[level-membership-for-neurology-category]
Chapter 17 Hypoxic-Ischemic Brain Injury in the Term Newborn
Scope of the Problem
The syndrome of neonatal encephalopathy in the term newborn occurs in up to 6 per 1000 live term births and is a major cause of neurodevelopmental disability, with one-quarter of survivors sustaining permanent neurological deficits [Volpe, 2008]. While the syndrome of neonatal encephalopathy may result from a number of causes, this chapter will focus on the etiology, pathophysiology, and management of hypoxic-ischemic brain injury in the term newborn. Hypoxic-ischemic brain injury is recognized clinically by a characteristic encephalopathy with either a lack of alertness or hyperalertness, abnormal tone, abnormal reflex function, poor feeding, compromised respiratory status, and seizures [Miller et al., 2004]. Brain imaging has revealed patterns of brain injury following a hypoxic-ischemic insult that are unique to the immature brain; they depend on the age at which it occurs and on the severity and duration of the insult (Figure 17-1) [Miller et al., 2005; Cowan et al., 2003]. The capacity to repeat brain imaging safely in the newborn following hypoxia-ischemia has enabled studies confirming the clinical and experimental observations that neonatal brain injury evolves over days, if not weeks [Ferriero, 2004]. These clinical investigations are consistent with the prolonged temporal evolution of brain injury in neonatal animal models [Ferriero, 2004]. This time-course “opens the door” for therapeutic interventions instituted not only hours, but also days, after the hypoxic-ischemic insult, with different interventions needed as the injury evolves [Ferriero, 2004].
Etiology of Brain Injury in the Term Newborn
There is increasing recognition that neonatal brain injury is related to antenatal, perinatal, and postnatal factors, and is not always the result of “birth asphyxia” [Badawi et al., 1998]. While hypoxia-ischemia accounts for a substantial fraction of neonatal brain injury, a documented hypoxic-ischemic insult is lacking in many term newborns with encephalopathy [Volpe, 2008; Ferriero, 2004]. Many risk factors for neonatal encephalopathy are clearly prenatal [Badawi et al., 1998], such as maternal hypotension, infertility treatment, and thyroid disease. A minority of cases have only intrapartum risk factors, such as breech extraction, cord prolapse, or abruptio placentae [Badawi et al., 1998]. However, recent cohorts evaluated with magnetic resonance imaging (MRI) studies demonstrate that brain injury actually happens at or near the time of birth [Cowan et al., 2003; Miller et al., 2005]. It is also important to recognize that postnatal causes may account for up to 10 percent of neonatal encephalopathy in the term infant. Therefore, metabolic abnormalities such as acute bilirubin encephalopathy and inborn errors of metabolism, infection, severe respiratory distress, and trauma should be considered in the clinical evaluation.
Clinical Syndromes and Natural History
Clinical Syndromes
Neonatal encephalopathy and seizures are the most overt manifestation of the severity of brain injury. Clinical signs and symptoms depend on the severity, timing, and duration of the insult, as well as the newborn’s maturity, even within ages considered “full term.” It is equally important to note that not all newborns with significant acquired brain injuries present with an easily recognizable encephalopathy or seizures. Some conditions, such as stroke and perinatal white matter injury, discussed in Chapters 18 and 19, may only have subtle manifestations that may not be recognized on clinical examination. This chapter will focus on the clinical syndrome of neonatal encephalopathy secondary to hypoxia-ischemia. The diagnosis and management of neonatal seizures is discussed in Chapter 16.
Neonatal Encephalopathy
The major components of the syndrome of “neonatal encephalopathy” include: alertness, tone, respiratory status, reflexes, feeding, and seizures (Table 17-1). The severity of brain injury, reflecting the duration and magnitude of the hypoxic-ischemic insult, determines the severity of evolution of this clinical encephalopathy. In newborns with moderate to severe encephalopathy, symptoms usually evolve over days, making serial detailed examinations important. As with any neurologically ill newborn, the baby’s gestational age must be considered in the interpretation of physical findings. As described by Volpe, there is a characteristic progression of signs in newborns with a severe hypoxic-ischemic insult [Volpe, 2008]. The affected neonate exhibits a depressed level of consciousness from the very first hours of life. Periodic breathing with apnea and bradycardia often heralds this initial presentation. Hypotonia with decreased movement is almost universally found. In the first day of life, the pupillary response is often preserved and abnormalities of eye movements are not detected. In severely injured newborns, seizures may be seen within 6–12 hours after birth. Seizures at this stage are often subtle, manifesting as ocular movements, lip smacking, apnea, or bicycling movements of the extremities. The nursing staff should be alerted to identify and document paroxysmal behaviors. Multifocal or focal clonic seizures may also occur, and often indicate focal cerebral infarction. In the latter hours of the first day of life, there may be a transient increase in the level of alertness that is not accompanied by other signs of neurological improvement. This should not be falsely reassuring, as this apparent increase in alertness is frequently accompanied by more seizures and apnea, a shrill cry, and jitteriness. With careful bedside examination, weakness in the proximal limbs and increased muscle stretch reflexes may be observed, but in the very severely injured newborn, diffuse weakness with absent movements and reflexes is common.
Documenting this clinical evolution is a critical component of the evaluation, and use of a simple encephalopathy score as a bedside tool can help the clinician standardize assessment of encephalopathic newborns, monitor the evolution of the clinical syndromes, and help in systematically identifying neonates who require therapeutic intervention (see Table 17-1) [Miller et al., 2004].
| Sign | Score = 0 | Score = 1 |
|---|---|---|
| Feeding | Normal | Gavage feeds, gastrostomy tube, or not tolerating oral feeds |
| Alertness | Alert | Irritable, poorly responsive, or comatose |
| Tone | Normal | Hypotonia or hypertonia |
| Respiratory status | Normal | Respiratory distress (need for continuous positive airway pressure or mechanical ventilation) |
| Reflexes | Normal | Hyperreflexia, hyporeflexia, or absent reflexes |
| Seizure | None | Suspected or confirmed clinical seizure |
(From Miller et al. Clinical signs predict 30-month neurodevelopmental outcome after neonatal encephalopathy. Am J Obstet Gynecol 2004;190(1):93–99.)
Subtle Neonatal Syndromes
Stroke and white matter injury in the term newborn may present with clinical signs that are so subtle that they remain undetected for months or years. Advances in brain monitoring (amplitude integrated EEG, prolonged video-EEG monitoring) and brain imaging (MRI) in the neonatal intensive care unit have increased recognition of these injuries. However, both stroke and white matter injury may present in the neonatal period with an overt encephalopathy [Li et al., 2009; Ramaswamy et al., 2004]. These conditions share many of the risk factors of “global” hypoxic-ischemic brain injury considered in this chapter.
Management of Neonatal Encephalopathy
Clinical Management
As many etiologies of neonatal encephalopathy have specific therapies, the clinician’s initial task is to determine the underlying etiology through careful history taking, neurological examination, and laboratory and brain imaging studies. The history should elicit indicators of intrauterine distress that may have contributed to decreased placental or fetal blood flow: fetal heart tracing abnormalities, passage of meconium, or a history of a difficulty in labor or delivery. The Apgar score will indicate who is at risk for mortality. The details of the delivery-room resuscitation, medications, and ventilatory support should be noted. Because the clinical signs of encephalopathy and seizures are often not specific for an etiology, laboratory tests are critical to exclude reversible causes of neonatal encephalopathy. The management of moderate or severe encephalopathy should occur in a neonatal intensive care unit in close collaboration with a neonatologist. Immediate management requires securing an appropriate airway and maintaining adequate circulation. Ventilatory support with mechanical ventilation or continuous positive airway pressure (CPAP) is often required. Metabolic complications, such as hypoglycemia, hypocalcemia, hyponatremia, and acidosis, frequently accompany hypoxic-ischemic encephalopathy (HIE), and should be identified and treated. Liver enzymes and serum creatinine levels should be performed to detect injury to other organs, while serum ammonia and lactate levels can screen for possible inborn errors of metabolism. Lumbar puncture to evaluate for intracranial infections should be performed if the history is not typical for HIE, or if a clinical suspicion of infection exists. If infection is suspected, ampicillin and gentamicin are started in addition to acyclovir if herpes simplex virus infection is a consideration. If the history, examination, or initial laboratory investigation points to an inborn error of metabolism, early treatment is crucial and a biochemical geneticist should be consulted. Additional diagnostic tests, such as serum amino acids and urine organic acids, as well as specific management strategies, are required (detailed in Chapter 20). The diagnosis of a severe intracranial hemorrhage should prompt consultation with a neurosurgeon to manage raised intracranial pressure from mass effect or hydrocephalus; platelet levels and coagulation function should be measured. Since the clinical syndrome evolves considerably over the first 72 hours of life, management of specific complications, such as respiratory compromise or seizures, can often be anticipated.
Investigation of term newborns with encephalopathy addresses three primary questions:
Addressing these questions is critical for the application of “neuroprotection” strategies, such as hypothermia [Eicher et al., 2005; Gluckman et al., 2005; Shankaran et al., 2005; Azzopardi et al., 2009]. To assess the encephalopathic term newborn, diagnostic tools available to the clinician include clinical features and biochemical and electrophysiological tests. However, the severity of brain injury in term asphyxiated newborns is not reliably predicted by clinical indicators commonly used during the first days of life, such as umbilical cord pH and Apgar scores [Shevell et al., 1999]. On the other hand, the severity of clinical encephalopathy is a strong predictor of neurodevelopmental outcome [Miller, 2004]. However, while the risk of motor and cognitive deficits appears to be minimal in mild encephalopathy, and pronounced at the severe end of the spectrum, it is inconsistent in neonates with moderate encephalopathy [Sarnat and Sarnat, 1976; Robertson et al., 1989; Dixon et al., 2002; Marlow et al., 2005]. Newborns with moderate encephalopathy are thus the most likely to benefit from the improved prognostic capabilities of brain imaging.
Brain Imaging of Newborns with Encephalopathy
Current guidelines for neuroimaging term newborns with encephalopathy suggest a noncontrast computed tomography (CT) scan to detect hemorrhage in infants with a history of significant birth trauma, and evidence of low hematocrit or coagulopathy [Ment et al., 2002]. If CT findings cannot explain the newborn’s clinical status, then MRI is recommended. For other encephalopathic term newborns, MRI is recommended between days 2 and 8 to assess the location and extent of injury [Ment et al., 2002]. In a recent study, the neuroimaging findings from a group of 48 term newborns with encephalopathy uniformly scanned with CT and MRI (T1 and T2-weighted images) with diffusion-weighted imaging (DWI) on the third day of life were compared [Chau et al., 2009]. On the third day of life, both CT and MRI with DWI reliably identify injury to the basal nuclei. However, DWI more readily detects cortical injury and focal and multifocal lesions, such as strokes and white matter injury, than either CT or conventional MRI. The choice of the best neuroimaging technique must balance the risk of transport and sedation involved in MRI against the risk of ionizing radiation with CT [Brenner et al., 2003; Berrington de Gonzalez and Darby, 2004; Hall et al., 2004]. With the development of MR-compatible incubators and monitoring equipment [Dumoulin et al., 2002; Bluml et al., 2004], as well as improved capacity to scan newborns without pharmacological sedation, MRI should now be the modality of choice when possible. MRI with DWI appears to be the most sensitive imaging study to detect abnormalities associated with other causes of neonatal encephalopathy, such as cerebral dysgenesis, infections, stroke, and metabolic disorders [Volpe, 2008; Cowan et al., 2003]. In order to confirm the diagnosis of hypoxic-ischemic brain injury and determine the extent of injury, MRI and DWI are optimally obtained between 3 and 5 days of life in term newborns with encephalopathy [Chau et al., 2009; Barkovich, 1997; Rutherford et al., 2004; Barkovich et al., 2006]. In newborns treated with hypothermia, further studies are needed to determine the optimal timing of MRI. The importance of rigorous imaging protocols with appropriate quality controls, and high quality neuroradiological review, must be emphasised.
Advanced MR Techniques
Advanced MR techniques, such as diffusion and spectroscopic imaging, allow us to observe the progression of neonatal brain injury. Diffusion MRI and MR spectroscopy (MRS) can be used to measure brain maturation and also provide important measures of brain microstructure and metabolism following injury. Given the widespread availability of these techniques on current MR scanners, their application will be discussed below. In addition to diffusion MRI and MRS, other advanced MR techniques are emerging. Computer-assisted morphometric techniques, including voxel-based and deformation-based morphometry, are used to correlate regional brain volumes in newborns, children, and adolescents with a history of neonatal encephalopathy with their neurodevelopmental outcomes [Maneru et al., 2003; Nishida et al., 2006; Srinivasan et al., 2007]. Moreover, diffusion tensor tractography, an extension of diffusion MRI, is providing new insights into recovery and resilience by measuring microstructural development of specific functional pathways [Glenn et al., 2007]. Together, these quantitative techniques are helping to identify injuries and abnormalities of subsequent brain development that may not be apparent on conventional MRI.
Magnetic resonance spectroscopy imaging
MRS can be used to measure changes in certain brain metabolites from a given brain region. Of the compounds measured by MRS at long echo times, N-acetylaspartate (NAA) and lactate are the most useful in assessing brain injury. NAA is an acetylated amino acid found in high concentrations in neurons of the central nervous system (CNS). NAA levels increase with advancing cerebral maturity [Barkovich, 2000; Novotny et al., 1998], and decrease with cerebral injury or impaired cerebral metabolism [Barkovich, 2000; Novotny et al., 1998]. Lactate is normally produced in the brain by astrocytes and used as fuel by neurons to replenish energy stores via oxidative phosphorylation [Pellerin and Magistretti, 2004]. Lactate levels are elevated with disturbed brain energy substrate delivery and oxidative metabolism, as seen with hypoxia-ischemia. Elevated lactate and reduced NAA levels are highly predictive of neurodevelopmental outcome following neonatal brain injury [Barkovich, 2000; Novotny et al., 1998]. Given this, lactate/NAA ratios are especially discriminatory of newborns with adverse outcomes [Shanmugalingam et al., 2006; Thayyil et al., 2010]. Myo-inositol, one of the major brain osmolytes, is best measured with MRS at short echo times, and is elevated with neonatal hypoxic-ischemic brain injury [Robertson et al., 2001].
Diffusion imaging
Diffusion-weighted MR imaging (DWI) detects alterations in free water diffusion. Diffusion tensor imaging (DTI) measures the amount (apparent diffusion coefficient [ADC] or average diffusivity) and directionality of water motion (fractional anisotropy [FA]). With brain maturation, ADC decreases in gray and white matter, presumably due to a reduction in water content and the development of cell membranes that restrict water diffusion [Mukherjee et al., 2002; Beaulieu, 2002, Coats et al., 2009]. Over this period, FA increases in white matter, even before myelin is evident on T1 and T2-weighted images [Miller et al., 2002b; Drobyshevsky et al., 2005; Prayer et al., 2001]. DWI and DTI also provide sensitive measures of brain injury [Barkovich et al., 2001; McKinstry et al., 2002]. With acute injury, intracellular water increases and water movement are “restricted” by the cell membrane when a diffusion gradient is applied. The DWI image will show an area of restricted diffusion as increased signal intensity that is a complicated product of T2* properties and restricted diffusion. The ADC or average diffusivity map (Dav) is a quantitative water diffusion map that shows restricted diffusion as areas of diminished signal intensity. Reduced ADC values in the posterior limb of the internal capsule are associated with a greater risk of adverse neurodevelopmental outcome in term newborns with encephalopathy [Hunt et al., 2004]. FA values in the white matter and basal nuclei are decreased with significant injury during the first week of life in term newborns with encephalopathy [Ward et al., 2006]. In newborns with brain injury, DTI also detects abnormalities of microstructural brain development remote from the primary injuries, in areas of the brain that are normal on T1 and T2-weighted images [Miller et al., 2002b].
