Chapter 15 Lactic acidosis
Lactic acidosis is defined by convention as the combination of an increased blood lactate concentration (> 5 mmol/l) and acidaemia (arterial blood pH of < 7.35).1,2 However hyperlactaemia occurs whenever the blood lactate concentration is above the normal range (< 2 mmol/l). Lactic acidosis may be masked by coincident metabolic or respiratory alkalosis. Critically ill patients with lactic acidosis usually have a high mortality3 and blood lactate concentrations > 8 mmol/l predict fatality.4 A prospective study reported 83% mortality in patients with blood lactate concentrations of 10 mmol/l.1 In each individual patient, however, prognosis is completely dependent on the underlying condition, with the initial degree of lactic acidosis being a clinically useful indicator of shock severity, and serial assessment allowing evaluation of response to therapy. Recently, blood lactate concentration has been identified as one marker of the degree of shock triggering treatment in septic patients who may benefit from early goal-directed therapy.5 It is important to remember that, in healthy athletes, severe lactic acidosis during exercise is a normal, self-limited observation.
PATHOPHYSIOLOGY
There is a continuous cycle of lactate production and metabolism. Lactate is produced at about 0.8 mmol/kg per h, while simultaneous metabolism in liver, kidneys, skeletal muscle, brain and red blood cells ensures that blood lactate concentrations are normally low (< 1 mmol/l). Lactic acidosis occurs when lactate production exceeds metabolic capacity, or when metabolic capacity is decreased by organ dysfunction. The liver has a key role in lactate homeostasis and many patients who develop lactic acidosis have decreased metabolic capacity due to liver disease.Glucose is derived from absorption, glycogen and gluconeogenesis. Rate of glycolysis is controlled by three unidirectional enzymes and the activity of 6
Formation and metabolism of lactate in cells are catalysed by lactate dehydrogenase (LDH):
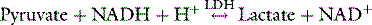
Lactate formation is in part dependent upon pyruvate concentrations and pyruvate is sourced from glycolysis (85%) and proteolysis (15%). one of these enzymes is increased by increasing intracellular pH. Acidosis therefore decreases (and alkalosis increases) glycolysis and pyruvate, and consequently lactate production. In oxygen excess, pyruvate is oxidised and lactate does not accumulate. Anaerobic metabolism however, causes lactate accumulation and an increased lactate/pyruvate ratio. The blood lactate/pyruvate ratio is however a poor indicator of mitochondrial concentrations, and is not of clinical use.
CLASSIFICATION (TYPES A AND B)
The Cohen and Woods classification of lactic acidosis7 defines two subgroups depending on the presence (type A) or absence (type B) of tissue hypoxia (Table 15.1). Type A lactic acidosis due to tissue hypoxia is common in critically ill patients. Type B (no tissue hypoxia) is much less common. Some patients with type B lactic acidosis have increased lactate generation (some malignancies and toxins impair oxygen utilisation by cells, despite adequate oxygen delivery), and other patients have decreased lactate clearance (liver disease). Patients with liver disease have greater hyperlactaemia during states of increased lactate production than those with normal liver function.6
| Type A | Shock |
| Very severe hypoxaemia | |
| Very severe anaemia | |
| Carbon monoxide poisoning | |
| Type B1 (underlying disease) | Sepsis |
| Liver failure | |
| Thiamine deficiency | |
| Malignancy | |
| Phaeochromocytoma | |
| Diabetes | |
| Type B2 (drug or toxin) | Epinephrine |
| Salbutamol | |
| Propofol | |
| Nucleoside analogue reverse transcriptase inhibitor | |
| Ethanol | |
| Methanol | |
| Paracetamol | |
| Nitroprusside | |
| Salicylates | |
| Ethylene (and propylene) glycol | |
| Biguanides | |
| Fructose | |
| Sorbitol | |
| Xylitol | |
| Cyanide | |
| Isoniazid | |
| Type B3 (rare inborn errors of metabolism) | Glucose-6-phosphatase deficiency |
| Fructose-1,6 diphosphatase deficiency | |
| Pyruvate carboxylase deficiency | |
| Deficiency of enzymes of oxidative phosphorylation |
COMBINED ABNORMALITIES (TYPES A AND B)
