Lens
Normal Anatomy
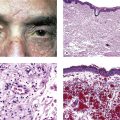
I. The lens (Fig. 10.1) is a transparent biconvex disc.
C. The equatorial diameter is approximately 6–6.5 mm at birth and 9–9.5 mm after 65 years of age.
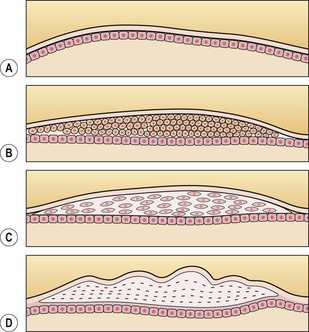
II. The lens is not only transparent but also has an inverted surface epithelium that grows inward at the equator.
A. New cells are formed constantly during life and are laid down externally to the older cells.
D. Normally, no blood vessels or nerves are present in or attached to the lens.
General Information
Congenital Anomalies
Introduction
Mittendorf’s Dot
See Fig. 12.2.
Congenital Aphakia
Fleck Cataract
Anterior Polar Cataract
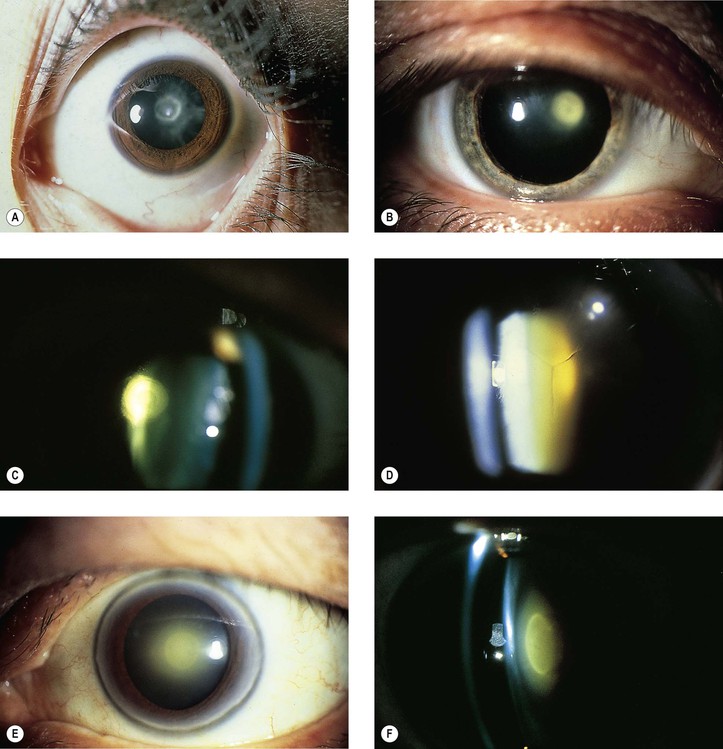
I. Anterior polar cataracts (Fig. 10.2) constitute approximately 3% of congenital cataracts.
Posterior Polar Cataract
I. Clinically and histologically, a posterior polar cataract appears similar to an acquired posterior subcapsular cataract (PSC: Fig. 10.3; see Fig. 10.2).
II. The cataract, rarer than its anterior counterpart, is usually stationary but may be progressive when associated with persistent hyperplastic primary vitreous (see Chapter 18). When this occurs, a dehiscence in the lens capsule may be seen posteriorly, with fibrovascular tissue within the posterior lens substance.
Anterior Lenticonus (Lentiglobus)
Posterior Lenticonus (Lentiglobus)
I. Posterior lenticonus (see Fig. 18.24), more properly called lentiglobus, consists of a spherical elevation or ridge on the posterior surface of the lens and is more common than its anterior counterpart. Clinically, an oil globule-like reflex is seen in the pupillary area of the lens.
