CHAPTER 6 Investigation and classification of anemia
Definition and causes of anemia
Anemia is defined as a reduction in the concentration of hemoglobin in the peripheral blood below the reference range for the age and gender of an individual (see Table 1.3 for reference ranges). It may be inherited or acquired and results from an imbalance between red cell production and red cell loss (Table 6.1). In general terms the causes of anemia are:
| Mechanism | Pathogenesis |
|---|---|
| Reduced or ineffective erythropoiesis | Decreased marrow erythropoiesis |
| Inadequately increased total erythropoiesis | |
| Increased ineffective erythropoiesis | |
| Increased red cell loss or reduced red cell life span | Acute or chronic blood loss |
| Increased red cell destruction | |
| Splenic pooling and sequestration | |
| Dilutional anemia | Plasma volume expansion |
Clinical features of anemia
A detailed clinical history is critical in determining the cause of anemia. Table 6.2 lists some of the important personal, dietary, drug and family history issues to be explored. The symptoms and signs of anemia result from decreased tissue oxygenation leading to organ dysfunction as well as from adaptive changes, particularly in the cardiovascular system.1,2 The nature and severity of symptoms is influenced by:
| History | Mechanism | Examples |
|---|---|---|
| Current illness | Acute hemorrhage | Epistaxis, menorrhagia, hematemesis, melaena |
| Chronic blood loss | Menorrhagia, melaena | |
| Infection | Parvovirus | |
| Hemolysis | Jaundice | |
| Past medical history | Anemia of chronic disease | Chronic infection Liver disease Renal impairment Hypothyroidism Malignancy |
| Malabsorption | Gastrectomy Gastric bypass Celiac disease Ileal surgery |
|
| Travel history | Intra-erythrocytic parasites | Malaria |
| Dietary history | Vegetarian or veganism | Vitamin B12 deficiency |
| Iron intake | Iron deficiency | |
| Excess alcohol | Liver disease | |
| Drugs | Antiplatelet agents | Aspirin, clopidogrel |
| Anticoagulants | Warfarin | |
| Oxidant drugs | Salazopyrin, dapsone | |
| Myelosuppressive agents | Methotrexate Cytotoxic chemotherapy |
|
| Exposure to toxins | Toxins or chemicals that interfere with erythropoiesis | Lead, aluminum |
| Family history | Inherited red cell abnormality | Hereditary spherocytosis G6PD deficiency Thalassemia Other hemoglobinopathy |
| Autoimmune disorders | Pernicious anemia Rheumatoid arthritis |
|
| Bleeding disorders | Hemophilia von Willebrand disease |
A moderate degree of chronic anemia is usually associated with only mild symptoms accompanied by slight increases in cardiac output at rest and slight decreases in mixed venous PO2. This is because there is a substantial shift of the oxygen dissociation curve to the right (see Chapter 1), mainly due to an adaptive increase in the levels of red cell 2,3-diphosphoglycerate. When the hemoglobin falls below 7–8 g/dl symptoms usually become more marked. The intra-erythrocytic adaptation cannot by itself maintain adequate oxygen delivery to the tissues and other compensatory mechanisms come into effect. These include:
The blood count and red cell indices in anemia
The mean cell volume (MCV) is the most useful red cell parameter for the assessment of the underlying cause of anemia. By using the MCV, anemias can be categorized by red cell size as microcytic (MCV <80 fl), normocytic (normal MCV) or macrocytic (MCV >100 fl). This provides a practical and rapid way of assessing possible causes and guiding further investigations (see below and Table 6.3). The mean cell hemoglobin (MCH) and mean cell hemoglobin concentration (MCHC) are generally of less value than the MCV in the assessment of anemia. The red cell distribution width (RDW), a quantitative measure of the degree of variation in red cell size, can be useful in the assessment of some types of anemia. Usually erythrocytes are of a standard size (6–8 µm) and the RDW is 12–14%. A high RDW indicates that there is variation in erythrocyte size and gives a quantitative measure of anisocytosis. For example, in microcytic anemias, a normal RDW is generally seen in thalassemias whereas in iron deficiency it is mildly elevated. The graphical depiction of red cell features on blood count histograms, such as red cell number versus MCV, may also give an indication of anisocytosis, or the presence of dimorphic populations of erythrocytes.
