98 mL for FVC in the thirty-three- to thirty-four-week gestation group and 121 mL for FEV1 and 65 mL for FVC in the twenty-five- to thirty-two-week gestation group. At fourteen to seventeen years, FEV1 and FVC were no longer significantly different in the thirty-three- to thirty-four-week gestation group as compared with term infants. Implied in this finding is the possibility that the lung function deficits observed early in life in these late preterm children could improve with time. In addition, the finding that both FEV1 and FVC were affected suggested that prematurity may be not only be associated with an obstructive respiratory defect but also with a restrictive one.
It is plausible to surmise that, as is the case for other young adults who do not reach their highest possible level of lung function in early adult life, adults born extremely premature should also be more susceptible to the effects of active cigarette smoking and other harmful exposures. Surprisingly, there are no studies assessing the specific effects of either smoking (passive or active) or air pollution on rates of decline in lung function among extremely premature children.
In summary, and given that lung function tracks markedly with age, it is likely that a large proportion of extremely premature infants, and especially those with BPD, will reach adult life with 10–20% lower levels of FEV1 as compared to their age peers. With the normal decline of lung function that occurs with aging, these children are thus at high risk of reaching the critical level of airflow limitation that would classify them as having COPD. There is some evidence that extreme prematurity and BPD may also be associated with a restrictive spirometric profile (33,35); such a profile, when present in adults, has been shown to increase subsequent risk for cardiovascular death (38). Mild to moderate prematurity is associated with modest changes in lung function, which may not have major impact on the risk for chronic respiratory impairment later in life.
Genetics of BPD
Although the association of extremely premature birth, and especially that associated with BDP, to subsequent impairment in lung function is clearly established, not all extremely premature infants go on to develop BPD. This raises the possibility that genetic factors may predispose to the development of BPD and, therefore, to the deleterious long-term respiratory outcomes associated with extreme prematurity. Comparisons of concordance for complex diseases such as BPD among monozygotic and dizygotic twins provide a strong tool to assess the role of genetic factors in the development of these diseases. Two studies (39, 40) that have addressed this issue yielded similar results: Concordance of BPD was significantly higher among monozygotic than among dizygotic twins, with a calculated heritability (i.e., the proportion of the variability of the trait that is explained by genetic factors) of between 53 and 80%. To determine which genetic variants may explain this striking role of hereditary factors on BPD, GWAS have been recently attempted. In these studies, the frequencies of hundreds of thousands of SNPs distributed across the whole genome are compared between affected subjects and appropriate controls. Hadchouel et al. (41) assessed a relatively small number of infants (n = 418) born after < 28 weeks gestation of European and African ancestry, of whom 22% developed BPD. Genetic variants in only one gene, SPOCK2 on chromosome 10q22.1, were found to be significantly associated with BPD in both ancestries, and results were replicated in a third population in Finland. Although the observed odds ratios for the most strongly associated SNP (rs1245560: 3.0 among Whites and 4.9 among Blacks, respectively) are high for these types of studies, the proportion of the total variance explained by this SNP was very low. SPOCK2, also called Testican-2, is a member of the testican group of extracellular chondroitin and heparan sulfate proteoglycans. Studies in rats showed that SPOCK2 is expressed in the developing rat lung and that mRNA levels changed with stages of lung development. SPOCK2 expression was low during the canalicular and saccular stages of rat lung development and increased significantly at the beginning of alveolarization, remaining high until the alveolarization was completed. It has been suggested that this pattern of expression may be consistent with the involvement of SPOCK2 in the control of septation (42). Interestingly, SPOCK2 was recently found to play an active role in the transdifferentiation of surfactant-producing pulmonary alveolar epithelial type II (AT2) cells into type I (AT1) cells (42). These results thus seemed to provide support to the contention that genetic studies could provide clues to the pathogenesis of BPD and also potential therapeutic targets for the prevention of BPD and of the deficits in lung function that are often a long-term sequelae of the disease.
A more recent GWAS, however, yielded disappointing results. The new study (43) included 1,726 very low-birth-weight infants with a gestational age of twenty-five to less than twenty-nine weeks who had a minimum of three days of intermittent positive pressure ventilation and were in the hospital at thirty-six weeks postmenstrual age. At that age, BPD cases (52%) required continuous supplemental oxygen and were defined as having BDP, whereas controls (48%) did not. Almost 1.8 million SNPs were successfully genotyped, and none showed significant genome-wide association with BPD status (preestablished at p ≤ 5 × 10–8). Moreover, SNPs identified in both candidate gene studies and GWAS, including those in SPOCK2, could not be replicated. There are many possible explanations for these discrepancies between studies, including (among others) differences in the definition of the phenotypes, insufficient power, and inclusion of subjects from different ethnic backgrounds. The negative study by Wang et al., for example, included mostly children born in the United States of Hispanic/Mexican origins. There is strong evidence that different sets of genetic variants and genes may determine susceptibility of the same phenotypes in Mexican-Americans, who as a group show evidence of European–Native American admixture, as compared with subjects of European origins (44,45). As has been the case for most other complex phenotypes, the genetic variants that explain most of the “hidden heritability” of BPD remain to be ascertained, and their identification may require large, whole genome sequencing studies.
