Chapter 14 Huntington disease
Introduction
George Huntington published his essay “On Chorea” in 1872 (Huntington, 1872), one year after graduating from Columbia College of Physicians and Surgeons while practicing general medicine with his father on Long Island. Soon afterward, the eponym Huntington’s chorea was adopted in the literature to draw attention to chorea (derived from the Latin choreus, meaning “dance” and Greek choros, meaning “chorus”) as the clinical hallmark of this neurodegenerative disorder. However, since many other manifestations of the disease exist and since chorea might not even be present, the term Huntington disease (HD) (OMIM 143100) is more appropriate (Penney and Young, 1998; Jankovic, 2006; Fahn and Jankovic, 2009). Although chorea (known as the dancing mania) has been recognized since the Middle Ages, the origin of the affected families described by Huntington was traced to the early seventeenth century in the village of Bures in southeast England. The inhabitants of this area later migrated to various parts of the world, accounting for the marked variation in the regional prevalence of HD.
This chapter will focus on HD; other diseases, such as Huntington disease-like type 1 (HDL1), HDL2, and HDL3, dentatorubral-pallidoluysian atrophy, and spinocerebellar atrophy type 17 (SCA17) that may present as HD-like phenotypes are discussed in Chapter 15 and other reviews (Toyoshima et al., 2004; Schneider et al., 2006; Schneider et al., 2007; Wild et al., 2008; Jankovic and Fahn, 2009; Fekete and Jankovic, 2010).
Epidemiology
While the estimated prevalence of HD in the United States is 2 to 10 per 100 000 population (Kokmen et al., 1994), which translates to about 30 000 people with HD and another 200 000 at risk of developing the disease, the prevalence is geographically heterogeneous, and in certain regions of the world it is as high as 560 per 100 000 (Moray Firth, Scotland) and 700 per 100 000 (Lake Maracaibo, Venezuela) (Harper, 1992). Americo Negrette, a Venezuelan physician, first observed the dancing mania of Maracaibo in the 1950s, and his findings later led to the discovery of the gene locus and gene mutation for HD (Okun and Thommi, 2004). Because many epidemiologic studies were done before the advent of genetic testing, the true prevalence of the HD gene is not known. DNA testing has largely replaced other tests, such as magnetic resonance imaging (MRI) of the brain, in the evaluation of patients with HD.
Clinical aspects
The rich repertoire of neurologic, behavioral, and cognitive manifestations of HD makes it one of the most intriguing of all neurodegenerative disorders. In a study involving 1901 patients with HD, the following were considered the most frequent presenting symptoms, in descending order: chorea, trouble walking, unsteadiness, irritability, depression, clumsiness, speech difficulty, memory loss, dropping of objects, lack of motivation, paranoia, intellectual decline, sleep disturbance, hallucination, weight loss, and sexual problems (Foroud et al., 1999). The Unified Huntington’s Disease Rating Scale (UHDRS) was developed to assess and quantify various clinical features of HD, specifically motor function, cognitive function, behavioral abnormalities, and functional capacity (Huntington Study Group, 1996; Siesling et al., 1998). A shortened version of the UHDRS has been validated (Siesling et al., 1997). In addition, assessment protocol has been developed to evaluate various neurologic and behavioral features in HD patients who are undergoing striatal grafting (CAPIT-HD Committee, 1996). When so-called “soft signs” are identified at the initial evaluation, the cumulative relative risk of HD diagnosis at 1.5 years is 4.68 times greater than in controls (Langbehn et al., 2007).
Patients with HD often have poor insight into their chorea and are more likely to report consequences of the movement disorder, such as dropping objects, than “twitching” or other features of chorea, and there is little or no relationship between direct experience or awareness of involuntary movement and actual chorea scores (Snowden et al., 1998). This may explain why the presence and degree of chorea seem to correlate poorly with functional decline (Feigin et al., 1995). Besides chorea, other motor symptoms that typically affect patients with HD include dystonia, postural instability, ataxia, dystonia, bruxism, myoclonus, tics and tourettism, dysarthria, dysphagia, and aerophagia (Vogel et al., 1991; Carella et al., 1993; Thompson et al., 1994; Ashizawa and Jankovic, 1996; Hu and Chaudhuri, 1998; Louis et al., 1999; Tan and Jankovic, 2000; Jankovic and Ashizawa, 2003; Walker, 2007) (Videos 14.1, 14.2, 14.3, and 14.4). A characteristic feature of HD is the inability to maintain tongue protrusion, representing motor impersistence (sometimes also referred to as “negative chorea”) (Videos 14.5 and 14.6). 
Chorea is clearly not merely a cosmetic or embarrassing symptom, but this cardinal feature of HD may affect fine and gross motor function, activities of daily living, gait and balance, eventually impacting on the quality of life, and likely contributing to weight loss, falls, social isolation, loss of employability, and increased morbidity and dependence on others, eventually leading to institutionalization (Wheelock et al., 2003). In one study, only the change in the UHDRS chorea score correlated with the change in the complex task performance, such as peg insertion (Andrich et al., 2007). One study compared 45 HD patients with a history of at least one fall to 27 healthy controls (Grimbergen et al., 2008). The fallers had more chorea, bradykinesia, aggression and cognitive decline. The HD patients had decreased gait velocity and a decreased stride length. In one study of 24 patients with HD, 14 (58.3%) reported at least two falls within the previous 12 months and the fallers had worse scores on Berg Balance Scale (BBS) and the Timed “Up & Go” (TUG) test (Busse et al., 2009).
Hung-up and pendular reflexes are also typically present in patients with HD (Video 14.6) (Brannan, 2003). Some patients can “camouflage” their chorea by incorporating the involuntary movements into semipurposeful activities, so-called “parakinesia” (Video 14.7). Rarely, patients with HD present with tics and other features suggestive of adult-onset Tourette syndrome (“tourettism”) (Jankovic and Ashizawa, 1995) (Video 14.8). 
In a study of 593 members of a large kindred in Venezuela, generation of fine motor movements and of rapid eye saccades was found to be impaired in about 50% of at-risk individuals (Penney et al., 1990). Since at-risk individuals with these findings were more likely than those without them to develop overt HD within several years, these abnormalities were thought to represent the earliest clinical manifestations of the disease. If the first examination was normal, there was only a 3% risk of developing symptomatic HD within 3 years. HD patients seem to have greater defects in initiating internally than externally generated saccades (Tian et al., 1991). In 215 individuals at risk for HD or recently diagnosed with HD, a high-resolution, video-based eye tracking system demonstrated three types of significant abnormalities while performing memory-guided and anti-saccade tasks: increased error rate, increased saccade latency, and increased variability of saccade latency (Blekher et al., 2006). In another study initiation deficits of voluntary-guided, but not reflexive saccades were found in individuals with preclinical HD (Golding et al., 2006). Voluntary-guided saccades in 25 presymptomatic HD individuals have been found to correlate with deficits in white matter tracts as determined by diffusion tensor MRI (Klöppel et al., 2008). In addition to abnormal eye saccades, patients with HD demonstrate other neuro-ophthalmologic abnormalities, such as increased blink rates (Karson et al., 1984; Xing et al., 2008), irregular elevations of eyebrows due to choreic contractions of the frontalis muscles and eye closures with irregular narrowing of palpebral fissures, rarely leading to frank blepharospasm, and apraxia of eyelid opening and closure (Bonelli and Niederwieser, 2002).
Using quantitative assessments, Siemers and colleagues (1996) found subtle but significant abnormalities in simple and choice movement time and reaction time in 103 truly presymptomatic carriers of the HD gene. These deficits correlated well with the number of cytosine-adenine-guanine (CAG) trinucleotide repeats (see later). In a follow-up longitudinal study of 43 at-risk individuals, Kirkwood and colleagues (1999) found that the following variables declined more rapidly among the presymptomatic gene carriers (n = 12) than among the noncarriers (n = 31): psychomotor speed (digit symbol subscale of the Wechsler Adult Intelligence Scale), optokinetic nystagmus, and rapid alternating movements. In contrast to UHDRS, which is not very sensitive in detecting early clinical signs of HD, careful gait and balance analysis has detected gait bradykinesia and dynamic balance impairment in presymptomatic HD gene carriers with otherwise normal neurologic examination (Rao et al., 2008). In the Predict-HD study, 505 at-risk individuals, 452 of whom had more than 39 CAG repeats, but had not yet met clinical criteria for the diagnosis of HD, the striatum MRI volume decreased from 17.06 cm3, at diagnostic confidence level of 0, to 14.89 cm3, at diagnostic confidence level of 3 (probable HD, with a mean CAG repeat number of 44 and mean motor UHDRS score of 16.92) (Paulsen et al., 2006). The study found that smaller striatal volume, reduced finger tapping speed and consistency, and impaired odor identification were among the best markers of “pre-clinical” HD (Paulsen et al., 2008). Additional analyses found that even subtle abnormality in finger tapping (bradykinesia), tandem gait, Luria test, saccade initiation, and chorea were associated with high probability of disease diagnosis (Biglan et al., 2009).
While hyperkinesia, usually in the form of chorea, is typically present in adult-onset HD, parkinsonism (an akinetic-rigid syndrome) is characteristic of juvenile HD, also termed Westphal variant. Bradykinesia, usually evident in patients with the rigid form of HD, when it coexists with chorea might not be fully appreciated on a routine examination (van Vugt et al., 1996; Thompson et al., 1988; Sánchez-Pernaute et al., 2000). Variability in isometric grip forces while grasping an object has been found to correlate well with UHDRS and with progressive motor deficits associated with HD (Reilmann et al., 2001). Assessing simple repetitive movements, such as tapping, correlates with UHDRS and may be used to follow progression of HD (Andrich et al., 2007). Fast simple wrist flexion movements were found to be significantly slower in 17 patients with HD compared to controls (Thompson et al., 1988). While bradykinesia was most pronounced in the rigid-akinetic patients, it was also evident in patients with the typical choreic variety of HD. When bradykinesia predominates, the patients exhibit parkinsonian findings, some of which can be subtle. Using a continuous wrist-worn monitor of motor activity, van Vugt and colleagues (1996) also provided evidence of hypokinesia in patients with HD, particularly when they were treated with neuroleptics. Micrographia may be one manifestation of underlying parkinsonism; when chorea predominates, the handwriting is characterized by macrographia (Phillips et al., 1994). Bradykinesia in HD may be an expression of postsynaptic parkinsonism as a result of involvement of both direct and indirect pathways. This might explain why a reduction in chorea with antidopaminergic drugs rarely improves overall motor functioning and indeed can cause an exacerbation of the motor impairment. In their excellent review, Berardelli and colleagues (1999) argue that “bradykinesia results from degeneration of the basal ganglia output to the supplementary motor areas concerned with the initiation and maintenance of sequential movements” and “may reflect failure of thalamocortical relay of sensory information.” Although bradykinesia associated with HD usually does not respond to dopaminergic therapy, late-onset levodopa-responsive HD presenting as parkinsonism has been well documented (Reuter et al., 2000).
Besides chorea, the other two components of the HD cardinal triad include cognitive decline and various psychiatric symptoms, particularly depression (Paulsen et al., 2001a). Depression, poor impulse control, and other behavioral and socioeconomic factors associated with HD contribute to the markedly increased risk of suicide in patients with HD. Suicide rate in patients with HD is 138/100 000 person-years as compared to 12–13/100 000 person-years in the general population. Thus, the risk of suicide is ten times greater in HD than in the general population (Bird, 1999; Almqvist et al., 1999). In a study of 506 individuals with documented or suspected HD, suicide represented the third most common cause of death, accounting for 12.7% of 157 ascertained deaths, after bronchopneumonia (31.8%) and heart disease (15.3%) (Schoenfeld et al., 1984). In a review of records of 452 deceased patients diagnosed with HD, 5.7% of deaths were attributed to suicide, 4 times the expected rate for the corresponding US white population (Farrer, 1986). In a cross-sectional study of 2835 patients with “definite” diagnosis of HD, with disease onset at age 20 years or later, mean age of 49.6 years, and duration of symptoms for a mean 7.6 years, 50.3% reported seeking treatment for depression and 10.3% reported at least one suicide attempt (Paulsen et al., 2005b). Patients endorsing current symptoms of depression were twice as likely to have attempted suicide in the past as those who deny symptoms of depression (15.7% vs. 7.1%). In another study, using UHDRS in 4171 patients with HD, the frequency of suicidal ideation doubled from 9.1% in at-risk persons with normal neurologic examination to 19.8% in at-risk individuals with soft neurologic signs and increased to 23.5% in persons with “possible” HD. In patients with diagnosed HD, the risk of suicidal ideation increased from 16.7% at stage 1 to 21.6% at stage 2, and decreased thereafter to 19.5%, 14.1%, and 9.8% in stages 3, 4, and 5, respectively (Paulsen et al., 2005a). Thus the most critical periods of suicide risk are immediately before receiving a formal diagnosis of HD and in stage 2 of the disease, when activities, such as driving and taking care of one’s finances are beginning to be restricted and patients are becoming more dependent on others. While some studies (Foroud et al., 1995) showed that cognitive deficits correlated with the number of CAG repeats in asymptomatic carriers of the HD gene, other studies found no correlation between cognitive decline and CAG repeats in symptomatic patients with HD (Zappacosta et al., 1996). The neurobehavioral symptoms typically consist of personality changes, agitation, irritability, anxiety, apathy, social withdrawal, impulsiveness, depression, mania, paranoia, delusions, hostility, hallucinations, psychosis, and various sexual disorders (Fedoroff et al., 1994; Litvan et al., 1998). In a study of 52 patients with HD, Paulsen and colleagues (2001a) found the following neuropsychiatric symptoms in descending order of frequency: dysphoria, agitation, irritability, apathy, anxiety, disinhibition, euphoria, delusions, and hallucinations (Video 14.9). Affective disorders, such as depression, often preceded the onset of motor symptoms. In one study of presymptomatic individuals at risk for HD, gene carriers were 1.74 times (95% CI 1–3.07) more likely to report depression than non-carriers, and the rate of depression increased the closer the subject was to the clinical onset (Julien et al., 2007). Behaviors such as agitation, anxiety, and irritability have been related to hyperactivity of the medial and orbitofrontal cortical circuitry (Litvan et al., 1998). This is in contrast to the apathy and hypoactive behaviors that are seen in progressive supranuclear palsy, attributed to a dysfunction in the frontal cortex and associated circuitry. Progressive decline in attention and executive function, consistent with frontostriatal pathology, has been found in early HD (Ho et al., 2003). Criminal behavior, closely linked to the personality changes, depression, and alcohol abuse, has been reported to be more frequent in patients with HD than in nonaffected first-degree relatives (Jensen et al., 1998). Such behavior might be a manifestation of an impulsive disorder as part of disinhibition, seen not only in HD but also in other frontal lobe–basal ganglia disorders, particularly Tourette syndrome (Brower and Price, 2001). 
Cognitive changes, manifested chiefly by loss of recent memory, poor judgment, and impaired concentration and acquisition, occur in nearly all patients with HD; but some patients with late-onset chorea never develop dementia (Britton et al., 1995). In one study, dementia was found in 66% of 35 HD patients (Pillon et al., 1991). Tasks requiring psychomotor or visuospatial processing, such as skills tested by the Trail Making B and Stroop Interference Test, are impaired early in the course of the disease and deteriorate at a more rapid rate than memory impairment (Bamford et al., 1995). In addition to deficits in visual and auditory perception, patients with HD have impaired recognition of emotional facial expression (Sprengelmeyer et al., 1996). The presence of apraxia and other “cortical” features has cast doubt on this classification (Shelton and Knopman, 1991). In one study, ideomotor apraxia was found in three of nine HD patients, and seven of nine made some apraxic errors. It could not be determined whether the apraxia was due to involvement of frontal cortex or to involvement of subcortical structures (Shelton and Knopman, 1991). Speech and language may be also affected in HD, possibly as a result of striatal degeneration (De Diego-Balaguer et al., 2008). One study showed that high levels of insulin-like growth factor I (IGFI) are associated with cognitive deficits in HD (Saleh et al., 2010).
Although neurobehavioral symptoms precede motor disturbances in some cases, de Boo and colleagues (1997) showed that motor symptoms are more evident than cognitive symptoms in early stages of HD. Symptoms of REM behavioral sleep disorders may precede the onset of chorea or other motor symptoms of HD by many years (Arnulf et al., 2008). Asymptomatic at-risk individuals who are positive for the HD gene or the marker do seem to differ in their cognitive performance from asymptomatic at-risk individuals who are negative for the HD gene or marker (Giordani et al., 1995; Foroud et al., 1995; Lawrence et al., 1998). Most studies have found that neuropsychological tests do not differentiate between presymptomatic individuals who are positive for the HD gene and those who are negative (Strauss and Brandt, 1990; de Boo et al., 1997), but some studies have found that cognitive changes might be the first symptoms of HD (Hahn-Barma et al., 1998). In a longitudinal study by the Huntington Study Group of 260 individuals who were considered to be at risk for HD, Paulsen and colleagues (2001b) found that this group had worse scores on the cognitive section of the UHDRS at baseline, an average of 2 years before the development of motor manifestations of the disease.
In addition to motor, cognitive, and behavioral abnormalities, most patients with HD lose weight during the course of their disease, despite increased appetite (Marder et al., 2009). Weight loss in HD increases with higher CAG repeats (Aziz et al., 2008). Weight loss is not unique for HD among the neurodegenerative disorders; for example, it is typically seen in patients with Parkinson disease (Jankovic et al., 1992). The pathogenesis of weight loss in HD is unknown, but one study showed that patients with HD have a 14% higher sedentary energy expenditure than that of controls and that this appears to be correlated with the severity of the movement disorder (Pratley et al., 2000). The lower body mass index in HD parallels the weight loss in transgenic mice, suggesting that it represents a clinical expression of the gene abnormality associated with HD (Djousse et al., 2002; Aziz et al., 2008). Weight loss, and alterations in sexual behavior and wake–sleep cycle, frequently found in patients with HD, have been attributed to involvement of the hypothalamus even in early stages of HD as demonstrated by positron emission tomography (PET) and postmortem studies (Politis et al., 2008). Juvenile HD is associated with higher CAG repeats; 54% have 60 or more CAG repeats (Ribai et al., 2007).
About 10% of HD cases have their onset before age 20. This juvenile HD is usually inherited from the father (father vs. mother inheritance for patients with onset before 10 years is 3 : 1) and it typically presents with the combination of progressive parkinsonism, dementia, ataxia, myoclonus, and seizures. Myoclonus is particularly common in patients with juvenile HD (Video 14.10) and progressive myoclonic epilepsy has been reported as the initial presentation of juvenile HD (Gambardella et al., 2001) (Videos 14.10, 14.11, and 14.12). 
Natural course
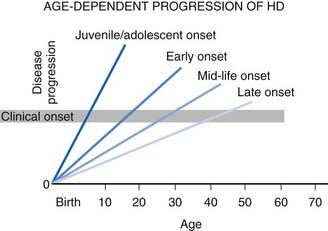
The natural course of HD varies; on the average, duration of illness from onset to death is about 15 years for adult HD, but it is about 4–5 years shorter for the juvenile variant. Patients with juvenile onset (<20 years) and with late onset (>50 years) of symptoms have the shortest duration of the disease (Foroud et al., 1999) (Fig. 14.1) (Videos 14.11 and 14.12). Using the Total Functional Capacity Scale, longitudinal follow-up of patients with HD showed a 0.72 unit/year rate of decline (Marder et al., 2000). Clinical-pathologic studies have demonstrated a strong inverse correlation between the age at onset and the severity of striatal degeneration (Myers et al., 1988). Striatal degeneration as measured by immunostaining with tyrosine hydroxylase (TH) is more pronounced in HD than in Parkinson disease (Huot et al., 2007). A review of clinical and pathologic data in 163 HD patients showed that patients with juvenile or adolescent onset had much more aggressive progression of the disease than patients with onset in middle and late life. Because of lack of efficacy, the clinical trial conducted by the Huntington Study Group that compared coenzyme Q10 (CoQ10) and remacemide, known as CARE-HD and involving 347 patients with documented HD, provides the best available data on the natural history of HD (Huntington Study Group, 2001). In this study, after 20 months, the Functional Assessment score decreased by 2.51 points, Total Functional Capacity score decreased by 1.95 points, and the Independence Scale score decreased by 8.58 points. Analysis of data collected by the Huntington Study Group on 1026 patients with HD, followed for a median of 2.7 years, concluded that the rate of progression was significantly more rapid with a younger age at onset and longer CAG repeats (Mahant et al., 2003). Although chorea and dystonia were not major determinants of disability, chorea was associated with weight loss. 
Progressive motor dysfunction, dementia, dysphagia, and incontinence eventually lead to institutionalization and death from aspiration, infection, and poor nutrition (Leopold and Kagel, 1985). Among 4809 HD subjects, 3070 of whom had a definite diagnosis of HD, 228 (7.4%) resided in a skilled nursing facility, and these residents had worse chorea, bradykinesia, gait abnormality, and imbalance, as well as more obsessions, compulsions, delusions, and auditory hallucinations, and they were more aggressive and disruptive than their counterparts living at home (Wheelock et al., 2003). In a study of 1642 patients with HD, 27.2% had some obsessive-compulsive features and nearly one-quarter received treatment for obsessive-compulsive disorder (Anderson et al., 2010). The HD patients with obsessive-compulsive disorder were older, had poorer functioning, and a longer duration of illness than those without these features, and more psychiatric comorbidities such as depression, suicidal ideation, aggression, delusions, and hallucinations. Quality of life is markedly affected by HD; the “work” and “alertness behavior” domains of the Sickness Impact Profile are affected the most (Helder et al., 2001). One study that assessed health-related quality of life (HrQoL) by SF-36 showed that depressive mood and functional disability are key factors in HrQoL for people with HD (Ho et al., 2009).
Neuroimaging
Caudate atrophy, as measured by the ratio of intercaudate to outer-table distances, has traditionally been used as an index of striatal atrophy in HD, and it has been demonstrated to correlate well with the degree of cognitive impairment in early HD (Bamford et al., 1995). Subsequent studies, however, showed that a reduction in the volume of putamen, measured by MRI, was a more sensitive index of neurologic dysfunction than caudate atrophy (Harris et al., 1992; Rosas et al., 2001). This suggests that the putamen is underdeveloped in HD or that it is one of the earliest structures to atrophy. Striatal volume loss correlates with length of CAG repeats (Rosas et al., 2001). Several MRI volumetric and single photon emission computed tomography (SPECT) blood flow studies have shown that basal ganglia volume and blood flow are reduced even before individuals become symptomatic with HD (Harris et al., 1999; Aylward et al., 2004; Paulsen et al., 2006; Leoni et al., 2008) and that widespread degeneration occurs in early and middle stages of HD (Rosas et al., 2003). Patients with the akinetic-rigid form of HD are more likely to show striatal hyperintensity on T2-weighted MRI than patients with the choreic form of HD (Oliva et al., 1993). Both neuroimaging and postmortem studies indicate that regional thinning of the cortical ribbon, particularly involving the sensorimotor region, is affected in early HD and “might provide a sensitive prospective surrogate marker for clinical trials of neuroprotective medications” (Rosas et al., 2002; Rosas et al., 2005). Altered cortex morphology with enlargement of gyral crowns and abnormally thin sulci were demonstrated using three-dimensional MRI, obtained in 24 subjects with preclinical HD and 24 matched healthy subjects (Nopoulos et al., 2007). The authors concluded that “These findings lend support to the notion that, in addition to the degenerative process, abnormal neural development may also be an important process in the pathoetiology of HD.”
