Chapter 90 Inflammatory Neuropathies
Although children and adults with peripheral neuropathies exhibit similar clinical and electrophysiologic features, the incidence and nature of the underlying disorders responsible for peripheral nerve diseases are widely divergent. Perhaps the most salient difference is that most peripheral nerve diseases in children are immune-mediated and potentially treatable. Table 90-1 lists the principal etiologies for neuropathy in a group of 249 children evaluated over 12 years at a tertiary pediatric referral center by one of the authors. Acquired inflammatory neuropathies were the most common peripheral nerve diseases in this group, followed by the genetically determined neuropathies. Table 90-2 illustrates a more specific classification in these patients of the acquired immune-mediated neuropathies. Acute and chronic inflammatory demyelinating neuropathies were the most common cause of neuropathy in the inflammatory subgroup. Some children with collagen vascular diseases developed neuropathy as a complication of rheumatoid arthritis, systemic lupus erythematosus, or mixed connective tissue disease. Necrotizing vasculitis selectively affecting the peripheral nervous system was also seen. Autoimmune neuropathies presumed to have developed as a consequence of other primary disorders were notable, and included neuropathy associated with graft-versus-host disease after bone marrow transplantation [Adams et al., 1995; Perry et al., 1994], and a chronic immune demyelinating polyneuropathy-like presentation in the course of Lyme disease. Although children with human immunodeficiency virus infection and acquired immune deficiency syndrome were frequently evaluated for neurologic complications, only one adolescent developed an associated peripheral neuropathy [Leger, 1992], presumably as a complication of treatment.
Table 90-1 Etiologies of Neuropathy in 249 Children (1980–1992)
| Category | Patients (%) |
|---|---|
| Immune and inflammatory disorders | 45 |
| Genetically determined disorders | 42 |
| Other causes of neuropathy | 13 |
(From unpublished observations of John T Sladky, MD.)
Table 90-2 Causes of Inflammatory and Immune Neuropathy in 112 Children
| Diagnosis | Patients (%) |
|---|---|
| Guillain-Barré syndrome | 58 |
| Chronic inflammatory demyelinating polyneuropathy | 31 |
| Cases associated with collagen vascular disease | 7 |
| Other immune and infectious disorders | 4 |
(Data from unpublished observations of John T Sladky, MD.)
Inflammatory neuropathies, because of their relative frequency and potential to respond to treatment, are important considerations in the evaluation of children with peripheral nerve disease. Guillain-Barré syndrome constitutes the most common category of acquired immune-mediated neuropathies in childhood [Koul et al., 2002]. The pathologic manifestations of Guillain-Barré syndrome are protean, with immunologic-targeted antigens directed toward both axolemmal and myelin-related epitopes [Hughes and Cornblath, 2005; Willison, 2005b; Willison et al., 2008]. The clinical manifestations of the disease are determined by the nature of the pathology and the populations of axons that come under immunologic attack. Although the salient clinical feature of Guillain-Barré syndrome is weakness, any organ system affected by peripheral nerve function may be involved. It is reasonable to conceptualize this syndrome as an inhomogeneous spectrum of clinical features with specific constellations of clinical characteristics clustering in distinct patterns, permitting definitions of subcategories of Guillain-Barré syndrome.
Acute Inflammatory Demyelinating Polyneuropathy
Epidemiology
Acute inflammatory demyelinating polyneuropathy (AIDP), one of several discrete varieties of Guillain-Barré syndrome, accounts for about 90 percent of the disease in North America and Western Europe. It is the most common paralytic illness affecting children in countries with established immunization programs. The incidence of Guillain-Barré syndrome has been estimated in population-based studies to be between 0.5 and 1.5 cases per 100,000 children younger than 16 years [Rantala et al., 1994]. Based on these estimates, about 400–600 children per year would be diagnosed with Guillain-Barré syndrome in the United States [Prevots and Sutter, 1997]. Similar data have been reported from Canada [McLean et al., 1994], UK, Europe, and the Scandinavian countries [Farkkila et al., 1991; Korinthenberg and Monting, 1996; Kollar et al., 2009; Korinthenberg et al., 2007; Ramirez-Zamora et al., 2009]; Latin America [Asbury and Cornblath, 1990; Hart et al., 1994; Korinthenberg and Monting, 1996; Molinero et al., 2003; Olive et al., 1997]; and the Far East [Hung et al., 1994; Barzegar et al., 2007, Korinthenberg et al., 2007; Ramirez-Zamora et al., 2009]. Both sexes are affected, although most pediatric series have noted a slight male preponderance.
Diagnostic Criteria
As part of a nationwide program to evaluate the possible relationship of swine-flu immunization to Guillain-Barré syndrome, diagnostic criteria were developed under the sponsorship of the National Institute of Neurological Disorders and Stroke and were later modified after analysis of data from several multicenter treatment trials [Asbury and Cornblath, 1990]. Boxes 90-1 and 90-2 provide summaries of the consensus statement describing the clinical characteristics necessary to diagnose Guillain-Barré syndrome, along with those features, including electrophysiologic testing and laboratory measurements, that mitigate for or against the diagnosis.
Box 90-1 Clinical and Laboratory Features in the Diagnosis of Guillain–Barré Syndrome


II. Strongly Supportive of the Diagnosis
 Progression – weakness may develop rapidly but cease to progress after 4 weeks; roughly 50% will plateau within 2 weeks, 80% by 3 weeks, and 90% after 4 weeks
Progression – weakness may develop rapidly but cease to progress after 4 weeks; roughly 50% will plateau within 2 weeks, 80% by 3 weeks, and 90% after 4 weeks






















(From Asbury AK, Cornblath DR: Assessment of current diagnostic criteria for Guillain-Barré syndrome, Ann Neurol 27:S21, 1990.)
Must demonstrate three of the following four features:
CMAP, compound motor unit action potential; LLN, lower limit of normal; ULN, upper limit of normal.(From Asbury AK, Cornblath DR: Assessment of current diagnostic criteria for Guillain-Barré syndrome, Ann Neurol 27:S21, 1990.)
Clinical Features
The clinical presentation of Guillain-Barré syndrome is similar in children and adults [Bradshaw and Jones, 1992; Korinthenberg and Monting, 1996; Rantala et al., 1991; Sakakihara and Kamoshita, 1991; Sarada et al., 1994; Sladky, 2004; Asbury, 2000; Barzegar et al., 2007; Gupta et al., 2008; Kollar et al., 2009; Korinthenberg et al., 2007; Vucic et al., 2009; Wu et al., 1999]. Although there is considerable variability in the nature of the initial symptoms, the overall pattern of the evolution of the clinical syndrome and the ultimate severity of disability usually conform to a triphasic model:
The initial phase is generally relentless and rapid. Between 50 and 75 percent of patients develop maximal weakness within 2 weeks, and 90–98 percent do so by 4 weeks [Asbury and Cornblath, 1990; Korinthenberg and Monting, 1996; Italian Guillain-Barré Study Group, 1996]. In several series describing the natural history in adults and children, the mean duration of the progressive phase was in the range of 10–12 days. A small number of patients continue to progress for longer than 4 weeks. This latter group overlaps to some degree with patients diagnosed with chronic inflammatory demyelinating polyneuropathy. The duration of the plateau phase is similar to that of the progressive phase, averaging 10–12 days but ranging from several days to 4 weeks [Italian Guillain-Barré Study Group, 1996]. Patients who develop severe axonal degeneration have a prolonged plateau phase and tend to have more severe residual weakness [Cornblath, 1990]. In a series primarily of adult patients, about half recovered within 6 months and more than 80 percent by 24 months [Italian Guillain-Barré Study Group, 1996].
A multicenter study of 175 children with Guillain-Barré syndrome has reported similar findings [Korinthenberg and Monting, 1996]. At the height of the disease, 26 percent of patients remained able to walk, but 16 percent required artificial ventilation. The median time from onset of symptoms to first recovery was 17 days; to walk unaided, 37 days; and to be free of symptoms, 66 days. The population in this study was heterogeneous, with many children receiving some form of treatment, including plasmapheresis, intravenous immune globulin, and corticosteroids. A large number of children have a relatively benign course, and a smaller group a more severe and protracted course. At 6 months’ follow-up evaluation, 98 of 106 children (92 percent) were free of symptoms and the others were able to walk unaided. Subsequent reports similarly have emphasized a favorable prognosis [Barzegar et al., 2007; Korinthenberg et al., 2007; Lee et al., 2008; Nagasawa et al., 2006; Ramachandran and Kuruvilla, 2004; Shafqat et al., 2006]. As in series of adult patients, the maximum degree of disability at clinical nadir appears to be the most powerful predictor of incomplete recovery.
In the past, there has been considerable discussion as to whether the natural history of Guillain-Barré syndrome in children is more benign or comparable to adults. The argument may ultimately be unresolvable because of the absence of large prospective controlled treatment trials in children compared with adults [Klegweg et al., 1989]. There are few case series that include descriptions of the natural course of this syndrome in untreated children with similar end-points. Two adult benchmark studies are the North American Treatment Trial and the French Plasmapheresis Treatment Trial.
Table 90-3 summarizes some of the presenting complaints seen in a series of 49 children hospitalized with Guillain-Barré syndrome. This cohort has proved to be typical of other reported pediatric series. Weakness was the most common initial complaint. Pain, particularly in the back and lower extremities, was also prominent and was near-universal in the most severely affected children. A history of difficulty with balance early in the course of the illness was elicited in almost half the children. The classic triad, characteristic of the Miller Fisher variant of Guillain-Barré syndrome (ataxia, ophthalmoparesis, and areflexia), was present in only two children. All children had evidence of progression. Maximal disability was quantitated according to the 1985 Guillain-Barré Syndrome Study Group grading scale (Table 90-4). At the time of maximal neurologic deficit, 25 percent of children were able to ambulate 5 meters without assistance (grade 2). Another 11 children (22 percent) reached a nadir of grade 3 (unable to walk 5 meters without assistance). The largest group of children (39 percent) were bed- or wheelchair-confined (grade 4), and 7 children (14 percent) required mechanical ventilatory assistance (grade 5). There were no fatalities. Autonomic dysfunction occurred in 14 children (28 percent), and included labile hypertension in 9 children and urinary or bowel incontinence in 6 children.
Table 90-3 Clinical Features in 49 Children Younger than 18 Years with Guillain-Barré Syndrome*
| Feature | Prevalence |
|---|---|
| Age | 7.1 years (mean) |
| Male-to-female ratio | 1.2:1 |
| Weakness | 73% |
| Pain | 55% |
| Ataxia | 44% |
| Paresthesias | 18% |
| Shortness of breath | 4% |
* Two patients had findings consistent with Miller Fisher syndrome.)
(Data from unpublished observations of John T Sladky, MD.
Table 90-4 Clinical Grading Scale from the U.S. Guillain-Barré Syndrome Plasmapheresis Trial
| Score | Clinical Criteria |
|---|---|
| 0 | Health state |
| 1 | Minor signs or symptoms |
| 2 | Able to walk 5 m without a walker or equivalent support |
| 3 | Able to walk 5 m with a walker or support |
| 4 | Bed- or chair-bound (unable to walk 5 m with a walker or support) |
| 5 | Requires assisted ventilation (for at least part of the day) |
| 6 | Dead |
(Adapted from the Guillain-Barré Syndrome Study Group: Plasmapheresis and acute Guillain-Barré syndrome, Neurology 35:1096, 1985.)
The duration of symptoms in children with Guillain-Barré syndrome requiring hospitalization varies in the reported studies. The incidence of admission to pediatric intensive care units for treatment of respiratory failure or other serious medical problems (e.g., infection) or for plasmapheresis ranges between 17 and 68 percent, with the average length of stay about 11 days [Jansen et al., 1993; Ramirez-Zamora et al., 2009; Yata et al., 2003]. The average duration for mechanical ventilation ranges from 17 to 22 days. The mean duration of hospitalization is also quite variable, ranging from 12 to 84 days [Jansen et al., 1993; Jones, 1996]. Time to independent walking reported in a multicenter study in Western Europe [Korinthenberg and Monting, 1996] was somewhat shorter than in other pediatric series, which reported durations of 43 days [Lamont et al., 1991], 52 days [Epstein and Sladky, 1990], and 113 days [Shahar and Leiderman, 2003]. The European study was a questionnaire-based survey that included children from community hospitals along with tertiary referral centers, which the authors thought may represent a less severely affected population. In addition, many of these children received therapies, including corticosteroids, intravenous immune globulin, and plasmapheresis.
Autonomic dysfunction has been reported in 12.5–25 percent of children with Guillain-Barré syndrome [Bradshaw and Jones, 1992; Hung et al., 1994; Lee et al., 2008; Shafqat et al., 2006] and is similar to that in adults [Ropper, 1994; Zochodne, 1994; Korinthenberg et al., 2007; Ryan, 2005; Shafqat et al., 2006]. Symptoms are usually intermittent and include postural hypotension, supraventricular tachycardia, bradycardia, and fluctuating blood pressure. Gastrointestinal complaints are numerous and related to impaired swallowing, gastroesophageal dysmotility, pseudo-obstruction, and constipation. Urinary retention also occurs, and the potential need for catheterization and bladder decompression should be considered in children with more severe symptoms during the acute phase. On rare occasions, cardiac arrest secondary to autonomic dysfunction has been observed in children [Bos et al., 1987; Briscoe et al., 1987].
Although AIDP is often regarded as an entity in which motor symptoms predominate, pain is a common symptom, occurring in up to 79 percent of children [Bradshaw and Jones, 1992; Nguyen et al., 1999; Sladky, 2004] and 72 percent of adult patients [Pentland and Donald, 1994]. The types of pain vary and include paresthesias, dysesthesias, axial and radicular pain, meningismus, myalgia, joint pain, and visceral discomfort. Pain intensity on admission appears to correlate poorly with the degree of initial neurologic disability and is not predictive of prognosis [Moulin et al., 1997]. Patients may also present with or develop pain in a variety of clinical settings, such as the intensive care unit or pediatric ward, or during the more chronic phase of the disease when in rehabilitation or at home. Back and leg pain usually resolves over the first 8 weeks, but dysesthetic extremity pain may persist longer in 5–10 percent of patients, despite motor recovery. Moderate to severe pain requires aggressive treatment. In a recent study of adult patients with Guillain-Barré syndrome, 75 percent required oral opioid analgesics, and 29 percent received parenteral analgesia to provide adequate pain relief [Hahn, 1996]. Despite aggressive therapy, most adults with grade 4 or 5 disease severity reported that their pain was inadequately treated. Few data regarding pain symptoms in children with this syndrome have been published. The principles of pain management for Guillain-Barré syndrome patients are well described [Moulin et al., 1997; Pentland and Donald, 1994].
Clinical Variants of Guillain-Barré Syndrome
Several clinical forms of Guillain-Barré syndrome have been characterized (Table 90-5). Some of the more clearly defined entities are discussed in this section.
Table 90-5 Variants of Immune-Mediated Acute and Chronic Demyelinating and Axonal Disorders Seen in Children
| Guillain-Barré Syndrome Subtype | Description | References* |
|---|---|---|
| Acute inflammatory demyelinating polyneuropathy (AIDP) | Acute onset of ascending weakness and hyporeflexia with elevated CSF protein and EMG showing demyelinating neuropathy; triphasic course, usually with good recovery | Hung et al. [1994]; Jones [1996, 2000]; Korinthenberg and Monting [1996]; Ensrud and Krivickas [2001]; Hughes and Cornblath [2005]; Korinthenberg et al. [2007]; Kushnir et al. [2008]; Kuwabara [2007]; Lu et al. [2000] |
| Acute motor and sensory axonal neuropathy (AMSAN) | Acute onset of ascending weakness and hyporeflexia with elevated CSF protein and EMG showing axonal involvement with reduction of CMAP; triphasic course, usually with poor or limited recovery | Alma et al. [1998]; Chowdhury and Arora [2001]; Currie et al. [1990]; Reisin et al. [1993]; Hiraga et al. [2005a]; Hiraga et al. [2005b]; Lu et al. [2000]; Vedanarayanan and Chaudhry [2000] |
| Acute motor axonal neuropathy (AMAN) | Clinical syndrome similar to AIDP or AMSAN; elevated CSF protein; electrophysiologic and histopathologic evidence of degeneration strictly limited to sensory axons | Griffin et al. [1995]; Ho et al. [1995, 1997b]; Lu et al. [2000]; Paradiso et al. [1999]; Chowdhury and Arora [2001]; Hiraga et al. [2005b]; Kuwabara [2007]; Lu et al. [2000]; Nachamkin et al. [2007]; Ortiz-Corredor et al. [2007]; Vedanarayanan and Chaudhry [2000]; Vucic et al. [2009]; Yuki and Kuwabara [2007] |
| Miller Fisher syndrome | Acute onset of ophthalmoplegia, hyporeflexia, and ataxia with elevated CSF protein and subsequent recovery | Hughes et al. [1999]; Jones [2000]; Sladky [2004]; Willison and O’Hanlon [1999]; Koga et al. [2005]; Overell and Willison [2005]; Susuki et al. [2001]; Willison [2001]; Yuki [2001]; Yuki and Koga [2006] |
| Polyneuritis cranialis | Acute onset of multiple cranial nerve palsies (usually bilateral VII and sparing of II) with elevated CSF protein, slowing of motor conduction velocities and recovery | McFarland [1976]; Morosini et al. [2003]; Polo et al. [1992]; MacLennan et al. [2004]; Murakami et al. [2006]; Onodera et al. [2002] |
| Acute sensory neuropathy | Acute onset of sensory loss, areflexia, elevated CSF protein, slowing of motor conduction velocities and recovery | Sahashi et al. [1985]; Seneviratne and Gunasekera [2002]; Wilmshurst et al. [1999]; Oh et al. [2001] |
| Acute pandysautonomia | Acute onset of multiple dysautonomic symptoms with limited or no motor involvement, CSF protein elevation, and good recovery | Chistiansen and Brodersen [2003]; Fagius et al. [1983]; Low [1994]; Nass and Chutorian [1982]; Zochodne [1994] |
| Chronic inflammatory demyelinating polyneuropathy | Subacute or indolent onset of weakness and hyporeflexia with elevated CSF protein and EMG showing demyelinating neuropathy; symptoms persist for >8 weeks and may remain chronic or become progressive | Connolly [2001]; Korinthenberg [1999]; Nevo [1998]; Ryan et al. [2000]; Hughes [2010]; Hughes et al. [2006]; Jo et al. [2010]; Lewis [2007]; Markowitz et al. [2008]; Rabie and Nevo [2009]; Vallat et al. [2010] |
| Chronic inflammatory relapsing demyelinating polyneuropathy | Acute or subacute onset of weakness and hyporeflexia with elevated CSF protein and EMG showing demyelinating neuropathy; symptoms persist for >8 weeks and follow a chronic relapsing course | Connolly [2001]; Gorson and Chaudhry [1999]; Hahn [1998]; Hahn et al. [1996a]; Hughes [1994]; Nevo [1998]; Ryan et al. [2000]; Said [2002]; Sladky et al. [1986]; Hughes et al. [2006]; Vallat et al. [2010] |
| Chronic inflammatory axonal polyneuropathy | Subacute or indolent onset of weakness and hyporeflexia with elevated CSF protein and EMG consistent with an axonal neuropathy; symptoms follow a chronic course; sural nerve biopsy is normal | Chin et al. [2004]; Feasby et al., [1990]; Hughes [1994]; Uncini et al. [1991]; Hughes et al. [2006]; Vallat et al. [2010] |
| Guillain-Barré syndrome with acute onset of weakness and hyporeflexia with encephalopathic features | Elevated CSF protein and EMG showing neuropathy. Accompanied by encephalopathic and brainstem symptoms and/or myelitis that may have a protracted course, usually with good recovery | Bradshaw and Jones [2001]; Brashear et al. [1985]; Feasby et al. [1990]; Maier et al. [1997]; Nadkarni and Lisak [1993]; Uncini et al. [1991] |
* References listed are for pediatric case series, if available.
