Life-Threatening Asthma
EPIDEMIOLOGY OF LIFE-THREATENING ASTHMA
PATHOPHYSIOLOGY AND IMMUNOLOGY
OBJECTIVE MEASUREMENT OF OBSTRUCTION
LABORATORY AND RADIOGRAPHIC DATA
NONTRADITIONAL THERAPY OF SEVERE BRONCHOSPASM
Epidemiology of Life-Threatening Asthma
Worldwide an estimated 300 million people suffer from asthma and this number is estimated to grow by more than 100 million by 2025. About 70% of asthmatics have allergies and 11% of cases are related to workplace conditions such as exposure to fumes, gases, or dust. Older asthmatic patients (≥60 years) tend to have severe or near-fatal asthma exacerbation compared to younger asthmatics (<60 years).1–4
Triggers of Acute Asthma
Common triggers for acute asthmatic attacks include air pollutants, respiratory tract infection, and allergen exposure. An association of panic-type anxiety and life-threatening asthma has been suggested. Box 38.1 contains a comprehensive list of precipitating factors.
Mortality Rates for Asthma
In the 1960s, a global alarm was sounded when sharp epidemic increases in asthma death rates were reported. Deaths approximately doubled in the United States from 1980 to 1995. After a long period of steady increase, asthma mortality and morbidity rates have continued to decline for the past decade.3,5–7
In patients 5 to 44 years old, death from asthma peaks in the summer months, although hospitalizations peak during the winter months. In older asthmatic patients, a different distribution is seen, with hospitalizations and mortality rates both peaking in the winter months. Older patients with asthma also have been shown to have fewer symptoms of dyspnea with methacholine-induced obstruction.8
Polynesians, African Americans, and black South Africans all have been reported to have higher asthma mortality rates. Likely reasons include genetic predisposition or poor management of severe asthma attacks because of reduced or delayed use of health care services and lower level of understanding. Self-medication also may play a role. Pendergraft and colleagues9 reported that among 29,430 admissions in the United States with a primary diagnosis of asthma, 10.1% were admitted to intensive care units (ICUs), and 2.1% were intubated. The risk of in-hospital death was significantly higher in patients who were intubated and who had comorbid conditions. Near-fatal events occur in 2% to 20% of patients admitted to ICUs with acute severe asthma and in 2% to 4% of those intubated for respiratory failure. Risk factors for death from asthma are previous severe exacerbations with ICU admission or intubation, two or more hospitalizations within the past year, three or more emergency department visits in the past month, use of more than two canisters of short-acting β-agonist in the past month, and difficulty in perceiving or articulating asthma symptoms. African-American ethnicity, low socioeconomic status, urban residence, substance abuse, cigarette-smoke exposure, psychological factors (anxiety and depression), and comorbid problems such as cardiovascular diseases, chronic pulmonary diseases, and chronic psychiatric illnesses are other potential risk factors for death from asthma.
The strongest predictor of mortality risk from asthma is a prior episode of near-fatal asthma, estimated to be 15% to 22%. Asthma death rates per 1000 persons with asthma were 30% higher for females than males, 75% higher for blacks than whites, and seven times higher for adults compared to children. Adults over 65 years old had the highest death rate of 0.58 per 1000 persons with asthma.9–13
Classification
About 5% of asthma patients have “difficult asthma” (asthma difficult to control with maximal recommended doses of inhaled medications, in particular, inhaled corticosteroids). Most of these patients meet the criteria for severe asthma or may have chronic mild or moderate disease with acute exacerbations. Two clinical patterns of life-threatening asthma have been reported. A more serious type is the slow onset of life-threatening asthma characterized by onset over days to weeks, copious amounts of mucoid secretions with intense eosinophilic infiltration, and resistance to bronchodilator therapy. This pattern is described as “slow onset–late arrival,” or type 1, and accounts for 80% to 85% of fatal asthma. Second is the sudden type of asthma characterized by onset over minutes to hours with acute bronchospasm, absence of large quantities of airway secretions with no mucous plugs but neutrophil infiltration of the submucosa, typically a marked response to bronchodilators, and quick recovery in most circumstances. This is described as “sudden asphyxic asthma,” or the type 2 scenario of asthma death. Sudden-type asthma accounts for about 15% to 20% of fatal asthma.14–16
Pathophysiology and Immunology
Manthous and Goulding studied the effect of intravascular volume status on deadspace fraction in mechanically ventilated patients with severe asthma. They noted a mean increase in deadspace ventilation of 4.2% in response to intravascular volume expansion with 250 to 500 mL of normal saline solution.17,18
Characteristic findings of fatal asthma are airways showing infiltration with neutrophils and eosinophils, degranulated mast cells, sub–basement membrane thickening, loss of epithelial cell integrity, occlusion of bronchial lumen by mucus, hyperplasia and hypertrophy of bronchial smooth muscle, and hyperplasia of goblet cells. Asthma is an inflammatory response evidenced by the presence of cytokines that mediate inflammation and chemotactic chemokines in bronchoalveolar lavage fluid and pulmonary secretions. Some cytokines initiate inflammatory response by activating transcription factors, which act on genes that encode inflammatory cytokines, chemokines, adhesion molecules, and other proteins that induce and perpetuate inflammation. Adhesion molecules provide a mechanism for the adhesion of inflammatory cells to the endothelium and migration of these cells from the circulation into the lamina propria, epithelium, and the airway lumen itself.19
Busse and Lemanske described the immunology of allergic inflammation in asthma. IgE antibodies are linked to the severity of asthma. The release of cytokines depends on cross-linking of IgE by allergen. IgE antibodies are synthesized and released by B cells; briefly circulate in the blood; and bind to high-affinity IgE receptors on the surface of mast cells in tissues and peripheral blood basophils and low-affinity IgE receptors on lymphocytes, eosinophils, platelets, and macrophages.20
The early phase of asthma (usually resolves within 1 hour) is characterized by an inhaled allergen precipitating acute constriction of smooth muscles by release of histamines and leukotrienes from mast cells. A prolonged late phase (4 to 6 hours later) occurs as a result of cytokines and chemokines generated by resident inflammatory cells (mast cells, macrophages, epithelial cells) and recruited inflammatory cells (lymphocytes, eosinophils) and causes further obstruction of airflow. Numerous cytokines regulate the function of eosinophils and other cells in asthma. Interferon-γ is elevated in severe asthma during the acute phase. Data also suggest that interferon-γ contributes to the activation of eosinophils and likely augments inflammation. There are two types of helper CD4+ T lymphocyte cells. Type 1 helper (TH1) T cells produce interleukin (IL) 2 and interferon-γ, which are essential for cellular defense mechanisms. Type 2 helper (TH2) T cells produce cytokines (IL-4, IL-5, IL-6, IL-9, and IL-13) that mediate allergic inflammation. Balance between the TH1 and TH2 type cytokine response contributes to the cause and evolution of atopic diseases, including asthma. The increasing prevalence of asthma in Western countries has led to the “hygiene hypothesis.” The immune system in newborns is primarily TH2 cells, and a timely and appropriate environmental stimulus is needed to create a balanced immune response. Alteration in the number of infections in early life, widespread use of antibiotics, adoption of the Western lifestyle, and repeated exposure to allergens may affect the balance between TH1-type and TH2-type cytokine responses and increase the likelihood of immune response by TH2 cells and lead to asthma. Mild intermittent asthma is thought to be a TH2 allergen-oriented reaction with adequate apoptosis and self-limiting inflammation, although severe persistent asthma is mediated by TH1 cytokines with progressive loss of apoptosis leading to longer exacerbations, expanded memory cells, and persistent inflammation. Evidence continues to underscore the importance of immune factors in the development of asthma and resulting inflammatory process. This particular strategy and insight into the mechanisms of these processes would be important for future treatment of acute severe asthma.19,21–25