Patterns of Brain Injury
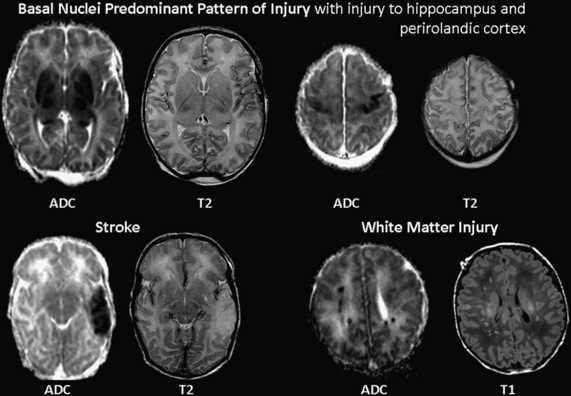
In a primate model of brain injury in the “term newborn,” the distribution of injury was associated with the duration and severity of ischemia. While acute-profound asphyxia produced injury in the basal ganglia and thalamus, partial asphyxia caused white matter injury [Myers, 1972, 1975]. Similar patterns of injury are found in term newborns following hypoxia-ischemia (see Figure 17-1). The basal ganglia-predominant pattern involves both the basal ganglia and thalamus, and perirolandic cortex [Miller et al., 2005; Sie et al., 2000; Chau et al., 2009]. The watershed pattern predominantly involves the vascular watershed, from the white matter and extending to the cerebral cortex [Miller et al., 2005; Sie et al., 2000]. Maximal injury in both the watershed region and basal nuclei results in the total pattern of brain injury [Miller et al., 2005; Sie et al., 2000]. Identifying the predominant pattern of brain injury is helpful to the clinician caring for a term newborn with encephalopathy, as the predominant pattern is more strongly associated with neurodevelopmental outcome than the severity of injury in any given region [Miller et al., 2005].
The final pattern of injury, increasingly recognized by MRI, is the “focal- or multifocal” pattern of injury: stroke or white matter injury (WMI). Recent data suggest that strokes (arterial or venous) are also associated with neonatal encephalopathy in the term newborn [Cowan et al., 2003]. Many newborns with stroke have multiple risk factors for brain injury, including intrapartum complications.
While WMI is the characteristic pattern of brain injury in premature newborns, it is increasingly recognized in term newborns with encephalopathy, identified in 23 percent in one series (see Figure 17-1) [Li et al., 2009]. In this series, WMI demonstrated restricted diffusion on ADC maps in almost all newborns, suggesting that these lesions were acquired near birth. As newborns with WMI had milder encephalopathy relative to other newborns in the cohort, these lesions may have been underdetected in the past. Lower gestational age at birth, within the range of term birth, was associated with an increasing severity of WMI, suggesting a role for brain maturation in the etiology of this injury pattern [Li et al., 2009]. In sequential studies of term newborns with encephalopathy, delayed white matter degeneration, extending past the first week of life, is also seen [Barkovich et al., 2006; Neil and Inder, 2006]. This delayed WMI might follow injury to the basal nuclei, just as Wallerian degeneration of the corticospinal tract is found following some middle cerebral artery strokes in the term newborn [De Vries et al., 2005; Kirton et al., 2007]. It should be noted that full-term infants with congenital heart disease also have a strikingly high incidence of WMI on MRI and at autopsy [McQuillen et al., 2007; Mahle et al., 2002; Galli et al., 2004; Gilles et al., 1973; Kinney et al., 2005]. Similar to premature newborns and term newborns with encephalopathy, those with congenital heart disease are at risk of impaired delivery of energy substrates due to ischemia, inflammation, and oxidative stress, particularly with cardiopulmonary bypass. Recent data from autopsy and brain imaging studies suggests that in utero brain development is delayed in newborns with some forms of serious congenital heart disease [Miller et al., 2007; Rosenthal, 1996; Licht et al., 2009]. These abnormalities in early brain development might explain the predominance of WMI in term newborns with congenital heart disease, as opposed to the more expected “term” predominance of injury to the basal nuclei or watershed regions predominantly.
Progression of Neonatal Brain Injury
Timing the onset and determining the progression of brain injury have been greatly facilitated with the use of diffusion MR techniques and MRS. Recent studies have shown that the reduction in ADC on diffusion imaging due to brain injury in term newborns evolves over the initial days of life, reaching their nadir by 2–4 days after injury [Barkovich et al., 2006; McKinstry et al., 2002]. Thus, MR diffusion images obtained prior to the nadir, as in the first day following an injury, may not show the full extent of injury. Importantly, diffusion abnormalities persist for 7–8 days in the newborn before returning towards normal values (pseudonormalization) and ultimately reflect increased diffusion [Barkovich et al., 2001; McKinstry et al., 2002; Coats et al., 2009]. In a proportion of patients, brain injury will progressively worsen over the first 2 weeks of life to involve new brain areas, particularly the white matter tracts [Barkovich et al., 2006]. It is critical to interpret diffusion imaging in the context of the time between injury and the acquisition of the scan [Barkovich et al., 2001; McKinstry et al., 2002]. MRS data in newborns mirror this time course of injury progression. In the first 24 hours following brain injury in the term newborn, lactate increases, followed by a decrease in NAA in the 3 days after injury [Barkovich, 2000; Barkovich et al., 2001; Novotny et al., 1998]. The prolonged progression of neonatal brain injury is consistent with the mechanisms of cell injury, discussed below, that persist for days following hypoxic-ischemic brain injury in the newborn. These observations also suggest that the opportunity to intervene to prevent or ameliorate brain injury may extend over days in the term newborn, if not weeks. These quantitative MR techniques now offer a dynamic measure of brain injury in the newborn that can be safely used to determine the short-term effects of novel intervention strategies.
Outcomes
Neurodevelopmental outcomes following neonatal encephalopathy depend on the pattern and severity of the brain injury. Neurodevelopmental deficits may involve motor, visual, and cognitive functions. Both genetic and postnatal variables such as socioeconomic factors (e.g., environmental exposures and parental education) likely modify an individual’s neurodevelopmental outcome following neonatal brain injury [Miller et al., 2002a; Robertson and Finer, 1993]. Given the broad spectrum of neurodevelopmental impairments following neonatal encephalopathy, follow-up of these newborns should include assessment of motor function, vision and hearing, cognition, behavior, and quality of life, through infancy and childhood. Epilepsy is identified in up to one-half of survivors from moderate to severe neonatal encephalopathy [Brunquell et al., 2002; Clancy and Legido, 1991], and is particularly common in those infants with cerebral palsy and developmental delay [Toet et al., 2005].
The American College of Obstetricians and Gynecologists (ACOG) Task Force on Neonatal Encephalopathy concluded that an acute intrapartum event could result in cerebral palsy of the spastic quadriplegic or dyskinetic type, but could not account for isolated cognitive deficits [ACOG, 2004]. Recently reviewed data [Gonzalez and Miller, 2006] indicate that cognitive deficits may feature prominently following term neonatal encephalopathy of presumed hypoxic-ischemic brain injury, even in the absence of cerebral palsy. This pattern of neurodevelopmental deficits follows an overt neonatal encephalopathy, often in the context of a critical illness, and is most commonly associated with the watershed pattern of injury and white matter damage, rather than the basal nuclei-predominant pattern of injury. A complete assessment of neurodevelopmental outcome must include aspects of cognition most readily assessed at school age: learning, executive function, behavior, and social competence. In addition, developmental coordination disorder, autism spectrum disorder, or specific language impairments should be considered as possibilities in follow-up of newborns with a history of encephalopathy [van Handel et al., 2007]. Finally, “quality of life,” an individual’s subjective perception of physical and psychological health, should be evaluated by the clinician assessing outcomes to tailor rehabilitation and monitoring services best.
Motor Function
In term survivors of hypoxic-ischemic brain injury, the risk of cerebral palsy or severe disability may involve more than one-third of affected newborns, and is most common in those with a severe encephalopathy [Volpe, 2008; Dixon et al., 2002; Barnett et al., 2002]. Spastic quadriparesis is the most common type of cerebral palsy, although athetoid or spastic hemiparesis also occurs. The diagnosis and management of cerebral palsy are addressed in Chapter 69. A complete assessment of neurosensory and cognitive functions is critical in children with cerebral palsy [Marlow et al., 2005]. Minor motor impairments that do not meet diagnostic criteria for cerebral palsy [Shevell et al., 1999] are diagnosed in more than one-third of children with moderate encephalopathy, and in more than one-quarter of children with mild encephalopathy [Van Kooij et al., 2008].
Vision and Hearing
Severe visual impairment occurs in up to one-quarter of children after moderate or severe encephalopathy [Robertson and Finer, 1985; Shankaran et al., 1991]. This may be due to injury to the posterior visual pathway, including the primary visual cortex, resulting in “cortical visual impairment” [Van Hof-van Duin and Mohn, 1984]. Injuries to the basal nuclei may also affect acuity, visual fields, or stereopsis (depth perception) [Mercuri et al., 1997]. Sensorineural hearing loss, likely secondary to brainstem injury, is also seen following neonatal encephalopathy [Robertson and Finer, 1985; Robertson et al., 1989], affecting 18 percent of survivors of moderate encephalopathy without cerebral palsy [Lindstrom et al., 2006].
Cognition
Overall, cognitive deficits are seen in 30–50 percent of childhood survivors of moderate neonatal encephalopathy [Dilenge et al., 2001]. Intellectual performance in children with severe encephalopathy without cerebral palsy is also affected [Marlow et al., 2005]. School-age survivors of moderate neonatal encephalopathy are more likely to have difficulties with reading, spelling, and arithmetic, or require additional school resources [Moster et al., 2002; Robertson et al., 1989]. Cognitive deficits, such as those in language and memory, may be seen, even when IQ scores are “normal.” [Marlow et al., 2005]. Behavioral difficulties, such as hyperactivity and emotional problems, should also be considered, even in individuals without motor disability [Marlow et al., 2005].
Brain Imaging and Outcome
The pattern of brain injury on neuroimaging conveys important prognostic information regarding the “pattern” of neurodevelopmental abnormalities. The basal nuclei pattern of injury and abnormal signal intensity in the posterior limb of the internal capsule are both predictive of severely impaired motor and cognitive outcomes [Rutherford et al., 1998; Miller et al., 2005]. The cognitive deficits associated with this pattern are not surprising, given the common involvement of the cerebral cortex [Miller et al., 2005] and cerebellum [Le Strange et al., 2004; Sargent et al., 2004]. In contrast, the watershed pattern is associated with cognitive impairments that are not necessarily accompanied by major motor deficits [Miller et al., 2005]. Importantly, the cognitive deficits following the watershed pattern may only be evident after 2 years of age [Miller et al., 2005]. In survivors of neonatal encephalopathy without functional motor deficits assessed at 4 years of age, the severity of watershed-distribution injury was most strongly associated with impaired language skills [Steinman et al., 2009]. While neurodevelopmental outcomes may be significantly better than anticipated, neurological deficits may also be found in some newborns whose brain imaging studies are normal [Bax et al., 2006]. More subtle brain injuries associated with later neurodevelopmental deficits, such as white matter injuries or hippocampal volume loss, may only be detectable with quantitative brain imaging techniques [Miller et al., 2002a; Nagy et al., 2005; Gadian et al., 2000].
Pathophysiology of Neonatal Hypoxic-Ischemic Brain Injury
In term infants with neonatal encephalopathy, perinatal hypoxia-ischemia predominates as the major cause of future neurologic disability. The adverse consequences of cerebral ischemia include deprivation of energy substrates and oxygen, and an inability to clear accumulated, potentially toxic metabolites. Although linear flow charts cannot accurately convey the complex cascade of interrelated molecular pathways that lead to hypoxic-ischemic neurodegeneration, Figure 17-2 highlights some of the critical mechanisms.
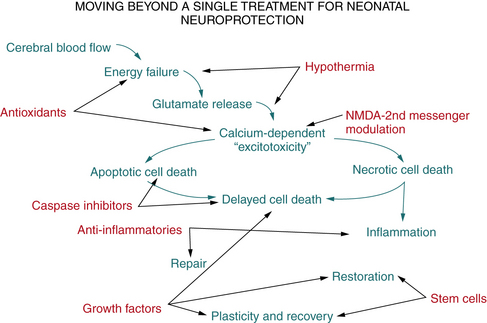
Over the past 20 years, considerable information has emerged about the cellular and molecular consequences of cerebral hypoxia-ischemia and the molecular events that lead to neuronal cell death. The underlying rationale for this scientific focus is the hope that a better understanding of the basic molecular mechanisms of neurodegeneration may provide ways to modulate these events pharmacologically to limit their adverse consequences and to protect the brain from irreversible damage. A complementary focus that has emerged – and will likely become particularly important in the setting of neonatal brain injury – is the delineation of the intrinsic neuronal mechanisms of adaptation and repair after hypoxic-ischemic brain injury. Despite the traditional view of greater resistance to CNS injury in the neonate because of lower metabolic demands and the greater plasticity of the developing CNS, at specific stages of brain maturation, susceptibility to hypoxia-ischemia may be amplified. Clinical and experimental data have demonstrated that specific brain structures and neural cells in the developing brain may be selectively vulnerable to hypoxic-ischemic injury [McQuillen and Ferriero, 2004]. One of the best-characterized examples is the increased vulnerability of immature oligodendroglia to hypoxic-ischemic injury, which is evident clinically in premature infants and which has been successfully demonstrated in fetal and neonatal animal models [Back et al., 2002; Segovia et al., 2008].
Risk of injury to the immature brain may be heightened by certain therapeutic interventions; this risk stems from the complex roles that pivotal molecular mediators of hypoxic-ischemic injury (e.g., glutamate, calcium) play in brain development. For example, some studies have provided compelling experimental evidence of maturational stage-dependent deleterious effects of several commonly used antiepileptic drugs [Ikonomidou and Turski, 2009].
This section reviews information about the pathophysiology of hypoxic-ischemic brain injury, integrates data obtained from experimental and clinical studies, and highlights mechanisms that are particularly relevant to understanding perinatal brain injury and repair. Several reviews can provide complementary perspectives [Gonzalez and Ferriero, 2008; Vexler and Yenari, 2009; Northington et al., 2005; Pediatric Neurology, 2009]. Please also see Part III of this book on emerging neuroscience concepts (Chapters 13, 14, and 15).
Cerebral Blood Flow and Energy Metabolism
Disruption of cerebrovascular autoregulation has been implicated as an important factor in the pathophysiology of neonatal hypoxic-ischemic brain injury. It is widely accepted that preterm infants have a “pressure-passive” cerebral circulation; however, term infants may remain at risk for impairment of cerebrovascular autoregulation [Boylan et al., 2000] and susceptibility to cerebral ischemia with fluctuations in systemic blood pressure. Several basic physiologic mechanisms may contribute to impaired autoregulation in the neonate. Increased expression of inducible and neuronal isoforms of nitric oxide synthase (iNOS and nNOS), as well as endothelial NOS, may narrow the autoregulatory window, and downregulation of prostaglandin receptors in response to high circulating prostaglandin levels may blunt the prostaglandin-mediated vasoconstrictive response to hypertension and thereby contribute to inappropriately increased cerebral blood flow [Chemtob et al., 1996]. After an ischemic insult, the neonate remains at high risk for further damage in the acute recovery phase because neonatal encephalopathy is often associated with blood pressure fluctuations.
An inadequate supply of glucose or alternate substrates plays a pivotal role in hypoxic-ischemic neuronal cell death. Although overall metabolic demands are lower in the neonatal than in the adult brain, during periods of rapid brain growth, particularly the perinatal period, metabolic needs rise. The pattern of injury after hypoxia-ischemia can be explained in part on the basis of this metabolic demand; brain regions most susceptible to hypoxic-ischemic injury in the term infant (e.g., subcortical gray matter structures such as the basal ganglia and thalamus) are the same regions that are most vulnerable to mitochondrial toxins. Brain development is associated with a transition from the ability to use glucose and ketones as energy substrates in the neonate to an absolute requirement for glucose in the adult. The immature brain can use lactate as an alternate fuel source to some degree, and the deleterious effects of lactate accumulation after hypoxia-ischemia therefore may be attenuated in the neonate compared with the adult. However, normal maturation is characterized by limitations in glucose transport capacity and increased use of these alternative fuels such as lactate. The inability to transport glucose across the blood–brain barrier threatens cerebral glucose utilization. These factors illustrate the importance of understanding the use of glucose, lactate, and ketones in the newborn brain under normal and pathologic conditions [Vannucci and Vannucci, 2000; Vannucci and Hagberg, 2004].
Excitotoxicity
Glutamate can activate a variety of excitatory amino acid receptors that are broadly classified based on their selective responses to specific agonists (e.g., NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid [AMPA], kainate) or their signaling mechanisms (i.e., ionotropic [ligand-gated ion channel] or metabotropic [G-protein-coupled]). Excitatory amino acid neurotransmission plays a pivotal role in brain development and in learning and memory. A substantial body of data has emerged over the past 30 years documenting the fact that overactivation of excitatory amino acid receptors (i.e., excitotoxicity) contributes to neurodegeneration in a broad range of acute and chronic neurologic disorders [Johnston, 2005].