III. The cause is unknown; the condition may occur in Lowe’s syndrome (see later).
Other Congenital Cataracts
I. Autosomal-dominant congenital cataract (ADCC)
A. ADCC is usually bilaterally symmetric but may be unilateral.
B. The lens opacity may be zonular, fetal nuclear pulverulent, nuclear, or sutural (or any combination; see Fig. 10.2).
III. Cataracts secondary to intrauterine infection
A. Anterior subcapsular cataract (see later in this chapter)
B. Posterior subcapsular cataract (see later in this chapter)
C. Rubella cataract (see Fig. 2.12)
IV. Galactosemia cataract (see later in this chapter)
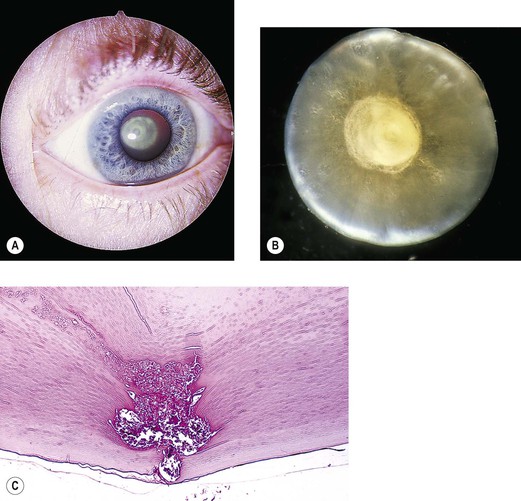
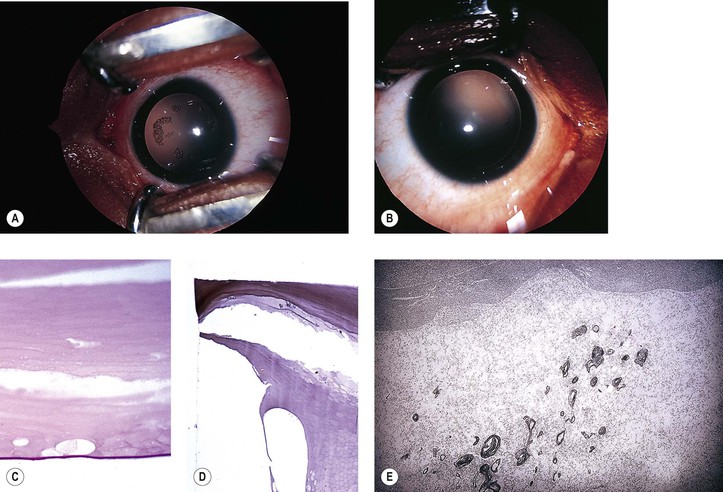
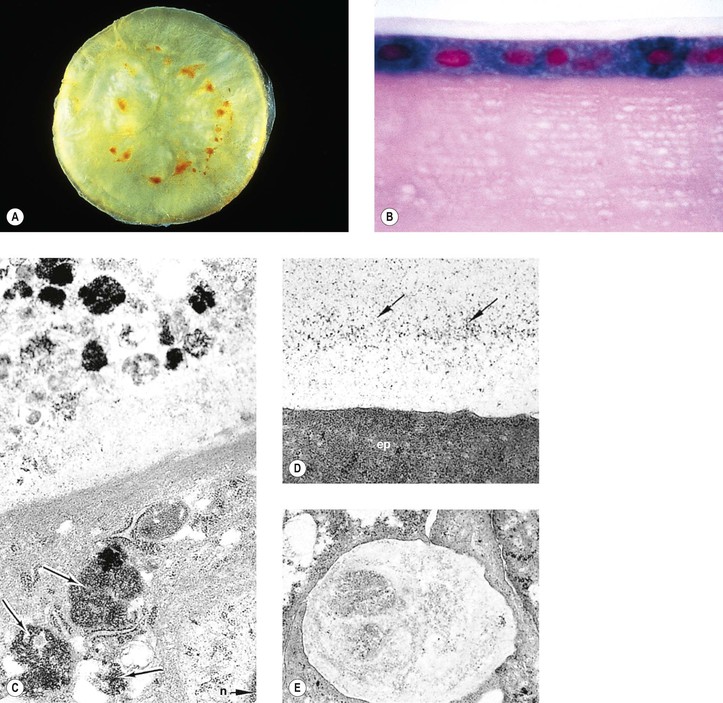
V. Transient neonatal lens vacuoles (Fig. 10.4)
C. Histologically, swelling of lens cortical cells in several lamellae of both anterior and posterior (mainly) cortex is seen.
1. Large, “watery” cortical vacuoles with a nonstaining content are found.