CMAP, compound motor unit action potentials; CSF, cerebrospinal fluid; EMG, electromyography.
Acute Motor and Sensory Axonal Neuropathy
Whether acute motor and sensory axonal neuropathy is distinct from the more common AIDP or part of a continuum of disease remains a topic for on-going discussion. The initial clinical presentations of these disorders are virtually identical; however, children and adults with acute motor and sensory axonal neuropathy with a severe extent of axonal degeneration are more likely to have incomplete recovery. The electrophysiologic hallmark of this syndrome is the decrement or absence of sensory and motor action potential amplitudes, with only minimal slowing of nerve conduction velocities commensurate with the degree of axonal loss. In most instances, pathologic evaluation of the peripheral nervous system from patients within the spectrum of Guillain-Barré syndrome reveals evidence of endoneurial inflammation with primary demyelination and axonal degeneration [Berciano et al., 1993]. Either process can occur in an individual patient, within a particular peripheral nerve or nerve segment [Berciano et al., 1997]. The mechanisms through which the immune system targets specific constituents of the peripheral nervous system remain poorly understood. It is clear that the immune response can be remarkably specific in affecting epitopes associated with myelin, those associated with axolemma, or both [Griffin et al., 1996a, 1996b; Hughes et al., 1999]. There have been a number of studies attempting to associate the presence of circulating immunoglobulins directed against specific ganglioside moieties with specific Guillain-Barré syndrome-related entities. For the most part, there has been only limited correlation between the targeted antigens and the syndrome phenotype. The strongest associations have been reported in the Miller Fisher syndrome with an immune response to the ganglioside GQ1B [Hughes et al., 1999; Saida, 1996; Kusunoki, 2008; Nishimoto, 2008; Asbury, 2000; Hughes and Cornblath, 2005; Yuki, 2000; van Doorn, 2009; Willison et al., 2002, 2008].
Reisin et al. [1993, 1996] reported a series of 44 children with an acute axonal form of Guillain-Barré syndrome. These patients initially had severe reduction in the amplitude of the compound motor action potentials (less than 10 percent of lower limit of normal) and, after 2 weeks of illness, had diffuse, severe denervation on needle electrode examination. Compared to patients with compound motor action potentials that were greater than 10 percent of normal, they were more likely to require assisted ventilation (60 versus 6.2 percent), were more frequently quadriplegic at the peak of their disability (80 versus 18.7 percent), and required longer periods to improve one functional grade (mean, 63.6 days versus 16.6 days) and to become ambulatory (mean, 156 versus 17.6 days).
Acute Motor Axonal Neuropathy
Although first recognized among patients from southern Mexico about 50 years ago as a distinct variant of typical Guillain-Barré syndrome on the basis of disease pathology, the first clear and detailed description of acute motor axonal neuropathy resulted from studies of clustered outbreaks in rural regions of northern China. The clinical presentation is similar to AIDP or acute motor and sensory axonal neuropathy, but is distinguished by the discrete involvement of motor axons with sparing of sensory axons [Lu et al., 2000]. Electrophysiologic hallmarks of the disease are normal sensory nerve conduction velocity studies and diminished compound motor action potential amplitudes, with normal or near-normal motor conduction velocities [McKhann et al., 1991]. Diffuse denervation is present during needle electromyography. Histopathologic examination reveals normal-appearing sensory nerves with degeneration of motor axons within motor nerve roots and motor fibers in the peripheral nervous system [Griffin et al., 1995, 1996a, b; Ho et al., 1995, 1997a; Lu et al., 2000; McKhann et al., 1993]. Just as AIDP and acute motor and sensory axonal neuropathy occur after gastrointestinal infection with Campylobacter jejuni, the association with acute motor axonal neuropathy is even stronger [Hadan and Gregson, 2001; Islam et al., 2010; Kalra et al., 2009b; Kuwabara et al., 2004; Liu et al., 2003; Nachamkin et al., 2007; Nishimoto et al., 2008a; Ogawara et al., 2000, 2003]. Patients who develop acute motor axonal neuropathy in northern China come from rural areas where the ability to purify communal drinking water is often limited or nonexistent [Wu et al., 1999]. Similar findings have been noted in children with Guillain-Barré syndrome who were treated in Buenos Aires, Argentina [Paradiso et al., 1999]. The patients with acute motor axonal neuropathy constituted 30 percent of children with Guillain-Barré syndrome with a high incidence of evidence of prior infection with C. jejuni. Although some authors have suggested that axonal forms of Guillain-Barré syndrome have a more protracted and less complete recovery than in acute inflammatory demyelinating polyneuropathy, most reports have noted that the long-term outcomes are equivalent, or nearly so [Barzegar et al., 2008; Gupta et al., 2008; Kalra et al., 2009a; Korinthenberg et al., 2007; Ramachandran and Kuruvilla, 2004; Ramirez-Zamora et al., 2009; Wierzba et al., 2008].
Miller Fisher Syndrome
Ophthalmoplegia, ataxia, and areflexia characterize this Guillain-Barré syndrome variant, described originally in adults in 1956 [Arakawa et al., 1993; Bradshaw and Jones, 1992; Eggenberger et al., 1993; Jones, 2000; Sladky, 2004; Wong, 1997]. The incidence of Miller Fisher syndrome is probably 2–4 percent in children with Guillain-Barré syndrome-like illnesses (see Table 90-5) [Bradshaw and Jones, 1992; Jones, 2000; Sladky, 2004; Jones, 2000; Korinthenberg et al., 2007; Ryan, 2005]. Cerebrospinal fluid protein elevation is seen in virtually all patients, and in most patients, complete recovery occurs. Brainstem auditory-evoked potentials have demonstrated peripheral and central auditory conduction defects in children [Wong, 1997]. Of interest are recent investigations demonstrating antibodies against the ganglioside GQ1b that recognize similar epitopes from specific C. jejuni strains [Chaudhry et al., 2006; Jacobs et al., 1997a; Jones, 2000, Korinthenberg et al., 2007; Ryan, 2005; Takahashi et al., 2005] and after Hemophilus influenzae infection [Jacobs et al., 2008; Koga et al., 2001; Nagashima, 2007; Nishimoto et al., 2004; Nishimoto, 2008a]. Cross-reactivity with the GQ1b ganglioside, which is present in cranial and other peripheral nerves, may account for the restricted involvement seen in patients with Miller Fisher syndrome [Willison et al., 1993; Willison and O’Hanlon, 1999; Willison 2005a, b, 2007]. This is in contrast to the classic form of Guillain-Barré syndrome, in which molecular mimicry has been described between anti-GM1 antibodies and C. jejuni [Ang et al., 2001a; Carpo et al., 1998; Kuroki et al., 2001; Hughes and Cornblath, 2005, Jacobs et al., 2008; Meyer Zu Horste et al., 2010; Nachamkin et al., 2007]. Successful treatment with intravenous immune globulin has been reported in a 3-year-old boy with Miller Fisher syndrome [Arakawa et al., 1993].
Polyneuritis Cranialis
Polyneuritis cranialis can occur in children, and manifests with the acute onset of multiple cranial nerve palsies (usually bilateral VII and sparing of II), with elevated cerebrospinal fluid protein, slowing of motor conduction velocities, and good recovery [Lyu and Chen, 2004; Polo et al., 1992, 2002]. Bilateral facial weakness, dysphonia, and dysphagia are typical symptoms. Evidence suggests some association with cytomegalovirus infection. One study of 20 cytomegalovirus-associated Guillain-Barré syndrome patients compared the findings with earlier established data of C. jejuni-related Guillain-Barré syndrome patients and found that the patients were significantly younger, initially had a severe course, indicated by a high frequency of respiratory insufficiency, and often developed cranial nerve involvement and severe sensory loss [Visser et al., 1996]. These observations were in contrast to patients with C. jejuni infection, which was more commonly associated with an AIDP phenotype [Hughes and Cornblath, 2005; Jacobs et al., 2008; Meyer Zu Horste et al., 2010; Nachamkin et al., 2007; Nagashima et al., 2004; Nagashima, 2007; Nishimoto et al., 2008a]. Enhancement of multiple cranial nerves seen with postcontrast magnetic resonance imaging (MRI) has also been documented, but clinical symptoms and electrodiagnostic studies reflected abnormalities caused by some, but not all, of the enhancing cranial nerves [Fulbright et al., 1995; Morosini et al., 2003].
Acute Pandysautonomia
Acute pandysautonomia is a relatively rare disorder, even within the group of diseases falling under the Guillain-Barré syndrome rubric. There are no large series of well-studied patients, and the most extensive reports cobble together a handful of subjects that have often been seen at institutions over a protracted time period. The collection of reports available in the literature, as might be anticipated, represents a heterogeneous patient population. The patients, by definition, have in common symptoms of autonomic failure manifestations that have a limited range of expression. Hence, dysautonomia provides the common thread that binds this group into a single entity without connoting a homogeneous underlying pathophysiology. Typically, such patients present with orthostatic hypotension, intestinal dysmotility, bladder dysfunction, syncope, vomiting, and difficulties with urination [Low, 1994]. They may also exhibit hypo- or hyperhidrosis, pupillary abnormalities, sensory deficits, and loss of reflexes. In addition, some patients have been described with symptoms or signs reflecting neurological abnormalities outside the confines of the autonomic nervous system and should probably be excluded from this diagnostic category. Even within this relatively homogeneous clinical group there is considerable heterogeneity in terms of its severity and outcome. In rare circumstances when the disorder has proven fatal, pathological data indicate that some patients have abnormalities quite distinctive from Guillain-Barré syndrome. There are a number of reports of patients who have developed an acute sensory neuronopathy or ganglionopathy that is usually associated with a poor recovery. Fortunately, most children who present with acute autonomic failure recover spontaneously, or respond to treatment with corticosteroids or intravenous immune globulin [Mericle and Triggs, 1997; Ramirez et al., 2004]. Acute dysautonomia most often is observed in the context of associated features that are typical of Guillain-Barré syndrome-like illnesses, making the diagnosis more straightforward. When children present with isolated acute autonomic failure, there is virtually no way to distinguish a Guillain-Barré syndrome variant from a ganglioneuronopathy early in the course of the illness. Empiric treatment is probably of limited risk and may be warranted when the dysautonomia is incapacitating. Most patients will improve relatively rapidly and fully recover.
Guillain-Barré Syndrome with Central Nervous System Manifestations
Although rare, Guillain-Barré syndrome with central nervous system (CNS) manifestations is reasonably well described in adults and children [Bradshaw and Jones, 2001; Maier et al., 1997; Nadkarni and Lisak, 1993; Okumura et al., 2002]. However, the incidence, severity, neuroimaging, and pathologic characterization of peripheral and central nervous system autoimmune disease in children remain elusive. One study reported postmortem findings in 13 adult patients with Guillain-Barré syndrome whose death ranged from 1 day to 12 months from the onset of neurologic symptoms [Maier et al., 1997]. CNS pathologic findings were predominantly secondary to injury to spinal nerve roots and cranial nerves. Evidence of primary inflammation was present, especially within the spinal cord, medulla, and pons, with clusters of mononuclear cells around small vessels. Although features of primary demyelination were identified in the peripheral nervous system in 12 of 13 patients, they were not detected in the CNS in any patients. Nadkarni and Lisak [1993] reported a 28-year-old patient with acute Guillain-Barré syndrome who developed bilateral optic neuritis and extensive CNS white-matter lesions on MRI. This illness was associated with Mycoplasma pneumoniae infection and suggested some degree of overlap with acute disseminated encephalomyelitis. It was hypothesized that certain infections might have a shared epitope that targets the peripheral and central nervous system. The clinical manifestations of CNS involvement in patients with Guillain-Barré syndrome-like illnesses range from nonspecific encephalopathy to more typical features of acute disseminated encephalomyelitis, transverse myelitis, optic neuritis, or brainstem/cerebellar syndromes [Bernard et al., 2008; Martens-Le Bouar and Korinthenberg, 2002]. The CNS component may be obscured by the peripheral nerve disease. The combination of mixed central and peripheral nervous system syndromes in children is rare, although not negligible. In one series, nearly 15 percent of children were thought to have evidence of central and peripheral nervous system disease; however, the actual frequency of Guillain-Barré syndrome-plus syndromes is probably lower. It is likely that such involvement in young children may be clinically difficult to detect, particularly if there is severe generalized weakness with or without respiratory failure.
Differential Diagnosis
A variety of other peripheral and occasionally CNS disorders can be confused with Guillain-Barré syndrome. These are outlined in Box 90-3, and the reader is referred to the specific chapters that discuss these conditions.





















Clinical Laboratory Evaluation
Cerebrospinal Fluid
A characteristic laboratory finding supporting the diagnosis of Guillain-Barré syndrome is albuminocytologic dissociation or a disproportionate elevation of cerebrospinal fluid protein in the absence of significant evidence of inflammation (i.e., >10 mononuclear cells/mm3 of cerebrospinal fluid) after the first week of symptoms of the disease, essentially regardless of the subtype. The presence of significant cerebrospinal fluid pleocytosis (>50 mononuclear cells/mm3) or the presence of polymorphonuclear leukocytes in the cerebrospinal fluid should raise doubt about the diagnosis [Asbury and Cornblath, 1990; Jones, 1996]. The elevated cerebrospinal fluid protein includes both albumin and immunoglobulins. This feature of the illness is due to breakdown of the blood–nerve barrier within the subarachnoid space surrounding spinal nerve roots. Cerebrospinal fluid protein elevation may not be detectable early in the course in about 20 percent of children. If sites of inflammation within the peripheral nervous system are distal to the intraspinal roots, protein elevation within cerebrospinal fluid may be minimal. In most reported pediatric series, the cerebrospinal fluid protein levels have been in the range of 80–200 mg/dL (normal being less than 40 mg/dL) [Briscoe et al., 1987; Epstein and Sladky, 1990; Jones, 1996, 2000; Rantala et al., 1991; Ryan, 2005; Sladky, 2004].
Electrophysiologic Testing
Electrophysiologic testing can help to confirm the diagnosis and also provide useful adjunctive information, including characterizing the pathology and disease subtype more precisely, and potentially contributing information concerning prognosis. The pathologic hallmark of AIDP is multifocal, immune-mediated demyelination within spinal roots and peripheral nerves [Asbury et al., 1969]. The electrophysiologic correlate is the presence of multifocal slowing of nerve conduction or conduction block [Alma et al., 1998; Cros and Trigges, 1996; Hadden et al., 1998; Vriesendorp et al., 1995; Hughes and Cornblath, 2005; van Doorn, 2005; van Doorn et al., 2008; Vucic et al., 2009]. In the case of acute inflammatory demyelinating polyneuropathy, the task of the clinical electrophysiologist is to demonstrate electrophysiologic evidence of multifocal demyelination in sensory and motor peripheral nerves indicative of an acquired inflammatory disorder. This task requires demonstrating asymmetric slowing of nerve conduction along the course of peripheral nerves, disparate conduction velocities among comparable nerve segments, or the presence of conduction block. Asymmetric slowing along the course of a peripheral nerve might be manifest as disproportionate prolongation of the distal motor latency compared with the degree of conduction slowing in a more proximal nerve segment. Alternatively, markedly prolonged F-wave or H-reflex latencies in the face of reasonably normal distal motor latency and nerve conduction velocity in the limb are indicative of slowing of conduction in proximal nerve segments or spinal roots. Conduction block and temporal dispersion of proximally evoked compound motor action potentials are also features of multifocal segmental demyelination in peripheral nerves and are indicative of an acquired inflammatory demyelinating neuropathy. Box 90-2 includes the somewhat dated but useful consensus criteria of electrodiagnostic features considered supportive of the diagnosis of Guillain-Barré syndrome [Asbury and Cornblath, 1990]. Application of a modified version of these criteria resulted in 60 percent of adult patients with Guillain-Barré syndrome fulfilling the criteria for polyneuropathy at the first examination (mean time interval, 6 days since disease onset) [Meulstee and van der Meche, 1995].
Magnetic Resonance Imaging
Because the differential diagnosis includes transverse myelitis, a possibility that is underscored when weakness is confined to the lower extremities and the child is complaining of back pain, spinal MRI studies occasionally may need to be acquired. Several observers have noted marked gadolinium enhancement of the cauda equina and lumbar nerves that is indicative of an inflammatory process confined to dorsal and ventral spinal nerve roots in individuals with Guillain-Barré syndrome [Baran et al., 1993; Bertorini et al., 1995; Crino et al., 1994]. Conspicuous nerve root enhancement in some studies appeared to correlate with pain, disability grade, and duration of recovery [Gorson et al., 1996]. Nerve root enhancement was also 83 percent sensitive in the diagnosis of acute Guillain-Barré syndrome and was present in 95 percent of typical cases; it is potentially useful in patients in whom the electrophysiologic abnormalities are equivocal.