Table 6.3 Practical classification of anemia based on mean cell volume
| Types | Mean cell volume | Conditions |
|---|---|---|
| Microcytic | <80 fl | Iron deficiency Anemia of chronic disease Hemoglobinopathies Hereditary sideroblastic anemia |
| Normocytic | Within reference range (80–100 fl) |
Blood loss Hemolysis Failure of erythropoiesis |
| Macrocytic | >80 fl | Deficiency of folate or vitamin B12 Myelodysplasia Liver disease Hypothyroidism |
Red cell morphology in anemia
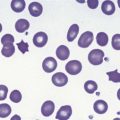
Blood film examination to review red cell morphology has a critical role in the investigation and diagnosis of anemia. The identification of red cell morphological abnormalities may lead to a definitive or differential diagnosis and guide further investigations (Fig. 6.1A–F). The film should be prepared from a freshly collected blood sample, well-stained and coverslipped. Blood stored for >6 hours in anticoagulant prior to the preparation of the film can result in artifacts (e.g. red cell crenation) that can interfere with interpretation of the true red cell morphology. Morphological artifacts can also result from the blood being stored at incorrect temperatures (hot or cold) prior to preparation of the blood film. The film should be examined in an area where only occasional red cells overlap. In such an area normal red cells are primarily round and show a central area of pallor which occupies less than a third of the diameter of the cell. The film should be assessed systematically for:
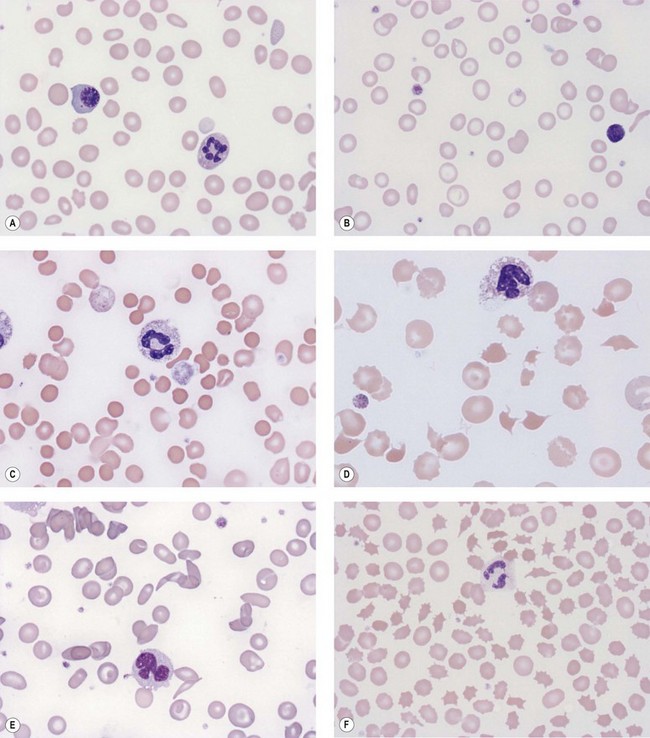
Some of the important diagnostic red cell morphological features are described together with their disease associations below and in Table 6.4:3,4
Table 6.4 Morphological abnormalities of red cells in anemia (see also Fig. 6.1)
| Morphological feature | Pathogenesis | Disorders (examples) |
|---|---|---|
| Microcytosis | Impaired synthesis of heme or globin | Iron deficiency, thalassemias, congenital sideroblastic anemia, congenital atransferrinemia, aluminum-induced anemia (dialysis patients) |
| Macrocytosis | Dyserythropoiesis or accelerated release of reticulocytes | Megaloblastic erythropoiesis e.g. vitamin B12 or folate deficiency, congenital dyserythropoietic anemia types I, III, non-megaloblastic (e.g. liver disease, alcohol, hypothyroidism) |
| Hypochromasia | Impaired synthesis of heme | Iron deficiency, anemia of chronic disease |
| Anisocytosis | Nonspecific evidence of a perturbation of erythropoiesis | Various |
| Target cells | Increased surface area relative to volume | Thalassemias, hemoglobinopathies, (HbAC, HbCC, HbEE), liver disease, obstructive jaundice, hyposplenism |
| Stomatocytes | Cation leak | Hereditary stomatocytosis, Rh null phenotype, alcohol, drugs |
| Spherocytosis | Abnormality of cell membrane | Hereditary spherocytosis, immune-hemolytic anemia |
| Elliptocytosis | Abnormality of cell membrane | Hereditary elliptocytosis |
| Acanthocytes | Membrane lipid imbalance | Liver disease, anorexia nervosa, hyposplenism, abetalipoproteinemia, McLeod phenotype |
