[level-membership-for-dermatology-category]
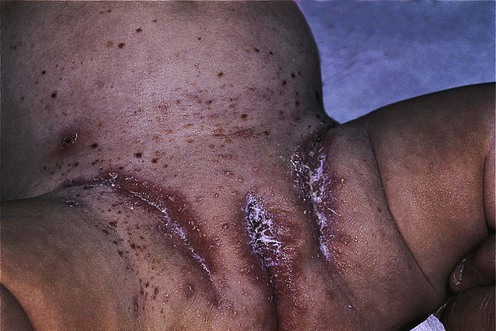
Langerhans cell histiocytosis
Specific investigations
 Biopsy of organ involved for staining for S100 and CD1a or electron microscopy for Birbeck granules. The presence of Birbeck granules may also be demonstrated by immunohistochemical staining of langerin (CD207)
Biopsy of organ involved for staining for S100 and CD1a or electron microscopy for Birbeck granules. The presence of Birbeck granules may also be demonstrated by immunohistochemical staining of langerin (CD207)
 Full blood count with differential
Full blood count with differential





 Whole body MRI is the most effective way to identify both skeletal and non-skeletal disease
Whole body MRI is the most effective way to identify both skeletal and non-skeletal disease
 Lung function tests in patients with lung disease
Lung function tests in patients with lung disease
 CT chest for all adult smokers or children with chest signs or symptoms
CT chest for all adult smokers or children with chest signs or symptoms
 Bronchoalveolar lavage for CD1a+ cells or open lung biopsy if evidence of lung involvement
Bronchoalveolar lavage for CD1a+ cells or open lung biopsy if evidence of lung involvement
 CT brain and pituitary fossa if signs of diabetes insipidus
CT brain and pituitary fossa if signs of diabetes insipidus
 Plasma and urine osmolality if signs of diabetes insipidus. A water deprivation test, if required
Plasma and urine osmolality if signs of diabetes insipidus. A water deprivation test, if required
 Full hormone screen if diabetes insipidus confirmed
Full hormone screen if diabetes insipidus confirmed
 MRI brain if lytic skull lesions, diabetes insipidus or symptoms suggestive of CNS involvement
MRI brain if lytic skull lesions, diabetes insipidus or symptoms suggestive of CNS involvement
 Panorthogram if gum involvement
Panorthogram if gum involvement
 Bone marrow biopsy if hematological abnormalities
Bone marrow biopsy if hematological abnormalities
 Abdominal ultrasound and liver biopsy if abnormal liver function
Abdominal ultrasound and liver biopsy if abnormal liver function
 Multiple bowel biopsies if evidence of malabsorption or failure to thrive in an infant
Multiple bowel biopsies if evidence of malabsorption or failure to thrive in an infant
First-line therapies
 Topical nitrogen mustard
Topical nitrogen mustard Phototherapy (PUVA, narrowband UVB)
Phototherapy (PUVA, narrowband UVB) Topical tacrolimus
Topical tacrolimus Vinblastine
Vinblastine Etoposide (VP16)
Etoposide (VP16)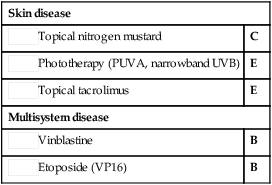
Second-line therapies
 Prednisolone
Prednisolone 6-Mercaptopurine
6-Mercaptopurine Thalidomide
Thalidomide Methotrexate
Methotrexate Cytosine arabinoside (Ara-C)
Cytosine arabinoside (Ara-C) 2-Chlorodeoxyadenosine (2-CdA)
2-Chlorodeoxyadenosine (2-CdA)Third-line therapies
 Radiotherapy
Radiotherapy Cyclosporine
Cyclosporine Bone marrow transplantation
Bone marrow transplantation Trimethoprim/sulfamethoxazole
Trimethoprim/sulfamethoxazole Interferon-α
Interferon-α 2-Deoxycoformycin
2-Deoxycoformycin Interleukin-2
Interleukin-2 Isotretinoin
Isotretinoin Acitretin
Acitretin Lenalidomide
Lenalidomide Photodynamic therapy
Photodynamic therapy Clofarabine
Clofarabine Mistletoe
MistletoeLenalidomide induced therapeutic response in a patient with aggressive multi-system Langerhans cell histiocytosis resistant to 2-chlorodeoxyadenosine and early relapse after high dose BEAM chemotherapy with autologous peripheral stem cell transplant.
Adam Z, Rehak Z, Koukalova R, Szturz P, Krejci M, Pour L, et al. Vnitr Lek 2012; 58: 62–71.
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Langerhans cell histiocytosis
Specific investigations
Biopsy of organ involved for staining for S100 and CD1a or electron microscopy for Birbeck granules. The presence of Birbeck granules may also be demonstrated by immunohistochemical staining of langerin (CD207)
Full blood count with differential
Whole body MRI is the most effective way to identify both skeletal and non-skeletal disease
Lung function tests in patients with lung disease
CT chest for all adult smokers or children with chest signs or symptoms
Bronchoalveolar lavage for CD1a+ cells or open lung biopsy if evidence of lung involvement
CT brain and pituitary fossa if signs of diabetes insipidus
Plasma and urine osmolality if signs of diabetes insipidus. A water deprivation test, if required
Full hormone screen if diabetes insipidus confirmed
MRI brain if lytic skull lesions, diabetes insipidus or symptoms suggestive of CNS involvement
Panorthogram if gum involvement
Bone marrow biopsy if hematological abnormalities
Abdominal ultrasound and liver biopsy if abnormal liver function
Multiple bowel biopsies if evidence of malabsorption or failure to thrive in an infant