Using voxel-based MRI morphometry to identify differences between presymptomatic carriers and gene-negative controls, Thieben and colleagues (2002) found significant reductions in the gray matter volume in the left striatum, bilateral insula, dorsal midbrain, and bilateral intraparietal sulcus in the gene carriers. Other techniques, such as real-time sonography, showed abnormalities not only in the caudate but also in the substantia nigra of patients with HD (Postert et al., 1999). Using tensor-based morphometry, Kipps and colleagues (2005) were able to demonstrated progression of gray matter atrophy in presymptomatic HD mutation carriers compared to controls. Certain MRI techniques focusing on the gray matter have allowed successful identification of presymptomatic HD gene mutation (Klöppel et al., 2009). Other imaging studies have suggested that myelin breakdown and changes in ferritin iron distribution underlie the regional toxicity and may be important in pathogenesis of HD (Bartzokis et al., 2007). Pre-HD individuals have been found to have lower gross gray matter and white matter volume, and voxel-wise analysis demonstrated local gray matter volume loss, most notably in regions consistent with basal ganglia-thalamocortical pathways, whereas pre-HD individuals showed widespread reductions in white matter integrity, probably due to a loss of axonal barriers (Stoffers et al., 2010). In another study, not only were the volumes of the caudate nucleus and putamen reduced in premanifest HD long before predicted onset (>10.8 years), but atrophy of the accumbens nucleus and pallidum was also apparent in premanifest HD (van den Bogaard et al., 2010). In addition, the 12-month whole-brain atrophy rates were greater in early HD individuals as well as premanifest gene-positive carriers (less than 10.8 years from predicted diagnosis compared with controls) (Tabrizi et al., 2011). All gene-positive groups also showed faster rates of caudate and putamen atrophy over 12 months compared with controls. The whole-brain and caudate atrophy rates were found to correlate with the UHDRS total functional capacity score and with cognitive and quantitative motor measures. While these studies are intriguing, it is, however, still too early to conclude that neuroimaging can be used as a surrogate marker in future trials of putative neuroprotective agents.
In addition to striatal atrophy shown by neuroimaging studies, a variety of techniques, such as 18F-2-fluoro-2-deoxyglucose PET and SPECT, have been used to demonstrate hypometabolism and reduced regional cerebral blood flow in the basal ganglia and the cortex (Kuwert et al., 1990; Hasselbach et al., 1992; Harris et al., 1999). PET studies of presymptomatic HD carriers have shown initial (compensatory) thalamic hypermetabolism with subsequent decline heralding the onset of HD symptoms and signs in a setting of steady decline of striatal raclopride binding (Feigin et al., 2007).
Abnormalities in striatal metabolism measured by PET scans may precede caudate atrophy (Grafton et al., 1990). Regional cerebral metabolic rate of glucose consumption has been found to be decreased by 62% in the caudate, by 56% in the lenticular nucleus, and by 17% in the frontal cortex (Kuwert et al., 1990). Bilateral reduction in the uptake of technetium-99m HM-PAO and iodine-123 IMP in the caudate and putamen has been demonstrated by SPECT in patients with HD (Nagel et al., 1991). Using PET and [11C]flumazenil binding to GABA receptors in the striatum, Künig and colleagues (2000) found marked reduction in the caudate (but not in the putamen) of HD patients compared to normal controls. The authors interpreted these findings as indicative of compensatory GABA receptor upregulation in the striatal (putamen) GABAergic medium spiny neurons projecting to the pallidum. As microglial activation has been found to be correlated with neuronal degeneration, techniques designed to image microglial activation, such as [11C](R)-PK11195 (PK) PET, show that microglia activation correlates with severity of the disease (e.g., UHDRS score) and is inversely related to D2 density as demonstrated by raclopride PET studies (Pavese et al., 2006). Similar findings were reported in presymptomatic HD gene carriers who had lower striatal raclopride binding than the controls but significantly higher striatal and cortical PK binding (Tai et al., 2007). This study provided strong evidence that widespread microglial activation occurs even in preclinical stages of HD and that this early pathologic change correlates with striatal neuronal dysfunction (see below).
Neuropathology and neurochemistry
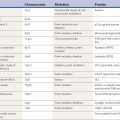
Pathologic studies have provided evidence for primary involvement of the basal ganglia–thalamocortical circuitry in HD (Joel, 2001). Postmortem changes in HD brains include neuronal loss and gliosis in the cortex and the striatum, particularly the caudate nucleus (Table 14.1). Morphometric analysis of the prefrontal cortex in HD patients reveals loss of cortical pyramidal cells, particularly in layers III, V, and VI (Sotrel et al., 1991). At the cortical level motor dysfunction seems to correlate with primary motor cortex cell loss whereas mood symptomatology is associated with cell loss in the cingulate cortex (Thu et al., 2010). Chorea seems to be related to the loss of medium spiny striatal neurons projecting to the lateral pallidum (GPe), whereas rigid-akinetic symptoms correlate with the additional loss of striatal neurons projecting to the substantia nigra compacta (SNc) and medial pallidum (GPi) (Albin et al., 1990b; Albin, 1995). Pathologic studies have suggested that the earliest changes associated with HD consist of degeneration of the striato-SNc neurons followed by striato-GPe and striato-SNr neurons and finally striato-GPi neurons (Hedreen and Folstein, 1995; Albin, 1995) (Fig. 14.2). There appears to be a correlation between the number of CAG repeats and the severity of pathology; longer trinucleotide repeat length is associated with greater neuronal loss in the caudate and putamen (Furtado et al., 1996). In addition, significant association between pronounced mood dysfunction and loss of the GABA(A) receptor marker in striosomes of the striatum has been found in an autopsy study of 35 HD brains and 13 controls (Tippett et al., 2007). Increased density of oligodendrogliocytes and intranuclear inclusions was found in the tail of the caudate nucleus among presymptomatic HD gene carriers, suggesting that pathologic changes occur long before the onset of symptoms (Gómez-Tortosa et al., 2001). In addition to neuronal loss in the basal ganglia, there is evidence of increased iron in this brain area as measured by MRI, which in part might contribute to the regional neurotoxicity (Bartzokis et al., 2007).
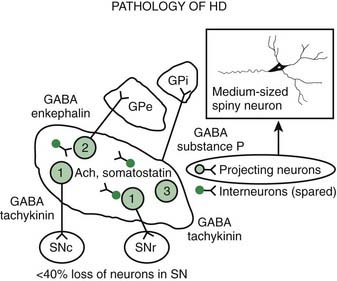
Using specific monoclonal antibodies against phosphorylated and nonphosphorylated neurofilaments and neuronal cell adhesion molecules, Nihei and Kowall (1992) found marked abnormalities in these cytoskeletal components in the striatal neurons of HD brains. The earliest degenerative changes, the loss of medium-sized spiny neurons containing calbindin, enkephalin, and substance P immunoreactivity, occur in the dorsomedial aspect of the caudate and putamen (Ferrante et al., 1991). The enkephalin neurons appear to die before the substance P neurons. In contrast, the spiny striatal cholinergic and somatostatin-containing interneurons are spared. As a result of these degenerative changes, the matrix zone of the striatum is reduced in size, while the patches (striosomes) remain unaltered. In one postmortem study, however, calbindin D28k mRNA in the striata of HD brains was normal, while the preproenkephalin neurons were markedly reduced in the HD caudate nucleus (Richfield et al., 1995). Another class of medium-sized (8–18 µm) striatal interneurons, characterized by intense immunostaining against calcium-binding protein calretinin, was found to be markedly increased in the striatum of brains of patients with HD compared to controls (Cicchetti and Parent, 1997). In contrast, the density of the larger, 17–44 µm, interneurons was markedly decreased. Since both populations of interneurons express calretinin, the implication of the findings of the study is that calretinin, for some as yet unknown reason, protects the medium-sized neurons but not the large neurons against HD-related degeneration.
Curtis and colleagues (2003) observed an interesting phenomenon in the brains of patients with HD. When they were compared to non-HD control brains, there was marked increase in cell proliferation in the subependymal layer, and the degree of cell proliferation increased with pathologic severity and increasing CAG repeats in the HD gene. These results suggest that progenitor cell proliferation and neurogenesis occur in the diseased adult human brain, providing further evidence for regeneration.
Huntingtin (Htt), the product of the HD gene (see below), is expressed throughout the brain in both affected and unaffected regions; therefore, its contribution to neurodegeneration in HD is unclear. Using quantitative in-situ hybridization methods, Landwehrmeyer and colleagues (1995) showed that the expression of Htt mRNA was not selectively increased in the neurons that were particularly susceptible to degeneration in HD. The authors suggested that this lack of correlation indicates that the gene mutation is not sufficient to produce the disease and that other factors probably also play a role in its pathogenesis. Since the mutated Htt is not limited to vulnerable neurons, other factors besides mutated Htt must play a role in the neuronal degeneration associated with HD (Gourfinkel-An et al., 1997). An immunohistochemical study showed that neuronal staining for Htt is reduced in the striatal medium-sized spiny neurons, but the large striatal neurons that are spared in HD retain normal levels of Htt (Sapp et al., 1997). Surprisingly, however, Htt staining was markedly reduced in both segments of the globus pallidus. This suggests a postsynaptic response to a reduction in striatal inputs; or it might indicate that the globus pallidus is for some yet unknown reason preferentially involved in HD.
N-terminal mutant Htt also binds to synaptic vesicles and inhibits their glutamate uptake in vitro (Li et al., 2000). Using a transgenic mouse model generated by introducing mutant polyQ tract into one of the mouse genes, Li and colleagues (2000) found aggregated Htt only in medium striatal neurons, neurons that are most susceptible to degeneration in HD. The aggregated Htt was first found in the neuronal nucleus and later in synaptic terminals. Scherzinger and colleagues (1999) found that the aggregation of Htt protein is time- and concentration-dependent, suggesting “a nucleation-dependent polymerization.” Aggregation was proportional to repeat length, and no aggregation could be induced for a repeat length of 27 or fewer glutamines. This might in part explain the inverse correlation of repeat length and age at onset. Since the probability of neural death in HD with time is constant and since the age at onset is inversely dependent on the length of repeats, it has been suggested that the polyglutamine aggregates are toxic when directed to the nucleus and cause the cell death in HD (Yang et al., 2002). This is supported by the physics of protein aggregation as a result of transition to a new intramolecular configuration (Perutz and Windle, 2001).
The finding of neuronal intranuclear inclusions and dystrophic neurites in the two regions that are most affected in HD, the cortex and striatum, has revolutionized our understanding of the genetic-pathologic mechanisms of this heredodegenerative disorder (DiFiglia et al., 1997). Using an antiserum against the NH2-terminus of Htt, DiFiglia and colleagues (1997) found intense labeling localized to neuronal intranuclear inclusions in brains of three juvenile and six adult cases of HD. These inclusions had an average diameter of 7.1 µm and were almost twice as large as the nucleolus. The neurons with these inclusions were found in all cortical layers and the medium-sized neurons of the striatum but not in the globus pallidus or the cerebellum. They were more frequent in juvenile HD, and in these cases, the inclusions were found not only in the nuclei but also in the cytoplasm. The inclusions were not found in the cortex of one subject who had the HD gene but was still asymptomatic but were found in his striatum, thus providing evidence that these changes preceded clinical onset and might be critical in the pathogenesis of the disease. In addition, the investigators found spherical and slightly ovoid dystrophic neurites in cortical layers 5 and 6. None of the control brains demonstrated this finding, and antibodies raised against the internal site of Htt failed to differentiate the HD brains from those of controls. Subsequent studies showed that the NH2-terminus of mutant Htt is cleaved by apopain and is ubiquitinated. Thus, ubiquitin staining can be also used to localize these inclusions. These inclusions are composed of granules, straight and tortuous filaments, and fibrils, and they are not membrane bound. Similar inclusions have been demonstrated in the HD transgenic mouse (Davies et al., 1997). Since the aggregates in the nucleus consist mostly of N-terminal fragments of the whole Htt protein, it has been postulated that the protein has to be cleaved before it enters the nucleus. Mutant Htt fragments accumulate in axon terminals and interfere with glutamate uptake (Li et al., 2000) and, as occurs in other polyglutamine disease, impair axonal transport (Szebenyi et al., 2003; Gunawardena and Goldstein, 2005). Using a yeast artificial chromosome transgenic mouse model of HD, Tang and colleagues (2005) found that repetitive application of glutamate elevates cytosolic Ca2+ levels in medium spiny neurons from the HD mouse but not from the wild-type or control mouse. They further found that the FDA-approved anticoagulant enoxaparin (Lovenox), a calcium blocker, the mitochondrial Ca2+ uniporter blocker ruthenium 360, and the dopamine depletor tetrabenazine (TBZ) (Tang et al., 2007) are neuroprotective. Thus, abnormal Ca2+ signaling seems to be directly linked with the degeneration of medium spiny neurons in the caudate nucleus in HD; therefore, calcium channel blockers could have a therapeutic potential for treatment of HD. Studies with the yeast artificial chromosome (YAC128) transgenic HD mouse model provide evidence for a connection between glutamate receptor activation, disturbed calcium (Ca2+) signaling, and apoptosis of HD striatal neurons (Tang et al., 2005, 2007). Furthermore, glutamate and dopamine signaling pathways acting via D1 receptors induce elevation of Ca2+ signals and cause TBZ-induced striatal cell loss in these mice, suggesting potential neuroprotective effects of this dopamine depletor. Further support and mechanistic insight comes from studies using Drosophila that express expanded full-length human huntingtin and elevated intracellular Ca2+ levels (Romero et al., 2008). The increased Ca leads to increased neurotransmitter release, motor dysfunction, and progressive neuronal degeneration. Partially blocking Ca channel activity, or partially impairing neurotransmission, restores the Ca levels and motor dysfunction and suppresses neurodegeneration. Since the findings in Drosophila of altered Ca homeostasis are in agreement with the early observations by Tang et al. (2007) in mammalian systems, Ca channel blockers may be useful in HD patients.
While some studies have shown a moderate (40%) degree of neuronal degeneration in the substantia nigra, particularly noticeable in the medial and lateral thirds (Oyanagi et al., 1989), this is probably not sufficient to cause bradykinesia in either choreic or rigid-akinetic HD patients (Albin et al., 1990a). Measurements of brain dopamine concentrations and cerebrospinal fluid (CSF) homovanillic acid levels have yielded conflicting results (Table 14.2). In one study, CSF homovanillic acid was normal in 51 patients with early HD, and there was no correlation between homovanillic acid levels and degree of parkinsonism (Kurlan et al., 1988). Dopamine, acetylcholine, and serotonin receptors are decreased in the striatum. Using PET imaging of dihydrotetrabenazine as a marker of striatal vesicular monoamine transporter (VMAT) type 2, Bohnen et al. (2000) found reduced binding especially in the posterior putamen, similar to Parkinson disease, particularly in patients with akinetic-rigid HD.
Table 14.2 Huntington disease: neurotransmitters, peptides, and receptors
CAT, choline acetyl transferase; DA, dopamine; GABA, gamma-aminobutyric acid; GAD, glutamic acid decarboxylase; NMDA, N-methyl-D-aspartate receptors; TRH, thyroid-releasing hormone.
Postsynaptic loss of dopamine receptors might be responsible for the parkinsonian findings in some HD patients (Sánchez-Pernaute et al., 2000). Dopamine D2 receptors, imaged with iodobenzamide–SPECT, have provided evidence of D2 receptor loss in HD (Brucke et al., 1991). Using PET to measure binding of specific D1 and D2 receptor ligands in the striatum, Weeks and colleagues (1996) found that these two receptors are markedly decreased in individuals who have the HD mutation but who are still presymptomatic. In another study, Andrews and colleagues (1999) showed that loss of striatal D1 and D2 binding as determined by PET was significantly greater in the known mutation carriers than in the combined at-risk and gene-negative patients. Although some studies found that the loss of D2 receptors correlates significantly with motor and cognitive slowing (Sánchez-Pernaute et al., 2000), other studies failed to show any correlation between clinical and psychological assessments and the loss of striatal D2 receptors (Pavese et al., 2003). Turjanski and colleagues (1995) showed that HD patients with rigidity had more pronounced reduction of these receptors (also measured by PET) compared with HD patients without rigidity. Since D2 binding was normal in a patient with chorea associated with systemic lupus erythematosus, the authors concluded that the presence of chorea is not determined by alterations in striatal dopamine receptor binding.
Loss of the medium-sized spiny neurons, which normally constitute 80% of all striatal neurons, is associated with a marked decrease in GABA and enkephalin levels. After examining two brains of presymptomatic individuals carrying the HD gene, Albin and colleagues (1992) concluded that striatal neurons projecting to the GPe and SN, but not those that project to the GPi, are preferentially involved in early stages of HD and even in the presymptomatic phase. Likewise, cannabinoid receptors are preferentially lost in the GPe (Blazquez et al., 2011). In contrast, the cholinergic and somatostatin striatal interneurons seem to be relatively spared in HD. Other neuropeptide alterations in HD include a decrease in substance P, cholecystokinin, and met-enkephalin and an increase in somatostatin, thyrotropin-releasing hormone, neurotensin, and neuropeptide Y. Prior to any clinical signs, in addition to the loss of cannabinoid receptors there is also loss of adenosine A2a receptor binding and increased GABA-A receptor binding in the GPe. As the disease progresses, there is typically loss of D1 receptors in the striatum as well as cannabinoid and D1 in the substantia nigra, eventually involving the rest of the basal ganglia. The preferential loss of enkephalinergic striatal neurons correlates with the appearance of chorea (Albin et al., 1992). Loss of substance P/dynorphin neurons correlates with the increase in dystonia in the later stages of disease (Table 14.2).
Evidence of immune activation not only of central microglia but also of the peripheral system is supported by the findings of increased levels of cytokines, such as interleukin 6 (IL-6) levels in HD gene carriers, with a mean of 16 years before the predicted onset of clinical symptoms (Björkqvist et al., 2008). The same study also found that monocytes from HD patients expressed mutant Htt. Autonomous immune activation was also noted in macrophages and microglia from HD mouse models, and in the CSF and striatum of HD patients. There are no reliable peripheral markers of HD, but 24S-hydroxycholesterol, previously found to be associated with neurodegeneration, has been found to be significantly decreased in patients in all stages of HD, although it was not different from controls among pre-manifest subjects, while caudate volume, as measured by MRI morphometry, was significantly decreased even in pre-HD subjects (Leoni et al., 2008). This is consistent with other MRI studies discussed earlier.
Although HD has been traditionally considered as a degenerative disease of the brain, mutant Htt is ubiquitously expressed throughout the body and HD is now recognized to be associated with abnormalities in peripheral tissues (van der Burg et al., 2009).
Genetics
For some time, HD has been regarded as truly an autosomal dominant disease in that homozygotes were thought to be no different from heterozygotes. However, subsequent studies have provided evidence that homozygotes (n = 8) have a more severe clinical course than that of heterozygotes (n = 75), even though the age at onset is similar (Squitieri et al., 2003). This suggests that the more rapid progression is a consequence of greater toxic effects because of doubling of mutated proteins and aggregate formation (see later). Linkage studies in HD families from various ethnic origins and countries have found that, despite the marked variability in phenotypic expression, genetic heterogeneity is unlikely. Localization of a gene marker near the tip of the short arm of chromosome 4 in 1983 by Gusella and colleagues (Gusella et al., 1983) initiated an intensive search for the abnormal gene, which was finally cloned 10 years later (Bates et al., 1991; Pritchard et al., 1991; Wexler et al., 1991; Huntington’s Disease Collaborative Research Group, 1993). The mutation that is responsible for the disease consists of an unstable enlargement of the CAG repeat sequence in the 5′ end of a large (210 kb) gene, huntingtin (Htt), also called IT15 (Huntington’s Disease Collaborative Research Group, 1993). This gene, located at 4p16.3, contains 67 exons and encodes a previously unknown 348 kDa protein, named Htt, without homology to known protein sequences (Fig. 14.3). The expanded CAG repeat, located in exon 1, alters Htt by elongating a polyglutamine segment near the NH2-terminus.
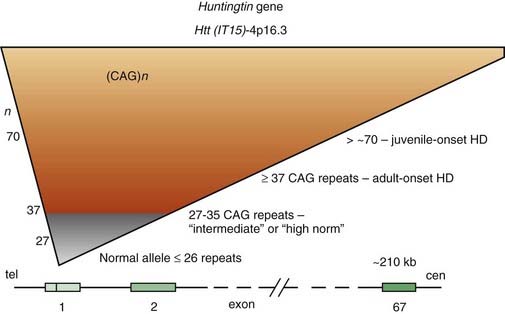
The study of HD, the prototype of trinucleotide repeat neurodegenerative diseases, has provided important insights into the pathogenesis of a growing number of disorders caused by accumulation of misfolded proteins (Everett and Wood, 2004). Whereas the number of repeats varies between 10 and 29 copies in unaffected individuals, the HD gene contains 36–121 of such repeats (Kremer et al., 1994). There are some alleles that do not cause HD in individuals carrying the allele but become unstable in the next generation. These alleles are termed intermediate, and their lower limit of CAG repeats might be as low as 27 (Nance, 1996). Of the 11 cases that lacked the triplet expansion, 5 had a prior history of Sydenham chorea during childhood, indicating the possibility of recurrence of autoimmune chorea. The intermediate-sized CAG repeats range from 27 to 35, but rare cases with CAG repeats in this range develop the full symptomatology of HD. One case, a 75-year-old man with 15-year history of the clinical manifestation of HD, was reported to have only 34 CAG repeats (Andrich et al., 2008). An autopsy-proven case of HD with only 29 CAG repeats has been reported (Kenney et al., 2007c; Kenney and Jankovic, 2008). We also studied a 50-year-old man with typical symptoms of HD since age 43 but without a family history of HD. His maternally derived allele had 17 CAG repeats, but his paternally derived allele had 45 CAG expands. His completely asymptomatic 80-year-old father and 76-year-old paternal uncle had 30 CAG repeats each. This patient indicates that a relatively short expansion (of 30 CAG repeats) might be unstable, particularly if it is of paternal origin, and might spontaneously expand in successive generations resulting in so-called sporadic HD (Alford et al., 1996). Since that time several cases of HD phenotype with less than 36 CAG repeats have been described and the clinical, genetic, and prognostic implications of the “intermediate” CAG repeat range is currently being re-examined. To assist with genetic counseling in these subjects, Hendricks et al. (2009) estimated that the probability that a male with intermediate or “high normal” (27–35) CAG repeats in one allele will have an offspring with an expanded penetrant allele ranges from 1/6241 to 1/951. Besides DNA CAG length, larger somatic CAG repeat length expansions (somatic instability) is associated with earlier disease onset and probably more rapid progression (Swami et al., 2009).
Approximately 11% of patients with clinically suspected HD exhibit no family history of HD, and some of these patients might have “new” mutations (Goldberg et al., 1993; Davis et al., 1994). In a study of 28 patients with clinically probable HD, but without a family history of HD, 25 (89%) patients were confirmed by DNA testing to have HD, and 5 of 16 (31%) patients with clinically doubtful HD had expanded triplet repeats, confirming the diagnosis of HD (Davis et al., 1994).
Several studies have demonstrated that the number of repeats inversely correlates with the age at onset (anticipation) (Duyao et al., 1993; Snell et al., 1993; Ashizawa et al., 1994; Gusela and MacDonald, 1995; Furtado et al., 1996; Brinkman et al., 1997; Nance et al., 1997; Maat-Kievit et al., 2002; Marder et al., 2002; Ravina et al., 2008). Brinkman and colleagues (1997) retrospectively examined the relationship between CAG length and age at onset and found a 50% probability of developing HD symptoms by age 65 when the CAG repeat length is 39 and by age 30 when the CAG repeat length is 50. The inverse relationship between age of onset and number of CAG repeats was confirmed in a Dutch cohort of 755 affected patients (Maat-Kievit et al., 2002). The correlation was stronger for paternal inheritance than for maternal inheritance. CAG repeat length also inversely correlates with late-stage outcomes, such as nursing home admission and placement of percutaneous endoscopic gastrostomy (Marder et al., 2002). One analysis, based on the data from the CARE-HD trial, modeled progression over 30 months on UHDRS and concluded that 10 additional CAG repeats were associated with a 7.7 (63%) increase on the UHDRS total motor score and 9.2 (81%) points in progression on the Independence Scale (Ravina et al., 2008). One study showed that increasing CAG repeat size in normal Htt gene diminishes the association between mutant CAG repeat size and disease severity and progression in HD (Aziz et al., 2009).
