Liver disorders
Features of liver disorders in children are:
• Prolonged (persistent) neonatal jaundice is the most common presentation of liver disease in the neonatal period.
• The earlier in life biliary atresia is diagnosed and treated surgically, the better the prognosis.
• Transmission of hepatitis B surface antigen-positive mothers is prevented by immunising their babies.
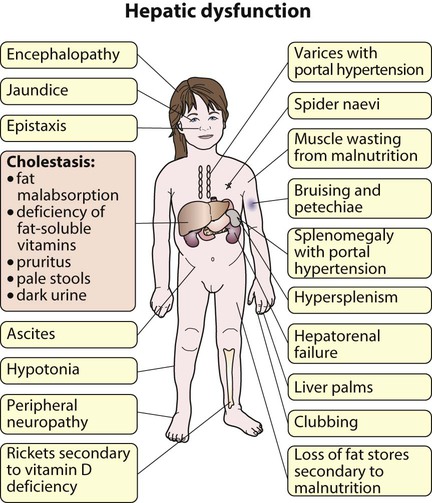
• Chronic liver disease (Fig. 20.1), cirrhosis and portal hypertension are uncommon and should be managed by, or in conjunction with tertiary or national centres.
• Liver transplantation is an effective therapy for acute or chronic liver failure, with a >80% 5-year survival rate; it is only performed at national centres.
Neonatal liver disease
Many newborn infants become clinically jaundiced. About 5–10% are still jaundiced at >2 weeks of age (3 weeks if preterm), when it is called ‘prolonged (or persistent) neonatal jaundice’. This is usually an unconjugated hyperbilirubinaemia, which resolves spontaneously (Box 20.1). Prolonged neonatal jaundice caused by liver disease is characterised by a raised conjugated bilirubin (>20 µmol/L) and is usually accompanied by:
 In prolonged (persistent) neonatal jaundice, check if it is conjugated hyperbilirubinaemia, as this is due to liver disease.
In prolonged (persistent) neonatal jaundice, check if it is conjugated hyperbilirubinaemia, as this is due to liver disease.Neonatal hepatitis syndrome
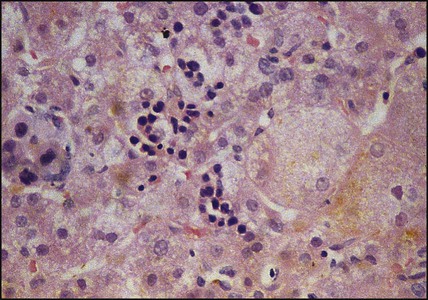
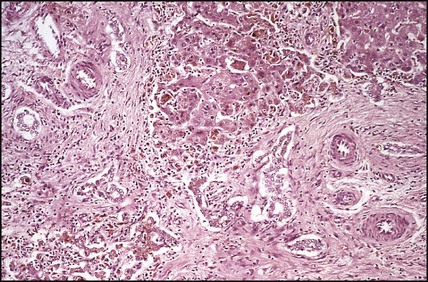
In neonatal hepatitis syndrome, there is prolonged neonatal jaundice and hepatic inflammation. Its causes are listed in Box 20.1, but often no specific cause is identified. In contrast to biliary atresia, these infants may have intrauterine growth restriction and hepatosplenomegaly at birth. Liver biopsy (Fig. 20.6) is often non-specific, demonstrating a giant cell hepatitis.
α1-Antitrypsin deficiency
Case History
• Prothrombin time – 28 s (normal range 10–13 s)
• Serum bilirubin 178 micromol/L – 140 micromol/L conjugated.
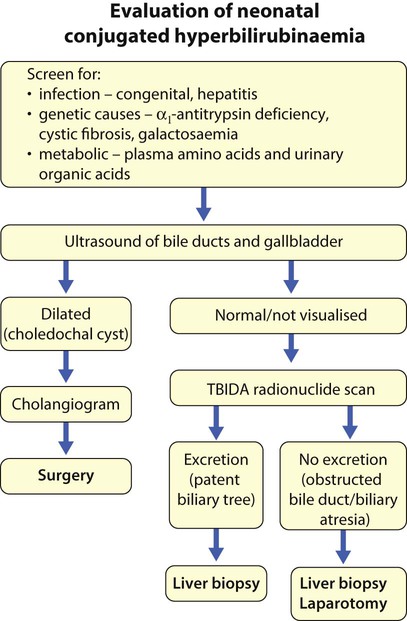
The investigation of conjugated hyperbilirubinaemia is shown in Figure 20.2. The TIBIDA radionuclide scan showed no excretion at 24 h (Fig. 20.3) and a liver biopsy suggested biliary atresia (Fig. 20.4). A hepatoportoenterostomy was performed at 6 weeks of age (Fig. 20.5).
