Chapter 27 Hydrocephalus and Arachnoid Cysts
Hydrocephalus
Definition
More recently, hydrocephalus has been more broadly defined as a disturbance of formation, flow, or absorption of CSF that leads to an increase in volume occupied by this fluid in the central nervous system (CNS) [Rekate, 2009]. This definition excludes other abnormalities of CSF dynamics, such as benign intracranial hypertension, in which the ventricles are not enlarged. It does not specify the source of production or absorption of CSF nor does it presuppose the mechanisms inherent in ventricular distension.
Classification
A number of classification systems for hydrocephalus have been suggested [Mori, 1995; Boaz and Edwards-Brown, 1999; Beni-Adani et al., 2006; Oi and Di Rocco, 2006; Rekate, 2009]. These include the following types of hydrocephalus:






The terms compensated and uncompensated hydrocephalus generally refer to whether an increase in ventricular size is associated with evidence of raised intracranial pressure. In compensated, or arrested, hydrocephalus, a gradual increase in ventricular size stabilizes by reaching a new equilibrium and the patient has no symptoms or signs of raised intracranial pressure. In contrast, uncompensated hydrocephalus is associated with clinical symptoms and signs of raised intracranial pressure and usually with progressive dilatation of the ventricles. This is the clinical situation where treatment is indicated. No classification is completely adequate for hydrocephalus at all ages. The classification of hydrocephalus into communicating or noncommunicating dates back to the early 1900s and is based on Walter Dandy’s experimental studies into the pathophysiology of hydrocephalus [Richards, 1990].
Hydrocephalus is also categorized as congenital, which is present at birth and often associated with developmental defects; and acquired, which occurs after development of the brain and ventricles [Mori, 1995; Chahlavi et al., 2001]. Hydrocephalus has also been classified based on the stage of development at the time that the ventricles became dilated [Oi and Di Rocco, 2006]. The various subtypes of fetal hydrocephalus are classified according to the mechanism of obstruction to the flow of CSF to include:
This classification is cross-referenced to the stage of fetal development (e.g., neuronal maturation, cell migration); it may prove useful in deciding when treatment may be futile if beyond the legal period for terminating a pregnancy, and in identifying potential candidates for early delivery or fetal surgery [Rekate, 2009].
Extraventricular obstructive hydrocephalus is now recognized to represent, almost universally, benign pericerebral collections of infancy that are usually familial, resolve with time, and almost never require treatment [Drake, 2008].
Benign extra-axial collections of infancy involve abnormal enlargement of the head and excessively large subarachnoid space. In infants whose neurological development is normal and in whom the enlargement of the subarachnoid space resolves by 24 months of age, no further work-up or treatment is required. In infants whose neurological development is abnormal or in whom the enlargement of the subarachnoid space does not resolve by 24 months of age, further investigations may be warranted. Several genetic conditions, such as certain mucopolysaccharidoses, achondroplasia, Sotos’ syndrome, and glutaric aciduria type I, can feature enlargement of the subarachnoid spaces as either an early or an associated finding on neuroimaging. In the case of the mucopolysaccharidoses, enlargement of the subarachnoid spaces may be a direct consequence of impairment of CSF absorption by the storage material. Thus, enlargement of the subarachnoid spaces may be an important clue to an early genetic diagnosis, which is important, as therapeutic options exist for many forms of mucopolysaccharidosis and for glutaric aciduria type I [Paciorkowski and Greenstein, 2007].
Epidemiology
In newborns, the overall incidence of hydrocephalus ranges from 0.3 to 4 per 1000 live births. Occurring as a single congenital disorder, the incidence of hydrocephalus has been reported as 0.9–1.5 per 1000 births [Milhorat, 1972; Serlo et al., 1986; El Awad, 1992; Blackburn and Fineman, 1994; Fernell and Hagberg, 1998]. It is estimated that approximately 125,000 persons are living with ventricular shunts and that 33,000 shunts are placed annually in the United States [Bondurant and Jimenez, 1995].
The incidence of pediatric hydrocephalus has declined in many developed countries [Drake, 2008]. Antenatal screening, genetic testing, and pregnancy termination have reduced the incidence of congenital malformations of the brain that cause hydrocephalus. The incidence of open neural tube defects has also decreased precipitously as a result of maternal folate supplementation, antenatal screening, and termination of pregnancy based on superior antenatal imaging with ultrasound and magnetic resonance imaging (MRI). The incidence of CSF shunting in open neural tube defects, formerly reported to be as high as 90 percent, has also declined, possibly as a result of a general, more conservative approach, and also the selection of lower-grade lesions for delivery with a lower need for shunting [Tulipan et al., 2003; Chakraborty et al., 2008]. The incidence of intraventricular hemorrhage (IVH) has also decreased as a result of better perinatal management of prematurity, and, as such, one of the major complications of post-hemorrhagic hydrocephalus (see Chapter 19) [Fernell and Hagberg, 1998].
CSF Production, Circulation, and Absorption
CSF is produced by two mechanisms: Most of the CSF (50–80 percent) is thought to be secreted by the choroid plexus within the cerebral ventricles. Extrachoroidal CSF production in subarachnoid sites and by way of a transependymal route has also been documented. About 20 percent or more of CSF is derived from brain extracellular fluid created as a byproduct of cerebral metabolism [Bering, 1962; Fishman, 1980; Rekate, 1997]. Normally, rates of production (0.35 mL per minute or approximately 400–500 mL/day) and absorption of CSF are equal. Total CSF volume is 65–140 mL in children, and 90–150 mL in adults. In one study of premature infants born at gestational age 32 ± 1.6 wk, CSF volume was estimated to be 23.33 ± 9.6 mL at birth and 40.66 ± 21.23 mL at term equivalent. Full-term infants have a CSF volume of 32.4 ± 11.1 mL at birth [Zacharia et al., 2006].
The process of CSF formation by the choroid plexus includes plasma ultrafiltration and secretion. Secretion, an energy-dependent process, is initiated by hydrostatic pressure in the choroidal capillaries and by active transport of sodium. The enzymes sodium-potassium adenosine triphosphatase (ATPase) and carbonic anhydrase partly regulate CSF secretion [Fishman, 1980]. CSF production has been reported to remain constant across the normal intracranial pressure range [Pollay, 1977], with CSF production decreasing when intracranial pressure approaches mean arterial pressure. There have been reports, however, of downregulation of CSF production in patients with chronic hydrocephalus [Silverberg et al., 2002].
By contrast, the process of CSF reabsorption is not an energy-dependent process [Rekate, 1997]. After formation, CSF exits from the lateral ventricles through the foramina of Monro into the third ventricle (Figure 27-1). CSF then traverses the aqueduct of Sylvius into the fourth ventricle, leaving though the foramina of Lushka and foramen of Magendie into the cisterna magna, from where it flows through the subarachnoid space around the cerebral hemispheres. Information gained from MRI analysis of CSF movement demonstrates pulsatile to-and-fro motion of CSF within the lateral ventricles, produced from a brain-pumping motion that ejects the CSF and causes a net downward flow [Feinberg and Mark, 1987].
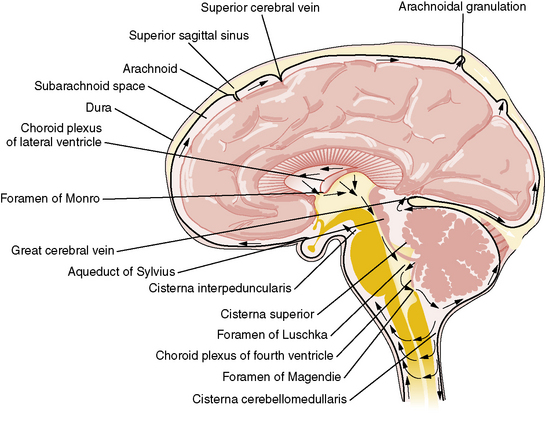
Historically, it has been held that CSF is absorbed into the vascular system mainly through the arachnoid villi within the arachnoid granulations covering the brain and spinal cord leptomeninges [Alksne and Lovings, 1972; Welch, 1975]. This process is thought to be passive and not energy-dependent. A layer of endothelium within the arachnoid villi separates the subarachnoid CSF space from the vascular system. Water and electrolytes pass freely across these arachnoid membranes. There is normally 5–7 mmHg difference in pressure between the dural venous sinuses and the subarachnoid space. This is presumed to be the hydrostatic force behind the absorption of CSF. Larger proteins and macromolecules cannot pass through intercellular junctions but are selectively transported across the cytoplasm of endothelial cells by an active process involving micropinocytosis [Welch, 1975]. Increased absorption through the arachnoid villi protects the brain from transient increases in intracranial pressure [Mann et al., 1978]. Newborn infants do not have visible arachnoid granulations, suggesting that the maximum capacity for reabsorption is less than in the adult, or that CSF is absorbed by different mechanisms in the neonate.
For some time, arachnoid granulations were thought to be the only CSF absorption pathway; however, other mechanisms have been identified, with evidence emerging that arachnoid granulations have only a secondary role. Olfactory nerves, the cribriform plate, and nasal lymphatics have been identified as important sites for CSF absorption [Johnston and Papaiconomou, 2002; Johnston, 2003; Johnston et al., 2004].
Absorption of CSF across brain tissue into capillaries has also been proposed [Greitz, 2004]. According to this theory, the distending force in the production of chronic hydrocephalus is an increased systolic pulse pressure in the brain tissue, due to decreased intracranial compliance.