The Huntington Study Group is conducting two studies to prospectively characterize the transition from health to illness (phenoconversion): (1) PHAROS (Pilot Huntington At Risk Observational Study) and (2) PREDICT (Predictors of Huntington Disease). Although some studies suggest a possible correlation between the length of CAG repeats and the rate of progression (Illarioshkin et al., 1994; Brandt et al., 1996; Antonini et al., 1998), other studies have found no correlation between the CAG repeats and progression (Ashizawa et al., 1994; Kieburtz et al., 1994; Claes et al., 1995) or between age at onset and progression of disease (Feigin et al., 1995). The rate of disease progression is generally faster in paternally transmitted HD independent of the CAG repeat length (Ashizawa et al., 1994). Because of the poor correlation between the number of CAG repeats and the rate of progression, some investigators have argued that the number of CAG repeats should not be disclosed to the patient or even to the physician. This may not, however, be practical and many patients insist on knowing the number of CAG repeats. We believe that (1) the CAG repeat size should be disclosed to appropriately informed physicians and counselors who take care of the patient, (2) appropriate training should be provided to inform physicians and counselors about the implications of the CAG repeat size in HD, and (3) information regarding the CAG repeat size should be disclosed to patients on the patient’s request, given that appropriate counseling is made available to the patient (Jankovic et al., 1995). When appropriate genetic counseling and a multidisciplinary approach are used in presymptomatic testing, the risk of adverse events such as psychological distress and depression requiring hospitalization or leading to attempted suicide may be as low as 2% (no difference between carriers and noncarriers) (Goizet et al., 2002). Noninvasive prenatal diagnosis of HD was confirmed in three of four at-risk fetuses in whom the father was affected with HD by direct analysis of the cell-free fetal DNA in maternal plasma (Bustamante-Aragones et al., 2008).
There is no difference in the mean number of repeats between patients presenting with psychiatric symptoms and those with chorea and other motor disorders, although, as expected, the rigid juvenile patients have the largest number of repeats (MacMillan et al., 1993; Nance and the US Huntington Disease Genetic Testing Group, 1997). The trinucleotide repeat is relatively stable over time in lymphocyte DNA but may be unstable in sperm DNA. This appears to account for the marked increase in the number of trinucleotide repeats to offspring by affected fathers, leading to a 10 : 1 ratio of juvenile HD when the affected parent is the father. However, after isolating X- and Y-bearing sperm of HD transgenic mice, Kovtun and colleagues (2004) found that the CAG distribution is the same as that in the founding fathers, suggesting that the “gender-dependent changes in CAG repeat length arise in the embryo.”
Analyzing DNA for the expansion of respective trinucleotide repeats utilizing polymerase chain reaction and Southern blotting has provided the means for a reliable diagnostic test that does not require participation of other family members. Such a test is helpful not only in confirming the diagnosis in index cases, but also in clarifying the diagnosis in atypical cases and asymptomatic at-risk individuals. In one study, 4 of 15 tests on presymptomatic individuals using linkage analysis were positive for the HD gene (Meissen et al., 1988). While all the individuals with positive results experienced transient symptoms of depression, none reported suicidal ideation. One year after the results of gene testing were disclosed, both increased-risk and the decreased-risk individuals had overall no increase in depression or deterioration in psychological well-being (Wiggins et al., 1992). A high rate of suicidal ideation, however, was found in a Swedish study of 13 HD carriers and 21 noncarriers (Robins Wahlin et al., 2000). In another study of 171 presymptomatic gene carriers compared to 414 noncarriers, Kirkwood and colleagues (2000) found that the carriers performed significantly worse on the digit symbol, picture arrangement, and arithmetic subscales of the Weschler Adult Intelligence Scale–Revised and various movement and choice–time measures. Other studies have addressed the natural history and progression of HD in the early and middle stages (Kirkwood et al., 2001). An adjustment to results of testing appears to depend more on the individual’s psychological makeup before the testing than on the testing itself (Meiser and Dunn, 2000). Because of potential psychological and legal implications of positive identification of a HD gene mutation in an asymptomatic, at-risk individual, predictive testing must be performed by a team of clinicians and geneticists who are not only knowledgeable about the disease and the genetic techniques, but also sensitive to the psychosocial and ethical issues associated with such testing. Although, as a result of the discovery of the HD gene, the cost of presymptomatic testing has been substantially reduced, the currently recommended extensive pretesting and posttesting counseling is still quite costly. An assessment of the cost–benefit ratio should be carried out. When the DNA test for HD became available, it was thought that demand for testing for HD in asymptomatic at-risk individuals would be high (Tyler et al., 1992), but recent studies have demonstrated a decline in the number of applicants (Maat-Kievit, et al., 2000). In one study only 3–4% of at-risk individuals have requested a presymptomatic test, and requests for prenatal diagnosis are rare (Lacone et al., 1999).
Important insights into the potential function of Htt have been gained by the study of various mouse models in which the HD homolog gene was inactivated (knock-out models) (Duyao et al., 1995) or expanded CAG repeats are introduced into the mouse hypoxanthine phosphoribosyltransferase gene (knock-in models) (Reddy et al., 1999). Homozygous inactivation resulted in embryonic death, suggesting that Htt is critical for early embryonic development. Since this model does not mimic adult HD and homozygote individuals apparently are indistinguishable from heterozygotes, it suggests that the HD gene mutation involves “gain” rather than “loss” in function. This hypothesis, however, has been challenged because the earlier studies suggesting no phenotypic difference between heterozygous and homozygous HD were based on linkage studies and focused predominantly on the age at onset (Cattaneo et al., 2001). Furthermore, homozygous transgenic mice expressing mutant Htt cDNA have a shorter lifespan than that of heterozygous mice. This suggests that either a double-dose of mutant Htt or loss of the normal allele (loss of function) contributes to the disease. Either wild-type or mutant Htt is needed for normal brain development. Huntingtin is associated with vesicle membranes and microtubules and as such probably has a role in endocytosis, intracellular trafficking, and membrane recycling. Thus, HD appears to result from a new toxic property of the mutant protein (gain of function) and loss of neuroprotective activity of the normal Htt (loss of function) (Cattaneo et al., 2001; Borrell-Pagès et al., 2006).
There are currently three types of HD transgenic mouse models: (1) mice expressing fragments, usually one or two exons of the human huntingtin gene that contain the polyglutamine expansion (R6/2 and N-171-82Q mice); (2) transgenic mice expressing the full-length human huntingtin gene with expanded polyglutamine tract (YAC128 mice); and (3) knock-in mice with pathogenic CAG repeats inserted into the existing CAG expansion (HdhQIII) (Hersch and Ferrante, 2004). Interestingly, the more genetically accurate the model, the more variable and subtle is the phenotype. Thus, the fragment models are used more frequently for therapeutic research, and these studies are then confirmed in the full-length models. Full-length Htt, when expressed in transgenic mice, does not appear to produce neurologic disease, but only the shortened or truncated form of the protein appears to be toxic to neuronal cells. Except for the mouse model that expresses the full-length Htt gene with 72 CAG repeats and displays selective striatal pathology at 12 months of age (Hodgson et al., 1999), all transgenic-mouse cell lines develop intranuclear inclusions and neurodegeneration in widespread areas of the brain. Using viral-vector-mediated expression, several investigators have been able to produce aggregates starting within 5 days after transduction and associated with striatal degeneration in rats and even in primates (Kirik and Bjorklund, 2003). Formation of inclusions correlated with longer CAG expansions and shorter protein fragments. Overexpression of a short fragment of Htt, carrying 82 glutamine repeats, in the putamen produced progressive dyskinesia and putaminal intranuclear aggregates and striatal degeneration (Regulier et al., 2003). Although intranuclear inclusions in HD are traditionally thought to be neurotoxic, recent studies provide evidence that they are actually neuroprotective (Arrasate et al., 2004). Using a robotic microscope imaging system that can follow the survival of individual neurons over several days, the authors found that neuronal death was related to the length of polyglutamine expansions in the Htt protein, not to the increase in size or number of inclusion bodies. Therefore, this study suggests that the inclusions protect neurons by reducing the levels of toxic diffuse forms of mutant Htt. Furthermore, compounds that promote the formation of inclusions lessen the pathology of HD (Bodner et al., 2006).
The R6/2 transgenic mouse line has been studied most extensively (Mangiarini et al., 1999; Ona et al., 1999). These mice develop normally until 5–7 weeks of age, when they start manifesting irregular gait, abrupt shuddering movements, and resting tremors, followed by epileptic seizures, muscle wasting, and premature death at 14–16 weeks. Striatal neurons show ubiquitinated neuronal inclusions, similar to those seen in the brains of patients with HD. Analysis of glutamate receptors in symptomatic 12-week-old R6/2 mice revealed decreases compared with age-matched littermate controls (Cha et al., 1998). Other neurotransmitter receptors that are known to be affected in HD were also decreased in R6/2 mice, including dopamine and muscarinic cholinergic, but not gamma-aminobutyric acid (GABA) receptors. D1-like and D2-like dopamine receptor binding was drastically reduced to one-third of control in the brains of 8- and 12-week-old R6/2 mice. Altered expression of neurotransmitter receptors precedes clinical symptoms in R6/2 mice and may contribute to subsequent pathology. Biochemical analysis at 12 weeks shows a marked reduction in striatal aconitase (Tabrizi et al., 2000), indicative of damage by superoxide (O2) and peroxynitrite (ONOO−), similar to human HD (Tabrizi et al., 1999). In addition, the R6/2 transgenic mouse has a marked reduction in striatal and cortical mitochondrial complex IV activity and increased immunostaining for inducible nitric oxide synthase and nitrotyrosine (Tabrizi et al., 2000). Since these changes occur before evidence of neuronal death, the described findings suggest that mitochondrial dysfunction and free radical damage play an important role in the pathogenesis of HD. Indeed, mutant Htt has been found to attach to the outer mitochondrial membrane, causing the mitochondria permeability transition pores to open, allowing an influx of calcium and releasing cytochrome c (Choo et al., 2004). N171 mice, in which the transgene is driven by the prion promoter, have been used also as models of HD, but they have not been subjected to as rigorous examination as the R6/2 mice. In another study mice in which the PGC-1a gene had been knocked out developed brain lesions in the striatum and PGC-1α levels in those particular neurons were much lower among mice with the HD mutation than in normal mice (Cui et al., 2006). Since PGC-1α is involved in energy metabolism, this study shows that transcriptional repression of PGC-1α leads to mitochondrial dysfunction and supports other studies suggesting that energy deficits contribute to neurodegeneration in HD (Ross and Thompson, 2006). One study of polymorphism in PGC-1a gene in over 400 individuals with HD concluded that PGC-1α (which is inhibited by mutant Htt) has a small modifying effect on the progression of HD (Taherzadeh-Fard et al., 2009). In addition to the mouse models, there are several Drosophila models (Zoghbi and Botas, 2002). These are now being utilized to better understand the pathogenic mechanisms of HD-related neurodegeneration and for testing drugs that might have a potential for favorably modifying the disease (Agrawal et al., 2005; Imarisio et al., 2008). For example, expanded full-length Htt (128QHtt) mutant Drosophila exhibit behavioral, neurodegenerative, and electrophysiologic phenotypes, thought to be caused by a Ca2+-dependent increase in neurotransmitter release (Romero et al., 2008). According to this study and in contrast to previous belief, the expanded protein does not have to be cleaved to be neurotoxic. Since partial loss of function in synaptic transmission (syntaxin, Snap, Rop) and voltage-gated Ca2+ channel genes suppresses both the electrophysiologic and the neurodegenerative phenotypes, it has been postulated that increased neurotransmission plays a key role in the neuronal degeneration caused by expanded CAG repeats in human HD.
In addition to the sub-primate models, progress is being made in developing a transgenic model of HD in a rhesus macaque monkey that expresses polyglutamine-expanded Htt (Yang et al., 2008; Wang et al., 2008a). The primate model was produced by injecting 130 mature rhesus oocytes with high titer of lentiviruses expressing Htt gene exon1 with 84 CAG repeats and green fluorescent protein (GFP) gene. After a transfer or 30 embryos into 6 surrogates, 5 liveborns were delivered at full term, including two sets of twins. Unfortunately, only one monkey survived one month and the others died at birth or shortly thereafter. The monkey that survived one month presumably demonstrated chorea and dystonia 2 days after birth, but these involuntary movements are difficult to characterize in infants. Brain autopsy showed abundant distribution of transgenic Htt in the cortex and striatum and nuclear inclusions and neuropil aggregates. Similar to the HD mouse models, which demonstrate more severe phenotypes, the transgenic monkeys expressing exon1 Htt with a 147-glutamine repeat (147Q) died early and showed abundant neuropil aggregates in swelling neuronal processes (Wang et al., 2008a).
Pathogenesis
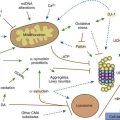
Multiple processes have been postulated to relate CAG expansion to neurodegeneration, including aberrant interaction between the mutant Htt and transcriptional factors and coactivators (Anderson et al., 2008; Ross and Tabrizi, 2011) (Fig. 14.4). For example, aberrant interaction with histone acetyltransferase interferes with acetylation and deacetylation of histones. Abnormal histone deacetylation, catalyzed by histone deacetylases, leads to tightly packed chromatin structure and transcriptional repression. Histone deacetylase inhibitors have been suggested as potential treatment strategies in HD and other neurodegenerative disorders (Chuang et al., 2009). Mutant Htt has been also shown to abnormally interact with histone acetyltransferases interfering with acetylation of histone proteins and also leading to transcriptional repression. Decreasing transcriptional activation by abnormal interaction between mutant Htt and the transcription factor cAMP-response element binding protein (CREB)-binding protein (CBP), a mediator of cell survival signals containing a short polyglutamine stretch, is another mechanism of cell death (Nucifora et al., 2001; Mantamadiotis et al., 2002). CBP is a coactivator for the transcription factor CREB linking DNA-binding proteins to RNA polymerase II transcription complex and functioning in histone acetyltransferase complex. By recruiting CREB into cellular aggregates, the mutant Htt prevents it from participating as a coactivator in CREB-mediated transcription, supporting the toxic gain-of-function mechanism of cell death in HD. The polyglutamine-containing domain of abnormal Htt protein directly binds the acetyltransferase domains of CBP and p300/CBP-associated factor (P/CAF) (Steffan et al., 2001). This reduces acetyltransferase activity and decreases acetylation of histones, proteins that package DNA. This in turn results in a decrease in gene transcription. Therefore, histone-deacetylase inhibitors could prevent HD-related neurodegeneration. Indeed, inhibition of histone deacetylase by sodium butyrate has been shown to partially protect HD R6/2 mice from neurodegeneration and against 3-nitropropionic acid neurotoxicity (Ferrante et al., 2003). According to current understanding, an inhibitor of κB kinase-mediated phosphorylation of the amino terminus of huntingtin targets mutant huntingtin into the nucleus, where it interferes with gene transcription (Krainc et al., 2010a, 2010b). When acetylated by CBP, mutant huntingtin is transported into autophagosomes for degradation, but mutant huntingtin that is not cleared accumulates in the nucleus and cytoplasm in the form of inclusions. Thus, recent neuroprotective strategies focus on increasing phosphorylation or acetylation of the mutant huntingtin to improve its clearance and reduce its neuronal toxicity. Several studies have demonstrated impaired autophagy in animal models of HD, suggesting that inefficient engulfment of cytosolic components by autophagosomes may contribute to the HD-related neurodegeneration (Martinez-Vicente et al., 2010).
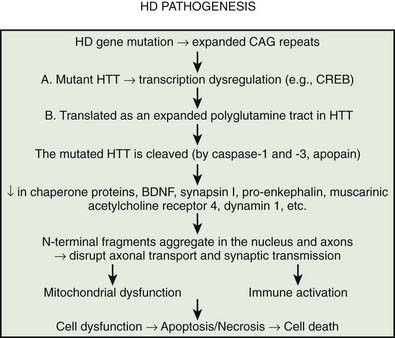
Transcriptional dysregulation as a pathogenic mechanism of neurodegeneration in HD has been also suggested by the study by Dunah and colleagues (2002), which showed that Htt interacts with the transcriptional activator Sp1 and coactivator TAFII130. Sp1, the first of many transcriptional activators isolated from human cells, binds to GC-box on the promoter region of DNA. It contains glutamine-rich activation domains that selectively bind to core components of the transcriptional machinery, such as the TAFII13 subunit of TFIID, which contains TATA-box-binding protein and multiple TATA-box-binding protein-associated factors. Therefore, one of the earliest steps in the development of HD may involve deregulation of specific transcriptional programs as suggested by Dunah and colleagues (2002), who demonstrated that mutant Htt inhibited Sp1 binding to DNA in postmortem brain tissues of both presymptomatic and affected HD patients. As a result, the RNA polymerase II is not able to locate the dopamine D2 receptor promoter region, and the gene cannot be transcribed (Freiman and Tjian, 2002).
It has been postulated that the common theme linking all the diseases pathogenetically with polyglutamine-tract expansion is that the various proteins (e.g., Htt, atrophin, androgen receptor, and ataxins), as a result of an increase in size of their intrinsic polyglutamine sequences, accumulate in the nucleus, forming insoluble amyloid-like fibrils and then somehow interfere with normal cellular metabolism (Davies et al., 1998). Proteolytic cleavage of a glutathione-S-transferase-Htt fusion protein results in spontaneous formation of insoluble aggregates. The aggregate formation is directly related to Htt and polyglutamine length; a polyglutamine tract of 51 glutamines or longer results in these aggregates (Scherzinger et al., 1997; Martindale et al., 1998). It has been hypothesized that after the expanded CAG repeat in the HD gene is transcribed in the Htt mRNA and then translated as an expanded polyglutamine tract in Htt, the mutated protein is cleaved (e.g., by caspase, apopain). This, the earliest step in the pathogenesis of HD, is followed by the liberation of polyglutamine-containing fragments, which are linked by transglutaminase to other proteins containing lysine residues, resulting in formation of perinuclear aggregates, eventually leading to neuronal apoptosis (Bates, 2003). The formation of inclusions, however, is not required for the initiation of cell death; in fact, neuronal intranuclear inclusions might have a protective role (Saudou et al., 1998). Kuemmerle and colleagues (1999) found that aggregate formation occurs predominantly in spared striatal interneurons rather than in the spiny neurons that are typically affected in HD. Thus, Htt aggregates do not predict neuronal death and might actually protect against polyglutamine-induced neurotoxicity. Several lines of evidence suggest that the expanded polyglutamine fragment of mutant Htt decreases protein degradation by the proteasome (Martin, 1999; Bence et al., 2001). The protein aggregates appear to be simultaneously inhibitors of the ubiquitin-proteasome system and the products that result from its inhibition. Arfaptin 2 has been shown to promote Htt protein aggregation, possibly by impairing proteasome function (Peters et al., 2002). Aggregating proteins besides Htt, including α-synuclein, tau, mutant SOD1, and mutant ubiquitin, inhibit proteasome function (Cookson, 2004). In addition to the brain, proteasomal dysfunction has been demonstrated in the skin fibroblasts of patients with HD (Seo et al., 2004). In the striatum, however, dysfunction of the proteasomal system correlated with neuronal pathology and decreased levels of brain-derived neurotrophic factor (BDNF), which is normally upregulated by wild-type Htt; decreased mitochondrial complex II/III activity; and increased levels of ubiquitin (Seo et al., 2004).
Abnormal clearance of mutant Htt due to impaired phosphorylation, which normally tags certain amino acids with phosphates for destruction by the cellular waste handling systems, proteasomes and lysosomes, is increasingly recognized as an important mechanism of cell loss in HD and in other neurodegenerative disorders. Htt is phosphorylated by the inflammatory kinase IKK, which regulates additional posttranslational modifications, including Htt ubiquitination, sumoylation, and acetylation, and increases Htt nuclear localization, cleavage, and clearance mediated by lysosomal-associated membrane protein 2A (LAMP-2A) and Hsc70 (Thompson et al., 2009). The mutant Htt interferes with proteasome activity and lysosomal degradation becomes impaired, partly as a result of age-related reduction of the LAMP-2A. Furthermore, impaired phosphorylation on serines 13 and 16 of the N-terminal 17 amino acids of Htt has been demonstrated to impair clearance of mutated Htt, but genetic engineering to either mimic phosphorylation (phosphomimetic) or resist it (phosphoresistant) in mice prevented formation of Htt clumps (Gu et al., 2009). Thus these and other studies show that uncleared mutant Htt and Htt fragments can accumulate and become toxic by interfering with cellular function and axonal transport.
The impairment of proteasomal degradation of mutant Htt leads to caspase-mediated apoptosis (Jana et al., 2001). Jana and colleagues (2001), for example, found “massive accumulation” of polyubiquitinated mutant, but not normal, Htt after 2 days of expression in mouse neural cells, indicating lack of proteasomal processing of the mutant protein. Decline of proteasomal activity with time in the soluble fraction of the cell and an increase in the insoluble portion correlated with increased accumulation of proteasomal components in cell aggregates. The expression of mutant Htt (or inhibition of proteasomes with lactacystin) was associated with activation of caspases and release of mitochondrial cytochrome c, indicators of apoptosis. Studies have demonstrated that wild-type Htt upregulates transcription of cortically derived BDNF (Zuccato et al., 2001). Thus, a decrease in BDNF appears to play an important role in the pathology and pathogenesis of HD by causing dysfunction of striatal enkephalinergic neurons (Canals et al., 2004; Cattaneo et al., 2005). Furthermore, since wild-type Htt has been found to transport BDNF, it is not surprising that mutated Htt interferes with intracellular transport of this trophic factor (Gauthier et al., 2004). This suggests that as a result of mutated Htt in HD, there is insufficient neurotrophic support for striatal neurons and that treatment with BDNF might possibly restore or rescue the damaged striatal neurons. Rapamycin, an antibiotic that acts as a specific inhibitor of mTOR, a kinase that regulates important cellular processes, has been found to attenuate Htt accumulation and cell death in cell models of HD, possibly by enhancing the clearance of proteins that abnormally accumulate and aggregate in the cytoplasm (Ravikumar et al., 2004). Therefore, drugs such as rapamycin might play a role also in other neurodegenerative diseases that result from accumulation of unwanted protein aggregates.
Htt also interacts with chaperone proteins that unfold the expanded mutant Htt protein, preventing the aggregate formation in a cell culture model (Jana et al., 2001). Studies have shown that processing of polyglutamine-containing proteins by proteases (e.g., caspases) liberates truncated fragments with expanded polyglutamine tracts that can form aggregates through hydrogen bonding or transglutaminase activity and may be toxic to the cell. Abnormalities in proteasome function are associated with altered expression of stress-response proteins or heat-shock proteins that function as chaperones. The chaperones normally maintain proteins in an appropriate conformation and denature misfolded proteins (aggregates) (Kobayashi and Sobue, 2001). Overexpression of these chaperone proteins protects cells from toxicity induced by Htt with expanded polyglutamines (Jana et al., 2001; Kobayashi and Sobue, 2001). It has been postulated that the mutant Htt with an expanded polyglutamine tract, which is resistant to proteasome-mediated protein processing, overloads the proteasome machinery, enhancing the pathogenic effects of mutant Htt. Since truncated proteins that contain the expanded polyglutamine tract appear to be more toxic than the full-length protein, blocking proteolytic processing, aggregation, and nuclear uptake might be reasonable therapeutic strategies.