2. Lipoidal degenerative products are also seen in the involved areas.
Capsule (Epithelial Basement Membrane)
General Reactions
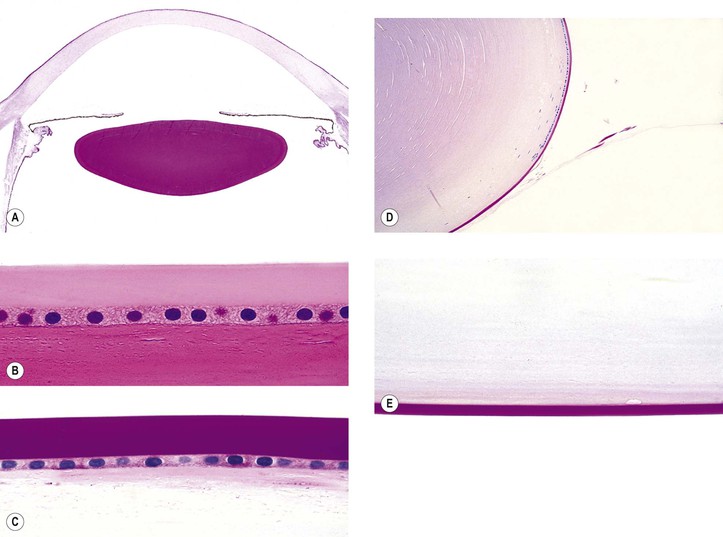
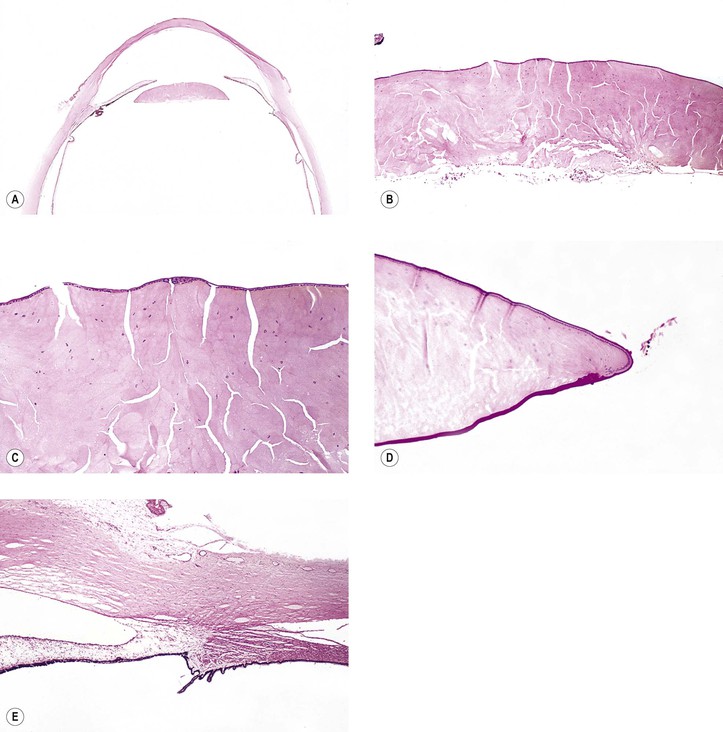
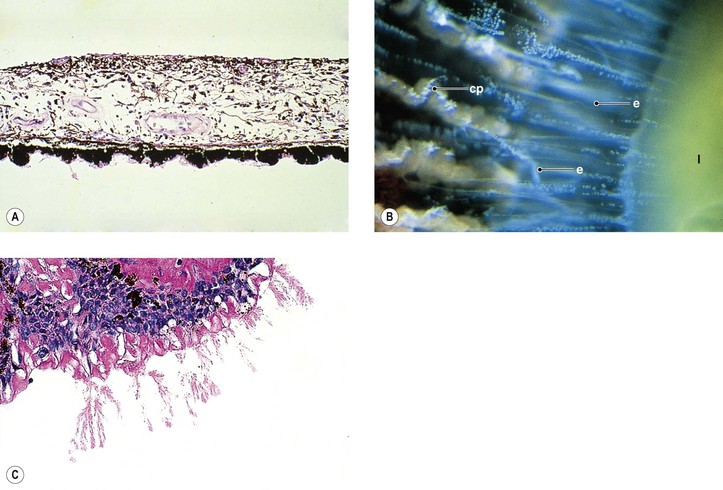
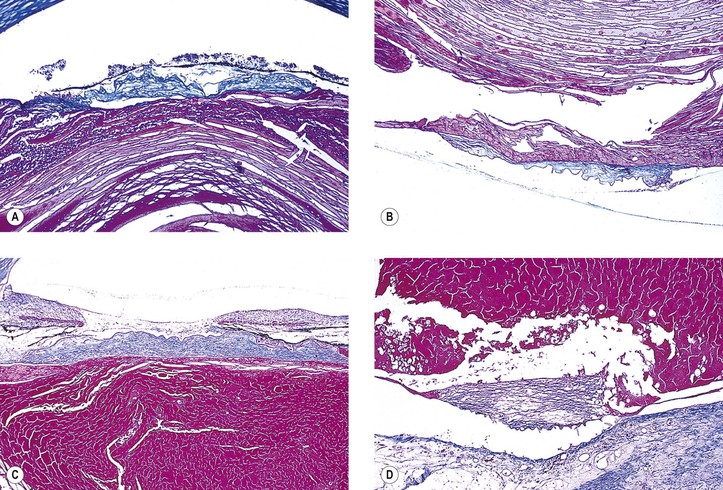
I. The lens capsule, which is the thickest basement membrane in the body, is elastic, easily molded, and resists rupture (Fig. 10.5). It is impermeable to the passage of most particulate matter (e.g., bacteria and inflammatory cells).
II. The lens capsule can undergo marked thinning, as in a mature cataract, or focal thickening, as in Lowe’s syndrome.
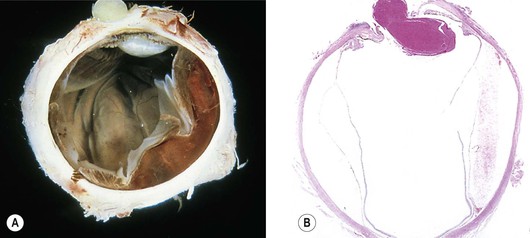
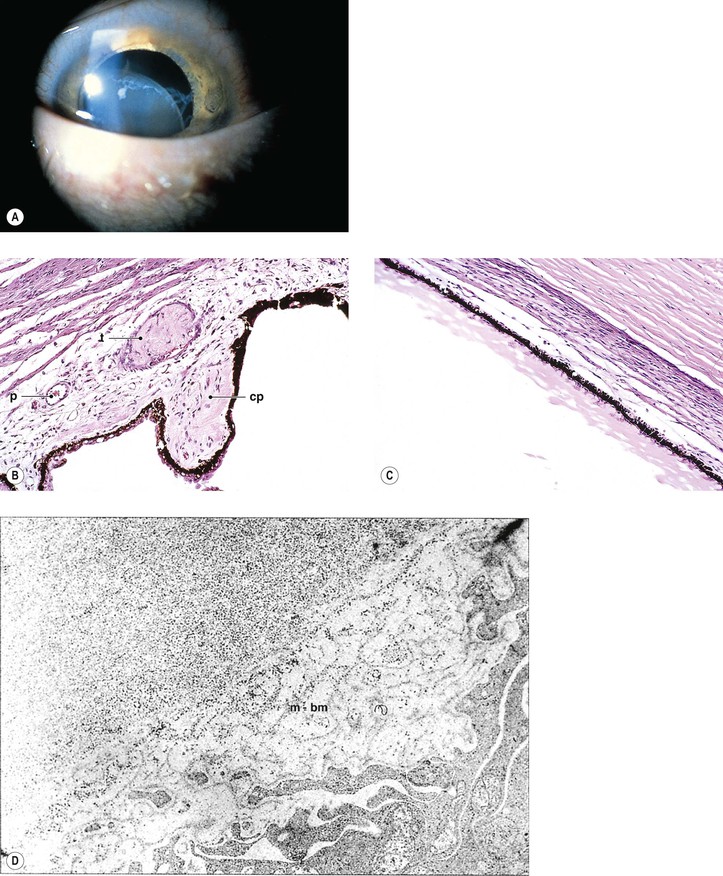
A. Oculocerebrorenal syndrome of Lowe (Fig. 10.6)
1. Lowe’s syndrome consists of systemic acidosis, organic aciduria, decreased ability to produce ammonia in the kidneys, renal rickets, generalized hypotonia, and buphthalmos.