Airflow limitation in asthma is caused by bronchoconstriction, airway edema, airway hyperresponsiveness, and airway remodeling. Permanent structural changes can occur with airway remodeling, leading to poor response to therapy, including thickening of sub–basement membrane and subepithelial fibrosis, airway smooth muscle hypertrophy and hyperplasia, blood vessel proliferation and dilation (angiogenesis), and mucus gland hyperplasia and hypersecretion.13
Asthma Genetics
Asthma and atopy are complex phenotypes that are influenced by genetic and environmental factors. About 79 genes have been associated with asthma or atopy phenotypes. The ADAM33 gene has been associated with asthma. A locus on the short arm of chromosome 20 has been linked to asthma and bronchial hyperresponsiveness. If further investigations confirm that ADAM33 is an asthma gene, future studies should enhance understanding of asthma and lead to new therapeutic targets. As Ober and Hoffjan described, such “molecular phenotyping” of patients with asthma and atopic diseases may generate informed decisions regarding treatment, laying the foundation for genomic medicine in the next decade.26,27
Symptoms and Signs
Wheezing may be expiratory and inspiratory and correlates with the degree of obstruction if adequate air movement is present. Absence of wheezing is an ominous finding in a severely distressed asthmatic patient because it implies minimal air movement and is a harbinger of respiratory arrest. Contraction of the sternocleidomastoid muscles and other accessory muscles indicates severe obstruction (FEV1 < 1 L). Intense inspiratory effort leads to large swings in intrathoracic pressure and to an accentuated pulsus paradoxus (representing a decreased stroke volume during inspiration). Pulsus paradoxus is often appreciated during routine blood pressure measurement in acute severe bronchospasm because systolic blood pressure decreases dramatically during inspiration (this decrease is <10 mm Hg in normal individuals). A decrease of more than 15 mm Hg in a patient in an acute asthma episode is associated with severe reduction in FEV1.18,28,29
Ominous signs and findings during an acute severe asthma episode include diaphoresis, inability to recline or talk, peak expiratory flow rate (PEFR) less than 60 L/minute, and use of accessory muscles. PEFR less than 25% predicted or personal best is defined as severe life-threatening asthma. In acute severe asthma, lung hyperinflation occurs secondary to increased expiratory airflow resistance, short expiratory time, high ventilatory demands, and increased postinspiratory activity of the inspiratory muscles. The presence of these factors in variable degrees does not allow the respiratory cycle to reach a static equilibrium volume at the end of expiration. Inspiration begins at a volume in which the respiratory system exhibits a positive elastic recoil pressure called intrinsic positive-end expiratory pressure (iPEEP), or auto-PEEP. This phenomenon is described as dynamic hyperinflation. Dynamic hyperinflation produces a significant decrease in systemic venous return to the heart, leading to a decrease in left ventricular diastolic filling. Also problematic is the increase in left ventricular afterload as a result of large negative intrathoracic pressure swings during inspiration. Pulmonary artery pressure also may be increased secondary to lung hyperinflation, resulting in increased right ventricular afterload. These events combine to produce pulsus paradoxus.15
Near-fatal asthma presents as raised PaCO2 requiring mechanical ventilation and is associated with high inflation pressures. Life-threatening asthma has any of the following features. Symptoms are altered mental status with confusion or coma, feeble respiratory effort, and exhaustion. Signs are cyanosis, silent chest, hypotension, bradycardia, oxygen saturation below 92% or PaO2 less than 60 mm Hg, PaCO2 higher than 60 mm Hg, and FEV1 below 30% predicted or personal best.30
All that wheezes is not asthma. Other entities to consider are upper airway obstruction and “cardiac asthma.” Upper airway obstruction should be considered in patients at risk (e.g., tracheal stenosis in patients who were previously intubated) and when there is no response to therapy in a patient without history of asthma. If the patient’s status would tolerate it, flow volume loops may be diagnostic. Likewise, wheezing that dissipates with intubation should make one suspect upper airway obstruction. Paradoxical vocal cord movement can stimulate asthma. Patients with acute left ventricular failure may wheeze as a result of interstitial fluid compression of bronchioles and edema-associated bronchiolar smooth muscle contraction.31,32
Objective Measurement of Obstruction
During an asthmatic attack, all indices of expiratory flow are significantly reduced, including FEV1; FEV1/FVC; PEFR; maximal expiratory flows at 75% (MEF75), 50% (MEF50), and 25% of vital capacity (MEF25); and maximal expiratory flow between 25% and 75% of the FVC (MEF25-75). With acute asthmatic crisis, high functional residual capacity, total lung capacity, and residual volume are observed.15
Although spirometry is the best objective measure of airway obstruction, a severely ill asthmatic patient is rarely able to perform the necessary full FVC maneuver. Objective assessment of airway obstruction in a severe asthmatic usually can be made by measuring the PEFR because this measurement requires patient cooperation only in the early part of the FVC maneuver. Because the greatest expiratory flow rates exist in early expiration, most patients are able to produce a reliable PEFR value. Normal expiratory flow rates vary considerably with age, sex, and height. In adults, a PEFR less than 100 to 125 L/minute implies severe obstruction to airflow. A severe exercebation of asthma is defined as an FEV1 less than 40% or peak expiratory volume, less than 40% predicted, or less than 1 L. Values less than 25% are consistent with life-threteaning asthma. Failure to improve PEFR significantly with initial aggressive bronchodilator therapy is the best predictor of morbidity in a patient with acute severe asthma.10
Laboratory and Radiographic Data
Asymmetrical breath sounds or chest pain should alert the physician to the possibility of pneumothorax and mandates an early chest radiograph. An increased white blood cell count may be produced by asthma alone in the absence of infection, β-receptor agonists, and theophylline shift of potassium intracellularly. Hypokalemia-induced dysrhythmias could occur after intensive bronchodilator therapy in elderly patients or in patients receiving other therapies that predispose to hypokalemia, such as steroidal and diuretic medications. Creatine phosphokinase (non-MB fraction) may be increased as a result of the strenuous activity of ventilatory muscles. Severe asthma may cause right-sided heart strain as shown on electrocardiogram; this resolves with clinical improvement. Arterial blood gas assessment adds little to the early management of acute asthma. The early stage of asthma usually reveals mild hypoxemia, hypocapnia, and respiratory alkalosis. A non–anion gap acidosis also may be observed in patients with severe asthma if several days of hyperventilation have led to renal compensation with bicarbonate wasting to compensate for the respiratory alkalosis. As the severity of obstruction increases, arterial carbon dioxide (PaCO2) normalizes and then increases as a sign of impending respiratory collapse. After initial therapy, arterial blood gases may be useful for decisions regarding hospital admission or tracheal intubation. Most asthma patients respond dramatically to initial therapy; arterial blood gases obtained when the patient is first seen are rarely predictive of outcome or useful clinically.33
Early attention should be directed toward aggressive therapeutic intervention. A normal PaCO2 level in a distressed asthmatic patient despite aggressive in-hospital therapy should alert the physician to respiratory fatigue and the danger of respiratory arrest. Respiratory acidosis may be preceded by a lactate-induced anion gap metabolic acidosis. This lactic acidosis is likely caused by a combination of failing inspiratory muscles, aggressive use of β-agonist therapy, and decreased liver perfusion resulting from increased intrathoracic pressure and blood flow diverted to the muscles of respiration. Lactic acidosis occurs more commonly in men and with administration of parenteral β-agonists.34–36
In patients with near-fatal asthma, high values for inflammation-related laboratory markers such as erythrocyte sedimentation rate (ESR), C-reactive proteins (CRP), and low nutritional status with low albumin levels were associated with poor prognosis. Exhaled nitric oxide is another biomarker of lung and airway inflammation. Elevated exhaled nitric oxide levels can be found in severe allergic asthma and may predict future exacerbations and steroid treatment response. Use of this biomarker is not recommended at this time and needs further evaluation.37,38
Inpatient Admission Decisions
Conditions typically requiring hospitalization for a patient with severe asthma are listed in Box 38.2.