Etiology and Pathophysiology
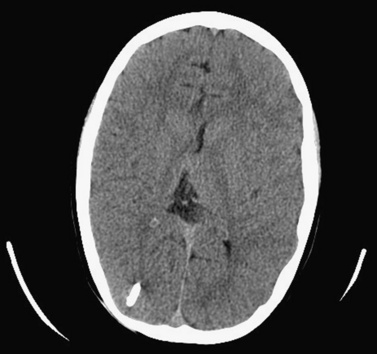
Hydrocephalus can be a symptom of a large number of disorders, and a list of conditions in which it has been reported is summarized in Box 27-1. It is associated with tumors and infections, and may be a complication of prematurity and trauma [Renier et al., 1988; Schrander-Stumpel and Fryns, 1998; Chi et al., 2005]. It is also seen in apparent isolation. High-resolution MRI of postnatal life has provided clues to the etiology of hydrocephalus, which in the past would have been labeled as idiopathic; some of these include IVH (Figure 27-2), aqueductal stenosis (Figure 27-3), and migrational abnormalities [Drake, 2008].
Box 27-1 Differential Diagnosis of Hydrocephalus



































































CRASH, corpus callosum agenesis, retardation, adducted thumbs, spastic paraparesis, hydrocephalus; VACTERL, vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula with esophageal atresia, renal abnormalities, limb abnormalities.
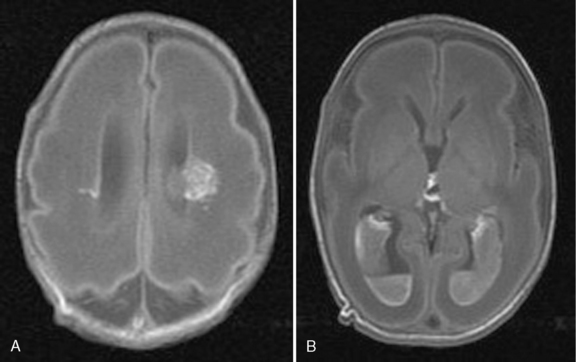
The etiologies of hydrocephalus in one series of pediatric patients are shown in Table 27-1 [Drake et al., 1998]. Hydrocephalus is due to either abnormal CSF reabsorption, or flow, or, rarely, overproduction. The main situation in which CSF production is increased enough to cause hydrocephalus is the presence of a choroid plexus papilloma. These tumors contain functional choroid epithelium and can produce very large amounts of CSF. However, even in the latter case, reabsorption is probably defective, as normal individuals can usually tolerate the elevated CSF production rate of these tumors. CSF accumulation, in turn, leads to raised intracranial pressure.
| Causes | Percentage |
|---|---|
| Intraventricular hemorrhage | 24.1 |
| Myelomeningocele | 21.2 |
| Tumor | 9.0 |
| Aqueductal stenosis | 7.0 |
| Infection | 5.2 |
| Head injury | 1.5 |
| Other | 11.3 |
| Unknown | 11.0 |
| Two or more causes | 8.7 |
(Modified from Drake JM, et al: Randomized trial of cerebrospinal fluid shunt valve design in pediatric hydrocephalus. Neurosurgery 43[2]:294–303, 1998; discussion 303–295.)
Congenital Causes in Infants and Children
Approximately 55 percent of all cases of hydrocephalus are congenital. Primary aqueductal stenosis accounts for approximately 5 percent of congenital hydrocephalus, whereas aqueductal stenosis secondary to neoplasm, infection, or hemorrhage accounts for another 5 percent [Chi et al., 2005] . Primary aqueductal stenosis usually presents in infancy. Its morphology may be that of “forking” of the aqueduct, an aqueductal septum, “true” narrowing of the aqueduct, or X-linked aqueductal stenosis. Bicker–Adams syndrome is an X-linked hydrocephalus accounting for 7 percent of cases in males. It is characterized by stenosis of the aqueduct of Sylvius, severe mental retardation, and, in 50 percent, an adduction-flexion deformity of the thumb. Secondary aqueductal stenosis is due to gliosis secondary to intrauterine infection or germinal matrix hemorrhage [Hill and Rozdilsky, 1984].
Anatomical malformations frequently observed with idiopathic congenital hydrocephalus are associated with abnormalities of hindbrain development (see Chapter 24), and include Chiari malformations, Dandy–Walker malformation (DWM), and others [Stoll et al., 1992; Schrander-Stumpel and Fryns, 1998]. DWM is associated with atresia of the foramina of Luschka and Magendie, and affects 2–4 percent of newborns with hydrocephalus. About 50 percent of all patients with DWM develop hydrocephalus. The dilated fourth ventricle does not communicate effectively with the subarachnoid space. In patients with Chiari malformations, hydrocephalus may occur, with fourth ventricle outlet obstruction in Chiari type 1 malformation; it is commonly associated with myelomeningocele in the Chiari type 2 malformation. Hydrocephalus occurs in approximately 80–90 percent of patients with myelomeningocele; of these cases, 50 percent are obvious at birth [Tuli et al., 2003; Tulipan et al., 2003].
Acquired Causes in Infants and Children
Post-hemorrhagic hydrocephalus (PHH) occurs following IVH and can be related to prematurity, head injury, or rupture of a vascular malformation. Communicating hydrocephalus post-subarachnoid hemorrhage is more common in adults and is rarely seen in children. Over the past two decades, there has been remarkable improvement in the survival of extremely low birth weight infants; however, the most immature of these infants remain at increased risk for neonatal complications that potentially affect long-term neurodevelopmental outcome, including IVH (see Chapter 19). The risk for severe IVH varies inversely with gestational age, with an overall incidence of 7–23 percent [Lemons et al., 2001; Ment et al., 2005; Wilson-Costello et al., 2005]. Approximately one-third of extremely low birth weight infants with an IVH develop PHH, 15 percent of whom will require shunt insertion [Dykes et al., 1989; de Vries et al., 2002; Kazan et al., 2005].
Hydrocephalus after IVH is usually ascribed to fibrosing arachnoiditis, meningeal fibrosis, and subependymal gliosis, which impair flow and resorption of CSF. Recent experimental studies have suggested that acute parenchymal compression and ischemic damage, and increased parenchymal and perivascular deposition of extracellular matrix proteins – probably due, at least partly, to upregulation of transforming growth factor-beta (TGF-β) – are further important contributors to the development of the hydrocephalus. IVH is associated with damage to periventricular white matter and the damage is exacerbated by the development of hydrocephalus; combinations of pressure, distortion, ischemia, inflammation, and free radical-mediated injury are probably responsible [Cherian et al., 2004].
Clinical Characteristics
The clinical features of hydrocephalus depend on the age of the child at presentation and the time of onset in relation to closure of the cranial sutures. With the current advances in antenatal monitoring, the majority of congenital cases of hydrocephalus are diagnosed early (Figure 27-4), allowing for planned cesarian delivery in the moderate to severe cases where cephalopelvic disproportion is expected.

Fig. 27-4 Antenatal MRI showing fetal hydrocephalus.
Symmetrical enlargement of the ventricular system is readily seen in the brain of this fetus.
Genetics
Although commonly considered a single disorder, hydrocephalus is a collection of heterogeneous complex and multifactorial disorders. A growing body of evidence suggests that genetic factors play a major role in its pathogenesis [Zhang et al., 2006].
Congenital hydrocephalus may occur alone (nonsyndromic) or as part of a syndrome with other anomalies (syndromic). It is estimated that about 40 percent of individuals with hydrocephalus have a genetic etiology [Haverkamp et al., 1999]. The isolated (nonsyndromic) form of congenital hydrocephalus is a primary and major phenotype caused by a specific faulty gene. In syndromic forms, it is difficult to define the defective gene because of the association with other anomalies. A discussion of the genetics of all syndromic forms of hydrocephalus is beyond the scope of this chapter and is reviewed elsewhere in this section on developmental malformations. This chapter will focus mainly on the genetic disorders associated with isolated forms of hydrocephalus.
Autosomal-recessive, autosomal-dominant, X-linked recessive [Castro-Gago et al., 1996] and X-linked dominant [Ferlini et al., 1995] forms of hydrocephalus are recognized (Table 27-2).
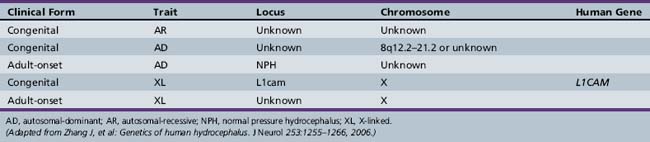
At least 43 gene mutations linked to hereditary hydrocephalus have been identified in animal models and humans. To date, nine genes associated with hydrocephalus have been identified in animal models, whereas only one such gene has been identified in humans: the hydrocephalus (X-linked) gene [Haverkamp et al., 1999]. X-linked hydrocephalus (HSAS1, OMIM) occurs in approximately 5–15 percent of congenital cases in which a genetic etiology is determined [Halliday et al., 1986; Haverkamp et al., 1999]. The gene responsible for X-linked human congenital hydrocephalus is at Xq28, encoding for L1CAM (L1 cell adhesion molecule) [Jouet et al., 1993]. Mutations are distributed over the functional protein domains. The exact mechanisms by which these mutations cause a loss of L1 protein function are still under investigation. L1CAM belongs to the immunoglobulin superfamily of neural cell adhesion molecules that is expressed in neurons and Schwann cells and appears to be essential for brain development and function. It plays a role in cell adhesion, axon growth and path-finding, and neuronal migration and myelination [Wong et al., 1995]. One of the possible mechanisms leading to the pathogenesis of hydrocephalus is the disruption of neural cell membrane proteins that play an important function during brain development.