| Echinocytes | Extrinsic effects | Uremia |
| Sickle cells | Abnormal globin chain | Sickle cell disease, HbS/β-thalassemia, HbSC disease, HbS/O-Arab, HbS/D-Punjab, HbS/Lepore |
| Schistocytes | Red cell fragmentation | Microangiopathic hemolytic anemias, hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, malignant hypertension, cardiac valve prostheses |
| Bite cells | Removal of oxidized hemoglobin | Oxidant stress, glucose-6-phosphate dehydrogenase deficiency, drugs (e.g. dapsone, salazopyrin, antimalarials) |
| Teardrop poikilocytes | Marrow fibrosis | Primary or secondary marrow fibrosis |
| Basophilic stippling | Ribosomes or RNA | Accelerated erythropoiesis, dyserythropoiesis, lead poisoning, thalassemias, pyrimidine 5′-nucleotidase deficiency |
| Pappenheimer bodies | Iron | Lead poisoning, sideroblastic anemias, hemolytic anemias, hyposplenism |
| Howell–Jolly bodies | Nuclear remnants | Hyposplenism, megaloblastic hemopoiesis |
| Polychromasia and nucleated red cells | Increased erythropoiesis and red cell release | Marrow erythroid response to anemia, especially hemolytic anemia and blood loss |
Target cells. These are abnormally thin red cells with a well-stained hemoglobinized zone in the middle of the usual central area of pallor (Fig. 6.1D). This morphology is due to a disproportionate increase in red cell membrane due to abnormal lipid content. They are seen in liver disease (especially cholestatic), hyposplenism and hemoglobinopathies such as hemoglobin C and E disease.
Spherocytes. Spherocytes, small, round, deeply-staining (hyperchromic) red cells without central pallor, have lost their biconcave shape and therefore have a spherical form. They occur due to loss of cell membrane and are a feature of hereditary spherocytosis, warm autoimmune hemolytic anemia and clostridial septicemia (Fig. 6.1C).
Schistocytes. These are fragmented red cells with sharp points and occur in fragmentation hemolysis as a result of their interaction with fibrin strands, diseased vessel walls or foreign surfaces (e.g. cardiac valve prostheses). Conditions in which they are seen include disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome and graft-versus-host disease (Fig. 6.1D).
Stomatocytes. These are red cells with a slit-like area of pallor across the center instead of the circular area of pallor. They are associated with hereditary stomatocytosis and Southeast Asian ovalocytosis (see Chapter 7).5,6 They can also be seen in alcoholic liver disease or as artifact on a poorly spread blood film.
Sickle cells. Sickle-shaped or crescentic red cells occur as a result of deoxygenation of hemoglobin S (Fig. 6.1E). Deoxygenated hemoglobin S is about 50 times less soluble than deoxygenated hemoglobin A and, under appropriate conditions, forms long fibers (tactoids) which deform the red cell. Sickle cells are found in hemoglobin S homozygotes and in double heterozygotes for hemoglobin S and β-thalassemia or other abnormal hemoglobins, such as hemoglobin C, E, O-Arab, D-Punjab or Lepore.