To examine the possible role of caspase-1, a cysteine protease that is important in regulating apoptosis, in the pathogenesis of cell death associated with HD, Ona and colleagues (1999) crossbred R6/2 with a transgenic mouse expressing dominant-negative mutant of caspase-1 in the brain. The inhibition of caspase-1 delayed the disease onset by 7.3 days and prolonged survival by 20.4 days. Furthermore, it significantly delayed the appearance of neuronal inclusions and neurotransmitter alterations. Although abnormal protein aggregation has been postulated to play an important role in the pathogenesis of HD, DiFiglia and colleagues (Kim et al., 1999) showed that aggregation was not intimately involved in cell death. While general inhibitors of caspases prolonged cell life, they did not alter aggregation, whereas specific caspase 3 inhibitors inhibited aggregation but had no effect on survival. Thus, continuous influx of the mutant protein is required to maintain inclusions. To determine whether caspase cleavage of Htt is a key event in the selective neurodegeneration in HD, Graham et al. (2006) found that mice expressing mutant Htt, resistant to cleavage by caspase-6 but not caspase-3, maintained normal neuronal function and did not develop striatal neurodegeneration and were protected against neurotoxicity induced by multiple stressors including NMDA, quinolinic acid, and staurosporine. These findings suggest that proteolysis of Htt at the caspase-6 cleavage site is critical for neurodegeneration in HD, which is enhanced by excitotoxicity. This and other studies provide strong evidence that caspases are important in the pathogenesis of cell death in HD and other neurodegenerative diseases.
The finding that N-methyl-D-aspartate (NMDA) receptors are markedly reduced in the putamen and cerebral cortex was the first clue that suggested an NMDA-mediated excitotoxicity as a mechanism of neuronal degeneration in HD (Young et al., 1988; Tabrizi et al., 1999). In addition to the 92% reduction in NMDA binding, phencyclidine binding was reduced by 67% and quisqualate by 55% in the same HD brains. The NMDA receptor density was decreased by 50% in the brain of a presymptomatic HD gene carrier, suggesting that the loss of the excitatory receptors occurred early in the course of the disease (Albin et al., 1990b). In contrast to the study by Young and colleagues (1988), no single excitatory amino acid receptor was selectively affected. Using quantitative in-vitro autoradiography, Dure and colleagues (1991) found a 50–60% reduction in binding sites of all the major excitatory receptors (NMDA, MK-801, glycine, kainate, and AMPA) in the caudate nucleus of HD brains. Furthermore, as was noted earlier, mutated Htt protein might interfere with vesicular glutamate uptake and thus contribute to excitotoxicity (Li et al., 2000).
In support of the excitotoxic theory is the observation that certain excitatory neurotoxins produce useful animal models of HD. Although the activity of 3-hydroxyanthranilic acid oxidase, the synthesizing enzyme for quinolinic acid, is markedly increased in the striatum of HD brains (Schwarcz et al., 1988), the concentration of quinolinic acid, an NMDA agonist, in the CSF and brains of patients with HD is normal (Heyes et al., 1991). Nevertheless, intrastriatal injections of quinolinic acid preferentially damage the medium-sized spiny neurons containing calbindin Dk28, enkephalin, and substance P, the very same neurons that degenerate in HD (Beal et al., 1991a; Ferrante et al., 1993). The striatal patch-matrix pattern closely resembles that seen in HD. Pretreatment with MK-801, a noncompetitive NMDA antagonist, protects the striatum from the quinolinic acid neurotoxicity. Despite neuronal loss and astrogliosis, the axons are spared, which might account for relatively normal dopamine, norepinephrine, and serotonin levels. GABA levels are markedly decreased in the GPe of these animals, similar to HD, indicating early loss of GABA afferents from the striatum to the GPe in both HD and the experimental model. In addition to pathologic and biochemical similarities between this animal model and HD, the animals also exhibit choreic movements. In addition to quinolinic acid, another excitotoxin that has been implicated in the pathogenesis of HD is glutamate. The observation that glutamate levels are decreased in the striatum of HD brains has been interpreted to be a result of chronic failure of the normal reuptake mechanism for glutamate released from the corticostriatal afferent terminals (Perry and Hansen, 1990). The resulting increase in concentration of glutamate at synapses might cause damage to the striatal neurons. The increased extracellular glutamate might also explain the finding of increased CSF glutamate in some patients with HD. Additional support for the excitotoxic theory of HD is provided by the observation that a lesion with the excitotoxin ibotenic acid, a glutaric acid agonist, in a striatum of the baboon produces behavioral and neuropathologic changes similar to those seen in patients with HD (Hantraye et al., 1990).
One hypothesis of mechanisms of cell death in HD is that a defect in mitochondrial energy metabolism makes certain neurons more vulnerable to the excitotoxic effects of endogenous glutamate (Beal, 1992; Bossy-Wetzel et al., 2008). It has been postulated that mutant Htt might cause mitochondrial dysfunction by either interfering with transcription of nuclear-encoded mitochondrial proteins or by causing damage directly to the mitochondria and interfering with Ca2+ buffering or mitochondria membrane potential (Bossy-Wetzel et al., 2008). Panov and colleagues (2002) showed that lymphoblast mitochondria from patients with HD and brain mitochondria from transgenic HD mice have a lower membrane potential and depolarize at lower calcium loads than mitochondria from controls. Since this defect preceded the onset of pathologic or behavioral abnormalities by months, the authors concluded that “mitochondrial calcium abnormalities occur early in HD pathogenesis and may be a direct effect of mutant Htt on the organelle.” Intrastriatal injections of inhibitors of oxidative phosphorylation, such as amino-oxyacetic acid or MPP+, or a systemic administration of 3-nitropropionic acid (3-NP), a mitochondrial poison, into animals produce even more striking resemblance to HD than when “pure” excitotoxins are used (Beal et al., 1991b; Storey et al., 1992; Brouillet et al., 1993; Beal et al., 1993). Studies have shown that 3-NP induces striatal neurodegeneration via c-Jun N-terminal kinase/c-Jun module (Garcia et al., 2002). Accidental ingestion of 3-NP has resulted in putaminal necrosis and delayed onset of dystonia and chorea in humans (Ludolph et al., 1991). The age-dependent neurotoxicity of 3-NP, demonstrated by increased striatal lactate levels in the older animals, appears to be related to the effect of 3-NP on energy metabolism. Severe defects have been demonstrated in the activities particularly of complexes II and III in the caudate nuclei of HD patients (Gu et al., 1996; Tabrizi et al., 1999). Using spectrophotometric techniques, Browne and colleagues (1997) found 29% and 67% reductions in the activity of the complex II to III in the caudate and putamen of HD brains, respectively. In addition, there was a 62% reduction in the complex IV activity. This finding provides further support for the potential role of oxidative damage in the pathogenesis of HD. The possibility that mitochondrial function is impaired in HD is supported by the observation of 5-fold to 11-fold increase in cortical mitochondrial DNA deletion in the brains of HD patients (Horton et al., 1995). Dichloroacetate, which stimulates pyruvate dehydrogenase complex, has been found to increase survival and improve motor function and prevent striatal atrophy in R6/2 and N171-82Q transgenic mouse models of HD (Andreassen et al., 2001). A “toxic-oxidation cycle” has been proposed as an important mechanism in neurodegeneration associated with HD (Kovtun et al., 2007). According to this hypothesis the accumulation of DNA oxidized bases, as a result of impaired function of the repair enzyme 7,8-dihydro-8-oxugunaine-DNA glycosylate (OGG1), overwhelms the repair machinery in the genome with generation of single-strand breaks (SSB), ultimately leading to further CAG expansion and neuronal death.
Defects in mitochondrial oxidative phosphorylation have been postulated to decrease cellular ATP production, resulting in a concomitant decrease in sodium-potassium ATPase activity and partial membrane depolarization. Under normal circumstances, the NMDA receptor-associated channels are blocked by a voltage-dependent Mg2+ system that pumps Mg2+ out of the cell. As a result of decreased ATPase activity, caused by the inhibition of oxidative phosphorylation, the normal resting potential cannot be maintained, resulting in the opening of the channel and influx of calcium. This triggers a cascade of events leading to a production of free radicals and associated oxidative damage to various cellular elements. Thus, as a result of inhibition of oxidative phosphorylation, even low levels of excitatory amino acids become toxic, a process that is referred to as slow excitotoxicity. More recent studies provide evidence that HD is associated with transcriptional dysregulation associated with interference in transcription mediated by Sp1 and CREB-binding protein (CBP) coupled with mitochondrial impairment due to reduced activity of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), which regulates the expression of many nuclear-encoded mitochondrial proteins and proteins that provide protection against reactive oxygen species (ROS) (Greenamyre, 2007).
It has been suggested that in HD, as a result of prolonged excitatory neurotransmission, certain neurons become “exhausted” and switch from aerobic to anaerobic glycolytic metabolism, leading to the production and accumulation of lactate. Increased lactate concentration in the cerebral cortex of patients and animal models of HD, postulated to be due to global cerebral energy failure, has been demonstrated by proton magnetic resonance spectroscopy (Jenkins et al., 1998). Subsequently 1H magnetic resonance spectroscopy was used to measure N-acetylaspartate, creatine, and choline, and a reduction in these markers correlated well with the progression of the disease and with striatal atrophy (Sánchez-Pernaute et al., 1999). Administration of agents that improve energy metabolism, such as CoQ10 and nicotinamide, might protect animals against toxicity produced by malonate, a complex II inhibitor (Beal et al., 1994). Using 31P magnetic resonance spectroscopy to measure lactate in the muscle and 1H magnetic resonance spectroscopy to measure lactate in the basal ganglia and the cortex, Koroshetz and colleagues (1997) showed that treatment with CoQ10 was associated with a significant decrease in cortical lactate concentrations in patients with HD. There is, however, no evidence that this biochemical change is in any way associated with clinical improvement or slowing of disease progression. Furthermore, increased lactate could not be confirmed by other studies (Hoang et al., 1998). A National Institutes of Health-sponsored multicenter study designed to evaluate the efficacy of CoQ10 and remacemide, an NMDA antagonist, is currently being conducted by the Huntington Study Group (see later).
The obvious question still to be answered is how the mutation in the HD gene leads to selective damage of certain neuronal populations. One explanation is that there are certain genes or proteins that are uniquely expressed in the striatum and cortex making these structures particularly vulnerable to neurodegeneration. For example, the CalDAG-GEFI gene has been found to be markedly downregulated in the brains of individuals with HD as well as in mouse models of the disease (Crittenden et al., 2010). By following mutant mice for up to 9 months, the investigators showed that this reduction occurs gradually, in parallel with the progression of the disease and when they blocked the expression of the gene using siRNA, the striatal neurons were protected from Htt-induced damage. Thus, the striking downregulation of CalDAG-GEFI in HD could be a protective mechanism against mutant Htt (mHtt), but even switching off of the gene is eventually unsuccessful, resulting in the striatal neurodegeneration.
There is a growing body of evidence to support the notion that neurodegeneration in HD is due to abnormal interaction between mutant Htt and transcription factors and other proteins interfering with normal transcriptional regulation (Paulson and Fischbeck, 1996; Roses, 1996; Anderson et al., 2008). One of the most provocative clues to the selective vulnerability of neuronal damage in HD was provided initially by Li and colleagues (1995). These investigators identified a protein, called Htt-associated protein (HAP1), that specifically binds to Htt. Subsequent studies have demonstrated that HAP1 and Htt are anterogradely transported in axons. HAP1 normally interacts with kinesin light chain, a subunit of the kinesin motor complex that drives anterograde transport along microtubules, and it links to transport proteins such as growth factor receptor tyrosine kinase (TrkA) (Rong et al., 2006). However, when mutant Htt is present HAP1 is unable to carry out its trafficking function, leading to degeneration of nerve terminals. Other Htt-interacting proteins have been identified, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH), HAP1, HIP14, apopain, α-adaptin, ubiquitin, calmodulin, and PSD95 (Reddy et al., 1999; Young, 2003; Yanai et al., 2006). The Htt protein might also interact with clathrin and adaptor protein-2, which are involved in endocytosis and other proteins (Li and Li, 2004). The small guanine nucleotide-binding protein, Rhes (Ras homolog enriched in striatum), which is localized to the striatum and to much lesser extent in the cortex, but not in the cerebellum, binds physiologically to mHtt and leads to selective cytotoxicity (Subramaniam et al., 2009). The mechanism of the cytotoxicity is not clear, but Rhes has been shown to markedly decrease aggregation of mHtt, presumably by Rhes-mediated sumoylation which involves binding of the small ubiquitin-like modifier (SUMO) to the mHtt. Thus, Rhes-elicited disaggregation of mHtt probably accounts for the localized neuropathology of HD in the striatum and to lesser extent in the cortex. Therefore, drugs that block the binding of Rhes to mHtt may exert neuroprotective effects in HD.
There are many other Htt interacting proteins, identified with high-throughput screening, that may modify polyglutatime neurotoxicity leading to neurodegeneration (Kaltenbach et al., 2007). The long tract of glutamines in mutant HD protein weakens the interaction with Htt-associated protein, which is then free to bind the protein Hippi and activate apoptosis through caspase-8 and caspase-3. The latter also cleaves Htt, producing fragments that eventually form intraneuronal inclusions (Gervais et al., 2002). Wild-type Htt has been found to partially protect cells against noxious stimuli, and removal of the caspase sites is beneficial for the cells (Young, 2003). Htt has been found to be normally palmitoylated at cysteine 214, which is essential for its trafficking and function and this process is regulated by the palmitoyl transferase Htt-interacting protein 14 (HIP14). Expansion of the polyglutamine tract of Htt results in reduced interaction between mutant Htt and HIP14 and consequently in a marked reduction in palmitoylation and increased formation of inclusions and neuronal toxicity (Yanai et al., 2006). These pathologic changes may be partly corrected by overexpression of HIP14.
The relationship of the abnormal interaction of Htt with other proteins and how this interaction contributes to the pathogenesis of HD is still unclear. It is possible that as the Htt polyglutamine tract expands and changes its tertiary configuration, it blocks the ATP-dependent import mechanisms for succinate dehydrogenase entry into the mitochondria in the striatum. This might lead to selective, localized energy depletion and pathologic changes that are typically associated with HD. As the expansion increases, the impairment of import mechanism becomes less selective, and the pathology becomes more widespread. Thus, in HD, in addition to the typical involvement of the striatum, the GPe and GPi, SNc, and cerebellum become involved, and the patient manifests additional features, such as rigidity, bradykinesia, and ataxia (Furtado et al., 1996). Morphometric analyses of structural MRI demonstrated marked cerebellar atrophy and white matter loss in patients with HD (Fennema-Notestine et al., 2004). Although activity of glyceraldehyde-3-phosphate dehydrogenase, an Htt-binding protein, is normal in the brains of patients with HD, the activity of aconitase, an Fe-S-containing tricarboxylic acid cycle enzyme, is reduced to 8% in the caudate, 27% in the putamen, and 52% in the cerebral cortex of HD brains (Tabrizi et al., 1999). Since this enzyme is particularly sensitive to inhibition by peroxynitrite ONOO− and superoxide O2·−, it is considered be a good marker of excitotoxic cell damage.
Htt may also contribute to the apoptotic cell death in HD; it has been found to be cleaved by apopain, an apoptosis-specific cysteine protease (Goldberg et al., 1996). It has been postulated that the aggregates of mutant Htt (DiFiglia et al., 1997) cannot be properly removed from the cell and therefore might interfere with the metabolic activities of the affected neurons. Since ubiquitin is important in protein degradation and this process requires energy, the cells that are most vulnerable to the accumulation of the ubiquitin-Htt aggregates are those whose metabolic function has been already compromised, possibly as a result of toxins that interfere with oxidative phosphorylation. This concept then might link this new finding of region-selective accumulation of Htt aggregates and inclusions (DiFiglia et al., 1997) with the theories of regional impairment of energy metabolism (Beal et al., 1994). More recently, the molecular pathogenesis of HD has been considered to result from a decrease in specific chaperone proteins, such as Hdj1, Hsp70, αSGT, and βSGT, as demonstrated in R6/2 mice (Hay et al., 2004). Antigliadin antibodies have been demonstrated in 44% of patients with HD, but this probably represents an epiphenomenon and a nonspecific finding, as it has been also demonstrated in ataxia and other neurodegenerative disorders (Bushara et al., 2004).
Recent findings, based largely on studies in fruit flies, suggest that neurodegeneration in polyglutamine diseases such as HD and SCA3 may be due to RNA toxicity (Li et al., 2008).
Treatment
HD is one of few neurodegenerative diseases in which the diagnosis can be made long before the onset of clinical symptoms. This offers an opportunity to intervene in the earliest stages of the neurodegenerative cascade, perhaps even before the disease-related cell loss is initiated. Thus, HD is an excellent model for testing early neuroprotective treatments (Yamamato et al., 2000; Beal and Ferrante, 2004; Leegwater-Kim and Cha, 2004; Ryu and Ferrante, 2005; Bodner et al., 2006; Adam and Jankovic, 2008; Imarisio et al., 2008; Hersch and Rosas, 2008; Phillips et al., 2008; Roze et al., 2008; Jankovic, 2009; Frank and Jankovic, 2010; Ross and Tabrizi, 2011). There is currently no treatment to stop or slow the progression of HD, but experimental therapeutics in transgenic mouse models promise to translate some of the pathogenesis-targeted strategies into clinical trials (Beal and Ferrante, 2004). These models, for example, have been used to demonstrate potential disease-modifying effects of caspase inhibitors (Ona et al., 1999; Chen et al., 2000; Yamamoto et al., 2000). In another study, 2% creatine has been found to increase survival from 97.7 days to 114.6 days, and this was associated with a reduction in the number of Htt-positive aggregates in the striatum of transgenic HD mice (Ferrante et al., 2000). In a pilot study, 10 g/day of creatine was well tolerated for 12 months by 13 patients with HD, except for transient nausea and diarrhea in 2 patients (Tabrizi et al., 2003). In a randomized, double-blind, placebo-controlled study of 64 patients with HD, 8 g/day of creatine administered for 16 weeks was well tolerated and was associated with an increase in serum and brain creatine and a reduction in serum 8-hydroxy-2′-deoxyguanosine, an indicator of oxidative injury to DNA (Hersch et al., 2006). In a placebo-controlled trial, using 5 g/day of creatine, Verbessem and colleagues (2003) showed no improvement in functional, neuromuscular, or cognitive status in patients with early to moderate HD. Although weight loss is very common in HD, paradoxically, food restriction (fasting) slows disease progression in transgenic mice (Duan et al., 2004). The same group also showed that paroxetine, a serotonin uptake inhibitor, improves survival in transgenic mice while preventing weight loss (Duan et al., 2004). Using the R6/1 HD mice, van Dellen and colleagues (2000) showed that the onset of neurologic deficit was significantly delayed if the mice were raised in an enriched and stimulating environment. Furthermore, environmental enrichment delayed the degenerative loss of peristriatal cerebral volume and rescued protein deficits, possibly through rescuing transcription or protein transport problems (Spires et al., 2004). Similarly, environmental stimulation was found to enhance the health and life expectancy in R6/2 mice that were transgenic for exon 1 (Carter et al., 2000). It is therefore possible that occupational therapy, based on the principle of environmental enrichment, could delay the onset of HD. These findings showing that environmental enrichment slows disease progression in HD mouse models have been confirmed by other studies (Hockly et al., 2002). In support of the role of environment in expression of HD symptoms is the observation of discordance after 7 years in monozygotic twins with HD (Friedman et al., 2005).
Knowledge of the natural history of HD is critical for the design of neuroprotective trials (Paulsen et al., 2006). Although several agents, such as CoQ10 and nicotinamide, are being tested in clinical trials to determine whether they exert a neuroprotective effect in humans, only symptomatic treatments have been found to be useful in HD patients at this time. CoQ10 (ubiquinone) carries electrons from complexes I and II to complex II of the mitochondrial electron transport chain (Jankovic and Ashizawa, 2003; Ryu and Ferrante, 2005). As such, it can act as an antioxidant or as a pro-oxidant, depending on the cell’s redox potential (Shults and Schapira, 2001). In the pilot study, CoQ10, at doses of 600 and 1200 mg/day, has been found to be well tolerated, although some patients may experience adverse effects, such as headache, heartburn, fatigue, and increased involuntary movements (Feigin et al., 1996). In a multicenter clinical trial conducted by the Huntington Study Group that compared CoQ10 and remacemide, an NMDA ion channel blocker, 347 patients with documented HD were randomized to receive CoQ10 300 mg twice daily, remacemide hydrochloride 200 mg three times daily, both, or placebo, the so-called CARE-HD study (Huntington Study Group, 2001). The patients were evaluated every 4–5 months for 30 months. Although patients who were treated with CoQ10 showed a trend toward slowing in total functional capacity decline (13%), this difference failed to reach statistical significance. Furthermore, CoQ10 was associated with higher frequency of stomach upset, and remacemide treatment was associated with higher frequency of nausea, vomiting, and dizziness. Because of these rather disappointing results, routine use of CoQ10 in patients with HD is not warranted. Likewise, remacemide, although relatively well tolerated at 200 and 600 mg/day (Kieburtz et al., 1996), cannot be recommended for the treatment of HD. In a 6-week open-label trial, another antiglutamatergic drug, riluzole (100 mg/day), was found to be well tolerated and to be associated with a 35% reduction in the chorea rating score (Rosas et al., 1999). The Huntington Study Group (2003) also found that riluzole at 200 mg/day significantly improved chorea scores but without improving functional capacity, and because of abnormalities in liver transaminase, this drug could not be recommended for routine use in HD. In a large, randomized, double-blind placebo-controlled trial, conducted by the European Huntington’s Disease Initiative Study Group, 537 genetically-confirmed HD patients were randomized 2 : 1 to receive riluzole 100 mg/day or placebo for 3 years, and assessed periodically with the UHDRS (Landwehrmeyer et al., 2007). No concomitant use of other anti-choreic medications was allowed (wash-out period was 1 month). A total of 379 patients completed the study; the main reasons for discontinuation were the introduction of additional anti-choreic treatment and consent withdrawal. Although well tolerated, riluzole had no significant effect on chorea, behavioral, cognitive, independence, and functional scores compared to the placebo group at 3 years or at any other time point during the study. The potentially useful role of riluzole in favorably modifying the progression of HD is supported by studies in animal models of HD (Schiefer et al., 2002). Schiefer and colleagues (2002) were able to significantly increase survival time of R6/2 HD transgenic mice treated with riluzole. Furthermore, they showed that striatal neuronal intranuclear inclusions were less ubiquitinated and were surrounded by ubiquitinated microaggregates in riluzole-treated animals compared to controls. Staining with antibodies directed against the mutated Htt revealed no significant difference in this component of neuronal intranuclear inclusions. Minocycline, a second-generation antibiotic with high levels of blood–brain barrier permeability, has been shown to delay disease progression in the mouse model R6/2 of HD, possibly by inhibiting caspase-1 and caspase-3 mRNA upregulation and decreasing inducible nitric oxide synthase (Chen et al., 2000). Thirty patients (19 women and 11 men) between the ages of 21 and 66 years with symptomatic and genetically confirmed HD were treated with minocycline at the Baylor College of Medicine Huntington’s Disease Center for at least 6 months (Thomas et al., 2003). Assessments included the Abnormal Involuntary Movements Scale, the UHDRS, and the Mini-Mental State Examination, performed at baseline and every 2 months throughout the study period. Laboratory studies at baseline and at 2-month intervals included complete blood and platelet counts and renal and liver function tests. In this pilot study, we showed that minocycline was well tolerated over the 6-month observation period, although no significant improvement was observed on any clinical assessment. No changes were noted in any of the laboratory tests. There was no evidence of any adverse interaction between minocycline and any of the concomitant medications. Our findings are similar to those reported by others (Bonelli et al., 2003). Future multicenter trials should focus not only on the long-term tolerability of minocycline but also, more important, on its potential disease-modifying effects. Interestingly, paroxetine, a commonly used serotonin reuptake inhibitor, has been found to slow the progression in Htt mutant mice (Duan et al., 2004). Early and sustained treatment with essential fatty acids has been demonstrated to protect against motor deficits in R6/1 transgenic mice expressing exon 1 and a portion of intron 2 of the huntingtin gene (Clifford et al., 2002).
