Hemoglobin Metabolism
After completion of this chapter, the reader will be able to:
1. Describe the primary structure of the globin chains found in hemoglobin.
2. Describe the quaternary structure of hemoglobin.
3. Describe the biosynthesis of heme and globin.
4. Differentiate steps in heme synthesis that occur in the mitochondria and the cytoplasm.
5. Identify the ontogeny of hemoglobin with emphasis on the hemoglobin of newborns and adults.
6. Identify the three types of normal hemoglobin in adults and their reference intervals.
7. Describe the regulatory effects of hemoglobin metabolism.
8. Identify the important role that hemoglobin plays in maintaining body functions.
9. Describe the mechanism by which hemoglobin carries oxygen to the tissue.
11. Explain the significance of the sigmoid shape of the oxygen dissociation curve.
12. Correlate right and left shifts in the oxygen dissociation curve with conditions that can cause shifts in the curve.
13. Identify the P50 value (amount of oxygen needed to saturate 50% of hemoglobin) as it pertains to a normal oxygen dissociation curve.
14. Differentiate T and R forms of hemoglobin.
15. Identify the source of production of 2,3-bisphosphoglycerate and describe its impact on hemoglobin oxygenation.
16. Identify the oxygen affinity of fetal hemoglobin.
17. Compare and contrast the composition of the chemically modified hemoglobins—methemoglobin, carboxyhemoglobin, and sulfhemoglobin—and their affinity for oxygen.
18. Compare and contrast oxygenated, deoxygenated, and oxidized hemoglobin and ferric versus ferrous iron.
19. Describe how hemoglobin is routinely measured in the laboratory.
20. Identify how different kinds of hemoglobins are identified by laboratory tests.
21. Identify the gene locations of the globins that make up the hemoglobin molecule, including the number of genes for each globin chain (for Hb A, A2, and F) and their general arrangement on chromosomes.
Case Study
1. Were these hemoglobin results within expected reference intervals?
2. Why were the mother’s and the newborn’s hemoglobin results so different?
3. What is the difference between a hemoglobin test and the hemoglobin electrophoresis test?
4. Why were the mother’s and newborn’s hemoglobin electrophoresis results so different?
Hemoglobin Structure
Hemoglobin (Hb) is the first protein whose structure was described using x-ray crystallography.1–4 The hemoglobin molecule is a conjugated globular protein consisting of four heme groups and two heterogenous pairs of polypeptide chains (Figure 10-1).
Heme Structure
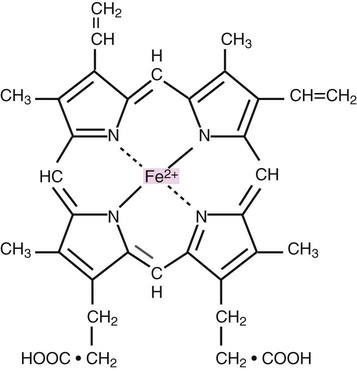
Heme consists of a ring of carbon, hydrogen, and nitrogen atoms called protoporphyrin IX with an atom of divalent ferrous iron (Fe2+) attached (ferroprotoporphyrin, Figure 10-2). Each of the four heme groups is positioned in a pocket of the polypeptide chain near the surface of the hemoglobin molecule. Each heme molecule combines reversibly with one oxygen molecule. Owing to its double bonds, heme renders blood red.