2. Ocular findings include congenital cataract, glaucoma, and miotic pupil.
3. Histology
b. Lens capsular excrescences, similar to those seen in trisomy 21 and Miller’s syndrome, may be found.
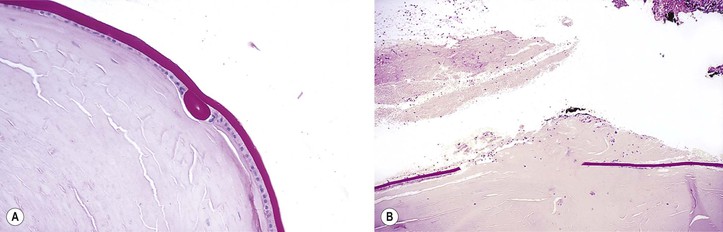
III. Rupture of the lens capsule (Fig. 10.7) may be sealed over by the underlying lens epithelium or by the overlying iris, if the rent is small enough.
Exfoliation of the Lens Capsule
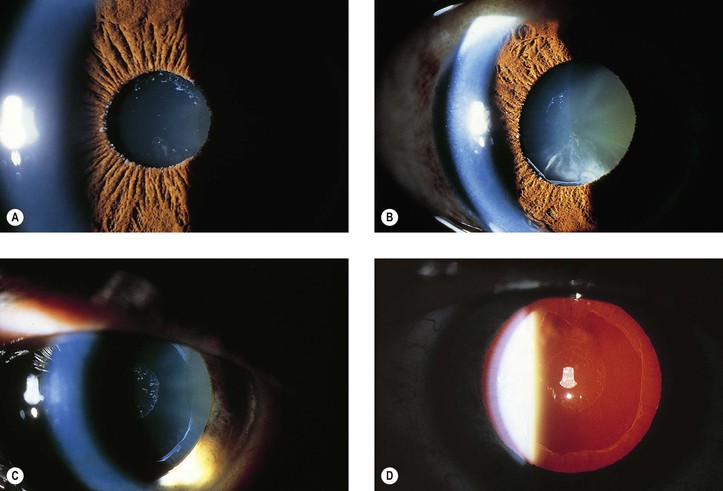
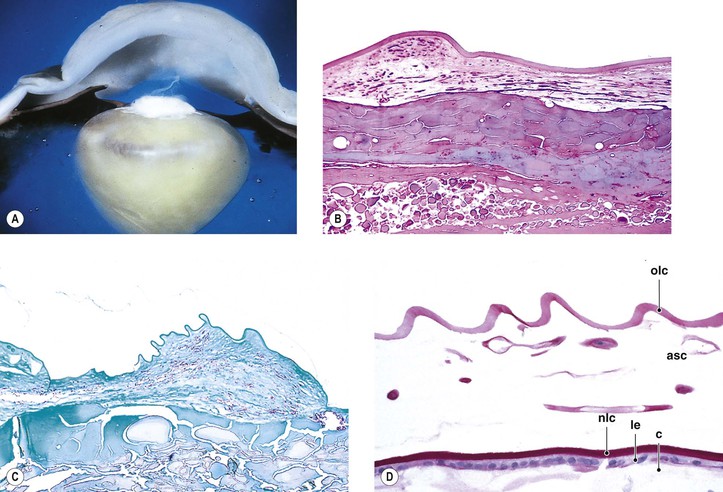
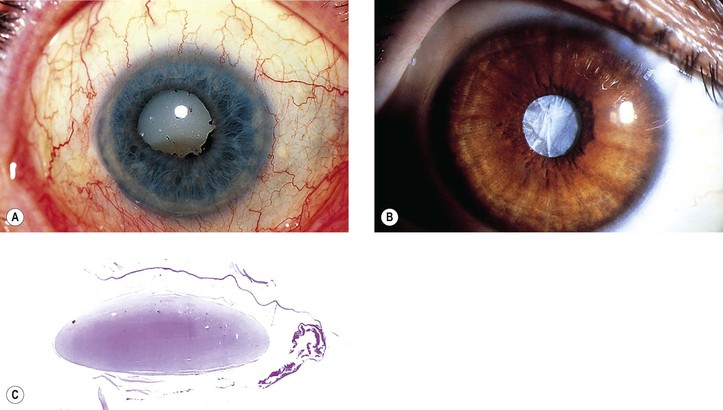
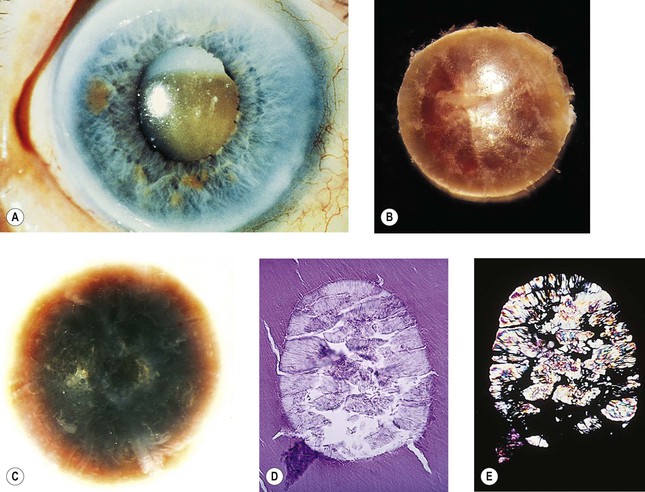
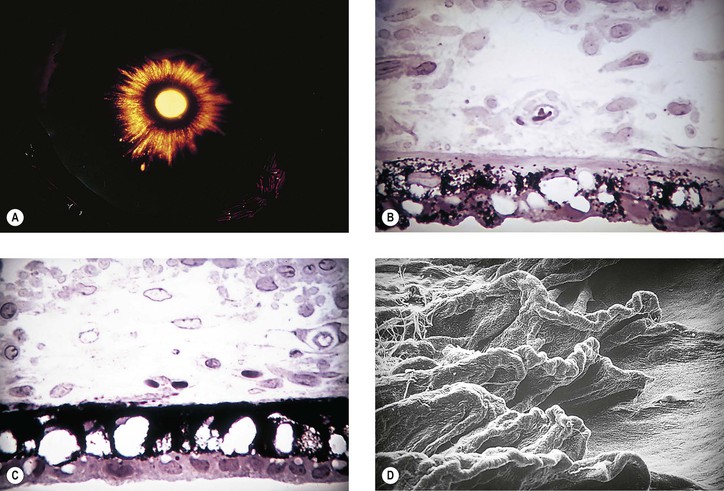
Pseudoexfoliation Syndrome (Pseudoexfoliation of Lens Capsule, Exfoliation Syndrome, Basement Membrane Exfoliation Syndrome, Fibrillopathia Epitheliocapsularis) (Figs. 10.8–10.11)
I. The pseudoexfoliation (PEX) syndrome has a worldwide distribution, but it seems to be most common in Scandinavian people (especially in Norway and Finland) and is quite rare in black people.
C. Increasing age and female gender are significant risk factors.
D. The ocular component appears to be the most dramatic part of a systemic disorder (see later).
III. In slightly more than 50% of people, the condition is bilateral.