The L1 disorders were initially described as different entities:
During the 1990s, it became clear that these disorders resulted from a mutation in a single gene, L1CAM [Fransen et al., 1995]. Some investigators have suggested that these separately named conditions be combined under the acronym CRASH (corpus callosum agenesis, retardation, adducted thumbs, shuffling gait, and hydrocephalus). The clinical phenotype is variable, even within a single family, but some genotype–phenotype correlations have been made [Fransen et al., 1998]. Hirschsprung’s disease (HSCR) is characterized by the absence of ganglion cells and the presence of hypertrophic nerve trunks in the distal bowel. There have been several reports of patients with X-linked hydrocephalus and HSCR with a mutation in the L1CAM gene. Decreased L1CAM may therefore be a modifying factor in the development of HSCR [Okamoto et al., 2004]. Congenital aqueductal stenosis can also be inherited as an autosomal-recessive disorder (OMIM 236635) [Lapunzina et al., 2002].
In general, the recurrence risk for congenital hydrocephalus excluding X-linked hydrocephalus is low. Empiric risk rates range from <1 to 4 percent [Burton, 1979], indicating the rarity of autosomal-recessive congenital hydrocephalus [Halliday et al., 1986; Chow et al., 1990; Haverkamp et al., 1999]. However, multiple human kindreds have been reported [Halliday et al., 1986; Teebi and Naguib, 1988; Haverkamp et al., 1999; Zhang et al., 2006]. The loci or genes for human autosomal-recessive congenital hydrocephalus have not yet been identified, but there is at least one locus for this trait. Furthermore, since there is heterogeneity among clinical phenotypes, there may be more genetic loci in human autosomal-recessive congenital hydrocephalus.
Two kindreds in which congenital hydrocephalus was transmitted in an autosomal-dominant fashion have been reported. One was associated with aqueductal stenosis but was not associated with mental retardation or pyramidal tract dysfunction, which is in contrast to X-linked or recessive congenital hydrocephalus with HSAS, in which these abnormalities are common [Verhagen et al., 1998]. The other kindred in which hydrocephalus developed had an 8q12.2–q21.2 microdeletion. This trait was also transmitted in an autosomal-dominant pattern [Vincent et al., 1994].
Hydrocephalus has been observed in many mammals [Zhang et al., 2006]. Animal hydrocephalus models have many histopathological similarities to humans and can be used to understand the genetics and pathogenesis of these disorders. It has been well documented that the majority of cases of congenital hydrocephalus in animal models occur on a genetic basis with specific mapping and identification of different loci. The development and progression of congenital hydrocephalus constitute a dynamic process that is not yet well understood. It is probably the consequence of abnormal brain development and perturbed cellular function, which emphasizes the important roles that congenital hydrocephalus genes play during brain development. It is thought that it may develop at an important and specific embryonic time period of neural stem-cell proliferation and differentiation [Zhang et al., 2006].
Hydrocephalus may also be caused by alterations in ependymal cell function [Takano et al., 1993; Wagner et al., 2003]. The protein of the axonemal heavy chain 5 gene (Mdnah5), dynein, is specifically expressed in ependymal cells, and is essential for ultrastructural and functional integrity of ependymal cilia. In Mdnah5-mutant mice, lack of ependymal flow causes closure of the aqueduct and subsequent formation of triventricular hydrocephalus during early postnatal brain development. The higher incidence of aqueductal stenosis and hydrocephalus formation in patients with disorders associated with ciliary defects supports the relevance of this novel mechanism in humans [Ibanez-Tallon et al., 2004].
Hydrocephalus may be caused by changes in the development and function of mesenchymal cells. In murine embryonic brain, the Msx1 gene is expressed along the dorsal midline. This regulatory gene is involved in epithelio-mesenchymal interactions in limb formation and organogenesis. In homozygous Msx1 mutants, there is absence or malformation of the posterior commissure and of the subcommissural organ, collapse of the cerebral aqueduct, and hydrocephalus. About one-third of the heterozygous mutants also develop hydrocephalus, suggesting that the phenotype may be determined by the Msx1 gene dosage during a critical developmental period [Ramos et al., 2004]. An example of this is the autosomal-recessive congenital hydrocephalus mouse model in which a truncated protein lacking the DNA-binding domain of the forkhead/winged helix gene, Mf1, was generated. Mesenchymal cells from Mf1lacZ embryos differentiate poorly into cartilage in micromass culture, and the differentiation of arachnoid cells in meninges of the mutant mice also was abnormal. Human patients with deletions in the region containing human Mf1 homolog FREAC3 were also found to develop multiple developmental disorders, including hydrocephalus [Kume et al., 1998].
Another mechanism for development of hydrocephalus may be associated with changes in growth factor signaling [Fukumitsu et al., 2000]. In mouse models, severe hydrocephalus has been observed in transgenic mice overexpressing TGF-β1, an important cytokine and growth-signaling molecule, in astrocytes [Galbreath et al., 1995]. In mouse models, fibroblast growth factor-2 (FGF-2) seems to play a predominant role in the proliferation of neuronal precursors and in neuronal differentiation in the developing cerebral cortex, even at relatively late stages of brain neurogenesis [Ohmiya et al., 2001].
Hydrocephalus may also be caused by disruption of the extracellular matrix (ECM). In the TGF-β1 overexpression mouse model, the changing expressions of a remodeling protein, matrix metalloproteinase-9 (MMP-9), and its specific inhibitor, tissue inhibitor of metalloproteinases-1 (TIMP-1), were found to be important factors in the spontaneous development of hydrocephalus by altering the ECM environment [Crews et al., 2004]. Furthermore, increased expression of cytokines such as TGF-β1 might reciprocally play an important role by disrupting vascular ECM remodeling, promoting hemorrhage and altering reabsorption of CSF. In another mouse model, ablation of the non-muscle myosin heavy chain II-B (NMHC-B) results in severe hydrocephalus with enlargement of the lateral and third ventricles. These defects may be caused by abnormalities in the cell adhesive properties of neuroepithelial cells, and suggest that NMHC-B is essential for early and late developmental processes in the mammalian brain [Tullio et al., 2001].
Other genetic mutations identified in animal models of hydrocephalus include those coding α-SNAP protein, which is essential for apical protein localization and cell fate determination in neuroepithelial cells [Chae et al., 2004]. α-SNAP plays a key role in a wide variety of membrane fusion events in eukaryotic cells, a function which is required for transport of molecules to inter- and intracellular compartments and for intercellular communication.
Neuroimaging
Cranial Ultrasound
In the presence of an open anterior fontanel in neonates and infants, transfontanellar ultrasound can visualize ventricular anatomy. This is by far the quickest, least expensive, and most convenient method to demonstrate ventricular enlargement. Ultrasonography is useful for the diagnosis of intrauterine hydrocephalus and communicating and noncommunicating PHH. Serial imaging after injury or in children with developmental malformations can be used to follow the evolution and possible development or progression of hydrocephalus (see Chapter 11).
Ventricular width measured from the midline to the lateral border of the lateral ventricle in the midcoronal view is the measurement with the least interobserver variability, and centiles for gestational age have been compiled [Levene, 1981]. Sedation is not required during acquisition of ultrasound images, and the procedure can be repeated frequently without any adverse effects. Limitations of cranial ultrasonography (see Chapter 11) are that it requires operator experience and often cannot establish a cause for the hydrocephalus.
Magnetic Resonance Imaging
There are no reliable measurement values that can confirm or exclude the presence of hydrocephalus in a single series. Serial imaging demonstrating an increase in ventricular size may be required in equivocal cases. Conversely, an apparently normal ventricular size cannot exclude active hydrocephalus in a patient with a pre-existing shunt. A number of methods have been used to attempt to define hydrocephalus quantitatively on CT or MRI studies [leMay and Hochberg, 1979]. Hydrostatic hydrocephalus is suggested when either:
Diagnosis
Modern ultrasonography and, since the late 1980s, fetal MRI have significantly improved the ability to detect ventricular enlargement as a result of hydrocephalus in utero. Antenatal ultrasound and MRI provide reasonably detailed fetal brain anatomy and can detect malformations (at 17–21 weeks) and fetal ventriculomegaly (at 8–21 weeks) [Benacerraf and Birnholz, 1987; Oi et al., 1998]. Normative data for ventricular size allow serial investigation during gestation [Rich et al., 2007].
Anatomical ventriculomegaly is not sufficient to diagnose hydrocephalus. When diagnosing hydrocephalus in neonates or infants, it is essential to establish that there is a truly abnormal rate of skull growth. Records of head circumference (HC) measurements and its comparison with body weight and length centile charts are an integral part of postnatal follow-up of any child. Head circumference must be recorded and plotted on an accepted growth curve chart, with the patient’s exact age (see Chapter 4 for standardized charts). In the presence of hydrocephalus, any of the following may be observed: HC more than 2 SD above normal or disproportionate to body length or weight, accelerated growth crossing centile curves, or continued head growth of more than 1.25 cm per week.
Differential Diagnosis
Benign extracranial hydrocephalus is a condition in infants and children with enlarged subarachnoid spaces accompanied by increasing head circumference with normal or mildly dilated ventricles. This condition is also known as “benign subdural collections of infancy” or “pericerebral CSF collections.” This disorder has been postulated by some to be a variant of communicating hydrocephalus, but tends to run a benign course and stabilize by 12–18 months of age [Ment et al., 1981; Barlow, 1984; Alvarez et al., 1986]. Close serial monitoring of head circumference and serial imaging with CT or MRI are recommended to monitor for ventriculomegaly. Shunting is rarely, if ever, required.
Pathology
The precise pathologic features of hydrocephalus vary, depending on the age of onset, the rate of ventricular enlargement, and the degree of ventriculomegaly. Typically, elevated CSF pressure initially enlarges the frontal horns of the lateral ventricles, followed by enlargement of the entire ventricular system above the site of obstruction. Hydrocephalus is associated with flattening and destruction of the ventricular ependymal lining, as well as edema and necrosis of the periventricular white matter [Weller and Shulman, 1972]. Periventricular glial cells proliferate, resulting in a layer of reactive gliosis. The pathologic findings may be a result of reduced blood flow to the white matter, causing hypoxic injury or toxicity to the white matter due to build-up of waste products not removed appropriately because of changes in the extracellular matrix [Del Bigio, 2004]. In PHH, high concentrations of proinflammatory cytokines [Savman et al., 2002], free iron, and hypoxanthine [Bejar et al., 1983; Savman et al., 2001], which can generate highly reactive radicals, have been measured in the CSF.