Drug Therapy (Table 38.1)
Oxygen
If pulse oximetry confirms the presence of hypoxemia, oxygen should be given to maintain oxygen saturation at greater than 92%. A transient decrease in arterial oxygen tension has been shown in some patients after initiation of β-adrenergic agonist therapy in severe asthma. Mechanisms of this decrease relate to some combination of β2-agonist-induced vasodilation in areas of decreased ventilation and increase in pulmonary blood flow resulting from a β1-adrenergic inotropic and chronotropic effect. Saturation may decrease initially during bronchodilator therapy with β-agonists, which produce vasodilation and may increase intrapulmonary shunting. Studies in children suggest that aerosolized salbutamol administration may cause hypoxemia during acute episodes of asthma if the drug is administered without oxygen. Most published data show that salbutamol does not have a clinically important effect on oxygenation in asthmatic adults. This seems to be true for stable and acute asthma; however, these studies in adults exclude the most severe exacerbations more likely to be associated with marked hypoxemia. Because inhaled β-agonist should be given in this circumstance, the only clinical response is to treat any worsening of oxygenation that occurs with additional oxygen. Hyperoxia may be harmful and may be associated with hypercarbia due to regional release of hypoxic pulmonary vasoconstriction during asthma exacerbations.5,39–42
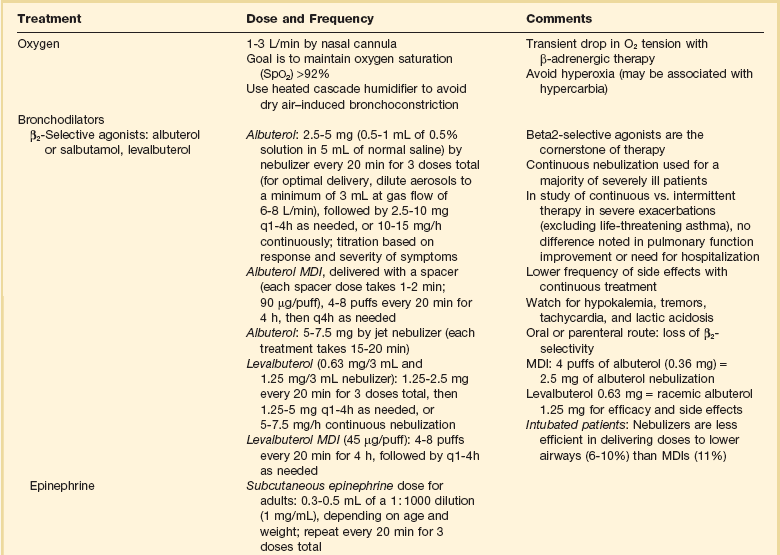


Moloney and colleagues showed that bronchoconstriction induced by dry air challenge can be prevented by humidifying inspired air. Humidification of inspired air should be achieved with a heated cascade humidifier. The use of heat and moisture exchangers is discouraged because they increase the deadspace and add to the expiratory airway resistance.43
β-Adrenergic Therapy
Inhaled β2-Selective Agonists (Albuterol or Salbutamol) and Short-Acting β-Agonists
Albuterol is the cornerstone of treatment for acute exacerbation in patients with acute asthma. Initial therapy in an acutely ill asthma patient, as recommended by the National Asthma Education and Prevention Program Update, is 2.5 to 5 mg of albuterol (0.5 to 1 mL of 0.5% solution in 5 mL of normal saline solution) by nebulization every 20 minutes for three doses (for optimal delivery, dilute aerosols to a minimum of 3 mL at gas flow of 6 to 8 L/minute), followed by 2.5 to 10 mg every 1 to 4 hours as needed, or 10 to 15 mg/hour continuously, with the titration based on response and severity of symptoms. Continuous nebulization should be considered in the most severe patients. Tachycardia and hypokalemia may occur with continuously nebulized albuterol. β2-Selective agents delivered parenterally or orally lose much of the β2 selectivity, which provides the rationale for inhalation treatment as the cornerstone of therapy.25,32,39,40,43,44
Adequate delivery of β-agonists can be accomplished by a metered-dose inhaler (MDI) with spacer during acute bronchospasm if proper technique is used and doses are increased. Four puffs of albuterol (0.36 mg) delivered with a spacer should be expected to be equipotent to 2.5 mg of albuterol by nebulization in patients with severe disease. It is advisable to deliver the β-agonist by nebulization in most acutely ill asthma patients because nebulization requires minimal coordination and cooperation of the patient and less bedside instruction and supervision by health care professionals. Many randomized controlled clinical studies over the last several decades have compared β-agonists delivered by MDIs or by nebulizer. Most studies show similar responses. Protocols typically include methods that ensure proper use of the MDI, however. Greater amounts of drug delivery are required with nebulized therapy to produce the same effect as that seen with an MDI with a spacer. To initiate therapy with nebulized albuterol and then switch to an MDI with spacer after the patient has improved and stabilized may be cost-effective. When aerosol β-agonists are delivered in intubated patients and patients receiving mechanical ventilatory support, much of the physiologic effect is lost as a result of deposition onto the endotracheal tube. Doubling the dose that would be used in a nonintubated patient is recommended.45–47
Aggressive inhaled selective β2-agonist therapy is preferred to intravenous albuterol because the same end point usually can be achieved with less risk for toxicity. Intravenous albuterol (if available) may be considered as an alternative when patients with life-threatening asthma have failed to respond to inhaled therapy. Oral β2-selective agents should not be used as primary treatment for patients with acute asthma because the therapeutic-to-toxicity ratio is less than with inhaled agents. Effects of corticosteroids and β2-agonists on airflow obstruction may be additive.48
Levalbuterol, 0.63 mg, is equivalent to racemic albuterol, 1.25 mg, for efficacy and side effects. Levalbuterol is available as 0.63 mg/3 mL and 1.25 mg/3 mL nebulizer solutions. The recommended adult dose of levalbuterol is 1.25 to 2.5 mg every 20 minutes for three doses, then 1.25 to 5 mg every 1 to 4 hours as needed, or 5 to 7.5 mg/hour continuous nebulization.25
In outpatient settings, monotherapy with inhaled long-acting β-agonists (LABA, salmeterol, Formoterol) has been shown to increase severe and life-threatening asthma exacerbations and asthma-related deaths.30
Subcutaneous β-Agonist Therapy (Epinephrine or Terbutaline)
Subcutaneous β-agonist therapy has a disadvantageous therapeutic-to-toxicity ratio compared with inhaled β2-selective agonists. Although there is no proven value of systemic therapy over aerosol therapy, rapid delivery of β-agonists to the airway may be beneficial in seriously ill asthmatic patients who are at imminent risk for respiratory arrest or in need of intubation and at low risk for β-agonist cardiac toxicity (young asthmatics). In this circumstance, a combination of inhaled and subcutaneously administered β-agonists may be useful. The subcutaneous epinephrine dose for adults is 0.3 to 0.5 mL of a 1 : 1000 dilution (1 mg/mL), depending on age and weight; it may be repeated in the initial management every 20 minutes for three times. An alternative subcutaneous β-agonist agent is subcutaneous terbutaline, 0.25 mg, which can be repeated every 20 minutes for three doses. When subcutaneous terbutaline is compared with subcutaneous epinephrine, equal cardiac side effects are seen. No clinical studies document benefit of subcutaneous terbutaline over subcutaneous epinephrine. Terbutaline is, however, the parenteral agent of choice in pregnancy. β1-Adrenergic stimulators are given subcutaneously with caution to the elderly and to patients with documented or suspected coronary artery disease.49,50
Anecdotal reports have suggested the success of epinephrine administration through the endotracheal tube after respiratory arrest from asthma. Prospective trials are needed in this area. Despite the lack of confirmatory studies, in an asthma patient with respiratory arrest, it may be considered.51
Corticosteroids
Corticosteroids are an essential part of in-hospital asthma therapy. The National Institutes of Health expert panel recommendation is intravenous methylprednisolone, 40 to 80 mg/day in one or two divided doses, until PEF reaches 70% of predicted or personal best. This is substantially lower dosage than the previous recommendations. Initial high-dose steroids with intravenous methylprednisolone 80 to 125 mg/day in divided doses (typically 40 mg every 6 hours) for the first 24 hours was recommended if the PEFR or FEV1 remains less than 50% in severe asthmatics.16 In hospitalized patients the use of higher than standard doses of prednisone (40-80 mg/day) offers no benefit. No differences in clinical effects between oral and intravenous forms of corticosteroid therapy have been proved. In acute severe asthma, intravenous steroids offer benefit with the possibility of early onset of action and peak effect. Some trials show improvement following initiation of steroids after a patient’s condition was refractory to initial therapy. Others show benefit when corticosteroid therapy is initiated early in the course of an acute asthma episode. Most corticosteroid benefit is thought to be delayed for approximately 6 hours, although a potential for earlier beneficial effect has been postulated. The delay in effect may reflect the time necessary for steroids to induce upregulation of new β2-receptors and reversal of β2-receptor desensitization and downregulation. Some patients show corticosteroid resistance. Inhaled corticosteroids can be started any time during the exercebation. Total course of steroids can be 3 to 10 days and less than 1 week; there is no need to taper the dose.13,16,52 Potential benefits of corticosteroids are listed in Box 38.3.
Inhaled Anticholinergic Therapy with Ipratropium
Although ipratropium achieves less bronchodilation at peak effect than β-agonists and less predictable clinical response, the effect is likely to be additive to albuterol. Most published evidence supports the addition of ipratropium to inhaled β-agonist therapy for acute asthma patients. It produces clinically modest improvement in lung function compared with albuterol alone. Addition of multiple high doses of ipratropium bromide in acute severe airflow obstruction in asthma patients in the emergency room has resulted in fewer hospital admissions. After admission to the hospital for acute severe asthma, clinical benefit from the addition of ipratropium is not detected in trials.13,42,53,54
The National Institutes of Health expert panel’s recommended dose of ipratropium is 0.5 mg by nebulizer every 20 minutes for three doses, then every 2 to 4 hours as needed. Onset of action is slow (20 minutes), with peak effectiveness at 60 to 90 minutes and no systemic side effects. A handheld mouthpiece nebulizer system should be used for nebulization because contamination of the ocular area with precipitation of narrow-angle glaucoma may occur in susceptible individuals if a facemask is used for delivery of an anticholinergic agent. Ipratropium may be combined with the nebulized albuterol dose. The deposition of ipratropium may be enhanced, however, when it follows albuterol-induced bronchodilation. In a patient with severe asthma, ipratropium may produce a clinically significant response within minutes of administration, as opposed to the longer delay to response in chronic obstructive pulmonary disease patients with chronic stable disease. If ipratropium is delivered by MDI (0.018 mg/puff), 4 to 8 puffs per treatment is recommended.13,40
Methyxanthines: Theophylline or Aminophylline
Although theophylline or aminophylline is an effective bronchodilator compared with placebo in patients with acute bronchospasm, inhaled β-agonists are accepted to be superior to theophylline as single agents for acute bronchospasm. Consensus opinion and meta-analyses support no significant additive clinical benefit with the addition of theophylline to a full course of inhaled β-agonists. Although a few studies have shown physiologic benefits evident at 24 or 48 hours, the addition of theophylline to high-dose inhaled β-agonist and corticosteroid therapy in patients with acute severe asthma seems to offer no clear-cut or substantial clinical benefit. Methylxanthines are infrequently used for acute asthma because of unpredictable pharmacokinetics and known side effects.40,55–58 Because theophylline toxicity is a potential problem, the use of theophylline should be limited to patients with life-threatening asthma who fail to respond to other therapy.