IV. Approximately 8% have glaucoma (glaucoma capsulare), and approximately 12% have ocular hypertension.
C. The cause of the glaucoma is unknown.
1. A suggested cause is the accumulation of the PEXM or pigment in the angle.
B. Delayed intraocular PEXM development
1. White, fluffy PEXM may appear on the anterior hyaloid and pupillary border years after intracapsular cataract extraction in eyes where no PEXM had been present before cataract surgery (see Fig. 10.9A).
C. Currently, the most appealing hypothesis is that PEXM is a product of abnormal metabolism of extracellular matrix, particularly abnormal basement membranes and elastic fibers.
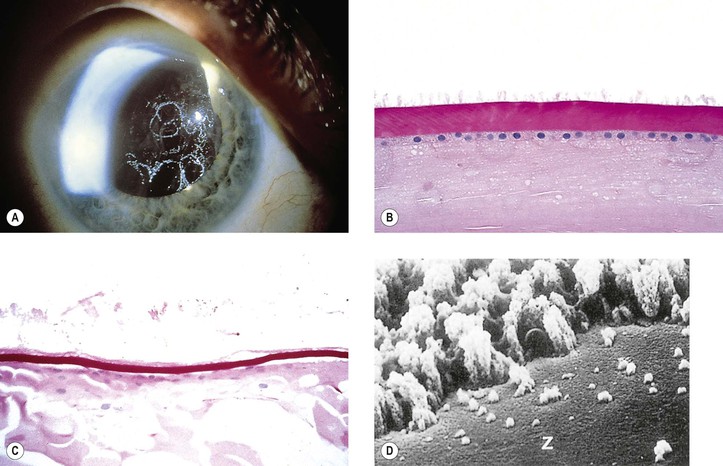
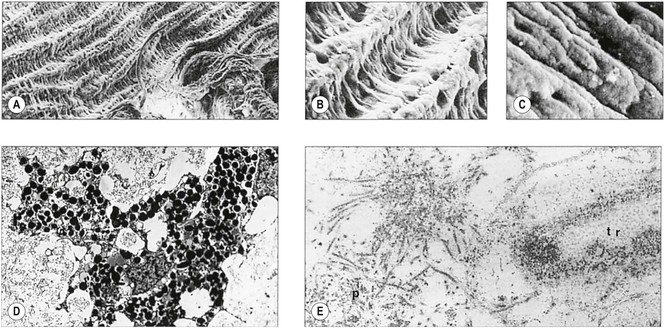
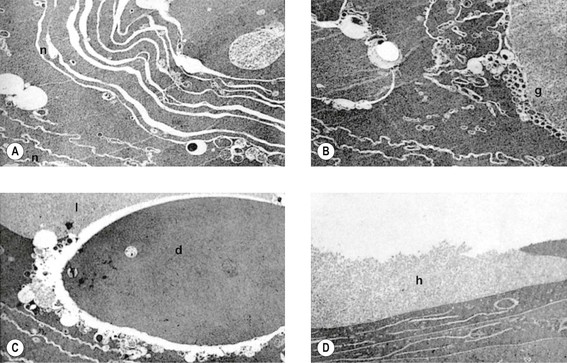
VI. Histologically, eosinophilic PEXM is found on the anterior surface of the lens, on the zonular fibers, on and in both surfaces of the iris and ciliary body, in the anterior chamber, on the corneal endothelium and incorporated in Descemet’s membrane, and in the trabecular meshwork.
C. Electron microscopically, a fibrogranular material is found in the deep (posterior) part of the anterior lens capsule.