A growing body of evidence supports the notion that preformed polyglutamine aggregates are highly toxic when directed to the cell nucleus; therefore, recent research has focused on pharmacologic intervention aimed at inhibiting aggregate formation (Bates, 2003; Sanchez et al., 2003; Bossy-Wetzel et al., 2004; Hay et al., 2004). One such strategy is to use histone deacetylase inhibitors, such as valproic acid, phenylbutyrate, suberoylanilide hydroxamic acid (SAHA), pyroxamide, vorinostat, and other molecules to arrest polyglutamine-dependent neurodegeneration (Marks et al., 2001; Steffan et al., 2001; Butler and Bates, 2006; Chuang et al., 2009). In Drosophila flies expressing 93Q Htt, the histone deacetylase inhibitors suberoylamide hydroxamic acid and sodium butyrate suppressed neuronal degeneration (Steffan et al., 2001; Kazantsev et al., 2002; Agrawal et al., 2005). Suberoylamide hydroxamic acid also ameliorates motor deficits in the R6/2 mouse model of HD (Hockly et al., 2003). Geldanamycin, a benzoquinone ansamycin antibiotic that binds to Hsp90 and activates the stress response, has been found in some in-vitro cell culture models of HD to prevent polyQ aggregation, presumably by activating a heat-shock response (Sittler et al., 2001). Geldanamycin and radicicol have been found to delay aggregate formation in cell cultures and to increase soluble exon 1 huntingtin at concentrations that are capable of inducing Hsp40 and Hsp70 expression, indicating that chaperone induction favorably changes the biophysical properties of the aggregates (Hay et al., 2004). These drugs do not cross the blood–brain barrier, however, and therefore might not be clinically useful. Intraperitoneal injections of cystamine, an amino acid derivative that competitively inhibits transglutaminase, into transgenic HD mice ameliorated tremor and gait abnormalities and prolonged survival from 92 to 103 days (Karpuj et al., 2002). Keene and colleagues (2002) reported that treatment with tauroursodeoxycholic acid, a hydrophilic bile acid, prevented neuropathology and associated behavioral deficits in the 3-nitropropionic acid rat model of HD and reduced striatal neuropathology in the R6/2 transgenic HD mouse. Inhibition of polyglutamine oligomerization by the azo dye Congo red interferes with the ability of expanded polyglutamine to induce cytotoxic events, thus facilitating the degradation of expanded polyglutamine by making it more accessible to proteasome (Sanchez et al., 2003). Congo red has also been found to inhibit the assembly of protofibrils into fibrils, thus preventing toxicity from mature fibrils (Poirier et al., 2002). Trehalose, a disaccharide composed of two glucose molecules, has been found to alleviate polyglutamine-mediated pathology in the R6/2 transgenic model of HD, including a 30% reduction in brain aggregates (Tanaka et al., 2004). Trehalose, which appears to bind directly to proteins with expanded polyglutamines, is highly soluble and therefore can be administered orally. Since it appears to have no toxicity, the drug is a promising therapeutic agent for various polyglutamine diseases, not just HD. Another strategy is to reduce the toxic effects of pathogenic fragment of Htt by small ubiquitin-like modifier (SUMO)-1, which at least partially prevents aggregation of Htt in vitro (Steffan et al., 2004). However, in a Drosophila model of HD, sumoylation of Htt fragment exacerbates neurodegeneration, whereas ubiquitination reduces HD pathology. These findings suggest that SUMO-1 blockers might delay the progression of HD. Some unorthodox attempts are under way to test various putative neuroprotective agents, such as CoQ10, creatine, cystamine, omega-3 fatty acids, trehalose, and blueberry extract (Couzin, 2004). A component of green tea polyphenol (−)−epigallocatechin-3-gallate (EGCG) has been found to potently inhibit the aggregation of mutant Htt exon 1 protein, and when fed to transgenic HD flies overexpressing a pathogenic Htt exon 1 protein, photoreceptor degeneration and motor function improved (Ehrnhoefer et al., 2006). Another approach, currently investigated in HD mouse models, is the use of an engineered virus to make an intracellular antibody or “intrabody” that will clear the cytoplasm of mutated Htt (Wang et al., 2008b). Improving clearance of the mutant protein is another strategy that is being explored in experimental models of HD. For example, increased acetylation of the mutant Htt at lysine residue 444 (K444) in a C. elegans model of HD facilitates trafficking of mutant Htt into autophagosomes and improves clearance of the mutant protein by macroautophagy (Jeong et al., 2009). Intrabody against the proline-rich domain of Htt (Happ1) has been found to improve motor and cognitive deficits as well as the neuropathology found in the lentiviral, R6/2, N171-82Q, YAC128, and BACHD models of HD (Southwell et al., 2009).
Transglutaminase inhibitors, such as cystamine and monodansyl cadaverine, reduce aggregate formation and, as such, might be potential therapeutic agents in diseases caused by CAG-repeat expansion (Igarashi et al., 1998). By inhibiting the HD mutant transgene with tetracycline, Yamamoto and colleagues (2000) were able to show that the neurodegeneration can be not only prevented but also reversed. Preliminary data based on pilot clinical trials suggest that minocycline is well tolerated and that it is not associated with serious adverse events or laboratory test abnormalities (Bonelli et al., 2003; Thomas et al., 2004). In a pilot safety and tolerability study of 30 patients with HD who were given minocycline over a 6-month period, minocycline was found to be well tolerated during this study period, and no serious adverse events were noted (Thomas et al., 2004). Minocycline was also found to be well tolerated in a larger, double-blind, placebo-controlled study of 60 HD patients (Huntington Study Group, 2004). It has been also found to have a potential neuroprotective role in other neurodegenerative disorders (Zemke and Majid, 2004). Smith and colleagues (2003), however, found no effect of minocycline or doxycycline on behavioral abnormalities or Htt aggregates in R6/2 mice.
Another caspase inhibitor with potential neuroprotective effects, currently being tested in patients with HD, is ethyl eicosapentaenoate (LAX-101, ethyl-EPA, Miraxion). This pure, highly unsaturated fatty acid, concentrated chiefly in fish oil, has been found to be effective in some pilot trials in HD and schizophrenia. In a small (n = 24) double-blind, placebo-controlled study of LAX-101, Vaddadi and colleagues (2002) showed possible improvement in both the orofacial and total components of the UHDRS after 6 months of treatment. In one double-blind, placebo-controlled study ethyl-EPA has been found to have a possible stabilizing effect on the progression of HD (Puri et al., 2005). Because of the encouraging results, a multicenter trial of ethyl-EPA, TREND-HD, was conducted in North America. A multicenter, randomized, double-blind, placebo-controlled trial of ethyl-EPA (1 g twice daily) for 6 months involving 316 HD patients, 190 of whom completed the 12-month trial, showed improvement in the Total Motor Score 4 (TMS-4) component of the UHDRS (Huntington Study Group, 2008). While ethyl-EPA has not been found to be effective for the movement symptoms of HD when taken over 6 months, after 12 months those subjects who received ethyl-EPA for 12 continuous months had no deterioration in their UHDRS Total Motor Score, while those who received active treatment for only 6 months worsened (P = 0.02) (Huntington Study Group, 2008). Thus, improvement in motor symptoms may require long-term treatment with ethyl-EPA. These important observations, of course, have obvious therapeutic implications and suggest that caspase inhibitors might play a role in the treatment of HD and related neurodegenerative disorders (Friedlander, 2000; Sanchez Mejia and Friedlander, 2001).
Latrepirdine (Dimebon), originally developed in Russia as an antihistamine drug, has been also found to inhibit butyrylcholinesterase and acetylcholinesterase as well as NMDA receptor, and may also act as a mitochondrial enhancer by inhibiting mitochondrial permeability transition pore opening (although the latter presumably requires higher dosages than those used in clinical trials). It has been also found to exert neuroprotective effects in HD models and in a phase II trial of Alzheimer disease. A phase II clinical trial conducted by the Huntington Study Group (HSG) showed that latrepirdine significantly improved mean MMSE scores compared with stable performance in the placebo group but there were no significant treatment effects on the UHDRS or the Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog) (Kieburtz et al., 2010).
Phenylbutyrate, which increases brain histone acetylation, decreases histone methylation levels, enhances the ubiquitin-proteasomal pathway, and downregulates caspases, has been found to exert significant neuroprotective effects in a transgenic mouse model of HD (Gardian et al., 2005), and is currently being investigated as a potential neuroprotective drug by the HSG. Lithium has been found to induce autophagy via inositol monophosphatase (IMPase) inhibition, a process independent of the mammalian target of rapamycin (mTOR) induced autophagy (Pan et al., 2008), but combining it with rapamycin may be particularly useful in the treatment of neurodegenerative diseases, such as HD. But, in contrast to enhancement of autophagy via IMPase inhibition, the inhibition of glycogen synthase kinase-3β (GSK-3β) by lithium has the opposite effect (Sarkar et al., 2008). Various drugs have been found to induce autophagy and as such have been proposed to be potentially useful in slowing neurodegeneration associated with HD. These included L-type Ca2+ channel antagonists, the K+ATP channel opener minoxidil, and the Gi signaling activator clonidine. These drugs appear to regulate autophagy through mechanisms other than the cyclical (mTOR) pathway, in which cAMP regulates inositol trisphosphate levels, influencing calpain activity, which completes the cycle by cleaving and activating Gs alpha, which regulates cAMP levels (Williams et al., 2008).
Until effective neuroprotective therapy is found, the management of patients with HD will focus primarily on relief of symptoms designed to improve their quality of life (Handley et al., 2006; Jankovic, 2006; Bonelli and Hofmann, 2007; Adam and Jankovic, 2008; Frank and Jankovic, 2010). In a European survey of 2128 registered HD patients, 84% were prescribed symptomatic treatment, 50% were treated for depression, and 28% received anti-choreic medications (Priller et al., 2008). Psychosis, one of the most troublesome symptoms, usually improves with neuroleptics, such as haloperidol, pimozide, fluphenazine, and thioridazine. These drugs, however, can induce tardive dyskinesia and other adverse effects; therefore, they should be used only if absolutely needed to control symptoms. Clozapine (Clozaril), an atypical antipsychotic drug that does not cause tardive dyskinesia, might be a useful alternative to the typical neuroleptics, but its high cost, risk of agranulocytosis, and other potential side effects might limit its use, particularly since it has anti-choreic effects only in relatively high doses (Bonuccelli et al., 1994; van Vugt et al., 1997). It is likely that the other atypical antipsychotics, such as olanzapine (Zyprexa) (Paleacu et al., 2002) and quetiapine fumarate (Seroquel), will also provide a beneficial effect. Anxiolytics and antidepressants also might be useful in some patients with psychiatric problems. Donepezil, a procholinergic drug that is used in the treatment of Alzheimer disease, was found to be ineffective in the treatment of cognitive impairment or chorea associated with HD (Fernandez et al., 2000).
One of the most effective drugs in the treatment of hyperkinetic movement disorders, including chorea, is TBZ (Jankovic and Beach, 1997; Kenney and Jankovic, 2006). This drug, approved under the brand name Xenazine by the Food and Drug Administration in 2008 for chorea associated with HD, is also marketed in other countries as Nitoman. A potent and selective depletor of dopamine and, to a lesser degree, norepinephrine and serotonin from nerve terminals, it has been shown to be effective in the treatment of a variety of hyperkinetic movement disorders. The effects of TBZ are largely restricted to the CNS, which differentiates it from another monoamine depletor, reserpine, an old antihypertensive drug that produces both central and peripheral depletion. By inhibiting the brain synaptic VMAT, TBZ impairs uptake of monoamines and serotonin into synaptic vesicles, causing them to remain in the cytoplasm, where they are rapidly degraded by monoamine oxidases (Fig. 14.5). There are two types of VMAT: type 1 (VMAT1) and type 2 (VMAT2), which are coded by two distinct genes, 8p21.3 and 10q25, respectively. In humans, VMAT2 is nearly exclusively expressed in the CNS neurons, whereas VMAT1 is present in the peripheral nerve terminals. TBZ exhibits a 10 000-fold higher affinity for human VMAT2 than for human VMAT1, reversibly binding to the intravesicular part of VMAT2. Reserpine, on the other hand, binds irreversibly to the cytoplasmic site of both VMAT1 (peripheral) and VMAT2 (central). These pharmacologic differences probably account for the absence of hypotension and gastrointestinal side effects with TBZ compared to reserpine. Also, the duration of action is much shorter with TBZ, about 12 hours versus several days. In contrast to reserpine, which depletes all the monoamines and serotonin equally, TBZ preferentially depletes dopamine. Finally, despite early reports of dopamine receptor inhibition, more recent studies suggest that since the affinity of TBZ for the dopamine D2 receptor is 1000-fold lower than its affinity for VMAT2, it is unlikely that the weak dopamine D2 receptor antagonism is involved in the therapeutic effects of TBZ. This might be one reason why tardive dyskinesia has never been reported to occur with TBZ, one of the chief advantages of this drug over the neuroleptic, dopamine receptor-blocking, drugs.
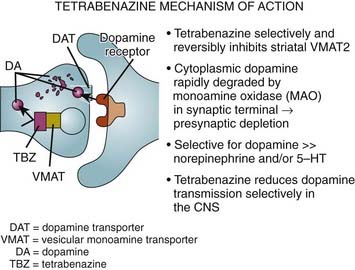
Although TBZ can cause or exacerbate depression, sedation, akathisia, and parkinsonism, in our experience it is clearly the most effective and safest anti-chorea drug (Jankovic and Beach, 1997; Ondo et al., 2002; Kenney and Jankovic, 2006; Kenney et al., 2007a, 2007b; Fasano and Bentivoglio, 2009; Jankovic, 2009) (Video 14.13). In one study of 15 patients who were treated for an average of 7 months at a mean dose of TBZ of 68 ± 27.1 mg/day (range: 25–150 mg/day), a “blinded” rating of videotapes showed improvement in motor scores in 12 (80%), and the mean score improved from 16 ± 3.6 to 13 ± 3.8 (Ondo et al., 2002). Adverse events included akathisia, insomnia, constipation, depression, drooling, and subjective weakness. Paradoxically, some patients with HD benefit from dopaminergic drugs, particularly when the disease is associated with parkinsonism (Racette and Perlmutter, 1998). In a multicenter, double-blind, placebo-controlled trial of TBZ involving 84 ambulatory patients with HD (TETRA-HD), TBZ treatment resulted in a reduction of 5.0 units in chorea severity compared with a reduction of 1.5 units on placebo treatment (adjusted mean effect size = −3.5 ± 0.8 UHDRS units (mean ± standard error); 95% confidence interval: −5.2 to −1.9; P < 0.0001) (Huntington Study Group, 2006) (Fig. 14.6). About 50% of TBZ-treated subjects had a 6-point or greater improvement compared with 7% of placebo-treated subjects. There was also a significant benefit on ratings of clinical global improvement. There were five study withdrawals in the TBZ group and five serious adverse events in four subjects (drowning suicide, complicated fall, restlessness/suicidal ideation, and breast cancer), compared with one withdrawal and no serious adverse events in the placebo group. This study concluded that TBZ, at adjusted dosages of up to 100 mg/day, was effective for the treatment of chorea in HD and was generally safe and well tolerated. In a retrospective study of 448 patients with hyperkinetic movement disorders (98 with chorea), treated with TBZ (mean dose at the last visit 60.4 ± 35.7 mg) for up to 21.6 years (mean 2.3 ± 3.4 years) at Baylor College of Medicine between 1997 and 2004, and evaluated with a clinical response scale (1: marked reduction in abnormal movements, 5: worsening), TBZ was found to be effective across the spectrum of the hyperactive movement disorders treated (Kenney and Jankovic, 2006). The percentage of patients with chorea that had a response of 1 or 2 at the last visit was 84.4%. The most common side effects, all dose related, were drowsiness (25%), parkinsonism (15.4%), depression (7.6%), akathisia (7.6%), with less common side effects including nausea/vomiting, nervousness/anxiety, and insomnia. Many patients are willing to tolerate side effects such as parkinsonism (sometimes effectively treated with amantadine, levodopa, dopamine agonists) because of TBZ’s beneficial effects on their hyperkinetic movement disorder. Even though this study was retrospective and open-label, it provides valuable information regarding the long-term tolerability and safety of the treatment with TBZ of hyperkinetic movement disorders. These observations have been confirmed by other, albeit smaller, longitudinal studies including one in which 68 patients with HD were followed for a mean period of 34.4 ± 25.2 months (Fasano et al., 2008a). An open-label, extension study following the TETRA-HD study showed that TBZ is a safe and effective drug (Frank et al., 2009). 
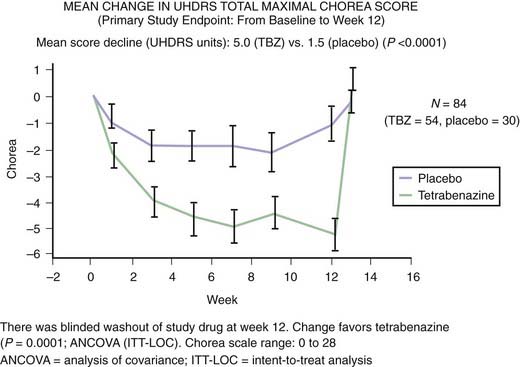
Figure 14.6 Results of placebo-controlled trial of tetrabenazine in HD.
From Huntington Study Group. A randomized, double-blind, placebo-controlled trial of tetrabenazine as antichorea therapy in Huntington disease. Neurology 2006;66:366–372.
TBZ is not only a dopamine depletor but may also decrease the brain serotonin and norepinephrine concentrations, with a potential for causing or worsening depression. To answer the question whether TBZ is contraindicated in depression, a retrospective study examined the effects of TBZ in 518 patients treated with TBZ for hyperkinetic movement disorders, of which 52.5% had a history of depression prior to the treatment (Kenney et al., 2006
). Of the patients without depression at the time of the treatment initiation, 11.4% were newly diagnosed with depression, and 18.4% of the patients with a previous diagnosis of depression at the time when the treatment with TBZ was started had a worsening of their depression. Overall 15.1% of patients experienced depression for the first time or had an exacerbation of the preexisting depression. On the other hand, there are patients whose depression improves with the TBZ treatment because of the marked benefit on the underlying hyperkinesia. Although depression is a known side effect of TBZ and may be potentially exacerbated by TBZ, a diagnosis of preexisting depression should not necessarily constitute an absolute contraindication for initiating the treatment with TBZ if the patient is otherwise a good candidate, but close monitoring of such patients is clearly warranted. There are no reports to this date of tardive dyskinesia induced by TBZ, this representing a major advantage of this medication over neuroleptics.The clinical pharmacokinetics of TBZ was studied in an open-label observational study (Kenney et al., 2007a). Ten patients with HD were assessed with serial motor UHDRS and Beck Depression Inventory at baseline and following their morning dose of TBZ, after at least 12 hours off from the last dose. There was an average of 42.4% reduction in chorea, with effect lasting between 3.2 and 8.1 hours (mean 5.4 ± 1.3 hours). Therefore, a minimum of three times a day dosing may be required in most patients to sustain the anti-chorea effects without wearing off. The best tolerated dose of TBZ, necessary to provide an adequate control of symptoms, varies greatly between individuals, from as low as 12.5 mg/day to as high as 400 mg/day. Although no blood levels or other pharmacokinetic data were obtained during this study, the findings suggest that the wash-out period of TBZ is short. This is also supported by a double-blind study of 30 HD patients treated with TBZ, randomly assigned to one of three groups: the first group of 12 patients stopped TBZ (which was replaced with a placebo) on day 1 (withdrawal group), the second group followed the same protocol on day 3 (partial withdrawal), while the third group continued the treatment with TBZ (non-withdrawal group) (Frank et al., 2008). The withdrawal group experienced reemergent chorea at day 3, with a difference in the UHDRS change of 5.33 units, compared to 2.94 units in the non-withdrawal group, which was in the hypothesized direction (P < 0.077). Post hoc analysis of the linear trend was positive for reemergent chorea (P = 0.0486) and provides further support for the effectiveness of TBZ in reducing chorea. TBZ has been found to be as effective as other neuroleptics, including “atypical” neuroleptics such as aripiprazole (Brusa et al., 2009), in reducing chorea with similar adverse effect profile, but it clearly has a lower risk of tardive syndrome. The Cochrane review, which examined 22 trials (1254 subjects) (Mestre et al., 2009a), concluded that based on available evidence, only TBZ showed a clear efficacy for the control of chorea.
Several guidelines on the treatment of HD have been published (Walker, 2007; Adam and Jankovic, 2008; Phillips et al., 2008; Jankovic, 2009; Mestre et al., 2009a, 2009b; Frank and Jankovic, 2010) (Fig. 14.7). Modulation of the dopaminergic system by presynaptically depleting dopamine, postsynaptically blocking dopamine receptors, or by activation of presynaptic dopamine receptors (autoreceptors) has been also suggested as a strategy to treat symptoms of HD. Apomorphine, a dopamine agonist that presumably acts by stimulating presynaptic D2 receptors and thus inhibiting dopamine release, has been found to be effective in five patients with HD, in whom it was continuously infused subcutaneously for 5 days (Vitale et al., 2007). A novel dopaminergic modulator, OSU6162, has been reported to improve chorea in a patient with HD (Tedroff et al., 1999), but further studies on the pharmacology and clinical efficacy of the drug are needed. Pridopidine (ACR16, Huntexil), a molecule similar to OSU162, is considered a “dopaminergic stabilizer” in that it stimulates or inhibits dopaminergic signaling depending on the dopaminergic tone and as such has been also classified as a partial dopamine receptor agonist or antagonist with preferential action on dopaminergic autoreceptors resulting in functional D2 antagonism (Rung et al., 2008). The drug may also decrease glutamatergic activity. Further studies by Carlsson and colleagues (Rung et al., 2008) have suggested that ACR16 has an “allosteric” effect on D2 receptors causing an enhanced response to dopamine and “orthosteric” effect, antagonizing dopamine. The plasma half-life increases from around 8 hours after a single dosing to about 15–20 hours at steady state. Although in a phase II trial in Europe the primary endpoint, the weighted cognitive score, did not significantly change and there was no significant improvement in chorea, the patients showed improvement in parkinsonian and gait scores and the drug was well tolerated (personal communication). Phase III (MermaiHD), which included 437 HD patients in 32 European centers with 92% compliance, failed to meet the primary endpoint, the modified Motor Score (mMS) (adopted from the UHDRS), after a 6-month trial of 45 mg twice daily. Further analysis showed that the significance value for the primary study endpoint of P = 0.042 did not meet the prespecified level of P < 0.025, but with inclusion of the clinically relevant CAG number adjustment, the P value is <0.02. There were no serious adverse effects detected. The NeuroSearch press release concluded that the drug “demonstrates a superior treatment effect in patients with an elevated CAG number score (considered a surrogate marker for rate of progression and disease prognosis) and that the significant improvements observed in mMS are driven primarily by positive effects on fine motor skills, gait and balance.” In contrast to the MermaiHD trial, in which about 40% of the patients were also receiving antipsychotics, the North American, phase IIb (HART) trial, involving 220 HD patients, currently under way, has excluded patients on antipsychotic or antidepressant drugs.