Globin Structure
The four globin chains comprising each hemoglobin molecule consist of two identical pairs of unlike polypeptide chains, 141 to 146 amino acids each. Variations in amino acid sequences give rise to different types of polypeptide chains. Each chain is designated by a Greek letter (Table 10-1).5
TABLE 10-1
| Symbol | Name | No. of Amino Acids |
| α | Alpha | 141 |
| β | Beta | 146 |
| γA | Gamma A | 146 (position 136: alanine) |
| γG | Gamma G | 146 (position 136: glycine) |
| δ | Delta | 146 |
| ε | Epsilon | Unknown |
| ζ | Zeta | 141 |
| θ | Theta | Unknown |
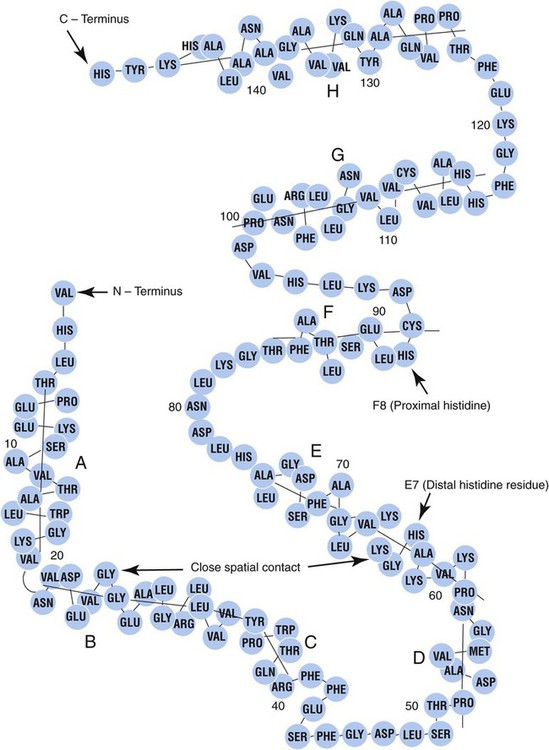
Each globin chain is divided into eight helices and seven nonhelical segments (Figure 10-3). The helices, designated A to H, contain subgroup numberings for the sequence of the amino acids in each helix and are relatively rigid and linear. The flexible nonhelical segments connect the helices, as reflected by their designations: NA for the sequence between the N-terminus and the A helix, AB between the A and B helices, and so forth with BC, CD, DE, EF, FG, GH, and finally HC between the H helix and the C-terminus.
Complete Hemoglobin Molecule
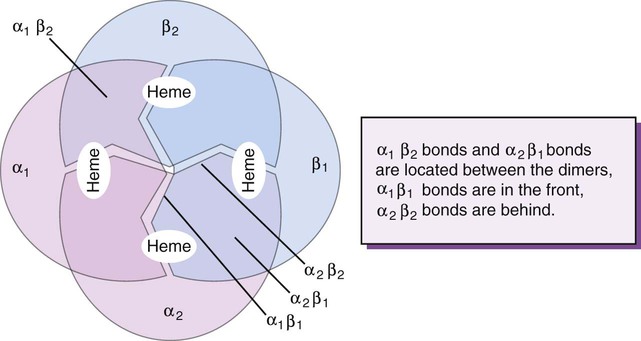
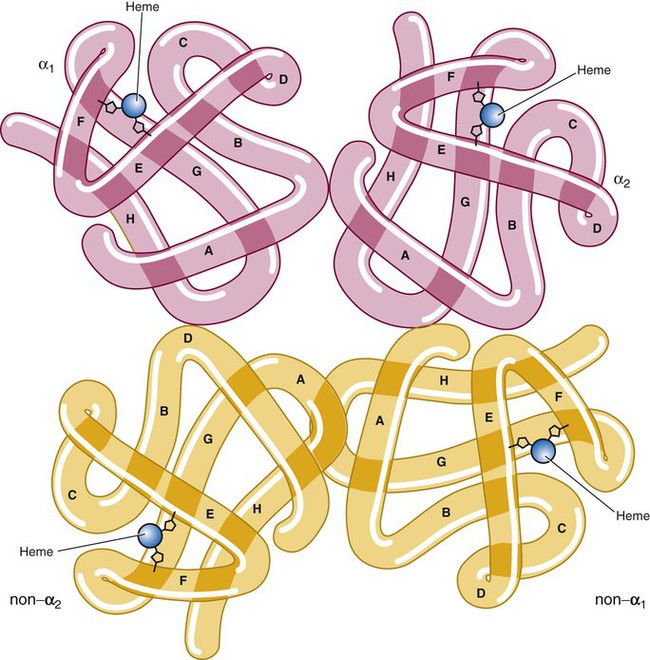
The quaternary structure of hemoglobin, also called a tetrameric molecule, describes the complete hemoglobin molecule. The complete hemoglobin molecule is spherical, has four heme groups attached to four polypeptide chains, and may carry four molecules of oxygen. It is composed of two α globin chains and two non-α globin chains. Each globin chain has a heme group attached. Each heme molecule is capable of carrying one molecule of oxygen. Strong α1,– non-α1 and α2,–non-α2 dimeric bonds hold the molecule in a stable form. Tetrameric α1–non-α2 and α2,–non-α1 bonds also contribute to the stability of the structure (Figure 10-4).6,7