 In persistent jaundice, always look to see if the stools are pale – suggests bile duct obstruction.
In persistent jaundice, always look to see if the stools are pale – suggests bile duct obstruction.Viral hepatitis
Hepatitis B
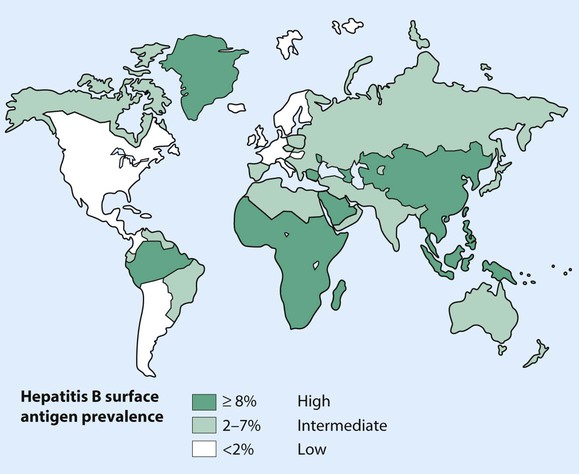
Hepatitis B virus (HBV) is a DNA virus which is an important cause of acute and chronic liver disease worldwide, with high prevalence and carrier rates in the Far East, sub-Saharan Africa and parts of North and South America (Fig. 20.7). HBV is transmitted by:
• Perinatal transmission from carrier mothers
• Transfusion of infected blood or blood products
Acute liver failure (fulminant hepatitis)
Acute liver failure in children is the development of massive hepatic necrosis with subsequent loss of liver function, with or without hepatic encephalopathy. The disease is uncommon, but has a high mortality. Most of the cases in childhood are attributed to paracetamol overdosage, non-A to G viral hepatitis and metabolic conditions (Table 20.1). The child may present within hours or weeks with jaundice, encephalopathy, coagulopathy, hypoglycaemia and electrolyte disturbance. Early signs of encephalopathy include alternate periods of irritability and confusion with drowsiness. Older children may be aggressive and unusually difficult. Complications include cerebral oedema, haemorrhage from gastritis or coagulopathy, sepsis and pancreatitis.
Table 20.1
Causes of acute liver failure in children
| Infection | Viral hepatitis A, B, C, non-A to G |
| Poisons/drugs | Paracetamol, isoniazid, halothane, Amanita phalloides (poisonous mushroom) |
| Metabolic | Wilson disease, tyrosinaemia |
| Autoimmune hepatitis | |
| Reye syndrome |
Management
• Maintaining the blood glucose (>4 mmol/L) with intravenous dextrose
• Preventing sepsis with broad-spectrum antibiotics and antifungals
• Preventing haemorrhage, particularly from the gastrointestinal tract with intravenous vitamin K, fresh frozen plasma or cryoprecipitate and H2-blocking drugs or proton pump inhibitors (PPIs)
• Treating cerebral oedema by fluid restriction and mannitol diuresis
Chronic liver disease
The causes of chronic liver disease are given in Box 20.2. The clinical presentation varies from an apparent acute hepatitis to the insidious development of hepatosplenomegaly, cirrhosis and portal hypertension with lethargy and malnutrition. The commonest causes of chronic hepatitis are hepatitis viruses (B or C) and autoimmune hepatitis, but Wilson disease should always be excluded. Histology may demonstrate varying degrees of hepatitis, with an inflammatory infiltrate in the portal tracts that spreads into the liver lobules.