Magnesium Sulfate
Magnesium has multifactorial actions relative to potential reversal of bronchoconstriction, which is based on characteristics of inhibition of the calcium channel and decreased acetylcholine release. Hashimoto and colleagues59 showed that 40% of asthmatic patients exhibited magnesium deficiency, and that low magnesium erythrocyte concentrations reflected decreased magnesium stores in patients with bronchial asthma.
Traditionally, 2 g of magnesium sulfate is administered over 20 minutes. Repeat doses, if used, require careful monitoring of magnesium levels and assessment for clinical manifestations of toxicity. Magnesium therapy should be avoided in the presence of renal insufficiency.59–61
Heliox
Heliox (mixture of helium and oxygen optimally effective at a 70 : 30 mix) has been shown to improve the delivery and deposition of nebulized albuterol. If a patient requires more than 30% oxygen, it cannot be used. Heliox may be useful in acute severe asthma refractory to conventional treatment. There are no data to support the use of heliox as the initial treatment for acute severe asthma. Heliox is available in mixtures of 60 : 40, 70 : 30, and 80 : 20. Helium is less dense than air and can be delivered through a tight-fitting nonrebreathing mask or, in an intubated patient, through the ventilatory circuit. Heliox results in decreased large airway resistance. One might anticipate that the role of heliox would be limited by the fact that heliox improves flow in large turbulent airways, and most of the obstruction in asthma is in the peripheral airways. Studies have nonetheless shown the ability of heliox to decrease inspiratory and expiratory resistance in severe asthma. Its potential to decrease PEEP not set on the ventilator (auto-PEEP) might be particularly useful. Heliox may augment carbon dioxide removal by facilitating carbon dioxide movement across the endothelial-epithelial barrier compared with the presence of a mixture of oxygen and nitrogen as the carrier gas. Heliox also has been shown to improve oxygenation, which may allow higher helium concentrations to be delivered.62–64
Other Agents
Antibiotics
Antibiotic therapy in an asthma patient is indicated only if bacterial infection is present. The Telithromycin, Chlamydophilia, and Asthma (TELECAST) study reported that, in patients presenting for unscheduled care because of an acute asthma exacerbation, treatment with telithromycin showed a significant improvement in FEV1 over placebo at the end of a 10-day treatment period.65 One of the primary efficacy end points, improvement in asthma symptom scores, was significantly greater in the telithromycin group than placebo. An editorial pointed out the possible anti-inflammatory effects of macrolides in the treatment of asthma but did not recommend this as standard therapy at this time.66
Fluids
If the patient is volume depleted, normal saline or lactated Ringer’s solution is used to reestablish adequate intravenous volume. There is no evidence that excess volume replacement liquefies or facilitates loosening of secretions. Chest physiotherapy and other maneuvers to mobilize secretions physically also are not recommended. Nebulization of acetylcysteine is not indicated and may irritate the airways. Saline solution and acetylcysteine have been successfully used as part of bronchial lavage with fiberoptic bronchoscopy in patients with severe asthma.67
Ketamine
Ketamine is an intravenous analgesic agent that has bronchodilator properties but may stimulate bronchial secretions and laryngospasm and may cause tachycardia, hypertension, delirium, dissociative state, and lowering of seizure threshold. So far no trials have been published to prove its effectiveness. When intubation of a severe acute asthmatic is required, in the absence of hypertension and known seizure disorder, ketamine seems to be an optimal induction agent. It also may be used in life-threatening situations when conventional therapy has failed.40,68
Leukotriene Antagonists
Leukotriene receptor antagonists (LTRAs: montelukast, zafirlukast) improve lung function and are often used in the management of chronic asthma, but their role in acute asthma is unclear. Significant improvement in pulmonary function within 10 minutes of intravenous administration of leukotriene antagonists was demonstrated in one study in patients with severe asthma exercebation.69
Omalizumab (Anti-IgE Antibody)
Recombinant anti-IgE antibody (omalizumab) improves asthma control in severely allergic asthmatics, reducing inhaled steroids and rescue medication requirement and improving asthma-related quality of life. Its role in acute severe asthma is unstudied. The delay in onset of effect likely makes its impact less likely.70
Strunk and Bloomberg reported that patients likely to benefit from omalizumab are patients with evidence of sensitization to perennial aeroallergens who require high doses of inhaled corticosteroids that have a potential for adverse effects, patients with frequent exacerbations of asthma, and patients with severe disease-related noncompliance. Total IgE levels should be measured in all patients, and the recommended dose, 0.016 mg/kg body weight per international unit of IgE every 4 weeks, should be administered subcutaneously at 2- or 4-week intervals. Strunk and Bloomberg recommended adding omalizumab in a compliant patient with severe asthma after a trial of leukotriene modifiers or extended-release theophylline proved ineffective.71
Nontraditional Therapy of Severe Bronchospasm (Table 38.2)
Table 38.2
Adjunct Therapies for Bronchospasm

IgE, immunoglobulin E; IV, intravenous; PEEP, positive end-expiratory pressure.
In a patient with refractory severe asthma who is receiving mechanical ventilatory support, anecdotal success with isoflurane or halothane anesthesia, intravenous thiopental, and rectally administered ether has been reported. Intravenous ketamine has the potential for administration in the ICU and is likely the best alternative for anesthetic therapy. Que and Lusaya anecdotally showed significant improvement when using sevoflurane induction for emergency cesarean section in a woman with severe life-threatening asthma. Maternal and neonatal outcome were good. Propofol has been reported to relax smooth muscle in arteries and veins, and a bronchodilator effect has been suggested. A case series report showed temporally related improvement in severe asthmatic bronchospasm after propofol infusion.72–80
Recent case reports suggest that extracorporeal carbon dioxide removal seems to be a valuable adjunct to mechanical ventilation for life-threatening asthma, and emergency extracorporeal life support (ECLS) as salvage therapy also has been reported.81–86
Acute Severe Asthma in Pregnancy
Asthma complicates 3% to 12% of pregnancies and is perhaps the most common serious medical problem to complicate pregnancy. Exacerbations occur in approximately 20% of all pregnant women with asthma, can occur any time during gestation, but tend to occur late in the second trimester. Acute attacks of asthma during labor are rare. Simultaneous management of the mother and the fetus is a challenge. The goal in approaching a pregnant patient with severe asthma is to prevent maternal hypoxemia.16,87–89
Studies of pregnancy-associated asthma reveal increased incidence of maternal and fetal complications, which include preeclampsia, perinatal fatality, low birth weight, and preterm infants. The risk of uncontrolled asthma is considered to outweigh any risk associated with the use of recommended medications for asthma. It is important that a physician skilled in the management of asthma be involved in the patient’s care. Uterine contractions are common during asthma exacerbation and usually do not progress to preterm labor. When more than one β-adrenergic tocolytic agent is being administered simultaneously (systemically and inhaled), the possibility of excessive systemic effects should be considered. This is particularly true as it pertains to maternal and fetal tachycardia. One study compared perinatal outcomes in 259 pregnant women treated with inhaled β2-adrenergic agonists with 101 pregnant women with asthma not treated with inhaled β2-agonists and with 295 pregnant women without asthma and found no difference in rates of perinatal mortality, congenital malformations, preterm delivery, or delivery of low-birth-weight infants. There also were no differences in Apgar scores, rates of complications of labor or delivery, and postpartum bleeding. Oral steroid use was associated with an increased risk of preterm delivery, although it is difficult to separate the effect of the medication from the effect of exacerbation.90,91
Asthma exacerbations have the potential to lead to severe problems for the fetus. During pregnancy, asthma exacerbations should be managed aggressively. During pregnancy, acute severe asthma is treated initially with inhaled short-acting β-agonist by MDI or nebulizer, one to three doses in the first hour; oxygen; and if no response, oral systemic corticosteroids. If there is severe exacerbation of asthma (PEFR <50%), β-agonist could be given continuously or every 20 minutes, combined with inhaled ipratropium bromide. Fetal assessment and monitoring should be done until the patient is stabilized. To control asthma, there are limited data using leukotriene receptor antagonists in humans during pregnancy, although reassuring animal data are available. Studies and clinical evidence confirm safety of theophylline at recommended doses (to serum concentrations of 5 to 12 µg/mL) during pregnancy. There were higher levels of reported side effects and discontinuation of the medication, however. The experimental animal studies confirm the association of high-dose theophylline and adverse pregnancy outcomes in animals.87–92
Mechanical Ventilation in Asthma Patients
Indications