Most information about mechanisms of excitotoxicity comes from experimental studies, but studies in the human neonate have provided some evidence to support the hypothesis that hypoxic-ischemic brain injury disrupts brain glutamate metabolism. Elevated levels of cerebrospinal fluid glutamate have been documented (by direct cerebrospinal fluid measurements and by proton MRS) in infants with severe hypoxic-ischemic injury [Pu et al., 2000]; cerebrospinal fluid levels of excitatory amino acids are directly proportional to the severity of clinical encephalopathy [Hagberg et al., 1997; Riikonen et al., 1992].
Two closely linked mechanisms contribute to ischemia-induced increases in synaptic glutamate: increased efflux from presynaptic nerve terminals and impaired reuptake by glia and neurons. The initial increase in efflux is mediated by a calcium-dependent process through activation of voltage- dependent calcium channels; later, calcium-independent efflux is thought to be mediated primarily by functional reversal of glutamate transporters. Removal of glutamate from the synaptic cleft depends primarily on energy-dependent glutamate transporters, which are predominantly glial. Any pathophysiologic process that depletes energy supply (e.g., hypoxia-ischemia, hypoglycemia, prolonged seizures) will disrupt these mechanisms and result in increased synaptic glutamate accumulation [Johnston, 2005].
The structure and function of the NMDA receptor channel complex are developmentally regulated. The receptor possesses multiple functional sites that recognize glutamate, co-agonists, modulatory molecules such as glycine, dissociative anesthetics, redox agents, steroids, zinc, magnesium (which blocks permeability to calcium), and a cation-selective ion channel that admits Na+, K+, and Ca2+. When the neurotransmitter recognition site is activated by glutamate, the ion channel allows influx of calcium and sodium. The resulting increase in intracellular calcium is the stimulus for a multitude of downstream events, including transcription factor modulation, cell cycle regulation, and DNA replication. The NMDA receptor is relatively overexpressed in the developing brain compared with the adult brain; in postnatal day 6–14 rats (which approximates to the term human neonate), the NMDA receptor is expressed at 150–200 percent of adult levels. In humans, receptor expression is significantly higher at term than in the adult [Johnston, 2005]. The particular combination of subunits determines the NMDA receptor’s functional state, and the predominating combination in the perinatal period seems to favor a more prolonged and pronounced calcium influx. In the setting of hypoxia-ischemia, NMDA receptor overactivation leads to massive Na+ and water influx, cell swelling, elevated intracellular calcium and its associated mitochondrial dysfunction, increased nitric oxide production, increased phospholipid turnover and accumulation of potentially toxic free fatty acids, and cell death by apoptotic or necrotic mechanisms. However, ischemia and energy failure also result in cation influx by non-NMDA-mediated mechanisms.
In experimental rodent models, there is compelling evidence that susceptibility to NMDA- and AMPA-mediated excitotoxicity peaks in the immature and that treatment with NMDA and AMPA receptor antagonists confers robust protection against neonatal hypoxic-ischemic brain injury [Johnston et al., 2001]. However, concerns have been raised that NMDA receptor antagonists might have specific risks in the immature brain; blockade of NMDA synaptic activity could disrupt critical neurodevelopmental processes. Whether AMPA antagonists, which can block neuronal and white matter hypoxic-ischemic damage in neonatal rodent models, have fewer potential risks in the immature brain than do NMDA antagonists is an important question for study [Silverstein and Jensen, 2007].
There has been considerable interest in evaluating the neuroprotective efficacy of magnesium sulfate in experimental and clinical models of neonatal brain injury. This interest stems from the intrinsic potentially neuroprotective properties of Mg2+ (including blockade of NMDA receptor activation), and the many years of clinical experience in safe use of magnesium sulfate in obstetric practice. Experimental studies provide evidence that pretreatment with magnesium sulfate can limit subsequent neonatal hypoxic-ischemic injury, but treatment after hypoxia-ischemia is of limited benefit [Greenwood et al., 2000; Turkyilmaz et al., 2002]. Magnesium sulfate has many effects, including blockade of NMDA receptors. It has been found give protection in some animal models of white matter damage [Turkyilmaz et al., 2002; Marret et al., 1995; Spandou et al., 2007], but did not affect cerebrospinal fluid levels of excitatory neurotransmitters in asphyxiated human neonates [Khashaba et al., 2006]. In addition, in a multicenter clinical trial of mothers treated with magnesium who were at risk for preterm delivery, no perinatal side effects were seen and there was some benefit in the neurodevelopment of survivors [Crowther et al., 2003]. A recent study, the BEAM trial, showed that exposure to magnesium sulfate before anticipated early preterm delivery did not reduce the combined risk of moderate or severe cerebral palsy or death, although the rate of cerebral palsy was reduced among survivors [Rouse et al., 2008].
Oxidative Stress
Oxidative stress describes the alterations in cellular milieu that result from an increase in free radical production as a result of oxidative metabolism under pathologic conditions. In the brain injured by hypoxia-ischemia, excitotoxicity and oxidative stress are inextricably linked. In cells with normally functioning mitochondria, more than 80 percent of available oxygen is reduced to energy equivalents (ATP) by cytochrome oxidase. The rest is converted to superoxide anions, which, under physiologic conditions, are reduced to water by enzymatic and nonenzymatic antioxidant mechanisms. An inevitable consequence of mitochondrial dysfunction is an accumulation of superoxide, and any process that depletes antioxidant defenses will result in the default conversion of superoxide to even more reactive species, such as the hydroxyl radical [Ferriero, 2001]. The concept of ischemia-reperfusion injury (i.e., progression of tissue injury with reoxygenation after ischemia) [Inder and Volpe, 2000] is fundamental to the understanding of oxidative stress. This mechanism is not limited to the brain, but excitotoxic mechanisms in the brain can amplify these processes. Excitotoxicity causes energy depletion, mitochondrial dysfunction, and cytosolic calcium accumulation, leading to the generation of free radicals, such as superoxide, nitric oxide derivatives, and the highly reactive hydroxyl radical.
With reoxygenation, mitochondrial oxidative phosphorylation is overwhelmed and reactive oxygen species accumulate. Intrinsic antioxidant defenses are depleted, and free radicals directly damage multiple cellular constituents (lipids, DNA, protein) and can activate pro-apoptotic pathways. The brain is particularly susceptible to free radical attack and lipid peroxidation, and vulnerability is magnified in the immature brain. Contributing factors include a high polyunsaturated fatty acid content, high level of lipid peroxidation (particularly in response to hypoxic stress), immaturity of antioxidant defense enzymes, and high free iron concentrations, compared with the adult brain [Ferriero, 2001]. The level of free iron, which catalyzes the production of various reactive oxygen species, is increased in the plasma and cerebrospinal fluid of asphyxiated newborns [Ogihara et al., 2003]. The deleterious effects of abundant iron and immaturity of the enzymatic oxidant defenses of the immature brain are tightly interrelated. Free radical scavengers (e.g., alpha-phenyl-n-tert-butyl-nitrone [PBN], a spin-trap agent that converts free radicals to stable adducts) and metal chelators (e.g., deferoxamine) can protect neurons from injury mediated by hydrogen peroxide in vitro and in vivo. These agents also protect neurons from NMDA-induced toxicity, providing complementary evidence of the pathophysiologic link between excitotoxicity and oxidative stress [Peeters-Scholte et al., 2003].
Nitric oxide metabolism provides another critical link between excitotoxicity and oxidative injury in the hypoxic-ischemic injured brain. Nitric oxide is produced constitutively in endothelium, astrocytes, and neurons in response to an increase in intracellular calcium. Hypoxic-ischemic increases in nitric oxide production have multiple potential beneficial and detrimental effects. Nitric oxide regulates vascular tone, influences inflammatory responses to injury, and directly modulates NMDA receptor function [Sorrentino et al., 2004]. In neonatal rodent brain, striatal neurons that express nNOS are selectively resistant to hypoxia-ischemia injury, providing a source for NMDA receptor-mediated regulation of nitric oxide production. Disruption of the nNOS gene and pharmacologic inhibition of nNOS (reviewed in Gonzalez and Ferriero) ameliorate neonatal hypoxia-ischemia injury [Gonzalez and Ferriero, 2008]. Other nitric oxide synthase isoforms contribute to injury via inflammation (iNOS) and may potentially provide another crosstalk for protection therapies. Early endothelial NO is protective by maintaining blood flow, but early neuronal NO and late inducible NO are neurotoxic by promoting cell death [Iadecola et al., 1997]. Brain iNOS is induced in multiple cell types during upregulation of the pro-inflammatory pathway after brain injury [Higuchi et al., 1998], enhancing excitotoxicity by modifying binding to NMDA receptors [Ishida et al., 2001].
Neuroprotection by selective inhibition of nNOS or iNOS has been demonstrated [van den Tweel et al., 2005]. Regions expressing nNOS correspond to those that express NMDA receptors and correlate with regions of neurotoxicity both in vivo and in vitro [Black et al., 1995; Ferriero et al., 1996; Dawson et al., 1993]. Destruction of neurons containing nNOS with local injections of quisqualate prior to hypoxia-ischemia results in lower injury scores and less infarction [Ferriero et al., 1995]. Likewise, neonatal mice with targeted disruption of the nNOS gene, when subjected to a similar hypoxic-ischemic insult, are markedly protected from injury [Ferriero et al., 1996], but nonspecific blockade of both nNOS and eNOS (endothelial NOS) in sheep with NG-nitro-L-arginine (L-NNA) was not protective [Marks et al., 1996]. Few studies have been performed in asphyxiated human newborns in relation to cerebral NO production. Cerebrospinal fluid NO levels increase with severity of HIE at 24–72 hours after asphyxia [Ergenekon et al., 1999], with increased NO and nitrotyrosine levels in spinal cord as well [Groenendaal et al., 2008]. Initial results in premature infants treated with inhaled NO for prevention of bronchopulmonary dysplasia showed reductions in ultrasound-diagnosed brain injury and improvements in neurodevelopmental outcomes at 2 years of age, but long-term results are pending [Schreiber et al., 2003; Ballard et al., 2006].
Several other antioxidant strategies that either block free radical production or increase antioxidant defenses have been studied. Melatonin is an indoleamine that is formed in higher quantities in adults and is a direct scavenger of reactive oxygen species and NO. It has been found to provide long-lasting neuroprotection in experimental hypoxia-ischemia and focal cerebral ischemic injury [Carloni et al., 2008; Koh, 2008]. Human neonates treated with melatonin were also found to have decreased proinflammatory cytokines [Gitto et al., 2004, 2005]. Allopurinol has mixed effects which have shown promise in animal and human studies. Xanthine oxidase-derived superoxide and H2O2 react with NO to form damaging RNS (reactive nitrogen species). Allopurinol functions by reducing free radical production via xanthine oxidase inhibition, while also scavenging hydroxyl radicals. Short-term benefits have also been seen in neonates undergoing cardiac surgery for hypoplastic left heart syndrome [Clancy et al., 2001]. Early allopurinol in asphyxiated infants improved short-term neurodevelopmental outcomes and decreased serum NO levels after administration; however, no improvement in long-term outcomes was seen for later treatment after birth asphyxia, perhaps because only a very brief window for benefit exists [Benders et al., 2006].
Inflammation
Nelson and colleagues first reported a link between elevated neonatal blood levels of a broad range of serum proinflammatory cytokines and subsequent diagnosis of spastic diplegia [Nelson et al., 1998]. Cytokines are polypeptides that act systemically and locally to mediate and regulate multiple components of inflammation. Cytokines that have been strongly implicated as mediators of brain inflammation in neonates include interleukin (IL)-1β, tumor necrosis factor (TNF)α, IL-6, and membrane co-factor protein-1 [Foster-Barber and Ferriero, 2002]. After an asphyxial episode, there are many potential sources of plasma cytokines, including injured endothelium and acutely injured organs, such as the brain by means of a disrupted blood–brain barrier. Measurements of cerebrospinal fluid and plasma levels of several cytokines in asphyxiated term infants suggest that the injured brain can be the source of acutely elevated cytokine levels [Silveira and Procianoy, 2003]. A more recent study links IL-6 with cerebral palsy, giving credence to the fact that there is an underlying genetic susceptibility in at-risk newborns [Wu et al., 2009].
The observed relation between maternal infection and neonatal brain injury could be explained by a number of mechanisms. Experimental studies provide support for the hypothesis that a proinflammatory milieu increases the susceptibility of the neonate to hypoxic-ischemic brain injury. In fetal or neonatal models, maternal infection is commonly simulated by injecting the potent proinflammatory agent, lipopolysaccharide (i.e., endotoxin derived from Escherichia coli). Systemically administered endotoxin amplifies neonatal hypoxic-ischemic brain injury in rodent and sheep models; some of these deleterious effects are mediated, at least in part, by systemic mechanisms (e.g., hypotension, hypoglycemia) [Lehnardt et al., 2003].
Drugs that block microglial activation and cytokine release protect the brain from excitotoxic damage [Dommergues et al., 2003]. Minocycline is a tetracycline derivative that crosses the blood–brain barrier and has been postulated to confer anti-inflammatory effects on microglia, as well as modulate immune cell activation, and cytokine and NO release, while also decreasing apoptosis. In the neonatal brain, minocycline appears to block hypoxia-ischemia-induced tissue damage and caspase-3 activation in rodents when given immediately before or after injury, but results are inconsistent [Arvin et al., 2002; Fox et al., 2005; Cai et al., 2006]. Minocycline attenuates lipopolysaccharide-induced brain injury and improves neurobehavioral outcomes in P5 rats via inhibition of microglial activation with resultant decreases in IL-1β and TNFα [Fan et al., 2005]. Low- and high-dose regimens were effective in reducing short-term hypoxia-ischemia-induced inflammation, protecting developing oligodendrocytes [Cai et al., 2006] and myelin content in neonatal rats [Carty et al., 2008], but this effect was only transient in another study of neonatal rodent stroke [Fox et al., 2005]. Following stroke, it decreases postischemic brain inflammation via inhibition of 5-LOX and enzymatic activation. Delayed therapy was found to decrease TNFα and matrix metalloproteinase (MMP)-12, but efficacy was lost when treatment was extended for a week after stroke [Wasserman et al., 2007]. These effects also appear to be species-dependent, with an increase in injury in developing C57B1/6 mice [Tsuji et al., 2004].
Apoptosis
Apoptosis is critical for normal brain development, but it is also an important component of injury following neonatal hypoxia-ischemia and stroke [Northington et al., 2005]. Activation of intrinsic or extrinsic apoptotic pathways leads to cleavage and activation of caspase 3, which is maximally produced in the neonatal period [Hu et al., 2000]. Pro-apoptotic Bax is present in high concentrations during the first 2 postnatal weeks [Lok and Martin, 2002]. While necrosis plays a major role in early neuronal death in both the immature and mature brain [Northington et al., 2001], there appears to be some aspect of apoptosis within the first 24 hours following neonatal hypoxia-ischemia [Portera-Cailliau et al., 1997], with a spectrum of cell death present that may result in heterogenous responses to anti-apoptotic therapies [Northington et al., 2005]. It is likely that apoptosis is not the cause of most acute cell death, but rather of delayed phases of injury and neurodegeneration. This delayed apoptotic cell death likely relates to target deprivation and loss of neurotrophic support [Northington et al., 2005]. Specific and nonspecific inhibition of caspases or cysteine proteases, which are highly activated after hypoxia-ischemia, has also been attempted with some success [Feng et al., 2003; Han et al., 2002; Blomgren et al., 2001; Ostwald et al., 1993]. See Chapter 14 for more details.
Brain Protection
Neuroprotection
A recent therapy that is becoming standard of care for selected newborns is the use of therapeutic hypothermia for brain injury. It is postulated to work by modifying apoptosis and interrupting early necrosis [Edwards et al., 1995], reducing cerebral metabolic rate and the release of excitotoxins, NO, and free radicals [Globus et al., 1995]. Multiple animal models of perinatal brain injury demonstrate histological and functional benefit of early initiation of hypothermia [Laptook et al., 1994, 1997; Thoresen et al., 1995; Gunn et al., 1997; Gunn et al., 1998b]. Brief hypothermia initiated early after injury provided partial neuroprotection [Laptook and Corbett, 2002; Towfighi et al., 1994], but prolonged moderate hypothermia to 32–34°C for 24–72 hours results in sustained improvement in behavioral performance in newborn and adult animals [Gunn et al., 1997, 1998b]. The only complications were transient reductions of heart rate and blood pressure [Thoresen and Whitelaw, 2000].