In clinical practice, separation of types A and B lactic acidosis is usually not helpful, because many critically ill patients have combined abnormalities. Increased lactate formation from tissue hypoxia and decreased lactate clearance often occur together. In patients with cancer, anaerobic glycolysis may be increased while hepatic lactate metabolism is impaired by tumour replacement. Diabetic patients may present with shock, but in non-insulin-dependent diabetes there may also be a defect in pyruvate oxidation, and in diabetic ketoacidosis ketones may also inhibit hepatic lactate uptake. Thiamine and biotin are essential cofactors for pyruvate dehydrogenase activity and for conversion of pyruvate to oxaloacetate. Malnutrition (beri-beri) and inadequate parenteral nutrition have therefore been associated with lactic acidosis due to deficiencies of these cofactors. In these cases, pyruvate accumulation increases lactate production. In alcoholics, ethanol oxidation increases the conversion of pyruvate to lactate and inhibits other pathways of pyruvate metabolism. Phenformin therapy is associated with lactic acidosis for several reasons: phenformin increases glycolysis in peripheral tissues, inhibits pyruvate oxidation, increases splanchnic lactate production and decreases hepatic lactate clearance. Phenformin was therefore used to induce lactic acidosis in older animal models until it was recognised that phenformin is also a potent cardiac depressant. These studies reporting the effects of lactic acidosis on cardiac function were flawed. Both endogenous and infused catecholamines cause hepatic vasoconstriction and impair hepatic lactate clearance, and epinephrine also increases hepatic glycogenolysis to lactate.8 Importantly, acidosis also stimulates adrenal catecholamine release, which may mask other cardiac effects of acidosis.
SEPSIS
There has been much recent debate9 whether the hyperlactaemia of sepsis results from net increased cellular production10 or reduced net clearance.11 However the potential for sepsis to derange lactate metabolism is clear. Both impaired regional microvascular and mitochondrial dysfunction have been implicated in the pathogenesis of lactic acidosis during sepsis. Excess catecholamines may impair hepatic lactate extraction (by reducing regional hepatic blood flow) and increase lactate production (increased glycogenolysis). At the same time lactate clearance is decreased because pyruvate dehydrogenase activity is reduced in both skeletal muscle and liver. Mitochondrial pyruvate oxidation is impaired.
Tissue hypoxia may not be a major mechanism for regional lactate production during sepsis: hyperlactaemia is thought to be linked to the severity of the septic cellular inflammatory response and hypermetabolic state.9,12 Net lactate production from the hepatosplanchnic bed is uncommon in septic patients13 and nuclear magnetic resonance spectroscopy suggests that hyperlactaemia may occur without tissue hypoxia.14
LUNG INJURY
The lung is a primary source of lactate production in patients with acute lung injury, with pulmonary release of lactate being directly related to the severity of lung injury,15,16 supporting the view that the primary contributors are the tissues with the most inflammation or injury. The increased lactate production by the injured lung is not only secondary to anaerobic metabolism in the hypoxic regions of the lung but also may be due to altered glucose metabolism and a direct effect of cytokines on pulmonary cells.16
Recent laboratory data suggest that both metabolic and respiratory acidosis protect the lung against injury whereas correction of acidosis compounded the injury.17,18 The two ventilator trials19,20 which demonstrated a positive impact on mortality in acute respiratory distress syndrome (ARDS) by limiting tidal volume and airway pressures differed widely on how they regarded the resultant hypercapnic acidosis. While Amato et al.19 allowed elevation of the PaCO2 (permissive hypercapnia) and resultant acidosis, the ARDSnet group20 in contrast aggressively corrected the hypercapnic acidosis by increasing the respiratory rate and allowing administration of sodium bicarbonate. There is growing evidence that not only may hypercapnic acidosis be beneficial in lung injury,17,21 but the ARDSnet interventions aimed at correcting the acidosis may also be deleterious.18,22 These findings have not only promoted a greater tolerance of acidosis in ARDS but also increased reluctance to buffer the acidosis exogenously towards ‘normal’ values.
ASTHMA
Lactic acidosis also often occurs in patients with acute severe asthma.23 Fatiguing respiratory muscles have been implicated, but severe lactic acidosis also occurs in sedated, paralysed mechanically ventilated patients who have no endogenous respiratory muscle activity.24 β-agonists, including salbutamol and epinephrine cause lactic acidosis by increasing gluconeogenesis, glycogenolysis, lipolysis and cyclic adenosine monophospate (AMP) activity. Clinical experience suggests that infusions of β-agonists are the primary cause of lactic acidosis in patients with asthma because decreasing intravenous salbutamol infusions to less than 10 mcg/min is usually associated with resolution of the acidosis. In asthma, lactic acidosis does not have specific prognostic implications.