Hemoglobin Biosynthesis
Heme Biosynthesis
Heme biosynthesis occurs in the mitochondria and cytoplasm of bone marrow RBC precursors, beginning with the pronormoblast (also known as proerythroblast) through the circulating polychromatic (also known as polychromatophilic) erythrocyte (see Chapter 8). As they lose their mitochondria and the citric/tricarboxylic acid cycle, mature RBCs can no longer make hemoglobin.
Heme biosynthesis begins with the condensation of glycine and succinyl coenzyme A (CoA) catalyzed by aminolevulinate synthase (ALAS) to form aminolevulinic acid (ALA). ALA dehydratase (also known as ALA dehydrase, porphobilinogen synthase) in the presence of ALA catalyzes the formation of porphobilinogen. Porphobilinogen deaminase, also known as hydroxymethylbilane synthase, in the presence of porphobilinogen catalyzes the formation of hydroxyl methylbilane. This pathway continues until, in the final step of production of heme, Fe2+ combines with protoporphyrin IX in the presence of ferrochelatase/heme synthase to make heme (Figure 10-5).6
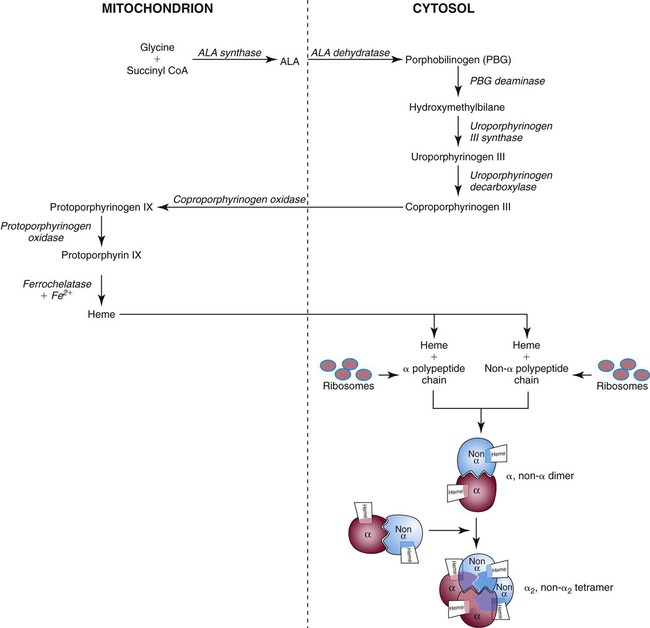
Globin Biosynthesis
The production of globin chains takes place in RBC precursors from the pronormoblast through the circulating polychromatic (polychromatophilic) erythrocyte, but not in the mature RBC.8,9 Globin proteins arise via transcription of the genetic code to messenger ribonucleic acid (mRNA) and translation of mRNA to the globin polypeptide chain. A slight excess of α-globin mRNA is present in pronormoblasts (proerythroblasts); however, β-globin mRNA is translated more efficiently than α-globin mRNA. This results in synthesis of sets of chains in approximately equal amounts. When synthesized, the chains are released from the ribosomes in the cytoplasm.7
Hemoglobin Assembly