Echinocytes (‘burr’ cells). Echinocytes are spiculated red cells with 10–30 short projections of similar length that are evenly distributed over the cell surface. They are seen in renal imapirment.7,8
Acanthocytes (‘spur’ cells). These are spiculated red cells with 5–10 projections of varying length and thickness that are irregularly spaced over the cell surface. These commonly lack central pallor (Fig. 6.1F). Acanthocytes are seen in hepatic failure, Zieve’s syndrome (‘spur’ cell anemia), malnutrition, abetalipoproteinemia and McLeod syndrome (inherited Kell blood group abnormality associated with hemolysis). Spiculated cells are also seen in pyruvate kinase deficiency.
Basophilic stippling. Fine or coarse basophilic stippling indicates the presence of ribosomes, generally within reticulocytes or young mature red cells, or RNA. Stippling indicates increased erythropoietic response to anemia or dyserythropoiesis (Fig. 6.1A).
Investigation of the cause of anemia
The further laboratory investigation of the cause of anemia should be guided by the MCV, red cell morphology and the reticulocyte count (Fig. 6.2).
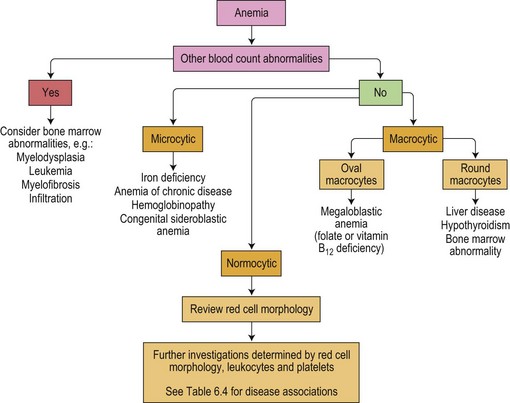
Fig. 6.2 Pathway for the investigation of anemia based on the mean cell volume and blood film morphology.
Microcytic anemias
Microcytic anemias are due to deficient synthesis of hemoglobin (Fig. 6.1B). This may be due to inadequate heme production (e.g. iron deficiency, anemia of chronic disease and hereditary sideroblastic anemia) or abnormalities of globin chain synthesis (i.e. hemoglobinopathies).9,10 The laboratory investigation should therefore include assessment of body iron status, i.e. ferritin, serum iron, transferrin and transferrin saturation (see Chapters 11 and 14). Evaluation of hemoglobin (e.g. high-performance liquid chromatography (HPLC); hemoglobin electrophoresis) may be required if a hemoglobinopathy is suspected (see Chapter 9). Bone marrow examination may be indicated especially to assess iron stores and its incorporation into sideroblasts.
Macrocytic anemias
Macrocytic anemias may be megaloblastic or non-megaloblastic, a distinction which can often be made on the blood film (see below and Chapter 12). The megaloblastic anemias are due to deficiencies of folate or vitamin B12 and cause a failure of DNA synthesis and resultant impaired cell division. Macrocytes in megaloblastic anemia tend to be oval with associated hypersegmented neutrophils and megaloblastic erythroid progenitors (Fig. 6.1A).11 In non-megaloblastic macrocytic anemias the macrocytes are round. There are many possible etiologies, which may be intrinsic to the marrow (e.g. myelodysplasia) or due to extrinsic causes (e.g. liver disease, hypothyroidism, drug therapy, reticulocytosis, myelodysplasia).12 Accompanying cytopenias, neutrophil morphological abnormalities and the presence of blast cells may suggest myelodysplasia. Serum and red cell folate and serum vitamin B12 levels should be measured in all cases. The requirement for analysis of hepatic and thyroid function and other biochemical analyses should be based on the clinical scenario. Bone marrow examination may be required if myelodysplasia is a consideration or the etiology cannot be determined following the above-mentioned investigations.