Wilson disease
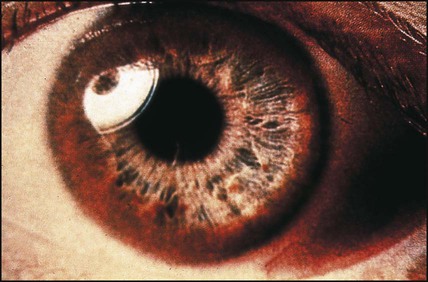
Wilson disease is an autosomal recessive disorder with an incidence of 1 in 200 000. Many mutations have now been identified (on chromosome 13). The basic genetic defect is a combination of reduced synthesis of caeruloplasmin (the copper-binding protein) and defective excretion of copper in the bile, which leads to an accumulation of copper in the liver, brain, kidney and cornea. Wilson disease rarely presents in children under the age of 3 years. In those presenting in childhood, a hepatic presentation is more likely. They may present with almost any form of liver disease, including acute hepatitis, fulminant hepatitis, cirrhosis and portal hypertension. Neuropsychiatric features are more common in those presenting from the second decade onwards and include deterioration in school performance, mood and behaviour change, and extrapyramidal signs such as incoordination, tremor and dysarthria. Renal tubular dysfunction, with vitamin D-resistant rickets, and haemolytic anaemia also occur. Copper accumulation in the cornea (Kayser–Fleischer rings) (Fig. 20.8) is not seen before 7 years of age.
Cirrhosis and portal hypertension
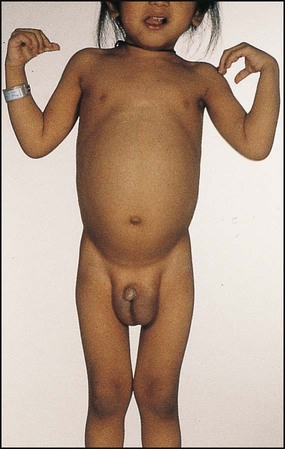
Cirrhosis is the end result of many forms of liver disease. It is defined pathologically as extensive fibrosis with regenerative nodules. It may be secondary to hepatocellular disease or to chronic bile duct obstruction (biliary cirrhosis). The main pathophysiological effects of cirrhosis are diminished hepatic function and portal hypertension with splenomegaly, varices and ascites (see Fig. 20.1). Hepatocellular carcinoma may develop.
Children with compensated cirrhosis may be asymptomatic if liver function is adequate. They will not be jaundiced and may have normal liver function tests. As the cirrhosis increases, however, the results of deteriorating liver function and portal hypertension become obvious (Fig. 20.9). Physical signs include palmar and plantar erythema and spider naevi, malnutrition and hypotonia. Dilated abdominal veins and splenomegaly suggest portal hypertension, although the liver may be impalpable.
• Screening for the known causes of chronic liver disease (see Box 20.2)
• Upper gastrointestinal endoscopy to detect the presence of oesophageal varices and/or erosive gastritis
• Abdominal ultrasound – may show a shrunken liver and splenomegaly with gastric and oesophageal varices
• Liver biopsy – may be difficult because of increased fibrosis but may indicate the aetiology (e.g. typical changes in congenital hepatic fibrosis, copper storage).
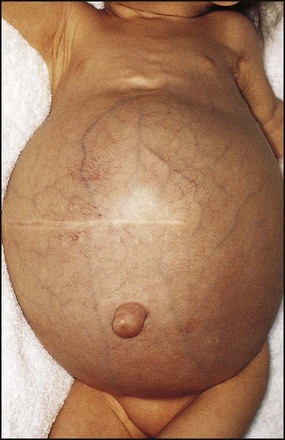
Ascites
Ascites is a major problem (Fig. 20.10). The pathophysiology of ascites is uncertain, but contributory factors include hypoalbuminaemia, sodium retention, renal impairment and fluid redistribution. It is treated by sodium and fluid restriction and diuretics. Additional therapy for refractory ascites includes albumin infusions or paracentesis.
Management of children with liver disease
Nutrition
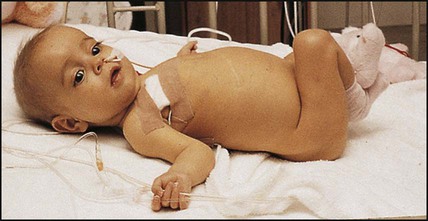
Treatment is to provide a high-protein, high-carbohydrate diet with 50% more calories than the recommended dietary allowance. In children with cholestasis, medium-chain triglycerides, which are absorbed by the portal circulation, will provide fat, but 20–40% long-chain triglycerides are required to prevent essential fatty acid deficiency. Many children will require nasogastric tube feeding or parenteral nutrition (Fig. 20.11).
Liver transplantation
The indications for transplantation in chronic liver failure are:
• Severe malnutrition unresponsive to intensive nutritional therapy
• Recurrent complications (bleeding varices, resistant ascites)