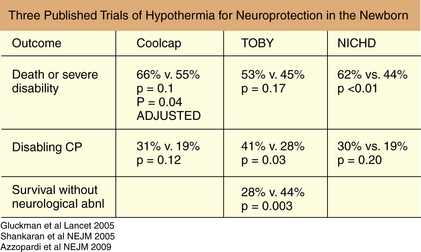
Review of human neonatal studies shows reduction in mortality and long-term neurodevelopmental disability at 12–24 months of age, with more pronounced effects in moderately encephalopathic infants [Gunn et al., 1998a; Eicher et al., 2005; Gluckman et al., 2005; Shankaran et al., 2005; Azzopardi et al., 2009]. Sustained protection does depend on the degree of hypothermia, with maximum benefit obtained with cooling to 33–34°C, as well as in minimizing delay time to treatment [Bona et al., 1998; Gunn et al., 1997]. Mild hypothermia to this level appears to be well tolerated without serious adverse effects if initiated within the first 6 hours of life [Gunn et al., 1998a; Azzopardi et al., 2000; Thoresen, 2000; Shankaran et al., 2002]. In selective head cooling, treatment benefited infants with moderate but not severe amplitude integrated EEG changes, improving survival without severe neurodevelopmental deficits or an increase in complications [Gluckman et al., 2005]. In addition to severity of encephalopathy, larger infants were more responsive to hypothermia and at higher risk for injury if hyperthermic at any point [Wyatt et al., 2007]. In a second multicenter trial, whole-body cooling to 33.5°C, initiated within 6 hours and continued for 72 hours, resulted in less death and severe disability at 18 months. In a piglet model, the degree of hypothermia altered the amount of protection in the cortical gray matter relative to the deep gray matter [Iwata et al., 2005]. Whole-body cooling may be more effective in reducing temperature in the deep brain structures [Van Leeuwen et al., 2000]. Most recently, another whole-body cooling trial was completed (TOBY) and the results were consistent with the other two major trials, with a significant number of treated babies showing survival without neurodevelopmental sequelae (Figure 17-3) [Azzopardi et al., 2009].
Xenon is approved for use as a general anesthetic in Europe, and has shown promise as a protective agent. It is an NMDA antagonist, preventing progression of excitotoxic damage. It appears to be superior to other NMDA antagonists, possibly through inhibition of AMPA and kainite receptors, reduction of neurotransmitter release, or effects on other ion channels [Ma and Zhang, 2003; Dinse et al., 2005; Gruss et al., 2004]. The combination of xenon and hypothermia initiated 4 hours after neonatal hypoxia-ischemia provided synergistic histological and functional protection when evaluated at 30 days after injury in rodents [Ma et al., 2005]. More recently, an additive effect was shown after hypoxia-ischemia in P7 rats that were cooled to 32°C and received 50 percent xenon, with improvement in long-term histology and functional performance that exceeded the individual benefit of either [Hobbs et al., 2008]. Studies on xenon use in human neonates are under way.
N-acetylcysteine (NAC) is a medication, approved for neonates, that is a scavenger of oxygen radicals and restores intracellular glutathione levels, attenuating reperfusion injury and decreasing inflammation and NO production. Adding NAC therapy to systemic hypothermia reduced brain volume loss at both 2 and 4 weeks after neonatal rodent hypoxia-ischemia, with increased myelin expression and improved reflexes [Jatana et al., 2006]. Inhibition of inflammation with MK-801 has also been effective when combined with hypothermia in neonatal rats post hypoxic-ischemic injury [Alkan et al., 2001]. In P7 rats who underwent hypoxia-ischemia, followed by early topiramate and delayed hypothermia, improved short-term histology and function was seen [Liu et al., 2004]. This may provide a window for protection if hypothermia is delayed, which is possible, given the difficulty in initiation of cooling if infants are born outside hospital or transport is delayed.
Neurotrophic Factors
EPO is a 34-kDa glycoprotein that was originally identified for its role in erythropoiesis, but has since been found to have a variety of other roles. The pleiotropic functions of this cytokine include modulation of the inflammatory and immune responses [Villa et al., 2003], and vasogenic and pro-angiogenic effects through its interaction with vascular endothelial growth factor (VEGF) [Wang et al., 2004; Chong et al., 2002], as well as effects on CNS development and repair. EPO and EPO receptors are expressed by a variety of different cell types in the CNS, with changing patterns during development [Juul et al., 1999]. EPO plays a vital role in neural differentiation and neurogenesis early in development, promoting neurogenesis in vitro and in vivo [Shingo et al., 2001].
Recent evidence suggests that exogenously administered EPO has a protective effect in a variety of different models of immature brain injury [Sola et al., 2005b]. In newborn rodents, pretreatment with EPO before injury has a protective effect [Sola et al., 2005b]. In addition, postinjury treatment protocols have demonstrated both short- and long-term histological and behavioral improvement. A single dose of exogenous EPO administered immediately after neonatal hypoxic-ischemic injury in rats significantly reduced infarct volume and improved long-term spatial memory after injury [Kumral et al., 2004]. Single- and multiple-dose treatment regimens of EPO following neonatal focal ischemic stroke in rats also reduced infarct volume [Sola et al., 2005a] and improved short-term sensorimotor outcomes [Chang et al., 2005], but there may be more long-term behavioral benefit in female rats [Wen et al., 2006]. EPO treatment that was delayed 24 hours after neonatal hypoxia-ischemia also attenuated brain injury [Sun et al., 2005]. In addition, EPO enhances neurogenesis and directs multipotential neural stem cells toward a neuronal cell fate [Shingo et al., 2001; Wang et al., 2004; Gonzalez et al., 2007]. Following transient ischemic stroke, there is temporary precursor cell proliferation in the rodent subventricular zone (SVZ), with this precursor cell proliferation and differentiation favoring gliogenesis [Plane et al., 2004]. EPO has been shown to enhance neurogenesis in vivo in the SVZ following stroke in the adult rat [Wang et al., 2004]. Neurogenesis has also been demonstrated following EPO treatment with an increase in newly generated cells from precursors [Wang et al., 2004; Lu et al., 2005; Shingo et al., 2001], and possibly also an effect on cell fate commitment [Wang et al., 2004; Shingo et al., 2001] in vitro.
In humans, EPO is safely used for treatment of anemia in premature infants [Aher and Ohlsson, 2006]. EPO for neuroprotection is given in much higher doses (1000–5000 U/kg/dose) than that given for anemia [Chang et al., 2005; Demers et al., 2005; McPherson and Juul, 2007] to enable crossing of the blood–brain barrier, with unknown pharmacokinetics in humans. Recently, extremely low birth weight infants tolerated doses between 500 and 2500 U/kg/dose [Juul et al., 2008]. A recent trial of EPO for neonatal asphyxia showed that repeated low-dose therapy (300–500 U/kg every other day for 2 weeks) reduced the risk of disability for infants with moderate HIE and no negative hematopoietic side effects were observed [Zhu et al., 2009].
VEGF is a regulator of angiogenesis that also promotes neuronal cell proliferation and migration [Zachary, 2005]. The endothelial microenvironment establishes a vascular niche to promote survival and proliferation of progenitor cells, which is tightly coordinated with angiogenesis [Palmer et al., 2000]. VEGF-A is the most important member of a family of growth factors that also includes placental growth factor (PLGF) and VEGFs B, C, and D. VEGF-A expression occurs in cortical neurons during early development, switching to mature glial cells near vessels during later development. Following exposure to hypoxia, there is increased neuronal and glial expression [Krum and Rosenstein, 1998], directing vascularization and stimulating proliferation of astrocytes, microglia, and neuronal cell types [Forstreuter et al., 2002; Mu et al., 2003; Jin et al., 2002]. VEGF also has chemotactic effects on neurogenic zones in the brain [Yang and Cepko, 1996], increasing migration of stem cells during anoxia [Bagnard et al., 2001; Maurer et al., 2003]. VEGF knockout mice have severe impairments in vascularization, neuronal migration, and survival [Raab et al., 2004].
VEGF appears to play an essential role in the beneficial and protective effects of hypoxic preconditioning in neonatal mice [Laudenbach et al., 2007]. VEGF-R2 inhibition increases tissue injury and cell death in a neonatal stroke model, as well as reducing endothelial cell proliferation in the injured core [Shimotake, 2010].
Other trophic factors have also shown promise in reducing brain injury but, given their role in normal neurodevelopment, the effects of treatment are not known. Insulin-like growth factor-1 (IGF-1) has prosurvival properties that can prevent perinatal hypoxic or excitotoxic injury [Johnston et al., 1996; Pang et al., 2007]. Brain-derived neurotrophic factor (BDNF) is a neurotrophin that also provides neuroprotection in neonatal hypoxia-ischemia [Cheng et al., 1997, 1998; Holtzman et al., 1996; Husson et al., 2005]. It prevents spatial memory learning impairments after insult, but its effectiveness is limited by the stage of development [Husson et al., 2005; Cheng et al., 1998]. While protective in mice at P5, it exacerbates excitotoxicity at P0 and has no effect at later time points [Husson et al., 2005].
Stem Cells
Neural stem cells (NSCs) are multipotent precursors that self-renew and retain the ability to differentiate into a variety of neuronal and non-neuronal cell types in the CNS. They reside in neurogenic zones throughout life, such as the SVZ and subgranular zone of the dentate gyrus in rodent models, and help maintain cell turnover at baseline and replace injured cells by migrating to penumbral tissue after injury. NSC transplantation has shown potential as a therapeutic strategy in adult animal models of stroke and hypoxia-ischemia. Implanted cells integrate into injured tissue [Park et al., 2002], decreasing volume loss [Hoehn et al., 2002; Park et al., 2006a, b] and improving behavioral outcomes [Capone et al., 2007; Hicks et al., 2007]. In neonatal models, intraventricular implantation of NSCs after hypoxia-ischemia results in their migration to areas of injury [Park et al., 2006a, b]. These stem cells differentiate into neurons, astrocytes, and oligodendrocytes, as well as undifferentiated progenitors. These cells not only promote regeneration, but non-neuronal phenotypes inhibit inflammation and scar formation, while promoting angiogenesis and neuronal cell survival in both rodent and primate models [Imitola et al., 2004; Mueller et al., 2006]. While no adverse effects have been noted, efficacy is dependent on time of implantation, and the therapeutic window is not known. More recent technology enables labeling of stem cells, which can then be tracked from their site of implantation through their migratory path into the ischemic tissue [Modo et al., 2004; Guzman et al., 2007; Rice et al., 2007; Obenaus et al., 2007], which will make tracking of these cells in humans possible [Ashwal et al., 2009].
Future Directions
A major challenge for future studies will be to establish meaningful links between experimental and clinical data, and to identify clinically important developmental stage-specific risks and benefits of specific treatment modalities that emerge. The use of advanced brain imaging may improve the application of newborn protective strategies by the early identification and stratification of injury severity, as well as providing a noninvasive method to monitor early responses to new treatments. Brain imaging may also provide important insight into how therapeutic hypothermia is acting so that its use can be optimized and synergistic therapies added. For example, in two recent studies of cooled newborns, one showed that systemic cooling reduced cortical injury [Inder et al., 2004], while a more recent one demonstrated a decrease of injury in the basal nuclei and white mater. Importantly, therapeutic hypothermia did not limit the predictive value of MRI for subsequent neurological impairment [Rutherford et al., 2010.] Studies are needed to determine whether applying a specific method of cooling to a defined pattern of injury will enable individualized treatment. Complementary investigations in neonatal animal models and in the neonatal intensive care unit should continue to provide insights about basic mechanisms of neurodegeneration and repair that are particularly important in the developing nervous system.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Pediatric Neurology March 2009, whole issue. Pediatr Neurol. 2009;40(3):143-244.
(ACOG) ACoOaG. Neonatal encephalopathy and cerebral palsy: executive summary. Obstet Gynecol. 2004;103(4):780-781.
Aher S., Ohlsson A. Late erythropoietin for preventing red blood cell transfusion in preterm and/or low birth weight infants. Cochrane Database Syst Rev. 3, 2006. CD004868
Alkan T., Kahveci N., Buyukuysal L., et al. Neuroprotective effects of MK 801 and hypothermia used alone and in combination in hypoxic-ischemic brain injury in neonatal rats. Arch Physiol Biochem. 2001;109(2):135-144.
Arvin K.L., Han B.H., Du Y., et al. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52(1):54-61.
Ashwal S., Obenaus A., Snyder S. Neuroimaging as a basis for stem cell therapy for neonatal hypoxic-ischemic brain injury. Pediatr Neurol. 2009;40(3):227-236.
Azzopardi D., Robertson N.J., Cowan F.M., et al. Pilot study of treatment with whole body hypothermia for neonatal encephalopathy. Pediatrics. 2000;106(4):684-694.
Azzopardi D.V., Strohm B., Edwards A.D., et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361(14):1349-1358.
Back S.A., Han B.H., Luo N.L., et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22(2):455-463.
Badawi N., Kurinczuk J.J., Keogh J.M., et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. Br Med J. 1998;317(7172):1554-1558.
Bagnard D., Vaillant C., Khuth S.T., et al. Semaphorin 3A-vascular endothelial growth factor-165 balance mediates migration and apoptosis of neural progenitor cells by the recruitment of shared receptor. J Neurosci. 2001;21(10):3332-3341.
Ballard R.A., Truog W.E., Cnaan A., et al. Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med. 2006;355(4):343-353.
Barkovich A.J. The encephalopathic neonate: choosing the proper imaging technique. Am J Neuroradiol. 1997;18(10):1816-1820.
Barkovich A.J. Pediatric Neuroimaging, ed 3. Philadelphia: Lippincott Williams & Wilkins; 2000.
Barkovich A.J., Westmark K.D., Bedi H.S., et al. Proton spectroscopy and diffusion imaging on the first day of life after perinatal asphyxia: preliminary report. Am J Neuroradiol. 2001;22(9):1786-1794.
Barkovich A.J., Miller S.P., Bartha A., et al. MR imaging, MR spectroscopy, and diffusion tensor imaging of sequential studies in neonates with encephalopathy. Am J Neuroradiol. 2006;27(3):533-547.
Barnett A., Mercuri E., Rutherford M., et al. Neurological and perceptual-motor outcome at 5–6 years of age in children with neonatal encephalopathy: relationship with neonatal brain MRI. Neuropediatrics. 2002;33(5):242-248.
Bax M., Tydeman C., Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296(13):1602-1608.
Beaulieu C. The basis of anisotropic water diffusion in the nervous system – a technical review. NMR Biomed. 2002;15(7–8):435-455.
Benders M.J., Bos A.F., Rademaker C.M., et al. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch Dis Child Fetal Neonatal Ed. 2006;91(3):F163-F165.
Berrington de Gonzalez A., Darby S. Risk of cancer from diagnostic X-rays: estimates for the UK and 14 other countries. Lancet. 2004;363(9406):345-351.
Black S.M., Bedolli M.A., Martinez S., et al. Expression of neuronal nitric oxide synthase corresponds to regions of selective vulnerability to hypoxia-ischaemia in the developing rat brain. Neurobiol Dis. 1995;2(3):145-155.
Blomgren K., Zhu C., Wang X., et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276(13):10191-10198.
Bluml S., Friedlich P., Erberich S., et al. MR imaging of newborns by using an MR-compatible incubator with integrated radiofrequency coils: initial experience. Radiology. 2004;231(2):594-601.
Bona E., Hagberg H., Loberg E.M., et al. Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pediatr Res. 1998;43(6):738-745.
Boylan G.B., Young K., Panerai R.B., et al. Dynamic cerebral autoregulation in sick newborn infants. Pediatr Res. 2000;48(1):12-17.
Brenner D.J., Doll R., Goodhead D.T., et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci USA. 2003;100(24):13761-13766.
Brunquell P.J., Glennon C.M., DiMario F.J.Jr, et al. Prediction of outcome based on clinical seizure type in newborn infants. J Pediatr. 2002;140(6):707-712.
Cai Z., Lin S., Fan L.W., et al. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137(2):425-435.
Capone C., Frigerio S., Fumagalli S., et al. Neurosphere-derived cells exert a neuroprotective action by changing the ischemic microenvironment. PLoS ONE. 2007;2(4):e373.