After their release from ribosomes, each globin chain binds to a heme molecule and they pair off. An α chain and a non-α chain combine to form heterodimers. Two heterodimers spontaneously combine to form tetramers. The tetrameric α1β2 and α2β1 bonds also contribute to the stability of the structure. This completes the hemoglobin molecule (see Figure 10-5).6,7
The combination of two α and two β chains along with the four heme molecules forms Hb A. This is the predominant hemoglobin in postnatal life. Hb A2 contains two α and two δ chains. The δ chains are inefficiently expressed; only small amounts of Hb A2 are found in the RBCs. Hb F contains two α and two γ chains. In adults, Hb F is distributed unevenly among RBCs; it is present in a few RBCs called F cells.7
The various globin chains affect the net negative charge of the hemoglobin molecule. In hemoglobin electrophoresis, hemoglobins under the influence of an electrical field exhibit varying mobilities, which allows differentiation of hemoglobin types. Various support media, buffer, and pH are employed for definitive identification (see Chapter 26).
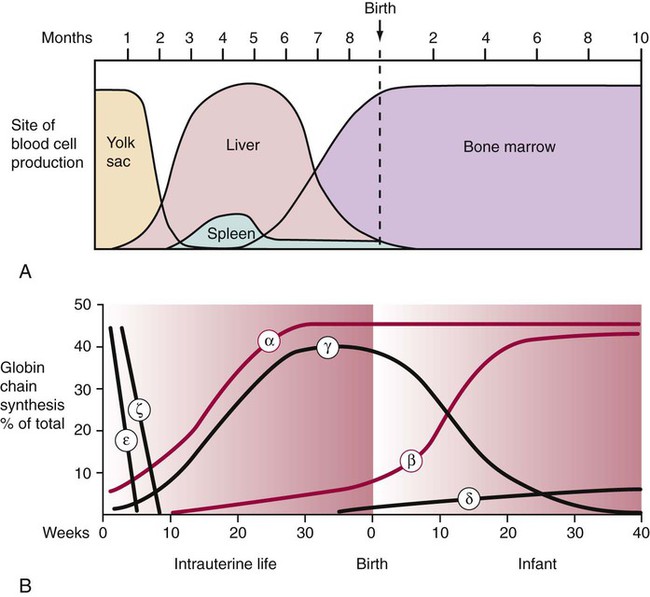
Hemoglobin Ontogeny
Hemoglobin composition differs with prenatal gestation time and postnatal age. Hemoglobin changes reflect changes in the activation and inactivation or switching of the globin genes, progressing from the ζ to the α gene on chromosome 16 and from the ε to the γ, δ, or β genes on chromosome 11. The ζ and ε genes normally appear only during the first 3 months of embryonic development. These two chains in addition to the α and γ chains are constituents of embryonic hemoglobins (Figure 10-6). At birth, Hb F is the predominant hemoglobin. Normal adult hemoglobin is predominately Hb A (α2β2) with small amounts of Hb A2 (α2δ2) and Hb F (α2γ2).7 Table 10-2 presents adult reference intervals.