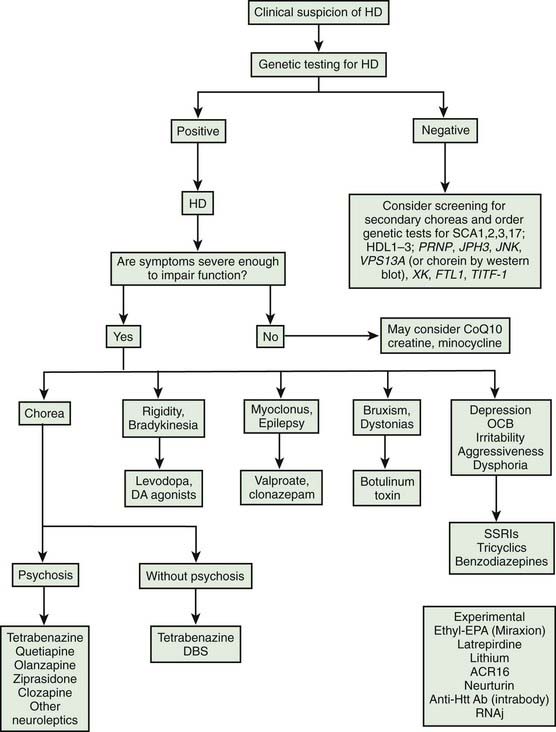
Baclofen, a putative GABA agonist, provides neither symptomatic nor protective effects in HD (Shoulson et al., 1989). Whether blocking glutamate release from the presynaptic terminals by drugs such as riluzole or lamotrigine (Brodie, 1992) or whether other antiglutamatergic drugs will be effective in slowing down the otherwise inexorable progression of the disease awaits further studies. In a double-blind, placebo-controlled trial, lamotrigine failed to slow HD progression but was found to improve symptoms and lessen chorea; 54% of patients on lamotrigine reported symptomatic improvement versus 15% of those on placebo, and the chorea subscale measure showed less deterioration for lamotrigine than for placebo (Kremer et al., 1999). Dystonia and bruxism, occasionally present in patients with HD, can be effectively treated with local injections of botulinum toxin (Tan et al., 2000). Pallidotomy, known to be effective in the treatment of levodopa-induced dyskinesias in patients with Parkinson disease, has been found also useful in the treatment of dystonia associated with HD (Cubo et al., 2000). Bilateral pallidal stimulation at 40 and 130 Hz improved chorea in one patient with HD (Moro et al., 2004).
Since NMDA supersensitivity might be responsible in part for chorea in patients with HD, selective NMDA antagonists might ameliorate this symptom. In a double-blind, controlled trial of 22 patients with HD, amantadine (400 mg/day) reduced extremity chorea by 36% (Verhagen Metman et al., 2002). In addition to symptomatic effect, amantadine might confer a neuroprotective effect. In another randomized placebo-controlled trial, amantadine at 300 mg/day had no effect on HD-associated chorea (O’Suilleabhain and Dewey, 2003). A 2-hour intravenous infusion of amantadine in a double-blind, randomized crossover study showed reduction in dyskinesias scores after both intravenous and oral administration (Lucetti et al., 2003). In a pilot trial of 9 patients with HD, memantine when titrated to the maximum dose of 20 mg/day, and followed for 3.8 ± 1.0 months, showed a significant decrease in total UHDRS motor scores (P = 0.008), mostly due to a significant (P = 0.008) decrease in the maximum chorea rating at their final visit (11.5 ± 6.3 to 4.8 ± 3.8) (Ondo et al., 2007). There was no significant change in their cognitive (P = 0.625) or behavioral (P = 0.258) ratings or total functional capacity (P = 0.078) and independence scale rating (P = 1.000). There were minimal adverse events, such as drowsiness and worsening of balance, speech, and social interaction.
Another group of medications used in the treatment of HD-related symptoms, such as depression, anxiety, and obsessive-compulsive disorder, are the selective serotonin uptake inhibitors (SSRIs). Fluoxetine, one of the SSRIs, has been also shown to stimulate CREB, increase production of BDNF, enhance glycogenolysis, block voltage-gated calcium and sodium channels, and decrease the conductance of mitochondrial voltage-dependent anion channels, and as such it may potentially exert neuroprotective effect in HD (Mostert et al., 2008). Nabilone, a synthetic analog of delta-9-tetrahydrocannabinol, the major psychoactive component of cannabis, has been found to exert modest symptomatic benefit in chorea associated with HD (Curtis et al., 2009).
In another Cochrane review of the literature, evaluating neuroprotective strategies, of 8 randomized, double-blind, placebo-controlled clinical trials (1366 subjects), no compounds studied thus have been found to provide disease-modifying effects although they were generally safe and well tolerated (Mestre et al., 2009b). Subjects were treated for a mean of 52 weeks (range 30–144 weeks) with one of the following compounds: vitamin E, idebenone, baclofen, lamotrigine, creatine, CoQ10 + remacemide, ethyl-eicosapentaenoic acid, and riluzole. All studies to date have used clinical scales to measure progression of disease, and future studies will likely need to use a more sensitive biomarker for disease onset and progression.
Palliative surgery, including pallidotomy and deep brain stimulation (DBS), has been tried in a few patients with severe chorea associated with HD but despite some improvement in chorea, the disease has continued to progress (Kang et al., 2011). When targeting the globus pallidus interna, DBS at a frequency of 40 Hz has been able to reduce chorea without producing parkinsonian features although the overall motor function and quality of life may not improve (Fasano et al., 2008b).
Experimental therapeutics
Following up on some encouraging results from animal studies, the delivery of trophic factors, such as nerve growth factor and ciliary neutrophic factor by genetically modified cells, into the striatum of patients with HD has become a reachable goal (Kordower et al., 1999; Mittoux et al., 2000; McBride et al., 2004). Implantation of nerve growth factor-producing fibroblasts into the rat striatum appeared to protect these animals against neurotoxic effects of the excitatory amino acids quinolate and quisqualate (Schumacher et al., 1991). Whether intrastriatal implantations of genetically engineered cells designed to produce trophic factors, such as ciliary neutrophic factor (Emerich et al., 1997; Kordower et al., 1999), or fetal cells will be useful in the treatment of HD awaits the results of further animal and clinical studies (Freeman et al., 2000; Bachoud-Levi et al., 2002; Peschanski et al., 2004; Reuter et al., 2008). The observation that implanted fetal neuronal cells survive in the HD brain and reconstitute damaged neuronal connections suggests that the host HD disease process does not affect the grafted tissue. No change in functional status or hyperkinetic movements was observed in 12 HD patients who were treated with transplantation of fetal porcine striatal cells (St Hillaire et al., 1998), but in another study (Bachoud-Levi et al., 2000), motor and cognitive improvement was noted in three of five patients after staged grafting of both striatal areas with human fetal neuroblasts. In their subsequent study, the investigators showed that the three patients who received fetal striatal allografts and demonstrated clinical improvement had a reduction of striatal and cortical hypometabolism as measured by F-deoxyglucose PET, whereas in the two patients who did not show clinical improvement, the striatal and cortical metabolism progressed (Gaura et al., 2004). Hauser and colleagues (2002) reported the results of an uncontrolled pilot study of transplantation of fetal striatal cells into the postcommissural putamen of seven patients with HD. There was no change in the primary outcome variable, the motor component of UHDRS, and three subjects suffered four subdural hemorrhages. Furthermore, the placebo effect, which was the same as the treatment effect (>20% improvement), lasted 18 months. Although the authors conclude that transplantation of human fetal striatal cells is feasible and that “a lack of significant worsening might reflect clinical benefit in a progressive neurodegenerative disease,” this study should be viewed as a negative study. It should, however, also provide a stimulus for future randomized, blind, and adequately controlled studies (Greenamyre and Shoulson, 2002). In five patients with HD followed for up to 6 years, the subjects were found to improve during the first 2 years and then their function again deteriorated (Bachoud-Levi et al., 2006). Autopsy studies, however, show that despite prolonged survival of the graft for up to 6 years or longer, there is poor integration with the host striatum, which probably accounts for the poor efficacy of this approach (Keene et al., 2007). Although fetal grafting in the treatment of HD is still being pursued in some, chiefly European, centers (Peschanski et al., 2004), it is unlikely that this approach will become an acceptable therapeutic strategy, especially with a recent report of symptomatic mass as a result of the graft overgrowth (Keene et al., 2009). The mechanisms of graft degeneration in HD are not well understood, but have been attributed to allograft immunoreactivity, microglial responses targeted to grafted cells and, similar to grafts in PD, cell-to-cell neurotoxicity (Cicchetti et al., 2011). Similarly, although adipose tissue stem cells implanted in the striata of R6/2 mice transgenic for HD showed some benefits, it is unlikely that this will translate into a clinically useful therapeutic strategy (Lee et al., 2009).
In addition, an initiative, Systemic Evaluation of Treatment for Huntington’s Disease, has been developed to systematically evaluate various strategies as potential treatments for HD (http://www.huntingtonproject.org; Walker and Raymond, 2004). One of the most promising approaches to the treatment of HD is neurturin, delivered via adeno-associated virus type 2 (AAV2) vector. Support for this statement is provided by an encouraging report using this approach in rats that received the neurotoxin 3NP (Ramaswamy et al., 2007).
Finally, RNA interference (RNAi) has been suggested as a promising therapeutic strategy, but further work is needed to determine whether this method can suppress the expression of only the mutated, but not the normal, Htt gene, for how long the suppression is required, and what the potential negative effects of knocking down mutated and wild-type Htt are (Harper, 2009). Furthermore, silencing of the wild-type and mutant Htt using small hairpin RNA administered via lentiform vectors and an amelioration of HD pathology was demonstrated in a rodent model of HD (Drouet et al., 2009).
A listing of available and experimental therapies for HD is provided in Table 14.3. In an evidence-based review of 218 publications on pharmacologic interventions in HD since 1965, Bonelli and Wenning (2006) found 20 level I, 55 level II, 54 level III trials, and 89 case reports. Chorea, the primary endpoint in nearly all studies, showed some improvement with haloperidol and fluphenazine, with less evidence for olanzapine. The analysis failed to result in any treatment recommendation of clinical relevance.
Adam O.R., Jankovic J. Symptomatic treatment of Huntington disease. Neurotherapeutics. 2008;5:181-197.
Agrawal N., Pallos J., Slepko N., et al. Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc Natl Acad Sci USA. 2005;102:3777-3781.
Albin R.L. Selective neurodegeneration in Huntington’s disease. Ann Neurol. 1995;38:835-836.
Albin R.L., Reiner A., Anderson K.D., et al. Striatal and nigral neuron subpopulations in rigid Huntington’s disease: Implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol. 1990;27:357-365.
Albin R.L., Reiner A., Anderson K.D., et al. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol. 1992;31:425-430.
Albin R.L., Young A.B., Penney J.B., et al. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington’s disease. N Engl J Med. 1990;332:1293-1298.
Alford R.L., Ashizawa T., Jankovic J., et al. Molecular detection of new mutations, resolution of ambiguous results and complex genetic counseling issues in Huntington’s disease. Am J Med Genet. 1996;66:281-286.
Almqvist E.W., Bloch M., Brinkman R., et al. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. Am J Hum Genet. 1999;64:1293-1304.
Anderson A.N., Roncaroli F., Hodges A., et al. Chromosomal profiles of gene expression in Huntington’s disease. Brain. 2008;131:381-388.
Anderson K.E., Gehl C.R., Marder K.S., et alHuntington’s Study Group. Comorbidities of obsessive and compulsive symptoms in Huntington’s disease. J Nerv Ment Dis. 2010;198:334-338.
Andreassen O.A., Ferrante R.J., Huang H.-M., et al. Dichloroacetate exerts therapeutic effects in transgenic models of Huntington disease. Ann Neurol. 2001;50:112-117.
Andrews T.C., Weeks R.A., Turjanski N., et al. Huntington’s disease progression. PET and clinical observations. Brain. 1999;122:2353-2363.
Andrich J., Arning L., Wieczorek S., et al. Huntington’s disease as caused by 34 CAG repeats. Mov Disord. 2008;23:879-881.
Andrich J., Saft C., Ostholt N., Muller T. Assessment of simple movements and progression of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:405-407.
Antonini A., Leenders K.L., Eidelberg D. [11C] Raclopride-PET studies of the Huntington’s disease rate of progression: Relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253-255.
Arnulf I., Nielsen J., Lohmann E., et al. Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol. 2008;65:482-488.
Arrasate M., Mitra S., Schweitzer E.S., et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805-810.
Ashizawa T., Jankovic J. Cervical dystonia as the initial presentation of Huntington’s disease. Mov Disord. 1996;11:457-459.
Ashizawa T., Wong L.-J.C., Richards C.S., et al. CAG repeat size and clinical presentation in Huntington’s disease. Neurology. 1994;44:1137-1143.
Aylward E.H., Sparks B.F., Field K.M., et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63:66-72.
Aziz N.A., Jurgens C.K., Landwehrmeyer G.B., et alEHDN Registry Study Group. Normal and mutant Htt interact to affect clinical severity and progression in Huntington disease. Neurology. 2009;73:1280-1285.
Aziz N.A., van der Burg J.M., Landwehrmeyer G.B., et al. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506-1513.
Bachoud-Levi A.C., Gaura V., Brugieres P., et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 2006;5:303-309.
Bachoud-Levi A.-C., Hantraye P., Peschanski M. Fetal neural grafts for Huntington’s disease: A prospective view. Mov Disord. 2002;17:439-444.
Bachoud-Levi A.-C., Remy P., Nguyen J.-P., et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356:1975-1979.
Bamford K.A., Caine E.D., Kido D.K., et al. A prospective evaluation of cognitive decline in early Huntington’s disease. Neurology. 1995;45:1867-1873.
Bartzokis G., Lu P.H., Tishler T.A., et al. Myelin breakdown and iron changes in Huntington’s disease: Pathogenesis and treatment implications. Neurochem Res. 2007;32:1655-1664.
Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642-1644.
Bates G.P., MacDonald M.E., Baxendale S., et al. Defined physical limits of the Huntington disease gene candidate region. Am J Hum Genet. 1991;49:7-16.
Beal M.F. Does impairment of energy metabolism result in excitotoxic neuronal degeneration in neurodegenerative diseases? Ann Neurol. 1992;31:119-130.
Beal M.F., Brouillet E., Jenkins B.G., et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitroproprionic acid. J Neurosci. 1993;13:4181-4192.
Beal M.F., Ferrante R.J. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat Rev Neurosci. 2004;5:373-384.
Beal M.F., Ferrante R.J., Swartz K.J., Kowall N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci. 1991;11:1649-1659.
Beal M.F., Henshaw D.R., Jenkins B.G., et al. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol. 1994;36:882-888.
Beal M.F., Swartz K.J., Hyman B.T., et al. Aminooxyacetic acid results in excitotoxin lesions by a novel indirect mechanism. J Neurochemistry. 1991;57:1068-1073.
Bence N.F., Sampat R.M., Kopito R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552-1555.
Berardelli A., Noth J., Thompson P.D., et al. Pathophysiology of chorea and bradykinesia in Huntington’s disease. Mov Disord. 1999;14:398-403.
Biglan K.M., Ross C.A., Langbehn D.R., et alPREDICT-HD Investigators of the Huntington Study Group. Motor abnormalities in premanifest persons with Huntington’s disease: the REDICT-HD study. Mov Disord. 2009;24:1763-1772.
Bird T.D. Outrageous fortune: the risk of suicide in genetic testing for Huntington disease. Am J Hum Genet. 64(5), 1999. Comment in: Am J Hum Genet. 1999;64(5):1289–1292
Björkqvist M., Wild E.J., Thiele J., et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205:1869-1877.
Blázquez C., Chiarlone A., Sagredo O., et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain. 2011;134(Pt 1):119-136.
Blekher T., Johnson S.A., Marshall J., et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology. 2006;67:394-399.
Bodner R.A., Outeiro T.F., Altmann S., et al. From the Cover: Pharmacological promotion of inclusion formation: A therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci USA. 2006;103:4246-4251.
Bohnen N.I., Koeppe R.A., Meyer P., et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54:1753-1759.
Bonelli R.M., Heuberger C., Reisecker F. Minocycline for Huntington’s disease: An open label study. Neurology. 2003;60:883-884.
Bonelli R.M., Hofmann P. A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother. 2007;8:141-153.
Bonelli R.M., Niederwieser G. Apraxia of eyelid closure in Huntington’s disease. J Neural Transm. 2002;109:197-201.
Bonelli R.M., Wenning G.K. Pharmacological management of Huntington’s disease: an evidence-based review. Curr Pharm Des. 2006;12:2701-2720.
Bonuccelli U., Ceravolo R., Maremmani C., et al. Clozapine in Huntington’s chorea. Neurology. 1994;44:821-823.
Borrell-Pagès M., Zala D., Humbert S., Saudou F. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642-2660.
Bossy-Wetzel E., Petrilli A., Knott A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31:609-616.
Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A. Molecular pathways to neurodegeneration. Nat Med. 2004;10(suppl 7):S2-S9.
Brandt J., Bylsma F.W., Gross H., et al. Trinucleotide repeat length and clinical progression in Huntington’s disease. Neurology. 1996;46:526-531.
Brannan T. The hung-up knee jerk in Huntington’s Disease. Parkinsonism Relat Disord. 2003;9:257-259.
Brinkman R.R., Mezei M.M., Theilmann J., et al. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60:1201-1210.
Britton J.W., Uiti R.J., Ahlskog J.E., et al. Hereditary late-onset chorea without significant dementia: Genetic evidence for substantial phenotypic variation in Huntington’s disease. Neurology. 1995;45:443-447.
Brodie M.J. Lamotrigine. Lancet. 1992;339:1397-1400.
Brouillet E., Jenkins B.G., Hyman B.T., et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochemistry. 1993;60:356-359.
Brower M., Price B. Neuropsychiatry of frontal lobe dysfunction in violent and criminal behavior: A critical review. J Neurol Neurosurg Psychiatry. 2001;71:720-726.
Browne S.E., Bowling A.C., MacGarvey U., et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646-653.
Brucke T., Podreka I., Wenger S., et al. Dopamine D2 receptor imaging with SPECT: Studies in different neuropsychiatric disorders. J Cereb Blood Flow Metab. 1991;11:220-228.
Brusa L., Orlacchio A., Moschella V., et al. Treatment of the symptoms of Huntington’s disease: preliminary results comparing aripiprazole and tetrabenazine. Mov Disord. 2009;24:126-129.
Bushara K.O., Nance M., Gomez C.M. Antigliadin antibodies in Huntington’s disease. Neurology. 2004;62:132-133.
Busse M.E., Wiles C.M., Rosser A.E. Mobility and falls in people with Huntington’s disease. J Neurol Neurosurg Psychiatry. 2009;80:88-90.
Bustamante-Aragones A., Trujillo-Tiebas M.J., Gallego-Merlo J., et al. Prenatal diagnosis of Huntington disease in maternal plasma: direct and indirect study. Eur J Neurol. 2008;15:1338-1344.
Butler R., Bates G.P. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784-796.
Canals J.M., Pineda J.R., Torres-Peraza J.F., et al. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci. 2004;24:7727-7739.
CAPIT-HD Committee. Core assessment program for intracerebral transplantation in Huntington’s disease (CAPIT-HD). Mov Disord. 1996;11:143-150.
Carella F., Scaioli V., Ciano C., et al. Adult onset myoclonic Huntington’s disease. Mov Disord. 1993;8:201-205.
Carter R.J., Hunt M.J., Morton A.J. Environmental stimulation increases survival in mice transgenic for Exon 1 of the Huntington’s disease gene. Mov Disord. 2000;15:925-937.
Cattaneo E., Rigamonti D., Coffredo D., et al. Loss of normal huntingtin function: New developments in Huntington’s disease research. Trends Neurosci. 2001;24:182-188.
Cattaneo E., Zuccato C., Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919-930.
Cha J.H., Kosinski C.M., Kerner J.A., et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480-6485.
Chen M., Ona V.O., Li M., et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of huntington disease. Nat Med. 2000;6:797-801.
Choo Y.S., Johnson G.V., MacDonald M., et al. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407-1420.
Chuang D.M., Leng Y., Marinova Z., et al. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591-601.
Cicchetti F., Parent A. Striatal interneurons in Huntington’s disease: Selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov Disord. 1997;11:619-626.
Cicchetti F., Soulet D., Freeman T.B. Neuronal degeneration in striatal transplants and Huntington’s disease: potential mechanisms and clinical implications. Brain. 2011;134(Pt 3):641-652.
Claes S., Zand K.V., Legius E., et al. Correlations between triplet repeat expansion and clinical features in Huntigton’s disease. Arch Neurol. 1995;113:749-753.
Clifford J.J., Drago J., Natoli A.L., et al. Essential fatty acids given from conception prevent topographies of motor deficit in a transgenic model of Huntington’s disease. Neuroscience. 2002;109:81-88.
Crittenden J., Dunn D.E., Merali F.I., et al. CalDAG-GEFI down-regulation in the striatum as a neuroprotective change in Huntington’s disease. Hum Mol Genet. 2010;19(9):1756-1765.
Cookson M.R. Roles of the proteasome in neurodegenerative disease: Refining the hypothesis. Ann Neurol. 2004;56:315-316.
Couzin J. Unorthodox clinical trials meld science care. Science. 2004;304:816-817.
Cubo E., Shannon K.M., Penn R.D., Kroin J.S. Internal globus pallidotomy in dystonia secondary to Huntington’s disease. Mov Disord. 2000;15:1248-1251.
Cui L., Jeong H., Borovecki F., et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59-69.
Curtis A., Mitchell I., Patel S., et al. A pilot study using nabilone for symptomatic treatment in Huntington’s disease. Mov Disord. 2009;24:2254-2259.
Curtis M.A., Penney E.B., Pearson A.G., et al. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc Natl Acad Sci USA. 2003;100(15):9023-9027.
Davies S.W., Beardsall K., Turmaine M., et al. Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet. 1998;351:131-133.
Davies S.W., Turmaine M., Cozens B.A., et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537-548.
Davis M.B., Bateman D., Quinn N.P., et al. Mutation analysis in patients with possible but apparently sporadic Huntington’s disease. Lancet. 1994;344:714-717.
de Boo G.M., Tibben A., Lanser J.B., et al. Early cognitive and motor symptoms in identified carriers of the gene for Huntington’s disease. Arch Neurol. 1997;54:1353-1357.
De Diego-Balaguer R., Couette M., Dolbeau G., et al. Striatal degeneration impairs language learning: evidence from Huntington’s disease. Brain. 2008;131:2870-2881.
DiFiglia M., Sapp E., Chase K.O., et al. Aggregation of Huntintgin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990-1993.
Djousse L., Knowlton B., Cupples L.A., et al. Weight loss in early stage of Huntington’s disease. Neurology. 2002;59:1325-1330.
Drouet V., Perrin V., Hassig R., et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65:276-285.
Duan W., Guo Z., Jiang H., et al. Paroxetine retards disease onset and progression in Huntingtin mutant mice. Ann Neurol. 2004;55:590-594.
Dunah A.W., Jeong H., Griffin A., et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238-2243.
Dure L.S., Young A.B., Penney J.B. Excitatory amino acid binding sites in the caudate nucleus and frontal cortex of Huntington’s disease. Ann Neurol. 1991;30:785-793.
Duyao M., Ambrose C., Myers R., et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. 1993;4:387-392.
Duyao M.P., Auerbach A.B., Ryan A., et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407-410.
Ehrnhoefer D.E., Duennwald M., Markovic P., et al. Green tea (-)-pigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum Mol Genet. 2006;15:2743-2751.
Emerich D.F., Winn S.R., Hantraye P.M., et al. Protective effect of encapsulated cells producing neurotrophic factor CNTF in a monkey model of Huntington’s diseease. Nature. 1997;386:395-399.
Everett C.M., Wood N.W. Trinucleotide repeats and neurodegenerative disease. Brain. 2004;127:2385-2405.
Fahn S., Jankovic J. Huntington disease. In: Rowland L.P., Pedley T., editors. Merritt’s Textbook of Neurology. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2009:723-726.