An increasing arterial PaCO2 despite aggressive therapy is an ominous sign. Although an elevated PaCO2 is not an indication for intubation, an increasing PaCO2 despite aggressive therapy typically calls for intubation and mechanical ventilation. Nowak and colleagues showed that an elevated PaCO2 correlates with FEV1 less than 25% of age-predicted values. Another sign of imminent respiratory failure is paradoxical breathing. Normally, as the diaphragm contracts, it moves caudad, and the abdomen moves out. When the diaphragm fails, it moves cephalad to fix the rib cage and assist the intercostal muscles of inspiration, and in that circumstance the abdomen moves in with inspiration. The intubation technique should be the one in which the operator feels most proficient. The largest endotracheal tube practical should be selected to decrease the degree of auto-PEEP.93
Aerosol Delivery
Aerosol delivery in an intubated patient receiving mechanical ventilatory support poses a significant problem because the nebulized agent reaching the lung parenchyma is markedly reduced, with most of the agent deposited in the endotracheal tube, probably because of its 90-degree curve. It is important to connect the ventilator circuit nebulizer system as close to the patient as possible and to consider increasing the amount of active agent in each treatment, usually double the dose used in a nonintubated patient. An MDI with a spacer is generally accepted as being as effective in delivering bronchodilator medication in patients receiving mechanical ventilation as nebulized therapy, and it costs less. Either method may give varying delivery based on technique and ventilator and patient variables.48
Neuromuscular Blockade
Some patients require neuromuscular blocking agents (NMBAs), especially early in their ventilatory course to control respiratory rate in the presence of life-threatening auto-PEEP and in some patients to facilitate intubation. Status asthmaticus patients receiving mechanical ventilation who are given corticosteroids and NMBAs are at risk for developing prolonged muscle weakness after discontinuation of NMBAs. This is particularly likely in patients with renal impairment, females, associated hypophosphatemia, and higher degrees of paralysis. Concomitant use of corticosteroids and NMBAs typically produces proximal and distal muscle weakness. Creatine kinase may be elevated, and myoglobinuria may be present. Steroid myopathy, by contrast, primarily involves proximal muscles, and the creatine kinase level is normal.94
If NMBAs are needed to initially stabilize the intubated, mechanically ventilated asthmatic (oxygenation, ventilation, and blood pressure), intermittent dosing should be used and they should be discontinued as soon as possible. In the rare patient who needs to be continuously paralyzed, infusions must be stopped every 4 to 6 hours to prevent accumulation and evaluation. A peripheral nerve stimulator should be used to limit paralysis to no less than a recording of one or two twitches in response to a train-of-four stimulus, as opposed to higher degrees of paralysis. The rationale for neuromuscular blockade is to control the respiratory rate, decrease chest wall stiffness, eliminate muscle loading from patient-ventilator dysynchrony and oxygen consumption, and lower the risk of barotrauma. These goals can often be achieved with a three-twitch or four-twitch response. If NMBAs are needed for longer than 24 hours, they should be discontinued daily to ensure patient arousal. Prolonged muscle paralysis may lead to a denervated state with an increased number of steroid receptors. The additive effect of NMBAs and steroids may be related to this finding. In patients with asthma, cisatracurium is the first-choice NMBA because it is eliminated by esterase degradation and spontaneous breakdown in the serum.16,95–97
Initiating Mechanical Ventilation
The strategy of mechanical ventilation is to avoid excessive airway pressures and reduce dynamic hyperinflation. To achieve this goal, it may be necessary to allow “controlled hypoventilation” or “permissive hypercapnia.” Recommended initial ventilator settings are tidal volume of 8 mL/kg ideal body weight and a frequency of 10 to 14 breaths per minute, minute ventilation less than 10 L/minute, and expiratory time increased and targeted to avoid auto-PEEP and to achieve arterial oxygen saturation greater than 90%. If volume ventilation is used, peak inspiratory flow rates should be set at 60 to 80 L/minute with a decelerating waveform. The use of noncompressible tubing facilitates lowering of inspiratory time and adds to expiration time. Total volume is reduced to 6 mL/kg ideal body weight or less to achieve inspiratory plateau pressure (IPP) of 30 cm H2O or less. The way to minimize hyperinflation is to minimize inspiratory time (inspiration-expiration ratio ≤ 1 : 3) while providing adequate oxygenation and ventilation, considering permissive hypercapnia in the latter case. Controlled mechanical ventilation may be required during initial ICU stay and requires heavy sedation and analgesia, or more typically sedation, analgesia, and muscle paralysis. Assisted controlled ventilation without heavy sedation or sedation/paralysis predisposes the patient to hyperinflation if the patient’s breathing rate is high. Heavy sedation or sedation/paralysis reduces carbon dioxide production, facilitates measurement of end-inspiratory and end-expiratory pressures, and facilitates mechanical ventilation of severely asthmatic patients. Pressure-control ventilation is discouraged because of fluctuating high airway resistance and intrinsic PEEP. Keeping the peak alveolar pressures low (IPP of ≤30 cm H2O) prevents overdistention of alveoli distal to the least obstructed airway.98,99
See Box 38.4 for a summary of general mechanical ventilation guidance in severe asthma with hyperinflation.
Auto–Positive End-Expiratory Pressure
Definition and Predisposing Factors
Auto-PEEP occurs when ventilator settings result in an inspiratory-to-expiratory ratio that does not allow adequate expiratory time for total exhalation of the delivered ventilator breath. Because airway obstruction increases expiratory time, mechanically ventilated asthma patients are at increased risk for auto-PEEP. After the first breath delivered in a setting conducive to auto-PEEP, the next breath is delivered before complete emptying of the first breath. With each subsequent breath that fails to empty completely, end-inspiratory and end-expiratory lung volumes increase, as do flow and pressure at end expiration. The increase in lung volume predisposes to the risk of barotraumas, and elevations of intrathoracic pressure decrease cardiac output.100
Increased Work of Breathing with Auto–Positive End-Expiratory Pressure
Auto-PEEP in the presence of assisted ventilation modes (assist control and synchronized intermittent mandatory ventilation) implies that the patient must initiate gas flow by producing an inspiratory effort equal to not only the sensitivity setting of inspiratory triggering, but also the level of auto-PEEP. Extrinsic PEEP normally has no indication, however, in acute severe asthma during controlled mechanical ventilation. If it is not advantageous or possible to eliminate auto-PEEP in the patient with assisted ventilation modes, applying or increasing ventilator-set PEEP to a level slightly below total PEEP would decrease patient effort necessary to trigger the ventilator breath. Inappropriately set ventilator PEEP offers additional risks to the patient, and this approach is recommended only by health care professionals well schooled in the intricacies of mechanical ventilation.98
Barotrauma
In a heavily sedated or sedated/paralyzed patient, IPP measurement is the best means for estimating peak alveolar pressure and the best indicator of hyperinflation and risk for barotraumas because closure of airways toward the end of expiration may lead to underestimation of hyperinflation. In the presence of normal chest wall and abdominal compliance factors, an IPP equal to or greater than 30 cm H2O puts the patient at risk for exceeding maximal alveolar size (total lung capacity). Decreases in IPP are usually accomplished by using low tidal volumes and minimizing auto-PEEP. In a paralyzed patient, the collection of the total exhaled volume of gas obtainable with 20 to 60 seconds of apnea allows the measurement of end-inspired volume above apnea functional residual capacity. This volume may be 3 L despite the use of small tidal volumes.101,102
Because high IPP is reduced by lowering tidal volume, a reduction in the tidal volume directly decreases the risk for barotraumas, as does any maneuver that decreases auto-PEEP. If, after heavy sedation or sedation/paralysis, the IPP remains 30 cm H2O or greater despite decreases in tidal volume to the lowest value that allows an acceptable pH, typically greater than or equal to 7.20, the use of bicarbonate infusion to allow acceptable pH with further reduction of tidal volume may be considered.103
The concept of reducing the tidal volume and accepting a higher PaCO2 and lower pH is called permissive hypercapnia. Permissive hypercapnia is relatively safe in asthma and well tolerated in the absence of contraindications, such as increased intracranial hypertension and pregnancy. Physicians are uncomfortable with allowing PaCO2 to become greater than 80 to 100 mm Hg, but several case reports described short durations of hypercapnia (>150 mm Hg) as well tolerated, and one report described PaCO2 of 200 mm Hg for 10 hours with no consequences reported. Permissive hypercapnia limits volutrauma and hemodynamic consequences of increased intrathoracic pressures. In normoxic states, acute hypercapnia has limited potential for inducing severe intracellular acidosis. If permissive hypercapnia results in pH less than 7.2, increased sedation and paralysis and methods of decreasing carbon dioxide production, such as reducing fever, overfeeding, and patient effort, should be considered. In the absence of tissue hypoperfusion or volume overload, iatrogenic compensation for acute respiratory acidosis with intravenous bicarbonate administration is appropriate. In severe acidosis, large amounts of bicarbonate may be necessary to elevate the pH substantially, potentially leading to volume overload.104–107