Increasing pressure and ventricular enlargement are associated with necrosis of brain parenchyma. Cerebral white matter is more vulnerable to destruction than gray matter in the presence of progressive hydrocephalus [Rubin et al., 1972]. The corpus callosum may also be preferentially affected, with evidence of transcallosal swelling, thickening, or demyelination [Spreer et al., 1996; Suh et al., 1997]. These effects do not appear to be associated with cognitive changes nor neuropsychologic evidence of callosal disconnection.
Management
Shunts
Ventriculoperitoneal (VP) shunting is the most accepted and most commonly used initial procedure. The lateral ventricle is the usual proximal location. The advantage of this shunt is that the need to lengthen the catheter with growth may be obviated by using a long peritoneal catheter. In patients with contraindications to ventriculoperitoneal shunting, such as the presence of peritonitis or peritoneal adhesions following abdominal surgery, ventriculopleural or ventriculoatrial shunts can be used. The latter is also called a “vascular shunt,” and shunts the cerebral ventricles through the jugular vein and superior vena cava into the right cardiac atrium. This shunt requires repeated lengthening in a growing child [Vernet et al., 1995].
Shunts cause CSF to flow unidirectionally under the aegis of a valve system. Pressures required to overcome valve resistance are preset and can be used in patients with different pressure requirements. The first spring, ball, and diaphragm valves were superseded by a number of innovative valve designs, including antisiphon devices [Portnoy et al., 1973], horizontal–vertical valves [Drake and Sainte-Rose, 1995], flow-controlled valves [Sainte-Rose et al., 1987], adjustable valves [Black et al., 1994], and others, developed with the hope that they would decrease shunt malfunction. Although each new valve was heralded as a significant advance that, functionally, was more physiological, with improved results in uncontrolled studies, prospective and randomized trials were all negative. Use of an antisiphon device developed in the 1970s improved the success rate of shunting by reducing overdrainage of CSF but was also associated with complications [Portnoy et al., 1973; DaSilva and Drake, 1991]. Orbis-Sigma valves or Delta valves have flow/pressure characteristics that reduce overdrainage [Drake and Kestle, 1996]. However, use of these systems did not reduce time to first shunt failure [Kestle et al., 2000]. When standard valves, a novel flow-controlled valve (Orbis Sigma, Cordis, Miami, Florida), and an updated antisiphon valve (Delta Valve; Medtronic PS Medical, Goleta, California) were compared in a prospective, randomized trial, there was no difference [Drake et al., 1998; Kestle et al., 2000]. Programmable valves are an alternative system that allows multiple pressure settings to adjust the valve for over- or underdrainage, avoiding a shunt revision for that reason; as the child grows, adjustments in flow can be made. Study of the time to shunt failure with these valves, however, did not show any difference from standard valves [Pollack et al., 1999]. A single-arm prospective study of an adjustable Delta valve, the Strata valve, suggested its failure rate was no different than any other previously studied valves [Kestle and Walker, 2005]. Furthermore, when ventricular size was measured pre- and postoperatively, comparing three very different valves, there was no difference over time, which indicates that the valves were not performing as the engineers had predicted [Tuli et al., 1999]. Studies on varying ventricular catheter position and endoscope-assisted catheter placement also did not demonstrate any improvement in outcome [Kestle et al., 2003].
Endoscopic Third Ventriculostomy
Endoscopic third ventriculostomy is an alternative to CSF shunting in appropriate patients [Drake, 2007; Jones et al., 1996; Barlow and Ching, 1997]. This creates an outlet in the floor of the third ventricle. A ventriculoscope is introduced into the lateral ventricle via a frontal horn approach, passed into the third ventricle through the foramen of Monro, and the floor of the third ventricle is then fenestrated just anterior to the mamillary bodies, allowing CSF to bypass any obstruction in the CSF pathway and be reabsorbed by the arachnoid villi.
Ventriculostomy is particularly useful in patients with congenital aqueductal stenosis that requires CSF diversion, and in selected patients with intracranial cysts or loculated cystic ventricular CSF collections [Drake, 2007]. The ventricles do not decrease in size to the degree they do with a ventriculoperitoneal shunt, so determining the function of a ventriculostomy by neuroimaging can be difficult [Kestle, 2003].
Complications of endoscopic third ventriculostomy are not infrequent and may be serious; they include as perforation of the basilar artery [McLaughlin et al., 1997]. CSF leak, meningitis, hypothalamic injury, and cranial nerve injury have all been reported [Teo et al., 1996]. The overall surgical complication rate appears to be approximately 10–15 percent. A rare and initially unrecognized complication is late rapid clinical deterioration. When this happens, patients initially appear to be doing well after the procedure, but begin to complain of headache and then rapidly deteriorate. Without immediate access to neurosurgery, they lapse into unconsciousness, and most die. A recent report compiled 15 such cases from the literature and around the world [Drake et al., 2006]. Thirteen patients died and, in all who had an autopsy or repeat endoscopic third ventriculostomy performed, the opening in the floor of the third ventricle was closed.
Management of Hydrocephalus in Preterm Infants
Shunting of these infants frequently is unnecessary and hazardous [Brockmeyer et al., 1989]. The course of the ventricular dilatation is variable; patients may demonstrate an arrest or reduction in ventriculomegaly with no treatment. Complications of shunting in this age group include a collapsed cortical mantle, subdural hemorrhage, marked cerebral conformational changes, intraparenchymal hemorrhage, or hardware erosion through the skin [Bass et al., 1995].
In view of the high morbidity for shunt placement in premature infants, multiple other interventions have been used. Studying the effectiveness of these interventions is challenging because of the variable course of post-hemorrhagic ventricular dilatation and the multiple other insults that occur in these infants. Acetazolamide and furosemide have been used in the past to reduce CSF output. Results of a larger, multicenter, randomized controlled trial, however, did not support the use of diuretics [Kennedy et al., 2001]. The principal secondary outcome measure of death in the first year of life or neurodevelopmental disability was significantly higher in infants receiving diuretic therapy. In addition, nephrocalcinosis developed in 25 percent of infants in the diuretic treatment group.
Repeated lumbar punctures to remove CSF are a common method of attempting to prevent ventricular dilatation after IVH, and as a therapy to manage symptomatic hydrocephalus. The theory is that removal of CSF containing blood and protein might allow the normal resorption of CSF to be restored. There have been two trials of repeated lumbar punctures in preventing development of hydrocephalus [Mantovani et al., 1980; Anwar et al., 1985], and two trials of infants with IVH and progressive ventricular dilatation that examined the effect of lumbar punctures [Dykes et al., 1989], or lumbar punctures or ventricular tapping [Ventriculomegaly Trial Group, 1990]. None of the studies demonstrated that CSF tapping decreased the need for shunting or decreased the likelihood of death or disability. The Ventriculomegaly Trial found a higher incidence of CSF infections in those infants who received CSF taps. A Cochrane review of this topic [Whitelaw, 2005] concluded that CSF tapping for infants at risk of or having developed PHH could not be recommended. However, the reviewers additionally stated that, despite the lack of evidence, CSF tapping should still be considered for symptomatic raised intracranial pressure. Likewise, despite the lack of evidence, some experts continue to recommend the use of CSF tapping in specific situations.
Intraventricular fibrinolytic agents to prevent the development of hydrocephalus after IVH have been found to be ineffective [Whitelaw, 2001]. Other options in patients with rapidly progressive ventriculomegaly who are too small for ventriculoperitoneal shunt placement include placement of an external ventricular drain, a subgaleal ventricular reservoir, or a subgaleal shunt. These devices can be used until the infant is large enough for permanent shunt placement. In addition, 25 percent of patients with these devices have arrest of their hydrocephalus, and do not require permanent shunt placement [Garton and Piatt, 2004].
Complications
The overall failure rate of shunts is 40 percent in the first year after placement [Drake et al., 1998; Pollack et al., 1999; Kestle et al., 2000]. Common reasons for failure are obstruction, infection, mechanical failure, overdrainage, loculated ventricles, and abdominal complications. Shunt malfunction caused by disconnection, kinking, or obstruction of the tubing results in typical signs and symptoms of increased intracranial pressure, or may present with more subtle symptoms. Fever may occur if infection is present, but shunt infection occasionally can be asymptomatic. In neonates, shunt malfunction manifests as alteration of feeding, irritability, vomiting, fever, lethargy, somnolence, and a bulging fontanel. Older children and adults present with headache, fever, vomiting, and meningismus. With VP shunts, abdominal pain may occur. Staphylococcus epidermidis and Staphylococcus aureus are the most common causes of shunt-related infection, which usually occurs within 2 months of shunt placement.
The infection rate varies but can be up to 10 percent [Drake, 2008]. Organisms that ordinarily are nonpathogenic may cause infection in the presence of shunt tubing in the ventricles, body cavities, or circulation. Whenever suppurative infection is suspected, appropriate blood and CSF specimens for culture must be obtained, the organism isolated, and sensitivities determined to facilitate effective antimicrobial therapy. Most shunts can be tapped to obtain CSF specimens and to assess CSF dynamics. If infection leads to impairment of the shunt mechanism, removal and replacement usually are required; temporary insertion of an external ventriculostomy may be necessary until the infection is controlled. In one series of patients whose shunts were tapped, normal CSF values correlated with an overall incidence of complications of 39.2 percent, whereas abnormal CSF values were correlated with a complication rate of 90.9 percent [Caldarelli et al., 1996]. Rates of infective complications were 2.7 percent in the patients with normal CSF and 77.3 percent in patients with abnormal CSF.