Carloni S., Perrone S., Buonocore G., et al. Melatonin protects from the long-term consequences of a neonatal hypoxic-ischemic brain injury in rats. J Pineal Res. 2008;44(2):157-164.
Carty M.L., Wixey J.A., Colditz P.B., et al. Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: a comparison of two different dose regimens. Int J Dev Neurosci. 2008;26(5):477-485.
Chang Y.S., Mu D., Wendland M., et al. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatr Res. 2005;58(1):106-111.
Chau V., Poskitt K.J., Sargent M.A., et al. Comparison of computer tomography and magnetic resonance imaging scans on the third day of life in term newborns with neonatal encephalopathy. Pediatrics. 2009;123(1):319-326.
Chemtob S., Li D.Y., Abran D., et al. The role of prostaglandin receptors in regulating cerebral blood flow in the perinatal period. Acta Paediatr. 1996;85(5):517-524.
Cheng Y., Gidday J.M., Yan Q., et al. Marked age-dependent neuroprotection by brain-derived neurotrophic factor against neonatal hypoxic-ischemic brain injury. Ann Neurol. 1997;41(4):521-529.
Cheng Y., Deshmukh M., D’Costa A., et al. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101(9):1992-1999.
Chong Z.Z., Kang J.Q., Maiese K. Angiogenesis and plasticity: role of erythropoietin in vascular systems. J Hematother Stem Cell Res. 2002;11(6):863-871.
Clancy R.R., Legido A. Postnatal epilepsy after EEG-confirmed neonatal seizures. Epilepsia. 1991;32(1):69-76.
Clancy R.R., McGaurn S.A., Goin J.E., et al. Allopurinol neurocardiac protection trial in infants undergoing heart surgery using deep hypothermic circulatory arrest. Pediatrics. 2001;108(1):61-70.
Coats J.S., Freeberg A., Pajela E.G., et al. Meta-analysis of apparent diffusion coefficients in the newborn brain. Pediatr Neurol. 2009;41(4):263-274.
Cowan F., Rutherford M., Groenendaal F., et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361(9359):736-742.
Crowther C.A., Hiller J.E., Doyle L.W., et al(ACTOMgSO4) ftACToMS. Effect of magnesium sulfate given for neuroprotection before preterm birth: a randomized controlled trial. JAMA. 2003;290(20):2669-2676.
Dawson V.L., Dawson T.M., Bartley D.A., et al. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13(6):2651-2661.
De Vries L.S., Van der Grond J., Van Haastert I.C., et al. Prediction of outcome in new-born infants with arterial ischaemic stroke using diffusion-weighted magnetic resonance imaging. Neuropediatrics. 2005;36(1):12-20.
Demers E.J., McPherson R.J., Juul S.E. Erythropoietin protects dopaminergic neurons and improves neurobehavioral outcomes in juvenile rats after neonatal hypoxia-ischemia. Pediatr Res. 2005;58(2):297-301.
Dilenge M.E., Majnemer A., Shevell M.I. Long-term developmental outcome of asphyxiated term neonates. J Child Neurol. 2001;16(11):781-792.
Dinse A., Fohr K.J., Georgieff M., et al. Xenon reduces glutamate-, AMPA-, and kainate-induced membrane currents in cortical neurones. Br J Anaesth. 2005;94(4):479-485.
Dixon G., Badawi N., Kurinczuk J.J., et al. Early developmental outcomes after newborn encephalopathy. Pediatrics. 2002;109(1):26-33.
Dommergues M.A., Plaisant F., Verney C., et al. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121(3):619-628.
Drobyshevsky A., Song S.K., Gamkrelidze G., et al. Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J Neurosci. 2005;25(25):5988-5997.
Dumoulin C.L., Rohling K.W., Piel J.E., et al. An MRI compatible neonate incubator. Concepts in Magnetic Resonance (Magnetic Resonance Engineering). 2002;15(2):117-128.
Edwards A.D., Yue X., Squier M.V., et al. Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post-insult hypothermia. Biochem Biophys Res Commun. 1995;217(3):1193-1199.
Eicher D.J., Wagner C.L., Katikaneni L.P., et al. Moderate hypothermia in neonatal encephalopathy: efficacy outcomes. Pediatr Neurol. 2005;32(1):11-17.
Ergenekon E., Gucuyener K., Erbas D., et al. Cerebrospinal fluid and serum nitric oxide levels in asphyxiated newborns. Biol Neonate. 1999;76(4):200-206.
Fan L.W., Pang Y., Lin S., et al. Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J Neurosci Res. 2005;82(1):71-82.
Feng Y., Fratkin J.D., LeBlanc M.H. Inhibiting caspase-8 after injury reduces hypoxic-ischemic brain injury in the newborn rat. Eur J Pharmacol. 2003;481(2–3):169-173.
Ferriero D.M., Sheldon R.A., Black S.M., et al. Selective destruction of nitric oxide synthase neurons with quisqualate reduces damage after hypoxia-ischemia in the neonatal rat. Pediatr Res. 1995;38(6):912-918.
Ferriero D.M., Holtzman D.M., Black S.M., et al. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis. 1996;3(1):64-71.
Ferriero D.M. Oxidant mechanisms in neonatal hypoxia-ischemia. Dev Neurosci. 2001;23(3):198-202.
Ferriero D.M. Neonatal brain injury. N Engl J Med. 2004;351(19):1985-1995.
Forstreuter F., Lucius R., Mentlein R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J Neuroimmunol. 2002;132(1–2):93-98.
Foster-Barber A., Ferriero D.M. Neonatal encephalopathy in the term infant: neuroimaging and inflammatory cytokines. Ment Retard Dev Disabil Res Rev. 2002;8(1):20-24.
Fox C., Dingman A., Derugin N., et al. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2005;25(9):1138-1149.
Gadian D.G., Aicardi J., Watkins K.E., et al. Developmental amnesia associated with early hypoxic-ischaemic injury. Brain. 2000;123(Pt 3):499-507.
Galli K.K., Zimmerman R.A., Jarvik G.P., et al. Periventricular leukomalacia is common after neonatal cardiac surgery. J Thorac Cardiovasc Surg. 2004;127(3):692-704.
Gilles F.H., Leviton A., Jammes J. Age-dependent changes in white matter in congenital heart disease. J Neuropathol Exp Neurol. 1973;32:179.
Gitto E., Reiter R.J., Cordaro S.P., et al. Oxidative and inflammatory parameters in respiratory distress syndrome of preterm newborns: beneficial effects of melatonin. Am J Perinatol. 2004;21(4):209-216.
Gitto E., Reiter R.J., Sabatino G., et al. Correlation among cytokines, bronchopulmonary dysplasia and modality of ventilation in preterm newborns: improvement with melatonin treatment. J Pineal Res. 2005;39(3):287-293.
Glenn O.A., Ludeman N.A., Berman J.I., et al. Diffusion tensor MR imaging tractography of the pyramidal tracts correlates with clinical motor function in children with congenital hemiparesis. Am J Neuroradiol. 2007;28(9):1796-1802.
Globus M.Y., Alonso O., Dietrich W.D., et al. Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem. 1995;65(4):1704-1711.
Gluckman P.D., Wyatt J.S., Azzopardi D., et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663-670.
Gonzalez F.F., Miller S.P. Does perinatal asphyxia impair cognitive function without cerebral palsy? Arch Dis Child Fetal Neonatal Ed. 2006;91(6):F454-F459.
Gonzalez F.F., McQuillen P., Mu D., et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29(4–5):321-330.
Gonzalez F.F., Ferriero D.M. Therapeutics for neonatal brain injury. Pharmacol Ther. 2008;120(1):43-53.
Greenwood K., Cox P., Mehmet H., et al. Magnesium sulfate treatment after transient hypoxia-ischemia in the newborn piglet does not protect against cerebral damage. Pediatr Res. 2000;48(3):346-350.
Groenendaal F., Vles J., Lammers H., et al. Nitrotyrosine in human neonatal spinal cord after perinatal asphyxia. Neonatology. 2008;93(1):1-6.
Gruss M., Bushell T.J., Bright D.P., et al. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol Pharmacol. 2004;65(2):443-452.
Gunn A.J., Gunn T.R., de Haan H.H., et al. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest. 1997;99(2):248-256.
Gunn A.J., Gluckman P.D., Gunn T.R. Selective head cooling in newborn infants after perinatal asphyxia: a safety study. Pediatrics. 1998;102(4 Pt 1):885-892.
Gunn A.J., Gunn T.R., Gunning M.I., et al. Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics. 1998;102(5):1098-1106.
Guzman R., Uchida N., Bliss T.M., et al. Long-term monitoring of transplanted human neural stem cells in developmental and pathological contexts with MRI. Proc Natl Acad Sci USA. 2007;104(24):10211-10216.
Hagberg H., Bona E., Gilland E., et al. Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatr Suppl. 1997;422(9):85-88.
Hall P., Adami H.O., Trichopoulos D., et al. Effect of low doses of ionising radiation in infancy on cognitive function in adulthood: Swedish population based cohort study. Br Med J. 2004;328(7430):19.
Han B.H., Xu D., Choi J., et al. Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J Biol Chem. 2002;277(33):30128-30136.
Hicks A.U., Hewlett K., Windle V., et al. Enriched environment enhances transplanted subventricular zone stem cell migration and functional recovery after stroke. Neuroscience. 2007;146(1):31-40.
Higuchi Y., Hattori H., Kume T., et al. Increase in nitric oxide in the hypoxic-ischemic neonatal rat brain and suppression by 7-nitroindazole and aminoguanidine. Eur J Pharmacol. 1998;342(1):47-49.
Hobbs C., Thoresen M., Tucker A., et al. Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke. 2008;39(4):1307-1313.
Hoehn M., Kustermann E., Blunk J., et al. Monitoring of implanted stem cell migration in vivo: a highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc Natl Acad Sci USA. 2002;99(25):16267-16272.
Holtzman D.M., Sheldon R.A., Jaffe W., et al. Nerve growth factor protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 1996;39(1):114-122.
Hu B.R., Liu C.L., Ouyang Y., et al. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab. 2000;20(9):1294-1300.
Hunt R.W., Neil J.J., Coleman L.T., et al. Apparent diffusion coefficient in the posterior limb of the internal capsule predicts outcome after perinatal asphyxia. Pediatrics. 2004;114(4):999-1003.
Husson I., Rangon C.M., Lelievre V., et al. BDNF-induced white matter neuroprotection and stage-dependent neuronal survival following a neonatal excitotoxic challenge. Cereb Cortex. 2005;15(3):250-261.
Iadecola C., Zhang F., Casey R., et al. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17(23):9157-9164.
Ikonomidou C., Turski L. Antiepileptic drugs and brain development. Epilepsy Res. 2009.
Imitola J., Raddassi K., Park K.I., et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA. 2004;101(52):18117-18122.
Inder T.E., Volpe J.J. Mechanisms of perinatal brain injury. Semin Neonatol. 2000;5(1):3-16.
Inder T.E., Hunt R.W., Morley C.J., et al. Randomized trial of systemic hypothermia selectively protects the cortex on MRI in term hypoxic-ischemic encephalopathy. J Pediatr. 2004;145(6):835-837.
Ishida A., Trescher W.H., Lange M.S., et al. Prolonged suppression of brain nitric oxide synthase activity by 7-nitroindazole protects against cerebral hypoxic-ischemic injury in neonatal rat. Brain Dev. 2001;23(5):349-354.
Iwata O., Thornton J.S., Sellwood M.W., et al. Depth of delayed cooling alters neuroprotection pattern after hypoxia-ischemia. Ann Neurol. 2005;58(1):75-87.
Jatana M., Singh I., Singh A.K., et al. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr Res. 2006;59(5):684-689.
Jin K., Zhu Y., Sun Y., et al. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA. 2002;99(18):11946-11950.
Johnston B.M., Mallard E.C., Williams C.E., et al. Insulin-like growth factor-1 is a potent neuronal rescue agent after hypoxic-ischemic injury in fetal lambs. J Clin Invest. 1996;97(2):300-308.
Johnston M.V., Jeon O.H., Pevsner J., et al. Neurobiology of Rett syndrome: a genetic disorder of synapse development. Brain Dev. 2001;23(Suppl 1):S206-S213.
Johnston M.V. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15(3):234-240.
Juul S.E., Yachnis A.T., Rojiani A.M., et al. Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr Dev Pathol. 1999;2(2):148-158.
Juul S.E., McPherson R.J., Bauer L.A., et al. A phase I/II trial of high-dose erythropoietin in extremely low birth weight infants: pharmacokinetics and safety. Pediatrics. 2008;122(2):383-391.
Khashaba M.T., Shouman B.O., Shaltout A.A., et al. Excitatory amino acids and magnesium sulfate in neonatal asphyxia. Brain Dev. 2006;28(6):375-379.
Kinney H.C., Panigrahy A., Newburger J.W., et al. Hypoxic-ischemic brain injury in infants with congenital heart disease dying after cardiac surgery. Acta Neuropathol (Berl). 2005;110(6):563-578.
Kirton A., Shroff M., Visvanathan T., et al. Quantified corticospinal tract diffusion restriction predicts neonatal stroke outcome. Stroke. 2007;38(3):974-980.
Koh P.O. Melatonin attenuates the focal cerebral ischemic injury by inhibiting the dissociation of pBad from 14-3-3. J Pineal Res. 2008;44(1):101-106.
Krum J.M., Rosenstein J.M. VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp Neurol. 1998;154(1):57-65.
Kumral A., Uysal N., Tugyan K., et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res. 2004;153(1):77-86.
Laptook A.R., Corbett R.J., Sterett R., et al. Modest hypothermia provides partial neuroprotection for ischemic neonatal brain. Pediatr Res. 1994;35(4 Pt 1):436-442.
Laptook A.R., Corbett R.J., Sterett R., et al. Modest hypothermia provides partial neuroprotection when used for immediate resuscitation after brain ischemia. Pediatr Res. 1997;42(1):17-23.
Laptook A.R., Corbett R.J. The effects of temperature on hypoxic-ischemic brain injury. Clin Perinatol. 2002;29(4):623-649. vi
Laudenbach V., Fontaine R.H., Medja F., et al. Neonatal hypoxic preconditioning involves vascular endothelial growth factor. Neurobiol Dis. 2007;26(1):243-252.
Le Strange E., Saeed N., Cowan F.M., et al. MR imaging quantification of cerebellar growth following hypoxic-ischemic injury to the neonatal brain. Am J Neuroradiol. 2004;25(3):463-468.
Lehnardt S., Massillon L., Follett P., et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100(14):8514-8519.
Li A.M., Chau V., Poskitt K.J., et al. White matter injury in term newborns with neonatal encephalopathy. Pediatr Res. 2009;65(1):85-89.
Licht D.J., Shera D.M., Clancy R.R., et al. Brain maturation is delayed in infants with complex congenital heart defects. J Thorac Cardiovasc Surg. 2009;137(3):529-536. discussion 536–527
Lindstrom K., Lagerroos P., Gillberg C., et al. Teenage outcome after being born at term with moderate neonatal encephalopathy. Pediatr Neurol. 2006;35(4):268-274.
Liu Y., Barks J.D., Xu G., et al. Topiramate extends the therapeutic window for hypothermia-mediated neuroprotection after stroke in neonatal rats. Stroke. 2004;35(6):1460-1465.
Lok J., Martin L.J. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J Neurotrauma. 2002;19(7):815-828.
Lu D., Mahmood A., Qu C., et al. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma. 2005;22(9):1011-1017.
Ma D., Hossain M., Chow A., et al. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann Neurol. 2005;58(2):182-193.
Ma J., Zhang G.Y. Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci Lett. 2003;348(3):185-189.
Mahle W.T., Tavani F., Zimmerman R.A., et al. An MRI study of neurological injury before and after congenital heart surgery. Circulation. 2002;106(12 Suppl 1):I109-I114.
Maneru C., Serra-Grabulosa J.M., Junque C., et al. Residual hippocampal atrophy in asphyxiated term neonates. J Neuroimaging. 2003;13(1):68-74.
Marks K.A., Mallard C.E., Roberts I., et al. Nitric oxide synthase inhibition attenuates delayed vasodilation and increases injury after cerebral ischemia in fetal sheep. Pediatr Res. 1996;40(2):185-191.
Marlow N., Rose A.S., Rands C.E., et al. Neuropsychological and educational problems at school age associated with neonatal encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2005;90(5):F380-F387.