Non-invasive Positive-Pressure Ventilation
Noninvasive positive-pressure ventilation (NPPV) is a safe treatment and can reduce the need for intubation in a selected group of patients with severe asthma and hypercapnia who fail to improve with initial medical management. Fernandez and colleagues further describe that the rationale for NPPV in severe asthma is its potential for improving alveolar ventilation, decreasing the risk of respiratory muscle fatigue. Mask continuous positive airway pressure (CPAP) produces bronchodilation and decreases the airway resistance, reverses atelectasis, and promotes removal of secretions. The work of the diaphragm and the inspiratory muscles is reduced, and intrinsic PEEP may be offset. In addition, CPAP decreases the adverse hemodynamic effects of large negative inspiratory swings in pleural pressure, which compromise right and left ventricular performance. NPPV potentially may enhance delivery of inhaled β-agonists. One study showed a small but statistically significant greater improvement in PEFR when NPPV was used for initial β-agonist delivery. A nonrandomized study by Meduri and associates reported a reduction in PaCO2 and improvement in dyspnea with the use of NPPV in 17 episodes of asthma with acute respiratory failure.108–111
CPAP, as opposed to NPPV, has been studied in asthmatic patients after induction of bronchospasm with aerosolized histamine. CPAP of 12 cm H2O increased the minimal pleural pressure and decreased swings in transdiaphragmatic pressure. Although ventilation increased, the inspiratory work per liter decreased significantly, as did the pressure time product for the inspiratory muscles. Functional residual capacity increased slightly. The authors concluded that CPAP produced a load on the inspiratory muscles, improving their efficiency and decreasing the energy cost of inspiration. Despite the potential for benefit of NPPV or CPAP in selected patients in severe acute asthma, it should be used with caution pending controlled clinical trials. Ram and colleagues112 also concluded in a Cochrane analysis that application of NPPV in status asthmaticus, despite some promising preliminary results, remains controversial. The need for endotracheal intubation in acute severe asthma has decreased after the introduction of NPPV as shown by a retrospective cohort study. If NPPV is used, initial setting should be an expiratory positive airway pressure (CPAP or PEEP) of about 5 cm H2O and inspiratory pressure (or pressure support) of approximately 8 cm H2O. If tidal volumes are shallow (<7 mL/kg), inspiratory pressure can be increased gradually by 2 cm H2O every 15 minutes, to a goal to reduce the respiratory rate to less than 25 breaths per minute. Peak pressures greater than 15 to 20 cm H2O rarely can be tolerated without mask leaks or discomfort or claustrophobia.19 See Box 38.5 for contraindications for NPPV.108–114
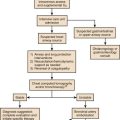
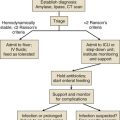
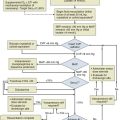
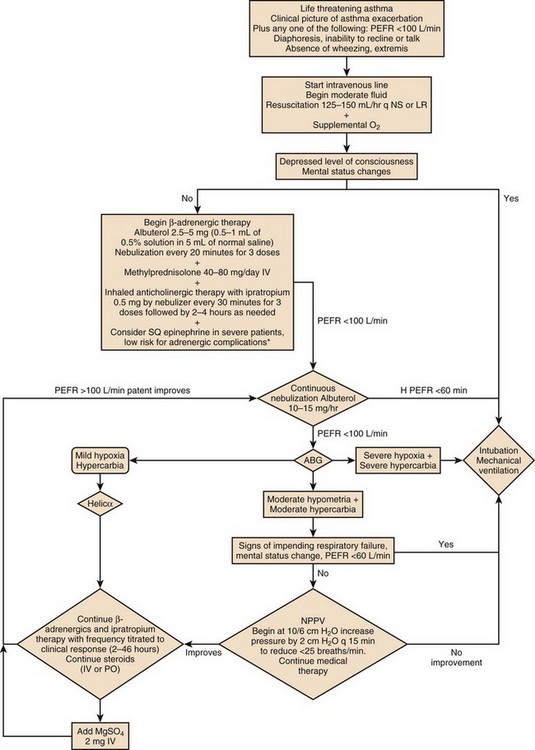
Approach to Acute Life-Threatening Asthma
Figure 38.1 is an algorithm that shows the approach to acute life-threatening asthma.
Key Points
• The cause of respiratory arrest in asthmatic patients is usually failure of the inspiratory muscles with ventilatory arrest.
• Ominous signs and findings in a patient with acute severe asthma, if not quickly reversed, include diaphoresis, inability to recline, inability to talk, increased PaCO2, PEF less than 60 L/minute or FEV less than 25% of personal best or predicted values, and use of accessory muscles.
• Pathophysiology of asthma consists of three key abnormalities: bronchoconstriction, airway inflammation, and mucous impaction.
• The ADAM33 gene is significantly associated with asthma.
• Inhaled β2-agonists are the cornerstone of medical therapy for hospitalized severe asthmatics. The addition of steroids is recommended.
• Adequate delivery of β-agonists can be accomplished by use of an MDI with a spacer during acute bronchospasm if proper technique is used and doses are increased. It is advisable to deliver the β-agonist by nebulization in most acutely ill asthma patients because nebulization requires minimal coordination and cooperation of the patient and less bedside instruction and supervision by health care professionals.
• Larger doses and more frequent dosing intervals for inhaled β-agonist therapy are needed in acute severe asthma because of decreased deposition at the site of action (low tidal volumes and narrowed airways), alteration in dose-response curve, and altered duration of activity. Continuous nebulization is an option.
• Theophylline is not recommended for general use in hospitalized patients with acute asthma (Table 38.3). Intravenous magnesium is recommended for the most severe asthma attacks.
Table 38.3
Treatments Not Recommended in Acute Severe Asthma

From National Heart, Lung, and Blood Institute and National Asthma Education and Prevention Program Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Full report. NIH Publication No. 08-5846. Bethesda, MD, National Institutes of Health, 2007.
• Although there is no proven value of systemic therapy over aerosol therapy, rapid delivery of β-agonists to the airway may be beneficial in seriously ill asthmatic patients who are at imminent risk for respiratory arrest or need intubation and who are at low risk for β-agonist cardiac toxicity (young asthmatics). In these patients, a combination of inhaled and subcutaneously administered β-agonists in this circumstance may be useful.
• If NMBAs are used, attempts to withdraw these agents over the first 24 to 48 hours after intubation are advised.
• Auto-PEEP is treated by decreasing total inspiratory time. Increasing expiratory time is best accomplished by decreasing rate. Decreasing tidal volume also is effective. Prolonging expiratory time is a diagnostic and a therapeutic maneuver.
• The concept of reducing the tidal volume to decrease alveolar inflation and accepting a higher PaCO2 and lower pH is called permissive hypercapnia and should be considered in asthma patients undergoing mechanical ventilation therapy with life-threatening asthma, severe hyperinflation, and high IPP.
References
1. Akinbami LJ, Moorman JE, Bailey C, et al: Trends in Asthma Prevalence, Health Care Use, and Mortality in the United States, 2001-2010, NCHS Data Brief, No. 94, May 2012.
2. American Lung Association. Trends in Asthma Morbidity and Mortality. New York: American Lung Association Epidemiology and Statistics Unit Research and Program Services; May 2005.
3. Bousquet J, Khaltaev N, eds. Global Surveillance, Prevention and Control of Chronic Respiratory Diseases: A Comprehensive Approach. Geneva: World Health Organization, 2007.
4. Oguzulgen, IK, Turktas, H, Mullaoglu, S, Ozkan, S. What can predict the exercebation severity in asthma? Allergy Asthma Proc. 2007; 28(3):344–347.
5. Rodrigo, GJ, Rodrigo, C, Hall, JB. Acute asthma in adults. Chest. 2004; 125:1081–1102.
6. Asthma fact sheet. accessed at http://www.epa.gov/asthma/pdfs/asthmafactsheeten.pdf, April 2011.
7. Trends in Asthma Morbidity and Mortality. accessed at http://www.lung.org/finding-cures/our-research/trend-reports/asthma-trend-report, July 2011.
8. Killian, KJ, Watson, R, Otis, J, et al. Symptom perception during acute bronchoconstriction. Am J Respir Crit Care Med. 2000; 162:490.
9. Pendergraft, TB, Stanford, RN, Beasley, R, et al. Rates and characteristics of intensive care unit admissions and intubations among asthma-related hospitalizations. Ann Allergy Asthma Immunol. 2004; 93:29–35.
10. Rogers, L, Reibman, J. Pharmacologic approaches to life-threatening asthma. Ther Adv Respir Dis. 2011; 5:397–408.
11. Rodriguez-Trigo, G, Picado, V, Sanchis, J. Management according to global initiative for asthma guidelines of patients with near-fatal asthma reduces morbidity and mortality. Arch Broncopneumol. 2008; 44:192–196.
12. Kolbe, J, Fergusson, W, Vamos, M, Garrett, J. Case control study of severe life threatening asthma (SLTA) in adults: Psychological factors. Thorax. 2002; 57:317–322.
13. NIH Publication No. 08-5846National Heart, Lung, and Blood Institute and National Asthma Education and Prevention Program Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma Full report. Bethesda, MD: National Institutes of Health, 2007.
14. Strek, ME. Difficult asthma. Proc Am Thorac Soc. 2006; 3:116–123.
15. Papiris, S, Kotanidou, A, Malagari, K, et al. Clinical review: Severe asthma. Crit Care. 2002; 6:30–44.
16. Rodrigo, GJ, Rodrigo, C, Hall, JB. Acute asthma in adults: A review. Chest. 2004; 125:1081–1102.
17. Rodriguez-Roisin, R. Acute severe asthma: Pathophysiology and pathobiology of gas exchange abnormalities. Eur Respir J. 1997; 10:1359.
18. Manthous, CA, Goulding, P. The effect of volume infusion on dead space in mechanically ventilated patients with severe asthma. Chest. 1997; 112:843.
19. Rosen, FS, MacKay, I. The immunology series comes to an end. N Engl J Med. 2001; 345:1343–1344.
20. Busse, WW, Lemanske, RF. Asthma. N Engl J Med. 2001; 344:350–362.
21. Sad, S, Marcotte, R, Mosmann, TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995; 2:27–29.
22. Strachan, DP. Hay fever, hygiene, and household size. BMJ. 1989; 299:1259–1260.
23. Mattes, J, Karmaus, W. The use of antibiotics in the first year of life and development of asthma: Which comes first? Clin Exp Allergy. 1999; 29:729–732.