Lung Structure and Function in Animal Models of Premature Birth and BPD
Preterm sheep and baboons have been used to develop experimental models of both BPD, associated with the requirement for neonatal mechanical ventilation, and preterm status per se, in the absence of respiratory support. In models of BPD, observed pathological changes are similar in preterm infants with BPD: Chronically ventilated preterm baboons and preterm lambs show alveolar simplification, persistent smooth muscle hypertrophy of both pulmonary arterioles and small airways (46). The main factor determining alveolar simplification is insufficient growth of secondary septa from the walls of the saccules, which in turn decreases the number of alveoli and the airspace surface area for gas exchange. Elastin synthesis plays a major role in septa growth, and it has been shown that elastin synthesis is continuously upregulated in chronically ventilated preterm baboons and preterm lambs (47). Decreased alveolarization in baboon models of BPD has been shown to persist up to thirty-three weeks, which corresponds to approximately two years of age in humans (48). In addition, these animals show stunted development of septal capillaries and thickened airspace walls, which could explain the restrictive spirometric pattern observed in some studies of adult with a history of BPD.
Baboon and lamb models of preterm birth followed by mechanical ventilation also shed light on the development of chronic airflow limitation that is observed in many survivors of BPD. Indeed, these animals have increased airway expiratory resistance and lower lung compliance. The main histopathological change associated with these functional impairments is increased accumulation of airway smooth muscle and a thickened airway wall (46).
Fewer studies have used animal models to study the association of prematurity per se in the absence of any ventilatory requirement, with the subsequent development of respiratory impairment. In sheep, these studies are limited to animals delivered two weeks premature (at 133 days gestation), the earliest gestational age at which mechanical ventilation is not required (49). Respiratory system compliance was not different at term equivalent age in preterm lambs as compared with term lambs, but was higher in preterm lambs at six weeks post-term-equivalent age. Pulmonary resistance was slightly, but not significantly, higher in preterm lambs than in controls at term-equivalent age, but this difference was no longer present at age six to eight weeks post-term age. Alveolar septa were 33% thicker and the blood–air barrier was 26% thicker in preterm lambs than in controls at term-equivalent age and remained thicker at six weeks post-term-equivalent age. In preterm lambs, the airway epithelium was also thicker at term-equivalent age and at six weeks post-term-equivalent age (50). These studies thus confirm the conclusion reached from studies in humans that mild to moderate preterm birth by itself is associated with only mild disruption of lung and airway structure (51), which does not seem to have clinically important long-term functional consequences during childhood and adult life.
Intrauterine Growth Retardation and Lung Function
The role of an adverse intrauterine environment on subsequent lung function and COPD risk has been an essential component of the “fetal origins” hypothesis first proposed by Barker (52). It is reasonable to assume that the mechanisms responsible for potential harmful effects on lung growth would be different between premature birth and intrauterine growth retardation (IUGR) associated with malnutrition or other noxious exposures occurring during pregnancy such as maternal smoking. In many studies addressing this issue, however, either gestational age was not available or birth weight was directly related to subsequent lung function, without attempting to specifically determine the separate roles of prematurity and IUGR. A meta-analysis (53) of nine studies among adults showed a positive association between birth weight and FEV1, with FEV1 increasing by 0.048 l (95% CI 0.026 to 0.070) for each increase of 1 kg birth weight. More recently, Hancox et al. (54) related birth weight to lung function at age thirty-two years in a birth cohort in Dunedin, New Zealand. Birth weight was not significantly associated with either FEV1 or FVC when the latter were adjusted for height and sex, although the coefficient for FEV1 was very similar to that reported in the previous meta-analysis (50 ml, 95% CI –8 to 120).
A few studies have specifically assessed lung function across the life span in small-for-gestational-age (SGA) children and compared results with those for their appropriate-for-gestational-age (AGA) peers. Results of a study of SGA (<10th percentile birth weight for gestational age) infants suggested that both flows and volumes derived from flow-volume loops measured with the raised volume technique were significantly lower than those of AGA infants after adjusting for relevant maternal and infant characteristics (55). In a study limited to premature children, mean airway resistance was found to be significantly higher at ages six to twenty-four months in thirty-one SGA infants with a mean gestational age of thirty-one weeks as compared to their AGA peers with a mean gestational age of twenty-eight weeks (38 vs. 34 cmH2O/L*s, respectively, p = 0.004) (56).