Normocytic anemias
If spherocytes are present investigation should be performed for a possible inherited (i.e. hereditary spherocytosis) or acquired (e.g. immune mediated hemolytic anemia) cause of spherocytic hemolytic anemia (see Chapter 10). Schistocytes (Fig. 6.1D) should prompt investigation for causes of possible microangiopathic hemolytic anemia (e.g. D-dimer, ADAMTS13). The presence of teardrop poikilocytes may require bone marrow examination to exclude underlying fibrosis. Inherited red cell enzyme defects, for example G6PD deficiency and pyruvate kinase deficiency, can also present with a normocytic anemia. The blood film may, however, show distinctive red cell abnormalities such as bite cells and spiculated cells, respectively. See Chapter 8 for the approach to the diagnosis of these conditions.
Normocytic anemia in the absence of any specific morphological abnormality or reticulocytosis may be due to acute or chronic infective or inflammatory conditions, hepatic, renal or endocrine conditions, red cell aplasia, dilution, or paroxysmal nocturnal hemoglobinuria (PNH). The clinical history, biochemical studies demonstrating the underlying abnormality and iron studies (changes associated with the anemia of chronic disease) may assist in establishing the cause. A bone marrow examination may be required in unresolved cases. PNH may require flow cytometry to demonstrate deficient expression of CD55 and CD59 antigens (see Chapter 10).
A normocytic anemia occurs following acute hemorrhage.13,14 The hemoglobin is initially normal but normocytic normochromic anemia occurs with plasma volume expansion; the hemoglobin is at its lowest 36–72 hours following blood loss. The reticulocyte count increases slightly after 1–2 days, reaches a peak with a few circulating normoblasts at 7–10 days and returns to normal by 2 weeks. Chronic blood loss eventually results in iron deficiency and a hypochromic microcytic anemia (see Chapter 11).
Assessment of the erythropoietic response to anemia
Overall erythropoietic activity is the total of ‘effective’ and ‘ineffective’ erythropoiesis (Table 6.5). Effective erythropoiesis is the rate of release of newly-formed red cells from the marrow. Ineffective erythropoiesis is the rate of loss of potential erythrocytes (as a result of apoptosis of progenitor cells) and phagocytosis of defective erythropoietic cells by bone marrow macrophages. In practice, the most readily measured parameters of the erythropoietic response are the peripheral blood reticulocyte count, and, in the bone marrow, the myeloid : erythroid ratio, morphology of erythropoiesis (for evidence of dyserythropoiesis) and phagocytosis of erythroblasts by macrophages. Serum transferrin receptor, a truncated soluble form of the surface receptor mainly produced by erythroblasts, can also be used. Serum transferrin receptor is increased in erythroid hyperplasia as well as in iron deficiency; in the absence of iron deficiency, it is a measure of total erythropoietic activity.15,16
| Erythropoietic activty | Measurement |
|---|---|
| Total erythropoiesis | Myeloid : erythroid (M : E) ratio in the bone marrow |
| Marrow erythropoiesis | |
| Serum transferrin receptor | |
| Plasma iron turnover | |
| Total marrow iron turnover | |
| Fecal urobilinogen excretion | |
| Effective erythropoiesis | Absolute reticulocyte count |
| Red cell turnover | |
| Red cell 59Fe utilization | |
| Red cell iron turnover | |
| Ineffective erythropoiesis | Difference between indices of total and effective erythropoiesis |
| Morphologic evidence of increased dyserythropoiesis | |
| Erythroblast phagocytosis by macrophages |
The classification of anemia
The pathophysiologic or mechanistic classification is based on the etiology of the anemia: whether it is ‘true’ (absolute) or ‘relative’ (dilutional), the underlying defect and whether the anemia is inherited or acquired (Table 6.1). These are listed as follows and detailed in Table 6.6:
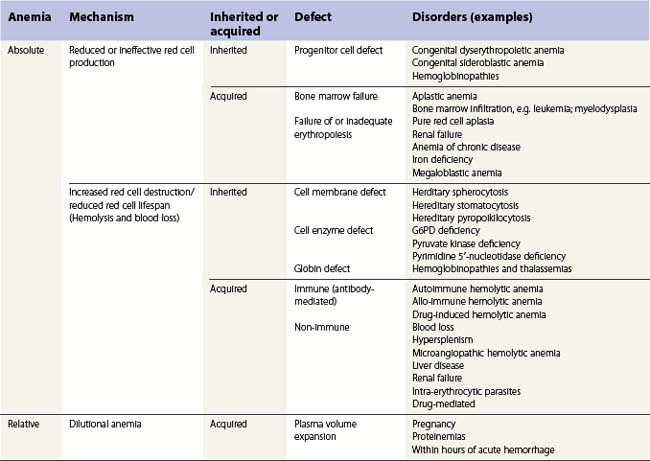
Detailed descriptions of each of these causes of anemia are in the following chapters.