24. Abdulamir, AS, Hafidh, RR, Abudbakar, F, Abbas, KA. Changing survival, memory cell compartment, and T-helper balance of lymphocytes between severe asthma and mild asthma. BMC Immunol. 2008; 9:73.
25. National Asthma Education and Prevention Program, Expert Panel Report: Guidelines for the Diagnosis and Management of Asthma: Update on Selected Topics 2002 NIH publication No. 02-5074. National Institutes of Health, Bethesda, MD, 2003.
26. Shapiro, SD, Owen, CA. ADAM-33 surfaces as an asthma gene. N Engl J Med. 2002; 347:936–938.
27. Ober, C, Hoffjan, S. Asthma genetics 2006: The long and winding road to gene discovery. Genet Immun. 2006; 7:95–100.
28. McFadden, ER, Jr., Kiser, R, DeGroot, WJ. Acute bronchial asthma: Relations between clinical and physiologic manifestations. N Engl J Med. 1973; 288:221.
29. Gerschke, GL, Baker, FJ, Rosen, P. Pulsus paradoxus as a parameter in treatment of the asthmatic. J Am Coll Emerg Physicians. 1977; 6:191.
30. Salpeter, SR, Buckley, NS, Ormiston, TM, Salpeter, EE. Meta-analysis: Effect of long acting beta agonists on severe asthma exercebations and asthma related deaths. Ann Intern Med. 2006; 144(12):904–912.
31. Murray, DM, Lawler, PG. All that wheezes is not asthma: Paradoxical vocal cord movement presenting as severe acute asthma requiring ventilatory support. Anesthesia. 1998; 53:1006.
32. Fishman, AP. Cardiac asthma—A fresh look at an old wheeze. N Engl J Med. 1989; 320:1346.
33. Blitz, M, Blitz, S, Beasely, R, et al. Inhaled magnesium sulfate in the treatment of acute asthma. Cochrane Database Syst Rev. 2005.
34. Appel, D, Rubenstein, R, Schrager, K, et al. Lactic acidosis in severe asthma. Am J Med. 1983; 75:580.
35. O’Connell, MB, Iber, C. Continuous intravenous terbutaline infusions for adult patients with status asthmaticus. Ann Allergy. 1990; 64:213.
36. Yanos, J, Wood, LD, Davis, K, et al. The effect of respiratory and lactic acidosis on diaphragm function. Am Rev Respir Dis. 1993; 147:616.
37. Kim, M, Cho, Y, Moon, H, Cho, S. Factors for poor prognosis of near-fatal asthma after recovery from a life threatening asthma attack. Korean J Intern Med. 2008; 23(4):170–175.
38. Harkins, MS, Fiato, KL, Iwamoto, GK. Exhaled nitric oxide predicts asthma exacerbation. J Asthma. 2004; 41(4):471–476.
39. Harris, L. Comparison of the effect on blood gases, ventilation, and perfusion of isoproterenol-phenylephrine and salbutamol aerosols in chronic bronchitis with asthma. J Allergy Clin Immunol. 1972; 49:63.
40. American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 10. 5: Near fatal asthma. Circulation. 2005; 112:1–142.
41. Inwald, D, Roland, M, Kuitert, L, et al. Oxygen treatment for acute severe asthma. BMJ. 2001; 323:98–100.
42. Rodrigo, GJ, Rodriquez Verde, M, Peregalli, V, Rodrigo, C. Effects of short-term 28% and 100% oxygen on arterial carbon dioxide tension and peak expiratory flow rate in acute athma: A randomized trial. Chest. 2003; 124:1312–1317.
43. Moloney, E, O’Sullivan, S, Hogan, T, et al. Airway dehydration: A therapeutic target in asthma? Chest. 2002; 121:1806–1811.
44. Lin, RY, Smith, AJ, Hergenroeder, P. High serum albuterol levels and tachycardia in adult asthmatics treated with high-dose continuously aerosolized albuterol. Chest. 1993; 103:221.
45. Rodrigo, C, Rodrigo, G. Salbutamol treatment of acute severe asthma in the ED: MDI versus hand-held nebulizer. Am J Emerg Med. 1998; 16:637.
46. Jasper, AC, Mohsenifar, Z, Kahan, S, et al. Cost-benefit comparison of aerosol bronchodilator delivery methods in hospitalized patients. Chest. 1987; 91:614.
47. Mandelberg, A, Chen, E, Noviski, N, et al. Nebulized wet aerosol treatment in emergency department—Is it essential? Chest. 1997; 112:1501.
48. Salmeron, S, Brochard, L, Mal, H, et al. Nebulized versus intravenous albuterol in hypercapnic acute asthma: A multicenter, double-blind, randomized study. Am J Respir Crit Care Med. 1994; 149:1466.
49. Appel, D, Karpel, P, Sherman, M. Epinephrine improves expiratory airflow rates in patients with asthma who do not respond to inhaled metaproterenol sulfate. J Allergy Clin Immunol. 1989; 84:90.
50. Amory, DW, Burnham, SC, Cheney, FW. Comparison of the cardiopulmonary effects of subcutaneously administered epinephrine and terbutaline in patients with reversible airway obstruction. Chest. 1975; 67:279.
51. Liebman, JB. Should epinephrine be administered exclusively by the endotracheal route in respiratory arrest secondary to asthma? Am J Emerg Med. 1997; 15:106.
52. Manser, R, Reid, D, Abramson, M. Corticosteroids for acute severe asthma in hospitalized patients. Cochrane Database Syst Rev. (1):2001.
53. Aaron, SD. The use of ipratropium bromide for the management of acute severe asthma exacerbation in adults and children: A systematic review. J Asthma. 2001; 38:521–530.
54. O’Driscoll, BR, Taylor, RJ, Horsley, MG, et al. Nebulized salbutamol with and without ipratropium bromide in acute airflow obstruction. Lancet. 1989; 24:1418.
55. Littenberg, B. Aminophylline in severe, acute asthma: A meta-analysis. JAMA. 1988; 259:1678.
56. Huang, D, O’Brien, RG, Harman, E, et al. Does aminophylline benefit adults admitted to the hospital for an acute exacerbation of asthma? Ann Intern Med. 1993; 119:1155.
57. Coleridge, J, Cameron, P, Epstein, J, et al. Intravenous aminophylline confers no additional benefit in acute asthma treated with intravenous steroids and inhaled bronchodilators. N Z Med J. 1993; 23:348.
58. Rodrigo, C, Rodrigo, G. Treatment of acute asthma: Lack of therapeutic benefit and increase of the toxicity from aminophylline given in addition to high doses of salbutamol delivered by metered-dose inhaler. Chest. 1994; 106:1071.
59. Hashimoto, Y, Nishimura, Y, Maeda, H, et al. Assessment of magnesium status in patients with bronchial asthma. J Asthma. 2000; 37:489.
60. Rowe, BH, Bretzlaff, JA, Bourdon, C, et al. Magnesium sulfate for treating exacerbations of acute severe asthma in the emergency department. Cochrane Database Syst Rev. 2000.
61. Silverman, RA, Osborn, H, Runge, J, et al. IV magnesium in the treatment of severe asthma: A multicenter randomized controlled trial. Chest. 2002; 122:489–497.
62. Rodrigo, GA, Rodrigo, C, Pollack, CV, et al. Use of helium-oxygen mixture in the treatment of acute asthma: A systematic review. Chest. 2003; 123:891–896.
63. Reuben, AD, Harris, AR. Heliox for asthma in the emergency department: A review of the literature. Emerg Med J. 2004; 21:131–135.
64. Schaeffer, EM, Pohlman, A, Morgan, S, et al. Oxygenation in status asthmaticus improves during ventilation with helium-oxygen. Crit Care Med. 1999; 27:2666.
65. Johnston, SL, Blasi, F, Black, PN, et al. The effect of telithromycin in acute exacerbations of asthma. N Engl J Med. 2006; 354:1589–1600.
66. Little, FF. Treating acute asthma with antibiotics—not quite yet. N Engl J Med. 2006; 354:1632–1634.
67. Corbridge, T, Hall, JB. State of the art: The assessment and management of adults with status asthmaticus. Am J Respir Crit Care Med. 1995; 151:1296.
68. Stather, DR, Stewart, TE. Clinical review: Mechanical ventilation in severe asthma. Crit Care. 2005; 9:581–587.
69. Camargo, CA, Jr., Smithline, HA, Malice, MP, et al. A randomized controlled trial of intravenous montelukast in acute asthma. Am J Respir Crit Care Med. 2003; 167(4):528–533.
70. Holgate, ST, Chuchalin, AG, Hebert, J, et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody in severe allergic asthma. Clin Exp Allergy. 2004; 34:632–638.
71. Strunk, RC, Bloomberg, GD. Omalizumab for asthma. N Engl J Med. 2006; 354:2689–2695.
72. Rosseel, P, Lauwers, LF, Baute, L. Halothane treatment in life-threatening asthma. Intensive Care Med. 1985; 11:241.
73. Schwartz, SH. Treatment of status asthmaticus with halothane. JAMA. 1984; 251:2688.
74. Robertson, CE, Sinclair, CJ, Steedman, D, et al. Use of ether in life-threatening acute severe asthma. Lancet. 1985; 1:187.