Pathogenesis
The pathologic findings of Guillain-Barré syndrome are confined predominantly to spinal nerve roots and peripheral nerves [Asbury et al., 1969]. The pathogenesis of the disorder is immune-orchestrated, with macrophage-mediated injury to sensory and motor axons within the peripheral nervous system [Hafer-Macko et al., 1996b; Hartung et al., 1996; Hughes et al., 1999]. In contradistinction to the traditional notion that Schwann cell/myelin-related constituents are the primary targets of the autoimmune process and that associated axonal damage is a nonspecific “bystander effect” of the inflammatory reaction, there is evidence that epitopes expressed exclusively on axolemma may also be singled out for immune attack and result in concomitant axonal degeneration [Hafer-Macko et al., 1996b; Hartung and Kieseier, 1999; Hughes et al., 1999; Asbury, 2000; Hughes and Cornblath, 2005; Jones, 2000; van Doorn, 2009; van Doorn et al., 2008]. It is likely that myelin/Schwann cell and axonal surface epitopes are immunologically targeted to variable degrees during the active phase of Guillain-Barré syndrome, resulting in the spectrum of clinical manifestations that are features of the syndrome. The typical pathogenic mechanism in Guillain-Barré syndrome involves macrophages insinuating themselves under the myelin sheath at the node of Ranvier or between myelin lamellae, stripping myelin from the underlying axon [Hafer-Macko et al., 1996b; Hartung et al., 1996, 2002; Hughes et al., 1992, 1999; Kiefer et al., 2001; Steck et al., 1998; Vucic et al., 2009; Willison et al., 2002, 2008; Willison, 2005b]. Documenting the early events in the immune-mediated attack on the peripheral nervous system is, for obvious reasons, difficult in humans. The early pathologic changes have been characterized in a laboratory model, experimental allergic neuritis (EAN), a disorder nearly homologous to acute inflammatory demyelinating polyneuropathy. Experimental allergic neuritis can be induced in laboratory rodents immunized with constituents of peripheral nerve myelin, or with transfer of activated T cells from animals with the disease (Figure 90-1) [Rostami, 1997]. A different pattern of injury has been described (Figure 90-2) in acute motor axonal neuropathy related to Campylobacter infection in children from northern China. In these patients, macrophages have been found to interrupt axoglial tight junctions at nodes of Ranvier and attack and destroy the motor axon, sparing the circumferential myelin, which then secondarily breaks down [Griffin et al., 1995, 1996a, b; Hafer-Macko et al., 1996a; McKhann et al., 1993]. This process results in a relatively pure motor syndrome without evidence of primary demyelination on histopathologic examination or electrophysiologic testing [Griffin et al., 1995, 1996a, b; Hafer-Macko et al., 1996a; McKhann et al., 1993; Hughes and Cornblath, 2005; Kuwabara et al., 2001; Lu et al., 2000; Vucic et al., 2009; Willison et al., 2002, 2008; Willison, 2005b].
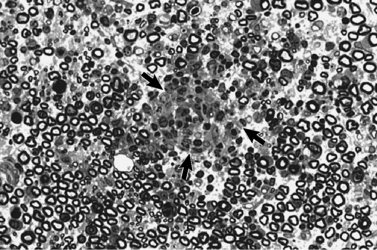
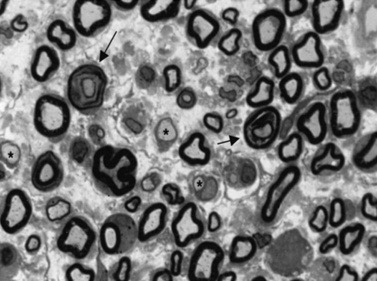
In the most directly relevant examples, there are recognizable homologies between the infectious agent and constituents of the nervous system that reinforce the notion of cross-reactivity underpinning immune-mediated neurological injury, a process that has been termed “molecular mimicry.” This case can best be made with C. jejuni, where epidemiological studies confirm a significant relation between antecedent Campylobacter enteritis and subsequent peripheral neuropathy [Hadden and Gregson, 2001; Kuwabara et al., 2004; Liu et al., 2003; Nachamkin et al., 2007; Takahashi et al., 2005; Wu et al., 1999]. Investigators also have established that infection with a relatively small group of specific C. jejuni serotypes accounts for the majority of cases of Guillain-Barré syndrome-like illness worldwide. The precise relation among serotypes, specific antibodies, and clinical phenotypes requires further study. There are compositional identities between oligosaccharides expressed on the lipo-oligosaccharide membrane of the bacterium and neural membrane gangliosides from which oligosaccharide moieties protrude, where they are recognizable by immune surveillance mechanisms. Antibodies directed toward these surface epitopes can be demonstrated in many patients and almost certainly play a role in disease pathogenesis [Hughes and Cornblath, 2005; van Doorn et al., 2008; Willison, 2005b; Willison and Plomp, 2008]. There are even stronger correlations between some phenotypical subtypes in which discrete populations of peripheral axons and Schwann cells are attacked by antibodies directed against specific gangliosides, as has been persuasively demonstrated with anti-GQ1b antibody expression in patients with the Miller Fisher syndrome variant [Akinci et al., 2007; Koga et al., 2001; Nagashima et al., 2004; Nagashima, 2007; Nishimoto et al., 2004; Takahashi et al., 2005]. Similarly, in the acute motor axonal variant of Guillain-Barré syndrome, acute motor axonal neuropathy, anti-GM1 or GD1a, ganglioside antibodies specifically target motor nerve axolemmal membranes.
The sequence of events that engenders autoimmune neuromuscular disorders such as Guillain-Barré syndrome is incompletely understood. Dalakas [1995] emphasized that several sequential steps are required to orchestrate autoimmune-mediated tissue injury:
The presence of proinflammatory and anti-inflammatory cytokines acts to modulate the intensity of the autoimmune attack on endoneurial contents through multiple pathways. Nerve fibers in peripheral nerves are relatively protected from systemic immune responses and inflammatory reactions by the specialized endoneurial, endothelial, and perineural barriers [Dalakas, 1995; Hartung et al., 2001, 2002; Hughes et al., 1999]. Compared with peripheral nerve trunks, there are more macrophage-like cells and more permeable blood vessels in the dorsal root ganglia and spinal roots that are preferred sites for the inflammatory response in experimental allergic neuritis. As noted earlier, experimental allergic neuritis can be induced by immunization with myelin proteins or fragments of the proteins. P0 and P2 proteins have been most consistently used to induce this model; a similar but less inflammatory, more chronic demyelinating neuropathy can be induced in rabbits with the glycolipid, galactocerebroside [Harvey et al., 1987; Ang et al., 2001b, 2002a, b; Willison, 2005b; Willison and O’Hanlon, 1999; Willison and Plomp, 2008]. In myelin protein-induced experimental allergic neuritis, CD4+ helper T-cell responses are sufficient to transfer full-blown disease, but antibodies of undefined specificity also may contribute to the disease process. In rabbit galactocerebroside neuritis, antibodies are probably more important and are directed predominantly against a haptenic group, including galactose. A small proportion of patients with Guillain-Barré syndrome and chronic immune demyelinating polyneuropathy show T-cell responses to P0 or P2, which may mark important pathogenetic reactions, or may simply represent an epiphenomenon. In addition, a fraction of patients with Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy have antibodies to gangliosides – in particular, the most abundant peripheral nerve ganglioside, GM1. There is an association between patients with acute severe Guillain-Barré syndrome and marked axonal degeneration and immunoglobin G1 antibodies to GM1 and GD1b; similarly, a strong correlation exists, linking Miller Fisher syndrome with antibodies to GQ1b and GD1a [Ang et al., 2001a, b, 2002a, b; Kusonoki et al., 2008; Nagashima et al., 2004; Nagashima, 2007; Nishimoto et al., 2004, 2008b; Willison, 2005b; O’Hanlon et al., 2001; Willison and Plomp, 2008]. In acute inflammatory demyelinating polyneuropathy, macrophage-mediated demyelination is probably orchestrated by autoantibody- and T-cell-mediated autoimmune responses. The nature of the autoantigen and the relative importance of T- versus B-cell responses are likely to vary, depending on the nature of the initial immunologic stimulus.
Antecedent events associated with the subsequent development of Guillain-Barré syndrome have included infection, immunization, drugs [Awong et al., 1996], trauma, or parturition. It is likely that many of the reported associations are purely coincidental. The relations between several common infections and immunizations and Guillain-Barré syndrome that seem relatively solid are summarized in Table 90-6. Some of the better-established infections include cytomegalovirus [Jacobs et al., 1997c], Epstein–Barr virus [Grose and Feorino, 1972; Jacobs et al., 1998] and other herpesviruses [Dowling and Cook, 1981], hepatitis [Tabor, 1987], and Mycoplasma spp. infections [Susuki et al., 2004; Thomas et al., 1993]. Of the acute viral infections implicated as possible antecedent events, cytomegalovirus and Epstein–Barr virus have been most consistently reported, using serologic markers, to be statistically related to subsequent Guillain-Barré syndrome illnesses [Jacobs et al., 1998; Winer et al., 1988; Hughes and Cornblath, 2005]. The subgroups of Guillain-Barré syndrome – acute inflammatory demyelinating polyneuropathy, acute motor and sensory axonal neuropathy, and acute motor axonal neuropathy – have all been associated with antecedent or coincident systemic infection with Campylobacter spp. [Hadden and Gregson, 2001; Hughes, 2004; Jacobs et al., 1997b; McCarthy and Giesecke, 2001; Melendez-Vasquez et al., 1997; Rees et al., 1995a; Toyka, 1999; Ang et al., 2000; Nachamkin et al., 2007; Ogawara et al., 2000; Schessl et al., 2006, 2007]. Controversy exists concerning the oral polio vaccine and its relation to Guillain-Barré syndrome. In 1993, the Institute of Medicine found that the evidence favored acceptance of a causal relationship between oral polio vaccine and Guillain-Barré syndrome [Ismail et al., 1998; Stratton et al., 1994]. Numerous subsequent epidemiologic studies have found no such correlation [Rantala et al., 1994; Hughes et al., 2006c; Kuitwaard et al., 2009]. This issue is discussed further in Chapter 104.
Table 90-6 Infectious Agents Associated with Guillain-Barré Syndrome
Treatment
Immunomodulating strategies for the treatment of Guillain-Barré syndrome can include agents to interrupt the autoimmune process at several stages from T-cell activation to macrophage-mediated tissue injury [Hahn, 1997; Hughes et al., 1999; Saida, 1996]. There is reasonable evidence that it is possible to blunt the severity of tissue injury by interfering with the autoimmune process at multiple levels, including interventions that:
The mechanisms by which plasmapheresis and intravenous immune globulin administration act in Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy are not entirely understood. It is likely that the primary effect of plasmapheresis is to remove circulating antibody directed toward peripheral nerve antigens. The predominant effect of intravenous immune globulin is speculative. High-dose intravenous immune globulin administration may act by binding anti-idiotypic antibodies, absorbing complement, or downregulating B-cell-mediated antibody production. Other actions that may also play a role include blockade of activated receptors, enhancement of suppressor-cell activity, and interference with lymphocyte proliferation [Thornton and Griggs, 1994]. Both of these therapies also downregulate production of proinflammatory cytokines, which play an important part in enhancing the severity of the immune-mediated injury [Aarli, 2003; Creange et al., 1996; Jander and Stoll, 2001; Lisak et al., 1997; Sharief et al., 1999; Stoll, 2002].
There are no definitive, prospectively derived data to indicate that treatment of children with Guillain-Barré syndrome using plasmapheresis or intravenous immune globulin is efficacious. Nevertheless, several limited studies indicate that both treatments may be effective for this disorder [Abd-Allah et al., 1997; Baskin et al., 1996; Epstein and Sladky, 1990; Gurses et al., 1995; Jansen et al., 1993; Kanra et al., 1997; Khatri et al., 1990; Lamont et al., 1991; McKhann, 1990; Shahar and Leiderman, 2003; Singhi et al., 1999; Sladky, 2004; Vajsar et al., 1994; Hughes et al., 2003b, 2004, 2006b, d, 2009; Korinthenberg et al., 2005]. Evidence in large prospective trials of plasmapheresis and intravenous immune globulin involving predominantly adults has confirmed that both treatment modalities reduce morbidity associated with Guillain-Barré syndrome and that they are comparable in efficacy [French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome, 1987; Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group, 1997; Hughes and Cornblath, 2005; Hughes et al., 2003b]. Both modalities appear to reduce the duration of the disease and improve the neurologic outcome [Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group, 1997]. In addition, the Guillain-Barré Syndrome Study Group documented that treatment with high-dose intravenous corticosteroids not only was ineffective, but also was likely to be harmful in adults due to the increase of adverse events in the steroid-treated cohort [Guillain-Barré Syndrome Steroid Trial Group, 1993; Hughes et al., 2010]. In a subsequent study in adults by these same investigators, plasmapheresis and intravenous immune globulin were comparable in efficacy, and these two treatments in combination were not superior to either treatment alone [Plasma Exchange/ Sandoglobulin Guillain-Barré Syndrome Trial Group, 1997]. Complementary studies have examined the benefit of performing two, four, or six plasmaphereses in patients with varying degrees of severity of Guillain-Barré syndrome [French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome, 1987]. The conclusions of this study recommended that patients with mild Guillain-Barré syndrome receive two cycles of plasmapheresis on admission, whereas patients with moderate or severe forms would benefit from two additional exchanges. More than four exchanges did not enhance the pace or completeness of recovery.
Plasmapheresis
Available data on treatment of children with Guillain-Barré syndrome are derived from small, nonrandomized series using, for the most part, historical controls. One of these studies, looking at treatment and control groups that were reasonably well matched in terms of demographic characteristics and disease severity, found that plasmapheresis significantly shortened the time required for nonambulatory patients to regain independent ambulation from 60 to 24 days [Epstein and Sladky, 1990]. This observation has been confirmed by other studies in at least 50 pediatric patients [Hammersjo, 1995; Jansen et al., 1993; Khatri et al., 1990; Lamont et al., 1991]. In adult studies, plasmapheresis was particularly effective when started within 7 days of the onset of symptoms. Similar observations have been noted in several other studies of children with severe Guillain-Barré syndrome [Jansen et al., 1993]. One study suggested that certain factors increase the risk for respiratory failure in children with Guillain-Barré syndrome and that, if these factors are present, early treatment with plasmapheresis or intravenous immune globulin is warranted. In this study, signs and symptoms predictive of respiratory failure included:
Plasmapheresis appears to be safe in children but is limited to some degree by size constraints of the patient and the availability of technically trained staff and appropriately sized equipment. Children as young as 8 months of age have been treated [Delgado, 1996], and the reported morbidity associated with plasmapheresis in children is minimal [Epstein and Sladky, 1990; Jansen et al., 1993]. Potential complications include hemorrhage, hypotension, transfusion reactions, transmitted infections, septicemia, hypocalcemia, arrhythmias, cardiac arrest, and local tissue injury [Kimoto et al., 2000]. There have been no reported deaths caused by plasmapheresis in children in any of these series.
Intravenous Immune Globulin
Similar results have also been reported in trials of intravenous immune globulin in children with Guillain-Barré syndrome [Abd-Allah et al., 1997; al-Qudah, 1994; Kanra et al., 1997; Reisin et al., 1996; Shahar et al., 1997]. Shahar and colleagues, in an open prospective multicenter study of 26 children with severe Guillain-Barré syndrome, concluded that intravenous immune globulin was effective and safe in severe childhood-onset disease and considered it the initial treatment of choice [Shahar et al., 1997]. They administered a total dose of 2 g/kg on 2 consecutive days and found marked and rapid improvement in 25 children (improvement by 1–2 disability grade scales within 2 weeks of treatment). Twenty were able to walk independently by 1 week, and 1 could be weaned off ventilatory support. Eighteen children recovered by 2 weeks. The rest recuperated in 4 months, including a child who was artificially ventilated for 4 weeks. Abd-Allah and colleagues compared a group of seven children treated with intravenous immune globulin with another cohort of eight children treated with plasmapheresis, and found that intravenous immune globulin was equally effective [Abd-Allah et al., 1997]. They reviewed data on 74 previously treated pediatric patients and recommended that intravenous immune globulin should be considered the initial choice of treatment. Despite strong sentiment in favor of more formal comparative studies of these therapies in children and the expenditure of considerable resources and energy, regulatory constraints have impeded efforts to address these issues systematically in the United States. Given these circumstances and the current economic environment, it is unlikely that persuasive data will be forthcoming in the near future that would permit evidence-based recommendations regarding an optimal treatment regimen. Unfortunately, there are insufficient reliable data available to assemble even a rudimentary treatment algorithm based on the clinical profile of a child with Guillain-Barré syndrome. As descriptive series have continued to suggest apparent efficacy for both plasmapheresis and intravenous immune globulin infusion, it seems reasonable for clinicians to adopt a pragmatic approach and choose intravenous immune globulin as a first-line therapy, based on ease of administration and side-effect profile, awaiting insights from the laboratory to provide new strategies for additional complementary therapeutic options [Goodhew and Johnson, 1996; Hughes 2008; Hughes et al., 2003a, b, 2006a, b, 2009; Korinthenberg et al., 2005; Koul and Alfutaisi, 2008; Latov 2004; Vallee et al., 1993].
Critical Care Management
Despite the dramatic therapeutic advances made during the past two decades, Guillain-Barré syndrome remains a serious disorder that requires careful management, particularly for patients who develop respiratory failure or lose the ability to swallow and maintain nutrition. The mortality rate for adults with Guillain-Barré syndrome remains at 5–8 percent [Hahn, 1996]. Within industrialized countries, the mortality from Guillain-Barré syndrome in children is virtually zero. For reasons incompletely understood, the mortality in countries with more limited resources remains in the range of 10 percent [Molinero et al., 2003]. Several articles have reviewed selected aspects of the management in the critical care setting, predominantly in adults and, to a lesser extent, in children [Fulgham and Wijdicks, 1997; Hahn, 1996; Henderson et al., 2003; Hughes, 1992, 1998; Hughes et al., 2003b; Kieseier et al., 2004; Ng et al., 1995; Ropper, 1994; Zochodne and Bolton, 1996].