Marret S., Gressens P., Gadisseux J.F., et al. Prevention by magnesium of excitotoxic neuronal death in the developing brain: an animal model for clinical intervention studies. Dev Med Child Neurol. 1995;37(6):473-484.
Maurer M.H., Tripps W.K., Feldmann R.E.Jr, et al. Expression of vascular endothelial growth factor and its receptors in rat neural stem cells. Neurosci Lett. 2003;344(3):165-168.
McKinstry R.C., Miller J.H., Snyder A.Z., et al. A prospective, longitudinal diffusion tensor imaging study of brain injury in newborns. Neurology. 2002;59(6):824-833.
McPherson R.J., Juul S.E. High-dose erythropoietin inhibits apoptosis and stimulates proliferation in neonatal rat intestine. Growth Horm IGF Res. 2007;17(5):424-430.
McQuillen P.S., Ferriero D.M. Selective vulnerability in the developing central nervous system. Pediatr Neurol. 2004;30(4):227-235.
McQuillen P.S., Barkovich A.J., Hamrick S.E., et al. Temporal and Anatomic Risk Profile of Brain Injury With Neonatal Repair of Congenital Heart Defects. Stroke. 2007;38(2):736-741.
Ment L.R., Bada H.S., Barnes P., et al. Practice parameter: neuroimaging of the neonate: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2002;58(12):1726-1738.
Mercuri E., Atkinson J., Braddick O., et al. Basal ganglia damage and impaired visual function in the newborn infant. Arch Dis Child Fetal Neonatal Ed. 1997;77(2):F111-F114.
Miller S.P., Newton N., Ferriero D.M., et al. Predictors of 30-month outcome after perinatal depression: role of proton MRS and socioeconomic factors. Pediatr Res. 2002;52(1):71-77.
Miller S.P., Vigneron D.B., Henry R.G., et al. Serial quantitative diffusion tensor MRI of the premature brain: Development in newborns with and without injury. J Magn Reson Imaging. 2002;16(6):621-632.
Miller S.P., Latal B., Clark H., et al. Clinical signs predict 30-month neurodevelopmental outcome after neonatal encephalopathy. Am J Obstet Gynecol. 2004;190(1):93-99.
Miller S.P., Ramaswamy V., Michelson D., et al. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146(4):453-460.
Miller S.P., McQuillen P.S., Hamrick S., et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357(19):1928-1938.
Modo M., Mellodew K., Cash D., et al. Mapping transplanted stem cell migration after a stroke: a serial, in vivo magnetic resonance imaging study. Neuroimage. 2004;21(1):311-317.
Moster D., Lie R.T., Markestad T. Joint association of Apgar scores and early neonatal symptoms with minor disabilities at school age. Arch Dis Child Fetal Neonatal Ed. 2002;86(1):F16-F21.
Mu D., Jiang X., Sheldon R.A., et al. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis. 2003;14(3):524-534.
Mueller F.J., Serobyan N., Schraufstatter I.U., et al. Adhesive interactions between human neural stem cells and inflamed human vascular endothelium are mediated by integrins. Stem Cells. 2006;24(11):2367-2372.
Mukherjee P., Miller J.H., Shimony J.S., et al. Diffusion-tensor MR imaging of gray and white matter development during normal human brain maturation. Am J Neuroradiol. 2002;23(9):1445-1456.
Myers R.E. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol. 1972;112(2):246-276.
Myers R.E. Four patterns of perinatal brain damage and their conditions of occurrence in primates. Adv Neurol. 1975;10:223-234.
Nagy Z., Lindstrom K., Westerberg H., et al. Diffusion tensor imaging on teenagers, born at term with moderate hypoxic-ischemic encephalopathy. Pediatr Res. 2005;58(5):936-940.
Neil J.J., Inder T.E. Detection of wallerian degeneration in a newborn by diffusion magnetic resonance imaging (MRI). J Child Neurol. 2006;21(2):115-118.
Nelson K.B., Dambrosia J.M., Grether J.K., et al. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44(4):665-675.
Nishida M., Makris N., Kennedy D.N., et al. Detailed semiautomated MRI based morphometry of the neonatal brain: preliminary results. Neuroimage. 2006;32(3):1041-1049.
Northington F.J., Ferriero D.M., Graham E.M., et al. Early Neurodegeneration after Hypoxia-Ischemia in Neonatal Rat Is Necrosis while Delayed Neuronal Death Is Apoptosis. Neurobiol Dis. 2001;8(2):207-219.
Northington F.J., Graham E.M., Martin L.J. Apoptosis in perinatal hypoxic-ischemic brain injury: how important is it and should it be inhibited? Brain Res Brain Res Rev. 2005;50(2):244-257.
Novotny E., Ashwal S., Shevell M. Proton magnetic resonance spectroscopy: an emerging technology in pediatric neurology research. Pediatr Res. 1998;44(1):1-10.
Obenaus A., Robbins M., Blanco G., et al. Multi-modal magnetic resonance imaging alterations in two rat models of mild neurotrauma. J Neurotrauma. 2007;24(7):1147-1160.
Ogihara T., Hirano K., Ogihara H., et al. Non-protein-bound transition metals and hydroxyl radical generation in cerebrospinal fluid of newborn infants with hypoxic ischemic encephalopathy. Pediatr Res. 2003;53(4):594-599.
Ostwald K., Hagberg H., Andiné P., et al. Upregulation of calpain activity in neonatal rat brain after hypoxic-ischemia. Brain Res. 1993;630(1–2):289-294.
Palmer T.D., Willhoite A.R., Gage F.H. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425(4):479-494.
Pang Y., Zheng B., Fan L.W., et al. IGF-1 protects oligodendrocyte progenitors against TNFalpha-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia. 2007;55(11):1099-1107.
Park K.I., Teng Y.D., Snyder E.Y. The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat Biotechnol. 2002;20(11):1111-1117.
Park K.I., Hack M.A., Ourednik J., et al. Acute injury directs the migration, proliferation, and differentiation of solid organ stem cells: evidence from the effect of hypoxia-ischemia in the CNS on clonal “reporter” neural stem cells. Exp Neurol. 2006;199(1):156-178.
Park K.I., Himes B.T., Stieg P.E., et al. Neural stem cells may be uniquely suited for combined gene therapy and cell replacement: Evidence from engraftment of Neurotrophin-3-expressing stem cells in hypoxic-ischemic brain injury. Exp Neurol. 2006;199(1):179-190.
Peeters-Scholte C., Braun K., Koster J., et al. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr Res. 2003;54(4):516-522.
Pellerin L., Magistretti P.J. Neuroscience. Let there be (NADH) light. Science. 2004;305(5680):50-52.
Plane J.M., Liu R., Wang T.W., et al. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16(3):585-595.
Portera-Cailliau C., Price D.L., Martin L.J. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997;378(1):70-87.
Prayer D., Barkovich A.J., Kirschner D.A., et al. Visualization of nonstructural changes in early white matter development on diffusion-weighted MR images: evidence supporting premyelination anisotropy. Am J Neuroradiol. 2001;22(8):1572-1576.
Pu Y., Li Q.F., Zeng C.M., et al. Increased detectability of alpha brain glutamate/glutamine in neonatal hypoxic-ischemic encephalopathy. Am J Neuroradiol. 2000;21(1):203-212.
Raab S., Beck H., Gaumann A., et al. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91(3):595-605.
Ramaswamy V., Miller S.P., Barkovich A.J., et al. Perinatal stroke in term infants with neonatal encephalopathy. Neurology. 2004;62(11):2088-2091.
Rice H.E., Hsu E.W., Sheng H., et al. Superparamagnetic iron oxide labeling and transplantation of adipose-derived stem cells in middle cerebral artery occlusion-injured mice. Am J Roentgenol. 2007;188(4):1101-1108.
Riikonen R.S., Kero P.O., Simell O.G. Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr Neurol. 1992;8(1):37-40.
Robertson C., Finer N. Term infants with hypoxic-ischemic encephalopathy: outcome at 3.5 years. Dev Med Child Neurol. 1985;27(4):473-484.
Robertson C.M., Finer N.N., Grace M.G. School performance of survivors of neonatal encephalopathy associated with birth asphyxia at term. J Pediatr. 1989;114(5):753-760.
Robertson C.M., Finer N.N. Long-term follow-up of term neonates with perinatal asphyxia. Clin Perinatol. 1993;20(2):483-500.
Robertson N.J., Lewis R.H., Cowan F.M., et al. Early increases in brain myo-inositol measured by proton magnetic resonance spectroscopy in term infants with neonatal encephalopathy. Pediatr Res. 2001;50(6):692-700.
Rosenthal G.L. Patterns of prenatal growth among infants with cardiovascular malformations: possible fetal hemodynamic effects. Am J Epidemiol. 1996;143(5):505-513.
Rouse D.J., Hirtz D.G., Thom E., et al. A randomized, controlled trial of magnesium sulfate for the prevention of cerebral palsy. N Engl J Med. 2008;359(9):895-905.
Rutherford M., Counsell S., Allsop J., et al. Diffusion-weighted magnetic resonance imaging in term perinatal brain injury: a comparison with site of lesion and time from birth. Pediatrics. 2004;114(4):1004-1014.
Rutherford M.A., Pennock J.M., Counsell S.J., et al. Abnormal magnetic resonance signal in the internal capsule predicts poor neurodevelopmental outcome in infants with hypoxic-ischemic encephalopathy. Pediatrics. 1998;102(2 Pt 1):323-328.
Rutherford M., Ramenghi L.A., Edwards A.D., et al. Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxi–ischaemic encephalopathy: a nested substudy of a randomised controlled trial. Lancet Neurol. 2010;9(1):39-45. Epub Nov 2009
Sargent MA, Poskitt K.J., Roland E.H., et al. Cerebellar vermian atrophy after neonatal hypoxic-ischemic encephalopathy. Am J Neuroradiol. 2004;25(6):1008-1015.
Sarnat H.B., Sarnat M.S. Neonatal encephalopathy following fetal distress. A clinical and electroencephalographic study. Arch Neurol. 1976;33(10):696-705.
Schreiber M.D., Gin-Mestan K., Marks J.D., et al. Inhaled nitric oxide in premature infants with the respiratory distress syndrome. N Engl J Med. 2003;349(22):2099-2107.
Segovia K.N., McClure M., Moravec M., et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63(4):520-530.
Shankaran S., Woldt E., Koepke T., et al. Acute neonatal morbidity and long-term central nervous system sequelae of perinatal asphyxia in term infants. Early Hum Dev. 1991;25(2):135-148.
Shankaran S., Laptook A., Wright L.L., et al. Whole-body hypothermia for neonatal encephalopathy: animal observations as a basis for a randomized, controlled pilot study in term infants. Pediatrics. 2002;110(2 Pt 1):377-385.
Shankaran S., Laptook A.R., Ehrenkranz R.A., et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574-1584.
Shanmugalingam S., Thornton J.S., Iwata O., et al. Comparative prognostic utilities of early quantitative magnetic resonance imaging spin-spin relaxometry and proton magnetic resonance spectroscopy in neonatal encephalopathy. Pediatrics. 2006;118(4):1467-1477.
Shevell M.I., Majnemer A., Miller S.P. Neonatal neurologic prognostication: the asphyxiated term newborn. Pediatr Neurol. 1999;21(5):776-784.
Shimotake J, Derugin N., Wendland M.F., et al. Vascular Endothelial Growth Factor Receptor2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke, Stroke Feb. 2010;41(2):343. 9
Shingo T., Sorokan S.T., Shimazaki T., et al. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21(24):9733-9743.
Sie L.T., van der Knaap M.S., Oosting J., et al. MR patterns of hypoxic-ischemic brain damage after prenatal, perinatal or postnatal asphyxia. Neuropediatrics. 2000;31(3):128-136.
Silveira R.C., Procianoy R.S. Interleukin-6 and tumor necrosis factor-alpha levels in plasma and cerebrospinal fluid of term newborn infants with hypoxic-ischemic encephalopathy. J Pediatr. 2003;143(5):625-629.
Silverstein F.S., Jensen F.E. Neonatal seizures. Ann Neurol. 2007;62(2):112-120.
Sola A., Rogido M., Lee B.H., et al. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57(4):481-487.
Sola A., Wen T.C., Hamrick S.E., et al. Potential for protection and repair following injury to the developing brain: a role for erythropoietin? Pediatr Res. 2005;57(5 Pt 2):110R-117R.
Sorrentino D.F., Fritz K.I., Haider S.H., et al. Nitric oxide-mediated modification of the glycine binding site of the NMDA receptor during hypoxia in the cerebral cortex of the newborn piglet. Neurochem Res. 2004;29(2):455-459.
Spandou E., Soubasi V., Papoutsopoulou S., et al. Neuroprotective effect of long-term MgSO4 administration after cerebral hypoxia-ischemia in newborn rats is related to the severity of brain damage. Reprod Sci. 2007;14(7):667-677.
Srinivasan L., Dutta R., Counsell S.J., et al. Quantification of deep gray matter in preterm infants at term-equivalent age using manual volumetry of 3-tesla magnetic resonance images. Pediatrics. 2007;119(4):759-765.
Steinman K.J., Gorno-Tempini M.L., Glidden D.V., et al. Neonatal watershed brain injury on magnetic resonance imaging correlates with verbal IQ at 4 years. Pediatrics. 2009;123(3):1025-1030.
Sun Y., Calvert J.W., Zhang J.H. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke. 2005;36(8):1672-1678.
Thayyil S., Chandrasekaran M., Taylor A., et al. Cerebral magnetic resonance biomarkers in neonatal encephalopathy: a meta-analysis. Pediatrics. 2010;125(2):e382-e395.
Thoresen M., Penrice J., Lorek A., et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res. 1995;37(5):667-670.
Thoresen M. Cooling the newborn after asphyxia – physiological and experimental background and its clinical use. Semin Neonatol. 2000;5(1):61-73.
Thoresen M., Whitelaw A. Cardiovascular changes during mild therapeutic hypothermia and rewarming in infants with hypoxic-ischemic encephalopathy. Pediatrics. 2000;106(1 Pt 1):92-99.
Toet M.C., Groenendaal F., Osredkar D., et al. Postneonatal epilepsy following amplitude-integrated EEG-detected neonatal seizures. Pediatr Neurol. 2005;32(4):241-247.
Towfighi J., Housman C., Heitjan D.F., et al. The effect of focal cerebral cooling on perinatal hypoxic-ischemic brain damage. Acta Neuropathol (Berl). 1994;87(6):598-604.
Tsuji M., Wilson M.A., Lange M.S., et al. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189(1):58-65.
Turkyilmaz C., Turkyilmaz Z., Atalay Y., et al. Magnesium pre-treatment reduces neuronal apoptosis in newborn rats in hypoxia-ischemia. Brain Res. 2002;955(1–2):133-137.
van den Tweel E.R., van Bel F., Kavelaars A., et al. Long-term neuroprotection with 2-iminobiotin, an inhibitor of neuronal and inducible nitric oxide synthase, after cerebral hypoxia-ischemia in neonatal rats. J Cereb Blood Flow Metab. 2005;25(1):67-74.
van Handel M., Swaab H., de Vries L.S., et al. Long-term cognitive and behavioral consequences of neonatal encephalopathy following perinatal asphyxia: a review. Eur J Pediatr. 2007;166(7):645-654.
Van Hof-van Duin J., Mohn G. Visual defects in children after cerebral hypoxia. Behav Brain Res. 1984;14(2):147-155.
Van Kooij B.J.M., Van Handel M., Uiterwaal C.S., et al. Corpus callosum size in relation to motor performance in 9- to 10-year-old children with neonatal encephalopathy. Pediatr Res. 2008;63(1):103-108.
Van Leeuwen G.M., Hand J.W., Lagendijk J.J., et al. Numerical modeling of temperature distributions within the neonatal head. Pediatr Res. 2000;48(3):351-356.
Vannucci R.C., Vannucci S.J. Glucose metabolism in the developing brain. Semin Perinatol. 2000;24(2):107-115.
Vannucci S.J., Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol. 2004;207(Pt 18):3149-3154.
Vexler Z.S., Yenari M.A. Does inflammation after stroke affect the developing brain differently than adult brain? Dev Neurosci. 2009;31(5):378-393.
Villa P., Bigini P., Mennini T., et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. 2003;198(6):971-975.
Volpe J.J. Neurology of the newborn, ed 5. Philadelphia: WB Saunders Company; 2008.
Wang L., Zhang Z., Wang Y., et al. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35(7):1732-1737.