TABLE 10-2
| Stage | Globin Chain | Hemoglobin |
| Intrauterine | ||
| Early embryogenesis (product of yolk sac erythroblasts) | ζ2 + ε2 | Gower-1 |
| α2 + ε2 | Gower-2 | |
| ζ2 + γ2 | Portland | |
| Begins in early embryogenesis; peaks during midgestation and declines rapidly just before birth | α2 + γ2 | F |
| Birth | ||
| α2 + γ2 | F, 60-90% | |
| α2 + β2 | A, 10-40% | |
| Adulthood | ||
| α2 + γ2 | F, 1-2% | |
| α2 + δ2 | A2, <3.5% | |
| α2 + β2 | A, >95% | |

A small percentage of Hb A is glycated. Glycation is a posttranslational modification formed by nonenzymatic binding of various sugars with globin chain amino groups. The most glycated hemoglobin is Hb A1c, in which glucose attaches to the N-terminal valine of the β chain. Normally, about 4% to 6% of Hb A circulates in the A1c form. In uncontrolled diabetes mellitus, the amount of A1c is increased. Other posttranslational modifications seem to be of little importance.7
Hemoglobin Production Regulation
Heme Regulation
The key rate-limiting step in heme synthesis is the initial reaction of glycine and succinyl CoA to form ALA, catalyzed by ALAS. Transcription of the ALAS gene is inhibited by heme, which leads to a decrease in heme production (a negative feedback mechanism). Other enzymes in the heme pathway inhibited by heme are ALA dehydrase and porphobilinogen deaminase/hydroxymethylbilane synthase. An increased demand for heme induces an increased synthesis of ALAS.9
Ferrochelatase (also known as heme synthase) also plays a regulatory role in heme biosynthesis. A negative feedback mechanism by heme or substrate inhibition by protoporphyrin IX is believed to inhibit the ferrochelatase/heme synthase enzyme.9
Globin Regulation
Globin production is regulated by the rate at which the deoxyribonucleic acid (DNA) is transcribed to mRNA. The amount of the specific globins synthesized is proportional in general to the content of their individual globin mRNAs. Hemin—the Fe3+ oxidation product of heme—is important in controlling the rate of globin synthesis in intact polychromatic erythrocytes and various cell-free systems, and in its absence, an inhibitor of globin synthesis accumulates. Balanced globin chain and heme synthesis is important, because excess components of hemoglobin—unpaired chains, protoporphyrin, and iron—reduce RBC survival. Normal mature RBCs contain only complete hemoglobin molecules; pools of free heme or globin chains are minute.7,9
Hemoglobin Function
The affinity of hemoglobin for oxygen depends on the partial pressure of oxygen (Po2), often defined in terms of the amount of oxygen needed to saturate 50% of hemoglobin, called the P50 value. The relationship is described by the oxygen dissociation curve of hemoglobin, which plots the percent oxygen saturation of hemoglobin versus the Po2 (Figure 10-7). The curve is sigmoidal, which indicates low hemoglobin affinity for oxygen at low oxygen tension and high affinity for oxygen at high oxygen tension.
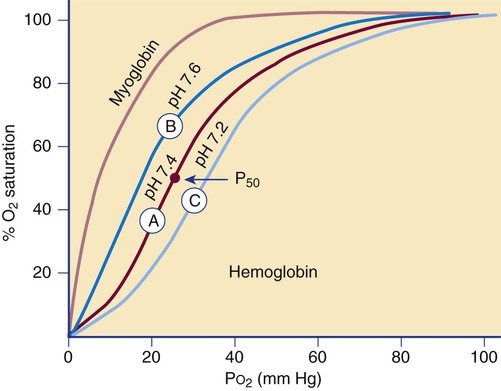
Cooperation among hemoglobin subunits contributes to the shape of the curve. Heme units do not undergo simultaneous oxygenation or deoxygenation; rather, the state of each heme unit in comparison with the other units influences further binding. That is, hemoglobin that is completely deoxygenated has little affinity for oxygen, but with each oxygen molecule that is bound, the avidity increases, and the hemoglobin molecule quickly becomes fully oxygenated.7 Shifts of the curve to the left or right occur if there are changes in the pH of the blood. This shift in the curve as a result of pH is termed the Bohr effect.
The reference interval for arterial oxygen saturation is 96% to 100%. If the oxygen dissociation curve shifts to the left, a patient with arterial and venous Po2 concentrations in the reference intervals (80 to 100 mm Hg arterial and 30 to 50 mm Hg venous) will have a higher percent oxygen saturation and a higher affinity for oxygen than a patient for whom the curve is normal. With a shift in the curve to the right, a lower oxygen affinity is seen. Hemoglobin absorbs less oxygen in the lungs but delivers more oxygen to the tissues than it normally would, as in the presence of Hb S (see Chapter 26).