By definition, children with Guillain-Barré syndrome who require admission to an intensive care unit are the most severely affected. In addition to paralysis and respiratory failure, attendant concerns include secondary infection, gastrointestinal dysfunction, urinary obstruction, and the risk of pulmonary embolism. Additional dysautonomic symptoms, including hypertension and hypotension, volume loss caused by excessive perspiration, and cardiac dysrhythmias may also complicate the course [Ropper, 1994; Zochodne, 1994]. Additional care must be taken with children concerning their fragile nutritional and hydration status. Intravenous hyperalimentation frequently is required, and occasionally, in long-term, severely compromised patients, percutaneous gastrostomy is necessary. Many severely affected children have marked dysesthetic symptoms and adequate pain control is essential. Attention to the stress of illness on the family and awareness of the problems attendant on the loss of verbal and nonverbal forms of communication, particularly in children, are critical for successful management [Khatri and Pearlstein, 1997; Manners and Murray, 1992; McDouall and Tasker, 2004; Mikati and DeLong, 1985; Moulin et al., 1997; Nguyen et al., 1999; Pentland and Donald, 1994; Roca et al., 1998; Ropper and Shahani, 1984]. In Guillain-Barré syndrome, as in other acute debilitating disorders, early implementation of a comprehensive rehabilitation program, preferably initiated while the patient is in the intensive care unit, will prevent many secondary disease complications.
Prognosis
Most case series indicate that the prognosis of Guillain-Barré syndrome in children is generally excellent and likely better than that for older age groups [Briscoe et al., 1987; Cole and Matthew, 1987; Kleyweg et al., 1989; Korinthenberg and Monting, 1996; Ortiz Corredor and Mieth Alviar, 2003; Rantala et al., 1991; Sladky, 2004; Vajsar et al., 2003; Korinthenberg et al., 2005, 2007; Koul and Alfutaisi, 2008; Kuitwaard et al., 2009; Ramachandran and Kuruvilla, 2004; Ramirez-Zamora et al., 2009; Ryan, 2005]. Opinion in the literature is not unanimous, and dissenting viewpoints argue that the clinical course and prognosis are similar [Kleyweg et al., 1989]. In the group of 49 children listed in Table 90-3, 80 percent achieved complete functional recovery within 6 months of the onset of illness. About 20 percent had mild residual neurologic deficits, including mild weakness in the facial or lower-extremity muscles and decreased deep tendon reflexes [Barbara Martin, MD, personal communication]. Similar findings, as reviewed earlier in this chapter, were reported from a retrospective international multicenter study of 175 children with Guillain-Barré syndrome [Korinthenberg and Monting, 1996].
A limitation in our ability to draw definitive conclusions regarding the prognosis of Guillain-Barré syndrome in children is the absence of prospectively defined, systematically collected clinical data. The number of children included in individual published studies is small and contributes to the variable fashion in which these limited observations are interpreted. The consensus in the literature is that children will do at least as well as, if not better than, older patients with Guillain-Barré syndrome [Jones, 2000; Korinthenberg and Monting, 1996; Sladky, 2004]. This opinion is reinforced by several nonprospective studies looking at treatment, in which children recover faster after plasmapheresis or intravenous immune globulin administration than published recovery rates in adults [Epstein and Sladky, 1990; Jansen et al., 1993; Jones, 2000; Korinthenberg and Monting, 1996; Lamont et al., 1991; Reisin et al., 1996; Sladky, 2004]. These studies are not directly comparable and need to be interpreted with caution; however, there is emerging consensus that the prognosis in childhood Guillain-Barré syndrome is good and may be enhanced as new translational initiatives bring novel treatment modalities to the bedside.
Chronic Inflammatory Demyelinating Polyneuropathy
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune-mediated polyneuropathy with a progressive phase of more than 2 months’ duration. Criteria for the diagnosis of this condition were developed in 1991 by an ad hoc subcommittee of the American Academy of Neurology. These criteria were developed for research purposes and proved to be somewhat restrictive for general clinical practice. A collaborative international task force issued a consensus paper in 2005 with guidelines for the diagnosis of CIDP that are more broadly applicable to clinical research and practice (Box 90-4). These have recently undergone revision [European Federation of Neurological Societies/Peripheral Nerve, 2005, 2010]. The disorder is recognized as having several distinct semiologies, which are included in Table 90-5.
Box 90-4 Diagnostic Criteria for the Diagnosis of Chronic Inflammatory Demyelinating Neuropathy











Axis II. Electrodiagnostic Criteria
1 Definite
At least one of the following:
2 Probable
 At least 30% amplitude reduction of the proximal negative-peak CMAP relative to distal, excluding the posterior tibial nerve, if distal negative-peak CMAP at least 20% of lower limit of normal values, in two nerves, or in one nerve and at least one other demyelinating parameter* in at least one other nerve
At least 30% amplitude reduction of the proximal negative-peak CMAP relative to distal, excluding the posterior tibial nerve, if distal negative-peak CMAP at least 20% of lower limit of normal values, in two nerves, or in one nerve and at least one other demyelinating parameter* in at least one other nerve
Axis III. Supportive Criteria







* Any nerve meeting any of the criteria (a–g). To apply these criteria, the median, ulnar (stimulated below the elbow), peroneal (stimulated below the fibular head), and tibial nerves on one side are tested. Temperatures should be maintained to at least 33°C at the palm and 30°C at the external malleolus (good practice points).
(Adapted from Joint Task Force of the EFNS and PNS: EFNS/PSN CIDP Guidelines, J Peripher Nerv Syst 10:220–228, 2005 and 15:1–9, 2010.)
Epidemiology
The true incidence of this disorder in children is unknown and is probably unlikely to be determined. Although it represents the most common acquired, potentially treatable chronic neuropathy in childhood, it none the less is rare [Iijima et al., 2008]. Ascertaining the incidence of this disease in children is further complicated by its propensity for mimicking genetically determined neuropathies, except that the often-anticipated history of dominant transmission is lacking. As many children affected with this disorder respond well to immunosuppressive therapy, it is critical to consider this disease in the differential diagnosis of any child with chronic neuropathy, particularly when there are features of demyelination and when evidence for a hereditary pattern cannot be elicited [Connolly, 2001; Korinthenberg, 1999; McLeod et al., 1999; Nevo, 1998; Rodriguez-Casero et al., 2003; Ryan et al., 2000; Sladky, 1996; Hughes et al., 2006; Markowitz et al., 2008; Vallat et al., 2010].
Clinical Features
The clinical features of CIDP are protean and diagnosis can be difficult. It can present as an indolently progressive, predominantly motor neuropathy and closely resemble hereditary motor and sensory neuropathy type I, including several genotypic variants [Carter et al., 2004]. Active demyelination may be confined to proximal nerve segments, and electrophysiologic testing may be most compatible with an axonal neuropathy with the disease masquerading as hereditary motor and sensory neuropathy type II. It has been reported to present in infancy with extremely slow nerve conduction velocity measurements resembling type III hereditary motor and sensory neuropathy [Sladky, 1996]. It may also present with features of sensory ataxia and can mimic Friedreich’s ataxia [Quan and Kleinschmidt-DeMasters, 2005]. Alternatively, the clinical manifestations may include any combination of these presentations. In some children, the disease presentation is more typical, with the distinct onset of neuropathic symptoms in a previously healthy child, a negative family history with normal examination of both parents, and confirmatory laboratory studies. This presentation is usually the exception and the likelihood of diagnosing children with more subtle presentations of CIDP is directly related to the persistence and expertise of the neurologist evaluating such patients. In summary, the disease may present in any age group, the rate of progression may be subacute to indolent, and it may affect selective populations of peripheral nerve axons and interrupt the task of any organ system that relies on peripheral nerve function for feedback or regulatory input.
There have been several series reporting the clinical, electrophysiologic, histopathological, and prognostic features in children with CIDP [Connolly, 2001; Hattori et al., 1998; Korinthenberg, 1999; Nevo et al., 1996; Simmons et al., 1997a, 1997b; Sladky, 1996; Markowitz et al., 2008; Rabie and Nevo, 2009; Rossignol et al., 2007]. As one might predict, there are divergent observations among individual reports, presumably due to the relatively small number of patients in each series. However, several common themes emerge. First, as in adults with CIDP, the clinical course and response to treatment are variable. Children may present with a subacute course or a more subtle indolent progression of symptoms. Lower-extremity weakness is the most common presenting complaint, whereas sensory symptoms are relatively uncommon. Areflexia is the rule. Second, the pace of progression of the initial symptoms may range from fulminant to subacute over 2–3 months [Rodriguez-Casero et al., 2005; Ruts et al., 2010] or may exhibit a more protracted course. In some individuals, the initial course may be so indolent as to mimic genetically determined neuropathies. Those individuals with a more rapid onset of symptoms may have a better likelihood of responding to treatment and a more benign outcome. Third, electrophysiologic testing is a useful diagnostic tool and will, in most cases, confirm the acquired nature of the neuropathy. These tests, however, have only limited predictive value regarding prognosis. Fourth, CIDP is an eminently treatable disease in children. Therapeutic outcomes fall roughly into three categories. Most children initially improve in response to treatment with corticosteroids or intravenous immune globulin. About half the children with CIDP have a monophasic course and are able to be weaned from their initial treatment, although, in many cases, over a protracted time period. The remaining patients become refractory to steroid treatment and require alternative immunosuppressant treatment or suffer one or more relapses. Most patients in this group will respond to other immunosuppressants and many achieve remission. A small fraction of children with CIDP may become refractory to aggressive immunotherapy and become dependent on chronic immunosuppression or sustain severe, widespread peripheral nerve injury with substantial residual neurologic disability. As in Guillain-Barré syndrome, the prognosis for children with CIDP is more favorable than in adults [Korinthenberg, 1999; Koul et al., 2002; Kuitwaard and van Doorn, 2009; Markowitz et al., 2008; Ryan et al., 2000].
Laboratory Evaluation
Although there are persuasive arguments to the contrary, it is sometimes useful to look at Guillain-Barré syndrome and CIDP as different ends of the temporal spectrum of the same disease. In that context, the similarities between the disorders are more striking than the differences. Like the children with the more fulminant Guillain-Barré syndrome, children with CIDP may show evidence of pathognomonic features of multifocal demyelination on electrophysiologic testing. These features have been reviewed extensively [Albers and Kelly, 1989; Connolly, 2001; Hattori et al., 1998; McCombe et al., 1987; Simmons et al., 1997b; Markowitz et al., 2008; Rabie and Nevo, 2009; Ruts et al., 2010].
Sensory and motor nerve conduction studies are the most useful and practical method to assess the physiologic properties of saltatory conduction in large-caliber myelinated fibers. Such data may be useful in distinguishing inherited from acquired disorders. However, none of the electrophysiologic measures is, in itself, an absolute discriminator. Apart from those rare kinships with steroid-responsive inherited demyelinating neuropathy [Bird and Sladky, 1991; Dyck et al., 1982c; Ginsberg et al., 2004], some children with typical hereditary motor and sensory neuropathy type I may demonstrate dispersion of proximal evoked compound motor action potentials and conduction block. Conversely, some children with CIDP may have electrophysiologic characteristics that suggest a genetically determined disease. When the clinical history is suggestive of an acquired disorder and electrophysiologic testing is compatible with an axonal neuropathy, cerebrospinal fluid examination may be helpful in demonstrating an elevated protein concentration typical of demyelinating neuropathies. Measurement of circulating antibodies to myelin protein constituents is rarely useful in children. Contrast-enhanced MRI may indicate blood–nerve barrier disruption caused by spinal nerve root inflammation [Bertorini et al., 1995; Crino et al., 1993; Kuwabara et al., 1997; Midroni et al., 1999]. Nerve biopsy may be necessary to assist in discriminating acquired from sporadic or recessively transmitted genetic disorders [Gabreels-Festen et al., 1993; Ginsberg et al., 2004; Hu et al., 2007; Schmidt et al., 1996; Vallat et al., 2003b]. Because de novo mutations can result in the appearance of a sporadic disorder, testing for an abnormality in the more common gene mutations associated with hereditary motor and sensory neuropathies phenotype may establish a diagnosis and define implications for prognosis and genetic counseling. Finally, when the clinical and laboratory data remain inconclusive, including clinical and electrophysiologic examination of family members, a therapeutic trial of immunosuppressive therapy may be warranted.
Pathologic Findings
The most characteristic histopathologic features on nerve biopsy of patients with CIDP are loss of myelinated axons, particularly larger-caliber axons; evidence of segmental demyelination and remyelination; and regional variability in the severity of fiber loss and inflammation [Baba et al., 1993; Gabreels-Festen et al., 1993; Matsumuro et al., 1994; Sladky et al., 1986]. The presence of inflammatory cells and selective fascicular variability in the expression of pathologic abnormalities are the only characteristics helpful in distinguishing an acquired from a genetically determined disorder [Gabreels-Festen et al., 1993]. The presence of thinly remyelinated internodes, segmental demyelination and remyelination, and onion bulb formations (indicative of chronicity) may be seen in any chronic demyelinating neuropathy. Specific features of inflammation (i.e., mononuclear cellular infiltrates) are present in a minority of biopsies from children and infants with CIDP. Most children with CIDP demonstrate evidence of subperineurial and endoneurial edema and an increased number of inflammatory cells dispersed within the endoneurium [Sladky et al., 1986]. Discrete perivascular collections of inflammatory cells around epineurial or endoneurial vessels are relatively rare. Teased fiber preparations demonstrating isolated demyelinated internodes or random variability in internodal length and myelin thickness are also suggestive of a multifocal inflammatory demyelinating process (Figure 90-3). Electron microscopy may confirm these features and demonstrate fragmentation of myelin sheaths, with splitting of myelin lamellae and intralamellar insinuation of macrophages ingesting internodal myelin. Immunocytochemical testing may be helpful to confirm activated lymphocytic subpopulations within the endoneurium and to demonstrate expression of class I and II major histocompatibility complex markers on Schwann cell surfaces in areas of active demyelination. Similar findings have been reported in Guillain-Barré syndrome [Asbury et al., 1969; Hughes et al., 1992; Midroni et al., 1999; van Doorn, 2009; van Doorn et al., 2008; Vucic et al., 2009; Willison, 2005b; Willison et al., 2008]. More recently, circulating antibodies to myelin-related gangliosides have been reported and associated to some degree with specific Guillain-Barré syndrome subtypes. However, antibodies to any of these candidate myelin autoantigens were not significantly more common in patients with CIDP than in controls or other groups studied. In addition, serologic evidence of C. jejuni or cytomegalovirus infection was not detected.

Treatment
Several therapeutic modalities have been found to be effective in the management of CIDP, including corticosteroids, plasmapheresis, intravenous immune globulin, and other immunosuppressive agents [Baba et al., 1993, 1996; Barnett et al., 1998; Berger et al., 2003; Brannagan, 2002; Brannagan et al., 2002; Choudhary et al., 2005b; Comi et al., 2003; Connolly, 2001; Dalakas, 2002, 2004a, b; Dyck et al., 1982a, b, 1985b, 1986, 1994; Dyck and Kurtzke, 1985a; Good et al., 1998; Gorson et al., 1998, 2004; Hahn et al., 1996a, b; Hodgkinson et al., 1990; Hughes et al., 2003a; Kissel, 2003; Mahattanakul et al., 1996; Mendell et al., 2001; Meriggioli and Rowin, 2000; Molenaar et al., 1997; Mowzoon et al., 2001; Vallat et al., 2003a; van Doorn and Ruts, 2004; Eftimov et al., 2009; Hughes, 2008, 2009; Hughes et al., 2009; Kieseier et al., 2008; Magy and Vallat, 2009; Midroni et al., 1999; van Doorn, 2009; van Doorn et al., 2008; Vucic et al., 2009; Willison, 2005b; Willison et al., 2008]. However, there are few data comparing the relative efficacy of these treatments [Choudhary and Hughes, 1995a; Kieseier et al., 2008; Kuitwaard and van Doorn, 2009; Lopate et al., 2005; Lunn and Willison, 2009]. It remains unclear which treatment should be used first. The subject is one of on-going and enthusiastic debate [Sladky, 2008; Teasley, 2008]. General consensus is for beginning with either corticosteroids or intravenous immune globulin. If initial favorable response to either treatment among naive patients is probably greater than 50 percent, and once efficacy can be confirmed for either regimen and the patient firmly established on a trajectory of improvement, undesirable aspects of either treatment can be addressed.
In patients with suspected or definite CIDP, initial treatment with oral methlyprednisolone at a dose of 2 mg/kg/day for 6–8 weeks has been successful. Most previously reported untreated children with the disease demonstrate substantial functional improvement within that timeframe [Baba et al., 1996; Connolly, 2001; Korinthenberg, 1999; Nevo, 1998; Sladky, 1996; Markowitz et al., 2008; Rossignol et al., 2007]. Lack of significant clinical improvement in this setting suggests that:
Patients nonresponsive to corticosteroids can be tapered off medication over several weeks. In steroid-responsive individuals, it is desirable to reduce the dosage schedule gradually over 6 months to an alternate-day dose of about 1 mg/kg/day, or a dose of 30 mg on alternate days in children weighing more than 30 kg. A cumulative daily dose of 0.5 mg/kg is considered to pose little significant risk for opportunistic infection, osteopenia, growth retardation, or cataract formation. This regimen can be slowly reduced over the next 4–6 months. About half the children will remain in remission after treatment is discontinued. There are scientific and empiric rationales for pulse corticosteroid regimens in CIDP. These have included oral and parenteral protocols using methylprednisolone or dexamethasone given at weekly or monthly intervals [Molenaar et al., 1997; Lopate et al., 2005; Eftimov and van Schaik, 2008; van Schaik et al., 2010].