1 Wickramasinghe SN, Weatherall DJ. The pathophysiology of erythropoiesis. In: Hardisty RM, Weatherall DJ, editors. Blood and its Disorders. 2nd ed. Oxford: Blackwell Scientific Publications; 1982:101.
2 Means RT, Glader B. Anaemia: general considerations. In Greer JP, Foerster J, Rodgers GM, et al, editors: Wintrobe’s Clinical Hematology, 12th ed, Philadelphia: Wolters Kluwer/Lippincott, Williams & Wilkins, 2009.
3 Pierre RV. Red cell morphology and the peripheral blood film. Clin Lab Haem. 2002;22(1):25-61.
4 Bain BJ. Morphology in the diagnosis of red cell disorders. Hematology. 2005;10(Suppl. 1):178-181.
5 Bruce LJ. Hereditary stomatocytosis and cation leaky red cells – recent developments. Blood Cells, Molecules and Diseases. 2009;42(3):216-222.
6 Wong P. A hypothesis of the stomatocytosis in individuals with the phenotype Rh(null). Medical Hypotheses. 2001;57:770-771.
7 Bessis M. Living blood cells and their ultrastructure. Berlin: Springer; 1973. p. 197
8 Brecher G, Bessis M. Present status of spiculed red cells and their relationship to the discocyte–echinocyte transformation: a critical review. Blood. 1972;40:333-344.
9 Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia. British Journal of Haematology. 2008;143(1):27-38.
10 Camaschella C. Hereditary sideroblastic anaemia: pathophysiology, diagnosis and treatment. Seminars in Hematology. 2009;46(4):371-377.
11 Wickramasinghe SN. Diagnosis of megaloblastic anaemias. Blood Reviews. 2006;20(6):299-318.
12 Morse EE. Mechanisms of hemolysis in liver disease. Annals of Clinical and Laboratory Science. 1990;20:169-174.
13 Adamson J, Hillman RS. Blood volume and plasma protein replacement following acute blood loss in normal man. Journal of the American Medical Association. 1968;205:609-612.
14 Wallace J, Sharpey-Shafer EP. Blood changes following controlled haemorrhage in man. Lancet. 1941;ii:393.
15 Beguin Y, Clemons GK, Pootrakul P, Fillet G. Quantitative assessment of erythropoiesis and functional classification of anaemia based on measurements of serum transferrin receptor and erythropoietin. Blood. 1993;81:1067-1076.
16 Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clinica Chimica Acta. 2003;329:9-22.
17 Dacie JV. The Haemolytic Anaemias, vol 1: The Hereditary Haemolytic Anaemias, part 1, 3rd ed. Edinburgh: Churchill Livingstone; 1985.
18 Dacie JV. The Haemolytic Anaemias, vol 2: The Hereditary Haemolytic Anaemias, part 2, 3rd ed. Edinburgh: Churchill Livingstone; 1988.
19 Dacie JV. The Haemolytic Anaemias, vol 3: The Auto-Immune Haemolytic Anaemias, 3rd ed. Edinburgh: Churchill Livingstone; 1992.
20 Dacie JV. The Haemolytic Anaemias, vol 4: Secondary or Symptomatic Haemolytic Anaemias, 3rd ed. New York: Churchill Livingstone; 1995.
21 Dacie JV. The Haemolytic Anaemias, vol 5: Drug- and Chemical-Induced Haemolytic Anaemias; Paroxysmal Nocturnal Haemoglobinuria; Haemolytic Disease of the Newborn, 3rd ed. New York: Churchill Livingstone; 1999.