Ward P., Counsell S., Allsop J., et al. Reduced fractional anisotropy on diffusion tensor magnetic resonance imaging after hypoxic-ischemic encephalopathy. Pediatrics. 2006;117(4):e619-e630.
Wasserman J.K., Zhu X., Schlichter L.C. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res. 2007;1180:140-154.
Wen T.C., Rogido M., Peng H., et al. Gender differences in long-term beneficial effects of erythropoietin given after neonatal stroke in postnatal day-7 rats. Neuroscience. 2006;139(3):803-811.
Wu Y.W., Croen L.A., Torres A.R., et al. Interleukin-6 genotype and risk for cerebral palsy in term and near-term infants. Ann Neurol. 2009;66(5):663-670.
Wyatt J.S., Gluckman P.D., Liu P.Y., et al. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119(5):912-921.
Yang X., Cepko C.L. Flk-1, a receptor for vascular endothelial growth factor (VEGF), is expressed by retinal progenitor cells. J Neurosci. 1996;16(19):6089-6099.
Zachary I. Neuroprotective role of vascular endothelial growth factor: signalling mechanisms, biological function, and therapeutic potential. Neurosignals. 2005;14(5):207-221.
Zhu C., Kang W., Xu F., et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics. 2009;124(2):e218-e226.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 17 Hypoxic-Ischemic Brain Injury in the Term Newborn
Scope of the Problem
The syndrome of neonatal encephalopathy in the term newborn occurs in up to 6 per 1000 live term births and is a major cause of neurodevelopmental disability, with one-quarter of survivors sustaining permanent neurological deficits [Volpe, 2008]. While the syndrome of neonatal encephalopathy may result from a number of causes, this chapter will focus on the etiology, pathophysiology, and management of hypoxic-ischemic brain injury in the term newborn. Hypoxic-ischemic brain injury is recognized clinically by a characteristic encephalopathy with either a lack of alertness or hyperalertness, abnormal tone, abnormal reflex function, poor feeding, compromised respiratory status, and seizures [Miller et al., 2004]. Brain imaging has revealed patterns of brain injury following a hypoxic-ischemic insult that are unique to the immature brain; they depend on the age at which it occurs and on the severity and duration of the insult (Figure 17-1) [Miller et al., 2005; Cowan et al., 2003]. The capacity to repeat brain imaging safely in the newborn following hypoxia-ischemia has enabled studies confirming the clinical and experimental observations that neonatal brain injury evolves over days, if not weeks [Ferriero, 2004]. These clinical investigations are consistent with the prolonged temporal evolution of brain injury in neonatal animal models [Ferriero, 2004]. This time-course “opens the door” for therapeutic interventions instituted not only hours, but also days, after the hypoxic-ischemic insult, with different interventions needed as the injury evolves [Ferriero, 2004].
Etiology of Brain Injury in the Term Newborn
There is increasing recognition that neonatal brain injury is related to antenatal, perinatal, and postnatal factors, and is not always the result of “birth asphyxia” [Badawi et al., 1998]. While hypoxia-ischemia accounts for a substantial fraction of neonatal brain injury, a documented hypoxic-ischemic insult is lacking in many term newborns with encephalopathy [Volpe, 2008; Ferriero, 2004]. Many risk factors for neonatal encephalopathy are clearly prenatal [Badawi et al., 1998], such as maternal hypotension, infertility treatment, and thyroid disease. A minority of cases have only intrapartum risk factors, such as breech extraction, cord prolapse, or abruptio placentae [Badawi et al., 1998]. However, recent cohorts evaluated with magnetic resonance imaging (MRI) studies demonstrate that brain injury actually happens at or near the time of birth [Cowan et al., 2003; Miller et al., 2005]. It is also important to recognize that postnatal causes may account for up to 10 percent of neonatal encephalopathy in the term infant. Therefore, metabolic abnormalities such as acute bilirubin encephalopathy and inborn errors of metabolism, infection, severe respiratory distress, and trauma should be considered in the clinical evaluation.
Clinical Syndromes and Natural History
Clinical Syndromes
Neonatal encephalopathy and seizures are the most overt manifestation of the severity of brain injury. Clinical signs and symptoms depend on the severity, timing, and duration of the insult, as well as the newborn’s maturity, even within ages considered “full term.” It is equally important to note that not all newborns with significant acquired brain injuries present with an easily recognizable encephalopathy or seizures. Some conditions, such as stroke and perinatal white matter injury, discussed in Chapters 18 and 19, may only have subtle manifestations that may not be recognized on clinical examination. This chapter will focus on the clinical syndrome of neonatal encephalopathy secondary to hypoxia-ischemia. The diagnosis and management of neonatal seizures is discussed in Chapter 16.
Neonatal Encephalopathy
The major components of the syndrome of “neonatal encephalopathy” include: alertness, tone, respiratory status, reflexes, feeding, and seizures (Table 17-1). The severity of brain injury, reflecting the duration and magnitude of the hypoxic-ischemic insult, determines the severity of evolution of this clinical encephalopathy. In newborns with moderate to severe encephalopathy, symptoms usually evolve over days, making serial detailed examinations important. As with any neurologically ill newborn, the baby’s gestational age must be considered in the interpretation of physical findings. As described by Volpe, there is a characteristic progression of signs in newborns with a severe hypoxic-ischemic insult [Volpe, 2008]. The affected neonate exhibits a depressed level of consciousness from the very first hours of life. Periodic breathing with apnea and bradycardia often heralds this initial presentation. Hypotonia with decreased movement is almost universally found. In the first day of life, the pupillary response is often preserved and abnormalities of eye movements are not detected. In severely injured newborns, seizures may be seen within 6–12 hours after birth. Seizures at this stage are often subtle, manifesting as ocular movements, lip smacking, apnea, or bicycling movements of the extremities. The nursing staff should be alerted to identify and document paroxysmal behaviors. Multifocal or focal clonic seizures may also occur, and often indicate focal cerebral infarction. In the latter hours of the first day of life, there may be a transient increase in the level of alertness that is not accompanied by other signs of neurological improvement. This should not be falsely reassuring, as this apparent increase in alertness is frequently accompanied by more seizures and apnea, a shrill cry, and jitteriness. With careful bedside examination, weakness in the proximal limbs and increased muscle stretch reflexes may be observed, but in the very severely injured newborn, diffuse weakness with absent movements and reflexes is common.
Documenting this clinical evolution is a critical component of the evaluation, and use of a simple encephalopathy score as a bedside tool can help the clinician standardize assessment of encephalopathic newborns, monitor the evolution of the clinical syndromes, and help in systematically identifying neonates who require therapeutic intervention (see Table 17-1) [Miller et al., 2004].
| Sign | Score = 0 | Score = 1 |
|---|---|---|
| Feeding | Normal | Gavage feeds, gastrostomy tube, or not tolerating oral feeds |
| Alertness | Alert | Irritable, poorly responsive, or comatose |
| Tone | Normal | Hypotonia or hypertonia |
| Respiratory status | Normal | Respiratory distress (need for continuous positive airway pressure or mechanical ventilation) |
| Reflexes | Normal | Hyperreflexia, hyporeflexia, or absent reflexes |
| Seizure | None | Suspected or confirmed clinical seizure |
(From Miller et al. Clinical signs predict 30-month neurodevelopmental outcome after neonatal encephalopathy. Am J Obstet Gynecol 2004;190(1):93–99.)
Subtle Neonatal Syndromes
Stroke and white matter injury in the term newborn may present with clinical signs that are so subtle that they remain undetected for months or years. Advances in brain monitoring (amplitude integrated EEG, prolonged video-EEG monitoring) and brain imaging (MRI) in the neonatal intensive care unit have increased recognition of these injuries. However, both stroke and white matter injury may present in the neonatal period with an overt encephalopathy [Li et al., 2009; Ramaswamy et al., 2004]. These conditions share many of the risk factors of “global” hypoxic-ischemic brain injury considered in this chapter.
Management of Neonatal Encephalopathy
Clinical Management
As many etiologies of neonatal encephalopathy have specific therapies, the clinician’s initial task is to determine the underlying etiology through careful history taking, neurological examination, and laboratory and brain imaging studies. The history should elicit indicators of intrauterine distress that may have contributed to decreased placental or fetal blood flow: fetal heart tracing abnormalities, passage of meconium, or a history of a difficulty in labor or delivery. The Apgar score will indicate who is at risk for mortality. The details of the delivery-room resuscitation, medications, and ventilatory support should be noted. Because the clinical signs of encephalopathy and seizures are often not specific for an etiology, laboratory tests are critical to exclude reversible causes of neonatal encephalopathy. The management of moderate or severe encephalopathy should occur in a neonatal intensive care unit in close collaboration with a neonatologist. Immediate management requires securing an appropriate airway and maintaining adequate circulation. Ventilatory support with mechanical ventilation or continuous positive airway pressure (CPAP) is often required. Metabolic complications, such as hypoglycemia, hypocalcemia, hyponatremia, and acidosis, frequently accompany hypoxic-ischemic encephalopathy (HIE), and should be identified and treated. Liver enzymes and serum creatinine levels should be performed to detect injury to other organs, while serum ammonia and lactate levels can screen for possible inborn errors of metabolism. Lumbar puncture to evaluate for intracranial infections should be performed if the history is not typical for HIE, or if a clinical suspicion of infection exists. If infection is suspected, ampicillin and gentamicin are started in addition to acyclovir if herpes simplex virus infection is a consideration. If the history, examination, or initial laboratory investigation points to an inborn error of metabolism, early treatment is crucial and a biochemical geneticist should be consulted. Additional diagnostic tests, such as serum amino acids and urine organic acids, as well as specific management strategies, are required (detailed in Chapter 20). The diagnosis of a severe intracranial hemorrhage should prompt consultation with a neurosurgeon to manage raised intracranial pressure from mass effect or hydrocephalus; platelet levels and coagulation function should be measured. Since the clinical syndrome evolves considerably over the first 72 hours of life, management of specific complications, such as respiratory compromise or seizures, can often be anticipated.
Investigation of term newborns with encephalopathy addresses three primary questions:
Addressing these questions is critical for the application of “neuroprotection” strategies, such as hypothermia [Eicher et al., 2005; Gluckman et al., 2005; Shankaran et al., 2005; Azzopardi et al., 2009]. To assess the encephalopathic term newborn, diagnostic tools available to the clinician include clinical features and biochemical and electrophysiological tests. However, the severity of brain injury in term asphyxiated newborns is not reliably predicted by clinical indicators commonly used during the first days of life, such as umbilical cord pH and Apgar scores [Shevell et al., 1999]. On the other hand, the severity of clinical encephalopathy is a strong predictor of neurodevelopmental outcome [Miller, 2004]. However, while the risk of motor and cognitive deficits appears to be minimal in mild encephalopathy, and pronounced at the severe end of the spectrum, it is inconsistent in neonates with moderate encephalopathy [Sarnat and Sarnat, 1976; Robertson et al., 1989; Dixon et al., 2002; Marlow et al., 2005]. Newborns with moderate encephalopathy are thus the most likely to benefit from the improved prognostic capabilities of brain imaging.
Brain Imaging of Newborns with Encephalopathy
Current guidelines for neuroimaging term newborns with encephalopathy suggest a noncontrast computed tomography (CT) scan to detect hemorrhage in infants with a history of significant birth trauma, and evidence of low hematocrit or coagulopathy [Ment et al., 2002]. If CT findings cannot explain the newborn’s clinical status, then MRI is recommended. For other encephalopathic term newborns, MRI is recommended between days 2 and 8 to assess the location and extent of injury [Ment et al., 2002]. In a recent study, the neuroimaging findings from a group of 48 term newborns with encephalopathy uniformly scanned with CT and MRI (T1 and T2-weighted images) with diffusion-weighted imaging (DWI) on the third day of life were compared [Chau et al., 2009]. On the third day of life, both CT and MRI with DWI reliably identify injury to the basal nuclei. However, DWI more readily detects cortical injury and focal and multifocal lesions, such as strokes and white matter injury, than either CT or conventional MRI. The choice of the best neuroimaging technique must balance the risk of transport and sedation involved in MRI against the risk of ionizing radiation with CT [Brenner et al., 2003; Berrington de Gonzalez and Darby, 2004; Hall et al., 2004]. With the development of MR-compatible incubators and monitoring equipment [Dumoulin et al., 2002; Bluml et al., 2004], as well as improved capacity to scan newborns without pharmacological sedation, MRI should now be the modality of choice when possible. MRI with DWI appears to be the most sensitive imaging study to detect abnormalities associated with other causes of neonatal encephalopathy, such as cerebral dysgenesis, infections, stroke, and metabolic disorders [Volpe, 2008; Cowan et al., 2003]. In order to confirm the diagnosis of hypoxic-ischemic brain injury and determine the extent of injury, MRI and DWI are optimally obtained between 3 and 5 days of life in term newborns with encephalopathy [Chau et al., 2009; Barkovich, 1997; Rutherford et al., 2004; Barkovich et al., 2006]. In newborns treated with hypothermia, further studies are needed to determine the optimal timing of MRI. The importance of rigorous imaging protocols with appropriate quality controls, and high quality neuroradiological review, must be emphasised.
Advanced MR Techniques
Advanced MR techniques, such as diffusion and spectroscopic imaging, allow us to observe the progression of neonatal brain injury. Diffusion MRI and MR spectroscopy (MRS) can be used to measure brain maturation and also provide important measures of brain microstructure and metabolism following injury. Given the widespread availability of these techniques on current MR scanners, their application will be discussed below. In addition to diffusion MRI and MRS, other advanced MR techniques are emerging. Computer-assisted morphometric techniques, including voxel-based and deformation-based morphometry, are used to correlate regional brain volumes in newborns, children, and adolescents with a history of neonatal encephalopathy with their neurodevelopmental outcomes [Maneru et al., 2003; Nishida et al., 2006; Srinivasan et al., 2007]. Moreover, diffusion tensor tractography, an extension of diffusion MRI, is providing new insights into recovery and resilience by measuring microstructural development of specific functional pathways [Glenn et al., 2007]. Together, these quantitative techniques are helping to identify injuries and abnormalities of subsequent brain development that may not be apparent on conventional MRI.
Magnetic resonance spectroscopy imaging
MRS can be used to measure changes in certain brain metabolites from a given brain region. Of the compounds measured by MRS at long echo times, N-acetylaspartate (NAA) and lactate are the most useful in assessing brain injury. NAA is an acetylated amino acid found in high concentrations in neurons of the central nervous system (CNS). NAA levels increase with advancing cerebral maturity [Barkovich, 2000; Novotny et al., 1998], and decrease with cerebral injury or impaired cerebral metabolism [Barkovich, 2000; Novotny et al., 1998]. Lactate is normally produced in the brain by astrocytes and used as fuel by neurons to replenish energy stores via oxidative phosphorylation [Pellerin and Magistretti, 2004]. Lactate levels are elevated with disturbed brain energy substrate delivery and oxidative metabolism, as seen with hypoxia-ischemia. Elevated lactate and reduced NAA levels are highly predictive of neurodevelopmental outcome following neonatal brain injury [Barkovich, 2000; Novotny et al., 1998]. Given this, lactate/NAA ratios are especially discriminatory of newborns with adverse outcomes [Shanmugalingam et al., 2006; Thayyil et al., 2010]. Myo-inositol, one of the major brain osmolytes, is best measured with MRS at short echo times, and is elevated with neonatal hypoxic-ischemic brain injury [Robertson et al., 2001].
Diffusion imaging
Diffusion-weighted MR imaging (DWI) detects alterations in free water diffusion. Diffusion tensor imaging (DTI) measures the amount (apparent diffusion coefficient [ADC] or average diffusivity) and directionality of water motion (fractional anisotropy [FA]). With brain maturation, ADC decreases in gray and white matter, presumably due to a reduction in water content and the development of cell membranes that restrict water diffusion [Mukherjee et al., 2002; Beaulieu, 2002, Coats et al., 2009]. Over this period, FA increases in white matter, even before myelin is evident on T1 and T2-weighted images [Miller et al., 2002b; Drobyshevsky et al., 2005; Prayer et al., 2001]. DWI and DTI also provide sensitive measures of brain injury [Barkovich et al., 2001; McKinstry et al., 2002]. With acute injury, intracellular water increases and water movement are “restricted” by the cell membrane when a diffusion gradient is applied. The DWI image will show an area of restricted diffusion as increased signal intensity that is a complicated product of T2* properties and restricted diffusion. The ADC or average diffusivity map (Dav) is a quantitative water diffusion map that shows restricted diffusion as areas of diminished signal intensity. Reduced ADC values in the posterior limb of the internal capsule are associated with a greater risk of adverse neurodevelopmental outcome in term newborns with encephalopathy [Hunt et al., 2004]. FA values in the white matter and basal nuclei are decreased with significant injury during the first week of life in term newborns with encephalopathy [Ward et al., 2006]. In newborns with brain injury, DTI also detects abnormalities of microstructural brain development remote from the primary injuries, in areas of the brain that are normal on T1 and T2-weighted images [Miller et al., 2002b].