Although most children with CIDP initially respond to corticosteroids, some become refractory or develop unacceptable side effects. Human intravenous immune globulin or plasmapheresis should then be considered [Barnett et al., 1998; Gay and Katz, 1986; Hahn, 2000; Hahn et al., 1996a, b; Kiprov and Hofmann, 2003; Tanaka, 1998; Vedanarayanan et al., 1991; Walk et al., 2004; Brannagan, 2009; Hughes et al., 2006d, 2009; Kieseier et al., 2008; Kuitwaard and van Doorn, 2009]. Intravenous immune globulin has the advantage of permitting treatment at home using visiting nursing teams, if the child has exhibited no untoward reactions to an initial course of treatment in the hospital or clinic. In older children who are large enough for adequate peripheral venous access for plasmapheresis to be established, the procedure can be initiated on an outpatient basis. The choice between therapies depends more on logistical and technical factors than on the inherent superiority of one or the other modality. Some adolescents strongly resent the forfeiture of autonomy that frequent visits to the clinic or sessions with a home health-care provider symbolize. Under these circumstances, the option of self-administered subcutaneous immunoglobulin can be explored.
A small number of children become refractory to these treatments and require more potent immunosuppressive agents to maintain remission. There are no data to guide the clinician as to which immunosuppressant will be best for an individual patient. It is probably wisest to begin with an agent with which the treating physician has the most experience [Brannagan, 2009; Hughes, 2008, 2009; Hughes et al., 2006d; Kuitwaard and van Doorn, 2009; Lunn and Willison, 2009; Magy and Vallat, 2009]. Clinicians have been inclined to avoid cyclophosphamide and azathioprine in the past because of the potential risk of developing future malignancies. However, there are probably only small differences in the risk for this late complication among different chemotherapeutic agents. Methotrexate is not commonly helpful in CIDP, but it is worth consideration in refractory cases because it can be given orally on a weekly basis and is associated with few complications when serum levels are carefully monitored. Cyclosporine can be useful in treating selected refractory cases [Barnett et al., 1998; Jongen et al., 1988; Mahattanakul et al., 1996; Ryan et al., 2000]. This agent is given orally at an initial dose of 3–5 mg/kg/day. It is generally a safe drug, if renal function and serum levels are carefully followed.
Prognosis
The prognosis in children is generally favorable [Ryan et al., 2000; Simmons et al., 1997b; Sladky, 1996; Sladky et al., 1986]. About 50 percent respond well to corticosteroids and remain in remission after treatment is discontinued. Another 25–30 percent respond to additional treatment modalities and require a prolonged course of treatment. The long-term prognosis in this group of patients remains uncertain. A small number of children respond poorly to all treatment strategies or, more typically, become refractory to treatment or develop significant side effects to protracted immunosuppression and suffer substantial neurologic disability [Connolly, 2001; Nevo, 1998; Nevo et al., 1996; Pollard, 1994, 2002; Ryan et al., 2000; Simmons et al., 1997a, b; Markowitz et al., 2008; Rabie and Nevo, 2009; Rossignol et al., 2007].
Other causes of Immune-Mediated Neuropathies in Children
Table 90-7 summarizes other causes of immune-mediated peripheral neuropathies observed in children. These occur infrequently and are associated with evidence of other systemic symptoms [Tan et al., 2003]. Although peripheral nerve disorders are described as being relatively frequent in patients with human immunodeficiency virus infection, many affected children have been identified based on electrodiagnostic criteria and often have little clinical evidence of peripheral nerve involvement [Floeter et al., 1997]. Many of these children are also receiving multiple drugs, and it is likely that peripheral neuropathy in this population is, in some cases, a side effect of treatment rather than a complication of infection. Inflammatory neuropathy, often with electrophysiologic evidence of demyelination, can occur in the course of graft-versus-host disease after bone marrow or stem cell transplantation [Adams et al., 1995; Amato et al., 1993; Imrie et al., 1994; Openshaw, 1997; Sladky, 1996; Khan et al., 2005; Lorenzoni et al., 2007; Zhang et al., 2008]. There are a number of reports associating Borrelia burgdorferi infection with peripheral neuropathy, especially CIDP. Most of these reports have involved older individuals. Chronic peripheral neuropathy including chronic inflammatory demyelinating polyneuropathy developing in the course of active Lyme disease is rare in childhood [Belman et al., 1993]. As in the case of many associations, a causal relation between CIDP and borreliosis may be inferred, but not proved. Finally, vasculitis confined to peripheral nerves and presenting as mononeuritis multiplex is extremely uncommon [Mok et al., 2001], but can occur in children and may respond well to corticosteroids or other immunosuppressant modalities [Bulun et al., 2001].
Table 90-7 Other causes of Immune-Mediated Neuropathies other than Guillain-Barré Syndrome and Chronic Immune Demyelinating Polyneuropathy in Children
| Immune Disorder | No. of Patients (Total of 10) |
|---|---|
| Bone marrow transplantation | 4 |
| Collagen vascular diseases (juvenile rheumatoid arthritis, lupus erythematosus, mixed connective tissue disease) | 3 |
| Human immunodeficiency virus infection | 1 |
| Lyme disease | 1 |
| Peripheral nerve vasculitis | 1 |
(Data from unpublished observations of John T Sladky, MD.)
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aarli J.A. Role of cytokines in neurological disorders. Curr Med Chem. 2003;10:1931.
Abd-Allah S.A., Jansen P.W., Ashwal S., et al. Intravenous immunoglobulin as therapy for pediatric Guillain-Barré syndrome. J Child Neurol. 1997;12:376.
Adams C., August C.S., Maguire H., et al. Neuromuscular complications of bone marrow transplantation. Pediatr Neurol. 1995;12:58.
Akinci G., Polat M., Tosun A., et al. Miller Fisher syndrome: a case with pattern of pure sensory polyneuropathy concomitant with anti-GQ1B antibody. Turk J Pediatr. 2007;49:109.
Albers J.W., Kelly J.J.Jr. Acquired inflammatory demyelinating polyneuropathies: Clinical and electrodiagnostic features. Muscle Nerve. 1989;12:435.
Aldrey J.M., Fernandez-Rial A., Lopez-Gonzalez F.J., et al. Guillain-Barré syndrome as first manifestation of typhoid fever. Clin Infect Dis. 1999;28:1171.
Al-Eissa Y.A., Al-Herbish A.S. Severe hypertension: An unusual presentation of Guillain-Barré syndrome in a child with brucellosis. Eur J Pediatr. 1996;155:53.
Alexander J.P.Jr, Baden L., Pallansch M.A., et al. Enterovirus 71 infections and neurologic disease – United States, 1977–1991. J Infect Dis. 1994;169:905.
Alma T.A., Chaudhry V., Cornblath D.R. Electrophysiological studies in the Guillain-Barré syndrome: Distinguishing subtypes by published criteria. Muscle Nerve. 1998;21:1275.
al-Qudah A.A. Immunoglobulins in the treatment of Guillain-Barré syndrome in early childhood. J Child Neurol. 1994;9:178.
Amato A.A., Barohn R.J., Sahenk Z., et al. Polyneuropathy complicating bone marrow and solid organ transplantation. Neurology. 1993;43:1513.
Ang C.W., Koga M., Jacobs B.C., et al. Differential immune response to gangliosides in Guillain-Barré syndrome patients from Japan and The Netherlands. J Neuroimmunol. 2001;121:83.
Ang C.W., De Klerk M.A., Endtz H.P., et al. Guillain-Barré syndrome- and Miller Fisher syndrome-associated Campylobacter jejuni lipopolysaccharides induce anti-GM1 and anti-GQ1b Antibodies in rabbits. Infect Immun. 2001;69:2462.
Ang C.W., Tio-Gillen A.P., Groen J., et al. Cross-reactive anti-galactocerebroside antibodies and Mycoplasma pneumoniae infections in Guillain-Barré syndrome. J Neuroimmunol. 2002;130:179.
Ang C.W., Laman J.D., Willison H.J., et al. Structure of Campylobacter jejuni lipopolysaccharides determines antiganglioside specificity and clinical features of Guillain-Barré and Miller Fisher patients. Infect Immun. 2002;70:1202.
Ang C.W., Tio-Gillen A.P., Groen J., et al. Cross-reactive anti-galactocerebroside antibodies and Mycoplasma pneumoniae infections in Guillain-Barré syndrome. J Neuroimmunol. 2002;130:179.
Ang C.W., van Doorn P.A., Endtz H.P., et al. A case of Guillain-Barré syndrome following a family outbreak of Campylobacter jejuni enteritis. J Neuroimmunol. 2000;111:229.
Arakawa Y., Yoshimura M., Kobayashi S., et al. The use of intravenous immunoglobulin in Miller Fisher syndrome. Brain Dev. 1993;15:231.
Asbury A.K. New concepts of Guillain-Barré syndrome. J Child Neurol. 2000;15:183.
Asbury A.K., Arnason B.G., Adams R.D. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine. 1969;48:173.
Asbury A.K., Cornblath D.R. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27:S21.
Awong I.E., Dandurand K.R., Keeys C.A., et al. Drug-associated Guillain-Barré syndrome: A literature review. Ann Pharmacother. 1996;30:173.
Baba M., Ogawa M., Matsunaga M. Treatment of chronic inflammatory demyelinating polyneuropathy (CIDP). Rinsho Shinkeigaku. 1996;36:1336.
Baba M., Takada H., Tomiyama M., et al. Chronic inflammatory demyelinating polyneuropathy in childhood. No To Shinkei. 1993;45:233.
Baran G.A., Sowell M.K., Sharp G.B., et al. MR findings in a child with Guillain-Barré syndrome. Am J Roentgenol. 1993;161:161.
Barnett M.H., Pollard J.D., Davies L., et al. Cyclosporin A in resistant chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 1998;21:454.
Barzegar M., Alizadeh A., Toopchizadeh V., et al. Association of Campylobacter jejuni infection and Guillain-Barre syndrome: a cohort study in the northwest of Iran. Turk J Pediatr. 2008;50:443.
Barzegar M., Dastgiri S., Karegarmaher M.H., et al. Epidemiology of childhood Guillan-Barre syndrome in the north west of Iran. BMC Neurol. 2007;7:22.
Baskin E., Turkay S., Icagasioglu D., et al. High-dose intravenous immune globulin in the management of severe Guillain-Barré syndrome. Turk J Pediatr. 1996;38:119.
Belman A.L., Iyer M., Coyle P.K., et al. Neurologic manifestations in children with North American Lyme disease. Neurology. 1993;43:2609.
Berciano J., Coria F., Monton F., et al. Axonal form of Guillain-Barré syndrome: Evidence for macrophage-associated demyelination. Muscle Nerve. 1993;16:744.
Berciano J., Figols J., Garcia A., et al. Fulminant Guillain-Barré syndrome with universal inexcitability of peripheral nerves: A clinicopathological study. Muscle Nerve. 1997;20:846.
Berciano J., Pascual J. Selective contrast enhancement of anterior spinal nerve roots on magnetic resonance imaging: A suggestive sign of Guillain-Barré syndrome and neurobrucellosis. J Peripher Nerv Syst. 2003;8:135.
Berger A.R., Bradley W.G., Brannagan T.H., et al. Guidelines for the diagnosis and treatment of chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst. 2003;8:282.
Berger J.R., Ayyar D.R., Kaszovitz B. Guillain-Barré syndrome complicating typhoid fever. Ann Neurol. 1986;20:649.
Bernard G., Riou E., Rosenblatt B., et al. Simultaneous Guillain-Barré syndrome and acute disseminated encephalomyelitis in the pediatric population. J Child Neurol. 2008;23:752.
Bertorini T., Halford H., Lawrence J., et al. Contrast-enhanced magnetic resonance imaging of the lumbosacral roots in the dysimmune inflammatory polyneuropathies. J Neuroimaging. 1995;5:9.
Bird S.J., Sladky J.T. Corticosteroid-responsive dominantly inherited neuropathy in childhood. Neurology. 1991;41:437.
Bos A.P., van der Meche F.G., Witsenburg M., et al. Experiences with Guillain-Barré syndrome in a pediatric intensive care unit. Intensive Care Med. 1987;13:328.
Bouma P.A., Carpay H.A., Rijpkema S.G. Antibodies to Borrelia burgdorferi in Guillain-Barré syndrome. Lancet. 1989;2:739.
Bradshaw D.Y., Jones H.R. Pseudomeningoencephalitic presentation of pediatric Guillain-Barré syndrome. J Child Neurol. 2001;16:505.
Bradshaw D.Y., Jones H.R.Jr. Guillain-Barré syndrome in children: Clinical course, electrodiagnosis, and prognosis. Muscle Nerve. 1992;15:500.
Brannagan T.H.3rd. Intravenous gammaglobulin (ivig) for treatment of CIDP and related immune-mediated neuropathies. Neurology. 2002;59(12 Suppl 6):S33.
Brannagan T.H.3rd, Pradhan A., Heiman-Patterson T., et al. High-dose cyclophosphamide without stem-cell rescue for refractory CIDP. Neurology. 2002;58:1856.
Brannagan T.H.3rd. Current treatments of chronic immune-mediated demyelinating polyneuropathies. Muscle Nerve. 2009;39:563.
Brashear H.R., Bonnin J.M., Login I.S. Encephalomyeloneuritis simulating Guillain-Barré syndrome. Neurology. 1985;35:1146.
Briscoe D.M., McMenamin J.B., O’Donohoe N.V. Prognosis in Guillain-Barré syndrome. Arch Dis Child. 1987;62:733.
Bulun A., Topaloglu R., Duzova A., et al. Ataxia and peripheral neuropathy: Rare manifestations in Henoch-Schönlein purpura. Pediatr Nephrol. 2001;16:1139.
Carpo M., Pedotti R., Lolli F., et al. Clinical correlate and fine specificity of anti-GQ1b antibodies in peripheral neuropathy. J Neurol Sci. 1998;155:186.
Carter G.T., England J.D., Chance P.F. Charcot-Marie-Tooth disease: Electrophysiology, molecular genetics and clinical management. IDrugs. 2004;7:151.
Chanmugam D., Waniganetti A. Guillain-Barré syndrome associated with typhoid fever. BMJ. 1969;1:95.
Chaudhry F., Gee K.E., Vaphiades M.S., et al. GQ1b antibody testing in Guillain-Barré syndrome and variants. Semin Ophthalmol. 2006;21:223-227.
Chin R.L., Latov N., Sander H.W., et al. Sensory CIDP presenting as cryptogenic sensory polyneuropathy. J Peripher Nerv Syst. 2004;9:132.
Choudhary P.P., Hughes R.A. Long-term treatment of chronic inflammatory demyelinating polyradiculoneuropathy with plasma exchange or intravenous immunoglobulin. QJM. 1995;88:493.
Choudhary P.P., Thompson N., Hughes R.A. Improvement following interferon beta in chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol. 1995;242:252.
Chowdhury D., Arora A. Axonal Guillain-Barré syndrome: a critical review. Acta Neurol Scand. 2001;103:267-277.
Christiansen I., Brodersen P. Pandysautonomia. Severe autonomic dysfunction accompanying polyneuropathy. Ugeskr Laeger. 2003;165:1366.
Coe C.J. Guillain-Barré syndrome in Korean children. Yonsei Med J. 1989;30:81.
Cole G.F., Matthew D.J. Prognosis in severe Guillain-Barré syndrome. Arch Dis Child. 1987;62:288.
Comi G., Quattrini A., Fazio R., et al. Immunoglobulins in chronic inflammatory demyelinating polyneuropathy. Neurol Sci. 2003;24(Suppl 4):S246.
Connolly A.M. Chronic inflammatory demyelinating polyneuropathy in childhood. Pediatr Neurol. 2001;24:177.
Cornblath D.R. Electrophysiology in Guillain-Barré syndrome. Ann Neurol. 1990;27:S17.
Creange A., Belec L., Clair B., et al. Circulating tumor necrosis factor (TNF)-alpha and soluble TNF-alpha receptors in patients with Guillain-Barré syndrome. J Neuroimmunol. 1996;68:95.
Crino P.B., Grossman R.I., Rostami A. Magnetic resonance imaging of the cauda equina in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 1993;33:311.
Crino P.B., Zimmerman R., Laskowitz D., et al. Magnetic resonance imaging of the cauda equina in Guillain-Barré syndrome. Neurology. 1994;44:1334.
Cros D., Triggs W.J. Guillain-Barré syndrome: Clinical neurophysiologic studies. Rev Neurol (Paris). 1996;152:339.
Currie D.M., Nelson M.R., Buck B.C. Guillain-Barré syndrome in children: Evidence of axonal degeneration and long-term follow-up. Arch Phys Med Rehabil. 1990;71:244.
Dalakas M.C. Basic aspects of neuroimmunology as they relate to immunotherapeutic targets: Present and future prospects. Ann Neurol. 1995;37(Suppl 1):S2.
Dalakas M.C. Advances in chronic inflammatory demyelinating polyneuropathy: Disease variants and inflammatory response mediators and modifiers. Curr Opin Neurol. 1999;12:403.
Dalakas M.C. Intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: Present status and practical therapeutic guidelines. Muscle Nerve. 1999;22:1479.
Dalakas M.C. Mechanisms of action of ivig and therapeutic considerations in the treatment of acute and chronic demyelinating neuropathies. Neurology. 2002;59(12 Suppl 6):S13.
Dalakas M.C. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291:2367.
Dalakas M.C. The use of intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: evidence-based indications and safety profile. Pharmacol Ther. 2004;102:177.
Delgado M.R. Guillain-Barré syndrome: A pediatric challenge. J Child Neurol. 1996;11:1.
Deretzi G., Pelidou S., Zou L., et al. Suppression of chronic experimental autoimmune neuritis by nasally administered recombinant rat interleukin-6. Immunology. 1999;97:69.
Donoso R. Guillain-Barré syndrome following typhoid fever [letter]. Ann Neurol. 1988;23:627.
Dowling P.C., Cook S.D. Role of infection in Guillain-Barré syndrome: Laboratory confirmation of herpes viruses in 41 cases. Ann Neurol. 1981;9(Suppl):44.
Duncan S., Will R.G., Catnach J. Mumps and Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1990;53:709.
Dyck P.J., Daube J., O’Brien P., et al. Plasma exchange in chronic inflammatory demyelinating polyradiculoneuropathy. N Engl J Med. 1986;314:461.
Dyck P.J., Kurtzke J.F. Plasmapheresis in Guillain-Barré syndrome. Neurology. 1985;35:1105.
Dyck P.J., O’Brien P., Swanson C., et al. Combined azathioprine and prednisone in chronic inflammatory-demyelinating polyneuropathy. Neurology. 1985;35:1173.
Dyck P.J., Litchy W.J., Kratz K.M., et al. A plasma exchange versus immune globulin infusion trial in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 1994;36:838.