Epithelium
Proliferation and Migration of Epithelium
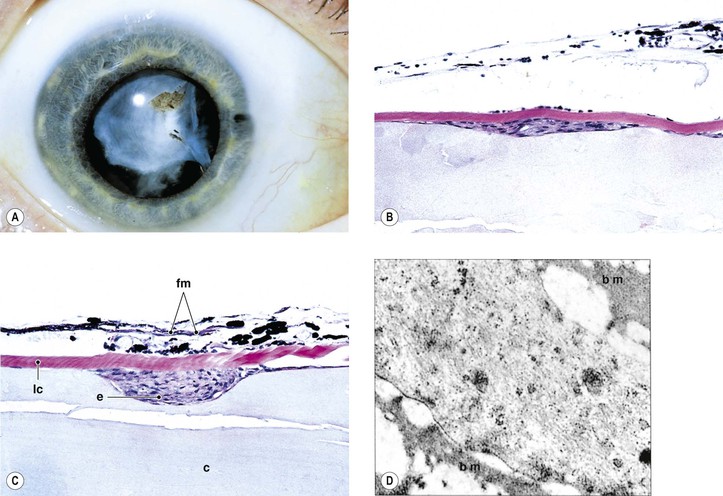
Anterior Subcapsular Cataract (ASC) (Figs. 10.12–10.15)
I. After an injury (traumatic or noxious, e.g., iritis or keratitis) to the anterior lens, the following sequence of events may take place and result in an ASC.
A. The lens epithelial cells in the region of the anterior pole of the lens become necrotic.
B. Adjacent cells migrate into the subcapsular area, proliferate, and form an epithelial plaque.
E. An ASC cataract is rarely seen clinically because an occluded pupil is most often superimposed.
II. Frequently, the same injury that initially caused the ASC also causes an anterior cortical cataract. The combination of an ASC cataract and an anterior cortical cataract is called a duplication cataract (see Fig. 10.14).
Posterior Subcapsular Cataract (PSC) (Figs. 10.16 and 10.17; See Fig. 10.15)
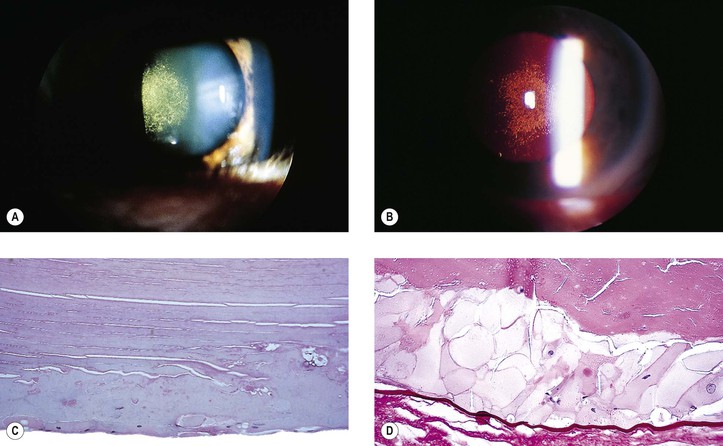
Elschnig’s Pearls (See Fig. 5.12)
Degeneration and Atrophy of the Epithelium
I. Degeneration and atrophy of the lens epithelium may occur as the result of aging or secondary to acute or chronic glaucoma, iritis or iridocyclitis, hypopyon, hyphema, chemical injury (especially alkali burn), noxious products in the aqueous (e.g., with anterior segment necrosis), or stagnation of the aqueous (e.g., with posterior synechiae and iris bombé).
C. Localized focal areas of epithelial necrosis, as often occur after an attack of closed-angle glaucoma, may result in multiple, discrete, stationary, permanent subcapsular opacities called glaukomflecken (cataracta disseminata subcapsularis glaucomatosa). Undoubtedly, stagnation of aqueous secondary to aqueous outflow blockage, along with a buildup of noxious materials in the aqueous (especially from iris necrosis), plays a role in the development of glaukomflecken (see Fig. 16.8).
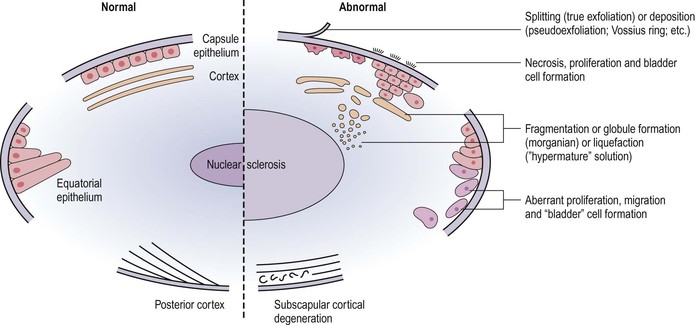
Cortex and Nucleus (Lens Cells or “Fibers”)
Cortex (“Soft Cataract”)
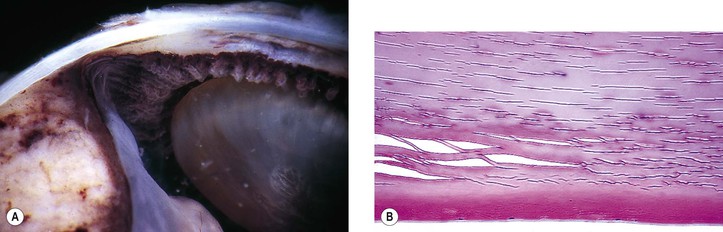
I. Biochemical changes in the lens cortex from any cause (congenital, inflammatory, traumatic) may result in clinically detectable opacities (i.e., cortical cataracts) (Figs. 10.18–10.22).