Patterns of Brain Injury
In a primate model of brain injury in the “term newborn,” the distribution of injury was associated with the duration and severity of ischemia. While acute-profound asphyxia produced injury in the basal ganglia and thalamus, partial asphyxia caused white matter injury [Myers, 1972, 1975]. Similar patterns of injury are found in term newborns following hypoxia-ischemia (see Figure 17-1). The basal ganglia-predominant pattern involves both the basal ganglia and thalamus, and perirolandic cortex [Miller et al., 2005; Sie et al., 2000; Chau et al., 2009]. The watershed pattern predominantly involves the vascular watershed, from the white matter and extending to the cerebral cortex [Miller et al., 2005; Sie et al., 2000]. Maximal injury in both the watershed region and basal nuclei results in the total pattern of brain injury [Miller et al., 2005; Sie et al., 2000]. Identifying the predominant pattern of brain injury is helpful to the clinician caring for a term newborn with encephalopathy, as the predominant pattern is more strongly associated with neurodevelopmental outcome than the severity of injury in any given region [Miller et al., 2005].
The final pattern of injury, increasingly recognized by MRI, is the “focal- or multifocal” pattern of injury: stroke or white matter injury (WMI). Recent data suggest that strokes (arterial or venous) are also associated with neonatal encephalopathy in the term newborn [Cowan et al., 2003]. Many newborns with stroke have multiple risk factors for brain injury, including intrapartum complications.
While WMI is the characteristic pattern of brain injury in premature newborns, it is increasingly recognized in term newborns with encephalopathy, identified in 23 percent in one series (see Figure 17-1) [Li et al., 2009]. In this series, WMI demonstrated restricted diffusion on ADC maps in almost all newborns, suggesting that these lesions were acquired near birth. As newborns with WMI had milder encephalopathy relative to other newborns in the cohort, these lesions may have been underdetected in the past. Lower gestational age at birth, within the range of term birth, was associated with an increasing severity of WMI, suggesting a role for brain maturation in the etiology of this injury pattern [Li et al., 2009]. In sequential studies of term newborns with encephalopathy, delayed white matter degeneration, extending past the first week of life, is also seen [Barkovich et al., 2006; Neil and Inder, 2006]. This delayed WMI might follow injury to the basal nuclei, just as Wallerian degeneration of the corticospinal tract is found following some middle cerebral artery strokes in the term newborn [De Vries et al., 2005; Kirton et al., 2007]. It should be noted that full-term infants with congenital heart disease also have a strikingly high incidence of WMI on MRI and at autopsy [McQuillen et al., 2007; Mahle et al., 2002; Galli et al., 2004; Gilles et al., 1973; Kinney et al., 2005]. Similar to premature newborns and term newborns with encephalopathy, those with congenital heart disease are at risk of impaired delivery of energy substrates due to ischemia, inflammation, and oxidative stress, particularly with cardiopulmonary bypass. Recent data from autopsy and brain imaging studies suggests that in utero brain development is delayed in newborns with some forms of serious congenital heart disease [Miller et al., 2007; Rosenthal, 1996; Licht et al., 2009]. These abnormalities in early brain development might explain the predominance of WMI in term newborns with congenital heart disease, as opposed to the more expected “term” predominance of injury to the basal nuclei or watershed regions predominantly.
Progression of Neonatal Brain Injury
Timing the onset and determining the progression of brain injury have been greatly facilitated with the use of diffusion MR techniques and MRS. Recent studies have shown that the reduction in ADC on diffusion imaging due to brain injury in term newborns evolves over the initial days of life, reaching their nadir by 2–4 days after injury [Barkovich et al., 2006; McKinstry et al., 2002]. Thus, MR diffusion images obtained prior to the nadir, as in the first day following an injury, may not show the full extent of injury. Importantly, diffusion abnormalities persist for 7–8 days in the newborn before returning towards normal values (pseudonormalization) and ultimately reflect increased diffusion [Barkovich et al., 2001; McKinstry et al., 2002; Coats et al., 2009]. In a proportion of patients, brain injury will progressively worsen over the first 2 weeks of life to involve new brain areas, particularly the white matter tracts [Barkovich et al., 2006]. It is critical to interpret diffusion imaging in the context of the time between injury and the acquisition of the scan [Barkovich et al., 2001; McKinstry et al., 2002]. MRS data in newborns mirror this time course of injury progression. In the first 24 hours following brain injury in the term newborn, lactate increases, followed by a decrease in NAA in the 3 days after injury [Barkovich, 2000; Barkovich et al., 2001; Novotny et al., 1998]. The prolonged progression of neonatal brain injury is consistent with the mechanisms of cell injury, discussed below, that persist for days following hypoxic-ischemic brain injury in the newborn. These observations also suggest that the opportunity to intervene to prevent or ameliorate brain injury may extend over days in the term newborn, if not weeks. These quantitative MR techniques now offer a dynamic measure of brain injury in the newborn that can be safely used to determine the short-term effects of novel intervention strategies.
Outcomes
Neurodevelopmental outcomes following neonatal encephalopathy depend on the pattern and severity of the brain injury. Neurodevelopmental deficits may involve motor, visual, and cognitive functions. Both genetic and postnatal variables such as socioeconomic factors (e.g., environmental exposures and parental education) likely modify an individual’s neurodevelopmental outcome following neonatal brain injury [Miller et al., 2002a; Robertson and Finer, 1993]. Given the broad spectrum of neurodevelopmental impairments following neonatal encephalopathy, follow-up of these newborns should include assessment of motor function, vision and hearing, cognition, behavior, and quality of life, through infancy and childhood. Epilepsy is identified in up to one-half of survivors from moderate to severe neonatal encephalopathy [Brunquell et al., 2002; Clancy and Legido, 1991], and is particularly common in those infants with cerebral palsy and developmental delay [Toet et al., 2005].
The American College of Obstetricians and Gynecologists (ACOG) Task Force on Neonatal Encephalopathy concluded that an acute intrapartum event could result in cerebral palsy of the spastic quadriplegic or dyskinetic type, but could not account for isolated cognitive deficits [ACOG, 2004]. Recently reviewed data [Gonzalez and Miller, 2006] indicate that cognitive deficits may feature prominently following term neonatal encephalopathy of presumed hypoxic-ischemic brain injury, even in the absence of cerebral palsy. This pattern of neurodevelopmental deficits follows an overt neonatal encephalopathy, often in the context of a critical illness, and is most commonly associated with the watershed pattern of injury and white matter damage, rather than the basal nuclei-predominant pattern of injury. A complete assessment of neurodevelopmental outcome must include aspects of cognition most readily assessed at school age: learning, executive function, behavior, and social competence. In addition, developmental coordination disorder, autism spectrum disorder, or specific language impairments should be considered as possibilities in follow-up of newborns with a history of encephalopathy [van Handel et al., 2007]. Finally, “quality of life,” an individual’s subjective perception of physical and psychological health, should be evaluated by the clinician assessing outcomes to tailor rehabilitation and monitoring services best.
Motor Function
In term survivors of hypoxic-ischemic brain injury, the risk of cerebral palsy or severe disability may involve more than one-third of affected newborns, and is most common in those with a severe encephalopathy [Volpe, 2008; Dixon et al., 2002; Barnett et al., 2002]. Spastic quadriparesis is the most common type of cerebral palsy, although athetoid or spastic hemiparesis also occurs. The diagnosis and management of cerebral palsy are addressed in Chapter 69. A complete assessment of neurosensory and cognitive functions is critical in children with cerebral palsy [Marlow et al., 2005]. Minor motor impairments that do not meet diagnostic criteria for cerebral palsy [Shevell et al., 1999] are diagnosed in more than one-third of children with moderate encephalopathy, and in more than one-quarter of children with mild encephalopathy [Van Kooij et al., 2008].
Vision and Hearing
Severe visual impairment occurs in up to one-quarter of children after moderate or severe encephalopathy [Robertson and Finer, 1985; Shankaran et al., 1991]. This may be due to injury to the posterior visual pathway, including the primary visual cortex, resulting in “cortical visual impairment” [Van Hof-van Duin and Mohn, 1984]. Injuries to the basal nuclei may also affect acuity, visual fields, or stereopsis (depth perception) [Mercuri et al., 1997]. Sensorineural hearing loss, likely secondary to brainstem injury, is also seen following neonatal encephalopathy [Robertson and Finer, 1985; Robertson et al., 1989], affecting 18 percent of survivors of moderate encephalopathy without cerebral palsy [Lindstrom et al., 2006].
Cognition
Overall, cognitive deficits are seen in 30–50 percent of childhood survivors of moderate neonatal encephalopathy [Dilenge et al., 2001]. Intellectual performance in children with severe encephalopathy without cerebral palsy is also affected [Marlow et al., 2005]. School-age survivors of moderate neonatal encephalopathy are more likely to have difficulties with reading, spelling, and arithmetic, or require additional school resources [Moster et al., 2002; Robertson et al., 1989]. Cognitive deficits, such as those in language and memory, may be seen, even when IQ scores are “normal.” [Marlow et al., 2005]. Behavioral difficulties, such as hyperactivity and emotional problems, should also be considered, even in individuals without motor disability [Marlow et al., 2005].
Brain Imaging and Outcome
The pattern of brain injury on neuroimaging conveys important prognostic information regarding the “pattern” of neurodevelopmental abnormalities. The basal nuclei pattern of injury and abnormal signal intensity in the posterior limb of the internal capsule are both predictive of severely impaired motor and cognitive outcomes [Rutherford et al., 1998; Miller et al., 2005]. The cognitive deficits associated with this pattern are not surprising, given the common involvement of the cerebral cortex [Miller et al., 2005] and cerebellum [Le Strange et al., 2004; Sargent et al., 2004]. In contrast, the watershed pattern is associated with cognitive impairments that are not necessarily accompanied by major motor deficits [Miller et al., 2005]. Importantly, the cognitive deficits following the watershed pattern may only be evident after 2 years of age [Miller et al., 2005]. In survivors of neonatal encephalopathy without functional motor deficits assessed at 4 years of age, the severity of watershed-distribution injury was most strongly associated with impaired language skills [Steinman et al., 2009]. While neurodevelopmental outcomes may be significantly better than anticipated, neurological deficits may also be found in some newborns whose brain imaging studies are normal [Bax et al., 2006]. More subtle brain injuries associated with later neurodevelopmental deficits, such as white matter injuries or hippocampal volume loss, may only be detectable with quantitative brain imaging techniques [Miller et al., 2002a; Nagy et al., 2005; Gadian et al., 2000].
Pathophysiology of Neonatal Hypoxic-Ischemic Brain Injury
In term infants with neonatal encephalopathy, perinatal hypoxia-ischemia predominates as the major cause of future neurologic disability. The adverse consequences of cerebral ischemia include deprivation of energy substrates and oxygen, and an inability to clear accumulated, potentially toxic metabolites. Although linear flow charts cannot accurately convey the complex cascade of interrelated molecular pathways that lead to hypoxic-ischemic neurodegeneration, Figure 17-2 highlights some of the critical mechanisms.
Over the past 20 years, considerable information has emerged about the cellular and molecular consequences of cerebral hypoxia-ischemia and the molecular events that lead to neuronal cell death. The underlying rationale for this scientific focus is the hope that a better understanding of the basic molecular mechanisms of neurodegeneration may provide ways to modulate these events pharmacologically to limit their adverse consequences and to protect the brain from irreversible damage. A complementary focus that has emerged – and will likely become particularly important in the setting of neonatal brain injury – is the delineation of the intrinsic neuronal mechanisms of adaptation and repair after hypoxic-ischemic brain injury. Despite the traditional view of greater resistance to CNS injury in the neonate because of lower metabolic demands and the greater plasticity of the developing CNS, at specific stages of brain maturation, susceptibility to hypoxia-ischemia may be amplified. Clinical and experimental data have demonstrated that specific brain structures and neural cells in the developing brain may be selectively vulnerable to hypoxic-ischemic injury [McQuillen and Ferriero, 2004]. One of the best-characterized examples is the increased vulnerability of immature oligodendroglia to hypoxic-ischemic injury, which is evident clinically in premature infants and which has been successfully demonstrated in fetal and neonatal animal models [Back et al., 2002; Segovia et al., 2008].
Risk of injury to the immature brain may be heightened by certain therapeutic interventions; this risk stems from the complex roles that pivotal molecular mediators of hypoxic-ischemic injury (e.g., glutamate, calcium) play in brain development. For example, some studies have provided compelling experimental evidence of maturational stage-dependent deleterious effects of several commonly used antiepileptic drugs [Ikonomidou and Turski, 2009].
This section reviews information about the pathophysiology of hypoxic-ischemic brain injury, integrates data obtained from experimental and clinical studies, and highlights mechanisms that are particularly relevant to understanding perinatal brain injury and repair. Several reviews can provide complementary perspectives [Gonzalez and Ferriero, 2008; Vexler and Yenari, 2009; Northington et al., 2005; Pediatric Neurology, 2009]. Please also see Part III of this book on emerging neuroscience concepts (Chapters 13, 14, and 15).
Cerebral Blood Flow and Energy Metabolism
Disruption of cerebrovascular autoregulation has been implicated as an important factor in the pathophysiology of neonatal hypoxic-ischemic brain injury. It is widely accepted that preterm infants have a “pressure-passive” cerebral circulation; however, term infants may remain at risk for impairment of cerebrovascular autoregulation [Boylan et al., 2000] and susceptibility to cerebral ischemia with fluctuations in systemic blood pressure. Several basic physiologic mechanisms may contribute to impaired autoregulation in the neonate. Increased expression of inducible and neuronal isoforms of nitric oxide synthase (iNOS and nNOS), as well as endothelial NOS, may narrow the autoregulatory window, and downregulation of prostaglandin receptors in response to high circulating prostaglandin levels may blunt the prostaglandin-mediated vasoconstrictive response to hypertension and thereby contribute to inappropriately increased cerebral blood flow [Chemtob et al., 1996]. After an ischemic insult, the neonate remains at high risk for further damage in the acute recovery phase because neonatal encephalopathy is often associated with blood pressure fluctuations.
An inadequate supply of glucose or alternate substrates plays a pivotal role in hypoxic-ischemic neuronal cell death. Although overall metabolic demands are lower in the neonatal than in the adult brain, during periods of rapid brain growth, particularly the perinatal period, metabolic needs rise. The pattern of injury after hypoxia-ischemia can be explained in part on the basis of this metabolic demand; brain regions most susceptible to hypoxic-ischemic injury in the term infant (e.g., subcortical gray matter structures such as the basal ganglia and thalamus) are the same regions that are most vulnerable to mitochondrial toxins. Brain development is associated with a transition from the ability to use glucose and ketones as energy substrates in the neonate to an absolute requirement for glucose in the adult. The immature brain can use lactate as an alternate fuel source to some degree, and the deleterious effects of lactate accumulation after hypoxia-ischemia therefore may be attenuated in the neonate compared with the adult. However, normal maturation is characterized by limitations in glucose transport capacity and increased use of these alternative fuels such as lactate. The inability to transport glucose across the blood–brain barrier threatens cerebral glucose utilization. These factors illustrate the importance of understanding the use of glucose, lactate, and ketones in the newborn brain under normal and pathologic conditions [Vannucci and Vannucci, 2000; Vannucci and Hagberg, 2004].
Excitotoxicity
Glutamate can activate a variety of excitatory amino acid receptors that are broadly classified based on their selective responses to specific agonists (e.g., NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid [AMPA], kainate) or their signaling mechanisms (i.e., ionotropic [ligand-gated ion channel] or metabotropic [G-protein-coupled]). Excitatory amino acid neurotransmission plays a pivotal role in brain development and in learning and memory. A substantial body of data has emerged over the past 30 years documenting the fact that overactivation of excitatory amino acid receptors (i.e., excitotoxicity) contributes to neurodegeneration in a broad range of acute and chronic neurologic disorders [Johnston, 2005].
[/not-level-membership-for-neurology-category]