Dyck P.J., O’Brien P.C., Oviatt K.F., et al. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann Neurol. 1982;11:136.
Dyck P.J., Pineda A., Swanson C., et al. The Mayo Clinic experience with plasma exchange in chronic inflammatory-demyelinating polyneuropathy (CIDP). Prog Clin Biol Res. 1982;106:197.
Dyck P.J., Swanson C.J., Low P.A., et al. Prednisone-responsive hereditary motor and sensory neuropathy. Mayo Clin Proc. 1982;57:239.
Eftimov F., van Schaik I.N. Immunotherapy of chronic inflammatory demyelinating polyradiculoneuropathy. Expert Opin Biol Ther. 2008;8:643-655.
Eftimov F., Winer J.B., Vermeulen M., et al. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2009. CD001797
Eggenberger E.R., Coker S., Menezes M. Pediatric Miller Fisher syndrome requiring intubation: A case report. Clin Pediatr (Phila). 1993;32:372.
Ensrud E.R., Krivickas L.S. Acquired inflammatory demyelinating neuropathies. Phys Med Rehabil Clin N Am. 2001;12:321-334. ix
Epstein M.A., Sladky J.T. The role of plasmapheresis in childhood Guillain-Barré syndrome. Ann Neurol. 1990;28:65.
Esselink R.A., Gerding M.N., Brouwers P.J., et al. Guillain-Barré syndrome associated with hantavirus infection. Lancet. 1994;343:180.
European Federation of Neurological Societies/Peripheral Nerve Society. Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst. 2005;10:220-228.
European Federation of Neurological Societies/Peripheral Nerve Society. Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–First Revision. J Peripher Nerv Syst. 2010;15:1-9.
Fagius J., Westerberg C.E., Olsson Y. Acute pandysautonomia and severe sensory deficit with poor recovery. A clinical, neurophysiological and pathological case study. J Neurol Neurosurg Psychiatry. 1983;46:725.
Farkkila M., Kinnunen E., Weckstrom P. Survey of Guillain-Barré syndrome in southern Finland. Neuroepidemiology. 1991;10:236.
Feasby T.E., Hahn A.F., Koopman W.J., et al. Central lesions in chronic inflammatory demyelinating polyneuropathy: An MRI study. Neurology. 1990;40:476.
Floeter M.K., Civitello L.A., Everett C.R., et al. Peripheral neuropathy in children with HIV infection. Neurology. 1997;49:207.
French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Efficiency of plasma exchange in Guillain-Barré syndrome: Role of replacement fluids. Ann Neurol. 1987;22:753.
Fulbright R.K., Erdum E., Sze G., et al. Cranial nerve enhancement in the Guillain-Barré syndrome. Am J Neuroradiol. 1995;16(4 Suppl):923.
Fulgham J.R., Wijdicks E.F. Guillain-Barré syndrome. Crit Care Clin. 1997;13:1.
Gabreels-Festen A.A., Gabreels F.J., Hoogendijk J.E., et al. Chronic inflammatory demyelinating polyneuropathy or hereditary motor and sensory neuropathy? Diagnostic value of morphological criteria. Acta Neuropathol (Berl). 1993;86:630.
Gabriel C.M., Gregson N.A., Redford E.J., et al. Human immunoglobulin ameliorates rat experimental autoimmune neuritis. Brain. 1997;120:1533.
Garcia T., Sanchez J.C., Maestre J.F., et al. Brucellosis and acute inflammatory polyradiculoneuropathy. Neurologia. 1989;4:145.
Gay T., Katz B. Plasma exchange as sole treatment for chronic inflammatory demyelinating polyneuropathy. Med J Aust. 1986;144:503.
Ghosh S. Guillain-Barré syndrome complicating mumps. Lancet. 1967;1:895.
Ginsberg L., Malik O., Kenton A.R., et al. Coexistent hereditary and inflammatory neuropathy. Brain. 2004;127:193-202.
Goodhew P.M., Johnston H.M. Immune globulin therapy in children with Guillain-Barré syndrome. Muscle Nerve. 1996;19:1490.
Good J.L., Chehrenama M., Mayer R.F., et al. Pulse cyclophosphamide therapy in chronic inflammatory demyelinating polyneuropathy. Neurology. 1998;51:1735.
Gorson K.C., Amato A.A., Ropper A.H. Efficacy of mycophenolate mofetil in patients with chronic immune demyelinating polyneuropathy. Neurology. 2004;63:715.
Gorson K.C., Chaudhry V.V. Chronic inflammatory demyelinating polyneuropathy. Curr Treat Options Neurol. 1999;1:251.
Gorson K.C., Ropper A.H., Clark B.D., et al. Treatment of chronic inflammatory demyelinating polyneuropathy with interferon-alpha 2a. Neurology. 1998;50:84.
Gorson K.C., Ropper A.H., Muriello M.A., et al. Prospective evaluation of MRI lumbosacral nerve root enhancement in acute Guillain-Barré syndrome. Neurology. 1996;47:813.
Griffin J.W., Ho T.W. The Guillain-Barré syndrome at 75: The Campylobacter connection. Ann Neurol. 1993;34:125.
Griffin J.W., Li C.Y., Ho T.W., et al. Guillain-Barré syndrome in northern China. The spectrum of neuropathological changes in clinically defined cases. Brain. 1995;118:577.
Griffin J.W., Li C.Y., Ho T.W., et al. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Ann Neurol. 1996;39:17.
Griffin J.W., Li C.Y., Macko C., et al. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol. 1996;25:33.
Grose C., Feorino P.M. Epstein-Barr virus and Guillain-Barré syndrome. Lancet. 1972;2:1285.
Guillain-Barré Syndrome Steroid Trial Group. Double-blind trial of intravenous methylprednisolone in Guillain-Barré syndrome. Lancet. 1993;341:586.
Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35:1096.
Gupta D., Nair M., Baheti N.N., et al. Electrodiagnostic and clinical aspects of Guillain-Barré syndrome: an analysis of 142 cases. J Clin Neuromuscul Dis. 2008;10:42.
Gurses N., Uysal S., Cetinkaya F., et al. Intravenous immunoglobulin treatment in children with Guillain-Barré syndrome. Scand J Infect Dis. 1995;27:241.
Hadden R.D., Cornblath D.R., Hughes R.A., et al. Electrophysiological classification of Guillain-Barré syndrome: Clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Ann Neurol. 1998;44:780.
Hadden R.D., Gregson N.A. Guillain-Barré syndrome and Campylobacter jejuni infection. Symp Ser Soc Appl Microbiol. 2001;30:145S.
Hafer-Macko C., Hsieh S.T., Li C.Y., et al. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann Neurol. 1996;40:635.
Hafer-Macko C.E., Sheikh K.A., Li C.Y., et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39:625.
Hahn A.F. Guillain-Barré syndrome: An evolving concept. Curr Opin Neurol. 1997;10:363.
Hahn A.F. Intravenous immunoglobulin treatment in peripheral nerve disorders-indications, mechanisms of action and side-effects. Curr Opin Neurol. 2000;13:575.
Hahn A.F. Management of Guillain-Barré syndrome (GBS). Baillieres Clin Neurol. 1996;5:627.
Hahn A.F. Treatment of chronic inflammatory demyelinating polyneuropathy with intravenous immunoglobulin. Neurology. 1998;51(6 Suppl 5):S16.
Hahn A.F., Bolton C.F., Pillay N., et al. Plasma-exchange therapy in chronic inflammatory demyelinating polyneuropathy. A double-blind, sham-controlled, cross-over study. Brain. 1996;119:1055.
Hahn A.F., Bolton C.F., Zochodne D., et al. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy. A double-blind, placebo-controlled, cross-over study. Brain. 1996;119:1067.
Hammersjö J.A. Plasma exchange in childhood Guillain-Barré syndrome. Acta Paediatr. 1995;84:355.
Hart D.E., Rojas L.A., Rosario J.A., et al. Childhood Guillain-Barré syndrome in Paraguay, 1990 to 1991. Ann Neurol. 1994;36:859.
Hartung H.P., Kieseier B.C. Antibody responses in the Guillain-Barré syndrome. J Neurol Sci. 1999;168:75.
Hartung H.P., Kieseier B.C. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol. 2000;107:140.
Hartung H.P., Kieseier B.C., Kiefer R. Progress in Guillain-Barré syndrome. Curr Opin Neurol. 2001;14:597.
Hartung H.P., Willison H.J., Kieseier B.C. Acute immunoinflammatory neuropathy: Update on Guillain-Barré syndrome. Curr Opin Neurol. 2002;15:571.
Hartung H.P., Zielasek J., Jung S., et al. Effector mechanisms in demyelinating neuropathies. Rev Neurol (Paris). 1996;152:320.
Harvey G.K., Pollard J.D., Schindhelm K., et al. Chronic experimental allergic neuritis. An electrophysiological and histological study in the rabbit. J Neurol Sci. 1987;81:215.
Hattori N., Ichimura M., Aoki S., et al. Clinicopathological features of chronic inflammatory demyelinating polyradiculoneuropathy in childhood. J Neurol Sci. 1998;154:66.
Henderson R.D., Lawn N.D., Fletcher D.D., et al. The morbidity of Guillain-Barré syndrome admitted to the intensive care unit. Neurology. 2003;60:17.
Hiraga A., Mori M., Ogawara K., et al. Recovery patterns and long term prognosis for axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2005;76:719-722.
Hiraga A., Kuwabara S., Ogawara K., et al. Patterns and serial changes in electrodiagnostic abnormalities of axonal Guillain-Barré syndrome. Neurology. 2005;64:856-860.
Hodgkinson S.J., Pollard J.D., McLeod J.G., et al. In the treatment of chronic demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 1990;53:327.
Horneff G., Huppertz H.I., Muller K., et al. Demonstration of Borrelia burgdorferi infection in a child with Guillain-Barré syndrome. Eur J Pediatr. 1993;152:810.
Ho T.W., Hsieh S.T., Nachamkin I., et al. Motor nerve terminal degeneration provides a potential mechanism for rapid recovery in acute motor axonal neuropathy after Campylobacter infection. Neurology. 1997;48:717.
Ho T.W., Li C.Y., Cornblath D.R., et al. Patterns of recovery in the Guillain-Barré syndromes. Neurology. 1997;48:695.
Ho T.W., Mishu B., Li C.Y., et al. Guillain-Barré syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain. 1995;118:597.
Hughes P.M., Wells G.M., Clements J.M., et al. Matrix metalloproteinase expression during experimental autoimmune neuritis. Brain. 1998;121:481.
Hughes R. Campylobacter jejuni in Guillain-Barré syndrome. Lancet Neurol. 2004;3:644.
Hughes R. Chronic inflammatory demyelinating polyradiculoneuropathy. J Clin Immunol. 2010. e-publication 4 April, 15, 2010
Hughes R. The role of ivig in autoimmune neuropathies: the latest evidence. J Neurol. 2008;255(Suppl 3):7.
Hughes R.A. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy: the ICE trial. Expert Rev Neurother. 2009;9:789.
Hughes R.A. Management of acute neuromuscular paralysis. J R Coll Physicians Lond. 1998;32:254.
Hughes R.A. The management of Guillain-Barré syndrome. Hosp Pract (Off Ed). 1992;27:107.
Hughes R.A. The spectrum of acquired demyelinating polyradiculoneuropathy. Acta Neurol Belg. 1994;94:128.
Hughes R.A., Allen D., Makowska A., et al. Pathogenesis of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2006;11:30-46.
Hughes R.A., Swan A.V., van Koningsveld R., et al. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006. CD001446
Hughes R.A., Raphael J.C., Swan A.V., et al. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006. CD002063
Hughes R.A., Charlton J., Latinovic R., et al. No association between immunization and Guillain-Barré syndrome in the United Kingdom, 1992 to 2000. Arch Intern Med. 2006;166:1301.
Hughes R.A., et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol. 2006;13:326.
Hughes R.A., Cornblath D.R. Guillain-Barré syndrome. Lancet. 2005;366:1653.
Hughes R.A., Dalakas M.C., Cornblath D.R., et al. Clinical applications of intravenous immunoglobulins in neurology. Clin Exp Immunol. 2009;158(Suppl 1):34.
Hughes R.A., Hadden R.D., Gregson N.A., et al. Pathogenesis of Guillain-Barré syndrome. J Neuroimmunol. 1999;100:74.
Hughes R.A., Raphael J.C., Swan A.V., et al. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2004. CD002063
Hughes R.A., Swan A.V., van Doorn P.A. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2, 2010. CD001446
Hughes R.A., Swan A.V., van Doorn P.A. Cytotoxic drugs and interferons for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 1, 2003. CD003280
Hughes R.A., Wijdicks E.F., Barohn R., et al. Practice parameter: Immunotherapy for Guillain-Barré syndrome. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736.
Hughes R., Atkinson P., Coates P., et al. Sural nerve biopsies in Guillain-Barré syndrome: Axonal degeneration and macrophage-associated demyelination and absence of cytomegalovirus genome. Muscle Nerve. 1992;15:568.
Hung K.L., Wang H.S., Liou W.Y., et al. Guillain-Barré syndrome in children: A cooperative study in Taiwan. Brain Dev. 1994;16:204.
Hu W., Janke A., Ortler S., et al. Expression of CD28-related costimulatory molecule and its ligand in inflammatory neuropathies. Neurology. 2007;68:277-282.
Iijima M., Koike H., Hattori N., et al. Prevalence and incidence rates of chronic inflammatory demyelinating polyneuropathy in the Japanese population. J Neurol Neurosurg Psychiatry. 2008;79:1040.
Imrie K.R., Couture F., Turner C.C., et al. Peripheral neuropathy following high-dose etoposide and autologous bone marrow transplantation. Bone Marrow Transplant. 1994;13:77.
Islam Z., Jacobs B.C., van Belkum A., et al. Axonal variant of Guillain-Barré syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010;74:581.
Ismail E.A., Shabani I.S., Badawi M., et al. An epidemiologic, clinical, and therapeutic study of childhood Guillain-Barré syndrome in Kuwait: Is it related to the oral polio vaccine? J Child Neurol. 1998;13:488.
Italian Guillain-Barré Study Group. The prognosis and main prognostic indicators of Guillain-Barré syndrome. A multicentre prospective study of 297 patients. Brain. 1996;119:2053.
Jacobs B.C., Endtz H.P., van der Meche F.G., et al. Humoral immune response against Campylobacter jejuni lipopolysaccharides in Guillain-Barré and Miller Fisher syndrome. J Neuroimmunol. 1997;79:62.
Jacobs B.C., Hazenberg M.P., van Doorn P.A., et al. Cross-reactive antibodies against gangliosides and Campylobacter jejuni lipopolysaccharides in patients with Guillain-Barré or Miller Fisher syndrome. J Infect Dis. 1997;175:729.
Jacobs B.C., van Doorn P.A., Groeneveld J.H., et al. Cytomegalovirus infections and anti-GM2 antibodies in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1997;62:641.
Jacobs B.C., Koga M., van Rijs W., et al. Subclass IgG to motor gangliosides related to infection and clinical course in Guillain-Barré syndrome. J Neuroimmunol. 2008;194:181.
Jacobs B.C., Rothbarth P.H., van der Meche F.G., et al. The spectrum of antecedent infections in Guillain-Barré syndrome: A case-control study. Neurology. 1998;51:1110.
Jander S., Stoll G. Interleukin-18 is induced in acute inflammatory demyelinating polyneuropathy. J Neuroimmunol. 2001;114:253.
Jansen P.W., Perkin R.M., Ashwal S. Guillain-Barré syndrome in childhood: Natural course and efficacy of plasmapheresis. Pediatr Neurol. 1993;9:16.
Jo H.Y., Park M.G., Kim D.S., et al. Chronic inflammatory demyelinating polyradiculoneuropathy in children: characterized by subacute, predominantly motor dominant polyeuropathy with a favorable response to the treatment. Acta Neurol Scand. 2010;121:342-347.
Jones H.R.Jr. Childhood Guillain-Barré syndrome: Clinical presentation, diagnosis, and therapy. J Child Neurol. 1996;11:4.
Jones H.R.Jr. Guillain-Barré syndrome: Perspectives with infants and children. Semin Pediatr Neurol. 2000;7:91.
Jongen P.J., Joosten E.M., Berden J.H., et al. Cyclosporine therapy in chronic inflammatory demyelinating polyradiculoneuropathy: A preliminary report of clinical results in two patients. Transplant Proc. 1988;20(3 Suppl 4):329.
Jung S., Toyka K.V., Hartung H.P. Soluble complement receptor type 1 inhibits experimental autoimmune neuritis in Lewis rats. Neurosci Lett. 1995;200:167.
Kalra V., Sankhyan N., Sharma S., et al. Outcome in childhood Guillain-Barré syndrome. Indian J Pediatr. 2009;76:795.
Kalra V., Chaudhry R., Dua T., et al. Association of Campylobacter jejuni infection with childhood Guillain-Barré syndrome: a case-control study. J Child Neurol. 2009;24:664.
Kanra G., Ozon A., Vajsar J., et al. Intravenous immunoglobulin treatment in children with Guillain-Barré syndrome. Eur J Paediatr Neurol. 1997;1:7.
Khan B., Hashmi K.U., Ahmed P., et al. Posttransplant Guillain Barre syndrome. J Coll Physicians Surg Pak. 2005;15:117-118.
Khatri A., Pearlstein L. Pain in Guillain-Barré syndrome. Neurology. 1997;49:1474.
Khatri B.O., Flamini J.R., Baruah J.K., et al. Plasmapheresis with acute inflammatory polyneuropathy. Pediatr Neurol. 1990;6:17.
Kiefer R., Kieseier B.C., Stoll G., et al. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109.
Kieseier B.C., Kiefer R., Gold R., et al. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30:131.
Kieseier B.C., Meyer Zu Horste G., Lehmann H.C., et al. Intravenous immunoglobulins in the treatment of immune neuropathies. Curr Opin Neurol. 2008;21:555.
Kimoto K., Odaka M., Yuki N., et al. [Complications with plasma exchange]. Rinsho Shinkeigaku. 2000;40:1044.
Kiprov D.D., Hofmann J.C. Plasmapheresis in immunologically mediated polyneuropathies. Ther Apher Dial. 2003;7:189.
Kissel J.T. The treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Semin Neurol. 2003;23:169.