CARDIAC SURGICAL PATIENTS
Hyperlactaemia during cardiopulmonary bypass is relatively frequent and is associated with an increased postoperative morbidity.25 Recent work has suggested that this ‘on-pump’ hyperlactaemia is secondary to inadequate peripheral oxygen delivery (DO2) which creates a condition similar to cardiogenic shock, leading to both direct lactate formation by dysoxic tissues and also to catecholamine release, insulin resistance and hyperglycemia-induced lactate production.
The use of epinephrine after cardiopulmonary bypass precipitates lactic acidosis in some patients.26 This phenomenon is probably β-agonist-mediated, is associated with increased whole-body blood flow and resolves after substitution of norepinephrine. However there is emerging evidence that the severity of lactic acidosis following cardiac surgery is related to certain genetic polymorphisms in tumour necrosis factor and interleukin-10 genes.27 Similar to asthma, lactic acidosis associated with the administration of epinephrine in this setting does not have the adverse implications of lactic acidosis associated with shock.
MESENTERIC ISCHAEMIA
The diagnosis of mesenteric ischaemia can be challenging to make in the critically ill due to a lack of clinical and diagnostic signs, difficulty transferring unstable patients for diagnostic imaging and concern about the deleterious effects of inappropriately administrating contrast agents. Animal models have shown that lactate increases within 1 hour of induced intestinal ischaemia. In addition, elevated lactate at the time of diagnosis of mesenteric ischaemia is a predictor of mortality.28 However, although plasma lactate is a very sensitive marker (100%) for detecting acute mesenteric ischaemia, the low specificity of this marker (42%) is a particular problem in the critically ill, who frequently have many plausible alternate diagnoses.
D-lactate is the isomer of lactate that is produced by intestinal bacterial and not by humans. Experimental work suggests that ischaemic bowel allows the translocation of D-lactate into the systemic circulation; as D-lactate is not eliminated by the liver, plasma levels may be more specific markers of mesenteric ischaemia.29 However, many issues need to be clarified, including the effect of antibiotic therapy on the intestinal bacteria, before D-lactate could be considered as a bedside diagnostic test.30
CLINICAL PRESENTATION
Patients present with clinical signs appropriate to their primary disorder. Their lactic acidosis is usually only evident after laboratory testing. In critically ill patients with shock, however, the severity of lactic acidosis can be a valuable monitor of the efficacy of resuscitation. Repeated measures of arterial blood gases and blood lactate concentrations are required. In hypovolaemic shock, resolving lactic acidosis along with the clinical signs of improving perfusion is one of several indicators of successful resuscitation. Conversely, failure of lactic acidosis to resolve in hypovolaemic shock suggests inadequate resuscitation or another undetected or unresolved clinical problem. In patients with severe lactic acidosis1 a blood lactate concentration of 5 mmol/l indicated a mortality approaching 80% and survival was best in patients whose hyperlactaemia resolved. In septic shock there are many contributors to lactic acidosis, so the time course of acidosis resolution in this setting is a less reliable indicator of the adequacy of shock resuscitation.
CARDIAC DYSFUNCTION – ASSOCIATION, CAUSE OR RESULT OF LACTIC ACIDOSIS
Cardiac dysfunction is common in shocked patients with lactic acidosis and it has often been assumed that therapies for lactic acidosis would improve cardiac function. However, cardiac dysfunction in these patients is very likely due to other factors, with cytokines (tumour necrosis factor-α, interleukins) being the major cause in septic shock, and with lactic acidosis a consequence or association rather than a cause of cardiac dysfunction. In lactic acidosis, some clinicians recommend normalisation of arterial pH based on two assumptions: (1) that acidosis causes cardiac dysfunction; and (2) that patients are better with ‘normal’ laboratory values.31 Both of these assumptions are incorrect in most critically ill patients. First, early research in isolated muscle, isolated heart preparations, animal models and clinical case reports supported the view that acidosis decreased cardiac function and decreased the haemodynamic response to catecholamines. However later large animal studies in which preload, afterload and heart rate were carefully controlled found only marginal effects of lactic acidosis on contractility.2,32 Also, laboratory reports of deceased cardiac function during acidosis studied an arterial pH much lower (pH 6.6–6.9) than that usually observed in critically ill patients.33 Increasing experience with permissive hypercapnia in ARDS and asthma has supported the view that patients with respiratory acidosis have fewer complications and better outcomes when normal values are not targeted. The major haemodynamic effect of acute hypercapnic acidosis in ARDS and asthma was increased cardiac output and vasodilatation, not cardiac depression.34 Therefore, targeting normal values in lactic acidosis is also unlikely in itself to be beneficial, and may be harmful.