Farrer L.A. Suicide and attempted suicide in Huntington disease: implications for preclinical testing of persons at risk. Am J Med Genet. 1986;24:305-311.
Fasano A., Cadeddu F., Guidubaldi A., et al. The long-term effect of tetrabenazine in the management of Huntington disease. Clin Neuropharmacol. 2008;31:313-318.
Fasano A., Mazzone P., Piano C., et al. GPi-DBS in Huntington’s disease: results on motor function and cognition in a 72-year-old case. Mov Disord. 2008;23:1289-1292.
Fasano A., Bentivoglio A.R. Tetrabenazine. Expert Opin Pharmacother. 2009;10:2883-2896.
Fedoroff J.P., Peyser C., Franz M.L., Folstein S.E. Sexual disorders in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6:147-153.
Feigin A., Kieburtz K., Bordwell K., et al. Functional decline in Huntington’s disease. Mov Disord. 1995;10:211-214.
Feigin A., Kieburtz K., Como P., et al. Assessment of coenzyme Q10 tolerability in Huntington’s disease. Mov Disord. 1996;11:321-323.
Feigin A., Tang C., Ma Y., et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain. 2007;130:2858-2867.
Fekete R., Jankovic J. Psychogenic chorea associated with family history of Huntington disease. Mov Disord. 2010;25:503-504.
Fennema-Notestine C., Archibald S.L., Jacobson M.W., et al. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. 2004;63:989-995.
Fernandez H.H., Friedman J.H., Grace J., Beason-Hazen S. Donepezil for Huntington’s disease. Mov Disord. 2000;15:173-176.
Ferrante R.J., Andreassen O.A., Jenkins B.G., et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neuroscience. 2000;204:389-397.
Ferrante R.J., Kowall N.W., Cipolloni P.B., et al. Excitotoxin lesions in primates as a model for Huntington’s disease: Histopatholgical and neurochemical characterization. Exp Neurol. 1993;119:46-71.
Ferrante R.J., Kowall N.W., Richardson E.P. Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877-3887.
Ferrante R.J., Kubilus J.K., Lee J., et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418-9427.
Foroud T., Gray J., Ivashina J., Conneally P.M. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry. 1999;66:52-56.
Foroud T., Siemers E., Kleindorfer D., et al. Cognitive scores in carriers of Huntington’s disease gene compared to noncarriers. Ann Neurol. 1995;37:657-664.
Frank S., Huntington Study Group TETRA-HD Investigators. Tetrabenazine as anti-chorea therapy in Huntington Disease: an open-label continuation study. BMC Neurol. 2009;9:62.
Frank S., Jankovic J. Advances in the pharmacological management of Huntington’s disease. Drugs. 2010;70:561-571.
Frank S., Ondo W., Fahn S., et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin Neuropharmacol. 2008;31:127-133.
Freeman T.B., Cicchetti F., Hauser R.A., et al. Transplanted fetal striatum in Huntington’s disease: Phenotypic development and lack of pathology. Proc Natl Acad Sci USA. 2000;97:13877-13882.
Freiman R.N., Tjian R. A glutamine-rich trail leads to transcription factors. Science. 2002;296:2149-2150.
Friedlander R.M. Role of caspase 1 in neurologic disease. Arch Neurol. 2000;57:1273-1276.
Friedman J.H., Trieschmann M.E., Myers R.H., Fernandez H.H. Monozygotic twins discordant for Huntington disease after 7 years. Arch Neurol. 2005;62:995-997.
Furtado S., Suchowersky O., Rewcastle N.B., et al. Relationship between trinucleotide repeats and neuropathological changes in Huntington’s disease. Ann Neurol. 1996;39:132-136.
Gambardella A., Muglia M., Labate A., et al. Juvenile Huntington’s disease presenting as progressive myoclonic epilepsy. Neurology. 2001;57:708-711.
Garcia M., Vanhoutte P., Pages C., et al. The mitochondrial toxin 3-nitropropionic acid induces striatal neurodegeneration via c-Jun N-terminal kinase/c-Jun module. J Neurosci. 2002;22:2174-2184.
Gardian G., Browne S.E., Choi D.K., et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280:556-563.
Gaura V., Bachoud-Levi A.C., Ribeiro M.J., et al. Striatal neural grafting improves cortical metabolism in Huntington’s disease patients. Brain. 2004;127:65-72.
Gauthier L.R., Charrin B.C., Borrell-Pages M., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127-138.
Gervais F.G., Singaraja R., Xanthoudakis S., et al. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95-105.
Giordani B., Berent S., Boivin M.J., et al. Longitudinal neuropsychological and genetic linkage analysis of persons at risk for Huntington’s disease. Arch Neurol. 1995;52:59-64.
Goizet C., Lesca G., Durr A. Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias. Neurology. 2002;59:1330-1336.
Goldberg Y.P., Kremer B., Andrew S.E., et al. Molecular analysis of new mutations for Huntington’s disease: Intermediate alleles and sex of origin effects. Nat Genet. 1993;5:174-179.
Goldberg Y.P., Nicholson D.W., Rasper D.M., et al. Cleavage of huntingtin by apopain, a proapoptotic cystein protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442-449.
Golding C.V., Danchaivijitr C., Hodgson T.L., et al. Identification of an oculomotor biomarker of preclinical Huntington disease. Neurology. 2006;67:485-487.
Gómez-Tortosa E., MacDonald M., Friend J.C., et al. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol. 2001;49:29-34.
Gourfinkel-An I., Cancel G., Trottier Y., et al. Differential distribution of the normal and mutated forms of huntingtin in the human brain. Ann Neurol. 1997;42:712-719.
Grafton S.T., Mazziotta J.C., Pahl J.J., et al. A comparison of neurological, metabolic, structural, and genetic evaluations in persons at risk for Huntington’s disease. Ann Neurol. 1990;28:614-621.
Graham R.K., Deng Y., Slow E.J., et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179-1191.
Greenamyre J.T. Huntinton’s disease – making connections. N Engl J Med. 2007;365:518-520.
Greenamyre J.T., Shoulson I. We need something better, and we need it now: Fetal striatal transplantation in Huntington’s disease? Neurology. 2002;58:675-676.
Grimbergen Y.A., Knol M.J., Bloem B.R., et al. Falls and gait disturbances in Huntington’s disease. Mov Disord. 2008;23:970-976.
Gu M., Gash M.T., Mann V.M., et al. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385-389.
Gu X., Greiner E.R., Mishra R. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828-840.
Gunawardena S., Goldstein L.S. Polyglutamine diseases and transport problems: Deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46-51.
Gusela J.F., MacDonald M.E. Huntington’s disease: CAG genetics expands neurobiology. Curr Opin Neurobiol. 1995;5:656-662.
Gusella J.F., Wexler N.S., Conneally P.M., et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306:234-238.
Hahn-Barma V., Deweer B., Dürr A., et al. Are cognitive changes the first symptoms of Huntington’s disease?: A study of gene carriers. J Neurol Neurosurg Psychiatry. 1998;64:172-177.
Handley O.J., Naji J.J., Dunnett S.B., Rosser A.E. Pharmaceutical, cellular and genetic therapies for Huntington’s disease. Clin Sci. 2006;110:73-88.
Hantraye P., Riche D., Maziere M., Isacson O. A primate model of Huntington’s disease: Behavioral and anatomical studies of unilateral excitotoxic lesions of the caudate putamen in the baboon. Exp Neurol. 1990;106:91-104.
Harper P.S. The epidemiology of Huntington’s disease. Hum Genet. 1992;89:365-376.
Harper S.Q. Progress and challenges in RNA interference therapy for Huntington disease. Arch Neurol. 2009;66(8):933-938.
Harris G.J., Codori A.M., Lewis R.F., et al. Reduced basal ganglia blood flow and volume in pre-symptomatic, gene-tested persons at-risk for Huntington’s disease. Brain. 1999;122:1667-1678.
Harris G.J., Pearlson G.D., Peyser C.E., et al. Putamen volume reduction on magnetic resonance imaging exceeds caudate changes in mild Huntington’s disease. Ann Neurol. 1992;31:69-75.
Hasselbach S.G., Oberg G., Sorensen S.A., et al. Reduced regional cerebral blood flow in Huntington’s disease studied by SPECT. J Neurol Neurosurg Psychiatry. 1992;55:1018-1023.
Hauser R.A., Furtado S., Cimino C.R., et al. Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology. 2002;58:687-695.
Hay D.G., Sathasivam K., Tobaben S., et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389-1405.
Hedreen J.C., Folstein S.E. Early loss of striosome neurons in Huntington’s disease. J Neuropathol Exp Neurol. 1995;54:105-120.
Helder D.I., Kaptein A.A., van Kempen G.M.J., et al. Impact of Huntington’s disease on quality of life. Mov Disord. 2001;16:325-330.
Hendricks A.E., Latourelle J.C., Lunetta K.L., et al. Estimating the probability of de novo HD cases from transmissions of expanded penetrant CAG alleles in the Huntington disease gene from male carriers of high normal alleles (27–35 CAG). Am J Med Genet. 2009;149A:1375-1381.
Hersch S.M., Ferrante R.J. Tanslating therapies for Huntington’s disease from genetic animal models to clinical trials. NeuroRx. 2004;1:298-306.
Hersch S.M., Gevorkian S., Marder K., et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′dG. Neurology. 2006;66:250-252.
Hersch S.M., Rosas H.D. Neuroprotection for Huntington’s disease: ready, set, slow. Neurotherapeutics. 2008;5:226-236.
Heyes M.P., Swartz K.J., Markey S.P., Beal M.F. Regional brain and cerebrospinal fluid quinolinic acid concentrations in Huntington’s disease. Neurosci Lett. 1991;122:265-269.
Ho A.K., Gilbert A.S., Mason S.L., et al. Health-related quality of life in Huntington’s disease: Which factors matter most? Mov Disord. 2009;24:574-578.
Ho A.K., Sahakian B.J., Brown R.G., et al. Profile of cognitive progression in early Huntington’s disease. Neurology. 2003;61:1702-1706.
Hoang T.Q., Bluml S., Dubowitz D.J., et al. Quantitative proton-decoupled 31P MRS and 1H MRS in the evaluation of Huntington’s and Parkinson’s disease. Neurology. 1998;50:1033-1040.
Hockly E., Cordery P.M., Woodman B., et al. Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. Ann Neurol. 2002;51:235-242.
Hockly E., Richon V.M., Woodman B., et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2003;100:2041-2046.
Hodgson J.G., Agopyan N., Gutekunst C.A., et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181-192.
Horton T.M., Graham B.H., Corral-Debrinski M., et al. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology. 1995;45:1879-1883.
Hu M.T.M., Chaudhuri K.R. Repetitive belching, aerophagia, and torticollis in Huntington’s disease: A case report. Mov Disord. 1998;13:363-365.
Huntington G. On chorea. Medical and Surgical Reporter. 1872;26:320-321.
Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and consistency. Mov Disord. 1996;11:136-142.
Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. 2001;57:397-404.
Huntington Study Group. Dosage effects of riluzole in Huntington’s disease: A multicenter placebo-controlled study. Neurology. 2003;61:1551-1556.
Huntington Study Group. Minocycline safety and tolerability in Huntington disease. Neurology. 2004;63:547-549.
Huntington Study Group. A randomized, double-blind, placebo-controlled trial of tetrabenazine as antichorea therapy in Huntington disease. Neurology. 2006;66:366-372.
Huntington Study Group TREND-HD Investigators. Randomized Controlled Trial of Ethyl-Eicosapentaenoic Acid in Huntington Disease: The TREND-HD Study. Arch Neurol. 2008;65:1582-1589.
Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971-983.
Huot P., Levesque M., Parent A. The fate of striatal dopaminergic neurons in Parkinson’s disease and Huntington’s chorea. Brain. 2007;130:222-232.
Igarashi S., Koide R., Shimohata T., et al. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA proteins with an expanded polyglutamine stretch. Nat Genet. 1998;18:111-117.
Illarioshkin S.N., Igarashi S., Onodera O., et al. Trinucleotide repeat length and rate of progression of Huntington’s disease. Ann Neurol. 1994;36:630-635.
Imarisio S., Carmichael J., Korolchuk V., et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191-209.
Jana N.R., Zemskov E.A., Wang G.-H., Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049-1059.
Jankovic J. Huntington’s disease in-chief. Noseworthy J., editor. Neurological Therapeutics: Principles and Practice, second ed. Abingdon, Oxon, UK: Informa Healthcare. 2006:2869-2881.
Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844-856.
Jankovic J., Ashizawa T. Tourettism associated with Huntington’s disease. Mov Disord. 1995;10:103-105.
Jankovic J., Ashizawa T. Huntington’s disease. In: Noseworthy J., editor. Neurological Therapeutics: Principles and Practice. London: Martin Dunitz; 2003:2550-2561.
Jankovic J., Beach J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology. 1997;48:358-362.
Jankovic J., Beach J., Ashizawa T. Emotional and functional impact of DNA testing on patients with symptoms of Huntington’s disease. J Med Genet. 1995;32:516-518.
Jankovic J., Fahn S. Choreas. In: Rowland L.P., Pedley T., editors. Merritt’s Textbook of Neurology. Philadelphia, PA: Lippincott Williams & Wilkins, 2009.
Jankovic J., Wooten M., Van der Linden C., Jansson B. Weight loss in Parkinson’s disease. South Med J. 1992;85:351-354.
Jenkins B.G., Rosas H.D., Chen Y.C.I., et al. 1H NMR spectroscopy studies of Huntington’s disease: Correlations with CAG repeat numbers. Neurology. 1998;50:1357-1365.
Jensen P., Fenger K., Bowling T.G., Sorensen S.A. Crime in Huntington’s disease: A study of registered offences among patients, relatives, and controls. J Neurol Neurosurg Psychiatry. 1998;65:467-471.
Jeong H., Then F., Melia T.J.Jr., et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60-72.
Joel D. Open interconnected model of basal ganglia-thalamocortical circuitry and its relevance to the clinical syndrome of Huntington’s disease. Mov Disord. 2001;16:407-423.
Julien C.L., Thompson J.C., Wild S., et al. Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:939-943.
Kaltenbach L.S., Romero E., Becklin R.R., et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoSGenetics. 2007:e82.
Karpuj M.V., Becher M.W., Springer J.E., et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8:143-149.
Kang G.A., Heath S., Rothlind J., Starr P.A. Long-term follow-up of pallidal deep brain stimulation in two cases of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2011;82:272-277.
Karson C.N., Burns R.S., LeWitt P.A., et al. Blink rates and disorders of movement. Neurology. 1984;34:677-678.
Kazantsev A., Walker H.A., Slepko N., et al. A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat Genet. 2002;30:367-376.
Keene C.D., Chang R.C., Leverenz J.B., et al. A patient with Huntington’s disease and long-surviving fetal neural transplants that developed mass lesions. Acta Neuropathol. 2009;117:329-338.
Keene C.D., Rodrigues C.M., Eich T., et al. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc Natl Acad Sci USA. 2002;99:10671-10676.
Keene C.D., Sonnen J.A., Swanson P.D., et al. Neural transplantation in Huntington disease: long-term grafts in two patients. Neurology. 2007;68:2093-2098.
Kenney C., Hunter C., Davidson A., Jankovic J. Short-term effects of tetrabenazine on chorea associated with Huntington’s disease. Mov Disord. 2007;22:10-13.
Kenney C., Hunter C., Jankovic J. Long-term tolerability of tetrabenazine in the treatment of hyperkinetic movement disorders. Mov Disord. 2007;22:193-197.
Kenney C., Hunter C., Mejia N., Jankovic J. Is history of depression a contraindication to treatment with tetrabenazine? Clin Neuropharmacol. 2006;29:259-264.
Kenney C., Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurotherapeutics. 2006;6:7-17.
Kenney C., Jankovic J. Reply: Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov Disord. 2008;23:1793-1796.
Kenney C., Powell S., Jankovic J. Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov Disord. 2007;22:127-130.
Kieburtz K., Feigin A., McDermott M., et al. A controlled trial of remacemide hydrocholoride in Huntington’s disease. Mov Disord. 1996;11:273-277.
Kieburtz K., MacDonald M., Shih C., et al. Trinucleotide repeat length and progression of illness in Huntington’s disease. J Med Genet. 1994;31:872-874.
Kieburtz K., McDermott M.P., Voss T.S., et aland The Dimebon in Subjects With Huntington Disease (DIMOND) Investigators of the Huntington Study Group. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol. 2010;67:154-160.
Kim M., Lee H.S., LaForet G., et al. Mutant huntingtin expression in clonal striatal cells: Dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci. 1999;19:964-973.
Kipps C.M., Duggins A.J., Mahant N., et al. Progression of structural neuropathology in preclinical Huntington’s disease: A tensor based morphometry study. J Neurol Neurosurg Psychiatry. 2005;76:650-655.
Kirik D., Bjorklund A. Modeling CNS neurodegeneration by overexpression of disease-causing proteins using viral vectors. Trends Neurosci. 2003;26:386-392.
Kirkwood S.C., Siemers E., Hodes M.E., et al. Subtle changes among presymptomatic carriers of the Huntington’s disease gene. J Neurol Neurosurg Psychiatry. 2000;69:773-779.
Kirkwood S.C., Siemers E., Stout J.C., et al. Longitudinal cognitive and motor changes among presymptomatic Huntington disease gene carriers. Arch Neurol. 1999;56:563-568.
Kirkwood S.C., Su J.L., Conneally P., Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol. 2001;58:273-278.
Klöppel S., Chu C., Tan G.C., et alPREDICT-HD Investigators of the Huntington Study Group. Automatic detection of preclinical neurodegeneration: presymptomatic Huntington disease. Neurology. 2009;72:426-431.
Klöppel S., Draganski B., Golding C.V., et al. White matter connections reflect changes in voluntary-guided saccades in pre-symptomatic Huntington’s disease. Brain. 2008;131:196-204.
Kobayashi Y., Sobue G. Protective effects of chaperons on polyglutamine diseases. Brain Res Bull. 2001;56:165-168.
Kokmen E., Özekmekci S., Beard C.M., et al. Incidence and prevalence of Huntington’s disease in Olmsted county, Minnesota (1950–1989). Arch Neurol. 1994;51:696-698.
Kordower J.H., Isacson O., Emerich D.F. Cellular delivery of trophic factors for the treatment of Huntington’s disease: Is neuroprotection possible? Exp Neurol. 1999;159:4-20.
Koroshetz W.J., Jenkins B.G., Rosen B.R., Beal M.F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann Neurol. 1997;41:160-165.
Kovtun I.V., Liu Y., Bjoras M., et al. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447-452.
Kovtun I.V., Spiro C., McMurray C.T. Triplet repeats and DNA repair: Germ cell and somatic cell instability in transgenic mice. Methods Mol Biol. 2004;277:309-320.
Krainc D. Huntington’s disease: tagged for clearance. Nat Med. 2010;16:32-33.
Krainc D. Clearance of mutant proteins as a therapeutic target in neurodegenerative diseases. Arch Neurol. 2010;67:388-392.
Kremer B., Clark C.M., Almqvist E.W., et al. Influence of lamotrigine on progression of early Huntington disease. Neurology. 1999;53:1000-1011.
Kremer B., Goldberg P., Andrew S.E., et al. A worldwide study of the Huntington’s disease mutation: The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330:1401-1406.
Kuemmerle S., Gutekunst C.-A., Klein A.M., et al. Huntingtin aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842-849.
Künig G., Leenders K.L., Sanchez-Pernaute R., et al. Benzodizepine receptor binding in Huntington’s disease: [11C]Flumazenil uptake measured using positron emission tomography. Ann Neurol. 2000;47:644-648.
Kurlan R., Goldblatt D., Zaczek R., et al. Cerebrospinal fluid homovanillic acid and parkinsonism in Huntington’s disease. Ann Neurol. 1988;24:282-284.
Kuwert T., Lange H.W., Langen K.-J., et al. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain. 1990;113:1405-1423.
Lacone F., Engel U., Holinski-Feder E., et al. DNA analysis of Huntington’s disease: Five years of experience in Germany, Austria, and Switzerland. Neurology. 1999;53:801-806.
Landwehrmeyer G.B., Dubois B., de Yebenes J.G. Riluzole in Huntington’s disease: a 3-year, randomized controlled study. Ann Neurol. 2007;62:262-272.
Landwehrmeyer G.B., McNeil S.M., Dure L.S., et al. Huntington’s disease gene: Regional and cellular expression in brain of normal and affected infividuals. Ann Neurol. 1995;37:218-230.
Langbehn D.R., Paulsen J.S., Huntington Study Group. Predictors of diagnosis in Huntington disease. Neurology. 2007;68:1710-1717.
Lawrence A.D.M., Godges J.R., Rosser A.E., et al. Evidence for specific cognitive deficits in preclinical Huntington’s disease. Brain. 1998;121:1329-1341.
Lee S.T., Chu K., Jung K.H., et al. Slowed progression in models of Huntington disease by adipose stem cell transplantation. Ann Neurol. 2009;66:671-681.
Leegwater-Kim J., Cha J.-H.J. The paradigm of Huntington’s disease: Therapeutic opportunities in neurodegeneration. NeuroRx. 2004;1:128-138.
Leoni V., Mariotti C., Tabrizi S.J., et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain. 2008;131:2851-2859.
Leopold N.A., Kagel M.C. Dysphagia in Huntington’s disease. Arch Neurol. 1985;42:57-60.
Li H., Li S.H., Johnston H., et al. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385-389.
Li L.B., Yu Z., Teng X., Bonini N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107-1111.
Li S.H., Li X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146-154.
Li X.J., Li S.H., Sharp A.H., et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398-402.
Litvan I., Paulsen J.S., Mega M.S., Cummings J.L. Neuropsychiatric assessment of patients with hyperkinetic and hypokinetic movement disorders. Arch Neurol. 1998;55:1313-1319.
Louis E.D., Lee P., Quinn L., Marder K. Dystonia in Huntington’s disease: Prevalence and clinical characteristics. Mov Disord. 1999;14:95-101.
Lucetti C., Del Dotto P., Gambaccini G., et al. IV amantadine improves chorea in Huntington’s disease. An acute randomized, controlled study. Neurology. 2003;60:1995-1997.
Ludolph A.C., He F., Spencer P.S., et al. 3-Nitropropionic acid: Exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 1991;18:492-498.
Maat-Kievit A., Losekoot M., Zwinderman K., et al. Predictability of age of onset in Huntington disease in the Dutch population. Medicine. 2002;81:251-259.
Maat-Kievit A., Vegter-van der Vlis M., Zoeteweij M., et al. Paradox of a better test for Huntington’s disease. J Neurol Neurosurg Psychiatry. 2000;69:579-583.
MacMillan J.C., Snell R.G., Tyler A., et al. Molecular analysis and clinical correlations of the Huntington’s disease mutation. Lancet. 1993;342:954-958.
Mahant N., McCusker E.A., Byth K., Graham S. Huntington’s disease: Clinical correlates of disability and progression. Neurology. 2003;61:1085-1092.
Mangiarini L., Sathasivam K., Bates G.P. Molecular pathology of Huntington’s disease: Animal models and nuclear mechanisms. Neuroscientist. 1999;5:383-391.
Mantamadiotis T., Lemberger T., Bleckmann S.C., et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47-54.
Marder K., Sandler S., Lechich A., et al. Relationship between CAG repeat length and late-stage outcomes in Huntington’s disease. Neurology. 2002;59:1622-1624.
Marder K., Zhao H., Eberly S., et alHuntington Study Group. Dietary intake in adults at risk for Huntington disease: analysis of PHAROS research participants. Neurology. 2009;73:385-392.
Marder K., Zhao H., Myers R.H., et al. Rate of functional decline in Huntington’s disease. Neurology. 2000;54:452-458.
Marks P.A., Richon V.M., Breslow R., Rifkind R.A. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477-483.
Martin J.B. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;24:1970-1980. 340
Martindale D., Hackman A., Wieczorek A., et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150-154.