Kleyweg R.P., van der Meche F.G., Loonen M.C., et al. The natural history of the Guillain-Barre syndrome in 18 children and 50 adults. J Neurol Neurosurg Psychiatry. 1989;52:853-885.
Koga M., Gilbert M., Li J., et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64:1605-1611.
Koga M., Yuki N., Tai T., et al. Miller Fisher syndrome and Haemophilus influenzae infection. Neurology. 2001;57:686.
Kollar K., Liptai Z., Rosdy B., et al. [Guillain-Barré syndrome in childhood]. Ideggyogy Sz. 2009;62:399.
Korinthenberg R. Chronic inflammatory demyelinating polyradiculoneuropathy in children and their response to treatment. Neuropediatrics. 1999;30:190.
Korinthenberg R., Monting J.S. Natural history and treatment effects in Guillain-Barré syndrome: A multicentre study. Arch Dis Child. 1996;74:281.
Korinthenberg R., Schessl J., Kirschner J. Clinical presentation and course of childhood Guillain-Barré syndrome: a prospective multicentre study. Neuropediatrics. 2007;38:10.
Korinthenberg R., Schessl J., Kirschner J., et al. Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barré syndrome: a randomized trial. Pediatrics. 2005;116:8.
Koul R., Chacko A., Javed H., et al. A profile of childhood neuropathies at a university hospital in Oman. Saudi Med J. 2002;23:450.
Koul R.L., Alfutaisi A. Prospective study of children with Guillain-Barré syndrome. Indian J Pediatr. 2008;75:787.
Kuitwaard K., Bos-Eyssen M.E., Blomkwist-Markens P.H., et al. Recurrences, vaccinations and long-term symptoms in GBS and CIDP. J Peripher Nerv Syst. 2009;14:310.
Kuitwaard K., van Doorn P.A. Newer therapeutic options for chronic inflammatory demyelinating polyradiculoneuropathy. Drugs. 2009;69:987.
Kuroki S., Saida T., Nukina M., et al. Three patients with ophthalmoplegia associated with Campylobacter jejuni. Pediatr Neurol. 2001;25:71.
Kushnir M., Klein C., Pollak L., et al. Evolving pattern of Guillain-Barré syndrome in a community hospital in Israel. Acta Neurol Scand. 2008;117:347-350.
Kusunoki S., Kaida K., Ueda M. Antibodies against gangliosides and ganglioside complexes in Guillain-Barre syndrome: new aspects of research. Biochim Biophys Acta. 2008;1780:441-444.
Kusunoki S., Shiina M., Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: Evidence of molecular mimicry. Neurology. 2001;57:736.
Kuwabara S. Guillain-Barré syndrome: epidemiology, pathophysiology and management. Drugs. 2004;64:597-610. 2007
Kuwabara S., Nakajima M., Matsuda S., et al. Magnetic resonance imaging at the demyelinative foci in chronic inflammatory demyelinating polyneuropathy. Neurology. 1997;48:874.
Kuwabara S., Ogawara K., Misawa S., et al. Does Campylobacter jejuni infection elicit “demyelinating” Guillain-Barré syndrome? Neurology. 2004;63:529.
Kuwabara S., Ogawara K., Mizobuchi K., et al. Mechanisms of early and late recovery in acute motor axonal neuropathy. Muscle Nerve. 2001;24:288.
Lamont P.J., Johnston H.M., Berdoukas V.A. Plasmapheresis in children with Guillain-Barré syndrome. Neurology. 1991;41:1928.
Latov N. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2004;62:1653.
Laura M., Gregson N.A., Curmi Y., et al. Efficacy of leukemia inhibitory factor in experimental autoimmune neuritis. J Neuroimmunol. 2002;133:56.
Lee J.H., Sung I.Y., Rew I.S. Clinical presentation and prognosis of childhood Guillain-Barré syndrome. J Paediatr Child Health. 2008;44:449.
Leger J.M. Involvement of the peripheral nervous system in HIV infection: Electromyographic study and nerve conduction velocity. Neurophysiol Clin. 1992;22:403.
Lewis R.A. Chronic inflammatory demyelinating polyneuropathy. Neurol Clin. 2007;25:71-87.
Lidin-Janson G., Strannegard O. Guillain-Barré syndrome after measles. BMJ. 1972;4:553.
Lisak R.P., Skundric D., Bealmear B., et al. The role of cytokines in Schwann cell damage, protection, and repair. J Infect Dis. 1997;176(Suppl 2):S173.
Liu G.F., Wu Z.L., Wu H.S., et al. A case-control study on children with Guillain-Barré syndrome in North China. Biomed Environ Sci. 2003;16:105.
Loly J.P., Rikir E., Seivert M., et al. Guillain-Barré syndrome following hepatitis E. World J Gastroenterol. 2009;15:1645-1647.
Lopate G., Pestronk A., Al-Lozi M. Treatment of chronic inflammatory demyelinating polyneuropathy with high-dose intermittent intravenous methylprednisolone. Arch Neurol. 2005;62:249.
Lopez de Munain A., Espinal Valencia J.B., Marti-Masso J.F., et al. Antibodies to Borrelia burgdorferi in Guillain-Barré syndrome. Lancet. 1990;335:1168.
Lorenzoni P.J., Scola R.H., Carsten A.L., et al. Chronic inflammatory demyelinating polyradiculoneuropathy in chronic graft-versus-host disease following allogeneic hematopoietic stem cell transplantation: case report. Arq Neuropsiquiatr. 2007;65:700-704.
Low P.A. Autonomic neuropathies. Curr Opin Neurol. 1994;7:402.
Lu J.L., Sheikh K.A., Wu H.S., et al. Physiologic-pathologic correlation in Guillain-Barré syndrome in children. Neurology. 2000;54:33.
Lunn M.P., Willison H.J. Diagnosis and treatment in inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2009;80:249.
Lyu R.K., Chen S.T. Acute multiple cranial neuropathy: A variant of Guillain-Barré syndrome? Muscle Nerve. 2004;30:433.
MacLennan S.C., Fahey M.C., Lawson J.A. Pharyngeal-cervical-brachial variant Guillain-Barré syndrome in a child. J Child Neurol. 2004;19:626-627.
Magy L., Vallat J.M. Evidence-based treatment of chronic immune-mediated neuropathies. Expert Opin Pharmacother. 2009;10:1741.
Mahattanakul W., Crawford T.O., Griffin J.W., et al. Treatment of chronic inflammatory demyelinating polyneuropathy with cyclosporin-A. J Neurol Neurosurg Psychiatry. 1996;60:185.
Maier H., Schmidbauer M., Pfausler B., et al. Central nervous system pathology in patients with the Guillain-Barré syndrome. Brain. 1997;120:451.
Mancardi G.L., Del Sette M., Primavera A., et al. Borrelia burgdorferi infection and Guillain-Barré syndrome. Lancet. 1989;2:985.
Manners P.J., Murray K.J. Guillain-Barré syndrome presenting with severe musculoskeletal pain. Acta Paediatr. 1992;81:1049.
Markowitz J.A., Jeste S.S., Kang P.B. Child neurology: chronic inflammatory demyelinating polyradiculoneuropathy in children. Neurology. 2008;71:e74.
Martens-Le Bouar H., Korinthenberg R. Polyradiculoneuritis with myelitis: a rare differential diagnosis of Guillain-Barré syndrome. Neuropediatrics. 2002;33:93.
Matsumuro K., Izumo S., Umehara F., et al. Chronic inflammatory demyelinating polyneuropathy: Histological and immunopathological studies on biopsied sural nerves. J Neurol Sci. 1994;127:170.
McCarthy N., Giesecke J. Incidence of Guillain-Barré syndrome following infection with Campylobacter jejuni. Am J Epidemiol. 2001;153:610.
McCombe P.A., Pollard J.D., McLeod J.G. Chronic inflammatory demyelinating polyradiculoneuropathy. A clinical and electrophysiological study of 92 cases. Brain. 1987;110:1617.
McDouall S.F., Tasker R.C. Are anticonvulsants a satisfactory alternative to opiate analgesia in patients experiencing pain with Guillain-Barré syndrome? Arch Dis Child. 2004;89:686.
McFarland H.R. Polyneuritis cranialis as the sole manifestation of the Guillain-Barré syndrome. Mo Med. 1976;73:227.
McKhann G.M. Guillain-Barré syndrome: Clinical and therapeutic observations. Ann Neurol. 1990;27:S13.
McKhann G.M., Cornblath D.R., Griffin J.W., et al. Acute motor axonal neuropathy: A frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333.
McKhann G.M., Cornblath D.R., Ho T., et al. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in northern China. Lancet. 1991;338:593.
McLean M., Duclos P., Jacob P., et al. Incidence of Guillain-Barré syndrome in Ontario and Quebec, 1983–1989, using hospital service databases. Epidemiology. 1994;5:443.
McLeod J.G., Pollard J.D., Macaskill P., et al. Prevalence of chronic inflammatory demyelinating polyneuropathy in New South Wales, Australia. Ann Neurol. 1999;46:910.
McMinn P., Stratov I., Nagarajan L., et al. Neurological manifestations of enterovirus 71 infection in children during an outbreak of hand, foot, and mouth disease in Western Australia. Clin Infect Dis. 2001;32:236.
Melendez-Vasquez C., Redford J., Choudhary P.P., et al. Immunological investigation of chronic inflammatory demyelinating polyradiculoneuropathy. J Neuroimmunol. 1997;73:124.
Mendell J.R., Barohn R.J., Freimer M.L., et al. Randomized controlled trial of ivig in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2001;56:445.
Merelli E., Sola P., Faglioni P., et al. Newest human herpesvirus (HHV-6) in the Guillain-Barré syndrome and other neurological diseases. Acta Neurol Scand. 1992;85:334.
Mericle R.A., Triggs W.J. Treatment of acute pandysautonomia with intravenous immunoglobulin. J Neurol Neurosurg Psychiatry. 1997;62:529.
Meriggioli M.N., Rowin J. Chronic inflammatory demyelinating polyneuropathy after treatment with interferon-alpha. Muscle Nerve. 2000;23:433.
Meseguer M.A., Aparicio M., Calvo A., et al. Mycoplasma pneumoniae antigen detection in Guillain-Barré syndrome. Eur J Pediatr. 1998;157:1034.
Meulstee J., van der Meche F.G. Electrodiagnostic criteria for polyneuropathy and demyelination: Application in 135 patients with Guillain-Barré syndrome. Dutch Guillain-Barré Study Group. J Neurol Neurosurg Psychiatry. 1995;59:482.
Meyer Zu Horste G., Heidenreich H., Lehmann H.C., et al. Expression of antigen processing and presenting molecules by Schwann cells in inflammatory neuropathies. Glia. 2010;58:80.
Midroni G., de Tilly L.N., Gray B., et al. MRI of the cauda equina in CIDP: clinical correlations. J Neurol Sci. 1999;170:36.
Mikati M.A., DeLong G.R. Childhood Guillain-Barré syndrome masquerading as a protracted pain syndrome. Arch Neurol. 1985;42:839.
Mok C.C., Lau C.S., Wong R.W. Neuropsychiatric manifestations and their clinical associations in southern Chinese patients with systemic lupus erythematosus. J Rheumatol. 2001;28:766.
Molenaar D.S., van Doorn P.A., Vermeulen M. Pulsed high dose dexamethasone treatment in chronic inflammatory demyelinating polyneuropathy: A pilot study. J Neurol Neurosurg Psychiatry. 1997;62:388.
Molinero M.R., Varon D., Holden K.R., et al. Epidemiology of childhood Guillain-Barré syndrome as a cause of acute flaccid paralysis in Honduras: 1989–1999. J Child Neurol. 2003;18:741.
Moran A.P., Annuk H., Prendergast M.M. Antibodies induced by ganglioside-mimicking Campylobacter jejuni lipooligosaccharides recognise epitopes at the nodes of Ranvier. J Neuroimmunol. 2005;165:179-185.
Mori M., Takagi K., Kuwabara S., et al. Guillain-Barré syndrome following hand-foot-and-mouth disease. Intern Med. 2000;39:503.
Morosini A., Burke C., Emechete B. Polyneuritis cranialis with contrast enhancement of cranial nerves on magnetic resonance imaging. J Paediatr Child Health. 2003;39:69.
Moulin D.E., Hagen N., Feasby T.E., et al. Pain in Guillain-Barré syndrome. Neurology. 1997;48:328.
Mowzoon N., Sussman A., Bradley W.G. Mycophenolate (cellcept) treatment of myasthenia gravis, chronic inflammatory polyneuropathy and inclusion body myositis. J Neurol Sci. 2001;185:119.
Murakami N., Tomita Y., Koga M., et al. An adolescent with pharyngeal-cervical-brachial variant of Guillain-Barré syndrome after cytomegalovirus infection. Brain Dev. 2006;28:269-271.
Murthy J.M. Guillain-Barré syndrome following specific viral infections: An appraisal. J Assoc Physicians India. 1994;42:27.
Nachamkin I., et al. Patterns of Guillain-Barré syndrome in children: results from a Mexican population. Neurology. 2007;69:1665.
Nadkarni N., Lisak R.P. Guillain-Barré syndrome (GBS) with bilateral optic neuritis and central white matter disease. Neurology. 1993;43:842.
Nagasawa K., Kuwabara S., Misawa S., et al. Electrophysiological subtypes and prognosis of childhood Guillain-Barré syndrome in Japan. Muscle Nerve. 2006;33:766.
Nagashima T. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64:1519.
Nagashima T., Koga M., Odaka M., et al. Clinical correlates of serum anti-GT1a IgG antibodies. J Neurol Sci. 2004;219:139.
Namiduru M., Karaoglan I., Yilmaz M. Guillain-Barré syndrome associated with acute neurobrucellosis. Int J Clin Pract. 2003;57:919.
Nass R., Chutorian A. Dysaesthesias and dysautonomia: A self-limited syndrome of painful dysaesthesias and autonomic dysfunction in childhood. J Neurol Neurosurg Psychiatry. 1982;45:162.
Nevo Y. Childhood chronic inflammatory demyelinating polyneuropathy. Eur J Paediatr Neurol. 1998;2:169.
Nevo Y., Pestronk A., Kornberg A.J., et al. Childhood chronic inflammatory demyelinating neuropathies: Clinical course and long-term follow-up. Neurology. 1996;47:98.
Ng K.K., Howard R.S., Fish D.R., et al. Management and outcome of severe Guillain-Barré syndrome. Q J Med. 1995;88:243.
Nguyen D.K., Agenarioti-Belanger S., Vanasse M. Pain and the Guillain-Barré syndrome in children under 6 years old. J Pediatr. 1999;134:773.
Nishimoto Y., Odaka M., Hirata K., et al. Usefullness of anti-GQ1b IgG antibody testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol. 2004;148:200-205.
Nishimoto Y., Susuki K., Yuki N. Serologic marker of acute motor axonal neuropathy in childhood. Pediatr Neurol. 2008;39:67.
Nishimoto Y., Odaka M., Hirata K., et al. Usefulness of anti-GQ1b IgG antibody testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol. 2008;148:200.
Ogawara K., Kuwabara S., Koga M., et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210:41.
Ogawara K., Kuwabara S., Mori M., et al. Axonal Guillain-Barré syndrome: relation to anti-ganglioside antibodies and Campylobacter jejuni infection in Japan. Ann Neurol. 2000;48:624.
O’Hanlon E.M., et al. Anti-EQ1b ganglioslide antibodies mediate complement-dependant distruction of the motor nerve terminal. Brain. 2001;124:893-906.
Oh S.J., LaGanke C., Claussen G.C. Sensory Guillain-Barré syndrome. Neurology. 2001;56:82-86.
Okumura A., Ushida H., Maruyama K., et al. Guillain-Barré syndrome associated with central nervous system lesions. Arch Dis Child. 2002;86:304.
Olive J.M., Castillo C., Castro R.G., et al. Epidemiologic study of Guillain-Barré syndrome in children <15 years of age in Latin America. J Infect Dis. 1997;175(Suppl 1):S160.
Onodera M., Mori M., Koga M., et al. Acute isolated bulbar palsy with anti-GT1a IgG antibody subsequent to Campylobacter jejuni enteritis. J Neurol Sci. 2002;205:83-84.
Ono S., Chida K., Takasu T. Guillain-Barré syndrome following fulminant viral hepatitis A. Intern Med. 1994;33:799.
Openshaw H. Peripheral neuropathy after bone marrow transplantation. Biol Blood Marrow Transplant. 1997;3:202.
Ormerod I.E., Cockerell O.C. Guillain-Barré syndrome after herpes zoster infection: A report of 2 cases. Eur Neurol. 1993;33:156.
Ortiz Corredor F., Mieth Alviar K.W. Prognostic factors for walking in childhood Guillain-Barré syndrome. Rev Neurol. 2003;36:1113.
Ortiz-Corredor F., Pena-Preciado M., Diaz-Ruiz J. Motor recovery after Guillain-Barré syndrome in childhood. Disabil Rehabil. 2007;29:883-889.
Overell J.R., Willison H.J. Recent developments in Miller Fisher syndrome and related disorders. Curr Opin Neurol. 2005;18:562-566.
Ozen H., Cemeroglu P., Ecevit Z., et al. Unusual neurologic complications of typhoid fever (aphasia, mononeuritis multiplex, and Guillain-Barré syndrome): A report of two cases. Turk J Pediatr. 1993;35:141.
Paradiso G., Tripoli J., Galicchio S., et al. Epidemiological, clinical, and electrodiagnostic findings in childhood Guillain-Barré syndrome: A reappraisal. Ann Neurol. 1999;46:701.
Pentland B., Donald S.M. Pain in the Guillain-Barré syndrome: A clinical review. Pain. 1994;59:159.
Perry A., Mehta J., Iveson T., et al. Guillain-Barré syndrome after bone marrow transplantation. Bone Marrow Transplant. 1994;14:165.
Phillips P.E. Guillain-Barré syndrome after measles. BMJ. 1972;4:50.
Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349:225.
Pollard J.D. Chronic inflammatory demyelinating polyradiculoneuropathy. Baillieres Clin Neurol. 1994;3:107.
Pollard J.D. Chronic inflammatory demyelinating polyradiculoneuropathy. Curr Opin Neurol. 2002;15:279.
Polo A., Manganotti P., Zanette G., et al. Polyneuritis cranialis: Clinical and electrophysiological findings. J Neurol Neurosurg Psychiatry. 1992;55:398.