Other complications of VP shunts include peritonitis, CSF ascites, inguinal hernia, intra-abdominal cysts, intracranial granulomas, gastrointestinal obstruction, migration of the shunt within the peritoneal cavity, headache, and perforation of abdominal viscera [Sgouros et al., 1995; Caldarelli et al., 1996]. Abdominal pseudocysts manifest with nausea, vomiting, and abdominal distention and pain; abdominal ultrasonography may demonstrate the cysts [Hann et al., 1985]. Thromboembolic phenomena, cardiac dysrhythmias, cardiovascular perforation, endocarditis, catheter embolization, pulmonary hypertension and thromboembolism, and immune complex shunt nephritis are special complications of ventriculoatrial shunts [Lam and Villemure, 1997]. Correction of long-standing hydrocephalus may cause subdural hematomas.
The “slit ventricle syndrome” (SVS; Figure 27-5) is defined as severe, life-modifying headaches in patients with shunts for the treatment of hydrocephalus and normal or smaller than normal ventricles [Rekate, 2008]. There are five potential pathophysiologies that may be involved in this condition. These pathologies are defined by intracranial pressure measurement as severe intracranial hypotension analogous to spinal headaches, intermittent obstruction of the ventricular catheter, intracranial hypertension with small ventricles and a failed shunt (normal-volume hydrocephalus), intracranial hypertension with a working shunt (cephalocranial hypertension), and shunt-related migraine [Rekate, 2008]. Management of SVS requires an understanding of the specific pathogenesis of the problem in individual patients, whether based on monitoring of intracranial pressure or observation at the time of shunt failure or symptoms. Overdrainage syndromes are managed with valve upgrades and the addition of devices that retard siphoning. Other management options used for this condition are subtemporal decompression, calvarial expansion or shunting devices that access the cortical subarachnoid space, such as lumboperitoneal shunts, or shunts involving the cisterna magna.
Prognosis
Before the 1950s, the outlook of untreated hydrocephalus was extremely poor. Some 49 percent of patients had died by the end of the 20-year observation period and only 38 percent of survivors had an IQ greater than 85 [Laurence and Coates, 1962]. The development of satisfactory shunting substantially improved the outlook of children with hydrocephalus but brought its own set of problems and complications. Most children with hydrocephalus will require multiple shunt revisions. Shunt dependence carries a 1 percent per year mortality [Sainte-Rose et al., 1991; Iskandar et al., 1998]. Another series of 907 patients reported a mortality rate of 12 percent at 10 years from the time of initial shunt insertion [Tuli et al., 2004], with the main risk factor for death being a history of shunt infection. Shunt-related complications, including death, have been reported to be greater in patients with myelomeningocele than in those who required shunt placement for the treatment of other conditions [Iskandar et al., 1998; Tuli et al., 2003].
The neurologic and intellectual disabilities among patients with hydrocephalus depend on many factors, including etiology and degree of hydrocephalus, thickness of the cortical mantle and corpus callosum [Fletcher et al., 1992], requirement for a shunt, and presence of other brain anomalies [Dennis et al., 1981]. Associated conditions, such as IVH, CNS infection [Tuli et al., 2004], and hypoxia, may dictate the ultimate prognosis more than the hydrocephalus.
A series of 233 patients with congenital hydrocephalus evaluated for longer than 20 years reported a mortality rate of 13.7 percent [Lumenta and Skotarczak, 1995]. The average number of shunt revisions was 2.7. In this series, 115 patients underwent psychological evaluation; approximately 63 percent showed normal performance, whereas 30 percent had mild retardation, and 7 percent had severe retardation. Another study found that children with congenital hydrocephalus were less likely to require special education placement (29 percent) than those in whom hydrocephalus was due to meningitis (52 percent) or IVH (60 percent) [Casey et al., 1997].
Epilepsy also is more prevalent in patients with hydrocephalus, and complications of shunt surgery appear to play a relatively minor role in its development [Piatt and Carlson, 1996].
Intellectual sequelae include significant scatter among Wechsler Intelligence Scale for Children-Revised (WISC-R) subtest scores, often with greater impairment of performance and motor tasks, as well as of nonverbal compared with verbal skills [Brookshire et al., 1995]. Normal intellectual function is present in 40–65 percent of patients who received appropriate treatment [Dennis et al., 1981]. The probability of normal intelligence is enhanced if shunts are placed early and proper function is maintained. A study of 99 children ranging in age from 6 to 13 years with shunted or arrested hydrocephalus demonstrated a close correlation between the area of the corpus callosum and nonverbal cognitive skills and motor abilities [Fletcher et al., 1996]. Behavioral problems also are more common in children with hydrocephalus, irrespective of etiology [Fletcher et al., 1995].
Intracranial Arachnoid Cysts
Definition
Intracranial arachnoid cysts are benign, nongenetic developmental cysts that contain spinal fluid and occur within the arachnoid membrane [Gosalakkal, 2002]. The mechanism of formation during embryogenesis is uncertain [Hirano and Hirano, 2004; Naidich et al., 1985–1986]. Several mechanisms could account for the enlargement of these cysts, including secretion by the cells forming the cyst walls, a unidirectional valve, or liquid movements secondary to pulsations of the veins [Gosalakkal, 2002]. The cysts occur in proximity to arachnoid cisterns, most often in the sylvian fissure (Table 27-3). Common neurologic features are headache, seizures, hydrocephalus, focal enlargement of the skull, and signs and symptoms of elevated intracranial pressure and developmental delay, as well as specific signs or symptoms resulting from neural compression. Some arachnoid cysts remain asymptomatic [Mason et al., 1997]. Progressive enlargement and intracystic or subdural hemorrhage are potential complications. Suprasellar arachnoid cysts may produce neuroendocrine dysfunction, hydrocephalus, and optic nerve compression. Posterior fossa cysts are now more frequently recognized with the use of MRI and CT, and frequently require surgical treatment [Domingo and Peter, 1996].
| Location | Percentage of Cysts |
|---|---|
| Sylvian fissure | 49 |
| Cerebellopontine angle | 11 |
| Quadrigeminal area | 10 |
| Vermian area and sellar-suprasellar area | 9 |
| Interhemispheric fissure | 5 |
| Cerebral convexity | 4 |
| Clival area | 3 |
(Modified from Rengachary SS, Watanabe I. Ultrastructure and pathogenesis of intracranial cysts. J Neuropathol Exp Neurol 40:61, 1981.)
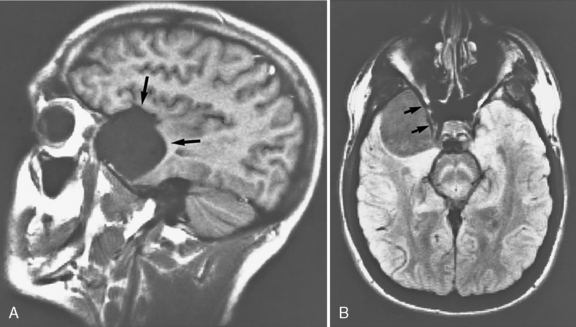
In a series of 61 children with arachnoid cysts, about 53 percent of cases were diagnosed before age 1 year; 42 percent were supratentorial and 46 percent infratentorial [Pascual-Castroviejo et al., 1991]. Macrocephaly was the presenting symptom in 72 percent, and associated features included cranial asymmetry in 39 percent, aqueductal stenosis in 16 percent, and agenesis of the corpus callosum in 13 percent. Developmental delay was a common finding. Skull radiographs may suggest the diagnosis; CT or MRI is the definitive diagnostic procedure [Weiner et al., 1987] (Figure 27-6). Injection of contrast medium into the cyst to document communication with the ventricular system is seldom necessary.
Clinical Characteristics
Sylvian Fissure/Middle Cranial Fossa
Nearly two-thirds of pediatric arachnoid cysts are located in the sylvian fissure/middle cranial fossa [Gosalakkal, 2002]. They may increase in volume, opening the fissure and exposing the middle cerebral artery. This exposure may result in compression and underdevelopment of the anterior superior surface of the temporal lobe. The origins of these cysts have been the subject of debate since they were first described. Controversy remains concerning whether they originate directly from the meninges adjacent to the temporal pole, or whether partial agenesis of the temporal lobe favors secondary formation of the cyst. Headaches are the most common presenting symptom; proptosis, contralateral motor weakness, and seizures also may occur. In 10 percent of children, developmental delay may be present. Treatment, as discussed later, depends on clinical symptoms. Recent studies have suggested some cognitive improvement after surgical treatment [Raeder et al., 2005]. Children with bitemporal arachnoid cysts also should be evaluated for the possibility of glutaricaciduria type 1 [Lutcherath et al., 2000]. Bitemporal arachnoid cysts also have been reported in children with neurofibromatosis [Martinez-Lage et al., 1993].
Sellar Region
Both suprasellar and intrasellar cysts can occur in children. Suprasellar cysts can cause third ventricular obstructive hydrocephalus at the level of the foramen of Monro, and may be associated with visual impairment and endocrine dysfunction [Mohn et al., 1999]. Progressive head enlargement, growth retardation, developmental delay, and bitemporal hemianopsia have all been described [Gosalakkal, 2002]. A bobble-head doll syndrome, with involuntary head movements secondary to increased pressure on the third ventricle and dorsomedial thalamic nuclei, is responsible [Fioravanti et al., 2004]. Endoscopic surgical approaches are now preferred.
Posterior Fossa
Arachnoid cysts of the posterior fossa are uncommon and must be differentiated from other cystic malformations of the posterior fossa, such as the Dandy–Walker malformation [Gosalakkal, 2002]. Macrocrania and raised intracranial pressure are frequently observed. Cerebellar cysts demonstrate nystagmus and other cerebellar signs. Other rare manifestations reported include cervical spinal cord compression, which may improve after posterior fossa cystoperitoneal shunting or endoscopic surgery. In such patients, gait disturbances and headache are commonly seen. Posterior fossa cysts may be very large [Lancon and Ellis, 2004] and also can occur in families [Sinha and Brown, 2004].