Martinez-Vicente M., Talloczy Z., Wong E., et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567-576.
McBride J.L., Behrstock S.P., Chen E.Y., et al. Human neural stem cell transplants improve motor function in a rat model of Huntington’s disease. J Comp Neurol. 2004;475:211-219.
Meiser B., Dunn S. Psychological impact of genetic testing for Huntington’s disese: An update of the literature. J Neurol Neurosurg Psychiatry. 2000;69:574-578.
Meissen G.J., Myers R.H., Mastromauro C.A., et al. Predictive testing for Huntington’s disease with use of linked DNA marker. N Engl J Med. 1988;318:535-542.
, Mestre, T., Ferreira, J., Coelho, M.M., et al., 2009a. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev (3), CD006456.
, Mestre, T., Ferreira, J., Coelho, M.M., et al., 2009b. Therapeutic interventions for disease progression in Huntington’s disease. Cochrane Database Syst Rev (3), CD006455.
Mittoux V., Joseph J.M., Conde F., et al. Restoration of cognitive and motor functions by ciliary neurotrophic factor in a primate model of Huntington’s disease. Hum Gene Ther. 2000;11:1177-1187.
Moro E., Lang A.E., Strafella A.P., et al. Bilateral globus pallidus stimulation for Huntington’s disease. Ann Neurol. 2004;56:290-294.
Mostert J.P., Koch M.W., Heerings M., et al. Therapeutic potential of fluoxetine in neurological disorders. CNS Neurosci Ther. 2008;14:153-164.
Myers R.H., Vonsattel J.P., Stevens T.J., et al. Clinical and neuropathologic assessment of severity in Huntington’s disease. Neurology. 1988;38:341-347.
Nagel J.S., Ichise M., Holman B.L. The scintigraphic evaluation of Huntington’s disease and other movement disorders using single photon emission computed tomography perfusion brain scans. Semin Nucl Med. 1991;21:11-23.
Nance M.A. Huntington’s disease: Another chapter rewritten. Am J Hum Genet. 1996;59:1-6.
Nance M.A., US Huntington Disease Genetic Testing Group. Genetic testing of children at risk for Huntington’s disease. Neurology. 1997;49:1048-1053.
Nihei K., Kowall N.W. Neurofilament and neural cell adhesion molecule immunocytochemistry of Huntington’s disease striatum. Ann Neurol. 1992;31:59-63.
Nopoulos P., Magnotta V.A., Mikos A., et al. Morphology of the cerebral cortex in preclinical Huntington’s disease. Am J Psychiatry. 2007;164:1428-1434.
Nucifora F.C., Sasaki M., Peters M.F., et al. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423-2428.
Okun M.S., Thommi N. Americo Negrette (1924 to 2003): Diagnosing Huntington disease in Venezuela. Neurology. 2004;63:340-343.
Oliva D., Carella F., Savoiardo M., et al. Clinical and magnetic resonance features of the classic and akinetic-rigid variants of Huntington’s disease. Arch Neurol. 1993;50:17-19.
Ona V.O., Li M., Vonsattel J.P.G., et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature. 1999;399:263-267.
Ondo W.G., Mejia N.I., Hunter C.B. A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Parkinsonism Relat Disord. 2007;13:453-454.
Ondo W.G., Tintner R., Thomas M., Jankovic J. Tetrabenazine treatment for Huntington’s disease-associated chorea. Clin Neuropharmacol. 2002;25:300-302.
O’Suilleabhain P., Dewey R.B.Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60:996-998.
Oyanagi K., Takeda S., Takashi H., et al. A quantitative investigation of the substantia nigra in Huntington’s disease. Ann Neurol. 1989;26:13-19.
Paleacu D., Anca M., Giladi N. Olanzapine in Huntington’s disease. Acta Neurol Scand. 2002;105:441-444.
Pan T., Kondo S., Le W., Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969-1978.
Panov A.V., Gutekunst C.A., Leavitt B.R., et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731-736.
Paulsen J.S., Hayden M., Stout J.C., et alPredict-HD Investigators of the Huntington Study Group. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63:883-890.
Paulsen J.S., Hoth K.F., Nehl C., Stierman L. Critical periods of suicide risk in Huntington’s disease. Am J Psychiatry. 2005;162:725-731.
Paulsen J.S., Langbehn D.R., Stout J.C., et alPredict-HD Investigators and Coordinators of the Huntington Study Group. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874-880.
Paulsen J.S., Nehl C., Hoth K.F., et al. Depression and stages of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2005;17:496-502.
Paulsen J.S., Ready R.E., Hamilton J.M., et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71:310-314.
Paulsen J.S., Zhao H., Staout J.C., et al. Clinical markers of early disease in persons near onset of Huntington’s disease. Neurology. 2001;57:658-662.
Paulson H.L., Fischbeck K.H. Trinucleotide repeats in neurogenetic disorders. Annu Rev Neurosci. 1996;19:79-107.
Pavese N., Andrews T.C., Brooks D.J., et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: A PET study. Brain. 2003;126:1127-1135.
Pavese N., Gerhard A., Tai Y.F., et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638-1643.
Penney J.B., Young A.B. Huntington’s disease. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. third ed. Baltimore: Williams and Wilkins; 1998:341-356.
Penney J.B., Young A.B., Snodgrass S.R., et al. Huntington’s disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov Disord. 1990;5:93-99.
Perry T.L., Hansen S. What excitotoxin kills striatal neurons in Huntington’s disease? Clues from neurochemical studies. Neurology. 1990;40:20-24.
Perutz M.F., Windle A.H. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature. 2001;412:143-144.
Peschanski M., Bachoud-Levi A.C., Hantraye P. Integrating fetal neural transplants into a therapeutic strategy: The example of Huntington’s disease. Brain. 2004;127:1219-1228.
Peters P.J., Ning K., Palacios F., et al. Arfaptin 2 regulates the aggregation of mutant huntingtin protein. Nat Cell Biol. 2002;4:240-245.
Phillips J.G., Bradshaw J.L., Chiu E., Bradshaw J.A. Characteristics of handwriting of patients with Huntington’s disease. Mov Disord. 1994;9:521-530.
Phillips W., Shannon K.M., Barker R.A. The current clinical management of Huntington’s disease. Mov Disord. 2008;23:1491-1504.
Pillon B., Dubois B., Ploska A., Agid Y. Severity and specificity of cognitive impairment in Alzheimer’s, Huntington’s, and Parkinson’s diseases and progressive supranuclear palsy. Neurology. 1991;41:634-643.
Poirier M.A., Li H., Macosko J., et al. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032-41037.
Politis M., Pavese N., Tai Y.F., et al. Hypothalamic involvement in Huntington’s disease: an in vivo PET study. Brain. 2008;131:2860-2869.
Postert T., Lack B., Kuhn W., et al. Basal ganglia alterations and brain atrophy in Huntington’s disease depicted by transcranial real time sonography. J Neurol Neurosurg Psychiatry. 1999;67:457-462.
Pratley R.E., Salbe A.D., Ravussin E., Caviness J.N. Higher sedentary energy expenditure in patients with Huntington’s disease. Ann Neurol. 2000;47:64-70.
Priller J., Ecker D., Landwehrmeyer B., Craufurd D. A Europe-wide assessment of current medication choices in Huntington’s disease. Mov Disord. 2008;23:1788.
Pritchard C., Cox D.R., Myers R.M. Invited editorial: The end in sight for Huntington disease? Am J Hum Genet. 1991;49:1-6.
Puri B.K., Leavitt B.R., Hayden M.R., et al. Ethyl-EPA in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology. 2005;65:286-292.
Racette B.A., Perlmutter J.S. Levodopa responsive parkinsonism in an adult with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1998;65:577-579.
Ramaswamy S., McBride J.L., Herzog C.D., et al. Neurturin gene therapy improves motor function and prevents death of striatal neurons in a 3-nitropropionic acid rat model of Huntington’s disease. Neurobiol Dis. 2007;26:375-384.
Rao A.K., Muratori L., Louis E.D., et al. Spectrum of gait impairments in presymptomatic and symptomatic Huntington’s disease. Mov Disord. 2008;23:1100-1107.
Ravikumar B., Vacher C., Berger Z., et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585-595.
Ravina B., Romer M., Constantinescu R., et al. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord. 2008;23:1223-1227.
Reddy P.H., Williams M., Tagle D.A. Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 1999;22:248-255.
Regulier E., Trottier Y., Perrin V., et al. Early and reversible neuropathology induced by tetracycline-regulated lentiviral overexpression of mutant huntingtin in rat striatum. Hum Mol Genet. 2003;12:2827-2836.
Reilmann R., Kirsten F., Quinn L., et al. Objective assessment of progression in Huntington’s disease: A 3-year follow-up study. Neurology. 2001;57:920-924.
Reuter I., Hu M.T.M., Andrews T.C., et al. Late onset levodopa responsive Huntington’s disease with minimal chorea masquerading as Parkinson plus syndrome. J Neurol Neurosurg Psychiatry. 2000;68:238-241.
Reuter I., Tai Y.F., Pavese N., et al. Long-term clinical and positron emission tomography outcome of fetal striatal transplantation in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2008;79:948-951.
Ribai P., Nguyen K., Hahn-Barma V., et al. Psychiatric and cognitive difficulties as indicators of juvenile Huntington disease onset in 29 patients. Arch Neurol. 2007;64:813-819.
Richfield E.K., Maguire-Zeiss K.A., Vonkeman H.E., Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington’s disease patients. Ann Neurol. 1995;38:852-861.
Robins Wahlin T.-B., Bäckman L., Haegermark A., et al. High suicidal ideation in persons testing for Huntington’s disease. Acta Neurol Scand. 2000;102:150-161.
Romero E., Cha G.H., Verstreken P., et al. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57:27-40.
Rong J., McGuire J.R., Fang Z.H., et al. Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J Neurosci. 2006;26:6019-6030.
Rosas H.D., Goodman J., Chen Y.I., et al. Striatal volume loss in HD as measured by MRI and the influence of CAG repeat. Neurology. 2001;57:1025-1028.
Rosas H.D., Hevelone N.D., Zaleta A.K., et al. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745-747.
Rosas H.D., Koroshetz W.J., Chen Y.I., et al. Evidence for more widespread cerebral pathology in early HD: An MRI-based morphometric analysis. Neurology. 2003;60:1615-1620.
Rosas H.D., Koroshetz W.J., Jenkins B.G., et al. Riluzole therapy in Huntington’s disease (HD). Mov Disord. 1999;14:326-330.
Rosas H.D., Liu A.K., Hersch S., et al. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology. 2002;58:695-701.
Roses A.D. From genes to mechanisms to therapies: Lessons to be learned from neurological disorders. Nat Med. 1996;2:267-269.
Ross C.A., Thompson L.M. Transcription meets metabolism in neurodegeneration. Nat Med. 2006;12:1239-1241.
Ross C.A., Tabrizi S.J. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83-98.
Roze E., Saudou F., Caboche J. Pathophysiology of Huntington’s disease: from huntingtin functions to potential treatments. Curr Opin Neurol. 2008;21:497-503.
Rung J.P., Rung E., Helgeson L., et al. Effects of (-)-OSU6162 and ACR16 on motor activity in rats, indicating a unique mechanism of dopaminergic stabilization. J Neural Transm. 2008;115:899-908.
Ryu H., Ferrante R.J. Emerging chemotherapeutic strategies for Huntington’s disease. Expert Opin Emerg Drugs. 2005;10:345-363.
Saleh N., Moutereau S., Azulay J.P., et al. High insulinlike growth factor I is associated with cognitive decline in Huntington disease. Neurology. 2010;75:57-63.
Sanchez I., Mahlke C., Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373-379.
Sanchez Mejia R.O., Friedlander R.M. Caspases in Huntington’s disease. Neuroscientist. 2001;7:480-489.
Sánchez-Pernaute R., Garcia-Segura J.M., Alba A.B., et al. Clinical correlation of striatal 1H MRS changed in Huntington’s disease. Neurology. 1999;53:806-812.
Sánchez-Pernaute R., Künig G., del Barrio Albe A., et al. Bradykinesia in early Huntington’s disease. Neurology. 2000;54:119-125.
Sapp E., Schwartz C., Chase K., et al. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol. 1997;42:604-612.
Sarkar S., Krishna G., Imarisio S., et al. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet. 2008;17:170-178.
Saudou F., Finkbeiner S., Devys D., Greenberg M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55-66.
Scherzinger E., Lurz R., Turmaine M., et al. Huntingtin-encoded polyglutamine expansions for amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:537-548.
Scherzinger E., Sittler A., Schweiger K., et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils. Implications for Huntington’s disease pathology. Proc Natl Acad Sci USA. 1999;96:4604-4609.
Schiefer J., Landwehrmeyer G.B., Luesse H.G., et al. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov Disord. 2002;17:748-757.
Schneider S.A., van de Warrenburg B.P., Hughes T.D., et al. Phenotypic homogeneity of the Huntington disease-like presentation in a SCA17 family. Neurology. 2006;67:1701-1703.
Schneider S.A., Walker R.H., Bhatia K.P. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol. 2007;3:517-525.
Schoenfeld M., Myers R.H., Cupples L.A., et al. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1984;47:1283-1287.
Schumacher J.M., Short M.P., Hyman B.T., et al. Intracerebral implantation of nerve growth factor-producing fibroblasts protects striatum against neurotoxic levels of excitatory amino acids. Neuroscience. 1991;45:561-570.
Schwarcz R., Okuno E., White R.J., et al. 3-hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc Natl Acad Sci USA. 1988;85:4079-4081.
Seo H., Sonntag K.C., Isacson O. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann Neurol. 2004;56:319-328.
Shelton P.A., Knopman D.S. Ideomotor apraxia in Huntington’s disease. Arch Neurol. 1991;48:35-41.
Shoulson I., Odoroff C., Oakes D., et al. A controlled trial of baclofen as protective therapy in early Huntington’s disease. Ann Neurol. 1989;25:252-259.
Shults C.W., Schapira A.H.V. A cue to queue for CoQ? Neurology. 2001;57:375-376.
Siemers E., Foroud T., Bill D.J., et al. Motor changes in presymptomatic Huntington disease gene carriers. Arch Neurol. 1996;53:487-492.
Siesling S., van Vugt J.P.P., Zwinderman K.A.H., et al. Unified Huntington’s Disease Rating Scale: A follow up. Mov Disord. 1998;13:915-919.
Siesling S., Zwinderman A.H., van Vugt J.P.P., et al. Shortened version of the motor section of the Unified Huntington’s Disease Rating Scale. Mov Disord. 1997:229-234.
Sittler A., Lurz R., Lueder G., et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307-1315.
Smith D.L., Woodman B., Mahal A., et al. Minocycline and doxycycline are not beneficial in a model of Huntington’s disease. Ann Neurol. 2003;54:186-196.
Snell R.G., MacMillan J.C., Cheadle J.P., et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. 1993;4:393-397.
Snowden J.S., Craufurd D., Griffiths H.L., Neary D. Awareness of involuntary movements in Huntington disease. Arch Neurol. 1998;55:801-805.
Sotrel A., Paskevich P.A., Kiely D.K., et al. Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology. 1991;41:1117-1123.
Southwell A.L., Ko J., Patterson P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J Neurosci. 2009;29:13589-13602.
Spires T.L., Grote H.E., Varshney N.K., et al. Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J Neurosci. 2004;24:2270-2276.
Sprengelmeyer R., Young A.W., Calder A.J., et al. Loss of disgust. Perception of faces and emotions in Huntington’s disease. Brain. 1996;119:1647-1665.
Squitieri F., Gellera C., Cannella M., et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946-955.
St Hillaire M., Shannon K., Schumacher J., et al. Transplantation of fetal porcine striatal cells in Huntington’s disease: Preliminary safety and efficacy results [abstract]. Neurology. 1998;50:A80-A81.
Steffan J.S., Agrawal N., Pallos J., et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100-104.
Steffan J.S., Bodai L., Pallos J., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739-743.
Stoffers D., Sheldon S., Kuperman J.M., et al. Contrasting gray and white matter changes in preclinical Huntington disease: an MRI study. Neurology. 2010;74:1208-1216.
Storey E., Hyman B.T., Jenkins B., et al. 1-Methyl-4-phenylpyridinium produces excitotoxic lesions in rat striatum as a result of impairment of oxidative metabolism. J Neurochem. 1992;58:1975-1978.
Strauss M.E., Brandt J. Are there neuropsychologic manifestations of the gene for Huntington’s disease in asymptomatic, at-risk individuals? Arch Neurol. 1990;47:905-908.
Swami M., Hendricks A.E., Gillis T., et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039-3047.
Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327-1330.
Szebenyi G., Morfini G.A., Babcock A., et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41-52.
Tabrizi S.J., Blamire A.M., Manners D.N., et al. Creatine therapy for Huntington’s disease: Clinical and MRS findings in a 1-year pilot study. Neurology. 2003;61:141-142.
Tabrizi S.J., Cleeter M.W.J., Xuereb J., et al. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25-32.
Tabrizi S.J., Workman J., Hart P.E., et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80-86.
Tabrizi S.J., Scahill R.I., Durr A., et al. The TRACK-HD investigators. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10:31-42.
Taherzadeh-Fard E., Saft C., Andrich J., et al. PGC-1alpha as modifier of onset age in Huntington disease. Mol Neurodegener. 2009;4:10.
Tai Y.F., Pavese N., Gerhard A., et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759-1766.
Tan E.-K., Jankovic J. Bruxism in Huntington’s disease. Mov Disord. 2000;15:171-173.
Tanaka M., Machida Y., Niu S., et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148-154.
Tang T.S., Slow E., Lupu V., et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci USA. 2005;102:2602-2607.
Tang T.S., Chen X., Liu J., Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J Neurosci. 2007;27:7899-7910.
Tedroff J., Ekesob A., Sonesson C., et al. Long-lasting improvement following (-)-OSU6162 in a patient with Huntington’s disease. Neurology. 1999;53:1605-1606.
Thieben M.J., Duggins A.J., Good C.D., et al. The distribution of structural neuropathology in pre-clinical Huntington’s disease. Brain. 2002;125:1815-1828.
Thomas M., Ashizawa T., Jankovic J. Minocycline in Huntington’s disease: A pilot study. Mov Disord. 2004;19:692-695.
Thomas M., Le W., Jankovic J. Minocycline and other tetracycline derivatives: A neuroprotective strategy in Parkinson’s disease and Huntington’s disease. Clin Neuropharmacol. 2003;26:18-23.
Thompson L.M., Aiken C.T., Kaltenbach L.S., et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187(7):1083-1099.
Thompson P.D., Berardelli A., Rothwell J.C., et al. The coexistence of bradykinesia and chorea in Huntington’s disease and its implications for theories of basal ganglia control of movement. Brain. 1988;111:223-244.
Thompson P.D., Bhatia K.P., Brown P., et al. Cortical myoclonus in Huntington’s disease. Mov Disord. 1994;9:633-641.
Thu D.C., Oorschot D.E., Tippett L.J., et al. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain. 2010;133:1094-1110.
Tian J.R., Zee D.S., Lasker A.G., Folstein S.E. Saccades in Huntington’s disease: Predictive tracking and interaction between release of fixation and initiation of saccades. Neurology. 1991;41:875-881.
Tippett L.J., Waldvogel H.J., Thomas S.J., et al. Striosomes and mood dysfunction in Huntington’s disease. Brain. 2007;130:206-221.
Toyoshima Y., Yamada M., Onodera O., et al. SCA17 homozygote showing Huntington’s disease-like phenotype. Ann Neurol. 2004;55:281-286.
Turjanski N., Weeks R., Dolan R., et al. Striatal D1 and D2 receptor binding in patients with Huntington’s disease and other choreas. A PET study. Brain. 1995;118:689-696.
Tyler A., Ball D., Craufurd D., et al. Presymptomatic testing for Huntington’s disease in the United Kingdom. BMJ. 1992;304:1593-1596.
Vaddadi K.S., Soosai E., Chiu E., Dingjan P. A randomized, placebo-controlled, double blind study of treatment of Huntington’s disease with unsaturated fatty acids. Neuroreport. 2002;13:29-33.
van Dellen A., Blakemore C., Deacon R., et al. Delaying the onset of Huntington’s in mice. Nature. 2000;404:721-722.
van den Bogaard S.J., Dumas E.M., Acharya T.P., et al. TRACK-HD Investigator Group. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 2011;258(3):412-420.
van der Burg J.M., Björkqvist M., Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8:765-774.
van Vugt J.P.P., van Hilten B.J., Roos R.A.C. Hypokinesia in Huntington’s disease. Mov Disord. 1996;11:384-388.
van Vugt J.P., Siesling S., Vergeer M., et al. Clozapine versus placebo in Huntington’s disease: a double blind randomized comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
Verbessem P., Lemiere J., Eijnde B.O., et al. Creatine supplementation in Huntington’s disease: A placebo-controlled pilot trial. Neurology. 2003;61:925-930.
Verhagen Metman L., Morris M.J., Farmer C., et al. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59:694-699.
Vitale C., Marconi S., Di Maio L., et al. Short-term continuous infusion of apomorphine hydrochloride for treatment of Huntington’s chorea: A double blind, randomized cross-over trial. Mov Disord. 2007;22:2359-2364.
Vogel C.M., Drury I., Terry L.C., Young A.B. Myoclonus in adult Huntington’s disease. Ann Neurol. 1991;29:213-215.
Walker F.O. Huntington’s disease. Lancet. 2007;369:218-228.
Walker F.O., Raymond L.A. Targeting energy metabolism in Huntington’s disease. Lancet. 2004;364:312-313.
Wang C.E., Tydlacka S., Orr A.L., et al. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum Mol Genet. 2008;17:2738-2751.
Wang C.E., Zhou H., McGuire J.R., et al. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol. 2008;181:803-816.
Weeks R.A., Piccini P., Harding A.E., Brooks D.J. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington’s disease. Ann Neurol. 1996;40:49-54.
Wexler N.S., Rose E.A., Housman D.E. Molecular approaches to hereditary diseases of the nervous system: Huntington’s disease as a paradigm. Ann Rev Neurosci. 1991;14:503-529.
Wheelock V.L., Tempkin T., Marder K., et al. Predictors of nursing home placement in Huntington disease. Neurology. 2003;60:998-1001.
Wiggins S., Whyte P., Huggins M., et al. The psychological consequences of predictive testing for Huntington’s disease. N Engl J Med. 1992;327:1401-1405.
Wild E.J., Mudanohwo E.E., Sweeney M.G., et al. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008;23:716-720.
Williams A., Sarkar S., Cuddon P., et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295-305.
Wu Y., Le W., Jankovic J. Preclinical biomarkers of Parkinson’s disease. Arch Neurol. 2011;68:22-30.
Xing S.C.L., Zhong P., Zeng J., Li J. Excessive blinking as initial manifestation of juvenile Huntington’s disease. Neurol Sci. 2008;29:275-277.
Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57-66.
Yanai A., Huang K., Kang R., et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824-831.
Yang S.H., Cheng P.H., Banta H., et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. 2008;453:921-924.
Yang W., Dunlap J.R., Andrews R.B., Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905-2917.
Young A.B. Huntingtin in health and disease. J Clin Invest. 2003;111:299-302.
Young A.B., Greenamyre J.T., Hollingsworth Z., et al. NMDA receptor losses in putamen from patients with Huntington’s disease. Science. 1988;241:981-983.
Zappacosta B., Monza D., Meoni C., et al. Psychiatric symptoms do not correlate with cognitive decline, motor symptoms, or CAG repeat length in Huntington’s disease. Arch Neurol. 1996;53:493-497.
Zemke D., Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 2004;27:293-298.
Zoghbi H.Y., Botas J. Mouse and fly models of neurodegeneration. Trends Genet. 2002;18:463-471.
Zuccato C., Ciammola A., Rigamonti D., et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493-498.