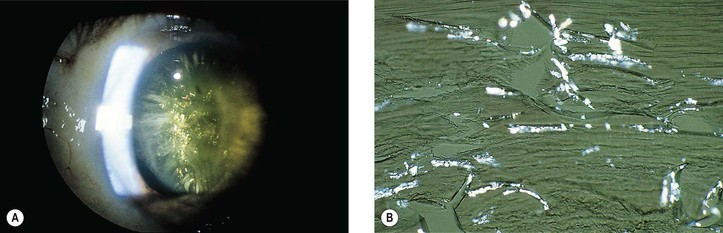
III. Histologically, the following pathologic changes may be found.
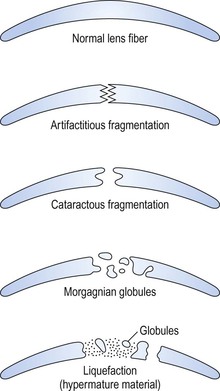
B. Cell fragments represent pieces of broken-up lens cortical cells.
C. Morgagnian globules represent small or large fragments of cortical cells that appear rounded from the increased liquidity of the cytoplasm.
3. A swollen (mainly in the anteroposterior diameter) intumescent cataract1 results.
4. The globules or abnormal protein may replace the entire cortex and result in a mature (morgagnian or liquefied) cataract. The nucleus then sinks, by gravity, inferiorly (see Fig. 10.20A). The whole lens looks clinically like a milk-filled sac.
During the process of cortical liquefaction, if the fluid is of sufficiently small molecular size, it may escape through the intact capsule and result in a smaller-than-normal lens with a wrinkled capsule (hypermature cataract; see Fig. 10.20B).
F. A break in the anterior capsule may result in cortical material becoming trapped in the equatorial region of the lens (i.e., a Soemmerring’s ring cataract; see Fig. 5.12). After an acquired break in the lens capsule, or congenitally, mesenchymal tissue may grow into the cataract, leading to bone formation (cataracta ossea) or the formation of adipose tissue (cataracta adiposa or xanthomatosis lentis).
Nucleus (“Hard Cataract”)
I. The increasing pressure of cell on cell, the breakdown of intercellular membranes in the lens nucleus, the slow conversion of soluble to insoluble protein, and the dehydration and accumulation of pigment (urochrome) all lead to optical and histologic densification of the nucleus and a nuclear cataract (Figs 10.23 and 10.24; see Fig. 10.20).
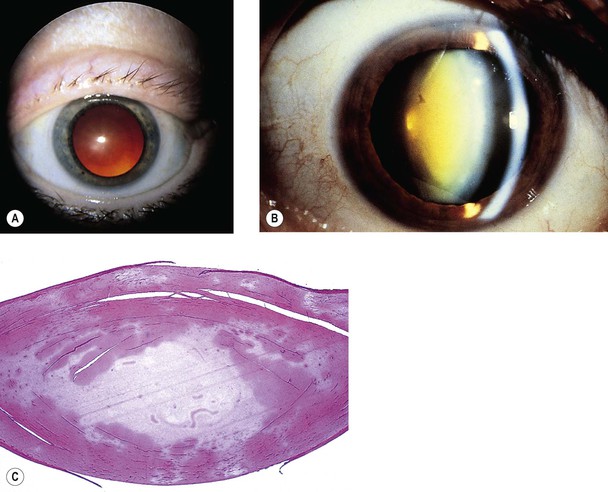
II. Histologically, the changes are usually subtle. Disappearance of the usual artifactitious nuclear clefts is noted.
A. The nucleus appears as an amorphous, homogeneous mass, with increased eosinophilia.
Age-Related (Senile) Cataracts
Secondary Cataracts
Intraocular Disease
I. Uveitis, malignant intraocular tumors (see Fig. 10.17), glaucoma, and retinitis pigmentosa (see Fig. 10.16C) can cause secondary cataracts.
II. Histologically, the lens changes are nonspecific and are the same as those described previously.
Trauma
See Chapter 5.
Toxic
I. Drugs such as steroids (topical, inhaled, and systemic), MER 29, phospholine iodide, Myleran, the phenothiazines and dinitrophenol, amiodarone, and allopurinol, along with toxic substances such as ergot or metallic foreign bodies [e.g., iron (Fig. 10.26; see also Figs 5.49 and 5.50) or copper (see Fig. 8.58)], can cause cataracts.
II. Histologically, the lens changes are nonspecific except in siderosis and hemosiderosis lentis, in which iron is present in lens epithelial cells (see Fig. 10.26), and in chalcosis, in which copper is deposited in the lens capsule.
Endocrine, Metabolic, and Others
I. Cataracts occur in conditions such as diabetes mellitus (see Chapter 15), galactosemia, hypoparathyroidism, hypothyroidism, aminoacidurias such as Lowe’s syndrome (see section Capsule, earlier), myotonic dystrophy (see Chapter 14), dermatologic disorders (e.g., atopic dermatitis, chronic eczema, and erythema multiforme), and chromosomal abnormalities.
A. Galactosemia
2. The cataract is usually noted a few days to a few months after birth.
B. Clinically and histologically, the lens changes tend to be nonspecific.
Complications of Cataracts
Glaucoma
I. Mechanical
A. An intumescent cataract may cause pupillary block and secondary angle closure.
B. A cataract may spontaneously dislocate anteriorly and cause a pupillary block directly (see Fig. 5.38), or it may dislocate posteriorly and cause a pupillary block indirectly by prolapsing vitreous into the pupil.