Complications
Epilepsy
Anecdotal studies suggest a relation between seizure reduction and removal of arachnoid cysts [D’Angelo et al., 1999]. The relation between the presence of arachnoid cysts and occurrence of seizures when the intracranial pressure is normal is uncertain, however, and differences in outcomes, whether patients are managed medically or surgically, are similar [Koch et al., 1995]. In addition, interictal and ictal EEG may not correspond to the site of the arachnoid cyst, raising the question of whether the presence of an EEG abnormality is incidental.
Subdural Hematoma and Hygroma
Subdural hematomas and hygromas are infrequently encountered complications of arachnoid cysts of the middle cranial fossa and are particularly rare with cysts in other regions [Donaldson et al., 2000]. Minor head trauma has been suggested to be a precipitating factor. Arachnoid cysts of the middle cranial fossa were found in 2.43 percent of patients with chronic subdural hematomas or hygromas in one report [Parsch et al., 1997].
Neuropsychiatric Disorders
Attention-deficit hyperactivity disorder, speech delay, and developmental delay have been found in association with arachnoid cysts, particularly in the temporal lobe, but a causal relation remains uncertain [Millichap, 1997]. Mental impairment and developmental delay have been associated with large arachnoid cysts [Gosalakkal, 2002], and the presence of cysts and developmental delay may be part of a common developmental process. Recent studies have suggested improvements in cognition after surgical treatment [Raeder et al., 2005]. The increased incidence of arachnoid cysts in conditions such as Down syndrome, mucopolysaccharidosis, schizencephaly, and neurofibromatosis suggests a higher incidence in children with underlying abnormalities of the brain [Gosalakkal, 2002]. Aphasia, including that of Landau–Kleffner syndrome, also has been associated with the presence of left sylvian arachnoid cysts. Even in patients in whom CT and MRI failed to reveal mass effect, positron emission tomography (PET) has demonstrated hypometabolism in speech areas. Postoperative improvement in PET studies corresponded to improvement in vocabulary [De Volder et al., 1994].
Management
When symptoms warrant, surgical intervention to decompress the cyst, including endoscopic management or shunting procedures, is required [Abbott, 2004; Germano et al., 2003; Godano et al., 2004; Raffel and McComb, 1988]. Arachnoid cysts may occur with or without hydrocephalus. The success rate of fenestration is higher in those patients without hydrocephalus (i.e., 73 percent required no additional treatment) than in hydrocephalus patients (32 percent) [Fewel et al., 1996]. About 12 percent of patients with hydrocephalus treated with fenestration alone may require a cystoperitoneal shunt. In general, cyst fenestration should be the primary procedure in patients without hydrocephalus. If hydrocephalus is present, cyst fenestration is still recommended, but a VP shunt should be placed if hydrocephalus is marked or after fenestration if the hydrocephalus is progressive [Fewel et al., 1996]. Congenital intraspinal cysts also can occur and require surgical evaluation [Chang, 2004].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abbott R. The endoscopic management of arachnoidal cysts. Neurosurg Clin N Am. 2004;15:9.
Alksne J.F., Lovings E.T. Functional ultrastructure of the arachnoid villus. Arch Neurol Clin. 1972;27:371.
Alvarez L.A., Maytal J., et al. Idiopathic external hydrocephalus: Natural history and relationship to benign familial macrocephaly. Pediatrics. 1986;77:901.
Anwar M., Kadam S., et al. Serial lumbar punctures in prevention of post-hemorrhagic hydrocephalus in preterm infants. J Pediatr. 1985;107:446.
Barlow C. CSF dynamics in hydrocephalus. Brain Dev. 1984;6(2):119-127.
Barlow P., Ching H.S. An economic argument in favour of endoscopic third ventriculostomy as a treatment for obstructive hydrocephalus. Minim Invasive Neurosurg. 1997;40:37.
Bass T., White L.E., et al. Rapid decompression of congenital hydrocephalus associated with parenchymal hemorrhage. J Neuroimaging. 1995;5:249.
Bejar R., Saugstad O., et al. Increased hypoxanthine concentrations in cerebrospinal fluid of infants with hydrocephalus. J Pediatr. 1983;103:44-48.
Benacerraf B., Birnholz J. The diagnosis of fetal hydrocephalus prior to 22 weeks. J Clin Ultrasound. 1987;15:531-536.
Beni-Adani L., Biani N., et al. The occurrence of obstructive vs absorptive hydrocephalus in newborns and infants: relevance to treatment choices. Childs Nerv Syst. 2006;22(12):1543-1563.
Bering E. Circulation of the cerebrospinal fluid. Demonstration of the choroid plexuses as the generator of flow of fluid and ventricular enlargement. J Neurosurg. 1962;19:405-413.
Blackburn B., Fineman R. Epidemiology of congenital hydrocephalus in Utah, 1940–1979: report of an iatrogenically related “epidemic”. Am J Med Genet. 1994;52:123-129.
Black P.M., Hakim R., et al. The use of the Codman-Medos Programmable Hakim valve in the management of patients with hydrocephalus: Illustrative cases. Neurosurgery. 1994;34:1110-1113.
Boaz J.C., Edwards-Brown M.K. Hydrocephalus in children: neurosurgical and neuroimaging concerns. Neuroimaging Clin N Am. 1999;9:73-91.
Bondurant C.P., Jimenez D.F. Epidemiology of cerebrospinal fluid shunting. Pediatr Neurosurg. 1995;23:254.
Brockmeyer D.L., Wright L.C., et al. Management of posthemorrhagic hydrocephalus in the low-birth-weight preterm neonate. Pediatr Neurosci. 1989;15:302.
Brookshire B.L., Fletcher J.M., et al. Verbal and nonverbal skill discrepancies in children with hydrocephalus: A five-year longitudinal follow-up. J Pediatr Psychol. 1995;20:785.
Burton B.K. Recurrence risks for congenital hydrocephalus. Clin Genet. 1979;16:47-53.
Caldarelli M., Di Rocco C., et al. Shunt complications in the first postoperative year in children with meningomyelocele. Childs Nerv Syst. 1996;12:748.
Casey A.T., Kimmings E.J., et al. The long-term outlook for hydrocephalus in childhood. A ten-year cohort study of 155 patients. Pediatr Neurosurg. 1997;27:63.
Castro-Gago M., Alonso A., et al. Autosomal recessive hydrocephalus with aqueductal stenosis. J Childs Nerv Syst. 1996;12:188.
Chae T.H., Kim S.H., et al. The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat Genet. 2004;36:264-270.
Chahlavi A., El-Babaa S.K., et al. Adult-onset hydrocephalus. Neurosurg Clin N Am. 2001;12:753-760.
Chakraborty A., Crimmins D., et al. Toward reducing shunt placement rates in patients with myelomeningocele. J Neurosurg Pediatr. 2008;1:361-365.
Chang I.C. Surgical experience in symptomatic congenital intraspinal cysts. Pediatr Neurosurg. 2004;40:165.
Cherian S., Whitelaw A., et al. The pathogenesis of neonatal post-hemorrhagic hydrocephalus. Brain Pathol. 2004;14(3):305-311.
Chi J., Fullerton H., et al. Time trends and demographics of deaths from congenital hydrocephalus in children in the United States: National Center for Health Statistics data, 1979 to 1998. J Neurosurg (Pediatrics 2). 2005;103:113-118.
Chow C.W., McKelvie P.A., et al. Autosomal recessive hydrocephalus with third ventricle obstruction. Am J Med Genet. 1990;35:310-313.
Crews L., Wyss-Coray T., et al. Insights into the pathogenesis of hydrocephalus from transgenic and experimental animal models. Brain Pathol. 2004;14:312-316.
D’Angelo V., Gorgoglione L., Catapano G. Treatment of symptomatic intracranial arachnoid cysts by stereotactic cyst-ventricular shunting. Stereotact Funct Neurosurg. 1999;72:62.
DaSilva M.C., Drake J.M. Complications of cerebrospinal fluid shunt antisiphon devices. Pediatr Neurosurg. 1991;17:304.
Del Bigio M.R. Cellular damage and prevention in childhood hydrocephalus. Brain Pathol. 2004;14:317.
Dennis M., Fitz C.R., et al. The intelligence of hydrocephalic children. Arch Neurol Clin. 1981;38:607.
De Volder A.G., Michel C., Thauvoy C., et al. Brain glucose utilisation in acquired childhood aphasia associated with a sylvian arachnoid cyst: Recovery after shunting as demonstrated by PET. J Neurol Neurosurg Psychiatry. 1994;57:296.
de Vries L.S., Liem K.D., et al. Early versus late treatment of post haemorrhagic ventricular dilatation: results of a retrospective study of five neonatal intensive care units in the Netherlands. Acta Paediatr. 2002;91(2):212-217.
Domingo Z., Peter J. Midline developmental abnormalities of the posterior fossa: Correlation of classification with outcome. Pediatr Neurosurg. 1996;24:111.
Donaldson J.W., Edwards-Brown M., et al. Arachnoid cyst rupture with concurrent subdural hygroma. Pediatr Neurosurg. 2000;32:137.
Drake J., Chumas P., et al. Late rapid deterioration after endoscopic third ventriculostomy: additional cases and review of the literature. J Neurosurg. 2006;105(Suppl 2):118-126.
Drake J.M. Endoscopic third ventriculostomy in pediatric patients: the Canadian experience. Neurosurgery. 2007;60(5):881-886. discussion 881–886
Drake J.M. The surgical management of pediatric hydrocephalus. Neurosurgery. 2008;62(Suppl 2):633-640. discussion 640–632
Drake J.M., Kestle J. Rationale and methodology of the Multicenter Pediatric Cerebrospinal Fluid Shunt Design trial. Pediatric Hydrocephalus Treatment Evaluation Group. Childs Nerv Syst. 1996;12:434.