Polo J.M., Alana-Garcia M., Cacabelos-Perez P., et al. Atypical Guillain-Barré syndrome: Multiple cranial neuropathy. Rev Neurol. 2002;34:835.
Prevots D.R., Sutter R.W. Assessment of Guillain-Barré syndrome mortality and morbidity in the United States: Implications for acute flaccid paralysis surveillance. J Infect Dis. 1997;175(Suppl 1):S151.
Quan D., Kleinschmidt-DeMasters B.K. A 71-year-old male with 4 decades of symptoms referable to both central and peripheral nervous system. Brain Pathol. 2005;15(4):369-370.
Rabaud C., May T., Hoen B., et al. Guillain-Barré syndrome associated with hantavirus infection. Clin Infect Dis. 1995;20:477.
Rabie M., Nevo Y. Childhood acute and chronic immune-mediated polyradiculoneuropathies. Eur J Paediatr Neurol. 2009;13:209.
Ramachandran R., Kuruvilla A. Guillain-Barré syndrome in children and adolescents–a retrospective analysis. J Indian Med Assoc. 2004;102:480.
Ramirez C., de Seze J., Stojkovic T., et al. [Pure subacute pandysautonomia: an assessment of treatment with intravenous polyvalent immunoglobulins.]. Rev Neurol (Paris). 2004;160:939.
Ramirez-Zamora M., Burgos-Ganuza C.R., Alas-Valle D.A., et al. [Guillain-Barré syndrome in the paediatric age: epidemiological, clinical and therapeutic profile in a hospital in El Salvador]. Rev Neurol. 2009;48:292.
Rantala H., Cherry J.D., Shields W.D., et al. Epidemiology of Guillain-Barré syndrome in children: Relationship of oral polio vaccine administration to occurrence. J Pediatr. 1994;124:220.
Rantala H., Uhari M., Cherry J.D., et al. Risk factors of respiratory failure in children with Guillain-Barré syndrome. Pediatr Neurol. 1995;13:289.
Rantala H., Uhari M., Niemela M. Occurrence, clinical manifestations, and prognosis of Guillain-Barré syndrome. Arch Dis Child. 1991;66:706. discussion, 708
Redford E.J., Smith K.J., Gregson N.A., et al. A combined inhibitor of matrix metalloproteinase activity and tumour necrosis factor-alpha processing attenuates experimental autoimmune neuritis. Brain. 1997;120:1895.
Rees J.H., Gregson N.A., Hughes R.A. Anti-ganglioside antibodies in patients with Guillain-Barré syndrome and Campylobacter jejuni infection. J Infect Dis. 1995;172:605.
Reisin R.C., Cersosimo R., Garcia Alvarez M., et al. Acute “axonal” Guillain-Barré syndrome in childhood. Muscle Nerve. 1993;16:1310.
Reisin R.C., Pociecha J., Rodriguez E., et al. Severe Guillain-Barré syndrome in childhood treated with human immune globulin. Pediatr Neurol. 1996;14:308.
Roca B., Mentero A., Simon E. Pain and opioid analgesics in Guillain-Barré syndrome. Neurology. 1998;51:924.
Rodriguez-Casero M.V., Shield L.K., Coleman L.T., et al. Childhood chronic inflammatory demyelinating polyneuropathy with central nervous system demyelination resembling multiple sclerosis. Neuromuscul Disord. 2003;13:158.
Rodriguez-Casero M.V., Shield L.K., Kornberg A.J. Subacute inflammatory demyelinating polyneuropathy in children. Neurology. 2005;64:1786.
Ropper A.H. Intensive care of acute Guillain-Barré syndrome. Can J Neurol Sci. 1994;21:S23.
Ropper A.H., Shahani B.T. Pain in Guillain-Barré syndrome. Arch Neurol. 1984;41:511.
Rossignol E., D’Anjou G., Lapointe N., et al. Evolution and treatment of childhood chronic inflammatory polyneuropathy. Pediatr Neurol. 2007;36:88.
Rostami A.M. P2-reactive T cells in inflammatory demyelination of the peripheral nerve. J Infect Dis. 1997;176(Suppl 2):S160.
Ruts L., Drenthen J., Jacobs B.C., et al. Distinguishing acute-onset CIDP from fluctuating Guillain-Barré syndrome. A prospective study. Neurology. 2010;74(21):1680-1686.
Ryan M.M. Guillain-Barré syndrome in childhood. J Paediatr Child Health. 2005;41:237.
Ryan M.M., Grattan-Smith P.J., Procopis P.G., et al. Childhood chronic inflammatory demyelinating polyneuropathy: Clinical course and long-term outcome. Neuromuscul Disord. 2000;10:398.
Sahashi K., Takahashi A., Ibi T., et al. Acute predominantly sensory neuropathy. Klin Wochenschr. 1985;63:319.
Saida K. The immunopathology of Guillain-Barré syndrome. Curr Opin Neurol. 1996;9:329.
Said G. Chronic inflammatory demyelinative polyneuropathy. J Neurol. 2002;249:245.
Sakakihara Y., Kamoshita S. Age-associated changes in the symptomatology of Guillain-Barré syndrome in children. Dev Med Child Neurol. 1991;33:611.
Sarada C., Tharakan J.K., Nair M. Guillain-Barré syndrome. A prospective clinical study in 25 children and comparison with adults. Ann Trop Paediatr. 1994;14:281.
Schessl J., Koga M., Funakoshi K., et al. Prospective study on anti-ganglioside antibodies in childhood Guillain-Barré syndrome. Arch Dis Child. 2007;92:48.
Schessl J., Luther B., Kirschner J., et al. Infections and vaccinations preceding childhood Guillain-Barré syndrome: a prospective study. Eur J Pediatr. 2006;165:605.
Schmidt B., Toyka K.V., Kiefer R., et al. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barré syndrome and chronic inflammatory demyelinating neuropathy. Muscle Nerve. 1996;19:474-487.
Seneviratne U., Gunasekera S. Acute small fibre sensory neuropathy: Another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry. 2002;72:540.
Shafqat S., Khealani B.A., Awan F., et al. Guillain-Barré syndrome in Pakistan: similarity of demyelinating and axonal variants. Eur J Neurol. 2006;13:662.
Shahar E., Leiderman M. Outcome of severe Guillain-Barré syndrome in children: Comparison between untreated cases versus gamma-globulin therapy. Clin Neuropharmacol. 2003;26:84.
Shahar E., Shorer Z., Roifman C.M., et al. Immune globulins are effective in severe pediatric Guillain-Barré syndrome. Pediatr Neurol. 1997;16:32.
Shapiro E.E. Guillain-Barré syndrome in a child with serologic evidence of Borrelia burgdorferi infection. Pediatr Infect Dis J. 1998;17:264.
Sharief M.K., Ingram D.A., Swash M., et al. I.V. immunoglobulin reduces circulating proinflammatory cytokines in Guillain-Barré syndrome. Neurology. 1999;52:1833.
Shubhakaran, Sharma C.M. Acute inflammatory demyelinating polyneuropathy with P. Falciparum malaria. J Assoc Physicians India. 2003;51:223-224.
Simmons Z., Wald J.J., Albers J.W. Chronic inflammatory demyelinating polyradiculoneuropathy in children: I. Presentation, electrodiagnostic studies, and initial clinical course, with comparison to adults. Muscle Nerve. 1997;20:1008.
Simmons Z., Wald J.J., Albers J.W. Chronic inflammatory demyelinating polyradiculoneuropathy in children: II. Long-term follow-up, with comparison to adults. Muscle Nerve. 1997;20:1569.
Singhi S.C., Jayshree M., Singhi P., et al. Intravenous immunoglobulin in very severe childhood Guillain-Barré syndrome. Ann Trop Paediatr. 1999;19:167.
Sladky J.T. Guillain-Barré syndrome in children. J Child Neurol. 2004;19:191.
Sladky J.T. Immune neuropathies in childhood. Baillieres Clin Neurol. 1996;5:233.
Sladky J.T. What is the best initial treatment for childhood chronic inflammatory demyelinating polyneuropathy: corticosteroids or intravenous immunoglobulin? Muscle Nerve. 2008;38:1638-1643.
Sladky J.T., Brown M.J., Berman P.H. Chronic inflammatory demyelinating polyneuropathy of infancy: A corticosteroid-responsive disorder. Ann Neurol. 1986;20:76.
Steck A.J., Schaeren-Wiemers N., Hartung H.P. Demyelinating inflammatory neuropathies, including Guillain-Barré syndrome. Curr Opin Neurol. 1998;11:311.
Stoll G. Inflammatory cytokines in the nervous system: Multifunctional mediators in autoimmunity and cerebral ischemia. Rev Neurol (Paris). 2002;158:887.
Stratton K.R., Howe C.J., Johnston R.B. Adverse events associated with childhood vaccines other than pertussis and rubella. Summary of a report from the Institute of Medicine. JAMA. 1994;271:1602.
Susuki K., Okada M., Mori M., et al. Acute motor axonal neuropathy after mycoplasma infection: Evidence of molecular mimicry. Neurology. 2004;62:949.
Susuki K., Yuki N., Hirata K. Features of sensory ataxic neuropathy associated with anti-GD1b IgM antibody. J Neuroimmunol. 2001;112:181-187.
Tabor E. Guillain-Barré syndrome and other neurologic syndromes in hepatitis A, B, and non-A, non-B. J Med Virol. 1987;21:207.
Takahashi M., Koga M., Yokoyama K., et al. Epidemiology of Campylobacter jejuni isolated from patients with Guillain-Barré and Fisher syndromes in Japan. J Clin Microbiol. 2005;43:335.
Tanaka M. Plasmapheresis in chronic inflammatory demyelinating polyradiculoneuropathy. Rinsho Shinkeigaku. 1998;38:853.
Tan M.J., Kandler R., Baxter P.S. Focal neuropathy in children with critical illness. Neuropediatrics. 2003;34:149.
Teasley J.E. Initial treatment of childhood chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2008;38:1640-1643.
Thomas N.H., Collins J.E., Robb S.A., et al. Mycoplasma pneumoniae infection and neurological disease. Arch Dis Child. 1993;69:573.
Thornton C.A., Griggs R.C. Plasma exchange and intravenous immunoglobulin treatment of neuromuscular disease. Ann Neurol. 1994;35:260.
Toerner J.G., Kumar P.N., Garagusi V.F. Guillain-Barré syndrome associated with Rocky Mountain spotted fever: Case report and review. Clin Infect Dis. 1996;22:1090.
Toyka K.V. Eighty three years of the Guillain-Barré syndrome: Clinical and immunopathologic aspects, current and future treatments. Rev Neurol (Paris). 1999;155:849.
Tsukada N., Koh C.S., Inoue A., et al. Demyelinating neuropathy associated with hepatitis B virus infection. Detection of immune complexes composed of hepatitis B virus surface antigen. J Neurol Sci. 1987;77:203.
Uncini A., Gallucci M., Lugaresi A., et al. CNS involvement in chronic inflammatory demyelinating polyneuropathy: an electrophysiological and MRI study. Electromyogr Clin Neurophysiol. 1991;31:365.
Usui T., Hamada Y., Arita M. A case of Guillain-Barré syndrome associated with Coxsackie b-5 virus infection. Tokushima J Exp Med. 1974;21:17.
Vajsar J., Fehlings D., Stephens D. Long-term outcome in children with Guillain-Barré syndrome. J Pediatr. 2003;142:305.
Vajsar J., Sloane A., Wood E., et al. Plasmapheresis vs intravenous immunoglobulin treatment in childhood Guillain-Barré syndrome. Arch Pediatr Adolesc Med. 1994;148:1210.
Vallat J.M., Hahn A.F., Leger J.M., et al. Interferon beta-1a as an investigational treatment for CIDP. Neurology. 2003;60(8 Suppl 3):S23.
Vallat J.M., Tabaraud F., Magy L., et al. Diagnostic value of nerve biopsy for atypical chronic inflammatory demyelinating polyneuropathy: evaluation of eight cases. Muscle Nerve. 2003;27:478-485.
Vallat J.M., Sommer C., Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010;9:402-412.
Vallee L., Dulac O., Nuyts J.P., et al. Intravenous immune globulin is also an efficient therapy of acute Guillain-Barré syndrome in affected children. Neuropediatrics. 1993;24:235.
van Doorn P.A. Treatment of Guillain-Barré syndrome and CIDP. J Peripher Nerv Syst. 2005;10:113.
Van Doorn P.A. What’s new in Guillain-Barré syndrome in 2007-2008? J Peripher Nerv Syst. 2009;14:72.
van Doorn P.A., Ruts L. Treatment of chronic inflammatory demyelinating polyneuropathy. Curr Opin Neurol. 2004;17:607.
van Doorn P.A., Ruts L., Jacobs B.C. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7:939.
van Schaik I.N., et al. Pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy (PREDICT study): a double-blind, randomised, controlled trial. Lancet Neurol. 2010;9:245-253.
Vedanarayanan V.V., Chaudhry V. Guillian Barre syndrome – recent advances. Indian J Pediatr. 2000;67:635-646.
Vedanarayanan V.V., Kandt R.S., Lewis D.V.Jr, et al. Chronic inflammatory demyelinating polyradiculoneuropathy of childhood: treatment with high-dose intravenous immunoglobulin. Neurology. 1991;41:828.
Visser L.H., van der Meche F.G., Meulstee J., et al. Cytomegalovirus infection and Guillain-Barré syndrome: the clinical, electrophysiologic, and prognostic features. Dutch Guillain-Barré Study Group. Neurology. 1996;47:668.
Vriesendorp F.J., Triggs W.J., Mayer R.F., et al. Electrophysiological studies in Guillain-Barré syndrome: Correlation with antibodies to GM1, GD1B and Campylobacter jejuni. J Neurol. 1995;242:460.
Vucic S., Kiernan M.C., Cornblath D.R. Guillain-Barré syndrome: an update. J Clin Neurosci. 2009;16:733.
Walk D., Li L.Y., Parry G.J., et al. Rapid resolution of quadriplegic CIDP by combined plasmapheresis and ivig. Neurology. 2004;62:155.
Wierzba T.F., et al. Campylobacter infection as a trigger for Guillain-Barré syndrome in Egypt. PLoS ONE. 2008;3:e3674.
Willison H. Gangliosides as targets for autoimmune injury to the nervous system. J Neurochem. 2007;103(Suppl 1):143-149.
Willison H.J. Ganglioslide complexes: new antibody targets in Guillian–Barre syndromes. Nat Clin Pract Neurol. 2005;1:2-3.
Willison H. The immunobiology of Guillain-Barré syndromes. J Peripher Nerv Syst. 2005;10:94.
Willison H.J. Fine specificity of anti-GQ1b antibodies and clinical features. J Neurol Sci. 2001;185:1-2.
Willison H.J., Halstead S.K., Beveridge E., et al. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain-Barré syndrome. J Neuroimmunol. 201, 2008.
Willison H.J., O’Hanlon G.M. The immunopathogenesis of Miller Fisher syndrome. J Neuroimmunol. 1999;100:3.
Willison H.J., Plomp J.J. Anti-ganglioside antibodies and the presynaptic motor nerve terminal. Ann N Y Acad Sci. 2008;1132:114-123.
Willison H.J., Veitch J., Paterson G., et al. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside. J Neurol Neurosurg Psychiatry. 1993;56:204.
Willison H., Stoll G., Toyka K.V., et al. Autoimmunity and inflammation in the peripheral nervous system. Trends Neurosci. 2002;25:127.
Wilmshurst J.M., Macleod M.J., Hughes E., et al. Acute sensory neuropathy in an adolescent girl following BCG vaccination. Eur J Paediatr Neurol. 1999;3:277.
Winer J.B., Hughes R.A., Anderson M.J., et al. A prospective study of acute idiopathic neuropathy. II. Antecedent events. J Neurol Neurosurg Psychiatry. 1988;51:613.
Wong V. A neurophysiological study in children with Miller Fisher syndrome and Guillain-Barré syndrome. Brain Dev. 1997;19:197.
Wu Z., et al. [A case-control study on Guillain-Barré syndrome in children of North China]. Zhonghua Yu Fang Yi Xue Za Zhi. 1999;33:279.
Yata J., Nihei K., Ohya T., et al. High-dose immunoglobulin therapy for Guillain-Barré syndrome in Japanese children. Pediatr Int. 2003;45:543.
Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barré and Fisher syndromes. Lancet Infect Dis. 2001;1:29.
Yuki N., Koga M. Bacterial infections in Guillain-Barré and Fisher syndromes. Curr Opin Neurol. 2006;19:451-457.
Yuki N., Kuwabara S. Axonal Guillain-Barré syndrome: carbohydrate mimicry and pathophysiology. J Peripher Nerv Syst. 2007;12:238-249.
Yuki N., Susuki K., Hirata K. Ataxic Guillain-Barré syndrome with anti-GQ1b antibody: relation to Miller Fisher syndrome. Neurology. 2000;54:1851-1853.
Yu S., Chen Z., Mix E., et al. Neutralizing antibodies to IL-18 ameliorate experimental autoimmune neuritis by counter-regulation of autoreactive Th1 responses to peripheral myelin antigen. J Neuropathol Exp Neurol. 2002;61:614.
Zhang L., Arrington S., Keung Y.K. Guillain-Barré syndrome after transplantation. Leuk Lymphoma. 2008;49:291-297.
Zochodne D.W. Autonomic involvement in Guillain-Barré syndrome: A review. Muscle Nerve. 1994;17:1145.
Zochodne D.W., Bolton C.F. Neuromuscular disorders in critical illness. Baillieres Clin Neurol. 1996;5:645.
Zou L.P., Abbas N., Volkmann I., et al. Suppression of experimental autoimmune neuritis by ABR-215062 is associated with altered Th1/Th2 balance and inhibited migration of inflammatory cells into the peripheral nerve tissue. Neuropharmacology. 2002;42:731.
Zou L.P., Deretzi G., Pelidou S.H., et al. Rolipram suppresses experimental autoimmune neuritis and prevents relapses in Lewis rats. Neuropharmacology. 2000;39:324.
Zou L.P., Ma M.H., Wei L., et al. IFN-beta suppresses experimental autoimmune neuritis in Lewis rats by inhibiting the migration of inflammatory cells into peripheral nervous tissue. J Neurosci Res. 1999;56:123.