The effect of 2,3-bisphosphoglycerate (2,3-BPG, formerly 2,3-diphosphoglycerate), on oxygen affinity is complex. The hemoglobin molecule is allosteric; that is, its function and structure are influenced by other molecules. In the presence of large amounts of 2,3-BPG, the hemoglobin molecule changes from a relaxed (R) oxygenated molecule to a tense (T) deoxygenated molecule. The T structure is stabilized by salt bridges, which become broken as the molecule switches to the R structure. When the salt bridges are broken, the hemoglobin molecule is able to bind to oxygen (Figure 10-8). Carbon dioxide, hydrogen ions, and chloride ions all decrease the affinity of hemoglobin for oxygen by strengthening the salt bridges that lock the molecule into its T conformation. Some abnormal hemoglobins with a high oxygen affinity and low P50 occur as a result of amino acid substitutions that lead to loss of bonds that would have stabilized the tetramer in the T conformation. Without these binding sites, the hemoglobin molecule holds on more tightly to oxygen—hence a higher oxygen affinity.7
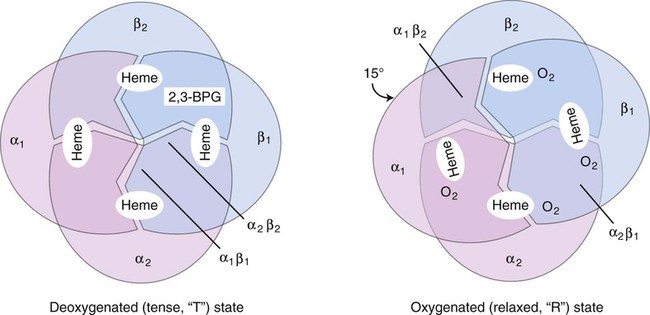
Clinical conditions that produce a shift to the left include a lowered body temperature due to external causes; multiple transfusions of stored blood with depleted 2,3-BPG; alkalosis; and the presence of methemoglobin, carboxyhemoglobin, or some other hemoglobin variants. Conditions producing a shift of the curve to the right include increased body temperature, increased 2,3-BPG concentrations, increased H+ concentration (decreased pH), and abnormal hemoglobins with a low affinity for oxygen. Clinical conditions that produce a right shift include a high fever, acidosis, and circumstances that produce hypoxia, such as high altitude, pulmonary insufficiency, congestive heart failure, severe anemia, and cardiac right-to-left shunt.7
The sigmoidal oxygen curve generated by normal hemoglobin contrasts with myoglobin’s hyperbolic curve (see Figure 10-7). Myoglobin, present in cardiac and skeletal muscle, is a 17,000-D monomeric oxygen-binding heme protein. It binds oxygen with greater affinity than hemoglobin. Its hyperbolic curve indicates that it releases oxygen only at very low partial pressures, which means it is not as effective as hemoglobin in releasing oxygen to the tissues at physiologic tensions. Myoglobin is released into the plasma when there is damage to the muscle in myocardial infarction, trauma, or severe muscle injury, called rhabdomyolysis. Myoglobin is normally excreted through the kidney, but levels may become elevated in renal failure. Testing for serum myoglobin aids in detecting myocardial infarction in patients who have no underlying trauma, rhabdomyolysis, or renal failure. An elevated myoglobin level generates a positive result on the urine hemoglobin dipstick test that must be differentiated from a response caused by hemoglobin. In contrast to myoglobinuria, hemoglobinuria is seen in intravascular hemolytic disorders.7
< ?xml:namespace prefix = "mml" />
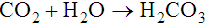
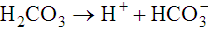
The first of these reactions is facilitated by the RBC enzyme carbonic anhydrase. The H+ from the second reaction binds deoxygenated hemoglobin. The bicarbonate diffuses across the RBC membrane, and a portion is exchanged with plasma Cl−; this is called the chloride shift. The plasma bicarbonate travels to the lungs, where it is expired. About 70% to 85% of tissue carbon dioxide is processed in this manner.10 Approximately 10% to 20% of carbon dioxide binds to the N-terminal amino group of each globin chain as carbaminohemoglobin. Carbaminohemoglobin has a lower affinity for oxygen than does hemoglobin in the absence of carbon dioxide.11
Variant Hemoglobins
Methemoglobin
Methemoglobin is a form of hemoglobin that contains iron in the oxidized or ferric state (Fe3+). Methemoglobin is continuously being formed by spontaneous oxidation, but fails to accumulate because several reducing enzyme systems restrict its concentration to less than 1% of total hemoglobin (see Chapter 9).