Deficits in lung function in SGA individuals have also been reported in two large studies in older children. Rona et al. (57) first suggested that children ages five to eleven who were SGA at birth had significantly lower lung function than AGA children. They estimated that the difference in expected FEV1 between two children with thirty-eight weeks of gestation (expected birth weight of 3,300 g), but one having a birth weight of 2,500 g and the other 3,500 g, would be 26 mL, or 1.7% of the expected FEV1. In the ALSPAC study quoted earlier (34), term children ages seven to eight years who had showed evidence of intrauterine growth retardation at birth had mean deficits of 50 mL for FEV1 and 40 mL for FVC, adjusted for height, age, and sex, when compared with AGA children. Observed deficits were statistically significant, consistent, but relatively modest in both studies. There are no reports that have specifically assessed the association between SGA status and adult lung function.
Of interest is the possibility that, as was suggested for prematurity, deficits in lung function associated with IUGR could improve with time. Catch-up growth does occur between birth and puberty in many SGA children, and it is possible that lung function may also improve in these children more than in those with no catch-up growth. In support of this contention, Suresh and coworkers compared lung function at age twenty-one years in subjects with IUGR at birth, that is, those in the lowest quintile for birth weight (58); no apparent adjustment was made for gestational age. Within the IUGR group, both FVC and FEV1 were significantly higher in children who showed catch-up growth than in those who did not, and this was true for catch-up growth assessed between birth and five years and between birth and fourteen years. This raises the possibility that aggressive nutritional care during childhood may reverse the significant effects of IUGR on long term-lung function up to adult life.
[level-membership-for-neonatal-and-perinatal-medicine-category]
Role of Malnutrition and Exposure to Tobacco Smoke
Two known causes of IUGR in humans and their potential role in susceptibility to chronic respiratory impairment have been thoroughly studied: maternal malnutrition, which still affects a significant proportion of the population in the developing world; and maternal smoking during pregnancy. In the case of maternal malnutrition, the Dutch famine of 1944–45 has provided the most important clues. The Dutch famine was a circumscribed episode of severe human starvation that occurred during a six-month period lasting from October 1944 to May 1945 (59). At the height of the famine, from December 1944 to April 1945, the official daily rations varied between 400 and 800 kcal. Studies using records from that period show that subjects who were exposed to the famine during late and midgestation had a lower birth weight, body length, and ponderal index (weight divided by the cube of length) and smaller heads. However, paradoxically, those exposed to famine in early gestation had a higher weight and body length at birth than nonexposed subjects (60). That there may be catch-up lung growth during malnourished pregnancies seems to be corroborated by experimental studies. Protein restriction during pregnancy in mice resulted in fetuses with impaired branch morphogenesis (day 12 of gestation) but by day 13, rapid recovery had already occurred (61).
In a study of survivors of the Dutch famine, lung function was successfully measured in 733 individuals age 50 whose mothers had been subjected to severe food restrictions during pregnancy. There was no difference in any spirometric parameter between subjects exposed to famine in utero and those not exposed (60). A more recent study assessed the impact on COPD of exposure to the Dutch famine during postnatal life (62). Women who reported to be exposed to moderate or severe famine were significantly more likely to have been hospitalized for COPD by a mean age of 60 years than their age peers not exposed to famine. These effects were particularly noticeable (and only significant) among women who were active smokers, suggesting that postnatal malnutrition may increase susceptibility to the deleterious effects of smoking.
The potential role of vitamin D deficiency during pregnancy on fetal lung growth and subsequent lung function has been the subject of intense scrutiny during the last decade. Vitamin D is known to play a role in alveolar growth in the embryo and fetus, and thus, vitamin D deficiency during pregnancy could plausibly affect lung function in the offspring. Older studies in rats (63) showed that offspring of mothers deprived of vitamin D during pregnancy had lower lung volumes and decreased lung compliance at fifty days postnatally when compared with offspring of mothers receiving usual doses of vitamin D. More recent studies showed increased bronchial responsiveness, decreased radial alveolar counts, and increased linear intercepts in offspring rats of vitamin D deficient pregnancies (64). In humans, no association was found between vitamin D levels in the mother at thirty-six weeks gestation and their offspring’s mean FEV1 at age six to seven years (65). Likewise, no association was reported between cord blood levels of vitamin D and lung function assessed repeatedly during the first seven years of life in a newborn cohort of children at high risk for asthma in Denmark (66). In the only clinical trial currently available, 180 pregnant women were randomized at twenty-seven weeks gestation to either no vitamin D, 800 IU ergocalciferol daily until delivery, or a single oral bolus of 200,000 IU cholecalciferol. Supplementation improved but did not optimize vitamin D status. There was no difference between groups in lung function, as assessed by impulse oscillometry, or in any other respiratory outcome at age three (67). Larger, ongoing randomized trials evaluating the effects of vitamin D supplementation during pregnancy are likely to provide more definitive answers in establishing the clinical significance of maternal vitamin D levels during pregnancy on lung function and asthma risk in their offspring(68).