MANAGEMENT
TREAT THE PRIMARY DISORDER
Specific therapies and supports must be directed at each underlying cause. In hypovolaemic and cardiogenic shock, restoration of an adequate global oxygen delivery is required. Vasoconstrictors may worsen tissue perfusion and should only follow adequate intravascular volume and appropriate cardiac supports. In septic shock, antibiotics appropriate to cover all likely sources of infection are a priority, and in patients with possible ischaemic gut, surgery may be required for both diagnosis and therapy. Postsurgical gastrointestinal leaks may sometimes be difficult to diagnose, may not be detectable on computed tomography and require early laparotomy. In status epilepticus, lactic acidosis is a result of muscle activity and rapid use of effective anticonvulsants is indicated. In diabetic ketoacidosis, insulin, appropriate fluid and treatment of precipitants enable resolution of all metabolic abnormalities, including associated lactic acidosis. In thiamine deficiency, highlighted during a nationwide American shortage of multivitamins for patients receiving total parenteral nutrition,35 high-dose intravenous thiamine corrected both the vasodilated shock and associated lactic acidosis. In acute severe asthma, lactic acidosis is commonly a result of high-dose intravenous β-agonist therapy, and salbutamol dose reduction usually resolves the problem. In vasodilated patients after cardiopulmonary bypass, lactic acidosis may also be related to β-agonist therapy26 and resolves after substitution of intravenous epinephrine with norepinephrine. In these cases, lactic acidosis is not related to decreased tissue perfusion and adverse effects upon prognosis have not been noted. Patients with human immunodeficiency virus (HIV) receiving nucleoside analogue reverse transcriptase inhibitor (NRTI) therapy have a high incidence of hyperlactaemia (8.3%) which can progress to a rapidly fatal metabolic lactic acidosis syndrome. These patients with NRTI-induced mitochondrial dysfunction require withdrawal of the therapy (if lactate > 5 mmol/l) and close monitoring.36 In symptomatic patients an elevated lactate level is associated with mortality .
BICARBONATE
Bicarbonate therapy for lactic acidosis is controversial.2,31,37 Correction of acidosis with bicarbonate might reverse depressed cardiac performance, but there is no evidence in critically ill patients that lactic acidosis depresses cardiac function, and more recent laboratory studies also report minimal depression in large animals.32,38 Further, not all studies have demonstrated a rise in pH after the administration of bicarbonate.39 Importantly, two randomised studies of bicarbonate therapy in critically ill patients with lactic acidosis and shock found no improvement in cardiac function or any other beneficial effects of pH correction.33,40 One reason for these findings is that sodium bicarbonate has adverse effects, including acute hypercapnia and ionised hypocalcaemia,33 which outweigh potential benefits in patients. Hypercapnia may increase intracellular acidosis (CO2 crosses cell membranes rapidly), and hypocalcaemia decreases myocardial contractility.41 Other side-effects of bicarbonate occur because bicarbonate is a hypertonic solution and include acute intravascular volume overload and cardiac depression. In addition, bicarbonate increases lactate production by increasing the activity of the rate-limiting enzyme phosphofructokinase, shifts the haemoglobin–oxygen dissociation curve, increases oxygen affinity of haemoglobin and thereby decreases oxygen delivery to tissues. Adverse effects of bicarbonate in patients can be reduced by using slow infusions in preference to rapid boluses, and by correcting side-effects, by increasing minute volume in ventilated patients and by correcting ionised hypocalcaemia.
Mechanical ventilation limits the ability to excrete the CO2 evolved from bicarbonate administration with the potential to attenuate the resultant increase in pH. In support of this, two prospective randomised controlled trials conducted in mechanically ventilated patients with lactic acidosis demonstrated that bicarbonate administration resulted in minimal improvements in pH.33,40 Furthermore, in the presence of severely limited ventilation such as a lung protection strategy, bicarbonate may lower arterial pH.42 The ARDSnet group supported administration of bicarbonate to limit the pH effects of hypercapnia,20 but emerging experimental work suggests that buffering of hypercapnic acidosis may be detrimental,18 and may not therefore be advisable during protective ventilator strategies.