Methemoglobin is brownish to bluish and does not revert to red upon oxygen exposure. Ferric iron cannot bind oxygen, but when one or more ferric ions are present the conformation of the molecule changes, and the oxygen affinity of the remaining heme groups increases.12,13 Increased methemoglobin produces a shift to the left in the oxygen dissociation curve, so that oxygen is not delivered efficiently to the tissues. If methemoglobin comprises more than 30% of total hemoglobin, patients present with hypoxia and cyanosis.5,14
Elevated levels of methemoglobin are seen when oxidants such as nitrites are present or when there is decreased activity of methemoglobin reductase, a genetic deficiency. It also is seen in patients who inherit Hb M disease, which is caused by an abnormality in the structure of the globin portion of the hemoglobin molecule (see Chapter 26).15
Methemoglobin is assayed by spectral absorption analysis instruments such as the CO-oximeter. Methemoglobin shows a peak in the range of 620 to 640 nm at a pH of 7.1. Acquired methemoglobinemia may be treated by removal of the offending substance or administration of ascorbic acid or methylene blue.15
Carboxyhemoglobin
Carboxyhemoglobin results from the combination of carbon monoxide with heme iron. Although carbon monoxide binds hemoglobin more slowly than oxygen, its affinity is 240 times that of oxygen and its release is 10,000 times slower. Carbon monoxide has been termed the silent killer because it is an odorless and colorless gas, and victims may quickly become hypoxic.5,13
Some carboxyhemoglobin is produced endogenously. The reference interval is 0.2% to 0.8%. Exogenous carbon monoxide is derived from the exhaust of automobiles and from industrial pollutants, such as coal, gas, charcoal burning, and tobacco smoke. In smokers, levels may vary from 4% to 20%.5,13 Exposure to carbon monoxide may be coincidental, accidental, or intentional (suicidal). Many deaths from house fires are the result of inhaling smoke, fumes, or carbon monoxide.16 Even when heating systems in the home are properly maintained, accidental poisoning with carbon monoxide may occur.17 Toxic effects, such as headaches and dizziness, are experienced at levels of 10% to 15%. Levels of more than 50% may cause coma and convulsions. Carboxyhemoglobin may be detected by spectral absorption instruments at 541 nm. It gives blood a cherry-red color, which is also imparted to the skin of victims. Hyperbaric oxygen therapy has been used for treatment.
Hemoglobin Measurement
The cyanmethemoglobin method is the reference method for hemoglobin assay.18 A lysing agent present in the cyanmethemoglobin reagent frees hemoglobin from RBCs. Free hemoglobin combines with potassium ferricyanide contained in the cyanmethemoglobin reagent, which converts the hemoglobin iron from the ferrous to the ferric state to form methemoglobin. Methemoglobin combines with potassium cyanide to form the stable pigment cyanmethemoglobin. The cyanmethemoglobin color intensity, which is proportional to hemoglobin concentration, is measured at 540 nm spectrophotometrically and compared with a standard (see Chapter 14). The cyanmethemoglobin method is performed manually but has been adapted for use in automated instruments.