75. Grunberg, G, Cohen, JD, Keslin, J, et al. Facilitation of mechanical ventilation in status asthmaticus with continuous intravenous thiopental. Chest. 1991; 99:1216.
76. Corssen, G, Gutierrez, J, Reves, JG, et al. Ketamine in the anaesthetic management of asthmatic patients. Anesth Analg. 1972; 51:588.
77. Que, JC, Lusaya, VO. Sevoflurane induction for emergency cesarean section in a parturient in status asthmaticus. Anesthesiology. 1999; 90:1475.
78. Bentley, GN, Gent, JP, Goodchild, CS. Vascular effects of propofol: Smooth muscle relaxation in isolated veins and arteries. Pharm Pharmacol. 1989; 41:797.
79. Gigarini, I, Bonnet, F, Lorino, AM, et al. Comparison of the effects of fentanyl on respiratory mechanics under propofol or thiopental anaesthesia. Acta Anaesth Scand. 1990; 34:253.
80. Pedersen, CM. The effect of sedation with propofol on postoperative bronchoconstriction in patients with hyperreactive airway disease. Intensive Care Med. 1992; 18:45.
81. Lang, DM, Simon, RA, Mathison, DA, et al. Safety and possible efficacy of fiberoptic bronchoscopy with lavage in the management of refractory asthma with mucous impaction. Ann Allergy. 1991; 67:324.
82. Franzen, D, Günther, H, Borberg, H, et al. Plasma exchange: An option for the treatment of life-threatening status asthmaticus in pregnancy. Eur Respir J. 1999; 13:938.
83. Wilber, ST, Wilson, JE, Blanda, M, et al. The bronchodilator effect of intravenous glucagon in asthma exacerbation: A randomized, controlled trial. Ann Emerg Med. 2000; 36:427.
84. Rishani, R, El-Khatib, M, Mroueh, S. Treatment of severe status asthmaticus with nitric oxide. Pediatr Pulmonol. 1999; 28:451.
85. Stuart, EC, Paramasivam, K, Oram, J, et al. Pumpless extracorporeal carbon dioxide removal for life threatening asthma. Crit Care Med. 2007; 35(3):945–948.
86. Mikkelsen, ME, Pugh, ME, Hansen-Flaschen, JH, et al. Emergency extracorporeal life support for asphyxic status asthmaticus. Respir Care. 2007; 52(11):1525–1529.
87. Venkataraman, MT, Shanies, HM. Pregnancy and asthma. J Asthma. 1997; 34:265.
88. Murphy, VE, Clifton, VL, Gibson, PG. Asthma exacerbations during pregnancy: Incidence and association with adverse pregnancy outcomes. Thorax. 2006; 61:169–176.
89. Schatz, M, Zeiger, RS, Hoffman, CP. Intrauterine growth is related to gestational pulmonary function in pregnant asthmatic women. Chest. 1990; 98:389.
90. Schatz, M, Zeiger, RS, Harden, KM, et al. The safety of inhaled beta-agonist bronchodilators during pregnancy. J Allergy Clin Immunol. 1988; 82:686.
91. Murphy, VE, Gibson, P, Talbot, PI, et al. Severe asthma exacerbations during pregnancy. Obstet Gynecol. 2005; 106(5 Pt 1):1046–1054.
92. NIH publication No. 05-3279Quick Reference from the Working Group Report on Managing Asthma during Pregnancy: Recommendations for Pharmacologic Treatment Update 2004. Bethesda, MD: National Institutes of Health, 2004.
93. Nowak, RM, Tomlanovich, MC, Sarker, DD, et al. Arterial blood gases and pulmonary function testing in acute bronchial asthma: Predicting patient outcomes. JAMA. 1983; 249:2043.
94. Layzer, R. Neuromuscular Manifestations of Systemic Disease. Philadelphia: Davis; 1985.
95. Papiris, S, Kotanidou, A, Malagari, K, Roussos, C. Clinical review: Severe asthma. Crit Care. 2002; 6(1):30–44.
96. Drachman, D, Stanley, E, Pestronik, A. Neural regulation of muscle properties. In: Serrantrice G, Cros D, Desnuelle C, et al, eds. Neuromuscular Diseases. New York: Raven Press, 1984.
97. DuBois, DC, Almon, RR. A possible role for glucocorticoids in denervation atrophy. Muscle Nerve. 1981; 4:370.
98. Oddo, M, Feihl, F, Schaller, MD, et al. Management of mechanical ventilation in acute severe asthma: Practical aspects. Intensive Care Med. 2006; 32:501–510.
99. Zimmerman, JL, Dellinger, RP, Shah, AN, et al. End tracheal intubation and mechanical ventilation in severe asthma. Crit Care Med. 1993; 21:1727.
100. Smyth, RJ. Ventilatory care in status asthmaticus. Can Respir J. 1998; 5:485.
101. Leatherman, JW, Ravenscraft, SA, Iber, C, et al. Does measured auto-PEEP accurately reflect the degree of dynamic hyperinflation during mechanical ventilation of status asthma? Am Rev Respir Dis. 1993; 147:877A.
102. Tuxen, DV, Williams, TJ, Scheinkestel, CD, et al. Use of a measurement of pulmonary hyperinflation to control the level of mechanical ventilation in patients with acute severe asthma. Am Rev Respir Dis. 1992; 146:1136.
103. Menitove, SM, Goldring, RM. Combined ventilator and bicarbonate strategy in the management of status asthmaticus. Am J Med. 1983; 74:898.
104. Bidani, A, Tzouanakis, AE, Cardenas, VJ, et al. Permissive hypercapnia in acute respiratory failure. JAMA. 1994; 272:957.
105. Tuxen, DV. Permissive hypercapnic ventilation. Am J Respir Crit Care Med. 1994; 150:870.
106. Mutlu, GM, Factor, P, Schwartz, DE, et al. Severe status asthmaticus: Management with permissive hypercapnea and inhalation anesthesia. Crit Care Med. 2002; 30:477–480.
107. Adnet, E, Plaisance, P, Borron, SW, et al. Prolonged severe hypercapnia complicating near fatal asthma in a 35-year-old woman. Intensive Care Med. 1998; 24:1335.
108. Fernandez, MM, Villagra, A, Blanch, L, et al. Non-invasive mechanical ventilation in status asthmaticus. Intensive Care Med. 2001; 27:486–492.
109. Martin, JG, Shore, S, Engel, LA. Effect of continuous positive airway pressure on respiratory mechanics and pattern of breathing in induced asthma. Am Rev Respir Dis. 1982; 126:817.
110. Pollack, CV, Fleisch, KB, Dowsey, K. Treatment of acute bronchospasm with β-adrenergic agonist aerosols delivered by a nasal bilevel positive airway pressure circuit. Ann Emerg Med. 1995; 26:552.
111. Meduri, G, Cook, TR, Turner, RE, et al. Noninvasive positive pressure ventilation in status asthmaticus. Chest. 1996; 110:767.
112. Ram, FS, Wellington, S, Rowe, B, et al. Non-invasive positive pressure ventilation for treatment of respiratory failure due to severe acute exacerbations of asthma. Cochrane Database Syst Rev. (3):2005.
113. Mannam, P, Siegel, MD. Analytic review: Management of life threatening asthma in adults. J Intensive Care Med. 2010; 25:3.
114. Murase, K, Tomii, K, Chin, K, et al. The use of noninvasive ventilation for life threatening asthma attacks: Changes in the need for intubation. Respirology. 2010; 15:714–720.
115. Lugogo, NL, MacIyntyre, NR. Life threatening asthma: Pathophysiology and management. Respir Care. 2008; 53(6):726–739.
116. Wieb, K, Rowe, BH. Nebulized racemic epinephrine used in the treatment of severe asthmatic exacerbation: A case report and literature review. CJEM. 2007; 9(4):304–308.
117. Adoun, M, Frat, JP, Doré, P, et al. Comparison of nebulized epinephrine and terbutaline in patients with acute severe asthma: A controlled trial. J Crit Care. 2004; 19(2):99–102.
118. Dhand, R, Mercier, EM. Effective inhaled drug administration to mechanically ventilated patients. Expert Opin Drug Delivery. 2007; 4:47–61.
119. Hull, JH, Castle, N, Knight, RK, Ho, TB. Nebulised DNase in the treatment of life threatening asthma. Resuscitation. 2007; 74(1):175–177.
120. Thompson, H, Harper, NJ, Parkes, A. Use of AnaconDa anesthetic delivery system to treat life threatening asthma. Anesthesia. 2007; 62:289–300.
121. Cygan, J, Trunsky, M, Corbridge, T. Inhaled heroin-induced status asthmaticus: Five cases and a review of the literature. Chest. 2000; 117:272.
122. Tan, WC. Viruses in asthma exacerbations. Curr Opin Intern Med. 2005; 4:178–183.
123. Hasler, G, Gergen, PJ, Kleinbaum, DG, et al. Asthma and panic in young adults. Am J Respir Crit Care Med. 2005; 171:1224–1230.