Importantly, however, despite decades of debate, bicarbonate has never been shown to be beneficial in any clinical trial and its use in patients with lactic acidosis is not now recommended, regardless of the degree of acidaemia.2
ALTERNATIVE THERAPIES
DICHLOROACETATE
Dichloroacetate (DCA) works by stimulating the phosphate dehydrogenase complex, the rate-limiting enzyme which regulates entry of pyruvate into the tricarboxylic acid cycle. DCA increases arterial pH and decreases lactate concentrations43 but nevertheless, a large multicentre randomised clinical trial in patients with lactic acidosis found no benefit for either haemodynamics or patient outcome.44 This study is really the best evidence currently available to support the view that correction of lactic acidosis in critically ill patients without improving the underlying primary disorder has no overall effect on patient outcome. DCA is not available commercially.
TRIS/THAM
Tris-hydroxymethyl aminomethane (THAM) is a commercially available weak alkali which is rarely used as a clinical therapy because of concerns about side-effects which include hyperkalaemia, hypoglycaemia, extravasation-related necrosis and neonatal hepatic necrosis. In acute lung injury, THAM has been demonstrated to be an effective buffer in ventilated patients, as it is not associated with an increased CO2 load and is capable of ameliorating some of the haemodynamic effects of hypercapnia.42,45 However, it is unclear whether buffering of hypercapnic acidosis in acute lung injury patients is of any benefit.
DIALYSIS/HAEMOFILTRATION
Peritoneal dialysis has been reported to be effective at removing lactate, but bicarbonate-buffered haemofiltration is ineffective, contributing to less than 3% of lactate clearance.46 Indeed, haemofiltration was so ineffective that it has been noted that lactate concentrations remain a useful clinical marker of disease progression in patients on bicarbonate-buffered haemofiltration.
However, in lactate-intolerant patients (i.e. those with shock-induced lactic acidosis and/or liver disease), the use of lactate-based dialysis fluid may overload the patient’s metabolic capacity for lactate, particularly if high-volume haemofiltration is employed.47 In these patients bicarbonate-based dialysis should be chosen.
1 Stacpoole PW, Wright EC, Baumgartner TG, et al. Natural history and course of acquired lactic acidosis in adults. DCA-Lactic Acidosis Study Group. Am J Med. 1994;97:47-54.
2 Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117:260-267.
3 Gunnerson KJ, Saul M, He S, et al. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care. 2006;10:R22.
4 Broder G, Weil MH. Excess lactate: an index of reversibility of shock in human patients. Science. 1964;143:1457-1459.
5 Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
6 Berry MN. The liver and lactic acidosis. Proc R Soc Med. 1967;60:1260-1262.
7 Cohen RD, Woods HF. Lactic acidosis revisited. Diabetes. 1983;32:181-191.
8 Stacpoole PW. Lactic acidosis. Endocrinol Metab Clin North Am. 1993;22:221-245.
9 Gutierrez G, Wulf ME. Lactic acidosis in sepsis: another commentary. Crit Care Med. 2005;33:2420-2422.
10 Chiolero RL, Revelly JP, Leverve X, et al. Effects of cardiogenic shock on lactate and glucose metabolism after heart surgery. Crit Care Med. 2000;28:3784-3791.
11 Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than overproduction. Am J Respir Crit Care Med. 1998;157:1021-1026.
12 Mizock BA. The hepatosplanchnic area and hyperlactatemia: a tale of two lactates. Crit Care Med. 2001;29:447-449.
13 De Backer D, Creteur J, Silva E, et al. The hepatosplanchnic area is not a common source of lactate in patients with severe sepsis. Crit Care Med. 2001;29:256-261.
14 Hotchkiss RS, Karl IE. Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. JAMA. 1992;267:1503-1510.
15 Kellum JA, Kramer DJ, Lee K, et al. Release of lactate by the lung in acute lung injury. Chest. 1997;111:1301-1305.
16 Iscra F, Gullo A, Biolo G. Bench-to-bedside review: lactate and the lung. Crit Care. 2002;6:327-329.
17 Laffey JG, Honan D, Hopkins N, et al. Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am J Respir Crit Care Med. 2004;169:46-56.
18 Laffey JG, Engelberts D, Kavanagh BP. Buffering hypercapnic acidosis worsens acute lung injury. Am J Respir Crit Care Med. 2000;161:141-146.
19 Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347-354.
20 Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301-1308.
21 Kregenow DA, Rubenfeld GD, Hudson LD, et al. Hypercapnic acidosis and mortality in acute lung injury. Crit Care Med. 2006;34:1-7.
22 Conrad SA, Zhang S, Arnold TC, et al. Protective effects of low respiratory frequency in experimental ventilator-associated lung injury. Crit Care Med. 2005;33:835-840.