Similar to vitamin D, vitamin A is known to influence alveolar and airway growth in fetal life. In a major study of vitamin A supplementation during pregnancy in Nepal, lung function was assessed at ages nine to thirteen years in children whose mothers had received weekly oral supplementation with either 7,000 μg retinol equivalents of vitamin A, 7000 μg retinol-equivalents of beta carotene, or placebo (69). Of note, this was a chronically undernourished population prior to initiation of the trial. Spirometry was performed in 1,371 offspring of the participating mothers. Children whose mothers had received vitamin A, but not those whose mothers had received beta carotene, had mean FEV1 and FVC levels that were significantly higher than those of children whose mothers had received placebo. The improvements in lung function (46 mL for both FEV1 and FVC) with vitamin A treatment amounted to a 2.6% and 2.9% improvement in FEV1 and FVC, respectively, which should be considered clinically meaningful.
The effects on lung function of exposure to tobacco smoke in utero and postnatally have been the matter of considerable research during the last thirty years. Experimental studies in animal models have convincingly shown that fetal exposure to nicotine negatively affects lung and airway development, reducing surface complexity of the lung parenchyma and increasing collagen deposition in the airways (70). In 1998, Cook and Strachan summarized the results of twenty-one studies comparing spirometric indices in children by exposure to parental smoking (71). They concluded that the reduction in FEV1 in children exposed to parental smoking compared with those not exposed was 1.4% (95% CI 1.0 to 1.9). These effects were considered “modest but statistically significant.” Studies that distinguished between maternal and paternal exposure indicated that most of the effects were due to exposure in utero and during the neonatal period. Studies performed after 1998 have, in general, confirmed that children exposed to tobacco smoke in utero do have alteration in lung function, but the nature of these alterations differs from study to study. Hollams and coworkers (72), for example, assessed lung function at age fourteen years in participants in a birth cohort in Perth, Australia. They found that offspring of mothers who smoked during pregnancy had similar FEV1 but larger FVC compared to those whose mothers did not smoke; as a consequence, diminished FEV1/FVC ratio was found in those exposed.
Whether effects of parental smoking persist into adult life has not been thoroughly explored. As part of the European Community Respiratory Health Survey, Svanes, et al. assessed lung function in 15,901 adults age twenty to forty-five who self-reported about the smoking habits of their parents (73). Adults who reported a history of maternal smoking during pregnancy had FEV1/FVC ratios that were 0.9% lower than those who did not (p < 0.001). Interestingly, they were also more likely to have chronic bronchitis and wheeze, but not asthma than their age peers not exposed to smoking during pregnancy. Guerra et al. (74) studied 519 adults enrolled in the Tucson Children’s Respiratory Study. They found no association between parental smoking during childhood, assessed longitudinally, and adult lung function up to age twenty-six years, but the study was not powered to detect the small effects reported in the much larger study described earlier by Svanes et al. However, they did notice that adults who had been exposed to parental smoking and were themselves active smokers had levels of lung function that were significantly lower than those of control subjects exposed to neither, whereas those who were either exposed to parental smoking or smoked themselves, but not to both, had levels of lung function that were not different from those of controls. This suggests that an important sequela of exposure to tobacco smoke in utero and during the growing years may be increased susceptibility to noxious exposures in adult life.
Conclusions and Future Directions
Solid evidence suggests that lung growth during fetal and early postnatal life is one of the strongest determinants of adult lung function. Genetic variation plays a critical role in determining maximal lung function reached in adult life. Factors that affect lung growth such as extreme prematurity with BPD, IUGR, exposure to tobacco smoke in utero and postnatally, and vitamin A and D deficiencies also play varying roles in determining lung function. However, catch-up growth seems to be able to reverse the negative effects of some of these conditions. Individuals who reach early adult life with lower levels of lung function are at increased risk of developing COPD during the decline phase of lung function after the third decade of life and may also be more susceptible to the deleterious effects of active cigarette smoking. Future studies are needed to better identify children at risk for an early onset of chronic lung disease as well as susceptibility factors that contribute to disease progression. Such insights may lead to novel interventional strategies to reduce the burden of chronic respiratory disorders in adult life.
References
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