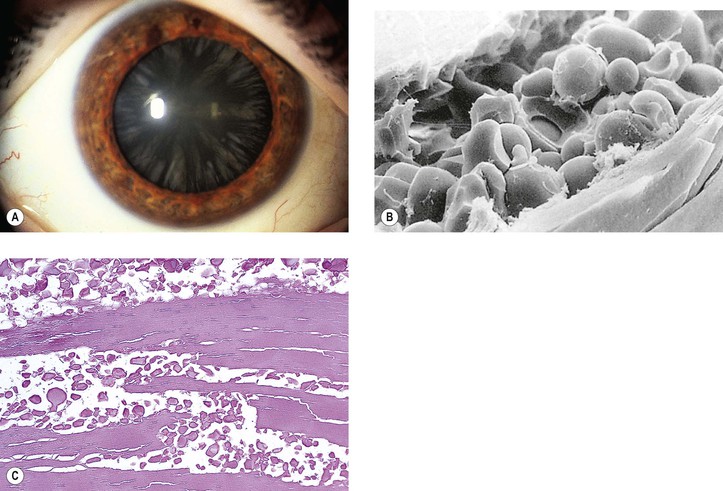
A. Phacolytic glaucoma (Fig. 10.27) is a secondary open-angle glaucoma characterized clinically by signs and symptoms of acute glaucoma, except that the anterior chamber angle is open, a white cataract is noted, and a milky material may be seen in the anterior chamber. The glaucoma may resemble open-angle glaucoma secondary to anterior uveitis, except that keratic precipitates are usually absent.
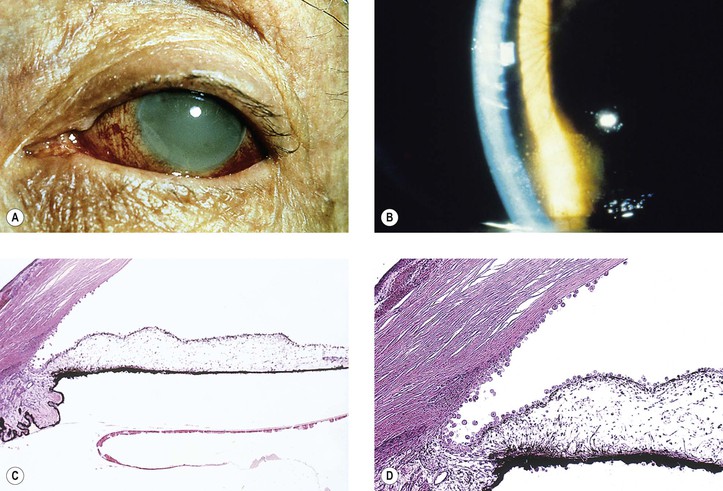
B. Phacolytic glaucoma occurs in an eye with a hypermature (white) cataract.
2. The lens material in the aqueous incites a macrophagic cellular response.
Phacoanaphylactic Endophthalmitis
See Chapter 4.
Ectopic Lens
Congenital
I. Congenital ectopia of the lens is usually bilateral and associated with generalized malformations or systemic disease such as homocystinuria, Marfan’s syndrome, or Weill–Marchesani syndrome, or less frequently with cutis hyperelastica (Ehlers–Danlos syndrome), proportional dwarfism, oxycephaly, Crouzon’s disease, Sprengel’s deformity, genetic spontaneous late subluxation of the lens, or Sturge–Weber syndrome. Only the first three are described.
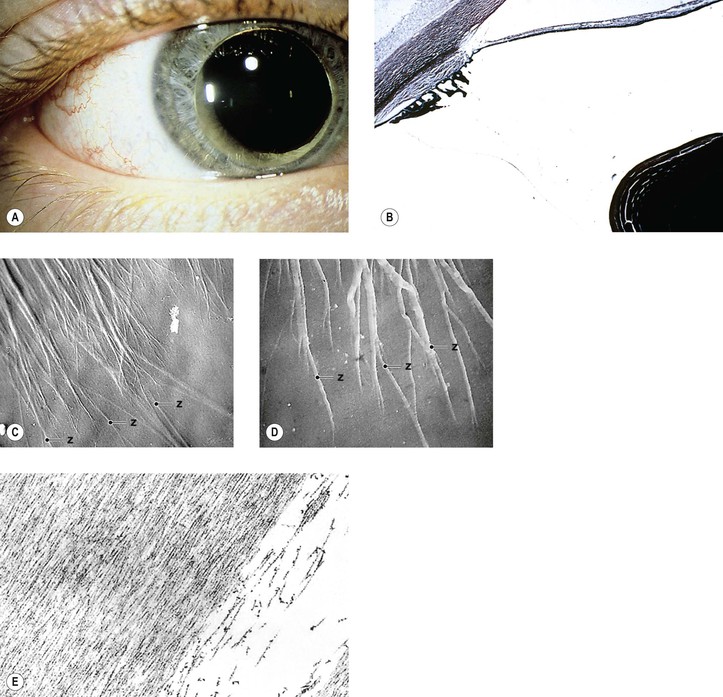
A. Homocystinuria (Fig. 10.28)
4. Histology
B. Marfan’s syndrome (arachnodactyly, dystrophia mesodermalis hypoplastica; Figs. 10.29 and 10.30)
1. Marfan’s syndrome consists of ocular, skeletal, and cardiovascular abnormalities.
5. Histology
a. The anterior chamber angle shows an immature configuration, but this is variable and nonspecific.
e. Qualitative abnormalities in fibrillin-1 staining can be seen in the conjunctiva.
3. It is inherited as an autosomal-recessive trait.
II. Congenital ectopia of the lens without associated systemic problems
2. Iridodonesis is often present.
3. It has an autosomal inheritance pattern, occasionally with decreased penetrance.
Access the complete reference list online at