23 Mountain RD, Heffner JE, Brackett NCJr, et al. Acid–base disturbances in acute asthma. Chest. 1990;98:651-655.
24 Manthous CA. Lactic acidosis in status asthmaticus: three cases and review of the literature. Chest. 2001;119:1599-1602.
25 Demers P, Elkouri S, Martineau R, et al. Outcome with high blood lactate levels during cardiopulmonary bypass in adult cardiac operation. Ann Thorac Surg. 2000;70:2082-2086.
26 Totaro RJ, Raper RF. Epinephrine-induced lactic acidosis following cardiopulmonary bypass. Crit Care Med. 1997;25:1693-1699.
27 Ryan T, Balding J, McGovern EM, et al. Lactic acidosis after cardiac surgery is associated with polymorphisms in tumor necrosis factor and interleukin 10 genes. Ann Thorac Surg. 2002;73:1905-1909. discussion 1910–11
28 Newman TS, Magnuson TH, Ahrendt SA, et al. The changing face of mesenteric infarction. Am Surg. 1998;64:611-616.
29 Murray MJ, Gonze MD, Nowak LR, et al. Serum D(–)-lactate levels as an aid to diagnosing acute intestinal ischemia. Am J Surg. 1994;167:575-578.
30 van der Voort PH. Diagnostic and scientific dilemma: the ischemic bowel. Crit Care Med. 2006;34:1561-1562.
31 Narins RG, Cohen JJ. Bicarbonate therapy for organic acidosis: the case for its continued use. Ann Intern Med. 1987;106:615-618.
32 Cooper DJ, Herbertson MJ, Werner HA, et al. Bicarbonate does not increase left ventricular contractility during L-lactic acidemia in pigs. Am Rev Respir Dis. 1993;148:317-322.
33 Cooper DJ, Walley KR, Wiggs BR, et al. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study. Ann Intern Med. 1990;112:492-498.
34 Thorens JB, Jolliet P, Ritz M, et al. Effects of rapid permissive hypercapnia on hemodynamics, gas exchange, and oxygen transport and consumption during mechanical ventilation for the acute respiratory distress syndrome. Intens Care Med. 1996;22:182-191.
35 Centers for Disease Control. Lactic acidosis traced to thiamine deficiency related to nationwide shortage of multivitamins for total parental nutrition. United States; 1997. MMWR Mortal Morbid Weekly Rep. 1997;46:523-528.
36 Claessens YE, Chiche JD, Mira JP, et al. Bench-to-bedside review: severe lactic acidosis in HIV patients treated with nucleoside analogue reverse transcriptase inhibitors. Crit Care. 2003;7:226-232.
37 Stacpoole PW. Lactic acidosis: the case against bicarbonate therapy. Ann Intern Med. 1986;105:276-279.
38 Walley K, Cooper DJ, Baile E, et al. Bicarbonate does not improve left ventiricular contractility during resuscitation from hypovolemic shock in pigs. J Crit Care. 1992;7:14-21.
39 Graf H, Leach W, Arieff AI. Metabolic effects of sodium bicarbonate in hypoxic lactic acidosis in dogs. Am J Physiol. 1985;249:F630-F635.
40 Mathieu D, Neviere R, Billard V, et al. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med. 1991;19:1352-1356.
41 Lang RM, Fellner SK, Neumann A, et al. Left ventricular contractility varies directly with blood ionized calcium. Ann Intern Med. 1988;108:524-529.
42 Kallet RH, Jasmer RM, Luce JM, et al. The treatment of acidosis in acute lung injury with tris-hydroxymethyl aminomethane (THAM). Am J Respir Crit Care Med. 2000;161:1149-1153.
43 Stacpoole PW, Harman EM, Curry SH, et al. Treatment of lactic acidosis with dichloroacetate. N Engl J Med. 1983;309:390-396.
44 Stacpoole PW, Wright EC, Baumgartner TG, et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N Engl J Med. 1992;327:1564-1569.
45 Weber T, Tschernich H, Sitzwohl C, et al. Tromethamine buffer modifies the depressant effect of permissive hypercapnia on myocardial contractility in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;162:1361-1365.
46 Benjamin E. Continuous venovenous hemofiltration with dialysis and lactate clearance in critically ill patients. Crit Care Med. 1997;25:4-5.
47 Naka T, Bellomo R. Bench-to-bedside review: treating acid–base abnormalities in the intensive care unit – the role of renal replacement therapy. Crit Care. 2004;8:108-114.