Drake J.M., Kestle J.R., et al. Randomized trial of cerebrospinal fluid shunt valve design in pediatric hydrocephalus. Neurosurgery. 1998;43(2):294-303. discussion 303–295
Drake J.M., Sainte-Rose C. The Shunt Book. New York: Blackwell Scientific; 1995.
Dykes F.D., Dunbar B., et al. Posthemorrhagic hydrocephalus in high-risk preterm infants: natural history, management, and long-term outcome. J Pediatr. 1989;114(4 pt 1):611-618.
El Awad M. Infantile hydrocephalus in the south-western region of Saudi Arabia. Ann Trop Paediatr. 1992;12:335-338.
Feinberg D.A., Mark A.S. Human brain motion and cerebrospinal fluid circulation demonstrated with MR velocity imaging. Radiology. 1987;163:793.
Ferlini A., Ragno M., et al. Hydrocephalus, skeletal anomalies, and mental disturbances in a mother and three daughters: A new syndrome. Am J Med Genet. 1995;59:506.
Fernell E., Hagberg G. Infantile hydrocephalus: declining prevalence in preterm infants. Acta Paediatr. 1998;87(4):392-396.
Fewel M.E., Levy M.L., McComb J.G. Surgical treatment of 95 children with 102 intracranial arachnoid cysts. Pediatr Neurosurg. 1996;25:165.
Fioravanti A., Godano U., Consales A., et al. Bobble-head doll syndrome due to a suprasellar arachnoid cyst: Endoscopic treatment in two cases. Childs Nerv Syst. 2004;20:770.
Fishman R.A. Cerebrospinal fluid in diseases of the nervous system. Philadelphia: WB Saunders; 1980.
Fletcher J.M., Bohan T.P., et al. Cerebral white matter and cognition in hydrocephalic children. Arch Neurol Clin. 1992;49:818.
Fletcher J.M., Bohan T.P., et al. Morphometric evaluation of the hydrocephalic brain: Relationships with cognitive development. Childs Nerv Syst. 1996;12:192.
Fletcher J.M., Brookshire B.L., et al. Behavioral adjustment of children with hydrocephalus: Relationships with etiology, neurological, and family status. J Pediatr Psychol. 1995;20:109.
Fransen E., Lemmon V., et al. CRASH syndrome: Clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur J Hum Genet. 1995;3:273.
Fransen E., Van Camp G., et al. Genotype-phenotype correlation in L1 associated diseases. J Med Genet. 1998;35:399.
Fukumitsu H., Ohmiya M., et al. Aberrant expression of neurotrophic factors in the ventricular progenitor cells of infant congenitally hydrocephalic rats. Childs Nerv Syst. 2000;16:516-521.
Galbreath E., Kim S.J., et al. Overexpression of TGF-beta 1 in the central nervous system of transgenic mice results in hydrocephalus. J Neuropathol Exp Neurol. 1995;54:339-349.
Garton H.J., Piatt J.H.Jr. Hydrocephalus. Pediatr Clin North Am. 2004;51(2):305-325.
Germano A., Caruso G., Caffo M., et al. The treatment of large supratentorial arachnoid cysts in infants with cyst-peritoneal shunting and Hakim programmable valve. Childs Nerv Syst. 2003;19:166.
Godano U., Mascari C., Consales A., et al. Endoscope-controlled microneurosurgery for the treatment of intracranial fluid cysts. Childs Nerv Syst. 2004;20:839.
Gosalakkal J.A. Intracranial arachnoid cysts in children: A review of pathogenesis, clinical features, and management. Pediatr Neurol. 2002;26:93.
Greitz D. Radiological assessment of hydrocephalus: new theories and implications for therapy. Neurosurg Rev. 2004;27(3):145-165. discussion 166–147
Halliday J., Chow C.W., et al. X linked hydrocephalus: a survey of a 20 year period in Victoria, Australia. J Med Genet. 1986;23:23-31.
Hann Y.S., Engelhard H., et al. Abdominal CSF pseudocyst: Clinical features and surgical management. Pediatr Neurosci. 1985;12:75.
Haverkamp F., Wolfle J., et al. Congenital hydrocephalus internus and aqueductal stenosis: aetiology and implications for genetic counselling. Eur J Pediatr. 1999;158:474-478.
Hill A., Rozdilsky B. Congenital hydrocephalus secondary to intra-uterine germinal matrix/intraventricular hemorrhage. Dev Med Child Neurol. 1984;26:509-527.
Hirano A., Hirano M. Benign cysts in the central nervous system: Neuropathological observations of the cyst walls. Neuropathology. 2004;24:1.
Ibanez-Tallon I., Pagenstecher A., et al. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet. 2004;13:2133-2141.
Iskandar B., Tubbs S., et al. Death in shunted hydrocephalic children in the 1990s. Pediatr Neurosurg. 1998;28:173-176.
Johnston M. The importance of lymphatics in cerebrospinal fluid transport. Lymphat Res Biol. 2003;1(1):41-44. discussion 45
Johnston M., Papaiconomou C. Cerebrospinal fluid transport: a lymphatic perspective. News Physiol Sci. 2002;17:227-230.
Johnston M., Zakharov A., et al. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1(1):2.
Jones R.F., Kwok B.C., et al. Third ventriculostomy for hydrocephalus associated with spinal dysraphism: Indications and contraindications. Eur J Pediatr Surg. 1996;1(Suppl):5.
Jouet M., Rosenthal A., et al. A missense mutation confirms the L1 defect in X-linked hydrocephalus (HSAS). Nat Genet. 1993;4:331.
Kazan S., Gura A., et al. Hydrocephalus after intraventricular hemorrhage in preterm and low-birth weight infants: analysis of associated risk factors for ventriculoperitoneal shunting. Surg Neurol. 2005;64(Suppl 2):S7-S81.
Kennedy C.R., Ayers S., et al. Randomized, controlled trial of acetazolamide and furosemide in posthemorrhagic ventricular dilation in infancy: Follow-up at 1 year. Pediatrics. 2001;108:597.
Kestle J., Drake J., et al. Long-term follow-up data from the Shunt Design Trial. Pediatr Neurosurg. 2000;33(5):230-236.
Kestle J.R. Pediatric hydrocephalus: current management. Neurol Clin. 2003;21(4):883-895. vii
Kestle J.R., Drake J.M., et al. Lack of benefit of endoscopic ventriculoperitoneal shunt insertion: a multicenter randomized trial. J Neurosurg. 2003;98(2):284-290.
Kestle J.R., Walker M.L. A multicenter prospective cohort study of the Strata valve for the management of hydrocephalus in pediatric patients. J Neurosurg. 2005;102(Suppl 2):141-145.
Koch C.A., Voth D., Kraemer G., et al. Arachnoid cysts: Does surgery improve epileptic seizures and headaches? Neurosurg Rev. 1995;18:173.
Kume T., Deng K.Y., et al. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985-996.
Lam C.H., Villemure J.G. Comparison between ventriculoatrial and ventriculoperitoneal shunting in the adult population. Br J Neurosurg. 1997;11:43.
Lancon J.A., Ellis A.L. Giant posterior fossa arachnoid cyst. Pediatr Neurosurg. 2004;40:151.
Lapunzina P., Delicado A., et al. Autosomal recessive hydrocephalus due to aqueductal stenosis: Report of a further family and implications for genetic counselling. J Matern Fetal Neonatal Med. 2002;12:64-66.
Laurence K.M., Coates S. The natural history of hydrocephalus. Detailed analysis of 182 unoperated cases. Arch Dis Child. 1962;37:345-362.
leMay M., Hochberg F.H. Ventricular differences between hydrostatic hydrocephalus and hydrocephalus ex vacuo by CT. Neuroradiology. 1979;17:191-195.
Lemons J.A., Bauer C.R., et al. Very-low-birth-weight outcomes of the NICHD Neonatal Research Network, January 1995 through December 1996. Pediatrics. 107(1), 2001.
Levene M. Measurement of the growth of the lateral ventricles in preterm infants with real time ultrasound. Arch Dis Child. 1981;56:900-940.
Lumenta C.B., Skotarczak U. Long-term follow-up in 233 patients with congenital hydrocephalus. Childs Nerv Syst. 1995;11:173.
Lutcherath V., Waaler P.E., Jellum E., et al. Children with bilateral temporal arachnoid cysts may have glutaric aciduria type 1 (GAT1); operation without knowing that may be harmful. Acta Neurochir (Wien). 2000;142:1025.
Mann J.D., Butler A.B., et al. Regulation of intracranial pressure in rat, dog, and man. Ann Neurol. 1978;3:156.
Mantovani J.F., Pasternak J., et al. Failure of daily lumbar punctures to prevent the development of hydrocephalus following intraventricular hemorrhage. J Pediatr. 1980;97:278.
Martinez-Lage J.F., Garcia Santos J.M., Poza M., et al. Bilateral temporal arachnoid cysts in neurofibromatosis. J Child Neurol. 1993;8:383.
Mason T.B.2nd, Chiriboga C.A., Feldstein N.A., et al. Intracranial arachnoid cyst in a developmentally normal infant: Case report and literature review. Pediatr Neurol. 1997;16:59.
McLaughlin M.R., Wahlig J.B., et al. Traumatic basilar aneurysm after endoscopic third ventriculostomy: Case report. Neurosurgery. 1997;41:1400-1404.
Ment L.R., Allan W.C., et al. Grade 3 to 4 intraventricular hemorrhage and Bayley scores predict outcome. Pediatrics. 2005;116(6):1597-1598.
Ment L.R., Duncan C., et al. Benign enlargement of the subarachnoid spaces in the infant. J Neurosurg. 1981;54(4):504-508.