Many instruments now use sodium lauryl sulfate (SLS) to convert hemoglobin to SLS-methemoglobin. This method does not generate toxic wastes (see Chapter 39).
Hemoglobin electrophoresis is used to separate the different types of hemoglobins such as Hb A, A2, and F (see Chapter 26).
Summary
• The hemoglobin molecule is composed of four heme groups (protoporphyrin IX + Fe2+) and two pairs of unlike polypeptide chains. Each heme molecule combines with one polypeptide chain.
• Hemoglobin, contained in RBCs, carries oxygen to the tissue bound to the ferrous iron in heme.
• Hemoglobin biosynthesis is regulated by hormones, oxygen tension in the kidneys, and enzymes in the heme synthesis pathway.
• 2,3-BPG produced by the glycolytic pathway facilitates the delivery of oxygen from hemoglobin to the tissues.
• The oxygenation dissociation curve of hemoglobin is sigmoid owing to cooperativity among the hemoglobin subunits.
• The Bohr effect is the influence of pH on the hemoglobin oxygen release mechanism.
• The three hemoglobins found in normal adults are Hb A, Hb A2, and Hb F. Hb A, composed of two αβ heterodimers, is the predominant hemoglobin of adults.
• Hemoglobin ontogeny describes which hemoglobins are produced by the body from the fetal period through birth to adulthood.
• Chemically modified hemoglobins do not transport oxygen to the tissues well, which results in cyanosis.
Review Questions
1. A hemoglobin molecule is composed of:
a. One heme molecule and four globin chains
b. Ferrous iron, protoporphyrin IX, and a globin chain
2. Normal adult Hb A contains which polypeptide chains?
3. A key rate-limiting step in heme synthesis is suppression of:
4. Which of the following forms of hemoglobin molecule has the lowest affinity for oxygen?
5. Using the normal hemoglobin oxygenation curve in Figure 10-7 for reference, predict the position of the oxygenation curve for methemoglobin.
6. The predominant hemoglobin found in a normal newborn is:
7. What is the distribution of normal hemoglobins in adults?
a. 80% to 90% Hb A, 5% to 10% Hb A2, 1% to 5% Hb F
b. 80% to 90% Hb A2, 5% to 10% Hb A, 1% to 5% Hb F
8. Which of the following is a description of the structure of oxidized hemoglobin?
a. Hemoglobin carrying oxygen on heme; synonymous with oxygenated hemoglobin
b. Hemoglobin with iron in the ferric state (methemoglobin) and not able to carry oxygen
c. Hemoglobin with iron in the ferric state so that carbon dioxide replaces oxygen in the heme structure
d. Hemoglobin carrying carbon monoxide; hence the “oxidized” refers to the single oxygen
9. In the quaternary structure of hemoglobin, the globin chains associate into:
a. α tetramers in some cells and β tetramers in others
10. How are the globin chain genes arranged?
a. With α genes and β genes on the same chromosome, including two α genes and two β genes
b. With α genes and β genes on separate chromosomes, including two α genes on one chromosome and one β gene on a different chromosome
c. With α genes and β genes on the same chromosome, including four α genes and four β genes
d. With α genes and β genes on separate chromosomes, including four α genes on one chromosome and two β genes on a different chromosome
11. The nature of the interaction between 2,3-BPG and hemoglobin is that 2,3-BPG:
a. Binds to the heme moiety, blocking the binding of oxygen
b. Binds simultaneously with oxygen to ensure that it stays bound until it reaches the tissues, when both molecules are released from hemoglobin
c. Binds to amino acids of the globin chain, contributing to a conformational change that inhibits oxygen from binding to heme
d. Oxidizes hemoglobin iron, diminishing oxygen binding and promoting oxygen delivery to the tissues