Milhorat T. Hydrocephalus and the cerebrospinal fluid. Baltimore: Williams & Wilkins; 1972.
Millichap J.G. Temporal lobe arachnoid cyst-attention disorder syndrome: Role of the electroencephalogram in diagnosis. Neurology. 1997;48:1435.
Mohn A., Schoof E., Fahlbusch R., et al. The endocrine spectrum of arachnoid cysts in childhood. Pediatr Neurosurg. 1999;3:316.
Mori K. Current concept of hydrocephalus: evolution of new classifications. Childs Nerv Syst. 1995;11:523-531.
Naidich T.P., McLone D.G., Radkowski M.A. Intracranial arachnoid cysts. Pediatr Neurosci. 1985–86;12:112.
Ohmiya M., Fukumitsu H., et al. Administration of FGF-2 to embryonic mouse brain induces hydrocephalic brain morphology and aberrant differentiation of neurons in the postnatal cerebral cortex. J Neurosci Res. 2001;65:228-235.
Oi S., Di Rocco C. Proposal of “evolution theory in cerebrospinal fluid dynamics” and minor pathway hydrocephalus in developing immature brain. Childs Nerv Syst. 2006;22(7):662-669.
Oi S., Honda Y., et al. Intrauterine high resolution magnetic resonance imaging in fetal hydrocephalus and prenatal estimation of postnatal outcomes with “perspective classification”. J Neurosurg. 1998;88:685-694.
Okamoto N., Del Maestro R., et al. Hydrocephalus and Hirschsprung’s disease with a mutation of L1CAM. J Hum Genet. 2004;49:334-337.
Paciorkowski A.R., Greenstein R.M. When is enlargement of the subarachnoid spaces not benign? A genetic perspective. Pediatr Neurol. 2007;37(1):1-7.
Parsch C.S., Krauss J., Hofmann E., et al. Arachnoid cysts associated with subdural hematomas and hygromas: Analysis of 16 cases, long-term follow-up, and review of the literature. Neurosurgery. 1997;40:483.
Pascual-Castroviejo I., Roche M.C., Martinez Bermejo A., et al. Primary intracranial arachnoidal cysts. A study of 67 childhood cases. Childs Nerv Syst. 1991;7:257.
Piatt J.H., Carlson C.V. Hydrocephalus and epilepsy: An actuarial analysis. Neurosurgery. 1996;39:722.
Pollack I.F., Albright A.L., et al. A randomized, controlled study of a programmable shunt valve versus a conventional valve for patients with hydrocephalus. Hakim-Medos Investigator Group. Neurosurgery. 1999;45(6):1399-1408. discussion 1408–1311
Pollay M. Review of spinal fluid physiology: production and absorption in relation to pressure. Clin Neurosurg. 1977;24:254-269.
Portnoy H.D., Schulte R.R., et al. Anti-siphon and reversible occlusion valves for shunting in hydrocephalus and preventing post-shunt subdural hematomas. J Neurosurg. 1973;38(6):729-738.
Raeder M.B., Helland C.A., Hugdahl K., et al. Arachnoid cysts cause cognitive deficits that improve after surgery. Neurology. 2005;64:160.
Raffel C., McComb J.G. To shunt or to fenestrate: Which is the best treatment for arachnoid cysts in pediatric patients? Neurosurgery. 1988;23:338.
Ramos C., Fernandez-Llebrez P., et al. Msx1 disruption leads to diencephalon defects and hydrocephalus. Dev Dyn. 2004;230:446-460.
Rekate H.L. A contemporary definition and classification of hydrocephalus. Semin Pediatr Neurol. 2009;16(1):9-15.
Rekate H.L. Recent advances in the understanding and treatment of hydrocephalus. Semin Pediatr Neurol. 1997;4:167.
Rekate H.L. Shunt-related headaches: the slit ventricle syndromes. Childs Nerv Syst. 2008;24(4):423-430.
Renier D., Sainte-Rose C., et al. Prenatal hydrocephalus: outcome and prognosis. Childs Nerv Syst. 1988;4:213-222.
Richards P. Walter Dandy. In: Ashwal S., editor. The Founders of Child Neurology. San Francisico: Norman Publishing; 1990:472-477.
Rich P., Jones R., et al. MRI of the foetal brain. Clin Radiol. 2007;62:303-313.
Rubin R.C., Hochwald G., et al. The effect of severe hydrocephalus on size and number of brain cells. Dev Med Child Neurol. 1972;27(Suppl):117.
Sainte-Rose C., Hooven M.D., et al. A new approach in the treatment of hydrocephalus. J Neurosurg. 1987;66:213-226.
Sainte-Rose C., Piatt J., et al. Mechanical complications in shunts. Pediatr Neurosurg. 1991;17:2-9.
Savman K., Blennow M., et al. Cytolike response in cerebrospinal fluid from preterm infants with posthaemorrhagic ventricular dilatation. Acta Paediatr. 2002;91:1357-1363.
Savman K., Nilsson U., et al. Non-protein-bound ion is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilatation. Pediatr Res. 2001;49(2):208-212.
Schrander-Stumpel C.T., Fryns J.P. Congenital hydrocephalus: nosology and guidelines for clinical approach and genetic counselling. Eur J Pediatr. 1998;157:355-362.
Serlo W., Kirkinen P., et al. Prognostic signs in fetal hydrocephalus. Childs Nerv Syst. 1986;2:93-97.
Sgouros S., Malluci C., et al. Long-term complications of hydrocephalus. Pediatr Neurosurg. 1995;23:127.
Silverberg G.D., Huhn S., et al. Downregulation of cerebrospinal fluid production in patients with chronic hydrocephalus. J Neurosurg. 2002;97:1271-1275.
Sinha S., Brown J.I. Familial posterior fossa arachnoid cyst. Childs Nerv Syst. 2004;20:100.
Spreer J., Ernestus R.I., et al. Lesions of the corpus callosum in hydrocephalic patients with ventricular drainage – a CT study. Acta Neurochir (Wien). 1996;138:174.
Stoll C., Alembik Y., et al. An epidemiologic study of environmental and genetic factors in congenital hydrocephalus. Eur J Epidemiol. 1992;8:797-803.
Suh D.Y., Gaskill Shipley M., et al. Corpus callosal changes associated with hydrocephalus: A report of two cases. Neurosurgery. 1997;41:488.
Takano T., Mekata Y., et al. Early ependymal changes in experimental hydrocephalus after mumps virus inoculation in hamsters. Acta Neuropathol (Berl). 1993;85:521-525.
Teebi A.S., Naguib K.K. Autosomal recessive nonsyndromal hydrocephalus. Am J Med Genet. 1988;31:467-470.
Teo C., Rahman S., et al. Complications of endoscopic neurosurgery. Childs Nerv Syst. 1996;12:248-253.
Tulipan N., Sutton L.N., et al. The effect of intrauterine myelomeningocele repair on the incidence of shunt-dependent hydrocephalus. Pediatr Neurosurg. 2003;38(1):27-33.
Tuli S., Drake J., et al. Long-term outcome of hydrocephalus management in myelomeningoceles. Childs Nerv Syst. 2003;19(5–6):286-291.
Tuli S., Tuli J., et al. Predictors of death in pediatric patients requiring cerebrospinal fluid shunts. J Neurosurg (Pediatrics). 2004;100(5):442-446.
Tuli S.M.D., O’Hayon B.M.D., et al. Change in Ventricular Size and Effect of Ventricular Catheter Placement in Pediatric Patients with Shunted Hydrocephalus. [Article]. Neurosurgery. 45(6), 1999.
Tullio A.N., Bridgman C., et al. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol. 2001;433:62-74.
Ventriculomegaly Trial Group. Randomised trial of early tapping in neonatal post haemorrhagic ventricular dilatation. Arch Dis Child. 1990;65:3.
Verhagen W., Bartels R., et al. Familial congenital hydrocephalus and aqueductal stenosis with probably autosomal dominant inheritance and variable expression. J Neurol Sci. 1998;158:101-105.
Vernet O., Campiche R., et al. Long-term results after ventriculo-atrial shunting in children. Childs Nerv Syst. 1995;11:176.
Vincent C., Kalatzis V., et al. A proposed new contiguous gene syndrome on 8q consists of Branchio-Oto-Renal (BOR) syndrome, Duane syndrome, a dominant form of hydrocephalus and trapeze aplasia; implications for the mapping of the BOR gene. Hum Mol Genet. 1994;3:1859-1866.
Wagner C., Batiz L., et al. Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J Neuropathol Exp Neurol Clin. 2003;62:1019-1040.
Weiner S.N., Pearlstein A.E., Eiber A. MR imaging of intracranial arachnoid cysts. J Comput Assist Tomogr. 1987;11:236.
Welch K. The principle of physiology of the cerebrospinal fluid in relation to hydrocephalus including normal pressure hydrocephalus. Adv Neurol. 1975;13:247.
Weller S., Shulman K. Infantile hydrocephalus: Clinical, histological and ultrastructural study of brain damage. Arch Dis Child. 1972;70:129-136.
Whitelaw A. Intraventricular streptokinase after intraventricular hemorrhage in newborn infants. Cochrane Database Syst Rev. (1):2001. CD000498
Whitelaw A. Repeated lumbar or ventricular punctures in newborns with intraventricular hemorrhage. Cochrane Database Syst Rev. (1):2005. CD000216
Wilson-Costello D., Friedman H., et al. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115(4):997-1003.
Wong E., Kenwrick S., et al. Mutations in the cell adhesion molecule L1 cause mental retardation. Trends Neurosci. 1995;18:168.
Zacharia A., Zimine S., Lovblad K.O., et al. Early assessment of brain maturation by MR imaging segmentation in neonates and premature infants. Am J Neuroradiol. 2006;27:972-977.
Zhang J., Williams M.A., et al. Genetics of human hydrocephalus. J Neurol. 2006;253(10):1255-1266.